

Abstract
Objective
NLRP3 inflammasome regulates T cell responses. This study examined the roles of NLRP3 inflammasome activation in the regulation of Tfh cells during humoral response to T dependent antigens and in systemic lupus erythematosus (SLE).
Methods
NLRP3 inflammasome activation of Tfh cells was studied in B6, MRL/lpr and NZM2328 mice and in SLE patients and healthy controls using a fluorescence-labeled caspase-1 inhibitor probe. MCC950, a selective inhibitor of NLRP3, was used to investigate the relation between NLRP3 inflammasome activation and germinal center (GC) reaction, Ab responses to immunization, and autoantibody production.
Results
NLRP3 inflammasome activation in Tfh cells after immunization was identified in B6 mice. MCC950 inhibited humoral responses to sRBC and NP-CGG with reduction of the GC reaction. B6 mice with lymphoid cell-specific deletion of NLRP3 or Casp1 mounted sub-optimal humoral responses with impaired GC formation and defective affinity maturation. In MRL/lpr and NZM2328 mice, inhibition of NLRP3 activation suppressed NLRP3 activated Tfh cell expansion as well as attenuated lupus-like phenotypes. Tfh cells with activated NLRP3 inflammasome exhibited increased expression of molecules for Tfh cell function and differentiation, and had greater ability to activate B cells. In SLE patients, disease activity was positively correlated with an increase in the activated NLRP3+ Tfh population and this population was markedly reduced in response to therapy.
Conclusions
The activation of NLRP3 inflammasome in Tfh cells is an integral part of responses to immunization. The activated NLRP3+ Tfh population is essential for optimal humoral responses, GC formation and autoimmunity.
Keywords: Autoantibodies, Autoimmunity, Systemic Lupus Erythematosus, T-Lymphocyte subsets, Inflammation
INTRODUCTION
T follicular helper (Tfh) cells, a subset of T helper cells that localize to B cell follicles, are essential for Germinal Center (GC) formation and the generation of high affinity B cells, (1, 2). Tfh cells provide help signals to GC B cells via a combination of secreted cytokines and expressed cell surface receptors, (1, 2). Although IL-1β has recently been shown to be made by T cells as a result of inflammasome activation, (3–5), the role of Tfh inflammasome activation in antibody responses and in GC formation remains largely undetermined.
The assembly of NLRP3 inflammasome leads to activation of caspase-1, which is required for the cleavage of the interleukins IL-1β and IL-18, (6) to their active secretory forms. The effects of NLRP3 inflammasome on adaptive immune response have been described mainly through the active IL-1β secreted by non-B and non-T cells, (7, 8). In the case of adaptive humoral responses, little information is available regarding the Tfh cell inflammasome expression and the effects of Tfh cell inflammasome activation on Ab formation. In view of recent studies showing that CD4+ T cells can also secrete IL-1β though inflammasome activations, (3–5), the role of Tfh NLRP3 inflammasome activation in humoral response is a timely area to be investigated.
We have been interested in the role of NLRP3 inflammasome activation in the pathogenesis of lupus nephritis, (9, 10) with emphasis on the small molecule inhibitor MCC950 specific for NLRP3. MCC950 was effective in modulating lupus nephritis with prolonged survival time. Further studies to be presented here show that anti-dsDNA Ab titers in both MCC950-treated MRL/lpr and NZM2328 mice were markedly diminished suggesting that MCC950 has inhibitory effects on autoantibodies formation in autoimmune prone strains of mice. It provides the rationale and impetus to determine whether NLRP3 activation is inherent in Tfh cells and whether this activation is essential in normal humoral responses and in autoimmunity. The experimental results further support the conclusion that Tfh cells are required for systemic autoimmunity, (11) with the important added information that Tfh cells with activated NLRP3 are essential for systemic autoimmunity.
METHODS
Patients
Seventy-one patients from the First Affiliated Hospital, Sun Yat-sen University fulfilling ACR criteria for SLE, (12) were screened with the SLEDAI scoring system, (13). Demographic and clinical characteristics of these SLE patients are shown in online supplementary material table S1. Twenty-five age and sex matched healthy donors were enrolled. This study was approved by the Ethics Committee for Clinical Research, the First Affiliated Hospital, Sun Yat-sen University. Written informed consent was obtained from all subjects.
Eleven SLE patients were followed longitudinally. They experienced a relapse and were treated again as inpatients. Blood samples were obtained before the initiation of treatment and at 4 weeks after treatment. The clinical characteristics of these patients are shown in online supplementary material table S2.
Human peripheral blood mononuclear cells purification, flow-cytometric analysis and immunofluorescence studies
Detailed methods are described in the online supplemental methods section.
Mice and treatments
Only female mice were used in this investigation. B6.NLRP3fl/fl mice were obtained from Taconic and B6.Casp1fl/fl mice were kindly provided by Dr. John Luckens at the University of Virginia. B6 and B6.CreLck with distal Lck promotor were obtained from Jackson Lab. (B6.CreLck × B6.NLRP3fl/fl)F2 were screened for B6.NLRP3fl/flCreLck and suitable mating pairs were set up for the generation of cohorts of B6.NLRP3fl/flCreLck. Similarly cohorts of B6.Casp1fl/flCreLck were generated for experiments described in the online supplementary figure S1 legend. Specific deletion in lymphoid cells was shown with isolated lymphocytes and macrophages by PCR with primers flanking the flox sites.
All MCC950 or vehicle treatments of MRL/lpr have been described, (9, 10). MRL/lpr mice were purchased from SLAC Laboratory Animal Company (Shanghai, China). NZM2328 mice, (14) were obtained from the University of Virginia. B6 mice were from the Experimental Animal Center at Sun Yat-sen University.
Animal studies were approved by the Institutional Animal Care and Use Committee (IACUC), Sun Yat-sen University (SYSU-IACUC-2018-000181) and by the University of Virginia Animal Care and Use Committee Protocol #1849.
NZM2328 mice (12 week-old) were injected with 1×107 particles of adenovirus-expressing interferon-α (adIFN-α) according to Dai et al, (15). One day after adIFNα, the mice were treated intraperitoneally with either MCC950 or vehicle. The mice were sacrificed 6 weeks after adIFNα injection.
Isolation of mouse spleen and lymph nodes lymphocytes, flow cytometry analysis and GC studies by splenic PNA staining.
Please see the online supplemental methods section.
Immunization and assays for specific Ab production
For sheep RBC (sRBC) immunization, 108 cells were injected ip without adjuvant. The same dose was given 14 days later. Sera were assayed by ELISA for anti-sRBC Abs 3 and 6 weeks later with sRBC lysate as the substrate. For NP33-CGG immunization, 100μg was given in the footpads without adjuvant. Sera at day 7 and day 14 were collected for assaying by ELISA with NP2-BSA and NP14-BSA as substrates for high affinity and low/moderate-affinity anti-NP Ab respectively, (16).
In vitro induction of B-cell activation by Tfh cells and IgG production by Tfh cells
Please see the online supplemental methods section.
Measurement of anti-dsDNA antibodies and cytokine levels, and evaluation of renal history and immune complex deposition.
These procedures have been described previously, (10).
Statistical analysis
Data are presented as the mean ± SD or as median ± interquartile range. Statistical analysis was performed using GraphPad Prism 5.0. Statistical differences were assessed by Student’s t-test, one-way analysis of variance (ANOVA) or non-parametric Mann-Whitney U-test. Spearman’s rank correlation test was used to determine correlation. Chi-square analysis was performed to compare the incidence of severe proteinuria between groups. A p-value < 0.05 was considered statistically significant.
RESULTS
Identification of act. NLRP3+ Tfh cells in normal B6 mice.
CD4+CXCR5+ Tfh cells that have act. caspase-1 can be readily identified in the B cell follicle and at the T-B border in the GC of the draining lymph nodes of the B6 mouse 5 days after immunization with NP33-CGG with alum in the high footpad (figure 1A). Act. caspase-1+ Tfh cells interacting with IgD+ B cells are shown in the insert of figure 1A. With an ImageStreamX MKII instrument (figure 1B), 7 cells that were act. caspase-1+CD4+ CXCR5+ expressed various amounts of IL-1β. It is apparent that the markers of interest expressed by these cells are very heterogeneous inferring that this population is changing continuously. To seek further support that Tfh cells make IL-1β with NLRP3 activation in response to immunizations, CD3+CXCR5+ cells with act. caspase-1+ making IL-1β were identified (figure 1C and 1D). This population was estimated to be 10% of Tfh cells. sRBC immunization increased splenic act. caspase-1+ Tfh from 2–3% to 7.5–10% (figure 2A). From the CD4+CXCR5+Foxp3− Tfh population, act. caspase-1+ and act. caspase-1− sub-populations were isolated. They were characterized by the ImageStreamX MKII imaging flow cytometer (figure 2B). The expression of mRNA for Rip3, Casp1, NLRP3, IL-1β, Bcl-6, IFNγ and CD40 were significantly increased in the act. caspase-1+ cells (figure 2C). However IL-10 was increased in the act. caspase-1− Tfh fraction. It is of note that IL-18 mRNA was not detected.
Figure 1. CXCR5+ CD4+ or CD3+ Tfh cells in the lymph node of a B6 mouse five days after the immunization with NP33-CGG with alum in the hind footpads express act. caspase-1+ and IL-1β.
