

Abstract
Aims
Several studies reported that astrocytes support neuronal communication by the release of gliotransmitters, including ATP and glutamate. Astrocytes also play a fundamental role in buffering extracellular glutamate in the synaptic cleft, thus limiting the risk of excitotoxicity in neurons. We previously demonstrated that extracellular tau oligomers (ex-oTau), by specifically targeting astrocytes, affect glutamate-dependent synaptic transmission via a reduction in gliotransmitter release. The aim of this work was to determine if ex-oTau also impair the ability of astrocytes to uptake extracellular glutamate, thus further contributing to ex-oTau-dependent neuronal dysfunction.
Methods
Primary cultures of astrocytes and organotypic brain slices were exposed to ex-oTau (200 nM) for 1 hour. Extracellular glutamate buffering by astrocytes was studied by: Na+ imaging; electrophysiological recordings; high-performance liquid chromatography; Western blot and immunofluorescence. Experimental paradigms avoiding ex-oTau internalization (i.e., heparin pre-treatment and amyloid precursor protein knockout astrocytes) were used to dissect intracellular vs. extracellular effects of oTau.
Results
Ex-oTau uploading in astrocytes significantly affected glutamate-transporter-1 expression and function, thus impinging on glutamate buffering activity. Ex-oTau also reduced Na-K-ATPase activity because of pump mislocalization on the plasma membrane, with no significant changes in expression. This effect was independent of oTau internalization and it caused Na+ overload and membrane depolarization in ex-oTau-targeted astrocytes.
Conclusions
Ex-oTau exerted a complex action on astrocytes, at both intracellular and extracellular levels. The net effect was dysregulated glutamate signalling in terms of both release and uptake that relied on reduced expression of glutamate-transporter-1, altered function and localization of NKA1A1, and NKA1A2. Consequently, Na+ gradients and all Na+-dependent transports were affected.
Keywords: tau oligomers, astrocytes, glutamate, GLT-1, NKA, Amyloid Precursor Protein
Graphical Abstract
• Extracellularly, tau oligomers cause NKAs mislocalization on the plasma membrane inducing intracellular Na+ overload in astrocytes leading to reduced Na+ gradient across the membrane;
• Intracellularly, tau oligomers downregulate GLT-1 expression;
• The net effects of extracellular tau oligomers is a reduction of Na+-driven uploading of extracellular glutamate from astrocytes.

Introduction
Several studies, including ours, have reported the detrimental effects of extracellular tau oligomers (ex-oTau) on neurons and astrocytes. The presence of tau oligomers in the extracellular medium may depend on activity-dependent secretion from neurons and/or release from dead cells [1]. Among the deleterious effects of ex-oTau in neuronal cells, there is an alteration of hippocampal synaptic transmission and plasticity [1–5] depending on the ability of tau oligomers to cross the plasma membrane and be accumulated intracellularly [1,4,5]. We and others also demonstrated that astrocytes efficiently upload oTau from the extracellular milieu [4,6,7] more rapidly and abundantly than neurons, probably because of a greater expression on the plasma membrane of attaching/entry sites for oTau, i.e., the heparan sulfate proteoglycans [4,8–10]. This ability renders astrocytes key players in the clearance of extracellular tau [6]. Nevertheless, intracellular oTau accumulation leads to alteration of Ca2+ signalling in astrocytes that affects Ca2+-dependent release of gliotransmitters (ATP, glutamate and D-serine), impinging on synaptic transmission [4]. Therefore, it is reasonable to hypothesize that astrocytes play a pivotal role in the disruption of synaptic function in tauopathies.
The concept of the tripartite synapse, indicating the active role played by astrocytes in communication between neurons, is now widely recognized. Specifically, astrocytes modulate synaptic transmission by releasing several neuroactive molecules, including (but not limited to) ATP, glutamate, and D-serine [11, 12]. In this complex neuron-astrocyte interplay, astrocytes also play a fundamental role in limiting the diffusion of extracellular glutamate released from neurons in the synaptic cleft [13, 14] by buffering it, thus lowering the risk of glutamate-induced toxicity (i.e., excitotoxicity). Such buffering action is assured by high-affinity glutamate transporters expressed on the plasma membrane of astrocytes [14–16]. Glutamate transporters, also named excitatory amino acid transporters (EAATs), exist in 5 different isoforms that are differentially expressed on neurons and astrocytes. In particular, EAAT1 and EAAT2 (also known as GLAST and GLT-1 in murine cells, respectively) are mainly expressed on astrocytes with GLT-1 being more expressed than GLAST, whereas EAAT3–5 are typically neuronal [17]. About 80% of extracellular glutamate is taken up by astrocytes through GLAST and GLT-1, and the remaining 20% of neurotransmitter returns to neurons [18]. Dysfunction of EAATs, and in particular GLT-1, is associated with neurodegenerative diseases including Alzheimer’s disease (AD) and other tauopathies [19–21]. GLT-1 works according to the following stoichiometry: the uptake of one molecule of glutamate is coupled to the influx of three Na+, one H+, and the efflux of one K+ ion. The driving force of this transport is due to Na+ gradients across the cell membrane, which strictly depends on the activity of the Na+/K+ ATPases (NKAs) [22]. Astrocytes express both the α1 (NKA1A1) and α2 (NKA1A2) isoforms of the NKA, even if α2 predominates and has a higher affinity for sodium [22]. EAATs and NKA1A2 interact at the plasma membrane together with other cellular components involved in energy metabolism [23]. In particular, Genda and co-workers [24] demonstrated that GLT-1 co-localizes with NKAs and mitochondria, providing a spatial energy system matching the glutamate-buffering capacity to the demands imposed by transport.
In this study we evaluated the impact of ex-oTau on the ability of astrocytes to buffer extracellular glutamate by focusing our attention on GLT-1 and NKA function and expression.
Material and Methods
Primary cultures of astrocytes and neurons from mouse hippocampus and cortex
Primary cultures of cortical astrocytes were obtained from E18 wild-type (WT) C57/bl6 mice and amyloid precursor protein (APP) knockout (KO) mice (B6.129S7-Apptm1Dbo/J) following procedures previously described [4,9]. Briefly, after the brain had been removed, neocortex was dissected under a stereomicroscope in Phosphate Buffered Saline (PBS) at 4°C and then incubated for 10 min at 37°C in PBS containing trypsin-ethylenediamine tetra-acetic acid 0.025%/0.01% w/v; (Biochrom AG, Berlin, Germany). After trypsin inactivation with fetal bovine serum (FBS, 1%), the tissue was centrifuged and suspended in a medium consisting of 97.8% minimum essential medium (MEM, Biochrom), 1% FBS, 1% glutamine (2 mM), 1% penicillin-streptomycin-neomycin antibiotic mixture (PSN, Thermo Fisher Scientific, Waltham, MA), and glucose (25 mM). The tissue was then mechanically dissociated with a fire-polished Pasteur pipette at room temperature (RT) and then centrifuged at 235 × g for 10 minutes at RT. Dissociated cells were suspended in a medium consisting of MEM with 3.7 g/L NaHCO3 and 1.0 g/L D-glucose (DMEM), 10% FBS and 1% PSN. Astrocytes were plated at density of 105 cells on 20-mm coverslips (for immunofluorescence [IF], Na+ imaging and electrophysiological measurements), or 106 cells on 35-mm six-well plates (for Western blot [WB] analysis). Both supports were pre-coated with poly-L-lysine (0.1 mg/mL, Sigma, St. Louis, MO).
Experiments were performed when confluence was reached, about 8–10 days after plating.
