

Abstract
CD19 and B-cell maturation antigen (BCMA) - directed CAR T cells have enabled unprecedented responses in a subset of refractory patients with B-cell and plasma cell malignancies, leading to their approval by the US Food and Drug administration (FDA) for the treatment of leukemia, lymphoma, and myeloma. These “living drugs” can become part of a synthetic immune system, persisting at least a decade in some patients. However, despite this tremendous impact, significant unmet treatment needs remain for patients with hematological malignancies and solid cancers. In this perspective, we highlight recent innovations that advance the field toward production of a more potent and universal cellular immunotherapy of the future.
Keywords: Synthetic, orthogonal, CAR T, CAR NK, genome editing
CD19 and beyond – Prospects for CAR T cells in hematologic malignancies
Chimeric antigen receptors (CAR) are synthetic receptors that re-direct T cells to target cancer cells in a major histocompatibility complex (MHC) independent manner. T cells engineered to express a CAR recognizing CD19 have led to unparalleled responses in a subset of otherwise refractory patients with B-cell malignancies (1). However, despite the tremendous impact of CD19-directed CAR T cell therapy, primary resistance or relapse is frequently observed with varying patterns and mechanisms of resistance across different entities.
Although the initial response rate is high, only approximately 50% of pediatric and young adult acute lymphoblastic leukemia (ALL) patients treated with CAR T cells will be alive and disease-free one year after treatment. For patients with higher disease burden at the time of treatment, the event-fee survival at 12 months drops down to 31% (2). The assessment of antigen status reveals CD19-negative or CD19-low disease in a high proportion of relapses (2,3). This pattern of resistance could be principally addressed by sequential administration of CAR T cells targeting a different antigen. For instance, ALL patients who were treated with anti-CD22 CAR T cells after relapse with CD19-directed therapy achieved a 73% complete response rate, but two third of these later relapsed with reduced CD22 surface density (4). Current approaches designed to block the escape of resistant tumor clones include upfront infusing of multi-specific CAR T cell products targeting several antigens simultaneously, either achieved by pooling of different CAR vectors, a single vector encoding two different CARs (bi-cistronic/dual CAR), or a single CAR construct (tandem CAR) incorporating two different single chain variable fragments (scFvs) (Fig. 1a). Early clinical studies demonstrated feasibility of such approaches, e.g., combining CD19/CD20 (5) or CD19/CD22 (6). However, such approaches come with the challenge to construct optimal CARs that retain efficacy for all targets. For example, Spiegel et al. lost potency against CD22 in their tandem construct indicated by undiminished CD22 expression in the relapse situation compared to baseline (6), which was not predicted in preclinical models (7) and contrasts with the observed immune pressure on CD22 with a monospecific construct (4). A potential strategy to address antigen-dim relapse relies on fine-tuning of the antigen density threshold for CAR mediated T cell activation. This can be achieved by adapting the scFv affinity of the CAR in accordance to a certain antigen-expression level, whereby a high-affinity CAR can principally detect a dim antigen (8), or by changing/altering domains of the CAR such as the transmembrane/hinge-region or the costimulatory domain, e.g., via manipulation of the number of immunoreceptor tyrosine-based activation motifs (ITAMs) (9–11). However, this threshold needs to be exquisitely adapted to the respective clinical scenario and carefully studied as CAR affinity modulation will potentially impact T cell effector function, kinetics of immunotoxicity and the threshold for on-target off-tumor toxicity for less specific antigens than CD19. More so, the superiority of a lower-affinity CD19 CAR above the clinically established FMC63 binder in an antigen abundant scenario has been suggested (12).
Fig. 1: Overview of next-generation CAR T cell approaches.
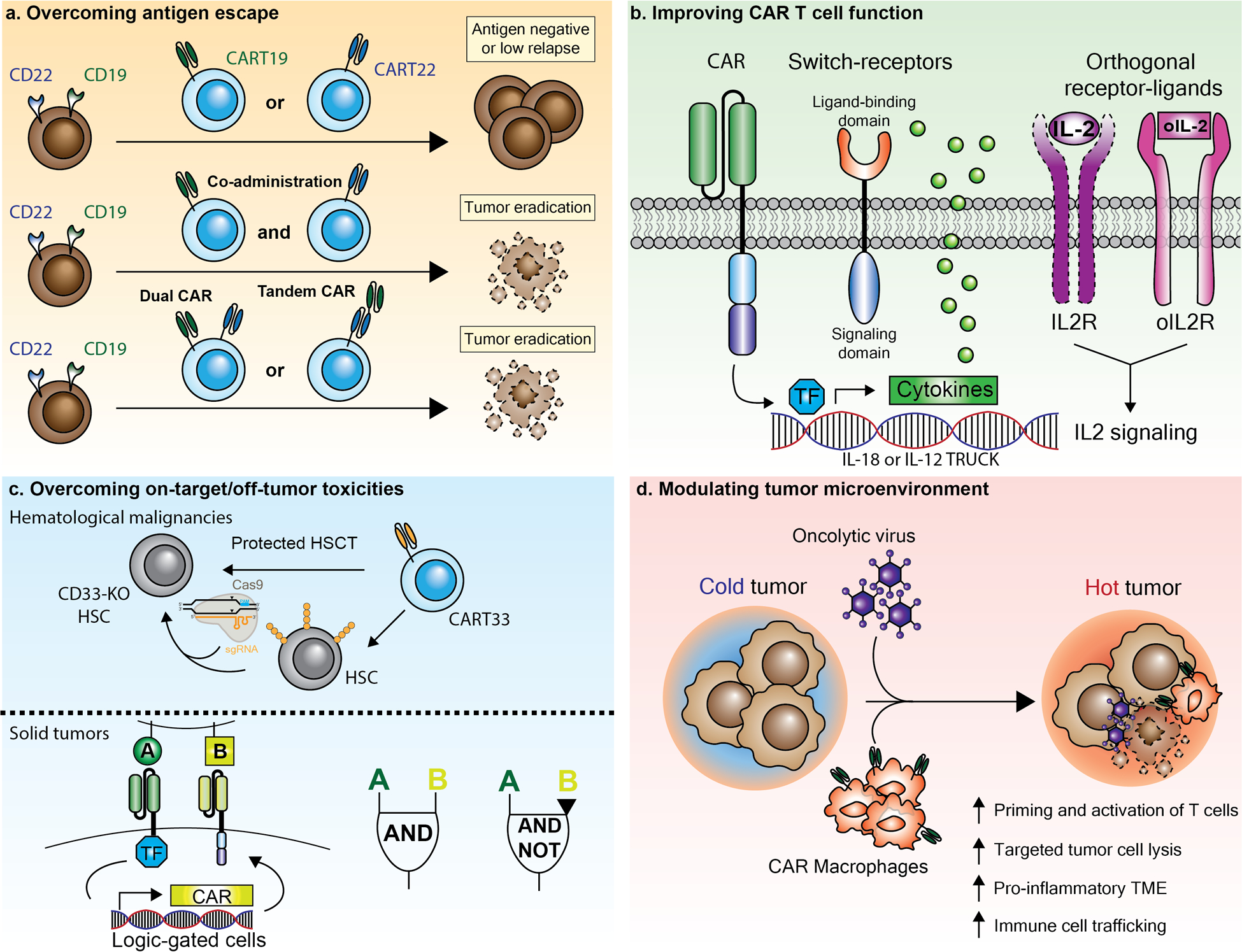
a) To reduce the risk of tumor relapse due to antigen-escape (e.g., loss or downregulation of CD19 or CD22), two CAR T cell products specific for different target antigens can be co-administered. The same T cell can also be transduced with two viral CAR vectors encoding for different constructs, or with one CAR construct with two single chain variable fragments (Dual or Tandem-CAR). b) CAR constructs with advanced functionalities include T cells redirected for antigen-unrestricted cytokine-initiated killing (TRUCK), which secrete cytokines upon activation (e.g., IL-18 or IL-12). In addition, CAR T cell function can be further enhanced by using switch-receptors (e.g., TGFβ), or through addition of orthogonal cytokine receptor system that act specifically on CAR-transduced cells (e.g., orthogonal IL2 receptor, oIL2R). TF= transcription factor. c) To overcome on-target / off-tumor toxicities in hematological malignancies, CAR T cells can be combined with hematopoietic stem cell transplantation (HSCT) that has been engineered to lack the CAR T cell target antigen (e.g., CD33 knock-out hematopoietic stem cells, CD33 KO HSC). For solid tumors, AND and NOT logic-gated CAR T cells have been developed that recognize combinatorial antigen expression to trigger or inhibit CAR T cell effector function. d) The additional administration of engineered viruses can effectively modulate the tumor microenvironment and engage innate / adaptive immunity. Also, CAR-macrophages can phagocytose tumor cells and induce anti-tumor bystander immune cells.