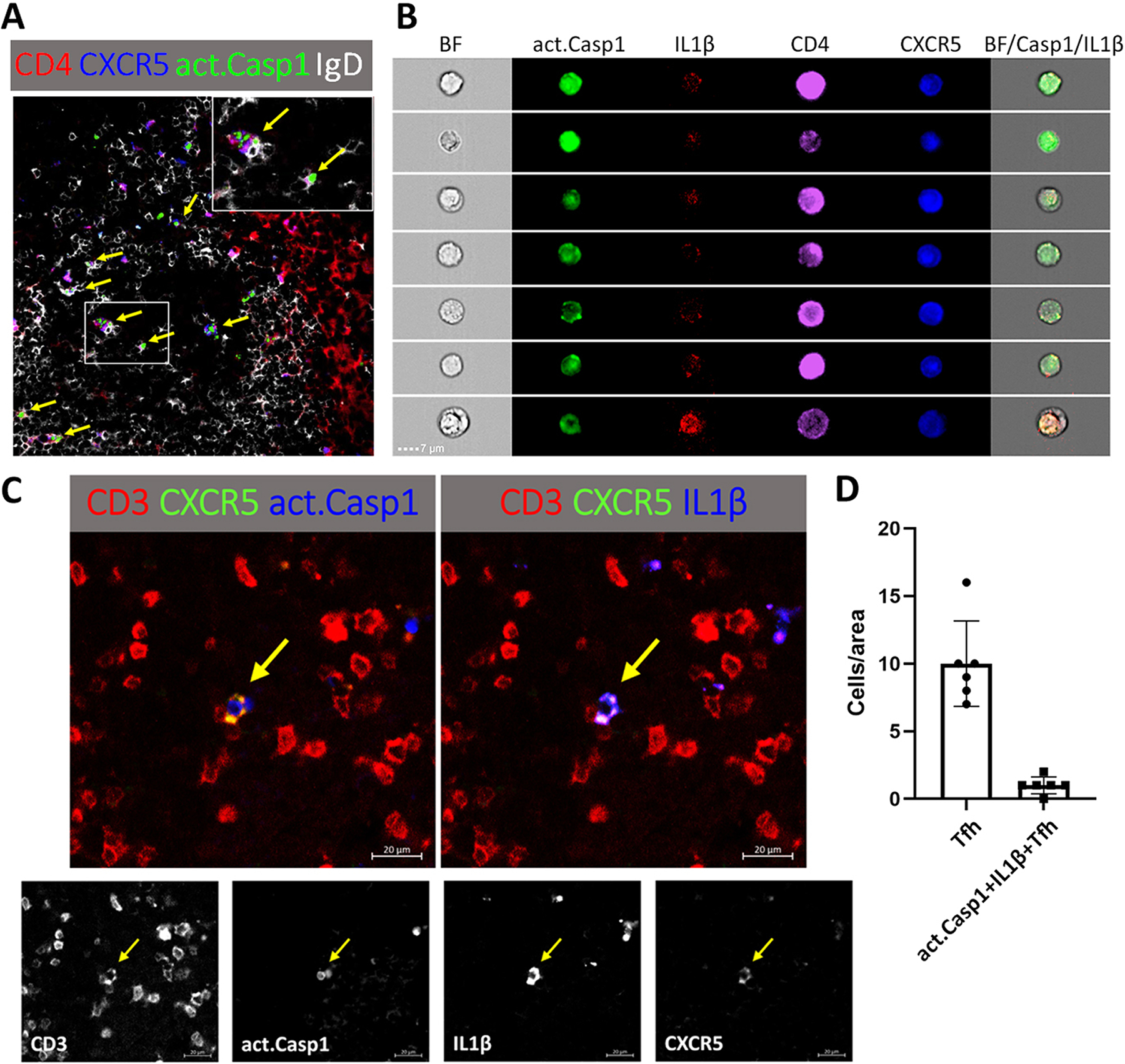
(A) Many of the act. caspase-1+ Tfh cells in the interface between T cell B cell zones and in the dark zone of the GC are identified by arrows. The inset shows interaction between act. caspase-1+ Tfh cells with IgD+ B cells. (B) CD4+CXCR5+ T cells as seen by the ImageStreamX MKII imaging flow cytometer with act. caspase-1 stained green, IL-1β red, CD4 purple and CXCR5 blue. (C) A CD3+CXCR5+ T cell stained for act. caspase-1 and IL-1β. (D) CD3+CXCR5+Tfh and caspase-1+ IL1β+ Tfh were counted in each 300 μm × 300 μm area.
Figure 2. Characterization of act. caspase-1+ Tfh cells.
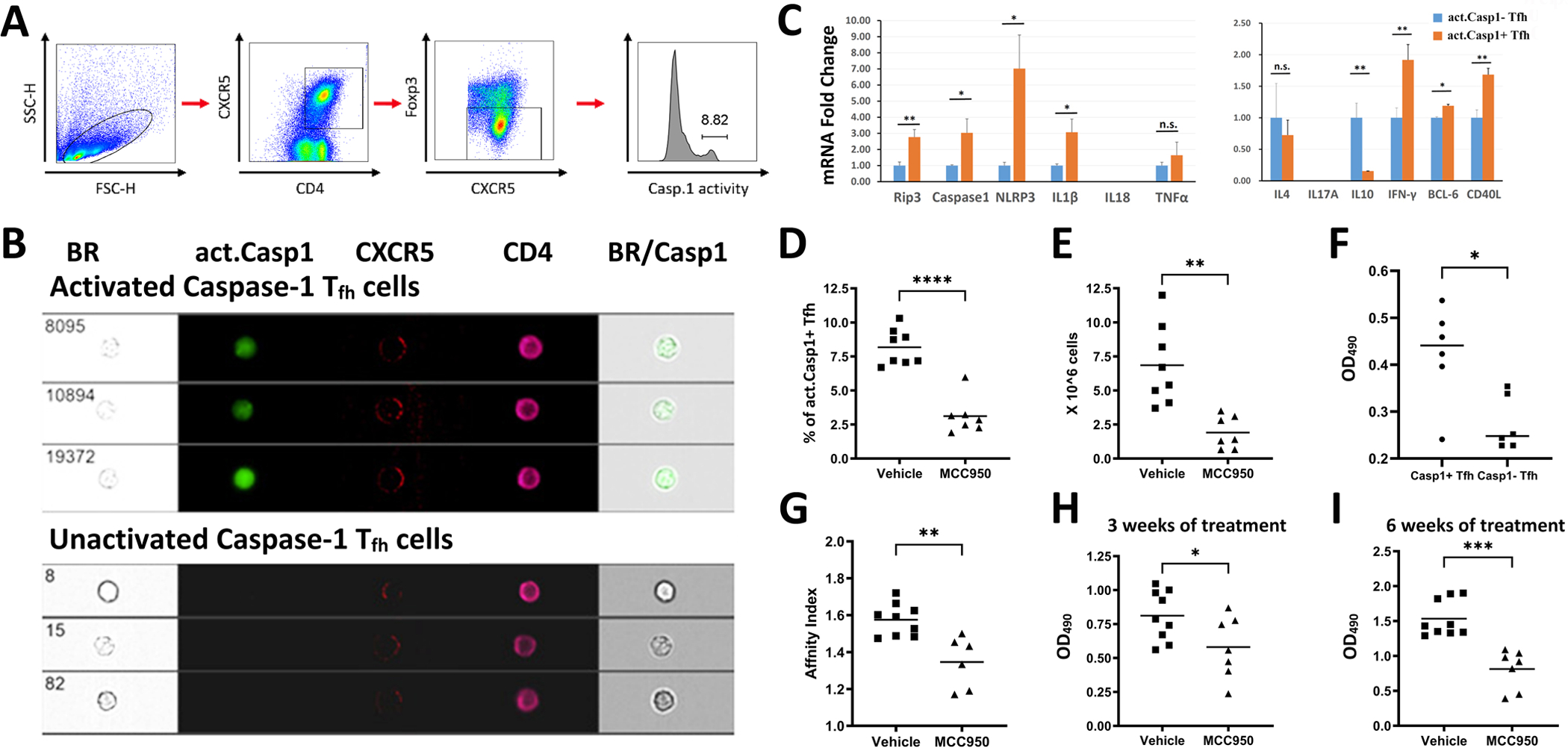
B6 mice were immunized with sheep Red Blood Cells (sRBC) as described in Methods. (A) Gating strategy for Tfh cells: Lymphocytes were gated by forward light scattering. CD4+CXCR5+ and FoxP3− cells were selected as Tfh cells. (B) The Tfh cells were sorted into act. caspase-1+ and act. caspase-1− cell populations. (C) mRNA expression of genes of interest in act. caspase-1+ Tfh and act. caspase-1− Tfh cells isolated from the spleens of immunized mice were interrogated by RT/PCR. (D) Act. caspase-1+ Tfh cells were elevated in the spleen of mice immunized with sRBC. This increase was inhibited by MCC950. (E) Total lymphocyte counts in the lymph nodes of immunized mice were decreased with MCC950. (F) Act. caspase-1+ Tfh cells provided better help to induce IgG secretion by B cells in comparison with act. caspase-1− Tfh cells. (G) MCC950 or vehicle treatment commenced five days after the second immunization, reduced affinity maturation to sRBC. Reduction of serum IgG anti-sRBC Ab in mice treated with MCC950 at three weeks (H) and at six weeks (I) after the secondary immunization. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
To demonstrate a role of NLRP3 activation in the humoral response MCC950, a small molecular NLRP3 inhibitor was given every other day for six weeks, five days after the second immunization. This treatment reduced act. caspase-1+ cells from ~8% to ~3% (figure 2D). MCC950 also reduced lymph node sizes (figure 2E).
In a five day in vitro experiment, act. caspase-1+ Tfh cells isolated 5 days after the second immunization induced significantly more IgG secretion by B cells with p<0.05 in comparison with act. caspase-1− Tfh cells (figure 2F). Sera were obtained three and six weeks after the second immunization. Affinity maturation indices as measured according to MacDonald et al. (17) were significantly reduced with p<0.01 (figure 2G). It is of note that affinity maturation indices were lower six weeks after treatment (data not shown). However, the differences were not statistically significant. MCC950 treatment significantly reduced anti-sRBC Ab when assayed at 1/100 dilution with p< 0.05 at three weeks and p<0.001 at six weeks (figures 2H and 2I).
MCC950 inhibits GC formation by the marked decrease in B cell division within the B cell zone of the GC.
B6 were immunized with NP33-CGG ip with alum as an adjuvant. Ki67 was used as a marker for proliferating cells. MCC950 inhibited B cell proliferation in the dark zone of the GC in the spleen (figure 3A). MCC950 significantly decreased Ki67 levels with p<0.001 as measured by the ImageJ software in comparison to GC of mice treated with vehicle (figure 3B). With NP2–BSA as the substrate, MCC950 inhibited the formation of high affinity IgG anti-NP Ab (figure 3C).
Figure 3. Inhibition of GC formation and reduction of NP-specific Ab production by MCC950.
