

Summary
Diabetic kidney disease (DKD) occurs in ~40% of patients with diabetes and causes kidney failure, cardiovascular disease and premature death. We analyzed the response of a murine DKD model to five treatment regimens using single cell RNA-sequencing (scRNA-seq). Our atlas of ~1 million cells revealed a heterogeneous response of all kidney cell types both to DKD and its treatment. Both monotherapy and combination therapies targeted differing cell types and induced distinct and non-overlapping transcriptional changes. The early effects of sodium-glucose cotransporter-2 inhibitors (SGLT2i) on the S1 segment of the proximal tubule suggest that this drug class induces fasting mimicry and hypoxia responses. Diabetes downregulated the spliceosome regulator serine/arginine-rich splicing factor 7 (Srsf7) in proximal tubule that was specifically rescued by SGLT2i. In vitro proximal tubule knockdown of Srsf7 induced a pro-inflammatory phenotype, implicating alternative splicing as a driver of DKD and suggesting SGLT2i regulation of proximal tubule alternative splicing as a potential mechanism of action for this drug class.
Keywords: Diabetes, single cell RNA-seq, T2D, kidney
eTOC blurb
Wu et al generate a single cell atlas of murine diabetic kidney disease and its treatment. They provide a useful resource for understanding the cell-specific kidney transcriptional responses to DKD and to five common treatment regimens.
Graphical Abstract

Introduction
Diabetes mellitus is estimated to affect over 10% of the U.S. population and DKD is the leading cause of end stage kidney disease (ESKD) worldwide (Oshima et al., 2021). Despite recent treatment advances, only a limited reduction in rates of ESKD from DKD have been observed (Brenner et al., 2001; Heerspink et al., 2020; Lewis et al., 2001; Perkovic et al., 2019; Wanner et al., 2016). While SGLT2i have emerged as a promising drug class for the treatment of DKD, their mechanism of action remains unknown (DeFronzo et al., 2021; Packer, 2020; Sen and Heerspink, 2021). Understanding the renoprotective effects of SGLT2i will inform the development of future therapeutics. For these reasons, deciphering the cellular and molecular drivers of DKD, and how best to target the main drivers of poor outcomes therapeutically, is a top priority.
The pathogenesis of DKD is multifactorial including endothelial dysfunction, glomerular hyperfiltration, hemodynamic effects and inflammation. Proteinuria often accompanies these changes, leading to tubular damage, interstitial fibrosis and glomerulosclerosis. Systemic hypertension occurs frequently in diabetics and worsens intraglomerular hypertension and kidney microvascular damage. Bulk transcriptomic studies have been informative in both experimental models and human DKD (Bansal et al., 2020; Fan et al., 2019; Komers et al., 2014; Woroniecka et al., 2011). These studies have revealed important insights but are also limited because signals from minority cell types are lost in the integrated dataset. scRNA-seq addresses this limitation, allowing construction of detailed cell atlases depicting cell type and state in health and disease. We recently applied scRNA-seq in early human diabetic nephropathy, revealing a distal nephron potassium secretory signature as well as a pro-angiogenic glomerular profile (Wilson et al., 2019). Given the multitude of cell types affected in DKD, we reasoned that scRNA-seq could provide further insight into the cellular and molecular drivers of DKD as well as cell-specific responses to different drug treatments including SGLT2 inhibition.
In this study, we utilized the db/db mouse model with uninephrectomy and renin-induced hypertension mouse model. We compared vehicle to ACE inhibitor, Rosiglitizone, the SGLT2i TA-1887 (Nomura et al., 2014), ACEi + Rosiglitizone and ACEi + SGLT2i at two time points (2 days and 2 weeks). We generated single nucleus RNA-seq (snRNA-seq) datasets from all groups comprising nearly 1 million cells. Analysis of the resulting atlas revealed that the different medications affected strikingly different cell types. Combination therapy had the largest effects, but these effects were also largely non-overlapping. By focusing on the earliest transcriptional effects of SGLT2 inhibitors on the S1 segment of the proximal tubule (PT), we infer that this drug class induces fasting mimicry and hypoxia responses. More specifically, diabetes downregulated the spliceosome regulator Srsf7 in PT and this was specifically rescued by SGLT2i. In vitro PT knockdown of Srsf7 triggered differential expression of ~2000 genes and induced a pro-inflammatory phenotype, suggesting a role for proximal tubule alternative splicing in driving DKD progression and of SGLT2i in rescuing it. This study highlights the cellular complexity of DKD and the effects of different classes of therapy. The results support the development of combination therapies since different drug classes target different cell types in kidney.
Results
Cellular heterogeneity in mouse kidneys
We induced diabetic DKD by adeno-associated virus delivery of renin (ReninAAV) to uninephretomized, female db/db mice (Figure 1A). This model mimics human progressive DKD and has been validated for use for evaluation of several drug treatments (Harlan et al., 2015, 2018a, 2018b). Physiological measurements confirmed that the ReninAAV model developed more severe proteinuria, hypertension and histopathological changes compared to the conventional db/db model (Figure 1B, C; Figure S1A). Our study design included six groups: db/m (control), db/db (vehicle), db/db + lisinopril (ACE inhibitor, ACEi), db/db + rosiglitazone (hypoglycemic drug, Rosi), db/db + JNJ-39933673 (SGLT2 inhibitor, SGLT2i) (Nomura et al., 2014), db/db + Lisinopril + Rosiglitazone (ACEi + Rosi) and db/db + Lisinopril + JNJ39933673 (ACEi + SGLT2i). Our histological data suggested that combination treatments achieve better outcomes in rescuing the kidney pathological changes compared to single treatments (Figure S1B). From the physiological data, we confirmed that after two weeks of treatment, mice that received ACEi either alone or in combination with either Rosi or SGLT2i, and mice that received Rosi alone experienced significant reductions in urinary albumin to creatinine ratio (UACR) (Figure 1D). Mice that received ACEi alone or in combination all had a significant reduction in blood pressure, whereas mice that received either Rosi or SGLT2i alone did not (Figure 1D). Finally, a significant reduction in serum glucose was achieved only in the Rosi + ACEi and SGLT2i treatment groups (Figure 1D). These results overall are consistent with expected treatment effects for each drug class.