Antigen-dim relapse has also been observed in multiple myeloma patients treated with anti-BCMA CAR T cells (13). Pharmacological modulation of target antigen expression levels is a promising strategy to combat antigen dim tumor relapse. For instance, small-molecule γ-secretase inhibitors can increase BCMA surface levels on myeloma cells (14) by blocking its cleavage and are currently under clinical investigation together with BCMA-directed CAR T cells (e.g., NCT03502577).
Unlike therapies with cytotoxic agents and small molecules, cell therapies are generally agnostic to tumor genotype, given expression of the selected CAR target antigen. However, within B cell tumors, despite similar target density of CD19, there is tumor type susceptibility to CD19-directed CAR T cells. Large B-Cell Lymphoma patients treated with anti-CD19 CAR T cells show lower complete response (CR) rates between 40–50% as compared to 80–90% with pre-B cell ALL. Responses in lymphoma patients tend to be durable, however approximately 30% of the relapsing patients demonstrate antigen downregulation (15). Reasons for failure to achieve initial response are complex and encompass associations with clinical factors such as high tumor burden (16) and T cell intrinsic deficits as well as features of the tumor microenvironment such as the accumulation of myeloid-derived suppressor cells (MDSCs) and interferon signaling (17).
While antigen escape is a major resistance mechanism in pre-B cell ALL, relapse is rare in the more mature B-cell derived malignancy Chronic Lymphocytic Leukemia (CLL). On the contrary, a high rate of initial resistance, linked to T cell intrinsic deficits in the patient, is observed. Durable responses can still be achieved in approximately 30% of CLL-patients (18). CD8+ T cells of CLL patients demonstrate impaired mitochondrial fitness and reduced glucose uptake upon stimulation (19). CLL patients who did achieve a complete remission were found to have enhanced mitochondrial biogenesis in correlation with better proliferation and persistence (19). Sustained remissions in CLL patients are furthermore associated with a higher frequency of memory-like T cells (CD27+CD45RO−CD8+) and STAT3 related gene signatures in the patient apheresis product (20).
Whenever a lack of T cell fitness is presumably the reason of CAR T cell treatment failure, two main strategies have been explored as possible solutions: It has been demonstrated for multiple myeloma patients that T cells collected early during the disease course with less exposure to cytotoxic treatment retain better fitness and proliferative capacity compared to those collected late from relapsed or refractory patients (21). Strategies to leverage these insights will incorporate manufacturing of autologous CAR T cell products from less dysfunctional T cells by either obtain apheresis products early during disease course or by shifting CAR T cell therapy into an earlier line of treatment. First examples of this strategy have already demonstrated an enhanced proliferative potential of CAR T cell products alongside promising response rates (e.g., ZUMA-12, NCT03761056) in patients with lymphoma (22). Alternatively, a potential source of healthy T cells is the use of allogenic donors with additional potential benefits as discussed below (23).
Another elegant way to improve CAR T cell function is to confer the ability to secrete active cytokines for beneficial immunomodulation, so called TRUCKs (T cells redirected for universal cytokine killing) (Fig. 1b). This way, robust T cell activation can become less dependent on an external ‘signal 3’. Examples are IL-12 and IL-18; improved antitumor effects of these engineered cytokines and receptors as demonstrated in xenograft and syngeneic mouse models (24,25). IL-18 not only benefits CAR T cells in an autocrine fashion but also modulates host immune cells via paracrine effects (26). These results have motivated several ongoing clinical trials. The recent characterization of IL-18 binding protein (IL-18BP) in the tumor microenvironment as a ‘secreted negative immune checkpoint’ highlights the potential for further refinement of this approach (27). In order to escape IL-18BP binding, CAR T cells could either be engineered to secrete a decoy-resistant IL-18 (DR-18) (27) or designed to activate IL-18 signaling independently of secreted IL-18 by utilizing a GM-CSF/IL-18 switch receptor (28).
Broadening the scope of CAR T cell therapies beyond CD19 (or BCMA) positive hematologic malignancies seems within reach, as there are several other potential surface antigens within the cluster of differentiation (CD) system with manageable potential for on-target off-tumor toxicity largely confined to hematopoietic cells, such as CD5, CD7 and CD30. For targets for which ablation of lineage antigen-bearing cell populations is not clinically tolerated, a tandem therapy combining targeted CAR T cells with a hematopoietic stem cell transplant genetically engineered to lack the target antigen has been proposed. For example, CD33 is highly expressed on most AML blasts, but is also expressed on myeloid precursor cells, foreshadowing unacceptable toxicity in the form of persistent myelosuppression if targeted with CAR T cells (29). Interestingly, CD33 is dispensable for myeloid function and by means of subtraction, a leukemia-specific antigen can therefore be created by combining CD33-directed CAR T cells with a CD33-deleted hematopoietic stem cell transplantation (HSCT) (30,31) (Fig. 1c).