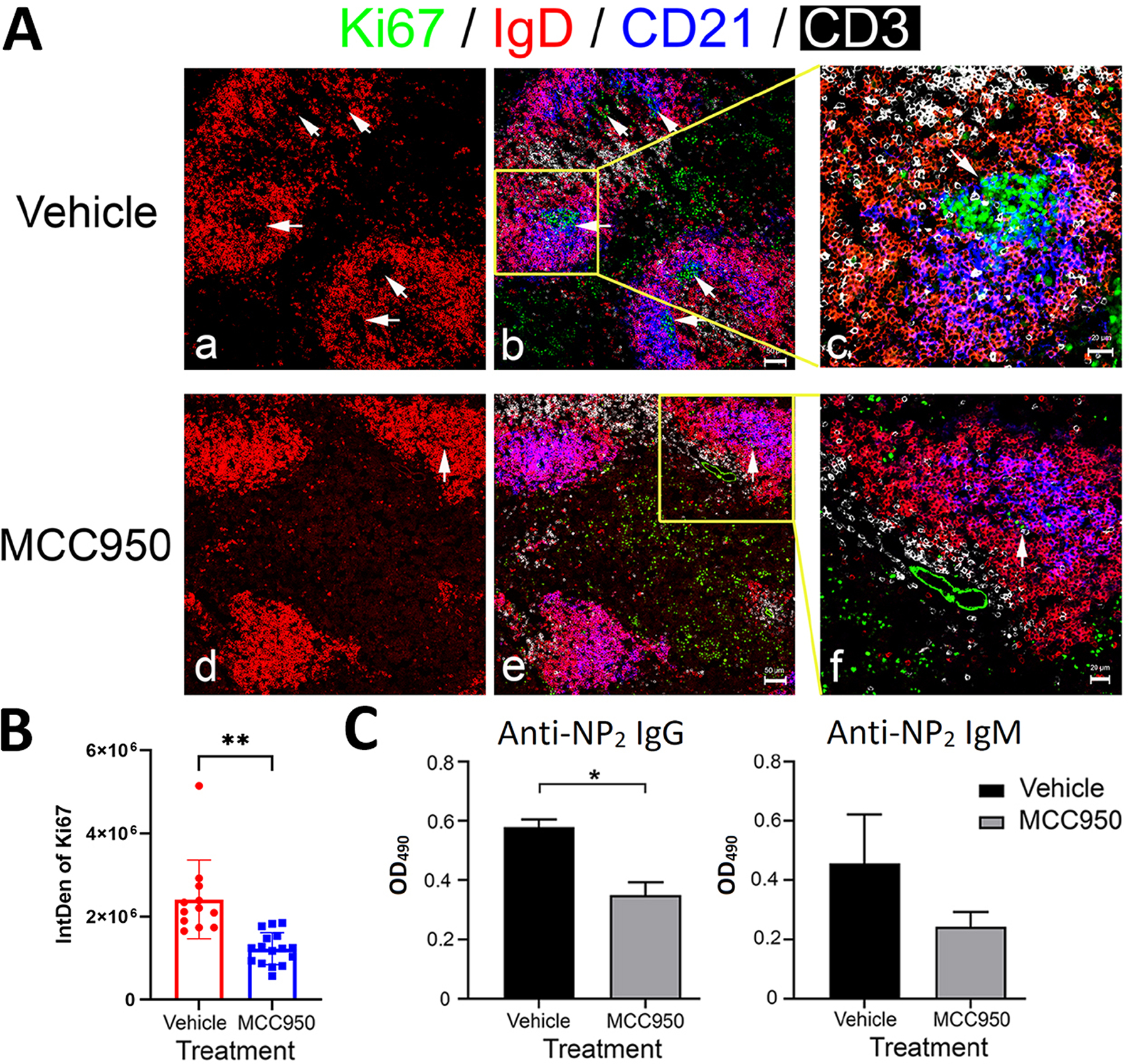
B6 female mice were immunized i.p. with 100 μg NP33-CGG and alum. They were treated i.p. daily with vehicle, MCC950 (20 mg/Kg). (A) Splenic GC were stained as labeled indicated by arrows. There is an absence of centroblast division in the spleen of MCC950 treated mice. (B) The integrated density (IntDen) of Ki67 staining in each GC was calculated by the ImageJ software. (C) Anti-NP IgM and IgG levels in serum samples 14 days after immunization were assayed by ELISA using NP2-BSA as coating Ag. Mean absorbance from 10,000-fold diluted samples of 5 mice per group is shown. *p < 0.05.
Lymphocyte-specific deletion of NLRP3 impeded Ab production to T dependent Ag with inhibition of GC formation.
With B6.NLRP3fl/fl from Taconic and B6.CreLck with distal promotor from Jackson Lab, B6.NLRP3fl/flCreLck were generated. B6.NLRP3fl/flCreLck and B6. NLRP3fl/fl mice were immunized with NP33-CGG without adjuvant. As shown in figure 4A, the only impaired IgM response was day 7 in B6.NLRP3fl/flCreLck mice with NLRP3 deletion in T cells in comparison with the response by B6.NLRP3fl/fl (p<0.05). Seven days after immunization, IgM responses of B6.NLRP3fl/flCreLck mice were impaired in comparison with those from B6.NLRP3fl/fl (p<0.05). IgG anti-NP2 and anti-NP14 Ab responses were impaired in mice with deletion of NLRP3 in their T cells with p<0.001 and p<0.01 respectively. At day 14, both IgG anti-NP2 and IgG anti-NP14 Ab responses were impaired in mice with deletion of NLRP3 in their T cells with p<0.05. We harvested the spleens of these two groups of mice. In mice with NLRP3 deletion in their T cells, GC formation was impaired with marked decrease in proliferation of centroblasts that stained for Ki67 in the dark zone (Fig. 4Be–4Bh vs 4Ba–4Bd), suggesting that activated NLRP3 is essential for high affinity Ab response and for GC formation.
Figure 4.
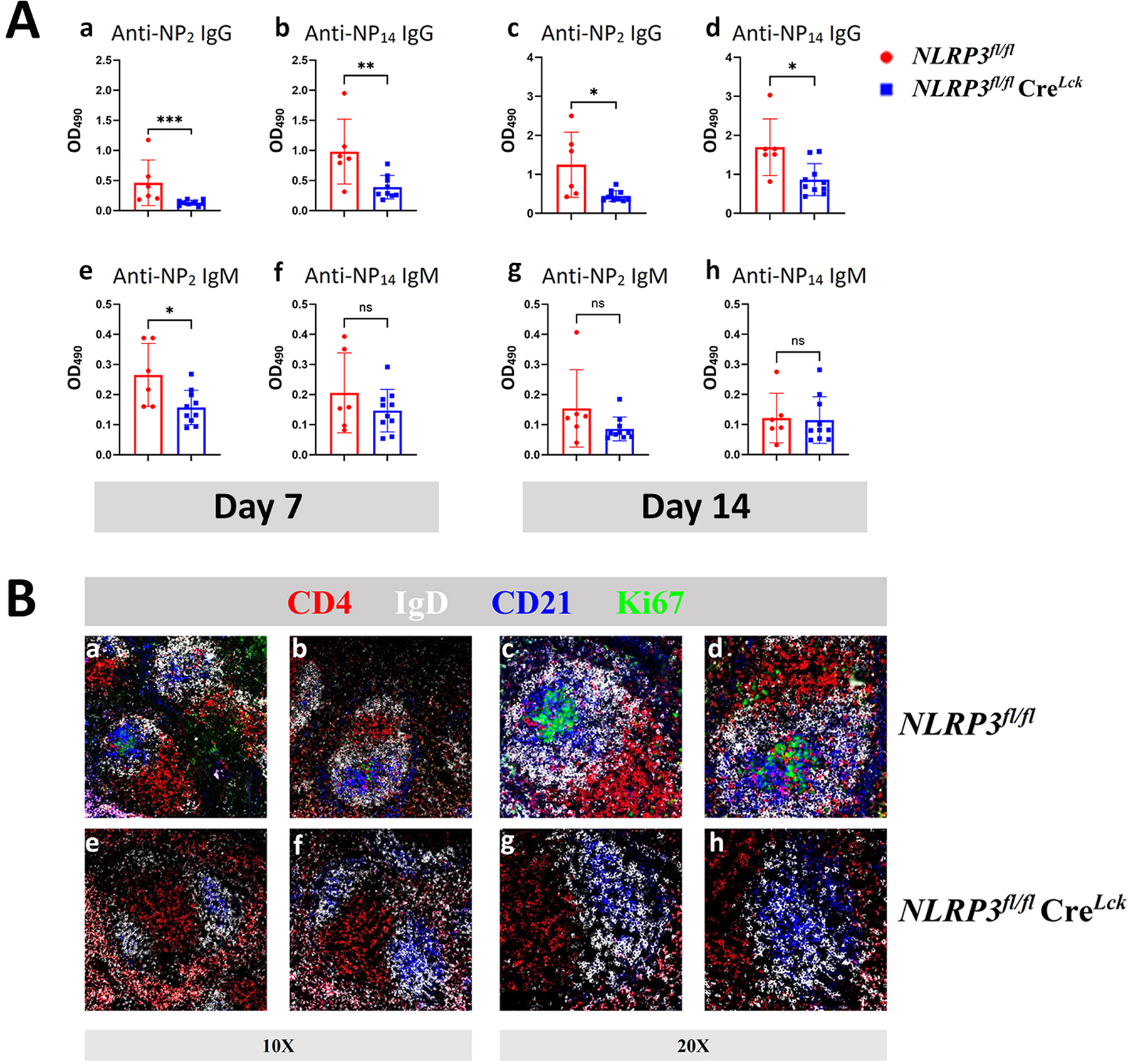
(A) B6.NLRP3fl/flCreLck mice showed reduced Ab response to soluble Ag. Groups of 6 WT (B6) and 10 mutant mice were immunized subcutaneously in the neck and in the footpad with NP33-CGG (100μg/mouse) without alum. Mice were bled on days 7 and 14 after immunization. Sera were diluted at 1:1000. Anti-NP Ab were quantified with ELISA using plate-bound NP14-BSA and NP2-BSA for low and high affinity Ab respectively. Results are expressed as mean ±SD. *p<0.05, **p<0.01 and ***p<0.001. (B) Lack of proliferating cells in the splenic germinal centers. (a-d) Two splenic germinal centers with Ki67+ cells in B6.NLRP3fl/fl (a&b 10X and c&d 20X) five days after immunization with NP33-CGG. (e-h) Absence of Ki67+ cells in the spleen of a B6.NLRP3fl/flCreLck mouse in that NLRP3 is specifically deleted from T cells.