Figure 1. Experimental plan, histology and physiologic readouts.
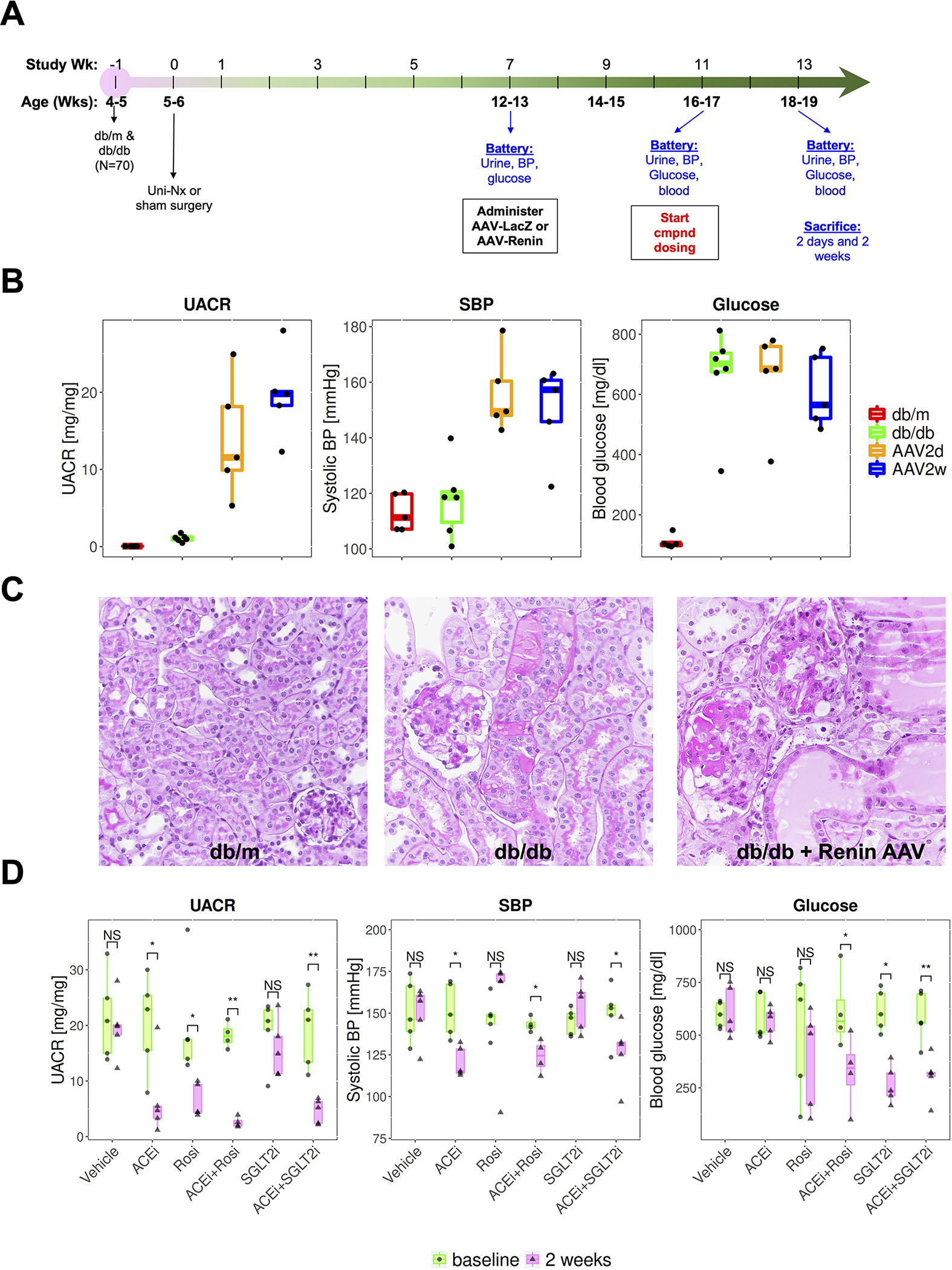
(A) Experimental scheme.
(B) Urinary albumin to creatinine ratio (UACR), systolic blood pressure (SBP) and glucose in control (db/m), db/db or db/db plus Renin-AAV at two days and two weeks.
(C) Hematoxylin and eosin (H&E) staining at two weeks across groups.
(D) Effect of treatment on UACR, SBP and glucose at baseline and two weeks.
* adj. p<0.05; ** adj.p < 0.01 (paired t-test, multiple comparisons with Benjamini-Hochberg correction).
We collected tissues at two early time points (2 days and 2 weeks post treatment) in order to identify the most proximate effects of these therapies and performed snRNA-seq on the kidneys of 70 mice from 14 groups using the 10x Chromium platform. We implemented several quality control steps during data processing, including the use of CellBender (Fleming et al., 2019) to remove ambient RNA contamination, DoubletFinder (McGinnis et al., 2019) to eliminate cell doublets, and Seurat data integration (Stuart et al., 2019) to correct for batch effect (Methods). We obtained a total of 946,660 high quality single cells which were classified into 18 major kidney cell types (Figure 2). These included the rare cell types such as 2,421 Macular Densa (MD) cells and 2,783 renin+ cells from the juxtaglomerular apparatus (JGA)(Figure 1). Every experimental group, mouse and timepoint contributed cells to every cluster, indicating the absence of dissociation bias across groups (Figure 2 and Figure S2A–C). Each cell type uniquely expressed expected cell-defining marker genes (Figure S2D), also reflecting the high quality of our data after quality control. Overall, we detected an average of 1,167 unique genes and 2,105 transcripts per cell (Figure S2E–G).
