

Abstract
Mechanically tunable hydrogels are attractive platforms for three-dimensional cell culture, as hydrogel stiffness plays an important role in cell behavior. Traditionally, hydrogel stiffness has been controlled through altering either the polymer concentration or the stoichiometry between crosslinker reactive groups. Here, we present an alternative strategy based upon tuning the hydrophilicity of an elastin-like protein (ELP). ELPs undergo a phase transition that leads to protein aggregation at increasing temperatures. We hypothesize that increasing this transition temperature through bioconjugation with azide-containing molecules of increasing hydrophilicity will allow direct control of the resulting gel stiffness by making the crosslinking groups more accessible. These azide-modified ELPs are crosslinked into hydrogels with bicyclononyne-modified hyaluronic acid (HA-BCN) using bioorthogonal, click chemistry, resulting in hydrogels with tunable storage moduli (100–1,000 Pa). Human mesenchymal stromal cells, human umbilical vein endothelial cells, and human neural progenitor cells are all observed to alter their cell morphology when encapsulated within hydrogels of varying stiffness. Taken together, we demonstrate the use of protein hydrophilicity as a lever to tune hydrogel mechanical properties. These hydrogels have tunable moduli over a stiffness range relevant to soft tissues, support the viability of encapsulated cells, and modify cell spreading as a consequence of gel stiffness.
Keywords: elastin-like protein, hyaluronic acid, three-dimensional culture, click chemistry, lower critical solution temperature
Graphical Abstract
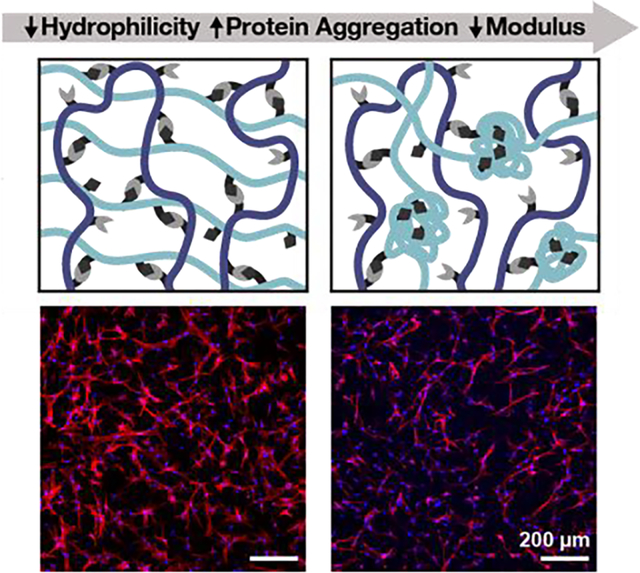
Matrix stiffness is a critical cue that affects cell phenotype and hence is an important variable in biomaterials design. We demonstrate a novel strategy to control the stiffness of protein-engineered biomaterials by tuning protein hydrophilicity. This strategy allowed synthesis of gels with storage moduli ranging from 100–1,000 Pa, resulting in variable cell spreading for a variety of cell types.
1. Introduction
Hydrogels are commonly used as biomimetic scaffolds for three-dimensional (3D) cell encapsulation and tissue engineering applications.[1] Because they are able to recapitulate key aspects of the biochemical and biomechanical microenvironment, hydrogels are an attractive platform for studying cell–matrix interactions in a controlled manner.[2, 3] A variety of natural and synthetic polymers have been utilized as cell-encapsulating materials, but potential limitations exist for both material classes. Natural biomaterials, such as collagen and reconstituted basement membranes, are degradable and contain natural cell-interactive components, but they can suffer batch-to-batch variability and exhibit limited tunability of mechanical properties.[4] On the other hand, synthetic materials like polyethylene glycol (PEG) offer well-defined, highly tunable systems, but they are bioinert and must be functionalized with bioactive molecules to permit cellular interaction.[5–7] Recombinant protein-based biomaterials offer an alternative that is reproducible, cell-interactive, and facilitates the independent tuning of biochemical and biomechanical properties, emerging as an advantageous 3D culture platform for a variety of cell types.[8–10]
Elastin-like proteins (ELPs) are a class of mechanically tunable, protein-engineered materials well-suited for studying encapsulated cell response to matrix-derived signaling cues.[11] Mechanically tunable hydrogels are particularly attractive for 3D cell culture, as hydrogel stiffness has been shown to play important roles in cell spreading, proliferation, and differentiation.[12–15] Traditionally, ELP hydrogel stiffness has been controlled through altering either the polymer concentration or the stoichiometry between crosslinker reactive groups.[9, 16, 17] While these strategies have been used successfully to modulate hydrogel stiffness, it is often technically cumbersome to modify mechanical properties without impacting other material properties such as adhesive ligand density or mesh size. Therefore, it would be beneficial to explore additional cell-compatible strategies for tuning mechanical properties of ELP hydrogels. Here, we present an alternative approach to modulating hydrogel stiffness that is based upon tuning ELP hydrophilicity.
ELPs exhibit lower critical solution temperature (LCST) behavior, defined as formation of protein-rich aggregates at temperatures above the transition temperature (Tt).[18–20] ELPs with increased hydrophilicity will have a higher Tt. Several approaches have been used to tune the Tt of ELPs, including changing the amino acid sequence,[21, 22] altering the pH of the gel precursor solution,[23] altering the pH at the time of gel crosslinking,[24] and adding salts or surfactants to alter the ionic strength.[25, 26] While a range of hydrogel stiffness is achievable through these approaches, materials for cell encapsulation must necessarily be cell compatible. Since cells are influenced by signaling from their surrounding microenvironment, alterations in pH and ionic strength that deviate from physiological conditions may affect cell viability and thus may not be suitable approaches for studying cell mechanobiology. While modifying amino acid residues offers a potential solution, this approach requires bacterial transformation, expression, and purification of multiple different proteins, increasing the complexity required for such studies. An ideal strategy would therefore involve tuning the hydrophilicity of ELP while maintaining physiological culture conditions and without requiring redesign of the amino acid sequence.
In this work, we have designed a bio-orthogonal, covalently-crosslinked, protein-engineered hydrogel wherein the hydrophilicity is tuned by the conjugation of pendant hydrophilic moieties. Bioconjugation of the ELP with azide molecules containing varying numbers of ethylene glycol repeats results in a shift in the Tt as the LCST behavior is perturbed. To initiate gelation, the different azide-functionalized ELPs are mixed with bicyclononyne-functionalized hyaluronic acid (HA) and allowed to crosslink via a strain-promoted azide-alkyne cycloaddition (SPAAC) reaction. While we have previously reported a hyaluronan–elastin-like protein (HELP) matrix formed via hydrazone-mediated crosslinking,[17, 27] SPAAC crosslinking presents a bio-orthogonal approach for creating a cell compatible hydrogel platform, termed HELP-SPAAC hydrogel. Increasing the hydrophilicity and the Tt of the azide-functionalized ELPs was found to offer a simple, cell-compatible strategy to tune HELP-SPAAC gel stiffness. These gels supported the viability and phenotypic maintenance of encapsulated human mesenchymal stromal cells (hMSCs), human umbilical vein endothelial cells (HUVECs), and human induced pluripotent stem cell-derived neural progenitor cells (hNPCs). Furthermore, we observed that tuning gel mechanics in this manner resulted in cell type-dependent morphological differences for all three cell types, supporting the future use of these materials in cell mechanobiology studies.
2. Results and Discussion
2.1. Synthesis and Characterization of Azide-Modified ELP
Recombinant proteins are uniquely suited biomaterials for 3D cell culture as they combine desirable traits from both natural and synthetic polymers, namely: biocompatibility, biodegradability, reproducibility, and customizability. These proteins can be engineered to mimic both biochemical and biomechanical properties that cells experience in the native extracellular matrix,[28, 29] and these signaling cues have been shown to be particularly important in influencing cell behavior in 3D cell culture.[30]
In this work, we use a previously reported, recombinantly expressed ELP that contains alternating repeats of amino acid sequences derived from the native human extracellular matrix proteins fibronectin and elastin.[29] Specifically, these two sequences include 1) an extended, cell-adhesive sequence derived from fibronectin that contains the integrin-binding peptide RGD and 2) an elastin-like sequence that provides elastic mechanical properties and sites for chemical modification (Figure 1A; Figure S1, Supporting Information). In particular, the elastin-like region is composed of fifteen repeats of the canonical elastin-like peptide VPGXG, where the ‘X’ residue position is designed to be 80% isoleucine and 20% lysine. This sequence results in an ELP with an LCST of ~32 °C, which allows for facile protein purification through alternating cycles of hot and cold centrifugation.[31] Additionally, the primary amine side chains of lysine residues represent a versatile reactive group for a variety of chemical modifications, including nucleophilic substitution and amidation coupling. [17, 32, 33]
Figure 1.