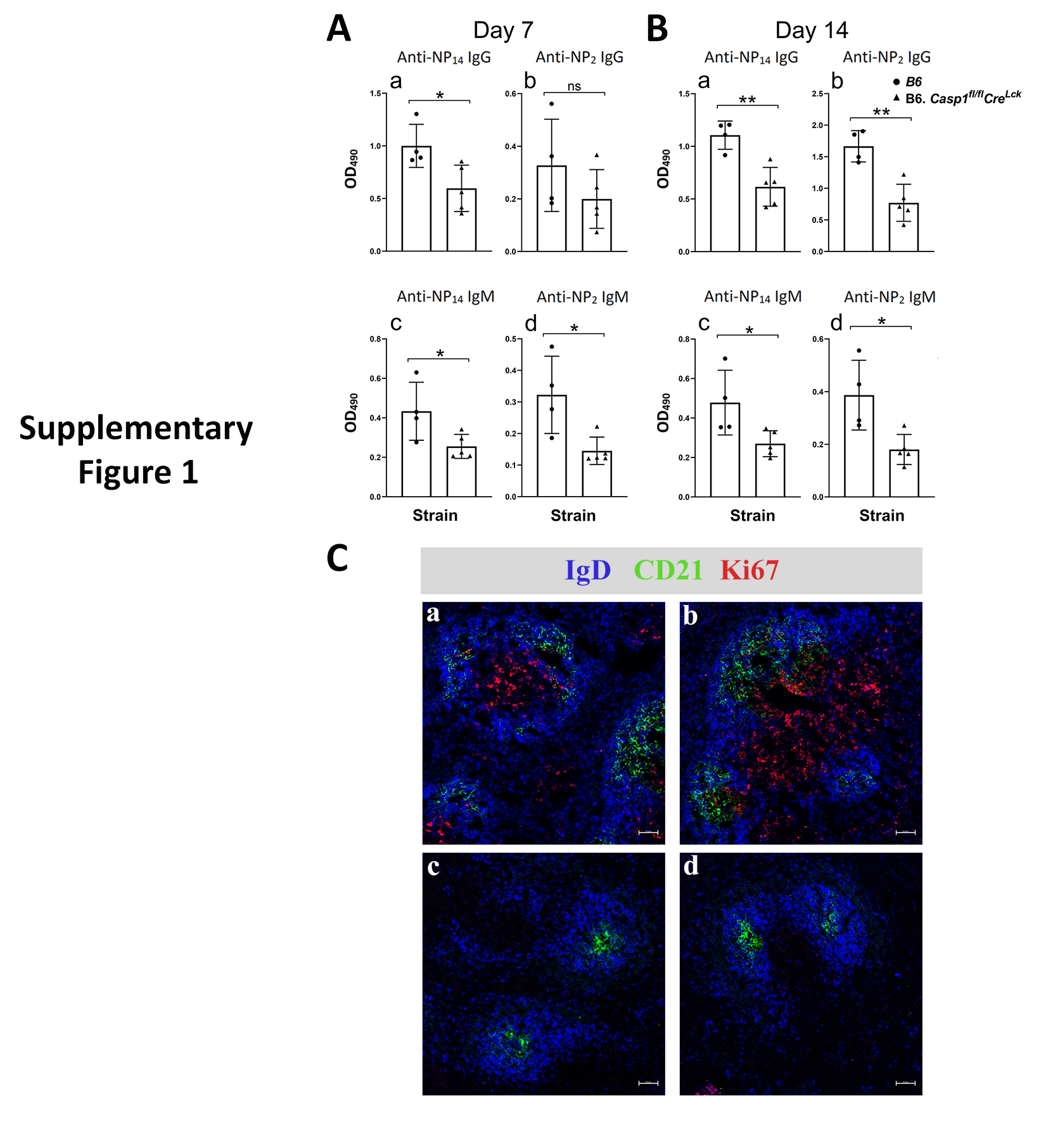
Similar results were obtained in a cohort of B6 mice with specific deletion of Casp-1 in their T cells (online supplementary figure 1).
Reduced GC formation and attenuated anti-dsDNA Ab in MRL/lpr and NZM2328 mice treated with MCC950.
In our work investigating the role of NLRP3 activation in MRL/lpr, it was noted that with MCC950 treatment that targets NLRP3 specifically (18), the spleen and lymph nodes were significantly smaller in the treated mice. The possibility that act. caspase-1+ Tfh may play a role in the pathogenesis of lymphoproliferation in MRL/lpr was explored.
By 2 months of ages MRL/lpr mice spontaneously had significantly more act. caspase-1+ Tfh in their spleen (~7% in figure 5A). Its congenic MRL/Mpl had ~2.5% such cells, which was similar to that in B6. With further aging, this Tfh population increased further (figure 5B). By week 16, approximately 20% of the Tfh cells were positive for act. caspase-1. MCC950 treatment given every other day from week 12 to week 16 diminished this population from ~20% to less than 10%. Circulating IL-1β was increased in 12 and 16 week old MRL/lpr mice (figure 5C). This increase was not seen in MCC950 treated mice. Treatment of MRL/lpr with MCC950 diminished splenic and lymph node weights (figures 5D and 5E). This treatment also diminished the titers of anti-dsdNA Ab (figure 5F). PNA staining revealed that MCC950 inhibited the proliferation of centroblasts resulting in defective GC formation (figure 5G).
Figure 5. The NLRP3 inflammasome activation and proliferation of Tfh cells in the spleens and lymph nodes were inhibited by MCC950 in MRL/lpr mice.
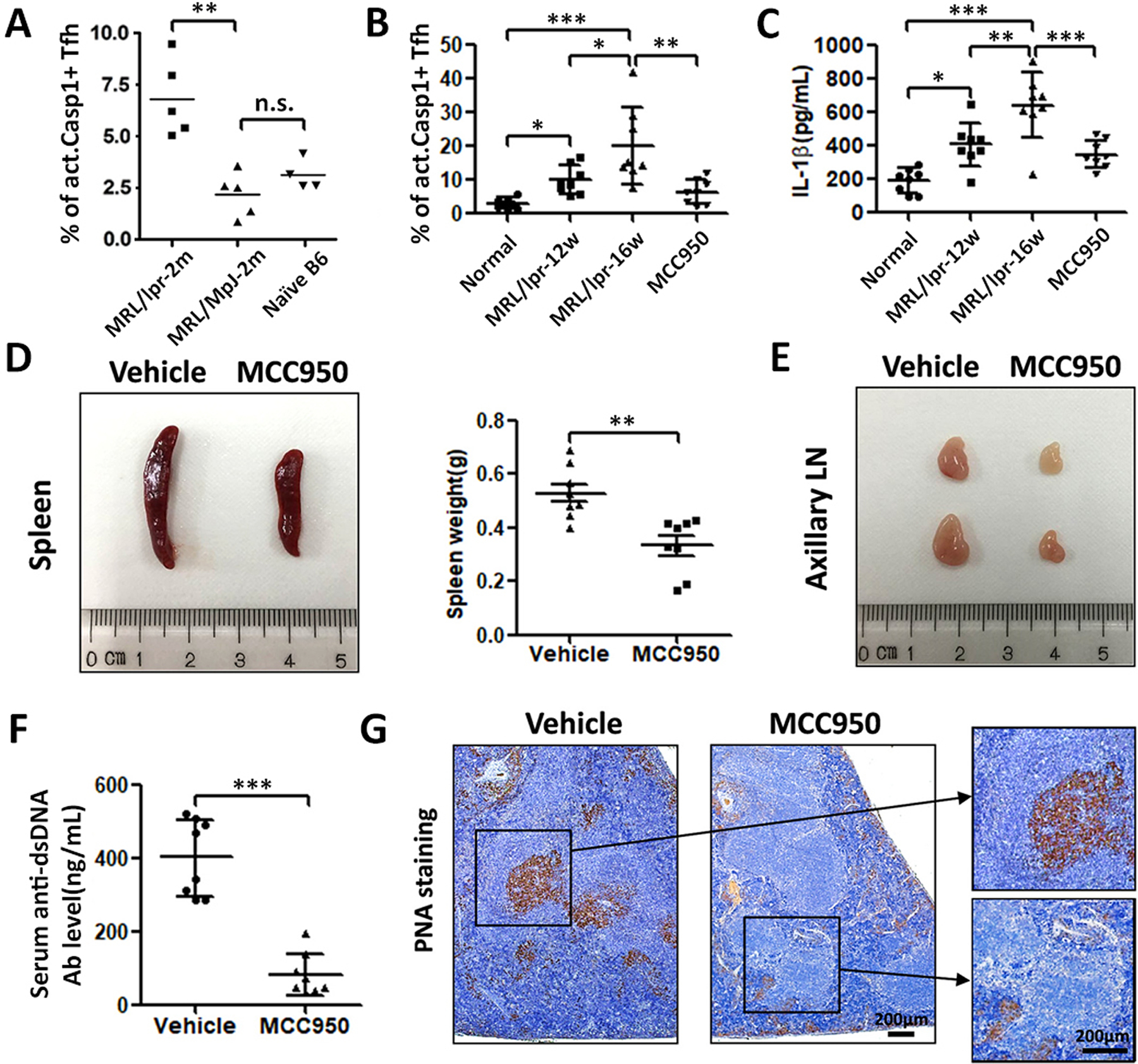
(A) At two months of age, MRL/lpr female mice had elevated act. caspase-1+ Tfh cells in their spleen in comparison with its congenic strain MRL/Mpl and B6. (B) Marked increases in act. caspase-1+ Tfh cells in the spleen in 12 and 16 weeks old MRL/lpr. MCC950 given every other day from week 10 to week 16 inhibited these increases. (C) Marked increases in circulating IL-1β in older MRL/lpr. The administration of MCC950 inhibited the increase in IL-1β. (D) MCC950 inhibited the increase in splenic weights. (E) MCC950 prevented the increases of lymph node sizes. (F) MCC950 inhibited the increases in anti-dsDNA Ab. (G) PNA staining revealed that MCC950 dramatically reduced proliferating B cells in the spleen. *p<0.05, **p<0.01 and ***p<0.001.
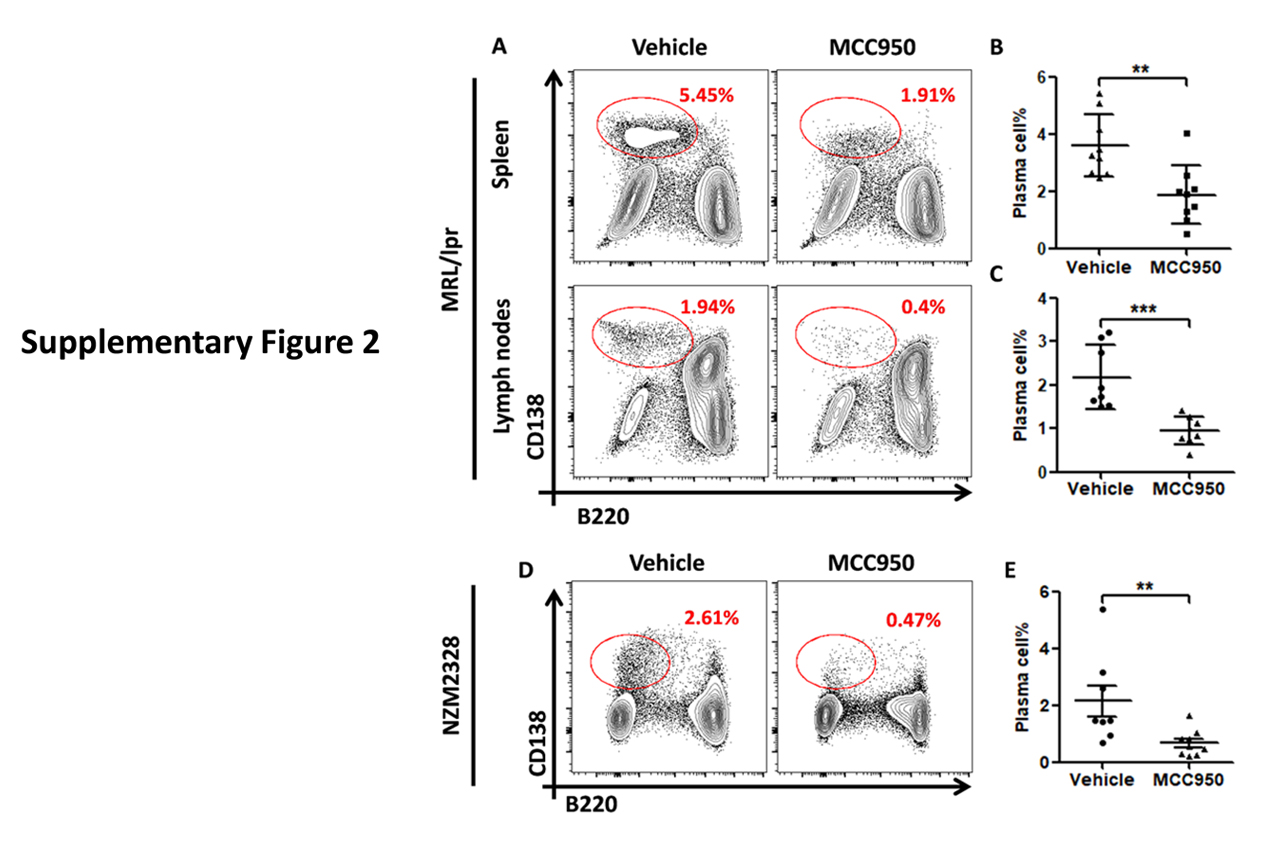
Act. caspase-1+ Tfh cells in MRL/lpr expressed significantly more IL-21, CD40L, pSTAT3 and Bcl-6 in comparison with act. caspase-1+ cells isolated from MCC950-treated MRL/lpr mice (figure 6). This suggests that MCC950 affects negatively the differentiations of Tfh cells with less B cell help capability. These cells co-cultured with act. NLRP3+ Tfh cells showed higher expression of the B cell activation marker GL-7 in comparison with those co-cultured with act. caspase-1− Tfh cells (data not shown). MRL/lpr mice had significant numbers of plasma cells (~3.8% in the spleen and ~2.20% in the lymph nodes (online supplementary figure 2A–2C). The plasma cells were reduced to ~1.9 % in the spleen and ~0.8% in the lymph nodes by the MCC950 treatment.