Figure 2. Single cell atlas of drug treatments in a mouse model of DKD.

A total of 946,660 high-quality cells from 70 mouse kidneys in 14 different groups are projected by UMAP plot. Colors indicate the major kidney cell types, and cluster boundaries are outlined by contour curve. The four corner insets show subclusters of endothelial cells, immune cells, fibroblasts and thick ascending limb of Loop of Henle (TAL). The axis outside the circular plot depicts the log scale of the total cell number for each cell class. The four colored tracks (from outside to inside) indicate class (colored as the central UMAP), group ID, mouse ID, and marker gene expression. Legends denote the group design (left) and marker genes (right) to define each cell class. The left legend shows the group ID/colors for the group ID track. The right legend shows marker genes/colors used for the marker track.
We observed substantial heterogeneity amongst endothelial cells, immune cells, the thick ascending limb (TAL) of the Loop of Henle and fibroblasts (Figure 2 and Figure S2 H–K). In particular, we could identify 6 endothelial subtypes including glomerular endothelial cells (Ehd3+) (Patrakka et al., 2007) (Figure 2 and Figure S2H) which were resolved by mapping the molecular signatures of the subtypes to those reported in a public endothelial cell single cell atlas (Kalucka et al., 2020). Unexpectedly, a total of 7 distinct subtypes were identified in TAL. These included three separate medullary TAL subclusters, three distinct cortical subclusters and Nos1+ MD cells, all expressing unique markers (Figure 2 and Figure S2I). Three fibroblast subclusters were identified, including an Acta2+ myofibroblast cluster, and two fibroblast subtypes expressing Dkk2 and Col8a1 that have also been found in other organs such as the bladder (Muhl et al., 2020) (Figure 2 and Figure S2J). Immune cells are relatively underrepresented in snRNA-seq datasets but despite this, we were able to identify T-cells, macrophages and dendritic cells (Figure 2 and Figure S2K). To corroborate our clustering results, we integrated our datasets against a separate reference (mouse E3019 instead of A3020) as well as using a separate integration tool (Harmony after log transformation) and confirmed similar clustering results (Figure S2L–P).
Kidney cell responses to DKD
To assess gene expression changes during progression of DKD in our model, we compared snRNA-seq profiles from control mice (db/m) to db/db, db/db + reninAAV at 2 days and 2 weeks. This analysis identified 2,422, 2,683, and 3,143 unique differentially expressed genes respectively (FDR<0.05, log fold change > 0.5). All cell types exhibited gene expression changes, but the number of differentially expressed genes varied dramatically – from over 500 in parietal cells to fewer than 25 in JGA (Figure 3A). The cell types with the highest number of differentially expressed genes during disease progression included PC, TAL, PEC, PT, and EC. The reduced number of differentially expressed genes in JGA, MD and immune cell clusters was smaller, possibly reflecting reduced statistical power in lower abundance cell types (cell fractions for JGA, MD and immune cells are 0.29%, 0.26% and 0.44%, respectively).
Figure 3. DN disease genes corrected according to treatment and heritability enrichment for kidney disease traits in DKD.
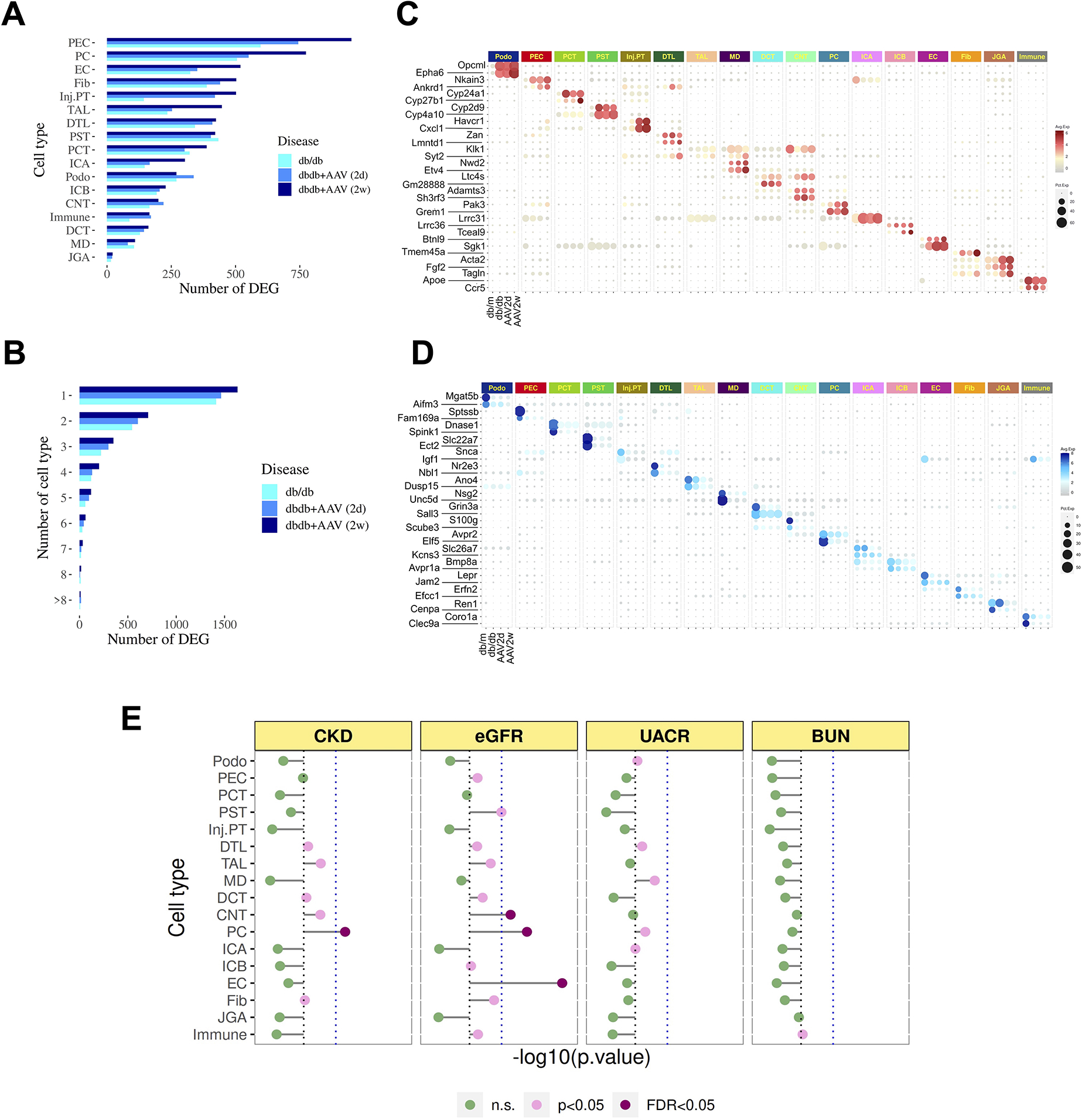
(A) Number of DE genes induced during DKD according to cell type. Bar colors indicate different comparisons: light blue: db/db vs db/m; medium blue: AAV 2d vs db/m; and dark blue: AAV 2w vs db/m.