For co-cultures of hippocampal neurons and astrocytes, hippocampal tissue from WT and APP KO mice was subjected to the same passages described above, but the pellet was suspended in the previously described medium with added 5% horse serum and 5% FBS. Cells were plated at a density of 105 cells on 20-mm coverslips (for IF, Na+ imaging, and electrophysiological measures) and 106 cells/well on 35-mm six-well plates (for WB analysis), pre-coated with poly-L-lysine (0.1 mg/ml). After 24 hours, culture medium was replaced with a mixture of 96.5% Neurobasal medium (Thermo), 2% B-27 (Thermo), 0.5% glutamine (2 mM) and 1% PSN. After 72 hours, this medium was replaced with a glutamine-free version of the same medium and cells were grown for 10 more days before carrying-out experiments.
Organotypic hippocampal brain slices preparation
Hippocampal organotypic slice cultures were prepared from postnatal day 10 C57/bl6 mice, without distinguishing between male and female, using a McIllwain tissue chopper as previously described in [25]. Slices (300 μm) were placed on semi-porous membranes (Merck Millipore, No. PCIMORG50, Burlington, MA) fed by tissue medium (for 1 L: 788 mL 1× MEM (Thermo -Gibco-, #11575–032, 7.16 g HEPES, 0.49 g NaHCO3, 4.8 g D-glucose, 50 μL ascorbic acid (25%), 50 μl insulin (10 mg/mL), 200 mL horse serum (Thermo –Gibco-, #16050–122), 2 mL MgSO4 (1M), 1 mL CaCl2 (1M). Slices were incubated at 35 °C in 5% CO2 and cultured for up to 4 days.
Preparation of recombinant oligomeric tau and human AD tau
Human recombinant tau 4R/2N was prepared as previously described [1,4,26]. After 20 hours of oligomerization by using 1 mM H2O2 at RT, the tau preparation was monitored through WB without reducing agent, as described in [27]. In a subset of experiments, tau obtained from prefrontal/frontal cortex of human AD brain specimens (human Tau - hTau) was used. Human tau preparation was obtained by pooling together post-mortem tissue samples from 4 AD patients: two men (68 and 72 years old) and two women (74 and 83 years old). All individuals were characterized for their Braak stage (VI for all patients), Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) Neuropsychological Battery score (B for the woman of 83 years old and C for the other three AD patients), and National Institute of Aging – Reagan Institute rating (NIA-R) (high for all patients). AD patients had no other diseases that might have contributed to the clinical deficits (see also Supplementary Table S1 of Ref. [1]). Human tissue was provided by the New York Brain Bank–The Taub Institute, Columbia University.
Both recombinant and human tau concentrations were determined by using a nanodrop spectrometer with the following parameters: absorbance at 280 nm, molecular weight of 45,900 kDa and extinction coefficient of 7450 cm−1 m−1, see note 24 of Ref. [27]).
Confocal Na+ imaging
Primary cultures of astrocytes were incubated for 30 min at 37°C with 5 μM CoroNa™ Green-AM (Thermo), a Na+ sensitive fluorescent dye, in Tyrode’s solution consisting of 150 mM NaCl, 4 mM KCl, 1 mM MgCl2, 10 mM glucose, 10 mM HEPES and 2 mM CaCl2 (pH adjusted to 7.4 with NaOH); then, cells were maintained in fresh Tyrode’s solution at RT for 20 min to allow dye de-esterification. Intracellular Na+ transients were elicited by exposing CoroNa Green–loaded cells to 100 mM L-glutamic acid (buffered at pH 7.4 with NaOH) for 20 s. The amplitude of Na+ signals was estimated for each cell in the imaged microscopic field in a semi-quantitative way by the following formula: ΔF/F = (Ft – Fpre)/(Fpre – Fbgnd). In this formula, Ft is the mean of fluorescence intensities measured in a region of interest (ROI) drawn around each cell body at a given time (t); Fpre is the basal fluorescence intensity in this ROI estimated as the mean value of fluorescence during 20-s prior glutamic acid perfusion; Fbgnd is background fluorescence intensity measured in an area lacking dye-filled cells.
For CoroNa Green probe we used the following calibration formula to estimate quantitative values of intracellular Na+ levels: [Na+]i = Kd × [Ft – Fmin] / [Fmax – Ft] [28, 29] where Kd represents the dissociation constant (80 mM for Na+), Ft is the fluorescence measured in a ROI at the given time “t”; Fmax are the maximum measured for each ROI after application of gramicidin; Fmin, the minimum fluorescence values (obtained in 0 mM Na+), were always undetectable and then set to zero. CoroNa Green was excited at 488 nm and its emission signal was collected between 500 and 550 nm with an inverted laser scanning confocal system Leica TCS-SP5 (Wetzlar, Germany).
Immunofluorescence
Wild-type astrocytes were treated with vehicle or 200 nM ex-oTau or ex-oTau + heparin (2 mg/mL) for 1 hour and then fixed with 10% formalin solution (pH 7.4 with NaOH) for 10 min at RT. As described in [30], cells were incubated with 0.3% Triton X-100 (Sigma) in PBS for 15 min to permeabilize cells and then placed in the blocking solution (0.3% BSA in PBS) for 20 minutes at RT, in order to avoid non-specific binding. Cells were then incubated overnight at 4°C with a pair of following antibodies in the blocking solution: mouse anti-GFAP (#3670; 1:500, Cell Signaling Technology Inc., Danvers, MA; RRID: AB_561049), and rabbit anti-EAAT2/GLT1 (#3838; 1:500, Cell Signaling; RRID: AB_2190743), or mouse anti-NKA1A1 (#C464.4 1:300; Santa Cruz Biotechnology, Dallas, Texas; RRID: AB_626713), or mouse anti-NKA1A2 (#Ab2871; 1:500, Abcam, Waltham, MA; RRID: AB_303373).
The next day, primary antibodies were removed and after PBS washing, cells were incubated for 90 min at RT with the proper secondary antibodies in the blocking solution: Alexa Fluor 546 donkey anti-rabbit (#A10040; Thermo; RRID: AB_2534016) and Alexa Fluor 488 goat anti-mouse (#A32723; Thermo; RRID: AB_2633275) were used at a dilution of 1:1,000. Finally, cells were incubated with 4’,6-diamidino-2-phenylindole (DAPI, Thermo), 0.5 mg/mL in PBS for 10 min at RT, in order to counterstain their nuclei. Cells were then coverslipped with ProLong Gold anti-fade reagent (Thermo) and studied by confocal microscopy as described below.
For some experiments, mitochondria were labelled by incubation of live cells with 10 nM MitoTracker® Orange CMTMRos (Thermo) for 30 minutes before fixation.
At least three independent experiments were carried out for each condition.
Immunoreactivity for GLT-1 and NKAs was quantified by drawing regions of interest (ROIs) in the acquired fields and expressing fluorescence as the sum of intensities (8-bit depth; 256 levels) measured for every pixel in the ROI divided by ROI area (~ 1,156±193 μm2). Co-localization analysis was performed using Image J software (FiJi version) [31].
Confocal stacks of images (512×512 or 1024×1024 pixels) were acquired at 40× or 60× magnification with a confocal laser scanning system Nikon A1MP (physical pixel size: 210–310 nm).