Figure 6. Decreased expression of IL-21, CD40L, pSTAT3 and Bcl-6 in splenic Tfh cells of MRL/lpr mice treated with MCC950.
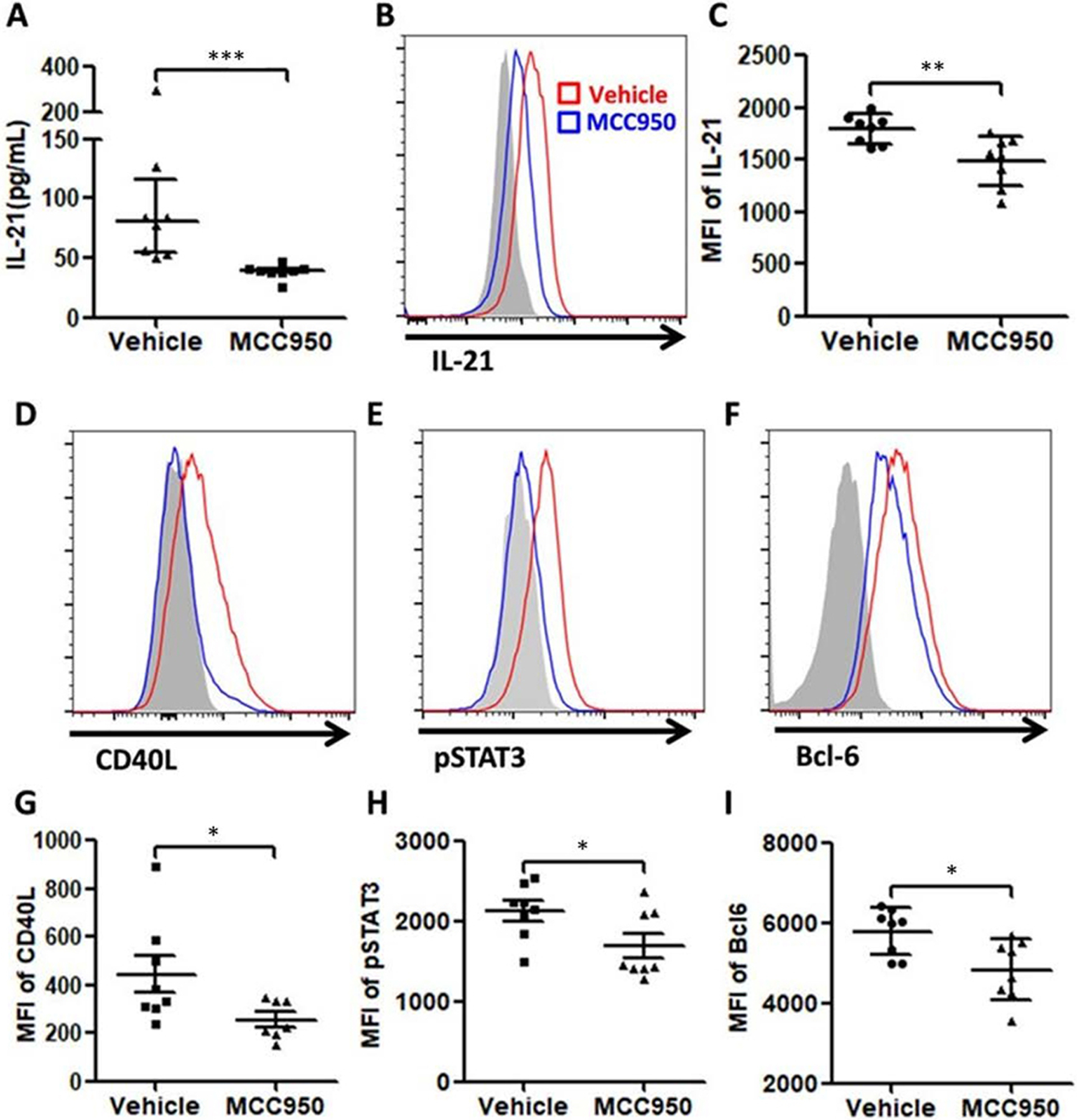
(A) Serum IL-21 level was significantly lower in MCC950-treated group than that in vehicle group. (B) and (C) The expressions of IL-21 in Tfh cells was also significantly lower in the MCC950-treated group. (D)–(I) Similar increases of CD40L, pSTAT3 and Bcl-6 expressions in Tfh cells were detected. These increases were inhibited by MCC950.
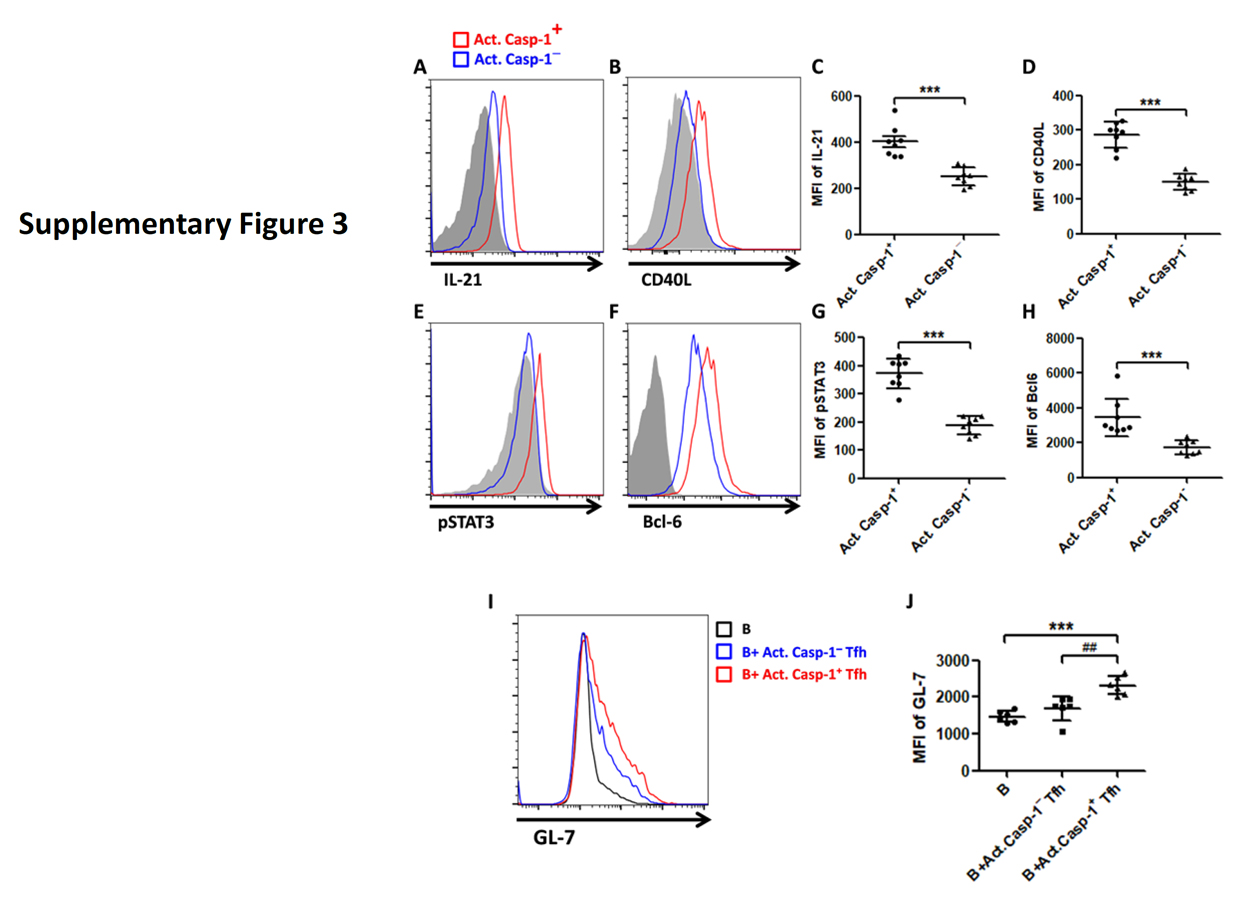
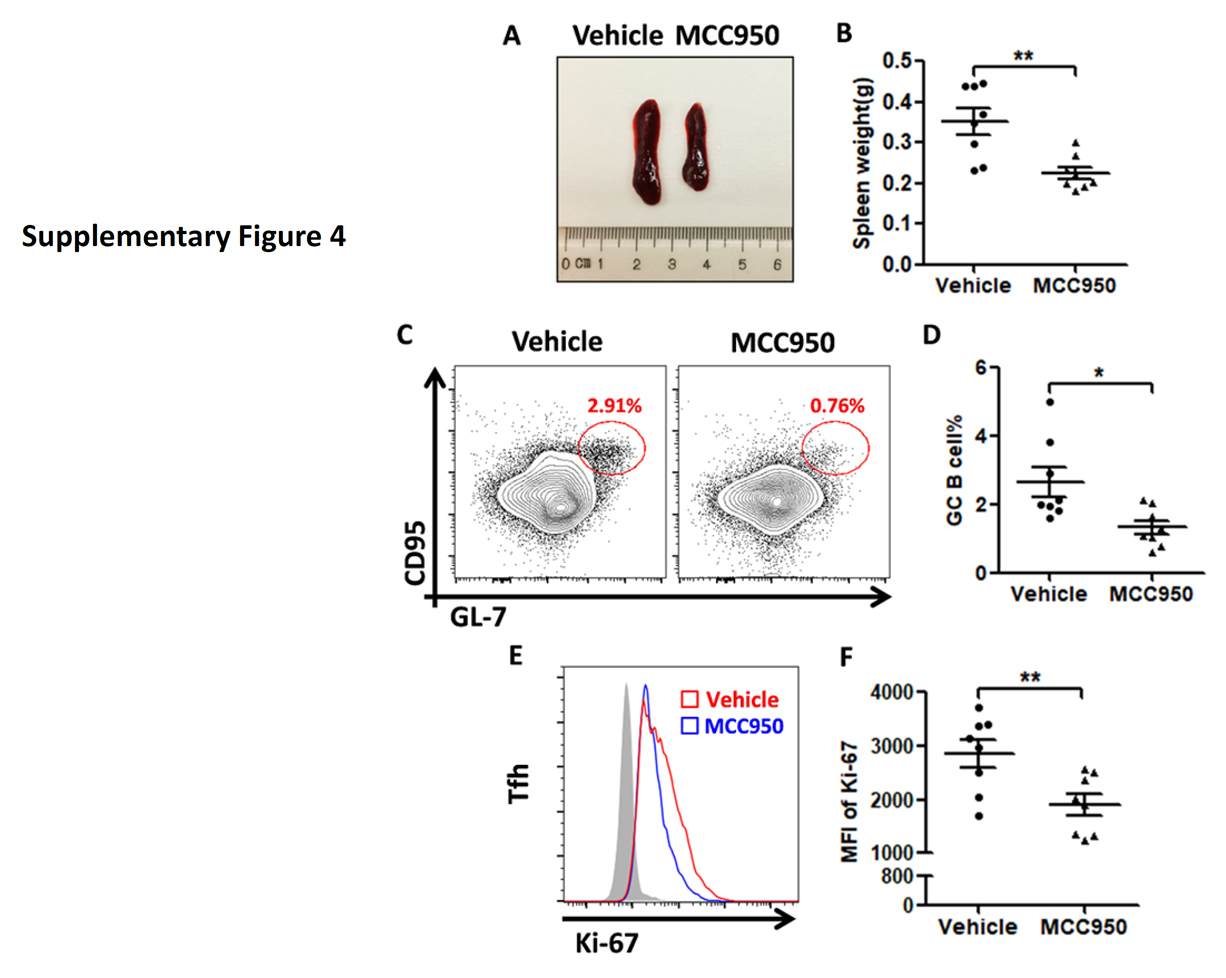
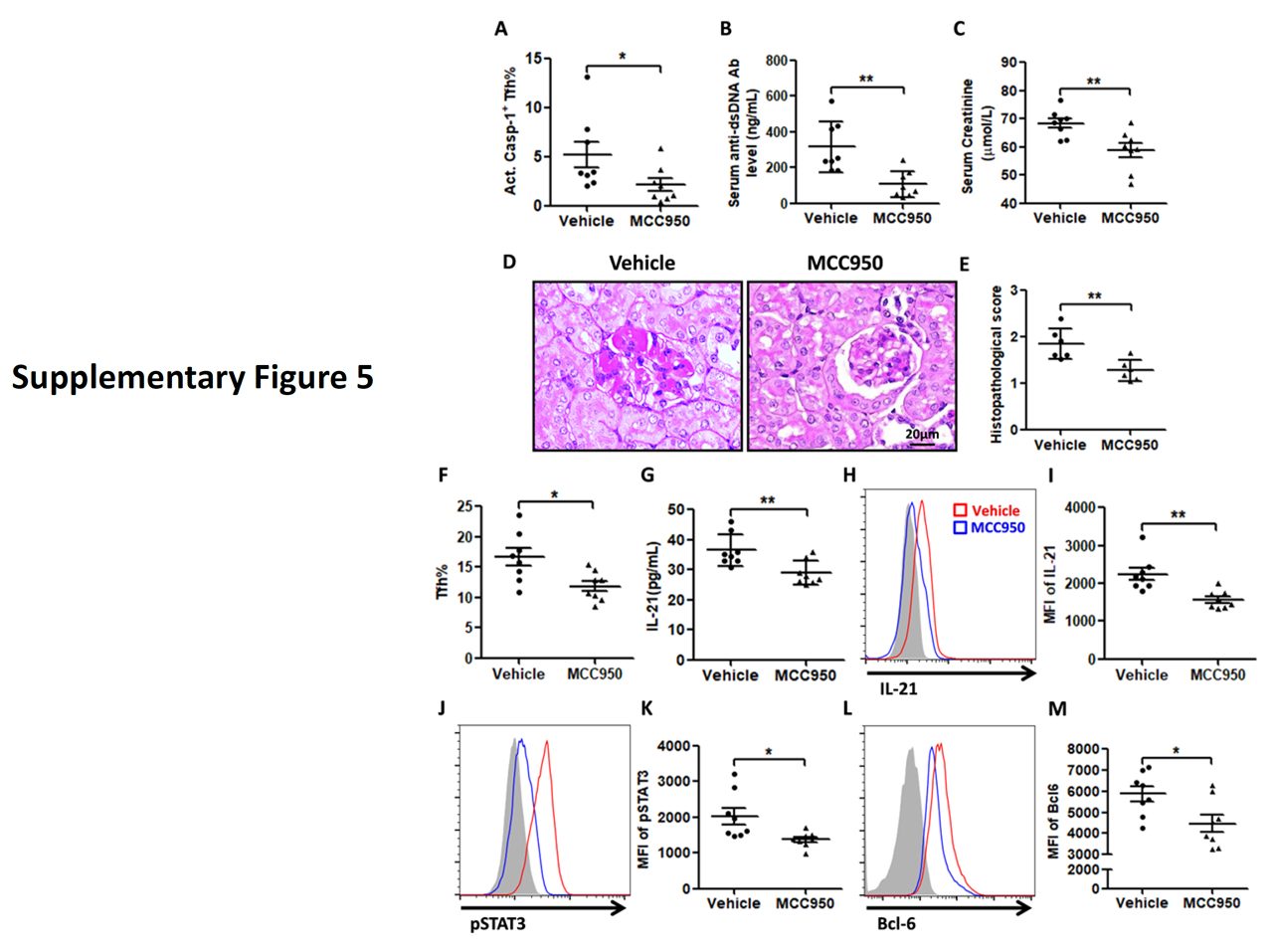
Similar results were obtained in NZM2328. Significant numbers of CD138+B220 low plasma cells were detected in their spleen (online supplementary figure 2D and 2E). Act. caspase-1+ Tfh cells were increased in the spleen. This population in comparison to the act. Caspase− Tfh expressed more IL-21, CD40L, pSTAT3 and Bcl-6 (online supplementary figure 3). MCC950 treatment targeted these parameters resulting in decreases in splenic weight, inhibition of expression of IL-21, CD40L, pSTAT3 and Bcl-6 in Tfh cells, reduction in activated B cell and plasma cell numbers, anti-dsDNA antibodies and renal pathology (online supplementary figures 3–5).
SLE patients had elevated circulating active NLRP3 + Tfh cells and this population of Tfh cells was responsive to pulse steroid plus immunosuppressive drugs.