(B) Number of genes detected in only one cell type vs. detected in multiple cell types.
(C) Dotplot depicting top two cell-specific, upregulated genes during DKD.
(D) Same plot depicting top two down-regulated genes during DKD.
(E) Gene hits from GWAS traits related to CKD, UACR and eGFR were mapped to DKD cell states at two weeks.
The dashed blue line denotes Bonferroni-adjusted significance (p=0.05/17).
More than half of the differentially expressed genes were only expressed in a single cell type, highlighting the cell-specific and heterogeneous response to diabetes (Figure 3B). In line with this, gene ontology (GO) terms enriched in each cell type also showed cell type specific patterns (Figure S3A). For example, the GO term glomerular basement membrane development was only significant in the podocyte, whereas the GO term for vasculature development was only enriched in EC. For cell-specific differentially expressed genes, about one third were unique to day 2, one third were unique to day 14 and one third were common to both time points (Figure S3A). This heterogeneity of responses between the two timepoints provides the rationale for using treatment control groups at both timepoints. We identified the top two cell-specific up- and down-regulated genes across the time course (Figure 3D, E). This analysis identified a number of genes that might serve as disease biomarkers. For example, the receptor tyrosine kinase EphA6 is specifically upregulated in podocytes, and its upregulation was also reported in the podocytes from other types of focal segmental glomerulosclerosis (FSGS) (Bukosza et al., 2020). AVPR2 was specifically downregulated in principal cells, suggesting a defect in water reabsorption in DKD.
Given cell-specific expression patterns for the majority of genes altered in DKD, we next asked whether CKD-associated common variants identified by genome-wide association studies (GWAS) also associate with specific kidney cell types. We used an approach similar to Li et al (Li et al., 2020) to relate heritability from GWAS of eGFR, proteinuria, and BUN to the disease program in each cell type using MAGMA (Methods). The analysis revealed that endothelial cells and collecting duct cells (PC and DCT) are significantly associated with eGFR (Figure 3E). For CKD variants, PC were significantly associated. It is striking that both PC and EC are also in the top three cell types with the most DEG in DKD. PC are not typically thought to drive the DKD phenotype, but we have previously reported a strong potassium secretory gene signature in this cell type in human DKD (Wilson et al., 2019). We compared these previous results to our mouse DKD atlas using the same informatic pipeline. This revealed consistent cell types between mouse and human but relatively few shared cell-specific DEGs (Figure S3C–E), likely reflecting the differing severity of disease in these two datasets, although mouse late distal tubule and collecting duct exhibited the highest magnitude of DKD-induced transcriptional changes, similar to our findings in human.
Cell-specific responses to treatment of DKD
We next examined the extent to which different therapeutic regimens rescued the gene expression changes induced by DKD in each cell type. Distinct cell types responded differently to each therapeutic regimen, both at day 2 and day 14. Transcriptional changes in some cell types occur early but not late (for example podocytes after ACEi+ Rosi treatment), whereas other cell types have greater responses later but not earlier (such as PEC and TAL after ACEi+ Rosi treatment, Zenodo data). We asked what percentage of genes altered by disease (up or down-regulated) were corrected by treatment both at day 2 and day 14. This revealed cell type and time-specific effects across therapeutic regimens (Figure 4A). For example, a higher fraction of disease genes were rescued at day 2 in podocytes, DCT and CNT compared to day 14, whereas 14 days of treatment rescued more disease genes in TAL and DTL compared to day 2 (Figure 4A). Intriguingly, Rosi had maximal therapeutic effect on PEC, TAL, PC and ICA at week 2 treatment. However, the actual genes and pathways being targeted by Rosi treatment in PEC, TAL, PC and ICA were quite distinct, and all of them were ameliorated by treatment (Figure 4B and Figure S4A). In line with a highly heterogeneous response, the actual genes and pathways altered by the different therapies differ substantially with very few genes shared by all therapeutic regimens (Figure S4B). For example, principle cells exhibited many more unique gene changes than conserved changes across treatment regimens (Figure S4C,D). These results indicate a distinct and heterogeneous transcriptional response of kidney cell types both to the diabetic milieu and to different treatment regimens.
Figure 4. Differentially expressed genes normalized by treatment according to cell type and cell-cell communication between injured proximal tubule and other cell types.