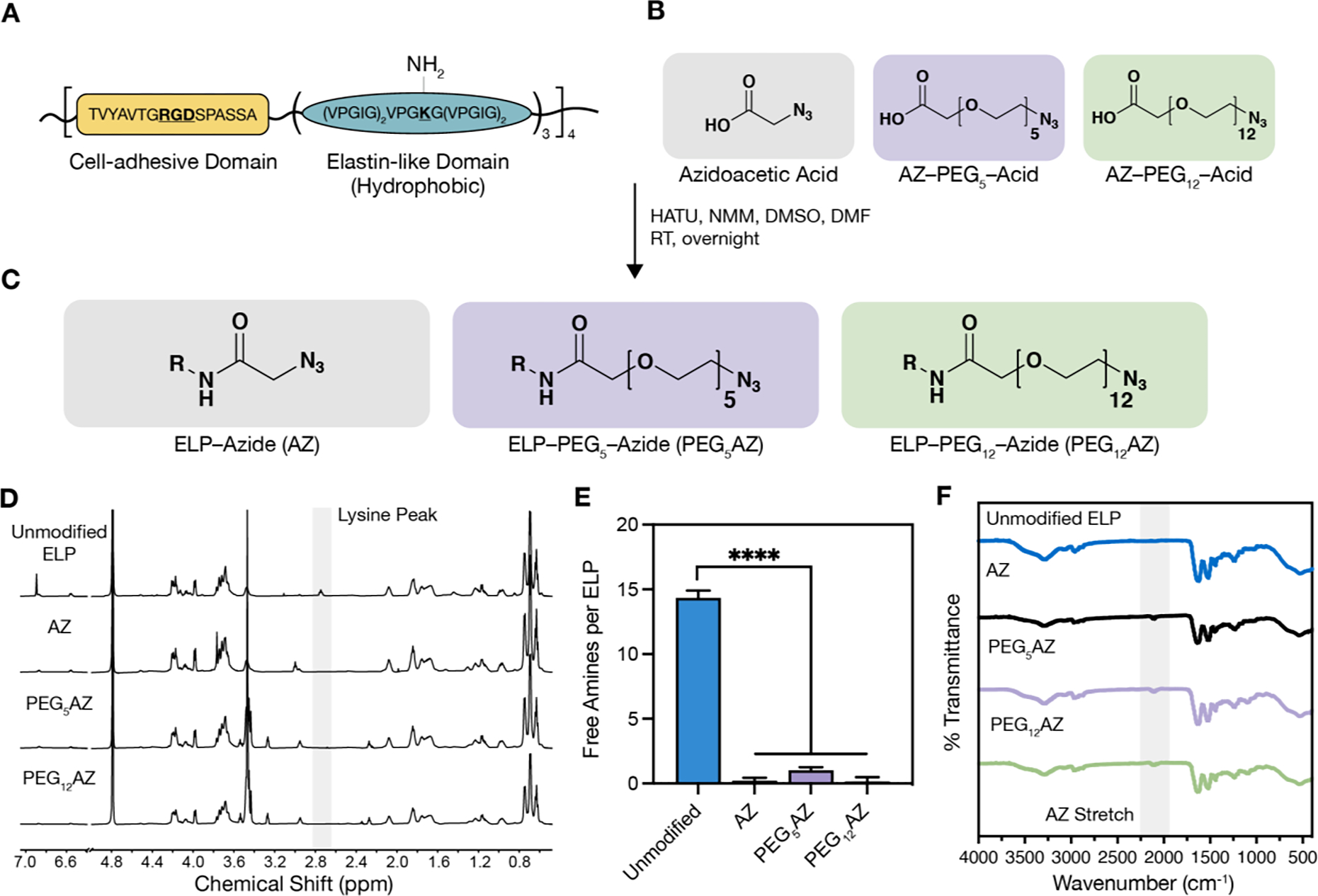
Schematic and characterization of azide-modified ELP. A) Amino acid sequence of ELP cell-adhesive domain and elastin-like domain, which contains the lysine amino acid (K) used for coupling to azide-containing molecules. B) Azide-containing molecules with increasing hydrophilicity from left to right. C) Reaction conditions and products for each of the resulting azide-modified ELPs; R = ELP. D) NMR spectra (600 MHz in D₂O) of azide-modified ELPs demonstrate a decreased peak at 2.75 ppm attributed to reaction of the primary amine side chain on lysine residues. E) Quantification of unmodified amines on ELP before and after coupling reaction (n = 3, data are averages ± standard deviation, ****p < 0.0001, one-way ANOVA with Tukey post hoc test). F) FTIR observation of azide functional group stretching (~2100 cm−1) before and after coupling reactions. HATU, hexafluorophosphate azabenzotriazole tetramethyl uronium; NMM, 4-methylmorpholine; DMSO, dimethyl sulfoxide; DMF, dimethylformamide; RT, room temperature.
Here, the lysine residues were bioconjugated with azide-containing molecules selected to have zero, five, or twelve ethylene glycol repeats (Figure 1B). We hypothesized that increasing the length of these short polyethylene glycol (PEG) linkers would increase the hydrophilicity of the ELP and hence increase the LCST. PEG has been extensively used to enhance protein and polymer solubility in a range of systems.[34–36]
The azide-containing molecules were conjugated to the ELP via a hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU)-mediated amidation reaction, resulting in three ELP variants: ELP–Azide (AZ), ELP–PEG5–AZ (PEG5AZ), and ELP–PEG12–AZ (PEG12AZ) (Figure 1C). Importantly, this reaction proceeds in a non-aqueous solvent, preventing any potential phase separation of the relatively hydrophobic ELP reactant and AZ product from occurring. This choice of reaction conditions also simplifies post-processing, as product purification and concentration is achieved through facile precipitation by addition of diethyl ether. Nuclear magnetic resonance (NMR) characterization shows disappearance of a peak at 2.75 ppm, which is assigned to the lysine residue,[37] indicating all three reactions were successful (Figure 1D). Further quantification of primary amines using a colorimetric trinitrobenzene sulfonic acid (TNBSA) assay demonstrated that we successfully modified the lysines with azide-containing molecules (Figure 1E). Additionally, Fourier-transform infrared (FTIR) spectroscopy indicated the appearance of a new azide stretch at ~2100 cm−1 (Figure 1F). These data confirm that all three azide-containing ELP variants were successfully synthesized.
2.2. ELP Undergoes an LCST-Driven Phase Transition
ELP exhibits an LCST behavior, which is characterized by a transition from a soluble phase to an insoluble protein-rich coacervation phase upon heating above the transition temperature (Tt) (Figure 2A). This protein aggregation is an entropy-driven event that is dependent on several factors including the amino acid sequence (i.e., hydrophobicity), protein concentration, number of repeating segments (i.e., molecular weight), buffer concentration, and pH. The temperature-triggered aggregation results in the association of hydrophobic side chains, obscuring them from the solvent and liberating water molecules.[38] The fifteen elastin-like peptide repeats of ELP are composed primarily of hydrophobic amino acids that decrease solubility. Our engineered ELP intersperses these hydrophobic elastin-like peptide repeats with a more hydrophilic, cell-adhesive sequence derived from fibronectin, resulting in a final Tt of ~32 °C (Figure 2B).
Figure 2.
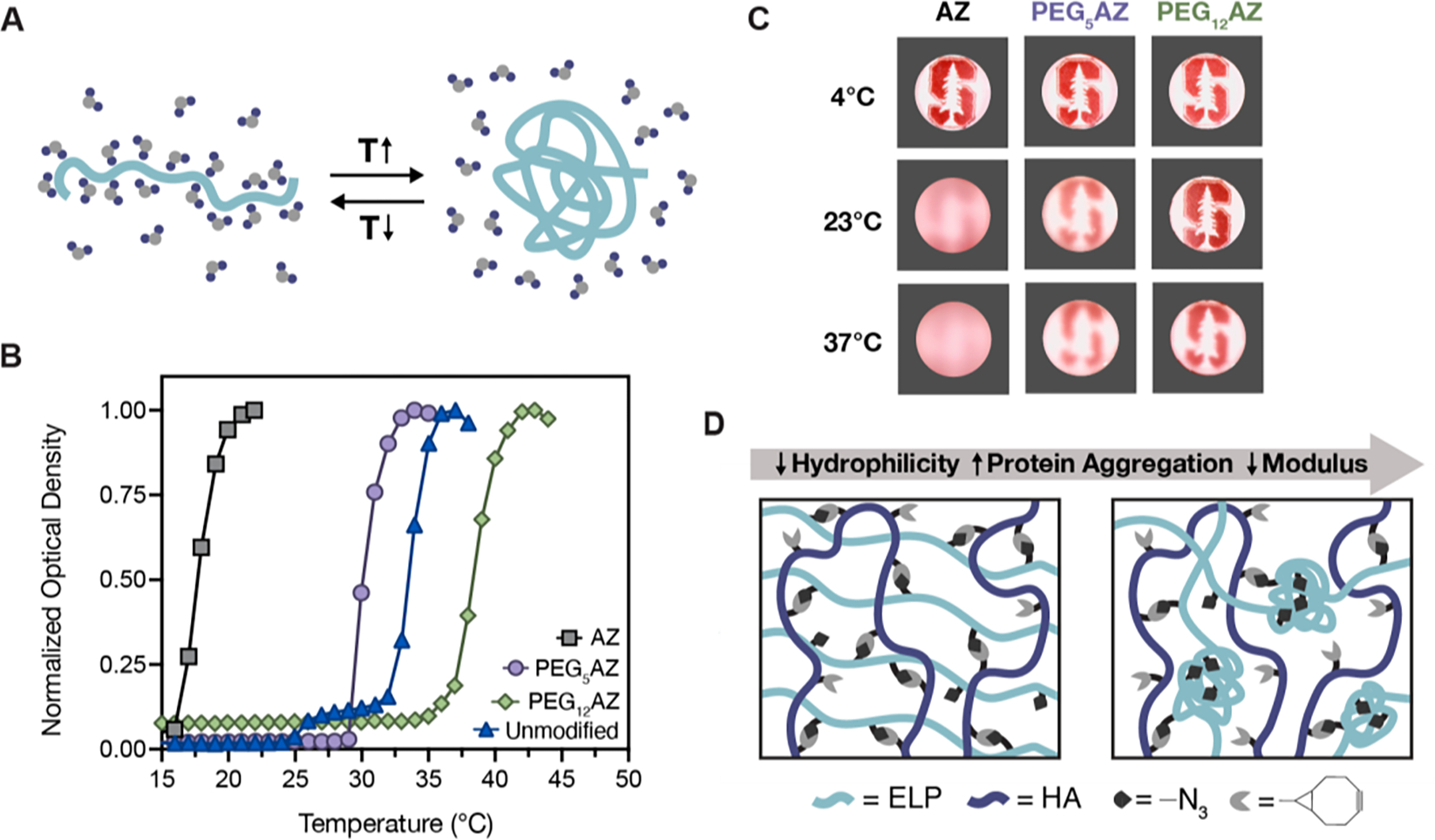
ELP displays a lower critical solution temperature (LCST) phase transition. A) Schematic depicting how temperature influences ELP hydrophobicity, leading to formation of polymer-rich aggregates above the LCST and increasing the translational entropy of associated solvent water molecules. B) Representative curves of unmodified ELP and ELPs modified with their respective azide molecules (1% w/v in PBS) present distinct LCST behaviors as measured by optical density (λ = 300 nm). C) ELP solutions (1% w/v in 10X PBS) modified with azide molecules exhibit observable changes in optical clarity in response to temperature increases. D) Schematic depicting how decreasing hydrophilicity leads to formation of protein aggregates, which obscures available crosslinking groups and reduces the overall modulus.
Protein hydrophilicity can also be tuned by modifying the protein after expression with an array of chemically active molecules. In this work, we postulate that by using increasingly hydrophilic, azide-containing molecules (AZ, PEG5AZ, and PEG12AZ), we can modulate the protein’s solubility and Tt. Observations of the three, chemically modified ELPs at distinct temperatures (4 °C, 23 °C, and 37 °C) revealed that their solutions were all transparent at 4 °C, demonstrating solubility at this temperature. At 23 °C and 37 °C, both the AZ and PEG5AZ solutions were turbid due to the formation of optically dense, protein-rich coacervates (Figure 2C). Uniquely, the PEG12AZ formulation showed no visible change in the solution’s optical properties at all tested temperatures. Quantification of the Tt through optical density measurements further demonstrated successful tuning of the Tt across a range of cell compatible temperatures: Tt = 20 °C, 30 °C, and 37 °C for AZ, PEG5AZ, and PEG12AZ, respectively (Figure 2B; Figure S2, Supporting Information). As expected, the more hydrophilic azide-containing molecules resulted in higher Tt.