Western blot
Wild-type and APP KO astrocytes were treated with 200 nM ex-oTau or with ex-oTau + heparin (2 mg/mL), or vehicle for 1 hour. Cells were then washed twice with PBS and scraped in RIPA buffer containing 1 mM phenylmethylsulfonyl fluoride, sodium fluoride, sodium orthovanadate and protease inhibitor mixture. Cell suspensions were sonicated and then centrifuged at 10,000 × g for 15 min at 4°C. The supernatants were collected and assayed to determine their protein concentration (by Bradford protein assay). An equivalent amount of protein (50 μg) for each sample was loaded onto 8% tris-glycine polyacrylamide gel for electrophoresis separation. Proteins were then electroblotted onto nitrocellulose membranes for WB analysis and then blocked with 5% non-fat dry milk in tris-buffered saline containing 0.1% Tween-20 for 1 hour at RT and then incubated with primary antibodies rabbit anti-EAAT2/GLT1 (Cell Signaling); rabbit anti-EAAT1/GLAST (#5684; Cell Signaling; RRID: AB_10695722); rabbit anti-NKA1A1 (Santa Cruz); mouse anti-NKA1A2 (Abcam) all used at dilution 1:1,000, overnight at 4°C. Housekeeping proteins, mouse anti-GAPDH (#ab8245; 1:5,000; Abcam; RRID: AB_2107448) and rabbit anti-β-actin (#8227; 1:2,000; Abcam; RRID: AB_2305186) The next day, membranes were incubated with appropriate secondary horseradish peroxidase-conjugated (HRP) antibodies (1:2,500; Cell Signaling; respectively, anti-rabbit #7074; RRID: AB_2099233 and anti-mouse #7076; RRID: AB_330924) for 1 hour at RT. Visualization was performed with WESTAR ECL (Cyanagen, Bologna, Italy), using UVItec Cambridge Alliance. Molecular weights for immunoblot analysis were determined through Precision Plus Protein™ Standards (BioRad, Hercules, CA). Densitometric analysis was carried out with UVItec software. Experiments were repeated at least three times.
Western blot of recombinant and human tau oligomers
The tau preparation was monitored through WB in which tau samples (prepared in nondenaturing/nonreducing conditions before loading) were resolved by Tris-Glycine SDS-PAGE and probed with anti-human tau monoclonal antibody Tau-13 (#sc-21796; 1:1,000; Santa Cruz Biotechnology; RRID: AB_628328). After incubation with above mentioned HRP-conjugated secondary antibodies visualization was performed with WESTAR ETA C ULTRA 2.0 using UVItec Cambridge Alliance. Molecular weights for immunoblot analysis were determined using Precision Plus Protein™ Standards.
Patch-clamp recordings
Synaptically activated Glu transporter currents
Synaptically activated Glu transporter current (STC) recordings were carried out as schematically described in Figure 1B. Organotypic hippocampal slices were constantly perfused with artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 3.2 KCl, 1.2 NaH2PO4, 4 MgCl2, 4 CaCl2, 26 NaHCO3, and 10 glucose, pH 7.4. ACSF was continuously bubbled with 95% O2/5% CO2. The extracellular solution also contained antagonists of AMPA receptors (10 μM NBQX), NMDA receptors (50 μM D-AP5) and GABAA receptors (20 μM (+)-bicuculline). Cells were approached under DIC with 5–8 MΩ pipettes pulled from borosilicate glass (Warner Instruments, Inc.) using a vertical Narishige PC-10 puller (Japan) and filled with an internal solution containing (in mM): 145 K-gluconate, 2 MgCl2, 10 HEPES, 0.1 EGTA, 2.5 Na-ATP, 0.25 Na-GTP, 5 phosphocreatine, pH adjusted to 7.2 with KOH. CA1 stratum radiatum astrocytes were identified by their small cell bodies with the aid of 40× water-immersion objectives on an upright microscope equipped with differential interference contrast optics under infrared illumination (BX5IWI; Olympus, Tokyo, Japan) and video observation (C3077–71 CCD camera; Hamamatsu Photonics, Japan). Studied astrocytes showed low input resistance (< 10 MΩ) and negative resting potential (~−90 mV). Glu transporter-mediated currents were evoked every 10 s through bipolar tungsten electrode (FHC, Neural microTargeting Worldwide) at least 200 μm away from the recorded astrocyte. During recordings, astrocytes were held at −90 mV). All recordings were made at 30°C. Data were collected with a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA), digitized at 10 kHz using the Digidata 1440B data acquisition system (Molecular Devices) and analysed using Clampfit software (Molecular Devices). Synaptically activated Glu transporter currents (STCs) were isolated by subtracting the residual current remaining in the presence of EAAT inhibitor DL-threo-beta-benzyloxyaspartate (DL-TBOA, 100 μM) from the total control current [32, 33]. One astrocyte per slice was studied.
Figure 1: ex-oTau treatment reduces EAAT-dependent, glutamate-induced Na+/ glutamate symport.

(A) Mean time-course of glutamic acid-induced intracellular Na+ transients in cultured astrocytes from C57/Bl6 mice treated with either vehicle (n=160 studied cells) or ex-oTau (200 nM, n=107 cells) for 1 hour. Insets show representative examples of confocal images of CoroNa Green-loaded astrocytes in the above-mentioned experimental conditions. Scale bar: 10 μm. (B) Picture schematizing Synaptically-activated Transporter-mediated Currents (STCs) recordings in astrocytes of the CA1 stratum radiatum of organotypic brain slices. (C) Representative traces of STCs recorded in organotypic brain slices treated for 1 hour with vehicle (n=13) or ex-Tau (200 nM, n=13). (D) Bar graph showing the mean peak-amplitude STCs in the two conditions analysed. (E) Bar graph showing the percent variation of intracellular glutamate levels evaluated by HPLC in cultured cortical astrocytes treated for 1 hour with vehicle (white bar), 200 nM ex-oTau (red bar) and ex-oTau+2 mg/mL heparin (grey bar) and exposed to L-glutamic acid (100 mM) for 5 min. Coloured lines represent the values of glutamate levels measured for the three conditions in every single experimental session (n=5). *p<0.05 and ***p<0.001 vs. vehicle; # p<0.05 vs. ex-oTau.
Resting membrane potential
Whole-cell patch-clamp recordings were performed with an Axopatch 700B amplifier (Molecular Devices). Stimulation and data acquisition were performed with the Digidata 1440 series interface and pCLAMP 11 software (Molecular Devices). All recordings were made at room temperature (RT: 23–25 °C). The external solution was the same Tyrode’s solution used for confocal Na+ imaging. Cells were approached under DIC with 3–5 MΩ pipettes and filled with an internal solution containing (in mM) 145 K-gluconate, 2 MgCl2, 0.1 ethylene glycol tetra-acetic acid (EGTA), 2 Na2ATP, 0.2 NaGTP, and 10 Hepes. In order to evaluate recording stability and cell health, access resistance and membrane capacity were monitored before and at the end of the experiments. Resting membrane potential was calculated in current-clamp experiments as previously described [34].
Determination of intracellular Glutamate concentrations by High-Performance Liquid Chromatography (HPLC)
For the determination of intracellular concentrations of glutamate, astrocytes were cultured in 30-mm wells and exposed for 5 min with 100 mM L-glutamic acid after 1-h treatment with vehicle, 200 nM ex-oTau or ex-oTau in the presence of heparin (2 mg/mL).