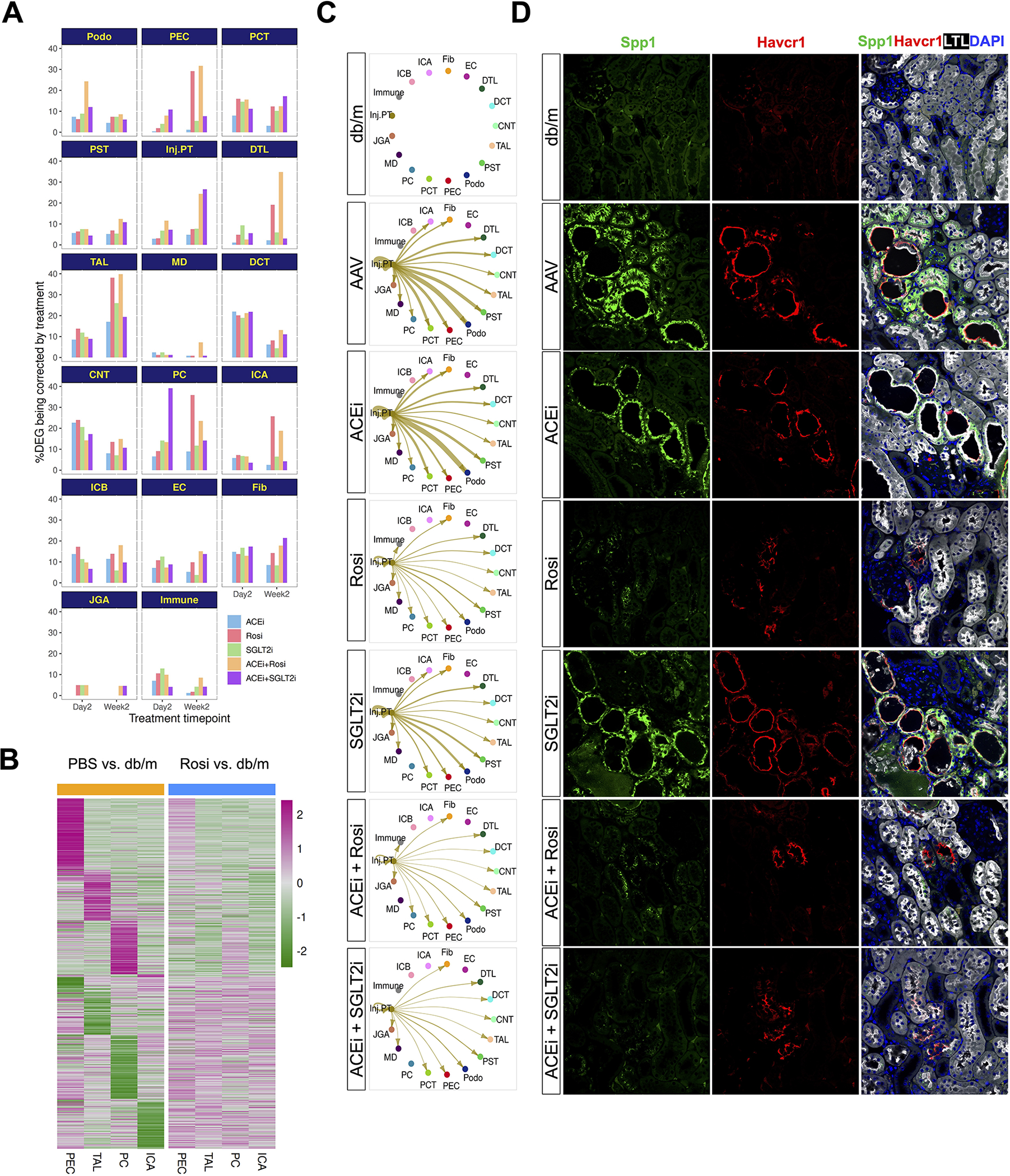
(A) Percentage of DEG induced by diabetes rescued by treatment according to cell type.
(B) Heatmap of gene expression changes in PEC, TAL, PC and ICA at week 2 in PBS (left) vs. Rosi (right).
(C) Cell communication network from injured proximal tubule to other cell types at two days, and amelioration of signaling by treatment. Cell-cell communication network quantifies signaling strength of injured proximal tubule-derived osteopontin (Spp1) to all other cell types according to treatment.
(D) Immunofluorescence analysis of Spp1 expression in disease and drug treatments (day 2). Cells were co-stained with PT (LTL, white) and Injured PT (Havcr1, red) markers.
To study patterns of intercellular communication and changes therein during disease and its treatment, we compared interaction strengths inferred by CellChat (Jin et al., 2021) between all kidney cell types. DKD induced substantial changes in cell-cell communication between all pairs of kidney cell types (Figure S4E). We calculated an incoming and outgoing signaling strength score for each cell type and compared the degree to which those scores changed in DKD compared to control kidney. Cell types for which diabetes induced the largest change in intercellular communication included podocytes, proximal tubule, DTL and TAL (Figure S4F). We then asked how each treatment regimen normalized the signaling score in podocytes, proximal tubule and TAL. This analysis revealed that ACEi had the least effect in all three cell types and SGLT2i and Rosi alone had similar effects (Figure S4G). For podocytes and Rosi, ACEi plus Rosi did the best job of normalizing the signaling strength score, whereas for PT, SGLT2i alone was best (Figure S4G). This analysis specifically identified osteopontin (Spp1) secreted by injured proximal tubule signaling with podocytes and fibroblasts (Figure 4C). Spp1 has already been shown to play an important role in mediating albuminuria through modulation of podocyte signaling in models of DKD (Lorenzen et al., 2008), and serum osteopontin levels strongly predict DKD and all-cause mortality in patients with type 1 diabetes mellitus (Gordin et al., 2014). Osteopontin intercellular signaling was best suppressed by combination therapy, either ACEi plus Rosi or ACEi plus SGLT2i, and this correlated with reduced expression of the injury marker Havcr1, further emphasizing the beneficial effects of combination therapies (Figure 4D).