Taken together we have demonstrated a method to modulate the hydrophilicity and hence, the Tt of an engineered ELP by the conjugation of pendant, azide-containing molecules, without requiring modification of the primary amino acid sequence. Upon bioconjugation with the azide functional group, the hydrophilic primary amine side chains of the lysine residues are replaced with the relatively more hydrophobic azide moiety, reducing the overall hydrophilicity of the ELP and resulting in a lower Tt compared to the unmodified ELP. The ethylene glycol oligomers present in PEG5AZ and PEG12AZ serve to solvate the hydrophobic ELP motifs and result in an increased Tt, with PEG12AZ achieving a Tt higher than that of unmodified ELP. The range of resulting Tt (20–37 °C) spans that commonly used in mammalian cell culture and cell manipulation protocols, suggesting these materials may be appropriate for further exploration as a cell-encapsulating matrix. Specifically, azide-modified ELP can be covalently crosslinked with bicyclononyne-modified hyaluronic acid (HA-BCN) to form a 3D hydrogel network using bioorthogonal SPAAC click chemistry. We hypothesize that the assembly of ELP aggregates above the Tt could obscure the azide-containing reactive groups, and thus be a straight-forward method to tune the crosslinking density and hence, control hydrogel stiffness (Figure 2D).
2.3. Bioorthogonal Crosslinking of HELP-SPAAC Gels and Tuning Stiffness with Hydrophilicity
To demonstrate our hypothesis that changing protein hydrophilicity will result in gels with varying stiffness due to thermally-triggered protein aggregation, we crosslinked each of the three ELPs with HA-BCN. Recombinant hyaluronic acid was chosen as the second major component of the gel as it is a polysaccharide commonly found in the extracellular matrix and has been reported to provide biochemical signals that enhance gel–cellular interactions in 3D culture.[39] The hyaluronic acid was modified with bicyclononyne as previously published,[40] at a relatively high degree of modification (~19%) (Figure S3, Supporting Information). The HA-BCN was used to form a gel network by mixing with the azide-containing ELPs via a biorthogonal SPAAC reaction (Figure 3A). This reaction was chosen as the click chemistry cycloaddition proceeds without catalyst or additives, and it is not temperature-dependent. Therefore, we can exclusively compare the effects of temperature-triggered aggregation that result in azide binding groups being obscured and this effect on the gel mechanical properties. Upon mixing the 19%-modified HA-BCN with the azide-modified ELP in a one-to-one mass ratio, we achieved a stoichiometric ratio of 1:1.7 for the AZ:BCN reactive groups, which ensures that all available azide groups can bind when accessible.
Figure 3.
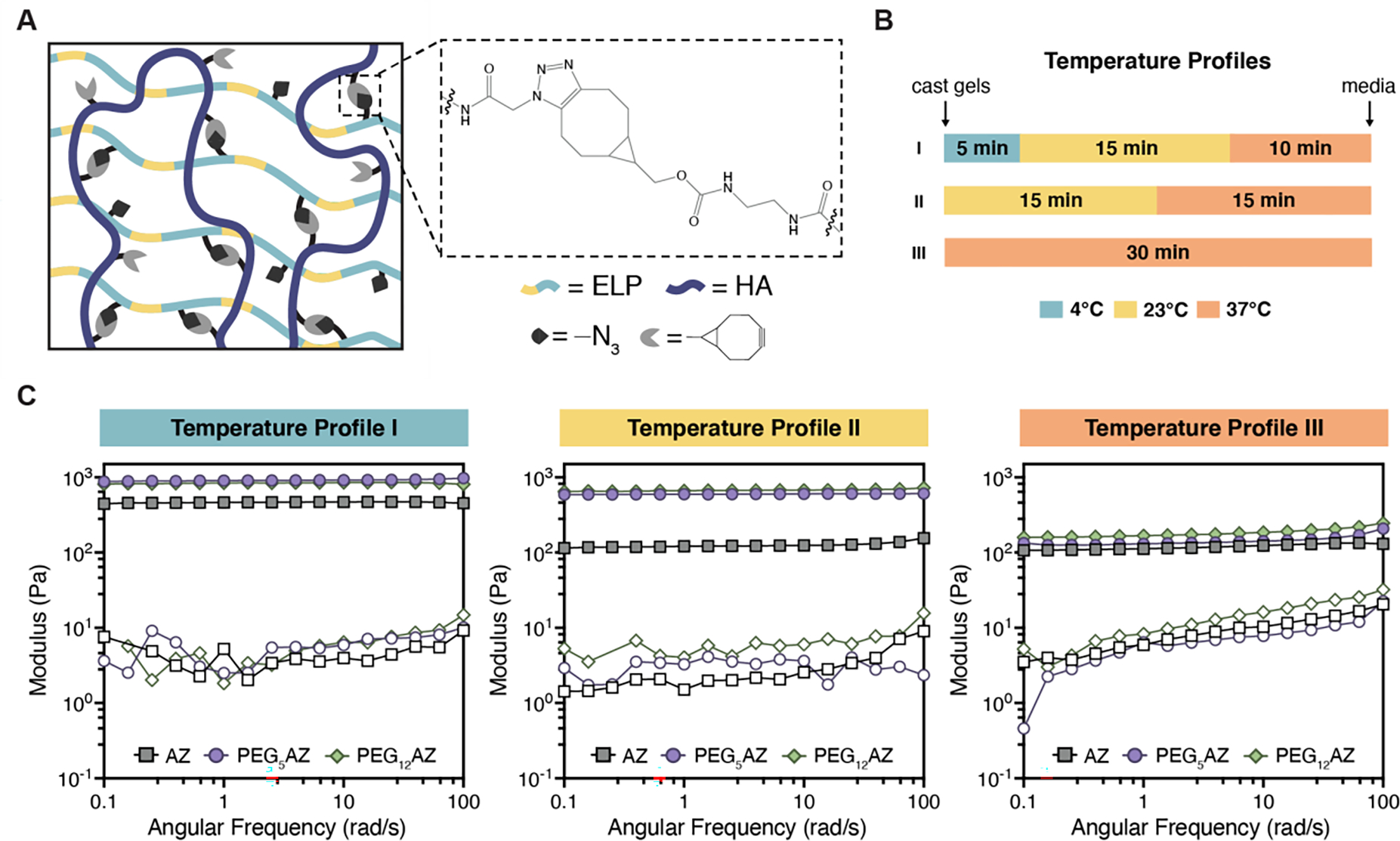
Protein hydrophilicity and gelation temperature regulate mechanical properties of bio-orthogonally crosslinked HELP-SPAAC gels. A) Schematic illustration of SPAAC-crosslinked hydrogels formed from azide-functionalized ELP and bicyclononyne-functionalized HA. B) Three distinct temperature profiles (I, II, and III) were used during gel crosslinking. C) Storage moduli (G’, filled symbols) and loss moduli (G”, open symbols) of HELP-SPAAC hydrogels during a frequency sweep at a fixed strain of 1% for the three temperature profiles and each of the HELP-SPAAC gel formulations.
We postulated that crosslinking the gels below the Tt would allow the greatest accessibility of pendant azides and hence form the maximum number of crosslinks possible before the ELP undergoes thermal aggregation. In contrast, if ELP thermal aggregation is allowed to occur at the same time as chemical crosslinking, we hypothesize that this will obscure the azides and prevent effective crosslinking, resulting in weaker gels. To test this, three different crosslinking temperature profiles were used, and the resulting storage and loss moduli, G’ and G”, respectively, were measured. The temperatures were chosen such that they are easily obtainable using common laboratory equipment. For each temperature profile, the overall gelation time was kept constant at 30 min, which was sufficient to reach a plateau modulus for all conditions (Figure S4, Supporting Information). Previous work has demonstrated that a range of cell types can tolerate a 30-minute encapsulation protocol prior to receiving nutrient-rich medium.[31, 33] The three crosslinking temperature profiles were: 5 min at 4 °C, 15 min at 23 °C, 10 min at 37 °C (Temperature Profile I); 15 min at 23 °C, 15 min at 37 °C (Temperature Profile II); and 30 min at 37 °C (Temperature Profile III) (Figure 3B). The specific times spent at each temperature were chosen to yield gels with distinct moduli while maintaining a constant gelation time. Rheological studies showed that crosslinking with temperature profile I (below the Tt) produced the stiffest gel (i.e., highest G’) for all three ELP variants (Figure 3C). At temperature profile II (crosslinking near the Tt), the modulus for all three gel formulations was lower compared to profile I. This decrease in modulus was most dramatic for the gel formulation containing AZ (the ELP variant with the lowest Tt). These results are consistent with the idea that azide reactive groups are obscured near the Tt and are no longer accessible for crosslinking. At temperature profile III, where we have surpassed the Tt for both AZ and PEG5AZ and are more closely approaching the Tt of PEG12AZ, we see that all moduli have drastically decreased compared to profiles I and II. These data suggest that changes in crosslinking temperature can lead to varying degrees of ELP aggregation, thereby potentially restricting accessibility to azide functional groups and altering the final crosslink density.
2.4. LCST Induces Protein Aggregation and Alters Gel Opacity
To evaluate the hypothesis that changes in gel stiffness are due to differences in ELP aggregation and hence altered azide accessibility, we explored how manipulating the thermal transition behavior can alter gel opacity. ELP-based hydrogels are typically optically dense, owing to the increased light scattering caused by hydrophobic ELP aggregates that are formed above the Tt.[41] Previous work reported that the optical density and microarchitecture of ELP-PEG hydrogels were strongly influenced by the crosslinking temperature, such that crosslinking above the Tt increased the optical density and resulted in the formation of ELP-rich protein domains.[42] Thus, we can use gel opacity as a simple and quick indicator of potential microscopic ELP aggregation within our HELP-SPAAC gels.
The gel optical properties were assessed at all three temperature profiles for all three ELP formulations (Figure 3B), both at the initial and final temperatures of the gelling process. Consistent with our hypothesis, we found that as the temperature was increased above the Tt for each condition, the gels became more optically dense as the ELP aggregates caused scattering of visible light (Figure 4A). With a Tt of 37 °C, PEG12AZ-containing gels were optically transparent at all temperature profiles. In contrast, gels formulated with the more hydrophobic PEG5AZ or AZ were opaque at the end of all three temperature profiles. To quantify the final gel optical properties, we measured the transmittance of visible light through all nine gels (three gel formulations crosslinked at three temperature profiles, Figure 4B). As expected, the final gel transmittance depended strongly on the hydrophilicity of the azide-containing ELP, with gels formulated from PEG12AZ exhibiting the greatest transmittance at all three temperature profiles. Evaluating the temporal change in optical density during the crosslinking process, transmittance through the gel was found to decrease as the temperature increased past the Tt, consistent with the idea that ELP aggregation is causing the change in opacity (Figure 4C). For example, in the case of gels formulated from PEG5AZ, a rapid decline in transmittance was observed with temperature profile III, whereas temperature profiles I and II yielded a slower decline in transmittance as the gels were allowed to crosslink at temperatures below the Tt (~30 °C) before transitioning to 37 °C.
Figure 4.