Characteristics of blood donors from 71 SLE patients from the rheumatology clinic at the First Affiliated Hospital of Sun Yat-sen University are listed in online supplementary table S1 and online supplementary figure 6. Twenty-five age- and sex- matched blood donors were recruited as controls. The percentage of act. caspase-1+ Tfh (defined as CD4+CXCR5+Foxp3− act. caspase-1+) was significantly higher in SLE patients than that in healthy controls (figures 7B and 7C), and showed a positive correlation with SLE disease activity as measured by SLEDAI (figure 7D). We also found increased IL-1β expression both in circulating Tfh cells (figures 7E and 7F) and the sera (figures 7G) of SLE patients.
Figure 7. Increased activation of NLRP3 inflammasome in circulating Tfh cells of SLE patients are correlated with SLEDAI and response to therapy.
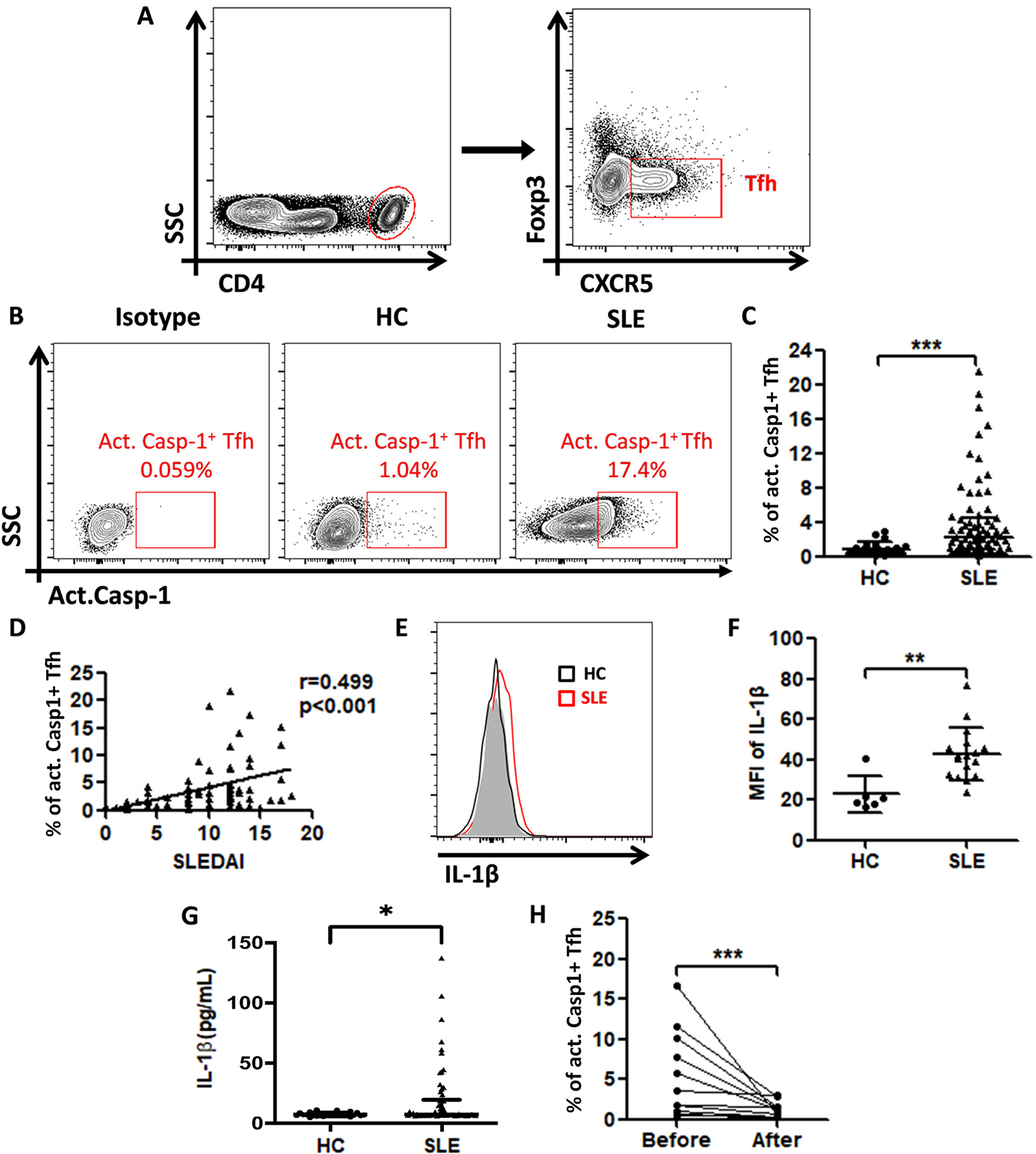
(A) Gating strategy to identify circulating Tfh cells (CD4+CXCR5+Foxp3−) in human blood. (B) Contour plots of act. caspase-1+ Tfh cells from a representative healthy control and a SLE patient. (C) Percentage of act.caspase-1+ Tfh cells among total Tfh cells in SLE patients (n=71) and healthy controls (n=25). (D) Correlation between the SLEDAI with the percentage of act. caspase-1+ Tfh cells in SLE patients. (E) Flow-cytometric histograms of IL-1β expression in Tfh cells of a representative healthy control (Black line) and a SLE patient. (Red line). (F) MFI of IL-1β on Tfh cells of HCs (n=6) and SLE patients (n=17). (G) Serum IL-1β levels of HCs (n=25) were compared with those in SLE patients (n=71). (H) Percentage of act. caspase-1+ Tfh cells before and after treatment (n=11). *p<0.05, **p<0.01 and ***p<0.001.