Similarly, targeting T cell malignancies with CAR T cells would threaten the healthy T cell compartment as a consequence of shared target gene expression. Even more prohibiting, CAR T cell expansion is prone to fail during manufacturing due to antigen-mediated self-killing or fratricide. However, successful production of fratricide-resistant CAR T cells targeting CD7 or CD3 and CD7 can be accomplished by knocking out these targets before in vitro expansion (32,33). An interesting byproduct of this strategy is that it also leads to self-enrichment for the desired edits in the final CAR T product by residual fratricide against unedited cells (33). As a limitation, such a treatment strategy would likely need to incorporate a subsequent stem cell transplantation in remission to restore the T cell compartment. Also, the therapy would likely rely on an allogeneic CAR T product to prevent contamination by malignant T cells.
Towards the “multiverse”: allogeneic cell therapies
Universal CAR T cells address a critical barrier in the field; healthy allogenic donor T cells with superior fitness have the potential to improve clinical responses. ‘Off-the-shelf’ allogeneic CAR T cell products also provide an opportunity to streamline manufacturing and allow treatment of rapidly progressing patients that are unable to bridge several weeks of manufacturing. By reducing costs, universal CAR T cells will potentially broaden patient access to this treatment modality. The major hurdles associated with allogeneic CAR T products are the risk for graft-versus-host disease (GvHD) mediated by the endogenous T cell receptor (TCR), and the risk of T cell graft rejection by the host immune system, resulting in limited persistence (23). A potential solution to tackle these hurdles is offered by advancements in genome editing, mostly based on CRISPR-Cas nucleases, and its technological successors (34). These technologies are also prone to address other challenges of the field such as the modulation of T cell exhaustion (35). Genome editing can be utilized in allogeneic CAR T cells to remove the endogenous TCR, eliminating the risk of GvHD, and Beta-2-Microglobulin (B2M) to ablate HLA class I molecule expression for preventing graft rejection mediated by host T cells. The feasibility of this concept has been already demonstrated in the two recently published UCART19 trials (36), but CAR T persistence was limited and only retained in patients with severe immunosuppression mediated by the anti-CD52 antibody alemtuzumab.
Clinical applications involving genome-editing come with the major concern for off-target editing, which is the unintended and uncontrolled modification of DNA loci beyond the intended edit that might alter cellular function (34). This risk is inherent to the mechanism of action by which CRISPR/Cas nucleases induces DNA double-strand breaks (DSBs), which poses a risk for translocations proportional to the number of targets in a multiplex setting, leading to theoretical concerns of malignant transformation (37). Also, DSBs likely imply negative effects on T cell proliferation and fitness (38,39). Recent techniques, such as prime and base editing, enable genome-editing without the creation of double strand breaks and their principal applicability for T cells have been demonstrated (40,41). Extensive work has been conducted to understand most of those off-target effects and to make them predictable and/or measurable (34) in order to mitigate their risks. In CAR T cell applications, the components of the editing machinery are usually only transiently exposed and applied ex vivo, contributing to safety. Further, in a pilot study, CRISPR-Cas9 modified T cells containing three targeted loci on three different autosomal chromosomes were infused into relapsed/refractory cancer patients and modified cells persisted for up to 9 months without any clinical toxicities or safety concerns (42).
In addition to overcoming host T cell mediated rejection of allogeneic cell products, it is likely that infused cells will require additional genome engineering to avoid rejection by the innate immune system. For example, in the mouse, host natural killer (NK) cells and macrophages rapidly destroy infused allogeneic T cells via distinct mechanisms (43–45). A major remaining challenge currently facing the field is to further elucidate the mechanisms of allogeneic T cell rejection in the human immune system.
Enlarging the toolset beyond T cells
Beyond T cells, NK cells offer a promising alternative approach for an allogenic cellular immunotherapy. NK cells are part of the innate lymphoid cell lineage and play a critical role in immune defense against tumors and pathogens (46). They can be adoptively transferred into patients as an allogeneic product without the need for gene editing to avoid the high risk of GvHD. Further if transduced with a CAR directed at a tumor-specific antigen, NK cells can attack tumor cells via antigen-dependent cytotoxicity, as well as through their activating NK receptors and thus are an attractive complement to allogeneic CAR T cell therapy. NK cells are generally less abundant in peripheral blood compared to T cells, posing challenges to clinical scale manufacturing. Cord blood, induced pluripotent stems cells (iPSCs), embryonic stem cells (ECs) and NK-92, an NK cell line, have been used as alternate sources of NK cells for adoptive cell transfer (ACT) (47). In addition, the proliferative capacity and persistence of NK cells is comparatively less than T cells, however treatment with cytokines such as IL-2, IL-15 and IL-21 can extend NK lifespan. In addition, the risk of toxicities like cytokine release syndrome or neurotoxicity may be reduced in comparison to CAR T cell therapies, since activated NK cells secrete low amounts of IL-6 or IL-1β (46). Currently, approximately 10 clinical trials in clinicaltrials.gov are recruiting patients for the treatment of cancers including various B-cell malignancies, Multiple Myeloma, AML, and solid tumors with CAR NK cell products. Notably, in a recent phase I/II clinical trial, patients with refractory non-Hodgkin’s lymphoma (NHL) or CLL were administered CD19 CAR-NK cells armored with IL-15 (48). 73% of patients experienced objective responses, however the durability of response could not be assessed because other therapies were delivered as early as 30 days after the infusion of CAR-NK cells (48). Patients experienced high-grade, transient myelotoxicity which may have been caused by the lymphodepleting chemotherapy, but not CRS, neurotoxicity, or GvHD. The product was manufactured from HLA-mismatched, and when possible, killer immunoglobulin-like receptor (KIR) ligand mismatched cord blood and was freshly infused after manufacturing (48). In the future, advancement of clinical-grade CAR-NK products that retain efficacy and persistence after cryopreservation and thaw will realize ‘off-the-shelf’ products for CAR-NK therapy, increasing treatment options for patients (49).