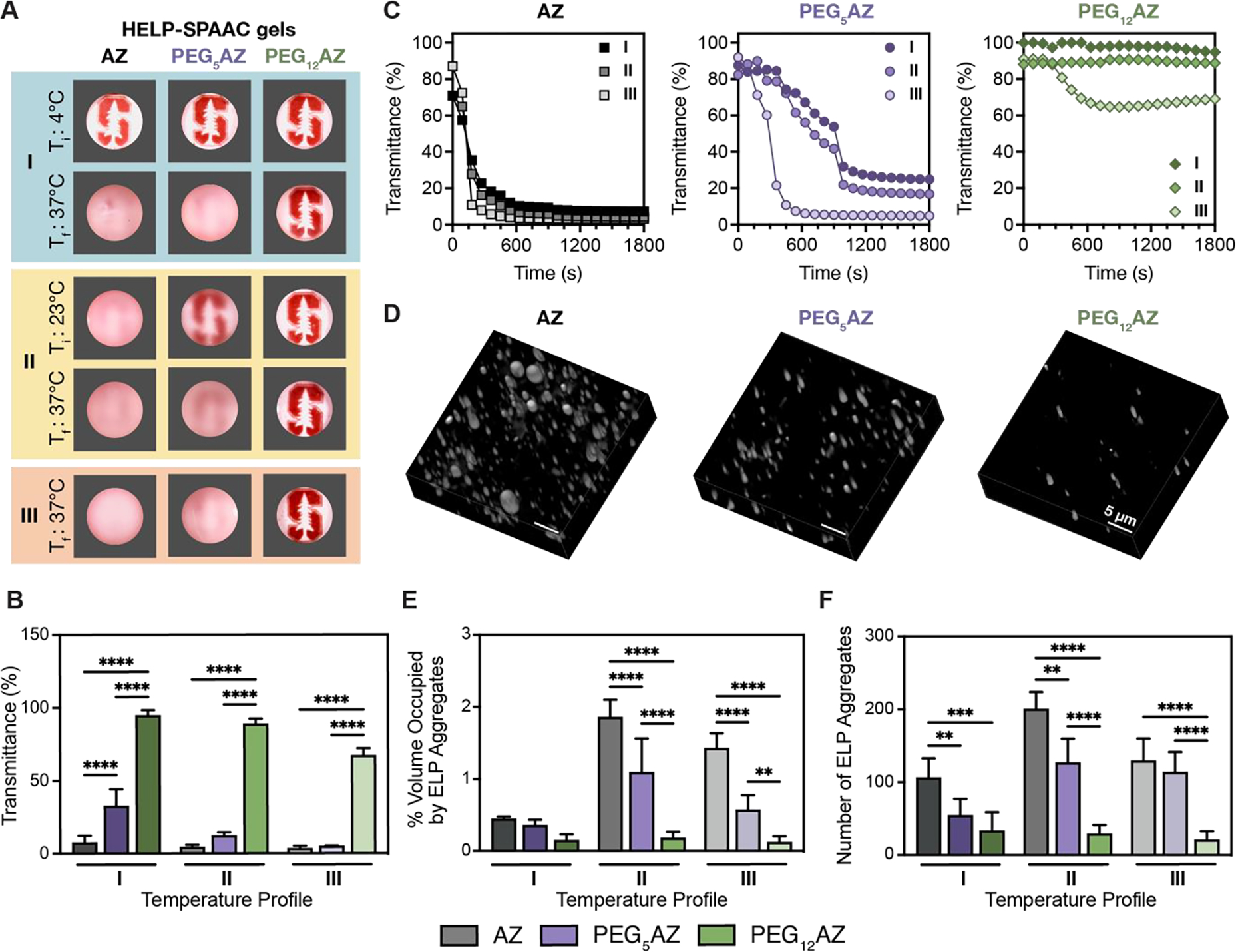
LCST induces ELP aggregation within HELP-SPAAC gels and alters gel opacity. A) Representative photographs demonstrating gel opacity at specific times for the three temperature profiles shown in Figure 3; initial temperature = Ti, final temperature = Tf. B) Final gel transmittance at Tf for all gel formulations crosslinked using the three temperature profiles (n = 4, data are averages ± standard deviation, ***p < 0.001, ****p < 0.0001, two-way ANOVA with Tukey post hoc test). C) Representative transmittance over time for the three temperature profiles and each of the three HELP-SPAAC gel formulations. D) 3D reconstruction of CARS images of HELP-SPAAC gels crosslinked with temperature profile II. Regions of bright intensity are protein-rich ELP aggregates. E) Quantification of CARS data showing volume percentage of gel occupied by ELP-rich aggregates for all gel formulations crosslinked using the three temperature profiles (n = 5, data are averages ± standard deviation, **p < 0.01, ****p < 0.0001, two-way ANOVA with Tukey post hoc test). F) Quantification of CARS data showing number of ELP-rich aggregates for all gel formulations crosslinked using the three temperature profiles (n = 5, data are averages ± standard deviation, **p < 0.01, ****p < 0.0001, two-way ANOVA with Tukey post hoc test).
To confirm that the change in optical density is due to formation of hydrophobic ELP aggregates, we used coherent anti-Stokes Raman scattering (CARS) microscopy[43] to observe the microarchitecture of HELP-SPAAC gels crosslinked with all three temperature profiles. We have previously validated CARS microscopy as a noninvasive method to characterize ELP aggregates within crosslinked gels.[9, 17, 23, 44, 45] CARS offers advantages over other imaging modalities by bypassing the need for sample dehydration so that the gel structure can be observed in its native, hydrated state. Furthermore, it probes inherent molecular vibrations, circumventing the need for fluorophores. Here, this was crucial as the Tt of ELP was found to be acutely sensitive to functionalization of crosslinking groups; hence, the addition of fluorophores may affect the thermal transition properties of our HELP-SPAAC gels. CARS imaging of samples crosslinked with temperature profile II corroborated the previously observed trend, with increased observation of ELP aggregates found in gels with lower hydrophilicity, and limited observation of ELP aggregates in the optically clear PEG12AZ-containing gels (higher hydrophilicity, Figure 4D). This trend is maintained in temperature profiles I and III as well (Figure S5, Supporting Information). Furthermore, quantification of CARS images showed statistically significant trends in both the overall volume percentage of ELP aggregates and the number of ELP aggregates as a consequence of ELP hydrophilicity (Figure 4E,F). While CARS micrographs showed that gels crosslinked with temperature profile III seem to have fewer ELP aggregates compared to those crosslinked with temperature profile II, the decrease in transmittance suggests that increased aggregation is still occurring in these gels. The reason for this inconsistency may be that the size of the hydrophobic aggregates in these samples was below the resolution limit of CARS, approximately 350 nm laterally.[23, 46] Overall, these data suggest that hydrogel hydrophilicity differentially regulates the degree of ELP aggregation and overall optical density of the gel.
2.5. Correlating Hydrogel Optical Properties with Mechanical Properties
Taken together, the data on gel mechanical properties (Figure 3) and optical properties (Figure 4) support our hypothesis that manipulation of ELP hydrophilicity can be used to tune the stiffness of HELP-SPAAC gels through altering the degree of ELP aggregation. Gels formulated with AZ readily form hydrophobic aggregates due to their decreased hydrophilicity, impairing the ability of crosslinks to form and yielding relatively compliant gels (Figure 5A). The addition of PEG linkers increases the hydrophilicity of the ELP, reducing the degree of ELP aggregation and increasing the accessibility of azide groups available for crosslinking, ultimately resulting in stiffer gels. While PEG spacers have also been shown to prevent hydrogel interactions through steric hindrance, the ethylene glycol linkers used here are relatively small (n = 5–12) compared to PEG spacers used for those applications (n > 100), suggesting that our resulting gel properties are primarily attributable to the increase in overall protein hydrophilicity rather than a disruption of protein interactions.[47] For each gel formulation, final gel transmittance—an indicator of ELP aggregation—increased with increasing gel stiffness (Figure 5B). For example, when gels are crosslinked with temperature profile I, all three gel formulations are allowed to form crosslinks below their Tt during the 4 °C incubation period, increasing accessibility to azide functional groups and resulting in limited ELP aggregation, yielding stiffer and more transparent gels. In contrast, gels formed with temperature profile III are crosslinked at or above their Tt, and the resulting ELP aggregation drives the formation of more compliant gels with limited transmittance, which we attribute to limited accessibility of the azide functional groups to participate in crosslinking. With temperature profile II, which uses an intermediate crosslinking temperature of 23 °C, an intermediate stiffness and transmittance is observed. Therefore, for each gel formulation, a range of hydrogel stiffness can be achieved by altering the temperature of gelation, consistent with our original hypothesis. Importantly, for each ELP variant, this control over gel stiffness is obtained without altering the gel chemistry, as cells are known to be exquisitely sensitive to the local chemical environment. Thus, these data demonstrate the utility of manipulating temperature profiles as a method for tuning ELP hydrogel stiffness for potential use in studies of cell-matrix biomechanics.
Figure 5.