Synergistic effect of combination drug treatments on injured PT
The integrated analysis of all groups identified a subset of proximal tubular cells that adopted an injured state (we annotated as “inj.PT”) expressing typical injury PT markers such as Havcr1, Vcam1, and C3 (Figure 2, Figure 5A). Combination drug treatments, but not single drug treatments, significantly attenuated the aberrant upregulation of the PT injury marker Havcr1 in DKD (Figure 5B, C). The number of inj.PT was significantly increased in our DKD models (Figure 5D, p = 0.038 for AAV2d versus db/m and p = 0.001 for AAV2w versus db/m, one-way ANOVA with post-hoc Tukey multiple test). We additionally performed bulk RNA-seq on our mouse samples, deconvoluted the fraction of inj.PT, and confirmed that combination therapy was most effective at reducing the proportion of these cells (Figure 5D). To generalize these results, we asked whether a similar population of injured PT is present in human DKD by deconvoluting cell type compositions from two public bulk RNA-seq datasets using the cell type specific molecular signatures identified from our dataset. In late stage DKD, we could detect a significant proportion of inj.PT in human DKD (Figure S5A, B).
Figure 5. Superiority of combination therapies in ameliorating proximal tubule injury responses.
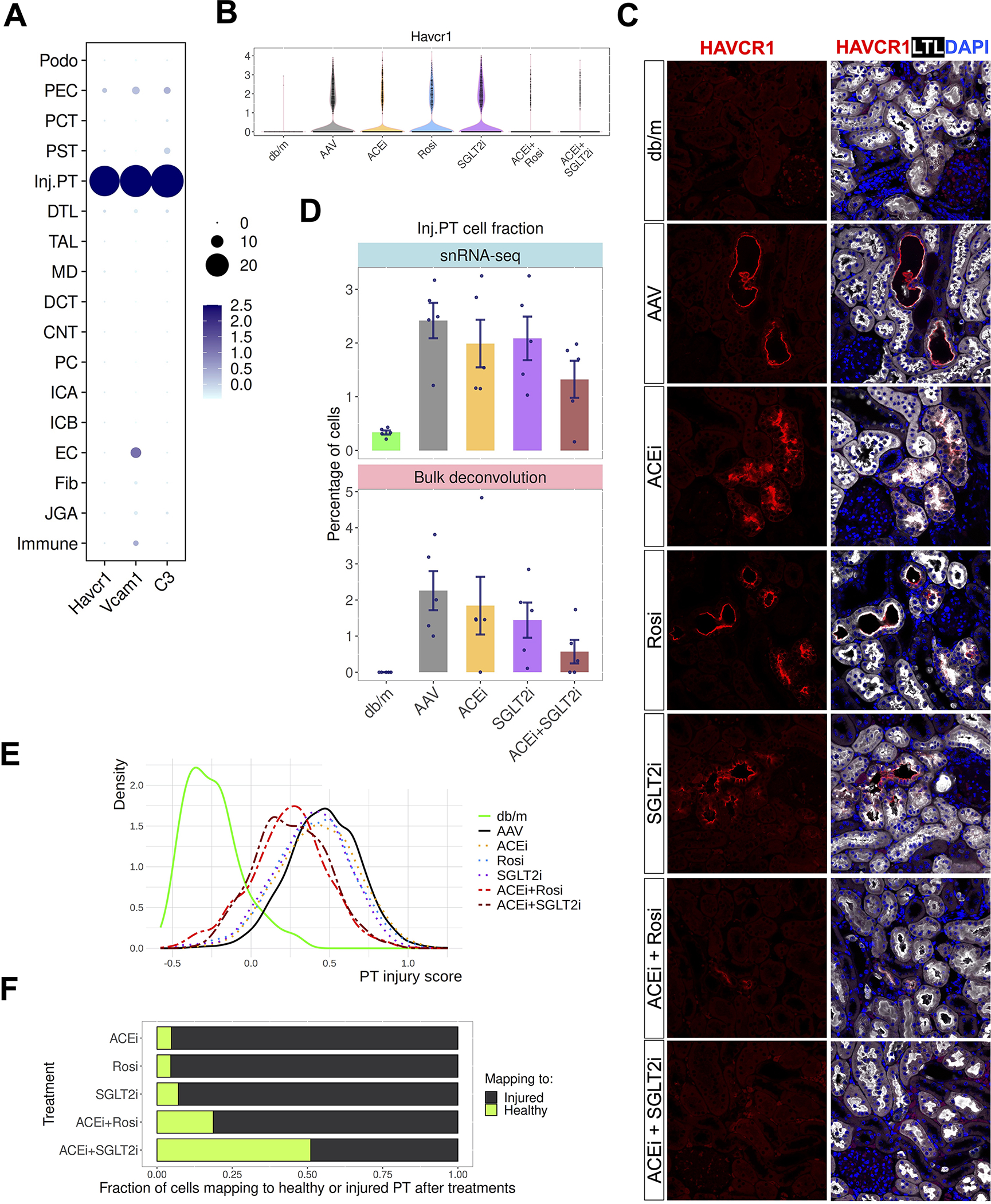
(A) Specific expression of injury genes Havcr1, Vcam1 and C3 in the injured PT subcluster.
(B) Expression of injured PT marker Havcr1 in disease and after week2 treatments.
(C) Validation of the Havcr1 expression by immunofluorescence staining. Cells were co-stained with PT marker (LTL, white).
(D) Percentage of injured PT across the week 2 groups by single cell quantification (upper) and bulk RNA-seq deconvolution. Each dot is an individual mouse.
(E) Density plot to show the injury scores across groups. Cells were scored by the top 50 upregulated genes from injured PT in comparing Renin-AAV vs db/m.
(F) Fraction of cells after treatment mapping to healthy or disease state as measured by scID.