Next-generation CAR T cell therapies for the treatment of solid tumors
Currently, the efficacy of CAR T cell therapies targeting solid tumors is restrained by the quantity, persistence, and functionality of transferred T cells and by the heterogeneity and immunosuppressive nature of the tumor microenvironment (TME) (50–69). Addressing these challenges is critical to future successes in ACT. Dose limiting toxicities (DLTs) associated with CAR T cell recognition of low level of target expression on normal tissue (‘on-target off-tumor’), and toxicities associated with strong CAR T cell efficacy pose barriers that limit therapeutic dosing of CAR T cells. For example, several ongoing clinical trials designed to treat mesothelin-expressing tumors have highlighted an association between anti-tumor efficacy and lung associated dose limiting toxicity. Interim results from the Phase 1 portion of a clinical trial administering anti-mesothelin directed TCR-T cells, gavo-cel (NCT03907852) for the treatment of mesothelin-expressing solid tumors reveal that, following lymphodepletion, patients infused with gavo-cel achieved a 25% objective response rate (ORR), however dose limiting pulmonary toxicities were reported. Likewise, in a phase 1 dose escalation trial targeting mesothelin-expressing refractory malignant cancers a mesothelioma patient experienced grade 5 (GR5) respiratory failure after infusion of fully human mesothelin-directed CAR T cells at a dose of 1–3×108 cells/m2 (NCT03054298). In both studies, these GR5 toxicities prompted a dose de-escalation and no serious events were reported at the lower dose. In patients with malignant pleural disease this lung-associated toxicity was bypassed by regional delivery of anti-mesothelin CAR T cells (0.3– 60 ×106 cells/kg) followed by PD-1 blockade resulting in SD for ≥ 6 months in 8/18 patients with 2 patients exhibiting a complete metabolic response on PET scan (NCT02414269) (70). Regional delivery of CAR T cells targeting mesothelin-expressing cancers is also under evaluation at other centers (NCT03054298; NCT03323944).
Further, when the efficacy of CAR T cell therapies targeting solid tumors reaches a therapeutic level, patients can experience severe toxicities. In particular, significant declines in prostate-specific antigen (PSA) have been observed in patients with Castration-Resistant Metastatic Prostate Cancer (CRPC) treated with Prostate specific membrane antigen (PSMA)-directed CAR T cells (NCT04249947) or TGFβ-insensitive PSMA-targeted CAR T cells (NCT03089203). In both studies, increases in CAR T cell doses led to significant anti-tumor activity, accompanied by patient mortality. The cause(s) of patient deaths are currently unclear, but may be associated with Macrophage Activation Syndrome (MAS) or immune effector cell-associated neurotoxicity (ICANS) (71,72), although ‘on-target off-tumor’ toxicity is theoretically possible. It is conceivable that new tocilizumab-like drugs will be needed to mitigate ICANS and MAS toxicity associated with robust CAR T cell therapy to allow solid tumor patients to tolerate an effective dose (73).
Another approach to bypassing on-target off-tumor related toxicities caused by aberrant CAR T cell destruction of normal tissues is the use of dual antigen-sensing strategies that permit more precise immune recognition of tumors. One such example is the synNotch receptor (Fig. 1c). This synthetic circuit is constructed to allow T cells to integrate combinatorial antigen expression data to discriminate normal and tumors cells. In the AND-gated version of a two-receptor circuit, activation of one receptor incudes the transcription of a second receptor (CAR or TCR), killing tumor cells that simultaneously express both antigens and sparing normal tissue that expresses only one antigen (74,75). In contrast, NOT-gated SynNotch circuits are designed to minimize CAR T cell recognition of normal cells by designing a circuit that combines a CAR recognizing the shared target antigen and an inhibitory CAR preventing killing when the second antigen, present only on normal bystander cells and not tumor, is present. Careful selection of antigen pairs specific to the CAR circuit and target disease permits the construction of CAR T cells with limited ‘on-target off-tumor’ toxicity in vitro and in vivo mouse models. The recent development of fully humanized circuits termed Synthetic Intramembrane Proteolysis Receptors (SNIPRs) that mirror the function of first-generation SynNotch receptors supports the clinical translation of CAR T cells with the capacity to sense multiple combinatorial antigens on a cancer cell by reducing potential immunogenicity. However, this approach does not abrogate the risk of tumor relapse due to antigen escape, including antigen loss or downregulation.
Unlike CAR T cells therapy directed against CD19, adoptively transferred CAR T cells directed against solid tumors undergo limited homeostatic expansion in the blood, and encounter tumor antigen in an immunosuppressive TME where the number of CAR T cells may be insufficient to eradicate disease (62). Local administration of CAR T cells is being investigated to address the limited number of CAR T cells reaching and expanding in the tumor (67,70,76,77). Additionally, in vivo antigen-specific CAR T cell expansion may enable dosing at sufficient levels to promote anti-tumor responses. One promising synthetic approach to selectively expand CAR T cells and omit systemic toxicity of native IL-2 administration is the engineering of IL-2 cytokine receptor orthogonal (ortho) pairs which interact with each other but not their native cytokine receptor pairs (27,78) (Fig. 1b). Nanoparticle RNA and peptide vaccines offer an alternate approach to enhance the in vivo expansion of CAR T cells targeting solid tumors. Both CAR antigen-encoding RNA-lipoplexes (RNA-LPX), and CAR T ligands bound to albumin-binding phospholipid polymers (amph-ligand) vaccines deliver CAR antigen to lymph nodes, decorating the surfaces of antigen-presenting cells (APCs) with the CAR ligand (79,80). Currently a Phase I/IIa dose escalation trial is open to evaluate the safety and preliminary efficacy of CLDN6 CAR T +/− CLDN6 RNA-LPX in patients with CLDN6-positive relapsed or refractory advanced solid tumors (NCT04503278).
When CAR T cells enter an immunosuppressive TME they acquire a dysfunctional or exhausted T cell state which limits the therapeutic efficacy of CAR T cells targeting solid cancers (81). One mechanism driving the dysfunction of CAR T cells in the solid tumor microenvironment is plasticity of the CD8 T cell state, where continuous antigen exposure promotes a CD8+ T-to-NK like T-cell transition (82). The identification and CRISPR-Cas9 mediated knock out of genes that restrain tumor immunity has enabled the production of next-generation CAR T cells (42,82–100). However, the extent to which the knockout of individual genes can prevent T cell dysfunction is unclear. Recent studies suggest that T cells undergo a transition from a plastic dysfunctional state in which therapeutic re-programmability is possible to a fixed dysfunction state that is resistant to reprograming (101). In particular, T cells exposed to chronic antigen stimulation are not able to fully recover after tumor clearance and acquire epigenetic changes or scars that can limit future immune responses (102,103). These data highlight the potential of therapies promoting epigenetic remodeling to permit sustained CAR T cell functionality. The possible therapeutic utility of epigenetic remodeling is further illustrated by mechanistic studies from a single CLL patient treated with CAR T cells targeting CD19 (104). Lentiviral integration of the CAR transgene disrupted the patient’s methylcytosine dioxygenease TET2 gene, resulting in enhanced potency. Another strategy aimed at maintaining CAR T functionality and limiting dysfunction associated epigenetic remodeling is to modulate CAR expression or antigen exposure (105,106). Unlike CAR T cells exhibiting constitutive CAR expression, regulated expression of the CAR by mechanisms such as transient rest, cessation of receptor signaling, or preventing CAR-mediated tonic signaling through synNotch-controlled expression (107,108) allows CAR T cells to avert exhaustion and maintain anti-tumor activity. In summary, future strategies aimed at mitigating the immunosuppressive effects of the TME will need to be multifactorial to target the complexity of cancer.