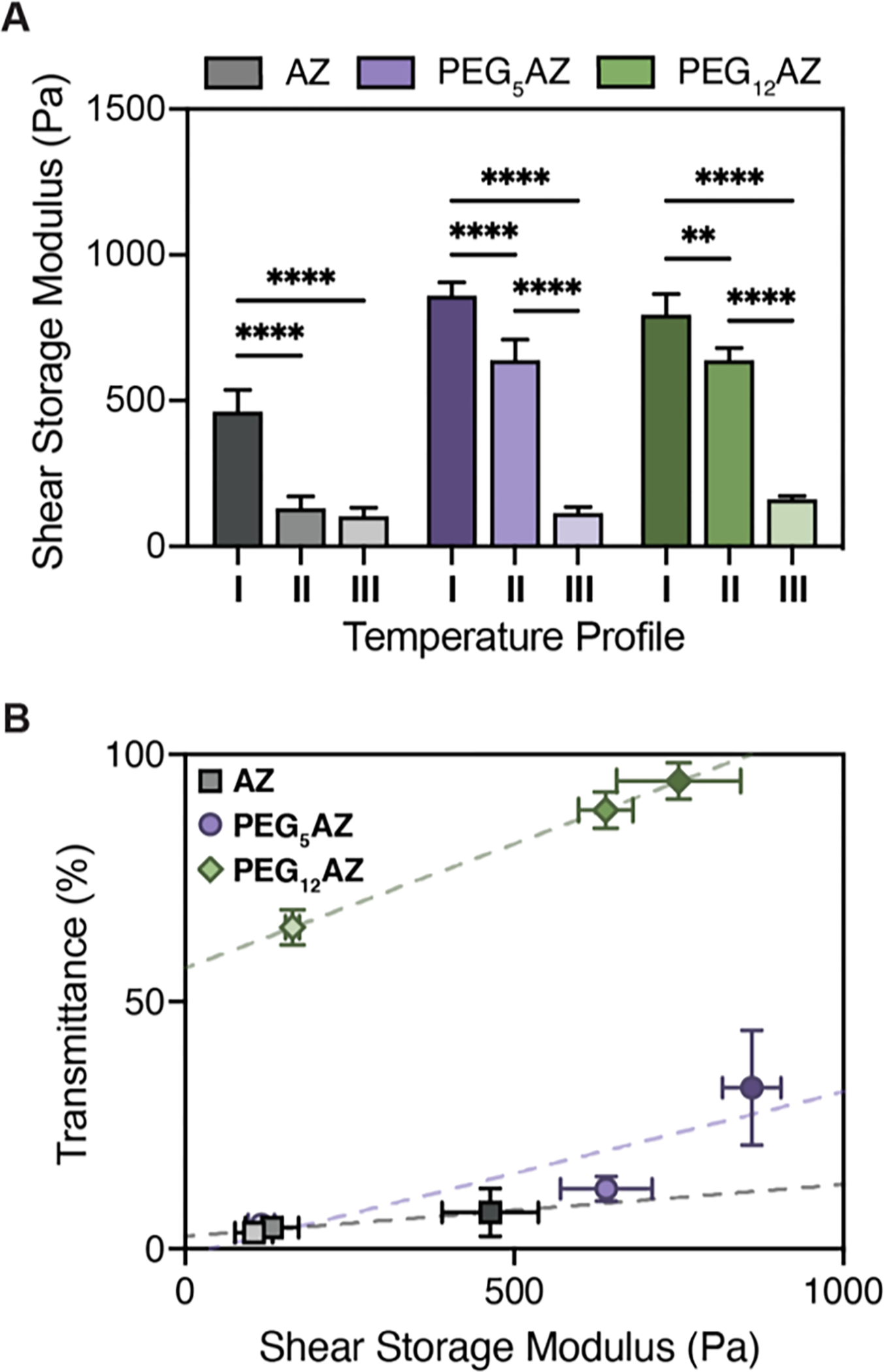
Gel transmittance and hydrogel storage modulus are correlated. A) Average shear storage moduli (G’) of HELP-SPAAC gels for all gel formulations crosslinked using the three temperature profiles (n = 3, data are averages ± standard deviation, **p < 0.01, ****p < 0.0001, two-way ANOVA with Tukey post hoc test). B) Final gel transmittance increases with increasing hydrogel stiffness for each gel formulation (n = 3, data are averages ± standard deviation). Dashed lines represent a fitted linear trend line to guide the eye.
2.6. Gel Mechanics Alters Cell Morphology in a Cell Type-Dependent Manner
To confirm that HELP-SPAAC gels support viability of a range of cell types, hMSCs, HUVECs, and hNPCs were encapsulated within PEG5AZ-containing HELP-SPAAC gels. This hydrogel formulation was chosen because PEG5AZ-containing HELP-SPAAC gels are able to achieve the widest range of stiffness by varying the temperature profile used for crosslinking while keeping the gel chemistry constant (Figure 5A). Ideal culture conditions for most cell types are 37 °C and 5% CO2, so it was particularly important to assess the viability of cells subjected to temperature profile I, which is the condition where cells are kept the longest in non-physiological culture conditions (total of 20 min). Using a Live/Dead cytotoxicity assay, short-term viability (24 h post-encapsulation) was ~96% for hMSCs, ~93% for HUVECs, and ~90% for hNPCs (Figure S6A, Supporting Information), indicating that the gelation procedure was well tolerated. We next demonstrated that the HELP-SPAAC gels support cell viability and phenotypic maintenance over longer times. All three cell types remained highly viable in the hydrogels 7 days after encapsulation, with viabilities of ~96% for hMSCs, ~96% for HUVECs, and ~91% for hNPCs (Figure S6A, Supporting Information). For all HELP-SPAAC gels in this study, the concentration of fibronectin-derived RGD peptide motifs was 1 × 10−3 M, which is known to promote cell adhesion and spreading.[48] After 1 week in culture, hMSCs exhibited a characteristic spread morphology and expressed the MSC surface marker CD44 (Figure S6B, Supporting Information), consistent with previous results which demonstrate MSC spreading in 3D cultures that enable cell adhesion and matrix remodeling.[33] Similarly, HUVECs retained expression of the endothelial marker CD31 and began to form multicellular structures after 1 week (Figure S6B, Supporting Information), both of which are indicative of endothelial cell phenotypic maintenance in vitro.[49] Lastly, hNPCs cultured for 1 week in HELP-SPAAC gels stained positive for the neuronal marker β-tubulin III and extended neuritic projections throughout the hydrogel (Figure S6B, Supporting Information), suggesting that the encapsulated hNPCs maintained their neuronal phenotype.[50] These results indicate that HELP-SPAAC gels support the in vitro culture and phenotypic maintenance of hMSCs, HUVECs, and hNPCs.
Next, we evaluated the cell response to changes in hydrogel mechanics by encapsulating the three cell types within PEG5AZ-containing HELP-SPAAC gels using the three temperature profiles described above. Crosslinking with temperature profiles I, II, and III, resulted in gels with storage moduli of ~1 kPa, ~600 Pa, and ~150 Pa, respectively (Figure 5A). The ability to independently control the mechanical properties of HELP-SPAAC hydrogels while keeping the gel chemistry (i.e., the total PEG content) constant makes this an attractive system for studies of 3D cell mechanobiology. Hydrogel stiffness in particular has been shown to regulate cellular morphology, behavior, and cell fate in 3D.[50–52] After 7 days in culture, hMSCs exhibited morphological differences across the three hydrogels, with statistically significant increased cell spreading in stiffer gels, while thinner, spindle-like morphology was observed in the more compliant gels (Figure 6A,B). Similarly, HUVECs also displayed increased spreading in stiffer gels along with increased formation of multicellular structures, while HUVECs in the lower stiffness gels remained as rounded, single cells (Figure 6C,D). Interestingly, in contrast to the behavior of hMSCs and HUVECs, hNPCs exhibited the opposite trend, with cell spreading and neurite outgrowth significantly increased in lower stiffness gels (Figure 6E,F). Taken together, these data demonstrate that changes in cell morphology mediated by matrix mechanics are cell type-dependent (Table S1, Supporting Information).
Figure 6.
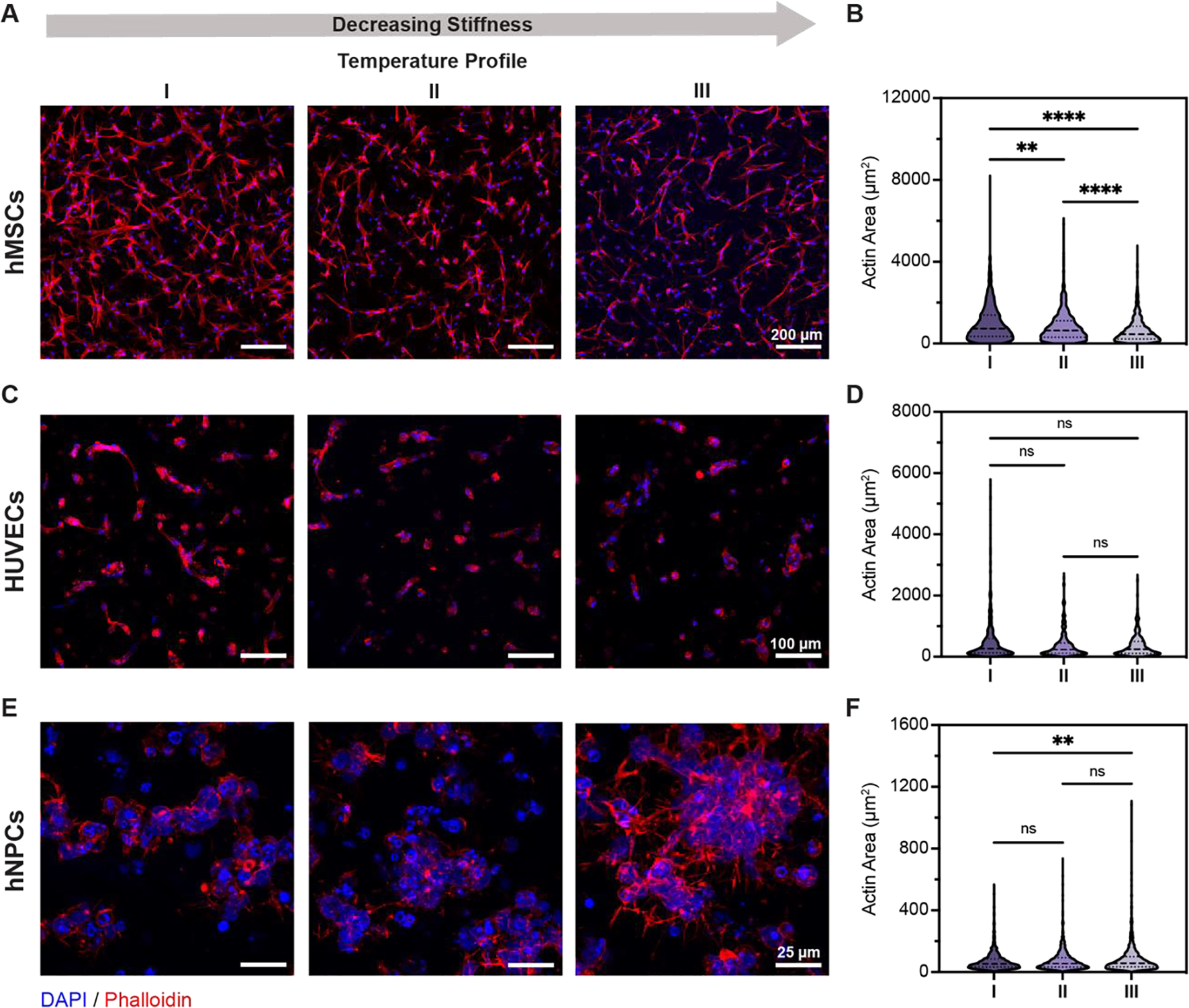
Mechanical properties of HELP-SPAAC hydrogels influence cell spreading and morphology of hMSCs, HUVECs, and hNPCs. A,C,E) Representative images of cell morphology (hMSCs, HUVECs, and hNPCs, respectively) within HELP-SPAAC hydrogels (formulated with PEG5AZ) with varying temperature profiles after 7 days. Nuclei staining with DAPI shown in blue, and F-actin staining with phalloidin shown in red. B,D,F) Quantification of actin area after 7 days in HELP-SPAAC hydrogels with varying temperature profiles for hMSCs, HUVECs, and hNPCs, respectively. In B) and F), actin area is normalized per cell using the DAPI counter-stain; while in D), actin area per multicellular cluster is reported (see Methods). Kruskal-Wallis with Dunn’s multiple comparisons test was used for data analysis: **p < 0.01, ****p < 0.0001. For B), n = 1,063, 823, and 899 (I, II, and III) cells examined over N = 3 independent gel samples. For D), n = 386, 448, and 503 (I, II, and III) cells examined over N = 3 independent gel samples. For F), n = 3,357, 3,013, and 3,067 (I, II, and III) cells examined over N = 3 independent gel samples.