After glutamate exposure, supernatants were discarded and cells were washed twice with large volumes of ice-cold phosphate-buffered saline (PBS, pH 7.4). Immediately after the second washing, cells were gently scraped in 250 μl of ice-cold 10 mM KH2PO4, pH 7.4. Cell suspensions were treated for the determination of intracellular compounds according to a method previously set up [35]. Briefly, 750 μl of HPLC-grade CH3CN were added to each cell suspension to create a precipitating solution composed by 75% CH3CN + 25% 10 mM KH2PO4, pH 7.4. After vigorous vortexing for 60 s, cell samples were centrifuged at 20,690 × g for 15 min at 4˚C to precipitate proteins. The protein-free supernatants were collected and subjected to 2 chloroform washes, in order to eliminate the organic solvent and obtain an upper aqueous phase cell extract that was directly injected onto a Hypersil C-18, 250 × 4.6 mm, 5-μm particle size column, provided with its own guard column (Thermo). The HPLC apparatus consisted of a Surveyor System (Thermo) with a highly sensitive diode array PDA detector (equipped with a 5 cm light path flow cell) set-up between 300 and 400 nm wavelengths. Data acquisition and analyses were automatically performed by a linked PC using the ChromQuest 5.0® software package provided by the HPLC manufacturer (Thermo). The identification and quantification of glutamate was performed in each sample of cell extracts, using an automated pre-column sample derivatization with a mixture of ortophtalaldehyde (OPA) and 3-mercaptopropionic acid (MPA), as described elsewhere [36]. Assignment and calculation of the OPA-Glutamate in chromatographic runs of cell extracts was carried out at 338 nm wavelengths by comparing retention time and area of the peak with that of chromatographic runs of freshly prepared ultra-pure true glutamate solutions with known concentrations. In cell extracts, the total amount of proteins was determined according to the Bradford method [37]. Glutamate concentrations were normalized for the total cell protein concentrations and expressed as fold change with respect to the control values.
Statistics
Statistical comparisons and analyses were carried out with SigmaPlot software 14.0. Data samples were subjected to normal distribution assay and then expressed as mean ± standard error of the mean (SEM). Two-tailed Student’s t-test and one-way ANOVA with Bonferroni’s post-hoc test were used for comparing two or many experimental groups, respectively. The Mann-Whitney (Wilcoxon) nonparametric statistic was used when experimental data were fewer than 10 observations (e.g., densitometric analysis of WB data). The level of significance (p) was set at 0.05.
Results
Extracellular oTau treatment affects EAAT-dependent, glutamate/Na+ symport in astrocytes due to reduced expression of GLT-1.
The stoichiometry of glutamate transport by GLT-1 causes a significant Na+ influx into astrocytes when glutamate uptake is stimulated by extracellular application of the neurotransmitter. By confocal Na+ imaging experiments we found that extracellular application of 100 mM L-glutamic acid induced rapid intracellular Na+ transients in cultured astrocytes, which reached a plateau after 12–14 s of continuous application (Figure 1A). In vehicle-treated cells, the mean peak amplitude of Na+ transients was 1.7±0.1 (expressed as ΔF/F; n = 160 cells). Once glutamic acid was removed by cell washout with Tyrode’s solution, intracellular Na+ levels returned to initial resting values with a time constant of 5.3±0.2 s. Treatment of astrocytes with 200 nM ex-oTau for 1 hour significantly reduced the amplitude of glutamic acid-induced Na+ transients. In fact, in this condition peak amplitude of Na+ transients was 1.0±0.1 (−43% vs. vehicle, n = 107 cells, p=7.5×10−11 assessed by Student’s t-test; Figure 1A).
The specificity of Na+ transients elicited in astrocytes after exposure to glutamic acid was confirmed by the EAAT blocker DL-TBOA (100 μM), which almost completely abolished signals, thus indicating that these transients were specifically due to Na+ entry via glutamate-induced EAAT activation. On the contrary, the blockade of AMPA and NMDA ionotropic glutamate receptors with NBQX (10 μM) and D-AP5 (50 μM), respectively, did not reduce the Na+ transients elicited in astrocytes after exposure to glutamic acid (Supplementary Figure S1A).
To further investigate the effects of ex-oTau on glutamate-induced EAAT-dependent inward Na+ currents in a more complex system than cultured astrocytes, we recorded synaptically-activated, transporter-mediated currents (STCs) in astrocytes of the CA1 stratum radiatum of organotypic hippocampal slices obtained from C57/Bl6 mice. After stimulation of Schaffer collaterals, hippocampal astrocytes in the CA1 stratum radiatum of vehicle-treated organotypic hippocampal slices exhibited a DL-TBOA-sensitive (Supplementary Figure S1B) inward current of −321.2±68.3 pA (n=13 slices), with a mean time-to-peak of 189.7±2.1 ms. One-hour lasting treatment of organotypic hippocampal slices with 200 nM ex-oTau significantly reduced current amplitude whose mean value was −162.7±40.6 pA (−49% vs. vehicle, n=13; p=0.049 assessed by Student’s t-test; Figure 1C–D). Moreover, a slight, even if not significant increment was also observed for time-to-peak in ex-oTau-treated astrocytes (217.5±44.2 ms).
Taken together, these results suggest that exposure to ex-oTau markedly affects EAAT activity in astrocytes.
The effects of ex-oTau on glutamate uptake were only indirectly estimated based on DL-TBOA-sensitive intracellular Na+ transients recorded after extracellular glutamate application (Figure 1A) and on inward Na+ currents after stimulated glutamate release (Figure 1C–D). To directly investigate the impact of ex-oTau on glutamate uptake by astrocytes we carried out HPLC experiments quantifying the amount of intracellular glutamate in vehicle- and ex-oTau-treated astrocytes after 5-min exposure to 100 mM L-glutamic acid. Considerable variability in the amount of intracellular glutamate was found in control cells (ranging from 452.5 to 1402.3 nmol/mg of proteins) as well as in tau-treated cells (329.5–846.1 nmol/mg). However, as expected, intracellular glutamate levels were significantly reduced in tau-treated astrocytes with respect to vehicle-treated ones (−28±7%; p=0.008, assessed by Mann Whitney Rank Sum test; Figure 1E), whereas no significant effects were detected between vehicle and oTau+heparin conditions (p=0.67).
Additionally, we checked if the observed decreases in inward glutamate-activated Na+ currents and intracellular glutamate in astrocytes were due to an ex-oTau-dependent alteration of EAAT expression and, in particular, of GLT-1. To address this issue, we performed IF and WB experiments. We found that the expression of GLT-1 in cultured astrocytes was significantly reduced after 1-h treatment with 200 nM ex-oTau compared with vehicle. In fact, with IF we observed a 45±7% reduction (p=0.020 ex-oTau vs. vehicle, assessed with Student’s t-test; Figure 2A,B,D), and this finding was confirmed by WB analysis that revealed a GLT-1 expression reduction by −29±2% (p=2.6×10−3 ex-oTau vs. vehicle assessed by paired t-test; Figure 2E–F). On the contrary, the expression of GLAST was not significantly affected by ex-oTau treatment (Supplementary Figure S2).
Figure 2: ex-oTau reduce GLT-1 expression dependently on their internalization.

(A-C) Representative examples of confocal images of WT cultured astrocytes treated with (A) vehicle, (B) 200 nM ex-oTau, or (C) 200 nM ex-oTau + heparin (2 mg/mL) for 1 hour and immunostained for the astrocytic marker GFAP and the glutamate transporter GLT-1. Scale bar: 10 μm. DAPI was used to identify cell nuclei. Insets in the panels A-C show GLT-1 staining only. (D) Bar graph showing the mean GLT-1 fluorescence intensity in the conditions represented in A-C (n=10, 11, and 13 fields, respectively). (E) Representative WB analysis of GLT-1 performed on WT astrocytes treated with either 200 nM ex-oTau or with vehicle for 1 hour (n=6 for each experimental condition). (F) Densitometric analysis of WB experiments carried out as in E. (G) Representative WB analysis of GLT-1 performed on APP KO astrocytes treated with either 200 nM ex-oTau or vehicle for 1 hour (n=3 for both). (H) Densitometric analysis of WB experiments carried out as in G. For all conditions, GAPDH was used as a loading control. * P<0.05 and *** P<0.001 vs. vehicle, # P<0.05 vs. ex-oTau. n.s. means not significant differences vs. vehicle.