Double hit: CAR T cells in combination with therapies targeting the TME
Engineered viruses, which specifically replicate in tumor cells without infecting healthy cells, have emerged as effective oncolytic agents in recent years. Although the molecular and cellular mechanisms are not fully understood, oncolytic viruses as a monotherapy are known to remodel the immunosuppressive TME through, e.g., inflammatory effects at injection site, but also to induce systemic innate and adaptive anti-tumor immunity (109). These viruses can, however, also be harnessed for combinational approaches in cancer immunotherapy due to their ability to specifically transduce cancer cells with immunomodulatory- or tumor suppressor genes (Fig. 1d). In 2015, the first therapy of this kind, talimogene laherparepvec (T-VEC), a herpes simplex virus (HSV) type 1-derived oncolytic agent, was approved (110). Response rates were further enhanced when T-VEC was combined with immune checkpoint blockade (ICB) antibodies, while increased side effects were not observed (111). Mechanistically, oncolytic viruses can improve clinical responses to ICB by influencing the presence of tumor infiltrating lymphocytes (TILs) within the TME, as well as the tumor mutational burden, which are known predictors of response to ICB (112,113). Clinical trials testing the safety and feasibility of CAR T cells combined with oncolytic viruses are currently ongoing (NCT03740256 and NCT05057715).
A novel approach to modify the TME is the use of CAR-engineered macrophages (Fig. 2), which have demonstrated efficacy in animal models (114). Macrophages are part of the innate immune system and actively participate in the immune response through phagocytosis and clearance of cellular debris (115). In addition, they are known to be potent antigen-presenting cells inducing innate and adaptive immune responses. Based on these promising preclinical data, a first-in-human clinical trial on CAR-macrophages targeting HER2-expressing tumors was recently started (NCT04660929) and several others have been announced. A limitation on the use of CAR-macrophages is their terminally differentiated state, as unlike CAR T, they do not proliferate after encountering their tumor target. While macrophages are difficult to transduce with clinically proven lentiviral or retroviral vectors, CAR-macrophages can be efficiently produced with clinically available Ad535 vectors, which are being used in the ongoing trials.
Fig. 2: Broadening product availability.
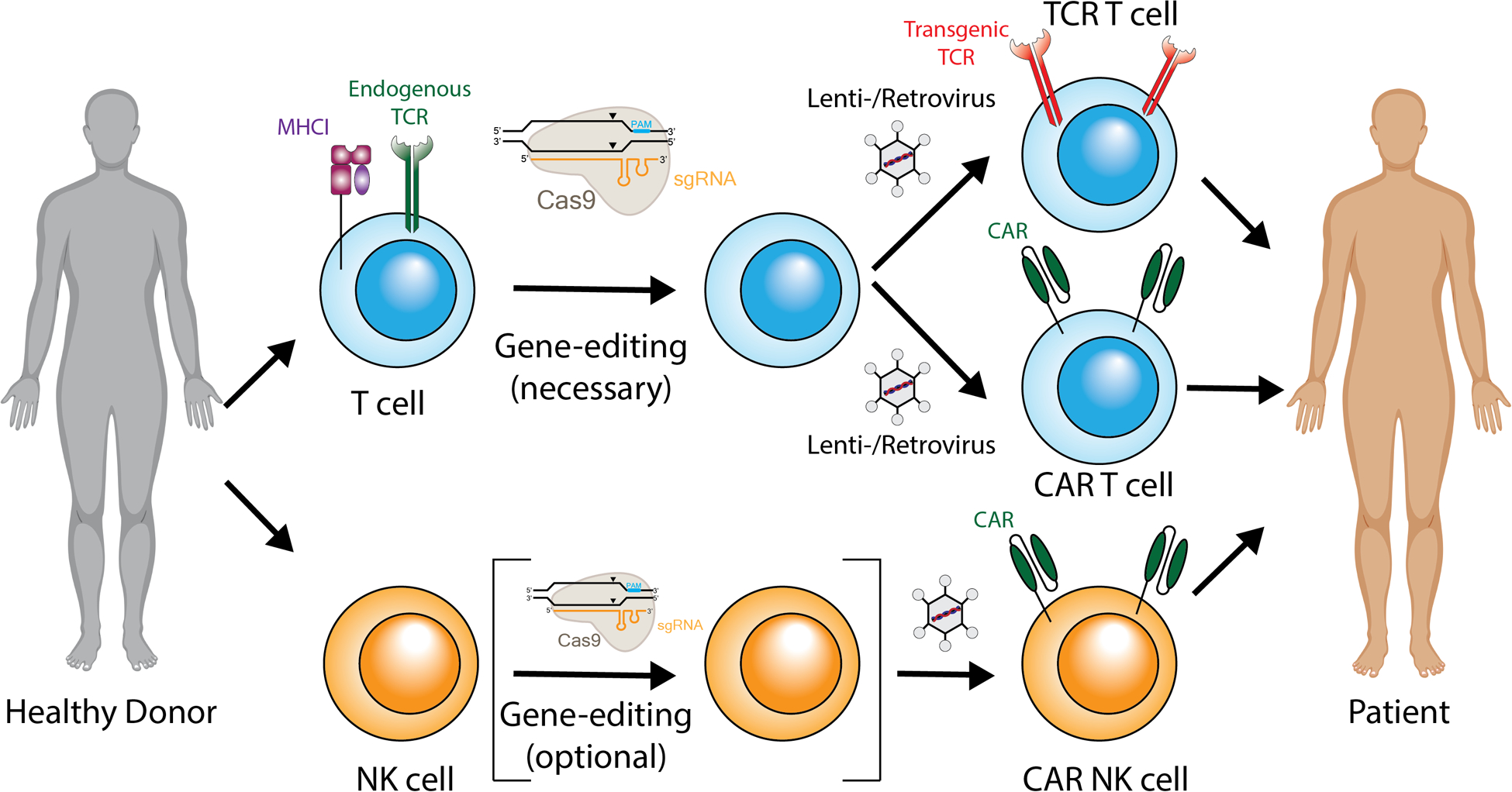
The use of gene-edited allogeneic (‘off-the-shelf’) TCR/CAR T cells could provide immediate availability of cryopreserved cells, increase product quality, and decrease costs by using an industrial process. Beyond T cells, natural killer (NK) cells could be used as an allogeneic cellular immunotherapy.