These trends in cell spreading are consistent with previously published observations of each cell type independently across a variety of biomaterials. When compared with other biodegradable gels of similar stiffness (~1–5 kPa), hMSCs have been reported to increase spreading in stiffer 3D substrates.[53] Similarly, studies of HUVEC spreading within biodegradable gels of similar stiffness (~180–1,200 Pa) have also reported increased spreading and a vasculature-like phenotype in stiffer gels.[54] It is important to note that cell mechanosensing and spreading of high tension-generating cells, such as hMSCs and HUVECs, within 3D gels typically requires that the cells be able to remodel the surrounding polymer network, often through incorporation of hydrolytically degradable elements.[48, 55, 56] The HELP-SPAAC gels used here can undergo enzymatic remodeling through the local action of both hyaluronidases and proteases.[9, 27, 57, 58] Neural-type cells are reported to generate much smaller stresses on their surrounding matrix.[59] While few studies have specifically investigated hNPCs within 3D gels, our observations parallel what was observed for murine NPCs and chick dorsal root ganglia, which show increased neurite extension in softer 3D matrices.[60–63] Overall, these results highlight the exquisite sensitivity and cell type-specific behaviors that cells exhibit in response to matrix mechanical cues. Precise control over and independent tuning of matrix mechanical properties is therefore crucial for modulating cell behavior, both for fundamental studies of cell biology and for clinical translation of biomaterials.
Hydrogel stiffness should be carefully considered when designing biomaterial platforms for cell culture. Previously reported methods for modulating stiffness of HELP gels include changing the degree of polymer functionalization or tuning the polymer weight percent, but these methods require either altering the gel chemistry or the gel composition.[17, 27] Within the HELP-SPAAC system, the LCST transition property of ELP is a useful design feature that can be leveraged to tune hydrogel stiffness without changing either gel chemistry or composition (Figure 5). Importantly, the temperatures required to achieve the range of stiffness in these studies are commonly used in other cell culture protocols and were well tolerated by a variety of cell types (Figure S6, Supporting Information). Specifically, these temperature transitions are prevalent in routine cell culture tasks, such as placing cells on ice after trypsinization, maintaining cells at room temperature during a passage, and transitioning to physiological temperature after placing cells in the incubator. The stiffness range reported here resulted in significant changes in cell morphology across three different cell types (Figure 6), demonstrating that this range of gel mechanics is biologically relevant. These data demonstrate that the tuning of ELP hydrophilicity, and hence LCST behavior and pendant-crosslinker accessibility, can control hydrogel mechanical properties and represents a promising strategy to prepare matrices for a range of biomaterial applications.
3. Conclusion
We hypothesized that covalently crosslinking the ELP and HA polymers together at temperatures below, at, or above the Tt would alter the accessibility of the azide reactive groups and hence the resulting gel stiffness. Consistent with this idea, gels formed between HA-BCN and azide-containing ELPs resulted in a stiffness (G’) range of 100–1,000 Pa under our crosslinking temperature profiles. Therefore, modulating polymer hydrophilicity and temperature profile obviates the need to increase stiffness through increased polymer weight percent or degree of polymer functionalization. To validate our hypothesis that this difference in gel stiffness is due to changes in ELP aggregation that alter azide accessibility, we evaluated the gels by CARS microscopy to measure the volume fraction of ELP aggregates and quantified the opacity by light absorbance. As ELP hydrophilicity increases, the volume fraction of protein aggregation was found to decrease for all three thermal profiles. As ELP aggregates cause scattering of visible light, we also observed increased light transmittance for gels crosslinked below the Tt, with PEG12AZ producing optically transparent hydrogels for all thermal profiles. Taken together, these data demonstrate that crosslinking at temperatures below the Tt increases azide accessibility and results in stiffer, more transparent gels.
Precise control over gel mechanical properties is crucial for modulating cell behavior. To demonstrate that our range of mechanical properties could alter cell behavior, we encapsulated hMSCs, HUVECs, and hNPCs in our HELP-SPAAC gels for 3D in vitro cell culture. Specifically, in gels formed using PEG5AZ, cells remained viable and expressed phenotypic markers for up to 7 days post-encapsulation. By using ELP hydrophilicity to modulate hydrogel stiffness, we observed that encapsulated hMSCs, HUVECs, and hNPCs exhibited differential cell type-dependent responses in cell morphology and spreading. Overall, this work presents a promising strategy for regulating gel mechanics by leveraging protein hydrophilicity as a design parameter, expanding the methods available for tailoring biomaterial properties.
Future directions could expand on this work by utilizing the unreacted BCN functional groups to conjugate additional cell-adhesion ligands or regulatory signals to drive specific cell behavior. Other promising approaches of this technology can offer a method to temporally control the stiffness of the resulting gel. For example, gels could be formed using temperature profile I and subsequently cooled to room temperature at designated time points. The lower temperature may reduce the ELP aggregates formed and make the azide functional groups available for further crosslinking, resulting in a stiffer modulus and further broadening the applications of this hydrogel platform.
4. Experimental Section
Materials:
Reagents were purchased from Sigma-Aldrich and used without further purification unless otherwise noted.
Expression and Purification of ELP:
ELP was produced via recombinant protein expression as described previously.[31] The amino acid sequence of ELP used in these experiments is shown in Figure S1 in the Supporting Information. Briefly, pET15b plasmids encoding the ELP sequence under control of the T7 promoter were transformed into BL21(DE3)pLysS Escherichia coli (Life Technologies). The bacteria were cultured in terrific broth to an OD600 of 0.8, at which point 1 × 10−3 M isopropyl β-D-1-thiogalactopyranoside (IPTG, Fisher) was added to induce protein expression. After 7 hours, the cells were pelleted, suspended in TEN buffer (10 × 10−3 M Tris (Fisher), 1 × 10−3 M EDTA (Fisher), and 100 × 10−3 M NaCl (Fisher), pH 8.0), and lysed via alternating freeze-thaw cycles. Cell lysate was treated with deoxyribonuclease (DNase) and 1 × 10−3 M phenylmethanesulfonyl fluoride (PMSF) to inhibit proteolysis. ELP was purified by inverse thermal cycling, dialyzed against deionized (DI) water for three days, and lyophilized to yield a white fibrous solid.
Synthesis of Azide-Functionalized ELP:
To modify ELP with azide functional groups, lyophilized ELP was dissolved in anhydrous dimethyl sulfoxide (DMSO) at 75 mg mL−1 and allowed to stir for ten min before adding an equal volume of anhydrous N,N-dimethylformamide (DMF) and allowed to stir for an additional ten min. In a separate flask, azidoacetic acid, azido-PEG5-acid (BroadPharm), or azido-PEG12-acid (BroadPharm) (2 eq. per ELP amine) was dissolved in the same volume of DMF used to dissolve the ELP. Once dissolved, hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU, 1.1 eq. per azide) and 4-methylmorpholine (2.5 eq. per azide) were quickly added and allowed to react for 10 min. Next, the azide solution was added to the ELP solution slowly over the course of ten min and left to react overnight. The reaction was then precipitated in ice-cold diethyl ether (Fisher), pelleted, and vacuum dried. The resulting protein was dissolved in DI water (1% w/v), dialyzed for three days, sterile filtered using a 0.22 μm filter, and lyophilized to yield a white fibrous solid.
Characterization of Azide-Functionalized ELP:
The ELP lysine modification was assessed with 1H NMR using a Varian Inova 600 Hz NMR spectrometer. ELP samples were dissolved at 10 mg mL−1 in deuterated water, and spectra were collected at 4 °C. The modification was verified by comparing the decrease in integrated signal of lysine protons (δ = 2.75). The modification was further confirmed by quantifying the number of free primary amines per ELP polymer using the trinitrobenzene sulfonic acid (TNBSA) assay (Pierce). Modified ELP was dissolved in 0.1 × 10−3 M sodium bicarbonate buffer (pH 8.0) at a concentration of 0.25 mg mL−1. A standard curve was prepared from serial dilutions of 0.5 mg mL−1 unmodified ELP dissolved in bicarbonate buffer. In each well of a 96-well plate, 140 μL of samples or standards were mixed with 70 μL of 0.01% w/v TNBSA in bicarbonate buffer, and the mixture was incubated at room temperature for 2 hours protected from light. The reaction was then quenched by adding 70 μL of 10% w/v sodium dodecyl sulfate (SDS) and 40 μL of 1 N HCl. Absorbance was measured at 335 nm using a Molecular Devices SpectraMax M2 plate reader. The number of free primary amines on the modified ELP was calculated by comparing to the standard curve generated from unmodified ELP. Incorporation of azides onto the ELP was demonstrated using FTIR spectroscopy to identify the presence of an azide stretch peak near 2100 cm−1. FTIR was performed using a Nicolet iS50 FT/IR Spectrometer in the Stanford Soft Materials Facility.
Lower Critical Solution Temperature Measurements:
Unmodified and modified ELP were dissolved in 1X PBS (Corning) at a concentration of 10 mg mL−1. 200 μL of each solution and a PBS blank were pipetted into a quartz cuvette with 1 mm path length. Absorbance readings at 300 nm were collected on a circular dichroism instrument with a temperature controller (Jasco J-815 Spectropolarimeter). The temperature was increased from 4 °C to 65 °C at a rate of 1 °C per minute. The temperature was allowed to stabilize for 10 seconds before each measurement.
Synthesis and Characterization of BCN-Functionalized HA:
100 kDa sodium hyaluronate (HA, Lifecore Biomedical) was modified with BCN-amine as described previously.[40] Briefly, HA was completely dissolved in 100 × 10−3 M MES buffer (pH 7.0) at 1% w/v. Once dissolved, 1-Hydroxybenzotriazole hydrate (HOBt, Fisher) (2 eq. to the HA dimer unit) was added to the HA and allowed to dissolve for 30 min. In a separate vial, endo-Bicyclo[6.1.0]non-4-yne-9-ethylenediamine (BCN-amine, Conju-Probe) (2 eq.) was dissolved in a 6 mL solution of acetonitrile (MeCN) and DI water (5:1 v/v) to which 1-ethyl-3-(−3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, Fisher) (2 eq.) was added and allowed to dissolve. The solution was then added over the course of 30 min into the dissolved HA solution and allowed to react overnight. The reaction mixture was dialyzed for two days against a 10% MeCN solution, followed by 3 days against DI water. The solution was then sterile filtered using a 0.22 μm filter and lyophilized to produce a white fibrous solid. The degree of modification on HA-BCN was quantified using 1H NMR by integration of the proton signal of the methylene group adjacent to the amine group (δ = 4.1, 2H) relative to that of the methyl groups on N-acetylglucosamine of the HA backbone (δ = 1.8, 3H).