Eleven SLE patients were in renal relapse during this investigation. They were studied when diagnosed to have renal relapse and again 4 weeks after initiation of treatments. After 4 weeks of treatment, the patients showed improved clinical symptoms with decreases in proteinuria, hematuria and anti-dsDNA Ab (online supplementary table S2) and decreased circulating act. caspase-1+ Tfh cells (figure 7H). All patients were treated with hydroxychloroquine and prednisone. In two patients, immunosuppressants were added (see footnote of online supplementary table S2).
DISCUSSION
IL-1β has been shown to be involved in Ab responses during normal immunization and in autoimmunity, (7). More importantly, IL-1β has been shown to act directly on CD4+ T cells to enhance their antigen-driven expansion and differentiation, (19). Despite playing a significant role in humoral responses, it remains unsettled whether IL-1β is produced by CD4+ T cells in normal humoral responses to immunization. The data from Arbore et al, (3) suggests that Th1 responses require complement driven NLRP3 activity in CD4+ T cells. In the present investigation, we have provided data showing that NLRP3 activation in CD4+ Tfh cells is an integral part of the humoral response to T dependent antigens. We have shown that NLRP3 activation as detected by act. caspase-1 provides evidence for an increase in the percentage of Tfh cells that have act. caspase-1 activity. This subpopulation of Tfh cells can be detected readily at the border between T and B cells within the GC. This population can provide superior helper activity. Thus it is concluded that CD4+ Tfh cells do make IL-1β in response to immunization. This IL-1β serves in an autocrine fashion to provide good humoral responses.
MCC950 has been considered to specifically block NLRP3 activity, (18). It inhibits humoral responses by inhibiting IL-1β activation from pro-IL-1β. Since it is a small molecule without cell specificity, the data from MCC950 treatment do not permit us to conclude that at least part of the action of MCC950 is exerted on Tfh cells. However, the experiments involving lymphoid specific deletion of NLRP3 and Casp-1 provides support for the conclusion that T cell activation of NLRP3 is required for optimal humoral responses. It is of note that our experiments do not rule out the contribution of IL-1β from CD8+ T cells although these cells may play a minor role in humoral response and from other cells such as dendritic cells, (20).
It remains to be determined what initiates the activation of NLRP3 as a response to immunization. In addition to the complement-driven activation of NLRP3, the possibility that TNFα may play a major role in this process should be considered. TNFα interacts with receptor interacting kinase 3 (Rip3), (21). This interaction activates both the necrosis pathway and the NLRP3 pathway, (22). It has been shown that TNFα is expressed during T and B cell activation, (23, 24,). The TNFα made by activated T cells may act in an autocrine manner while that made by activated B cells may act in a paracrine fashion. Our preliminary data supports this hypothesis. GSK872, a Rip3 specific inhibitor that was shown to be effective in treating nephritis in NZM2328, (9) was able to inhibit activation of NLRP3 in Tfh cells. It is also effective in reducing high affinity Ab and inhibiting GC formation. More definitive genetic experiments involving TNFα deletion in T cells are needed.
Anakinia is an IL-1 receptor antagonist and has been approved for the treatment of rheumatoid arthritis. From the results of our study, it is surprising that Anakinia is not effective in lupus trials. The reason for this failure is complex including trial design, heterogeneity of lupus patients and strength of Anakinia as a IL-1 antagonist. Perhaps these reagents could best be used as one of the agents in a multi-targeted drug trial.
We have been interested in the pathogenesis of SLE with studies of MRL/lpr and NZM2328. Generation of pathogenic autoantibodies is a hallmark of SLE, (11, 25). Autoantibodies are the products of autoreactive B cells which are derived from GCs, (26). Several studies on lupus mice revealed that IL-1β is involved in generating autoantibodies, (27–29). Caspase-1 is a key component of NLRP3 inflammasome and gene knockout of caspase-1 resulted in a reduction of autoantibody production in the pristane-induced lupus mouse model, (30) suggesting that NLRP3 inflammasome contributes to the generation of autoantibodies. In the present study, inhibition of NLRP3 inflammasome activation reduced GC formation, B cell frequency, and autoantibody production in lupus-prone mice. These effects are accompanied by the reduction of pathological changes and prolong survival. These results are congruent with the studies cited. Thus targeting NLRP3+ Tfh cells may be a promising therapeutic approach.
Supplementary Material
Supplementary Figure 1. Specific deletion of Casp1 in lymphoid cells in B6 cause reduction in Ab responses to NP33-CGG. (A) Deletion of Casp1 in lymphoid cells in B6 caused reduction of high and low affinity IgG Ab responses to NP2-BSA and NP14-BSA, respectively. At day 7 the IgG Ab NP2-BSA reduction did not reach statistical significance. IgM Ab were also reduced in both day 7 and day 14 responses. (B) Presence of dividing cells in the germinal centers of a wild-type B6 mouse five days after immunization with NP33 –CGG (a&b). Lack of dividing cells as stained by anti-Ki67 mAb in the germinal centers of the spleen of a similarly immunized B6 mouse with deletion of Casp1 in their lymphoid cells (c&d).
{kind=link}
Supplementary Figure 2. The numbers of plasma cells were reduced by MCC950 in both spleen and lymph nodes of MRL/lpr (A-C). This reduction was also observed in NZM2328 (D, E).
{kind=link}
Supplementary Figure 3. In NZM2328, act. caspase-1+ Tfh cells in comparison with act. caspase-1− expressed more IL-21 (A and C), CD40L (B and D), pSTAT3 (E and G) and Bcl-6 (F and H). This act. caspase-1+ Tfh population induced more GL-7 expression on B cells when they were incubated with B cells (I and J).
{kind=link}
Supplementary Figure 4. (A) and (B) MCC950 when administered from weeks 20 to 26 of age reduced spleen sizes of NZM2328. (C) and (D) GC B cells in the spleen of NZM2328 were reduced by the administration of MCC950, (E) and (F) The intensity of Ki67+ staining of splenocytes of NZM2328 was reduced by MCC950.
{kind=link}
Supplementary Figure 5. MCC950 reversed the increase in act. caspase-1+ Tfh cells and modulated the development of immune complex-mediated nephritis in NZM2328. Twelve week old NZM2328 female mice were injected intravenously with 1×107 particles of adenovirus-expressing IFNα. Two groups of mice were treated either with vehicle (saline) or MCC950 every other day until the termination of the experiment at 18 weeks of age. (A) Reduction of act. caspase-1+ Tfh in MCC950-treated NZM2328. Treatment with MCC950 reduced anti-dsDNA Ab titers (B), serum creatinine (C), reduced histopathological scores for nephritis (D and E), reduction in % of Tfh cells (F), reduction of circulating IL-21 and (G), reduction of expression of IL-21(H and I), pSTAT3 (J and K) and Bcl6 (L and M). *p<0.05 and **p<0.01.
{kind=link}
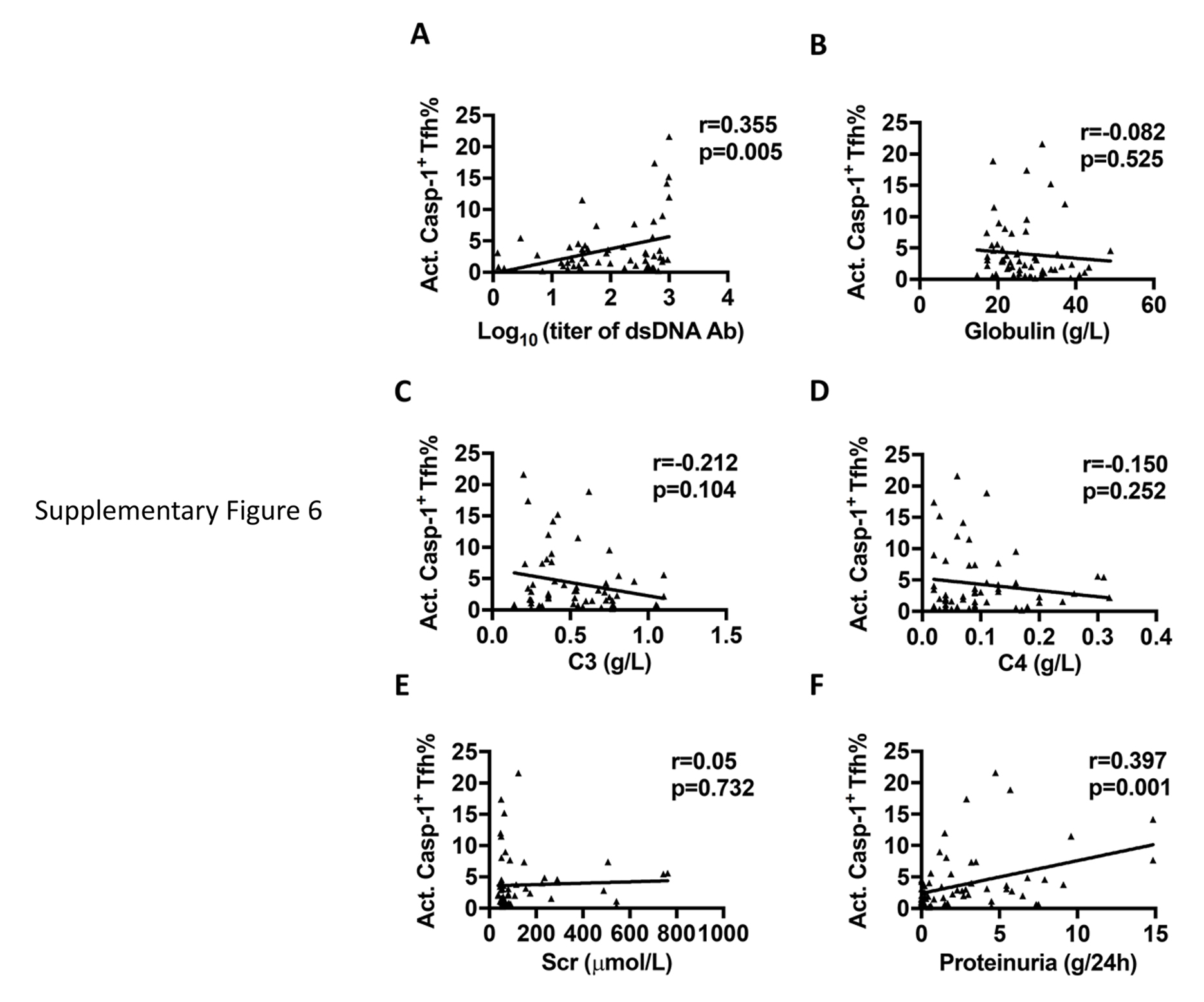
Supplementary Figure 6. Correlation between the percentage of act. caspase-1+ Tfh cells and clinical parameters in SLE patients. The correlation between the percentages of act. caspase-1+ Tfh cells and serum anti-dsDNA antibody (A), globulin (B) Complement C3 (C), Complement C4 (D), serum creatinine level (E) and 24 hour-urinary protein excretion (F) in SLE patients.