The effects of ex-oTau on glutamate-induced intracellular Na+/glutamate symport depend on their uploading in cultured astrocytes.
In a previous paper, we demonstrated that cultured astrocytes abundantly uploaded ex-oTau and, once internalized, oTau affected astrocytes’ functions including the ability to generate intracellular Ca2+ transients and to release gliotransmitters [4]. To determine the contribution of oTau internalization to the ex-oTau-induced alteration of GLT-1-dependent buffering of extracellular glutamate, we applied ex-oTau (200 nM) in experimental conditions hindering oTau uploading by cells, namely: i) in the presence of 2 mg/mL heparin, that it is known to block oTau internalization by interacting with heparan sulfate proteoglycans mediating tau entry into the cells [8, 38]; ii) in astrocytes obtained from amyloid precursor protein (APP) knockout (KO) mice. This second experimental paradigm is based on our previous findings indicating that APP expression is necessary for oTau internalization and the subsequent alteration of Ca2+-dependent gliotransmitter release [4, 5].
We found that the impairment of the EAAT-driven Na+-Glu uptake depended on oTau internalization in astrocytes. In fact, heparin impeded the ex-oTau-induced reduction in intracellular glutamate levels, measured by HPLC, after 5-min L-glutamic-acid application to astrocytes (p=0.032 vs. ex-oTau, assessed by Mann Whitney Rank Sum test; Figure 1E) and no significant reduction in the GLT-1 expression was observed in both experimental paradigms avoiding oTau internalization mentioned above, i.e., heparin (Figure 2C,D), and APP KO cells (Figure 2G,H). Accordingly, in heparin-treated astrocytes (Figure 3A) and APP KO astrocytes (Figure 3B) ex-oTau were unable to reduce the amplitude of Na+ transients induced by 20-s lasting glutamate application. Noteworthy, a significant difference in the amplitude of Na+ transients was observed between 10 and 14 s of glutamate application among vehicle-treated and oTau-treated cells unable to internalize oTau (heparin-treated, Figure 3A, p=0.03; and APP KO, Figure 3B, p=0.0042]; both assessed by Student’s t-test).
Figure 3: Treatment with ex-oTau affects NKA function independently on oTau internalization.

(A) Mean time-course of glutamic acid-induced intracellular Na+ transients in mouse cultured WT astrocytes treated with ex-oTau (200 nM; n=107 cells), ex-oTau (200 nM) + heparin (2 mg/ml; n=102 cells) or vehicle (n=160 cells) for 1 hour. (B) Mean time-course of glutamic acid-induced intracellular Na+ transients in mouse cultured APP KO astrocytes treated with either ex-oTau (200 nM; n=56 cells) or vehicle (n=75 cells) for 1 hour. Pink shadowed areas in A and B highlight intracellular Na+ level recovery time in the various experimental conditions. Dotted red boxes indicate the time points in which there is a significant difference between vehicle-treated cells and cells treated with ex-oTau+Hep for WT astrocytes (panel A, * p<0.05) and oTau for APP KO astrocytes (panel B, ** p<0.001).
Collectively, these results suggest that the astrocyte-specific, EAAT-dependent Na+-Glu uptake is impaired by ex-oTau treatment and mainly depends on tau oligomers’ internalization even if an internalization-independent mechanism is also involved.
Treatment with ex-oTau affects NKA function independently of oTau internalization.
Analysis of the time-course of glutamate-induced Na+ transients revealed that the time needed to return to resting values was longer in ex-oTau-treated astrocytes than in controls (see the pink-shadowed area in Figure 3A). In fact, the time constant of intracellular Na+ fall was 11.3±0.9 s in ex-oTau-treated cells and 5.3±0.2 s in control ones (p=6.5×10−12, assessed by Student’s t-test). Such difference was even more evident if the two curves were normalized to the same maximum value (e.g.: 1; Supplementary Figure S2A,B, pink-shadowed area). Removal of the Na+ excess from the intracellular side of plasma membrane is mainly mediated by Na+/K+ ATPase (NKA) pumps. Notably, neither heparin nor the absence of APP spared astrocytic NKA function from the action of ex-oTau, the time constants being significantly different from that of vehicle (8.9±0.5 s and 12.8±1.1 s, respectively; Figure 3A–B, shadowed areas; one-way ANOVA followed by Dunnett’s post-hoc test: F3,349=24.71, p<0.001). We then checked if ex-oTau affected NKA expression, but no significant changes were observed between vehicle- and ex-oTau-treated WT astrocytes for both α1 (Figure 4A–D) and α2 isoforms (Figure 4E–H). Moreover, 1-h treatment with the specific NKA blocker ouabain (1 mM), which does not affect NKA expression, had the same effects of ex-oTau (Supplementary Figure S3C,D). Collectively, these findings suggest that the detrimental action of oTau on intracellular Na+ removal is mainly exerted at extracellular level, and it consists in inhibiting NKA function.
Figure 4: Treatment with ex-oTau affects NKA localization on plasma membrane.

(A-B) Representative examples of confocal images of WT cultured astrocytes treated with vehicle (A) or 200 nM ex-oTau (B), immunostained for the α1 isoform of Na+/K+ ATPase pump (NKA1A1; A1, B1) and the astrocytic marker GFAP (A2, B2). DAPI was used to stain cell nuclei. Merged images are shown in A3 and B3. (C) Representative WB analysis of NKA1A1 performed on WT astrocytes treated as in A-B (n=3). (D) Densitometric analysis of WB experiments carried out as in C. (E-F) Representative examples of confocal images of WT astrocytes cultures treated with vehicle (E) or 200 nM ex-oTau (F), immunostained for the α2 isoform of Na+/K+ ATPase pump (NKA1A2; E1, F1) and the astrocytic marker GFAP (E2, F2). DAPI was used to stain cell nuclei. Merged images are shown in E3 and F3. (G) Representative WB analysis of NKA1A2 performed on WT astrocytes treated in E-F (n=7). (H) Densitometric analysis of WB experiments carried out as in G. For all conditions, β-actin was used as a loading control. Scale bars: 10 μm. n.s. means no significant differences.
It is known that NKAs are tau-interacting partners [39], and that they co-localize with mitochondria and EAATs [23,24]. Therefore, to investigate how ex-oTau affected NKA function, we studied their localization on the plasma membrane and association with mitochondria. As shown in Figure 4A, B, E, F, 1-h ex-oTau treatment significantly affected the distribution of NKAs on the cell membrane that resulted accumulated in specific “hot spots”, especially located on cell edges. In particular, we observed a significant mislocalization of NKAs with respect to mitochondria, identified by mitotracker (Supplementary Figure S4). The correlation coefficient between NKA1A1 and mitochondria labelling was, in fact, significantly reduced from 0.68±0.06 in vehicle-treated cells to 0.41±0.09 in ex-oTau-treated ones (p=0.03; assessed by Student’s t-test).