Formation of HELP-SPAAC Hydrogels:
Lyophilized HA-BCN and ELP-azide were separately dissolved in 10X isotonic PBS (81 × 10−3 M sodium phosphate dibasic, 19 × 10−3 M sodium phosphate monobasic, 60 × 10−3 M sodium chloride (Fisher) in DI water, pH 7.4) at a stock concentration of 2% w/v overnight at 4 °C. The final solutions were kept on ice until use. An equal volume of HA and ELP were mixed in a tube, and 10 μL of the resulting mixture were pipetted into each silicone mold (4 mm diameter, 0.8 mm height, plasma bonded to a 12 mm circular #2 coverglass, Electron Microscopy Sciences). The gels were allowed to crosslink using the 3 different temperature profiles described in Figure 3B.
Hydrogel Rheological Characterization:
Gels were crosslinked in situ on an ARG2 rheometer (TA instruments) with a 20 mm, 1° cone geometry and a solvent trap to prevent sample dehydration. To determine gelation time and confirm gel formation, a time sweep was performed at 1% strain and 1 rad s−1 oscillatory frequency following the three temperature conditions described in Figure 3B. This protocol was followed by a frequency sweep from 0.1 to 100 rad s−1 at a fixed 1% strain. The storage and loss moduli were taken to be the values at 1 rad s−1 from these measurements.
Characterization with CARS Microscopy:
ELP aggregation was visualized and quantified within gels by CARS microscopy using an inverted microscope (Nikon Ti2-E with a C2 confocal scanning head and a Nikon CFI Apochromat TIRF 100XC oil immersion objective). The carbon–hydrogen (C–H) vibrations were coherently driven by overlapping two near-infrared laser beams in time and space, generated by a picosecond-pulsed laser system (APE picoEmerald S, 2 ps pulse length, 80 MHz repetition rate, and 10 cm−1 bandwidth) composed of a 1031 nm mode-locked Nd:YVO4 laser and an optical parametric oscillator (OPO) tunable between 700–960 nm (pumped by the second harmonic of the 1031 nm laser). The OPO wavelength was set to 791.8 nm to drive the symmetric stretching vibration of CH3 at 2930 cm−1. The quadratic dependence of the CARS signal on the number density of the probed C–H vibrational group, inherently present in the ELP polymer structure, provides sharp contrast for the polymer-dense regions without the need for external labels or other disruptive sample preparations. A photomultiplier tube (Hamamatsu, R6357) was used to detect the CARS signal produced by simultaneously scanning the two excitation beams over the sample pixel-by-pixel. The input power at the sample position was 30 mW for the pump (OPO) beam and 10 mW for the Stokes (1031 nm) beam.
For each of the nine experimental conditions, five image stacks at different positions in the same sample were acquired. Each stack consisted of 21 slices at a resolution of 512 × 512 pixels (31.82 × 31.82 μm) per image with a dwell time of 10.3 μs/pixel and 2x frame averaging. Slices were spaced 0.4 μm apart for a total imaging depth of 8 μm. All images were analyzed using the Fiji distribution package of ImageJ.[64] Each stack was processed using east shadow correction, Gaussian blur, background subtraction, and Fourier bandpass filtering. Particles were thresholded using the method of Rényi entropy, segmented with the “Disconnect Particles” command, and quantified using the “3D Objects Counter” command.[65]
Optical Density Characterization:
HELP-SPAAC gels of 40 μL were formed at the bottom of a 96-well plate and optical density was measured with a temperature-controlled plate reader (BioTek Synergy Neo2 Hybrid Multi-Mode Reader). For all gels, precursor solutions were kept on ice prior to mixing. To characterize the optical density of gels formed using temperature profile I, the 96-well plate was incubated at 4 °C for 5 min before being placed in the plate reader, after which it was allowed to crosslink according to the temperature profiles described in Figure 3B. For temperature profiles II and III, the plate was immediately placed in the plate reader and allowed to crosslink using the temperature profiles described in Figure 3B. Absorbance was measured at 500 nm. Once the plate was placed inside the plate reader, measurements were taken every 30 seconds over the course of the 30 min gelation process. Final transmittance was taken to be the values after 30 min of these measurements.
Cell Culture and Encapsulation:
hMSCs from a 25 year old male donor were purchased from Lonza (PT-2501), expanded in Dulbecco’s modified Eagle medium (DMEM, Fisher) plus 10% fetal bovine serum (FBS, Fisher), and used between passage 3–6. HUVECs from pooled donors were purchased from Lonza (C2519A), expanded in EGM-2 (Lonza), and used between passage 3–5. As previously reported,[66] human induced pluripotent stem cells isolated from at 27 year old male were differentiated in N3 media consisting of DMEM/F12 (Fisher), Neurobasal (Fisher), 1% N-2 Supplement (Fisher), 2% B-27 Supplement (Fisher), 1% GlutaMax (Fisher), 1% MEM NEAA (Fisher), and 2.5 μg mL−1 human recombinant insulin (Fisher). For the first 11 days, N3 media was further supplemented with 5 × 10−6 M SB-431542 (Tocris) and 100 × 10−9 M LDN-193189 (Stemgent). At Day 12, the cells were dissociated with Cell Dissociation Solution and plated onto plates coated with 50 μg mL−1 Poly-D-Ornithine and 5 μg mL−1 Laminin (Roche). hiPSC-derived NPCs were then cultured in N3 media without SB-431542 or LDN-193189 until Day 16 when they were dissociated and encapsulated. hiPSC-derived NPCs were used between passage 18–22.
Prior to encapsulation in HELP-SPAAC hydrogels, cells were dissociated, counted, and pelleted by centrifugation. Gel solution was prepared by mixing a 2% w/v ELP-azide solution with a 2% w/v HA-BCN solution in an Eppendorf tube. For all gels, precursor solutions were kept on ice prior to mixing. Cell pellets were resuspended in the gel solution at the desired final cell densities (final densities: 1 × 106 cells mL−1 for hMSCs, 7.5 × 106 cells mL−1 for HUVECs, and 3 × 107 cells mL−1 for hNPCs), which are consistent with densities used in other 3D culture methods.[9, 33, 53] The cell suspensions were mixed thoroughly, and 10 μL gels were cast in 4 mm diameter, 0.8 mm height silicone molds, which were then placed into 24-well plates for culture. The gels were allowed to crosslink according to the temperature profiles described in Figure 3B and then were covered with the appropriate expansion medium. hMSC and HUVEC media were replenished every other day, and hNPC media was replenished daily.
Cell Viability Measurements:
On Day 1 and Day 7 of culture, cell viability was characterized using Live/Dead staining (Life Technologies). Briefly, live (calcein AM) and dead (ethidium homodimer-1) stains were diluted in DPBS according to the manufacturer’s protocol, and gels were allowed to incubate for 10 min at 37 °C in the diluted solution. After incubation, samples were inverted onto a glass coverslip and imaged using a Leica SPE confocal microscope. Fiji was used to measure the number of cells with intact or damaged membranes, and results were plotted in GraphPad Prism 9.0.
Immunocytochemistry:
Cells are fixed and stained within the hydrogel. To prepare samples for fixation, each well was washed with PBS. Samples were fixed by incubating with 4% paraformaldehyde (PFA) at 37 °C for 30 min. Fixation solution was then aspirated and three 10 min washes of PBS were performed. Cells were permeabilized for 1 hour with 0.25% v/v Triton X-100 in PBS (PBST), then blocked for 3 hours in PBS with 5% w/v bovine serum albumin (BSA, Roche), 5% v/v goat serum (Gibco), and 0.5% v/v Triton X-100. Primary antibody dilutions were prepared in PBS with 2.5% w/v BSA, 2.5% v/v goat serum, and 0.5% v/v Triton X-100 (Antibody Dilution Solution) using the following antibodies as appropriate: mouse anti-HCAM (1:100, Santa Cruz Biotechnology no. sc-7297), mouse anti-CD31 (1:200, Abcam no. ab24590), and mouse anti-β3-Tubulin (1:200, Cell Signaling no. 4466). Primary antibody incubation was performed overnight at 4 °C. Antibody solutions were removed, and three 30 min washes in PBST were performed. Secondary antibody staining was performed using goat anti-mouse Alexa Fluor 488 (1:500; Invitrogen, no. A-11001) diluted in Antibody Dilution Solution. DAPI (1 μg mL−1) and phalloidin-TRITC (0.2 μg mL−1) were included to stain for nuclei and F-actin, respectively. Secondary antibody incubation was performed overnight at 4 °C. Secondary antibody solution was then removed and three 20 min washes in PBST were performed. Samples were then mounted onto a no. 1 coverslip using ProLong Gold Antifade mounting medium (Fisher) and allowed to cure for 24 hours before imaging on a Leica SPE confocal microscope. Images were taken at a depth at least ~50 μm away from the coverglass and were presented as maximum intensity projections (100 μm z-stack depth).
Image Analysis:
Actin spread area was analyzed from fluorescent actin images using CellProfiler.[67] For each cell, nuclei were identified using the “IdentifyPrimaryObjects” command with a “Minimum Cross-Entropy” thresholding method. For hMSCs and hNPCs, actin area was normalized on a per cell basis. To do so, the “IdentifySecondaryObjects” command with a “Propagation” method was used, which identifies actin objects using the nuclei as a starting point. This method will find dividing lines between clumped objects where the secondary object shows a change in staining, such as a dimmer or brighter line, using local image similarity to identify boundaries between cells. For HUVECs, actin area is reported per multicellular cluster, where the “IdentifyPrimaryObjects” command was used to identify distinct actin objects. The area of each actin object was plotted using GraphPad Prism 9.0.
Statistical Analysis:
Statistical analysis and plotting were performed using GraphPad Prism 9.0. In vitro experiments had three independent gel samples in each experiment. Statistical significance was assessed using a one-way ANOVA with Tukey post-hoc test, unless otherwise noted. No outlier tests were performed, and all data points were included in the analyses. All errors are reported as the standard deviation of error (SD).
Supplementary Material
Acknowledgements
R.S.N. and M.S.H. contributed equally to this work. The authors acknowledge Prof. Theo Palmer (Stanford Neurosurgery) for providing the iPSCs; Prof. Eric Appel (Stanford Materials Science & Engineering) for use of plate-reading equipment; Dr. Patrik Johansson for guidance during CARS microscopy measurements; and Lucia Brunel for helpful discussion on biorthogonal click-chemistry and rheological characterization. Part of this work was performed at the Stanford Nano Shared Facilities (SNSF), supported by the National Science Foundation under award ECCS-1542152. The authors acknowledge financial support from the US National Institutes of Health (R21 NS114549, R01 HL142718, R01 HL151997, and R01 EB027171) and the US National Science Foundation (CBET 2033302 and DMR 2103812) to S.C.H.; the National Science Foundation Graduate Research Fellowship Program (DGE-1656518) to M.S.H., J.G.R., and C.M.L.; the Stanford ChEM-H O’Leary-Thiry Graduate Fellowship to M.S.H.; the Stanford Smith Family Graduate Fellowship to J.G.R.; and the Stanford Bio-X Fellowship to C.M.L.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Renato S. Navarro, Department of Materials Science & Engineering, Stanford University, 476 Lomita Mall, McCullough Room 246, Stanford, CA 94305, USA.