Combining stimulator of interferon genes (STING) agonists with CAR T cells could be another strategy to modify TME and to activate the innate/adaptive immunity, thereby improving overall CAR T cell response. STING agonists trigger the expression of type I interferons and inflammatory genes, which leads to the activation of the innate immune defense and to the induction of the adaptive immune response. In a recently published preclinical study, the STING agonist DMXAA promoted CAR T cell trafficking and persistence in a solid tumor model through alterations in the balance of innate and adaptive immunity as well as cytokine milieu within the TME (116). In another study, CAR T cells producing the RNA agonist RN7SL1 not only enhanced CAR T cell function and expansion, but also restricted the development of immune-suppressive cells and activated anti-tumor endogenous immunity. This resulted in control of different tumors, including tumors who had lost the CAR-targeted antigen (117).
Conclusion
Advances in next generation CAR T cell therapies such as exploiting innovations in synthetic biology and orthogonal receptors to create novel therapeutic signaling networks and utilizing combinatorial treatment approaches to increase response will broaden the impact of CAR T cell therapy in the next decade. Many of the strategies discussed in this perspective are ready to be tested in clinical trials and have the potential to improve patient responses. Additionally, as the field’s mechanistic and molecular understanding of CAR T cell resistance expands, these new concepts will increase therapeutic possibilities. The combinational use of different strategies described in this article hold promise for superior CAR T cell therapies designed to expand patients’ options for cancer treatment.
Significance:
Next generation CAR T cells will incorporate advances in gene engineering and synthetic biology to enhance functionality and persistence, and reduce treatment associated toxicities. The combination of autologous CAR T cells with various allogeneic cell treatment strategies designed to target the immunosuppressive tumor microenvironment will broaden the impact of future CAR T cell therapies.
Acknowledgments:
The authors thank Dr. Neil Sheppard for helpful discussions.
Financial support (including grants):
C.H.J is supported by NIH grants; P01CA214278 and U54CA244711. R.M.Y. and C.H.J. are supported by SU2C-Lustgarten Foundation Translational Cancer Research Team Grant and the Parker Institute for Cancer Immunotherapy. U.U. is supported by a Mildred-Scheel-Postdoctoral Fellowship of the German Cancer Aid.
Footnotes
Conflicts of Interest disclosure statement:
R.M.Y. and C.H.J. are inventors on patents and/or patent applications licensed to Novartis Institutes of Biomedical Research and receive license revenue from such licenses. R.M.Y. is an inventor on patents and/or patent applications licensed to Tmunity Therapeutics and receives license revenue from such licenses. C.H.J is a scientific founder of Tmunity Therapeutics and Capstan Therapeutics, and is a member of the scientific advisory boards of AC Immune, BluesphereBio, Cabaletta, Carisma, Cartography, Cellares, Cellcarta, Celldex, Danaher, Decheng, ImmuneSensor, Poseida, Verismo, Viracta, WIRB-Copernicus and Ziopharm. N.W.E., U.U., and N.W. have no conflicts of interest to declare.
References:
- 1.June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med 2018;379(1):64–73 doi 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schultz LM, Baggott C, Prabhu S, Pacenta HL, Phillips CL, Rossoff J, et al. Disease Burden Affects Outcomes in Pediatric and Young Adult B-Cell Lymphoblastic Leukemia After Commercial Tisagenlecleucel: A Pediatric Real-World Chimeric Antigen Receptor Consortium Report. J Clin Oncol 2021:JCO2003585 doi 10.1200/JCO.20.03585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med 2018;378(5):439–48 doi 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2018;24(1):20–8 doi 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med 2020;26(10):1569–75 doi 10.1038/s41591-020-1081-3. [DOI] [PubMed] [Google Scholar]
- 6.Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med 2021;27(8):1419–31 doi 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, et al. Preclinical Development of Bivalent Chimeric Antigen Receptors Targeting Both CD19 and CD22. Mol Ther Oncolytics 2018;11:127–37 doi 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res 2015;75(17):3596–607 doi 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov 2020;10(5):702–23 doi 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen XK M; Lakhani A; Meng X; Salvestrini E; Chen l.C.; Shafer A; Alag A; Ding Y; Nicolaou D; Park JO; Chen YY. Rational Tuning of CAR Tonic Signaling Yields Superior T-Cell Therapy for Cancer. bioRxiv 2020. doi 10.1101/2020.10.01.322990. [DOI] [Google Scholar]
- 11.Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell 2022;40(1):53–69 e9 doi 10.1016/j.ccell.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med 2019;25(9):1408–14 doi 10.1038/s41591-019-0549-5. [DOI] [PubMed] [Google Scholar]
- 13.Mikkilineni L, Kochenderfer JN. CAR T cell therapies for patients with multiple myeloma. Nat Rev Clin Oncol 2021;18(2):71–84 doi 10.1038/s41571-020-0427-6. [DOI] [PubMed] [Google Scholar]
- 14.Pont MJ, Hill T, Cole GO, Abbott JJ, Kelliher J, Salter AI, et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019;134(19):1585–97 doi 10.1182/blood.2019000050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plaks V, Rossi JM, Chou J, Wang L, Poddar S, Han G, et al. CD19 target evasion as a mechanism of relapse in large B-cell lymphoma treated with axicabtagene ciloleucel. Blood 2021;138(12):1081–5 doi 10.1182/blood.2021010930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dean EA, Mhaskar RS, Lu H, Mousa MS, Krivenko GS, Lazaryan A, et al. High metabolic tumor volume is associated with decreased efficacy of axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv 2020;4(14):3268–76 doi 10.1182/bloodadvances.2020001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jain MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, et al. Tumor interferon signaling and suppressive myeloid cells are associated with CAR T-cell failure in large B-cell lymphoma. Blood 2021;137(19):2621–33 doi 10.1182/blood.2020007445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frey NV, Gill S, Hexner EO, Schuster S, Nasta S, Loren A, et al. Long-Term Outcomes From a Randomized Dose Optimization Study of Chimeric Antigen Receptor Modified T Cells in Relapsed Chronic Lymphocytic Leukemia. Journal of Clinical Oncology 2020;38(25):2862–71 doi 10.1200/jco.19.03237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Bruggen JAC, Martens AWJ, Fraietta JA, Hofland T, Tonino SH, Eldering E, et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8(+) T cells and impede CAR T-cell efficacy. Blood 2019;134(1):44–58 doi 10.1182/blood.2018885863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med 2018;24(5):563–71 doi 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garfall AL, Dancy EK, Cohen AD, Hwang WT, Fraietta JA, Davis MM, et al. T-cell phenotypes associated with effective CAR T-cell therapy in postinduction vs relapsed multiple myeloma. Blood Adv 2019;3(19):2812–5 doi 10.1182/bloodadvances.2019000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neelapu SS, Dickinson M, Ulrickson ML, Oluwole OO, Herrera AF, Thieblemont C, et al. Interim Analysis of ZUMA-12: A Phase 2 Study of Axicabtagene Ciloleucel (Axi-Cel) as First-Line Therapy in Patients (Pts) With High-Risk Large B Cell Lymphoma (LBCL). Blood 2020;136(Supplement 1):49- doi 10.1182/blood-2020-134449. [DOI] [Google Scholar]