To extend our findings in the context of human AD pathology, we performed a subset of experiments that evaluated the effects of human AD tau (i.e., tau derived from the brain of AD patients) on extracellular glutamate handling. Specifically, we repeated confocal Na+-imaging, STC, HPLC, and WB for GLT-1 and NKA1A2 experiments. As reported in the Supplementary Figure S5, the effects of hTau were similar to those observed with the 4R/2N tau isoform. In fact, we found that 1-h extracellular application of hTau induced: i) reduction in the peak amplitude of glutamate-induced Na+ transients (−23.7% vs. vehicle; p=5.4×10−11, assessed by Student’s t-test) and increase of the time constant of intracellular Na+ fall (+33%; p=6.6×10−8; Student’s t-test); ii) decrease of the amplitude of glutamate-induced EAAT-dependent inward Na+ currents (STCs) (−56% vs. vehicle; p=0.0355; Student’s t-test); iii) reduced levels of intracellular glutamate evaluated by HPLC (−25% vs. vehicle; p=0.03); iv) lower levels of GLT-1 expression vs. vehicle (−35%; p=0.008, assessed by Mann Whitney test) and no effects on ATP1A2 levels.
Intracellular Na+ levels and resting membrane potentials in cultured astrocytes are altered by treatment with ex-oTau.
Having found that ex-oTau treatment disrupts NKA function, we checked if basal intracellular Na+ levels in cultured WT astrocytes treated for 1 hour with 200 nM ex-oTau were accordingly affected. We found that in control cells the basal intracellular Na+ concentration, studied by confocal Na+ imaging, was 11.4±1.8 nM (n=36). This value raised to 36.0±2.9 nM in ex-oTau-treated astrocytes (n=54; p=6.9×10−9 vs. vehicle [Student’s t-test]; Figure 5A–C). These differences were paralleled by a significant change of resting membrane potential in WT astrocytes: from −69.9±0.9 (n=18) in vehicle-treated to −66.0±1.1 mV (n=17) in ex-oTau-treated ones (p=6.2×10−2 vs. vehicle assessed by Student’s t-test; Figure 5D). Very similar results were found in APP KO astrocytes, thus confirming the extracellular effect of ex-oTau on NKA function. Indeed, resting Na+ levels were 11.2±0.1 vs. 37.3±0.2 mM in vehicle- and ex-oTau-treated APP KO astrocytes, respectively (Figure 5E–G) and resting membrane potential was affected accordingly: −69.6±0.3 mV (n=6; vehicle) and −59.1±0.9 mV (n=7; ex-oTau; p<1×10−4 assessed by Student’s t-test; Figure 5H).
Figure 5: Treatment with ex-oTau affects intracellular Na+ levels and resting membrane potential of cultured astrocytes and neurons.
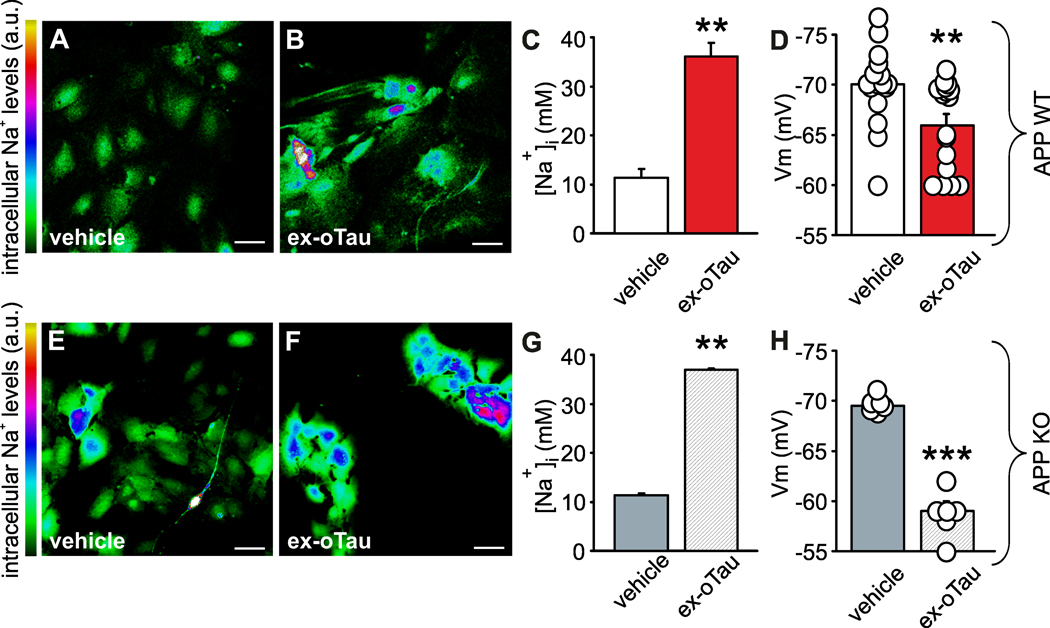
(A-B) and (E-F) Representative examples of confocal images showing intracellular Na+ levels at rest in WT (A-B) and APP KO (E-F) cultured astrocytes loaded with CoroNa green probe and treated for 1 hour with either vehicle (A, E; n=36) or 200 nM ex-oTau (B, F; n=54). Scale bars: 50 μm. (C-D) and (G-H) Bar graphs representing the mean values of intracellular Na+ levels at rest (C, G) and the mean resting membrane potential (D, H) in ex-oTau- and vehicle-treated WT (C, D; n=17 and 18, respectively) and APP KO (G, H; n=7 and 6, respectively) astrocytes. **p<0.005; ***p<0.001 vs. vehicle.
Discussion
In the present study we investigated the effects of ex-oTau application on the handling of extracellular glutamate by astrocytes. One-hour of ex-oTau treatment (sufficient for allowing tau internalization in astrocytes but not in neurons [4]) exerted a synaptotoxic action consisting in a depression of synaptic transmission, which was attributed to a significant reduction of gliotransmitters release (including glutamate) from astrocytes.
In the present paper we extended our study of tau action on astrocytic glutamate handling focusing on mechanisms regulating the neurotransmitter uploading from the extracellular space. We found that ex-oTau impaired the ability of astrocytes to upload glutamate from the synaptic cleft by acting on both the glutamate transporter GLT-1 and the Na+/K+ ATPases.
Of note, the effect of tau oligomers on GLT-1 required their entry into the cells and consisted of a reduction of protein expression, whereas the tau-induced alterations of NKA function relied on an extracellular action resulting in pump mislocalization with no changes in its expression. In particular, our findings suggest that: i) 1-h treatment with 200 nM ex-oTau reduces the expression of the glutamate transporter GLT-1 in cultured astrocytes, thus affecting the uptake of glutamate from the extracellular medium; ii) ex-oTau interact extracellularly with NKAs (ATP1A1 and ATP1A2), determining their delocalization on plasma membrane that alters their ability to extrude Na+ from the cells. As a consequence, ex-oTau-targeted astrocytes exhibited increased intracellular Na+ concentrations, membrane depolarization, and a reduced transmembrane Na+ gradient affecting Na+-dependent transports including the Na+-glutamate symport by GLT-1, known to depend on NKAs [24].
In order to discern between the intra- and extracellular effects of tau oligomers, we used experimental paradigms suitable to impede the internalization of oTau into the cells (e.g., either cell pre-treatment with heparin [8] or the use of APP KO cells [4,5]). Notably, GLT-1 downregulation was not observed in experimental conditions hindering oTau build-up in astrocytes, whereas NKA dysfunction was still present in experimental conditions in which ex-oTau entry into astrocytes was prevented.