To further characterize these drug responses, we compared the gene expression changes in the injured PT from the diabetic kidneys between the combo treatment groups (ACEi + Rosi and ACEi + SGLT2i) versus their corresponding single treatment groups (ACEi, Rosi, and SGLT2i). To quantify PT injury in an unbiased way, we applied a similar algorithm developed by Melms et al (Melms et al., 2021) to score each individual PT cell using the top 50 upregulated genes from the comparison of vehicle treated versus control (db/m). This analysis revealed no drastic change in the injury score after single drug treatments, but substantial decrease in the scores after two weeks of combination therapy (Figure 5E). As a complementary analysis, we applied a computational algorithm (scID) (Boufea et al., 2020) to measure the fraction of cells after treatment mapping to the cells from healthy vs. diabetic kidneys (Methods). Consistent with the injury score analysis, a greater proportion of injured proximal tubule cells mapped to the cells from healthy kidney after either combination therapy (Figure 5F). In addition, we observed that the change of injured proximal tubule gene regulatory networks as well as the dysregulated pathways such as fatty acid oxidation in diabetes was also corrected by combination drug treatments to a greater degree compared to single drug treatments (Figure S5C–E). Together, these results emphasize that combination therapies have stronger transcriptional effects than monotherapy.
Drug induced glomerular responses
The size of our atlas allowed a detailed analysis of glomerular cell types and responses in DKD and its treatment. We identified glomerular cell types based on a score developed using marker genes identified from a recent glomerulus scRNA-seq atlas (He et al., 2021) (Methods). This allowed identification of podocyte, mesangial cells (MC), glomerular endothelial cells (GEC) and PEC (Figure 6A, B). For example, Ehd3, a regulator of capillary fenestration was strongly expressed in the gEC cluster (Figure 6B). Itga8 and Gata3, two putative mesangial cell genes, are strongly expressed in the MC cluster (Figure 6B). Diabetes induced substantial cell-type-specific transcriptional changes in glomerular cell types at both time points with fewer than 20% of these genes shared across cell types (Figure S6A, B). As observed before, drug treatments target non-overlapping genes across glomerular cell types (figure S6C, D).
Figure 6. Glomerular communication networks in diabetes and its treatment.
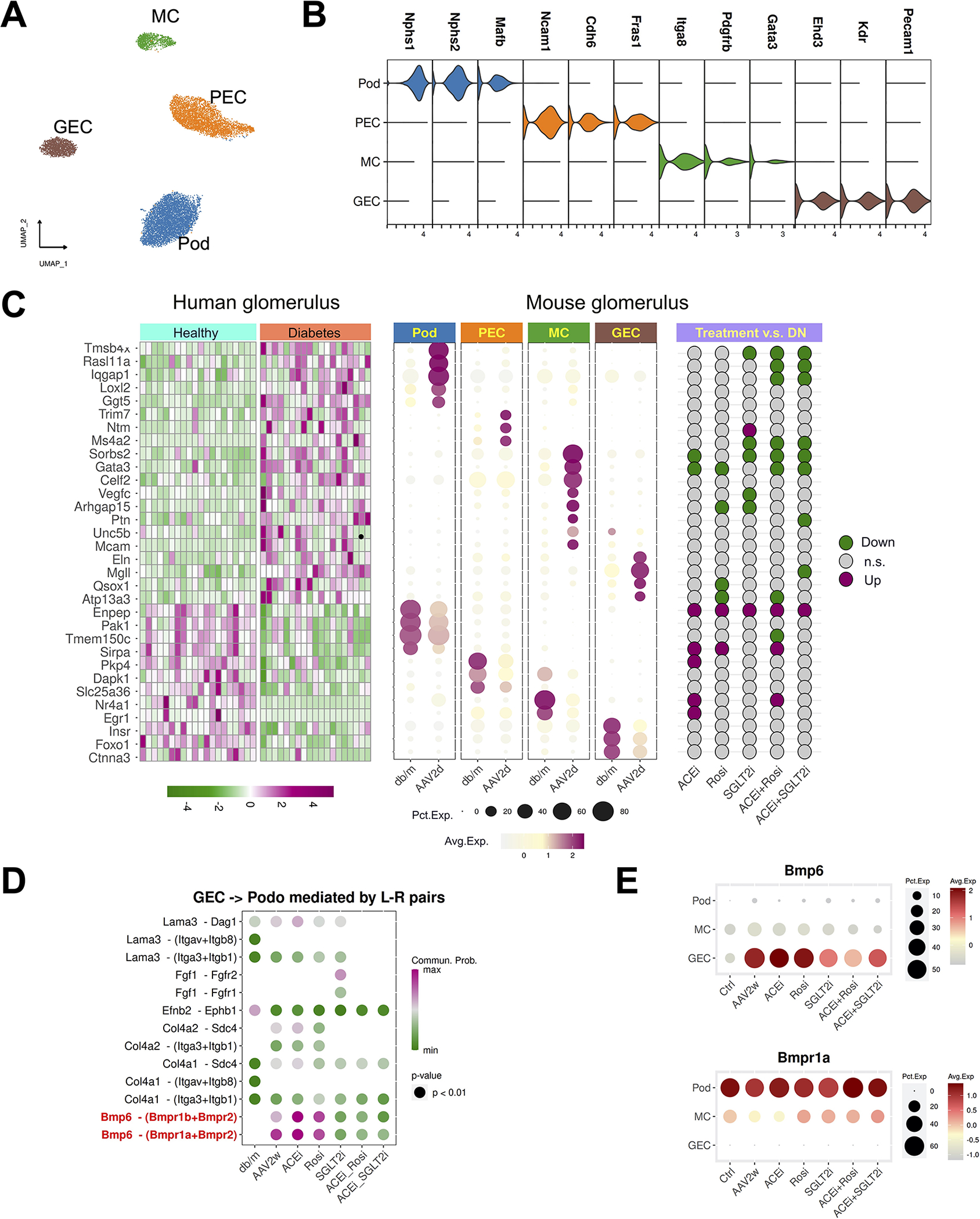
(A) Reclustering of the glomerular cells.