- 23.Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov 2020;19(3):185–99 doi 10.1038/s41573-019-0051-2. [DOI] [PubMed] [Google Scholar]
- 24.Hu B, Ren J, Luo Y, Keith B, Young RM, Scholler J, et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep 2017;20(13):3025–33 doi 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol Ther Oncolytics 2018;8:41–51 doi 10.1016/j.omto.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep 2017;21(11):3205–19 doi 10.1016/j.celrep.2017.11.063. [DOI] [PubMed] [Google Scholar]
- 27.Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 2020;583(7817):609–14 doi 10.1038/s41586-020-2422-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lange S, Sand LGL, Bell M, Patil SL, Langfitt D, Gottschalk S. A Chimeric GM-CSF/IL18 Receptor to Sustain CAR T-cell Function. Cancer Discov 2021;11(7):1661–71 doi 10.1158/2159-8290.CD-20-0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015;29(8):1637–47 doi 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018;173(6):1439–53 e19 doi 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A 2019;116(24):11978–87 doi 10.1073/pnas.1819992116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018;32(9):1970–83 doi 10.1038/s41375-018-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Georgiadis C, Rasaiyaah J, Gkazi SA, Preece R, Etuk A, Christi A, et al. Base-edited CAR T cells for combinational therapy against T cell malignancies. Leukemia 2021;35(12):3466–81 doi 10.1038/s41375-021-01282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anzalone AV, Koblan LW, Liu DR. Genome editing with CRISPR–Cas nucleases, base editors, transposases and prime editors. Nature Biotechnology 2020;38(7):824–44 doi 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 35.Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol 2019;19(11):665–74 doi 10.1038/s41577-019-0221-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. The Lancet 2020;396(10266):1885–94 doi 10.1016/s0140-6736(20)32334-5. [DOI] [PubMed] [Google Scholar]
- 37.Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology 2018;36(8):765–71 doi 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haapaniemi E, Botla S, Persson J, Schmierer B, Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 2018;24(7):927–30 doi 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 39.Ihry RJ, Worringer KA, Salick MR, Frias E, Ho D, Theriault K, et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 2018;24(7):939–46 doi 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 40.Webber BR, Lonetree CL, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun 2019;10(1):5222 doi 10.1038/s41467-019-13007-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petri K, Zhang W, Ma J, Schmidts A, Lee H, Horng JE, et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat Biotechnol 2021;40(2):189–93 doi 10.1038/s41587-021-00901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020;367(6481) doi 10.1126/science.aba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davenport C, Haile A, Kumar V, Bennett M. Hybrid and allogeneic resistance to T cell grafts mediated by murine NK and CD8+ T cells. J Immunol 1995;154(6):2568–77. [PubMed] [Google Scholar]
- 44.Liu W, Xiao X, Demirci G, Madsen J, Li XC. Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol 2012;188(6):2703–11 doi 10.4049/jimmunol.1102997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kanda Y, Takeuchi A, Ozawa M, Kurosawa Y, Kawamura T, Bogdanova D, et al. Visualizing the Rapid and Dynamic Elimination of Allogeneic T Cells in Secondary Lymphoid Organs. J Immunol 2018;201(3):1062–72 doi 10.4049/jimmunol.1700219. [DOI] [PubMed] [Google Scholar]
- 46.Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov 2020;19(3):200–18 doi 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- 47.Zhang C, Hu Y, Xiao W, Tian Z. Chimeric antigen receptor- and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell Mol Immunol 2021;18(9):2083–100 doi 10.1038/s41423-021-00732-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med 2020;382(6):545–53 doi 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kundu S, Gurney M, O’Dwyer M. Generating natural killer cells for adoptive transfer: expanding horizons. Cytotherapy 2021;23(7):559–66 doi 10.1016/j.jcyt.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 50.Majzner RG, Ramakrishna S, Yeom KW, Patel S, Chinnasamy H, Schultz LM, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature 2022. doi 10.1038/s41586-022-04489-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hegde M, Joseph SK, Pashankar F, DeRenzo C, Sanber K, Navai S, et al. Tumor response and endogenous immune reactivity after administration of HER2 CAR T cells in a child with metastatic rhabdomyosarcoma. Nat Commun 2020;11(1):3549 doi 10.1038/s41467-020-17175-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi D, Shi Y, Kaseb AO, Qi X, Zhang Y, Chi J, et al. Chimeric Antigen Receptor-Glypican-3 T-Cell Therapy for Advanced Hepatocellular Carcinoma: Results of Phase I Trials. Clin Cancer Res 2020;26(15):3979–89 doi 10.1158/1078-0432.CCR-19-3259. [DOI] [PubMed] [Google Scholar]
- 53.Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018;155(1):29–32 doi 10.1053/j.gastro.2018.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol 2017;3(8):1094–101 doi 10.1001/jamaoncol.2017.0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med 2016;375(26):2561–9 doi 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J Immunother 2019;42(4):126–35 doi 10.1097/CJI.0000000000000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther 2019;27(11):1919–29 doi 10.1016/j.ymthe.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol Ther 2017;25(9):2214–24 doi 10.1016/j.ymthe.2017.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: A phase I clinical trial. Cytotherapy 2020;22(10):573–80 doi 10.1016/j.jcyt.2020.04.088. [DOI] [PubMed] [Google Scholar]
- 60.Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011;118(23):6050–6 doi 10.1182/blood-2011-05-354449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 2010;18(4):843–51 doi 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9(399) doi 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol 2021;14(1):118 doi 10.1186/s13045-021-01128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stuber T, Monjezi R, Wallstabe L, Kuhnemundt J, Nietzer SL, Dandekar G, et al. Inhibition of TGF-beta-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer 2020;8(1) doi 10.1136/jitc-2020-000676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang X, Liu F, Liu Z, Cao Y, Zhang Z, Wang Y, et al. Bioactivity and safety of B7-H3-targeted chimeric antigen receptor T cells against anaplastic meningioma. Clin Transl Immunology 2020;9(6):e1137 doi 10.1002/cti2.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 2017;66(11):1425–36 doi 10.1007/s00262-017-2034-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Kunkele A, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med 2021;27(9):1544–52 doi 10.1038/s41591-021-01404-8. [DOI] [PubMed] [Google Scholar]