Our findings are in agreement with previous studies demonstrating the interaction of neuronal NKA (1A3) with proteins able to oligomerize/aggregate, including α-synuclein [40], amyloid β (Aβ) [41] and tau [42]. Moreover, a recent proteomic study identified NKA1A1 and NKA1A2 as interacting partners of tau [39]. Melone and co-authors recently reported that GLT-1 and NKA isoforms (α1–3) are coupled [23]. In particular, they demonstrated that the interaction between GLT-1 and NKAs is more efficient in astrocytes (i.e., GLT-1/α2 at the perisynaptic processes) than in neurons. We hypothesize that oTau, by binding NKA extracellularly, causes their delocalization and detachment from GLT-1 thus allowing GLT-1 to be targeted by intracellular tau oligomers and determining their degradation/clearance. This interpretation is in agreement with data obtained with Aβ oligomers reportedly inhibiting glutamate reuptake from astrocytes via altered GLT-1 diffusion [43] and its reduced expression on plasma membrane [44].
We also documented that ex-oTau induce delocalization of NKAs on plasma membrane. Our results are in line with those obtained by Shrivastava and collaborators who reported that α-synuclein and fibrillary tau assemblies affect the neuronal isoform of NKA by determining its sequestration and redistribution, thus altering transmembrane Na+ gradients [38, 40]. Also, Ohnishi et al. demonstrated that Na+/K+ ATPase α3 is a target of Aβ assembly [39]. Tau-dependent NKA redistribution is also associated with a mislocalization between NKA and mitochondria, which are normally spatially associated [24]. We did not directly investigate the effects of oTau on mitochondria, but it was recently reported that tau overexpression in astrocytes affects mitochondria localization and function [45].
Collectively, our data demonstrate that tau oligomers exert a double detrimental action on GLT-1 function: i) extracellularly, they reduce transmembrane Na+ gradient, thus affecting GLT-1-dependent Na+-driven glutamate uptake; ii) intracellularly, they inhibit GLT-1 expression. The net effect of ex-oTau is a marked impairment of astrocytes’ ability to uptake glutamate from the extracellular space. Data reported here, along with our previous findings demonstrating an oTau-dependent alteration of glutamate release from astrocytes, point to a complex dysregulation of glutamate signalling involving all the actors regulating the astrocytic glutamate handling that include GLT-1, NKAs and mitochondria, Na+ gradients and all Na+-dependent transports, in addition to intracellular Ca2+ signalling. Finally, our findings add a novel layer to the molecular mechanisms underlying the impact of tauopathies on brain health and cognitive function.
In fact, the glutamate dynamics at the hippocampus are of particular interest in terms of synaptic transmission and plasticity, and tau-induced glutamate handling dysregulation may be directly associated with the cognitive decline characterizing the early stages of AD. Moreover, the altered cell membrane excitability induced by the extracellular action of oTau may also impact on synaptic strength that underlies the pathogenesis of tauopathies [46].
Supplementary Material
Acknowledgments
Supported by: Italian Ministry of Health, Ricerca Corrente - Fondazione Policlinico Universitario A. Gemelli IRCCS and PRIN #2020AMLXHH to C.G.; NIH funds to O.A. (R01NS110024); Università Cattolica del Sacro Cuore contributed to funding this research publication (D1–2020 intramural funds to R.P.).
List of abbreviations used
- APP KO
amyloid precursor protein knockout
- DL-TBOA
DL-threo-beta-benzyloxyaspartate
- EAATs
excitatory amino acid transporters
- ex-oTau
extracellular tau oligomers
- ex-hTau
extracellular tau derived from AD patients
- GLT-1
glutamate transporter-1
- GLAST
Glutamate-aspartate Transporter
- HPLC
High-performance liquid chromatography
- mEPSCs
miniature excitatory postsynaptic currents
- NCX
Na+/Ca2+ exchanger
- NKA1A1
Na+/K+ ATPase α1
- NKA1A2
Na+/K+ ATPase α2
- NKAs
Na+/K+ ATPases
- STCs
synaptically activated Glu transporter currents
- WT
wild type
Footnotes
Ethics approval and consent to participate
All animal procedures were approved by the Ethics Committee of Università Cattolica and were fully compliant with Italian (Ministry of Health guidelines, Legislative Decree No. 116/1992) and European Union (Directive No. 86/609/EEC) legislation on animal research (Authorization n° 63/2019-PR). Animals were supplied by the Division of Animal Resources of Università Cattolica.
Tissue for human tau isolation was provided by the New York Brain Bank–The Taub Institute, Columbia University. Postmortem Tissue and Specimens were de-identified (Protocol: IRB-AAAB0192(Y20M00)).
Consent for publication
All authors qualified the authorship and approved the publication of this study.
Availability of data and materials
The data used and /or analysed during the current study are available from the corresponding author on reasonable request.
Conflict of interests
All the authors declare that they have no conflict of interest.
References
- 1.Fá M, Puzzo D, Piacentini R, Staniszewski A, Zhang H, Baltrons MA, et al. Extracellular Tau Oligomers Produce An Immediate Impairment of LTP and Memory. Sci Rep. 2016; 20: 19393; 10.1038/srep19393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerrero-Muñoz MJ, Gerson J, Castillo-Carranza DL. Tau Oligomers: The Toxic Player at Synapses in Alzheimer’s Disease. Front Cell Neurosci. 2015; 2: 464; 10.3389/fncel.2015.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hill E, Karikari TK, Moffat KG, Richardson MJE, Wall MJ. Introduction of Tau Oligomers into Cortical Neurons Alters Action Potential Dynamics and Disrupts Synaptic Transmission and Plasticity. eNeuro. 2019; 6: ENEURO.0166–19.2019; 10.1523/ENEURO.0166-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piacentini R, Li Puma DD, Mainardi M, Lazzarino G, Tavazzi B, Arancio O, et al. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia. 2017; 65: 1302–1316; 10.1002/glia.23163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puzzo D, Piacentini R, Fá M, Gulisano W, Li Puma DD, Staniszewski A, Zhang H, Tropea MR, Cocco S, Palmeri A, Fraser P, D’Adamio L, Grassi C, Arancio O. LTP and memory impairment caused by extracellular Aβ and Tau oligomers is APP-dependent. Elife. 2017; 6: e26991; 10.7554/eLife.26991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martini-Stoica H, Cole AL, Swartzlander DB, Chen F, Wan YW, Bajaj, et al. TFEB enhances astroglial uptake of extracellular tau species and reduces tau spreading. J Exp Med. 2018; 215: 2355–2377; 10.1084/jem.20172158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P, Ye Y. Filamentous recombinant human Tau activates primary astrocytes via an integrin receptor complex. Nat Commun. 2021; 12: 95; 10.1038/s41467-020-20322-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013; 110: E3138–47; 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Puma DD, Marcocci ME, Lazzarino G, De Chiara G, Tavazzi B, Palamara AT, et al. Ca2+ -dependent release of ATP from astrocytes affects herpes simplex virus type 1 infection of neurons. Glia. 2021; 69: 201–215; 10.1002/glia.23895. [DOI] [PubMed] [Google Scholar]
- 10.Puangmalai N, Bhatt N, Montalbano M, Sengupta U, Gaikwad S, Ventura F, et al. Internalization mechanisms of brain-derived tau oligomers from patients with Alzheimer’s disease, progressive supranuclear palsy and dementia with Lewy bodies. Cell Death Dis. 2020; 11: 314; 10.1038/s41419-020-2503-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006; 7: 423–36; Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006; 7: 423–36; 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zorec R, Verkhratsky A, Rodríguez JJ, Parpura V. Astrocytic vesicles and gliotransmitters: Slowness of vesicular release and synaptobrevin2-laden vesicle nanoarchitecture. Neuroscience. 2016; 323: 67–75; 10.1016/j.neuroscience.2015.02.033. [DOI] [PubMed] [Google Scholar]