Michelle S. Huan, Department of Chemical Engineering, Stanford University, Stanford, CA 94305, USA.
Julien G. Roth, Institute for Stem Cell Biology & Regenerative Medicine, Stanford University School of Medicine, Stanford, CA 94305, USA
Kelsea M. Hubka, Maternal & Child Health Research Institute, Stanford University School of Medicine, Stanford, CA 94305, USA
Chris M. Long, Department of Materials Science & Engineering, Stanford University, 476 Lomita Mall, McCullough Room 246, Stanford, CA 94305, USA
Annika Enejder, Department of Materials Science & Engineering, Stanford University, 476 Lomita Mall, McCullough Room 246, Stanford, CA 94305, USA.
Sarah C. Heilshorn, Department of Materials Science & Engineering, Stanford University, 476 Lomita Mall, McCullough Room 246, Stanford, CA 94305, USA
References
- [1].Lee KY, Mooney DJ, Chem. Rev. 2001, 101, 1869. [DOI] [PubMed] [Google Scholar]
- [2].Kyburz KA, Anseth KS, Ann. Biomed. Eng. 2015, 43, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Madl CM, Heilshorn SC, Annu. Rev. Biomed. Eng. 2018, 20, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hughes CS, Postovit LM, Lajoie GA, Proteomics 2010, 10, 1886. [DOI] [PubMed] [Google Scholar]
- [5].Burdick JA, Anseth KS, Biomaterials 2002, 23, 4315. [DOI] [PubMed] [Google Scholar]
- [6].Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA, Adv. Mater. 2003, 15, 888. [Google Scholar]
- [7].Morton LD, Hillsley A, Austin MJ, Rosales AM, J. Mater. Chem. B 2020, 8, 6925. [DOI] [PubMed] [Google Scholar]
- [8].Ravi S, Krishnamurthy VR, Caves JM, Haller CA, Chaikof EL, Acta Biomater. 2012, 8, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Madl CM, LeSavage BL, Dewi RE, Dinh CB, Stowers RS, Khariton M, Lampe KJ, Nguyen D, Chaudhuri O, Enejder A, Heilshorn SC, Nat. Mater. 2017, 16, 1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sun F, Zhang W-B, Mahdavi A, Arnold FH, Tirrell DA, Proc. Natl. Acad. Sci. USA 2014, 111, 11269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Varanko AK, Su JC, Chilkoti A, Annu. Rev. Biomed. Eng. 2020, 22, 343. [DOI] [PubMed] [Google Scholar]
- [12].Discher DE, Janmey P, Wang YL, Science 2005, 310, 1139. [DOI] [PubMed] [Google Scholar]
- [13].Hadjipanayi E, Mudera V, Brown RA, J. Tissue. Eng. Regen. Med. 2009, 3, 77. [DOI] [PubMed] [Google Scholar]
- [14].Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS, Cell Stem Cell 2009, 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gilbert PM, Weaver VM, Semin. Cell Dev. Biol. 2017, 67, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Holloway JL, Ma H, Rai R, Burdick JA, J. Control. Release 2014, 191, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang H, Zhu D, Paul A, Cai L, Enejder A, Yang F, Heilshorn SC, Adv. Funct. Mater. 2017, 27, 1605609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Urry DW, Okamoto K, Harris RD, Hendrix CF, Long MM, Biochemistry 1976, 15, 4083. [DOI] [PubMed] [Google Scholar]
- [19].Urry DW, Gowda DC, Parker TM, Luan C-H, Reid MC, Harris CM, Pattanaik A, Harris RD, Biopolymers 1992, 32, 1243. [DOI] [PubMed] [Google Scholar]
- [20].Catherine C, Oh SJ, Lee K-H, Min S-E, Won J-I, Yun H, Kim D-M, Biotechnol. Bioprocess Eng. 2015, 20, 417. [Google Scholar]
- [21].Urry DW, Luan CH, Parker TM, Gowda DC, Prasad KU, Reid MC, Safavy A, J. Am. Chem. Soc. 1991, 113, 4346. [Google Scholar]
- [22].Ribeiro A, Arias FJ, Reguera J, Alonso M, Rodríguez-Cabello JC, Biophys. J. 2009, 97, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang H, Paul A, Nguyen D, Enejder A, Heilshorn SC, ACS Appl. Mater. Interfaces 2018, 10, 21808. [DOI] [PubMed] [Google Scholar]
- [24].Hollingshead S, Liu JC, Macromol. Biosci. 2020, 20, 1900369. [DOI] [PubMed] [Google Scholar]
- [25].Van Durme K, Rahier H, Van Mele B, Macromolecules 2005, 38, 10155. [Google Scholar]
- [26].Reguera J, Urry DW, Parker TM, McPherson DT, Rodríguez-Cabello JC, Biomacromolecules 2007, 8, 354. [DOI] [PubMed] [Google Scholar]
- [27].Hunt DR, Klett KC, Mascharak S, Wang H, Gong D, Lou J, Li X, Cai PC, Suhar RA, Co JY, LeSavage BL, Foster AA, Guan Y, Amieva MR, Peltz G, Xia Y, Kuo CJ, Heilshorn SC, Adv. Sci. 2021, 8, 2004705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haider M, Cappello J, Ghandehari H, Leong KW, Pharm. Res. 2008, 25, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Straley KS, Heilshorn SC, Soft Matter 2009, 5, 114. [Google Scholar]
- [30].Roth JG, Huang MS, Li TL, Feig VR, Jiang Y, Cui B, Greely HT, Bao Z, Paşca SP, Heilshorn SC, Nat. Rev. Neurosci. 2021, 22, 593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].LeSavage BL, Suhar NA, Madl CM, Heilshorn SC, J. Vis. Exp. 2018, e57739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chung C, Lampe KJ, Heilshorn SC, Biomacromolecules 2012, 13, 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Madl CM, Katz LM, Heilshorn SC, Adv. Funct. Mater. 2016, 26, 3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Veronese FM, Biomaterials 2001, 22, 405. [DOI] [PubMed] [Google Scholar]
- [35].Harris JM, Chess RB, Nat. Rev. Drug Discov. 2003, 2, 214. [DOI] [PubMed] [Google Scholar]
- [36].Nischan N, Hackenberger CPR, J. Org. Chem. 2014, 79, 10727. [DOI] [PubMed] [Google Scholar]
- [37].Madl CM, Heilshorn SC, Chem. Mater. 2019, 31, 8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li NK, Quiroz FG, Hall CK, Chilkoti A, Yingling YG, Biomacromolecules 2014, 15, 3522. [DOI] [PubMed] [Google Scholar]
- [39].Wolf KJ, Kumar S, ACS Biomater. Sci. Eng. 2019, 5, 3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Selegård R, Aronsson C, Brommesson C, Dånmark S, Aili D, Sci. Rep. 2017, 7, 7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Meyer DE, Trabbic-Carlson K, Chilkoti A, Biotechnol. Prog. 2001, 17, 720. [DOI] [PubMed] [Google Scholar]
- [42].Meco E, Lampe KJ, Biomacromolecules 2019, 20, 1914. [DOI] [PubMed] [Google Scholar]
- [43].Camp CH Jr, Cicerone MT, Nat. Photonics 2015, 9, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang H, Cai L, Paul A, Enejder A, Heilshorn SC, Biomacromolecules 2014, 15, 3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Paul A, Stührenberg M, Chen S, Rhee D, Lee WK, Odom TW, Heilshorn SC, Enejder A, Soft Matter 2017, 13, 5665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Downes A, Mouras R, Elfick A, J. Raman Spectrosc. 2009, 40, 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H.-p., Lippens E, Duda GN, Mooney DJ, Nat. Mater 2016, 15, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick JA, Nat. Mater. 2013, 12, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen D-HT, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS, Nat. Mater. 2012, 11, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kapr J, Petersilie L, Distler T, Lauria I, Bendt F, Sauter CM, Boccaccini AR, Rose CR, Fritsche E, Adv. Healthcare Mater. 2021, 10, 2100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali OA, Bencherif SA, Rivera-Feliciano J, Mooney DJ, Nat. Mater. 2010, 9, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pek YS, Wan ACA, Ying JY, Biomaterials 2010, 31, 385. [DOI] [PubMed] [Google Scholar]
- [53].Caliari SR, Vega SL, Kwon M, Soulas EM, Burdick JA, Biomaterials 2016, 103, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bordeleau F, Mason BN, Lollis EM, Mazzola M, Zanotelli MR, Somasegar S, Califano JP, Montague C, LaValley DJ, Huynh J, Mencia-Trinchant N, Negrón Abril YL, Hassane DC, Bonassar LJ, Butcher JT, Weiss RS, Reinhart-King CA, Proc. Natl. Acad. Sci. USA 2017, 114, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schultz KM, Kyburz KA, Anseth KS, Proc. Natl. Acad. Sci. USA 2015, 112, E3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Humphrey JD, Dufresne ER, Schwartz MA, Nat. Rev. Mol. Cell Biol. 2014, 15, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chung C, Anderson E, Pera RR, Pruitt BL, Heilshorn SC, Soft Matter 2012, 8, 10141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].DiMarco RL, Dewi RE, Bernal G, Kuo C, Heilshorn SC, Biomater. Sci. 2015, 3, 1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Shi P, Shen K, Ghassemi S, Hone J, Kam LC, Cell Mol. Bioeng. 2009, 2, 464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Banerjee A, Arha M, Choudhary S, Ashton RS, Bhatia SR, Schaffer DV, Kane RS, Biomaterials 2009, 30, 4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Seidlits SK, Khaing ZZ, Petersen RR, Nickels JD, Vanscoy JE, Shear JB, Schmidt CE, Biomaterials 2010, 31, 3930. [DOI] [PubMed] [Google Scholar]
- [62].Lampe KJ, Antaris AL, Heilshorn SC, Acta Biomater. 2013, 9, 5590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zustiak SP, Pubill S, Ribeiro A, Leach JB, Biotechnol. Prog. 2013, 29, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Nat. Methods 2012, 9, 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kapur JN, Sahoo PK, Wong AKC, Comput. Graphics Image Process. 1985, 29, 273. [Google Scholar]
- [66].Shi Y, Kirwan P, Smith J, Robinson HPC, Livesey FJ, Nat. Neurosci. 2012, 15, 477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, Wiegraebe W, Singh S, Becker T, Caicedo JC, Carpenter AE, PLoS Biol. 2018, 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.