- 68.Wang Y, Chen M, Wu Z, Tong C, Dai H, Guo Y, et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology 2018;7(7):e1440169 doi 10.1080/2162402X.2018.1440169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res 2017;5(12):1152–61 doi 10.1158/2326-6066.CIR-17-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adusumilli PS, Zauderer MG, Riviere I, Solomon SB, Rusch VW, O’Cearbhaill RE, et al. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti-PD-1 Agent Pembrolizumab. Cancer Discov 2021;11(11):2748–63 doi 10.1158/2159-8290.CD-21-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sandler RD, Tattersall RS, Schoemans H, Greco R, Badoglio M, Labopin M, et al. Diagnosis and Management of Secondary HLH/MAS Following HSCT and CAR-T Cell Therapy in Adults; A Review of the Literature and a Survey of Practice Within EBMT Centres on Behalf of the Autoimmune Diseases Working Party (ADWP) and Transplant Complications Working Party (TCWP). Front Immunol 2020;11:524 doi 10.3389/fimmu.2020.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sheth VS, Gauthier J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant 2021;56(3):552–66 doi 10.1038/s41409-020-01134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med 2020;383(23):2255–73 doi 10.1056/NEJMra2026131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 2016;164(4):780–91 doi 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 2016;164(4):770–9 doi 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Donovan LK, Delaidelli A, Joseph SK, Bielamowicz K, Fousek K, Holgado BL, et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat Med 2020;26(5):720–31 doi 10.1038/s41591-020-0827-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 2020;26(5):712–9 doi 10.1038/s41591-020-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science 2018;359(6379):1037–42 doi 10.1126/science.aar3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020;367(6476):446–53 doi 10.1126/science.aay5967. [DOI] [PubMed] [Google Scholar]
- 80.Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 2019;365(6449):162–8 doi 10.1126/science.aav8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell 2018;33(4):547–62 doi 10.1016/j.ccell.2018.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021;184(25):6081–100 e26 doi 10.1016/j.cell.2021.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmidt R, Steinhart Z, Layeghi M, Freimer JW, Bueno R, Nguyen VQ, et al. CRISPR activation and interference screens decode stimulation responses in primary human T cells. Science 2022;375(6580):eabj4008 doi 10.1126/science.abj4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 2019;576(7787):471–6 doi 10.1038/s41586-019-1821-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019;178(5):1189–204 e23 doi 10.1016/j.cell.2019.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 2018;175(7):1958–71 e15 doi 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen J, Lopez-Moyado IF, Seo H, Lio CJ, Hempleman LJ, Sekiya T, et al. NR4A transcription factors limit CAR T cell function in solid tumours. Nature 2019;567(7749):530–4 doi 10.1038/s41586-019-0985-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Behrens G, Edelmann SL, Raj T, Kronbeck N, Monecke T, Davydova E, et al. Disrupting Roquin-1 interaction with Regnase-1 induces autoimmunity and enhances antitumor responses. Nat Immunol 2021;22(12):1563–76 doi 10.1038/s41590-021-01064-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 2020;37(6):818–33 e9 doi 10.1016/j.ccell.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res 2018;78(16):4692–703 doi 10.1158/0008-5472.CAN-18-0030. [DOI] [PubMed] [Google Scholar]
- 91.LaFleur MW, Nguyen TH, Coxe MA, Miller BC, Yates KB, Gillis JE, et al. PTPN2 regulates the generation of exhausted CD8(+) T cell subpopulations and restrains tumor immunity. Nat Immunol 2019;20(10):1335–47 doi 10.1038/s41590-019-0480-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019;576(7786):293–300 doi 10.1038/s41586-019-1805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res 2017;23(9):2255–66 doi 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 2020;181(3):728–44 e21 doi 10.1016/j.cell.2020.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 2017;7(1):737 doi 10.1038/s41598-017-00462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Seo H, Chen J, Gonzalez-Avalos E, Samaniego-Castruita D, Das A, Wang YH, et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8(+) T cell exhaustion. Proc Natl Acad Sci U S A 2019;116(25):12410–5 doi 10.1073/pnas.1905675116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020;5(4) doi 10.1172/jci.insight.133977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wiede F, Lu KH, Du X, Liang S, Hochheiser K, Dodd GT, et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J 2020;39(2):e103637 doi 10.15252/embj.2019103637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med 2017;11(4):554–62 doi 10.1007/s11684-017-0543-6. [DOI] [PubMed] [Google Scholar]
- 100.Zhao H, Liu Y, Wang L, Jin G, Zhao X, Xu J, et al. Genome-wide fitness gene identification reveals Roquin as a potent suppressor of CD8 T cell expansion and anti-tumor immunity. Cell Rep 2021;37(10):110083 doi 10.1016/j.celrep.2021.110083. [DOI] [PubMed] [Google Scholar]
- 101.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017;545(7655):452–6 doi 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yates KB, Tonnerre P, Martin GE, Gerdemann U, Al Abosy R, Comstock DE, et al. Epigenetic scars of CD8(+) T cell exhaustion persist after cure of chronic infection in humans. Nat Immunol 2021;22(8):1020–9 doi 10.1038/s41590-021-00979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Abdel-Hakeem MS, Manne S, Beltra JC, Stelekati E, Chen Z, Nzingha K, et al. Epigenetic scarring of exhausted T cells hinders memory differentiation upon eliminating chronic antigenic stimulation. Nat Immunol 2021;22(8):1008–19 doi 10.1038/s41590-021-00975-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018;558(7709):307–12 doi 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 2021;372(6537) doi 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017;543(7643):113–7 doi 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med 2021;13(591) doi 10.1126/scitranslmed.abd8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med 2021;13(591) doi 10.1126/scitranslmed.abe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015;14(9):642–62 doi 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J Clin Oncol 2015;33(25):2780–8 doi 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 111.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017;170(6):1109–19 e10 doi 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014;515(7528):563–7 doi 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med 2017;377(25):2500–1 doi 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol 2020;38(8):947–53 doi 10.1038/s41587-020-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther 2021;6(1):127 doi 10.1038/s41392-021-00506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu N, Palmer DC, Robeson AC, Shou P, Bommiasamy H, Laurie SJ, et al. STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J Exp Med 2021;218(2) doi 10.1084/jem.20200844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Johnson LR, Lee DY, Eacret JS, Ye D, June CH, Minn AJ. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell 2021;184(19):4981–95 e14 doi 10.1016/j.cell.2021.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]