- 13.Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells. 2019; 8: 184; 10.3390/cells8020184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rose CR, Felix L, Zeug A, Dietrich D, Reiner A, Henneberger C. Astroglial Glutamate Signaling and Uptake in the Hippocampus. Front Mol Neurosci. 2018; 10: 451; 10.3389/fnmol.2017.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG, et al. Glial cells in (patho)physiology. J Neurochem. 2012. 121: 4–27; 10.1111/j.1471-4159.2012.07664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schousboe A, Scafidi S, Bak LK, Waagepetersen HS, McKenna MC. Glutamate metabolism in the brain focusing on astrocytes. Adv Neurobiol. 2014; 11: 13–30; 10.1007/978-3-319-08894-5_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001; 65: 1–105; 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 18.López-Bayghen E, Ortega A. Glial glutamate transporters: new actors in brain signaling. IUBMB Life. 2011; 63: 816–23; 10.1002/iub.536. [DOI] [PubMed] [Google Scholar]
- 19.Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011; 226: 2484–93; 10.1002/jcp.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nilsen LH, Rae C, Ittner LM, Götz J, Sonnewald U. Glutamate metabolism is impaired in transgenic mice with tau hyperphosphorylation. J Cereb Blood Flow Metab. 2013; 33: 684–691; 10.1038/jcbfm.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener. 2020; 15: 40; 10.1186/s13024-020-00391-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirischuk S, Héja L, Kardos J, Billups B. Astrocyte sodium signaling and the regulation of neurotransmission. Glia. 2016; 64: 1655–66; 10.1002/glia.22943. [DOI] [PubMed] [Google Scholar]
- 23.Melone M, Ciriachi C, Pietrobon D, Conti F. Heterogeneity of Astrocytic and Neuronal GLT-1 at Cortical Excitatory Synapses, as Revealed by its Colocalization With Na+/K+-ATPase α Isoforms. Cereb Cortex. 2019; 29: 3331–3350; 10.1093/cercor/bhy203. [DOI] [PubMed] [Google Scholar]
- 24.Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, et al. Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci. 2011; 14: 18275–88; 10.1523/JNEUROSCI.3305-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spinelli M, Fusco S, Mainardi M, Scala F, Natale F, Lapenta Ret al. Brain insulin resistance impairs hippocampal synaptic plasticity and memory by increasing GluA1 palmitoylation through FoxO3a. Nat Commun. 2017; 8: 2009; 10.1038/s41467-017-02221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puzzo D, Argyrousi EK, Staniszewski A, Zhang H, Calcagno E, Zuccarello E, et al. Tau is not necessary for amyloid-β-induced synaptic and memory impairments. J Clin Invest. 2020; 130: 4831–4844; 10.1172/JCI137040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Argyrousi EK, Staniszewski A, Nicholls RE, Arancio O. Preparation of tau oligomers after the protein extraction from bacteria and brain cortices. Methods Mol Biol. 2018; 1779: 85–97; 10.1007/978-1-4939-7816-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santoro M, Piacentini R, Perna A, Pisano E, Severino A, Modoni A, et al. Resveratrol corrects aberrant splicing of RYR1 pre-mRNA and Ca2+ signal in myotonic dystrophy type 1 myotubes. Neural Regen Res. 2020; 15: 1757–1766; 10.4103/1673-5374.276336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier SD, Kovalchuk Y, Rose CR. Properties of the new fluorescent Na+ indicator CoroNa Green: comparison with SBFI and confocal Na+ imaging. J Neurosci Methods. 2006; 155: 251–259; 10.1016/j.jneumeth.2006.01.009. [DOI] [PubMed] [Google Scholar]
- 30.Li Puma DD, Piacentini R, Leone L, Gironi K, Marcocci ME, De Chiara G, et al. Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-β Protein Accumulation. Stem Cells. 2019, 37: 1467–1480; 10.1002/stem.3072. [DOI] [PubMed] [Google Scholar]
- 31.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012, 9: 676–682; 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scimemi A, Diamond JS. Deriving the time course of glutamate clearance with a deconvolution analysis of astrocytic transporter currents. J Vis Exp. 2013; 78: 50708; 10.3791/50708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Ascenzo M, Podda MV, Fellin T, Azzena GB, Haydon P, Grassi C. Activation of mGluR5 induces spike after depolarization and enhanced excitability in medium spiny neurons of the nucleus accumbens by modulating persistent Na+ currents. J Physiol. 2009; 587(Pt 13):3233–50; 10.1113/jphysiol.2009.172593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ripoli C, Piacentini R, Riccardi E, Leone L, Li Puma DD, Bitan G, et al. Effects of different amyloid β-protein analogues on synaptic function. Neurobiol Aging. 2013; 34: 1032–1044; 10.1016/j.neurobiolaging.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 35.Ciccarone F, Di Leo L, Lazzarino G, Maulucci G, Di Giacinto F, Tavazzi B, et al. Aconitase 2 inhibits the proliferation of MCF-7 cells promoting mitochondrial oxidative metabolism and ROS/FoxO1-mediated autophagic response. Br J Cancer. 2020; 122: 182–193, 10.1038/s41416-019-0641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amorini AM, Lazzarino G, Di Pietro V, Signoretti S, Lazzarino G, Belli A, et al. Severity of experimental traumatic brain injury modulates changes in concentrations of cerebral free amino acids. J Cell Mol Med. 2017; 21: 530–542; 10.1111/jcmm.12998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding, Anal. Biochem. 1976. May 7; 72: 248–54; 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 38.Mirbaha H, Holmes BB, Sanders DW, Bieschke J, Diamond MI. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J Biol Chem. 2015; 290: 14893–903; 10.1074/jbc.M115.652693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinsky J, Majerova P, Kovac A, Kotlyar M, Jurisica I, Hanes J. Physiological Tau Interactome in Brain and Its Link to Tauopathies. J Proteome Res. 2020; 19: 2429–2442; 10.1021/acs.jproteome.0c00137. [DOI] [PubMed] [Google Scholar]
- 40.Shrivastava AN, Redeker V, Fritz N, Pieri L, Almeida LG, Spolidoro M, et al. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015; 34: 2408–23; 10.15252/embj.201591397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohnishi T, Yanazawa M, Sasahara T, Kitamura Y, Hiroaki H, Fukazawa Y, et al. Na, K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc Natl Acad Sci U S A. 2015; 112: E4465–74; 10.1073/pnas.1421182112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shrivastava AN, Redeker V, Pieri L, Bousset L, Renner M, Madiona K, Mailhes-Hamon C, Coens A, Buée L, Hantraye P, Triller A, Melki R. Clustering of Tau fibrils impairs the synaptic composition of α3-Na+/K+-ATPase and AMPA receptors. EMBO J. 2019; 38: e99871; 10.15252/embj.201899871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zott B, Simon MM, Hong W, et al. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science. 2019; 365: 559–565; 10.1126/science.aay0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG. Amyloid-β1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci. 2013; 33: 5312–5318; 10.1523/JNEUROSCI.5274-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richetin K, Steullet P, Pachoud M, et al. Tau accumulation in astrocytes of the dentate gyrus induces neuronal dysfunction and memory deficits in Alzheimer’s disease. Nat Neurosci. 2020; 23: 1567–157; 10.1038/s41593-020-00728-x. [DOI] [PubMed] [Google Scholar]
- 46.Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013; 70: 1158–1166; 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.