{kind=link}
Key messages
What is already known about this subject?
NLRP3 activation in CD4+ T cells has been shown in several Th1 response models.
What does this study add?
A population of Tfh cells with active NLRP3 exhibiting potent helper activity has been identified.
Blocking NLRP3 activation with MCC950 inhibits optimal humoral responses with delayed affinity maturation and impaired germinal center formation. Specific deletions of NLRP3 or Casp1 in T cells with the impaired humoral responses provides evidence to support the hypothesis that NLRP3 activations in Tfh is an integral part of the humoral response.
MCC950 inhibited autoantibody formation and modulated autoimmune manifestations.
How might this impact on clinical practice?
The activated NLRP3+ Tfh cell population appears to play an important role in the pathogenesis of human lupus suggesting that targeting this population may have therapeutic potentials.
ACKNOWLEDGEMENT OF GRANT FUNDING
This work was supported by the National Natural Science Foundation of China (81701595, 81671593, 81971519 and 82171770) and Guangdong Natural Science Foundation (2021A1515012072). S. M. Fu was supported in part by NIH grants (R01 AR-047988 and R01 AI-148231) and a grant from the Alliance for Lupus Research (TIL332635).
Footnotes
COMPETING INTEREST
All the authors have no interests to declare.
RESEARCH ETHICS APPROVAL
Human: Ethics Committee for Clinical Research, First Affiliated Hospital, Sun Yat-sen University (81701595). The study was exempted because the participants were de-identified peripheral blood and urine.
- Sun Yat-sen University First Affiliated Hospital Institutional Animal Care and Use Committee (IACUC) (2018-000181)
- University of Virginia Animal Care and Use Committee Protocol # 1849
REFERENCES
- 1.Crotty S T follicular helper cell differentiation, function, and roles in disease. Immunity 2014;41(4):529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol 2015;16(2): 142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arbore G, West EE, Spolski R, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science 2016;352(6292):aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin BN, Wang C, Zhang CJ, et al. T cell-intrinsic ASC critically promotes T(H)17-mediated experimental autoimmune encephalomyelitis. Nat Immunol 2016;17:583–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doitsch G, Galloway NL, Geng X, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014;505:509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 2014;1319:82–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ritvo TG, Klatzmann D. Interleukin-1 in the Response of Follicular Helper and Follicular Regulatory T Cells. Front Immunol 2019;10:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen M, Wang H, Chen W, et al. Regulation of adaptive immunity by the NLRP3 inflammasome. Int Immunopharmacol 2011;11:549–54. [DOI] [PubMed] [Google Scholar]
- 9.Guo C, Fu R, Zhou M, et al. Pathogenesis of lupus nephritis: RIP3 dependent necroptosis and NLRP3 inflammasome activation. J Autoimmun 2019;103:102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fu R, Guo C, Wang S, et al. Podocyte Activation of NLRP3 Inflammasomes Contributes to the Development of Proteinuria in Lupus Nephritis. Arthritis Rheumatol 2017;69:1636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linterman MA, Rigby RJ, Wong RK, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med 2009;206:561–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 1997;40(9):1725. [DOI] [PubMed] [Google Scholar]
- 13.Bombardier C, Gladman DD, Urowitz MB, et al. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992; 35:630–40. [DOI] [PubMed] [Google Scholar]
- 14.Waters ST, Fu SM, Gaskin F, et al. NZM2328: a new mouse model of systemic lupus erythematosus with unique genetic susceptibility loci. Clin Immunol 2001;100(3):72–83. [DOI] [PubMed] [Google Scholar]
- 15.Dai C, Wang H, Sung SS, et al. Interferon alpha on NZM2328.Lc1R27: enhancing autoimmunity and immune complex-mediated glomerulonephritis without end stage renal failure. Clin Immunol 2014;154: 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Le TV, Kim TH, Chaplin DD. Intraclonal competition inhibits the formation of high-affinity antibody-secreting cells. Immunol 2008;181:6027–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macdonald RA, Hosking CS, Jones CL. The measurement of relative antibody affinity by ELISA using thiocyanate elution. J Immunol Methods 1988; 106:191–4. [DOI] [PubMed] [Google Scholar]
- 18.Coll RC, Robertson AA, Chae JJ, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015;21:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ben-Sasson SZ, Hu-Li J, Quiel J, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA 2009; 106:7119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koide SL, Inaba K, Steinman RM. Interleukin 1 enhances T-dependent immune responses by amplifying the function of dendritic cells. J Exp Med 1987;165:515–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137:1100–11. [DOI] [PubMed] [Google Scholar]
- 22.Lawlor KE, Khan N, Mildenhal A, et al. RIPK3 promotes cell death and NLRP3 inflammsome activation in the absence of MLKL. Nat Commun 2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sung SS, Bjorndahl JM, Wang CY, et al. Production of tumor necrosis factor/cachectin by human T cell lines and peripheral blood T lymphocytes stimulated by phorbol myristate acetate and anti-CD3 antibody. J Exp Med 1988;167(3):937–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sung SS, Jung LK, Walters JA, et al. Production of tumor necrosis factor/cachectin by human B cell lines and tonsillar B cells. J Exp Med 1988;168(5):1539–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blanco P, Ueno H, Schmitt N. T follicular helper (Tfh) cells in lupus: Activation and involvement in SLE pathogenesis. Eur J Immunol 2016;46:281–90. [DOI] [PubMed] [Google Scholar]
- 26.Vinuesa CG, Sanz I, Cook MC. Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol 2009; 9(12):845–57. [DOI] [PubMed] [Google Scholar]
- 27.Voronov E, Dayan M, Zinger H, et al. IL-1 beta-deficient mice are resistance to induction of experimental SLE. Eur Cytokine Netw 2006;17(2):109–16. [PubMed] [Google Scholar]
- 28.Lebedeva TV, Singh AK. Increased responsiveness of B cells in the murine MRL/lpr model of lupus nephritis to interleukin-1 beta. J Am Soc Nephrol 1995;5(7):1530–4. [DOI] [PubMed] [Google Scholar]
- 29.Sing AK, Lebedeva TV. Interleukin-1 contributes to high level IgG production in the murine MRL/lpr lupus model. Immunol Invest 1994;23(4–5):281–92. [DOI] [PubMed] [Google Scholar]
- 30.Kahlenberg JM, Yalavarthi S, Zhao W, et al. An essential role of caspase 1 in the induction of murine lupus and its associated vascular damage. Arthritis Rheumatol 2014;66(1):152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Specific deletion of Casp1 in lymphoid cells in B6 cause reduction in Ab responses to NP33-CGG. (A) Deletion of Casp1 in lymphoid cells in B6 caused reduction of high and low affinity IgG Ab responses to NP2-BSA and NP14-BSA, respectively. At day 7 the IgG Ab NP2-BSA reduction did not reach statistical significance. IgM Ab were also reduced in both day 7 and day 14 responses. (B) Presence of dividing cells in the germinal centers of a wild-type B6 mouse five days after immunization with NP33 –CGG (a&b). Lack of dividing cells as stained by anti-Ki67 mAb in the germinal centers of the spleen of a similarly immunized B6 mouse with deletion of Casp1 in their lymphoid cells (c&d).
Supplementary Figure 2. The numbers of plasma cells were reduced by MCC950 in both spleen and lymph nodes of MRL/lpr (A-C). This reduction was also observed in NZM2328 (D, E).
Supplementary Figure 3. In NZM2328, act. caspase-1+ Tfh cells in comparison with act. caspase-1− expressed more IL-21 (A and C), CD40L (B and D), pSTAT3 (E and G) and Bcl-6 (F and H). This act. caspase-1+ Tfh population induced more GL-7 expression on B cells when they were incubated with B cells (I and J).
Supplementary Figure 4. (A) and (B) MCC950 when administered from weeks 20 to 26 of age reduced spleen sizes of NZM2328. (C) and (D) GC B cells in the spleen of NZM2328 were reduced by the administration of MCC950, (E) and (F) The intensity of Ki67+ staining of splenocytes of NZM2328 was reduced by MCC950.
Supplementary Figure 5. MCC950 reversed the increase in act. caspase-1+ Tfh cells and modulated the development of immune complex-mediated nephritis in NZM2328. Twelve week old NZM2328 female mice were injected intravenously with 1×107 particles of adenovirus-expressing IFNα. Two groups of mice were treated either with vehicle (saline) or MCC950 every other day until the termination of the experiment at 18 weeks of age. (A) Reduction of act. caspase-1+ Tfh in MCC950-treated NZM2328. Treatment with MCC950 reduced anti-dsDNA Ab titers (B), serum creatinine (C), reduced histopathological scores for nephritis (D and E), reduction in % of Tfh cells (F), reduction of circulating IL-21 and (G), reduction of expression of IL-21(H and I), pSTAT3 (J and K) and Bcl6 (L and M). *p<0.05 and **p<0.01.
Supplementary Figure 6. Correlation between the percentage of act. caspase-1+ Tfh cells and clinical parameters in SLE patients. The correlation between the percentages of act. caspase-1+ Tfh cells and serum anti-dsDNA antibody (A), globulin (B) Complement C3 (C), Complement C4 (D), serum creatinine level (E) and 24 hour-urinary protein excretion (F) in SLE patients.