(B) Cell specific marker genes define glomerular subtypes.
(C) Human glomerulus DKD genes mapped to mouse glomerulus cell types. The left heatmap shows bulk glomerulus expression in healthy vs. DKD. The dot plot in the middle assigns those same genes to mouse glomerulus cell types. The dot plot on the right shows that some of these genes are normalized by therapy.
(D) Ligand-receptor scoring for glomerular endothelial cell signaling to podocytes.
(E) Bmp6 and its receptor Bmpr1a expression in glomerulus during diabetes and its treatment.
In recent work, human healthy and diabetic glomeruli underwent bulk transcriptional profiling by microdissection (Levin et al., 2020). We sought to assign the relevant cell type to a manually curated list of genes that were in common in both human and mouse DKD. For example, thymosin β4 (Tmsb4x) (Vasilopoulou et al., 2016) and the GTPase Rasl11a were both exclusively upregulated in podocytes (Figure 6C). Sorbs2, which is involved in the formation of stress fibers (Anekal et al., 2015) and has been associated with DKD by proteomic analysis (Nakatani et al., 2011), was upregulated only in mesangial cells (Figure 6C). We could additionally assess the effect of different treatment regimens on expression of these human DKD-relevant genes. Once again, this revealed a heterogeneous response. For example, the upregulation of Tmsb4x and Rasl11a was attenuated by ACEi + Rosi or ACEi + SGLT2i treatment (Figure 6C) but not by monotherapy. We then used CellChat (Jin et al., 2021) to identify the top changes in intercellular glomerular communication in diabetes and after therapy (Methods). During progression of diabetes, signals sent from GEC to podocytes were upregulated in DKD at both timepoints, and these were normalized only by combination therapies but not by any monotherapy (Figure S6E). We next asked which ligand-receptor pairs mediate GEC to podocyte signaling. This revealed that endothelial-derived Bmp6 signaling was strongly induced by diabetes, and this signaling was most strongly attenuated by Sglt2i alone, or either combination therapy (Figure 6D). Unlike Bmp6, which was specifically upregulated in glomerular endothelial cells by diabetes, its receptor Bmpr1a is constitutively expressed in podocytes (Figure 6E).
Inferring a mechanism for SGLT2i by comparing effects on S1 vs. S2 proximal tubule
SGLT2 inhibitors target the sodium – glucose cotransporter Slc5a2 which is exclusively expressed on the apical membrane of the S1 segment of the proximal tubule (Ghezzi et al., 2017). We reasoned that since the S1 and S2 segments are similar transcriptionally and adjacent to each other in the nephron, but since SGLT2 inhibitors have no direct effect on the S2 segment, that effects specific to the S1 segment would represent direct drug effects. In this way, we might dissociate direct drug effects from systemic effects such as glycosuria, hemodynamic changes and local inflammation. We first confirmed that Slc5a2 expression was specific to the proximal convoluted tubule (containing both S1 and S2 segments) and not expressed in other segments (Figure S7A). We then re-clustered the proximal convoluted tubule into S1 and S2 segments and confirmed that Slc5a2 expression was exclusive to the S1 segment (Figure 7A, B). We next asked how each treatment, both alone and in combination, altered gene expression in the S1 vs. S2 segments at two weeks. This revealed a striking difference in the number of unique genes (e.g., not shared with other therapies) for SGLT2i in the S1 vs. S2 segments (67 vs. 21, respectively, Figure 7C, Figure S7B). This is consistent with the S1 segment as the direct target for SGLT2 inhibition.
Figure 7. Effects of SGLT2 inhibitors on the S1 vs. the S2 segment.

(A) Reclustering of the PCT into S1 and S2 segments.
(B) Density plot showing that Slc5a2 expression is limited to the S1 segment.
(C) Upset plot showing the diabetes-specific DEGs that are rescued by each treatment, divided according to whether they are unique to each treatment or shared across treatments.
(D) Expression of Srsf7 in S1 and S2 across the day2 groups.
(E and F) Knockdown efficiency of siRNA as corroborated by qPCR and western blot.
(G) Volcano plot showing the differential genes in the comparison of Srsf7 siRNA and scramble.
(H) Enriched pathways from gene ontology analysis. NES: normalized enrichment score.
(I and J) Expression of the selected genes in RPTECs and mouse model. All selected genes are DEGs identified by comparing Srsf7 siRNA versus scramble in RPTECs, AAV versus db/m and drug treatments versus AAV in mouse PT-S1.
We next performed gene ontology analysis on the full set of DEGs in S1 vs. S2. Terms related to starvation and hypoxia responses were specifically rescued by SGLT2i in the S1 segment but not the S2 segment (Figure S7C, D). By contrast, terms related to fatty acid oxidation were rescued by treatment in both segments (Figure S7E, F). These results suggest that SGLT2i directly induce both starvation mimicry and hypoxia responses in the S1 segment, which has been one of several previously proposed mechanisms of action for this drug class (Sen and Heerspink, 2021) (Packer, 2020). Terms that were normalized in both segments, however, may reflect secondary systemic effects (such as lower glucose levels, glycosuria and hemodynamic effects) of SGLT2 inhibition.
To generate hypotheses for how SGLT2 inhibition may induce protective starvation mimicry and hypoxia responses in the S1 segment, we inspected the individual DEGs in the S1 vs. S2 dataset (Table S1). We reasoned that changes in gene expression at day 2 that are specific to the S1 segment might reflect the most proximate transcriptional effects of SGLT2 inhibition and could shed light on the molecular mechanism for their protective effects. There were 53 upregulated and 36 downregulated S1-specific genes in SGLT2i vs. PBS at day 2. The second most highly upregulated gene is Srsf7 which encodes a member of the serine/arginine (SR) spliceosome protein family that regulates mRNA splicing. Srsf7 has been shown in mice to promote a juvenile transcriptome gene program by regulating the alternative splicing of genes that promote anabolism and inhibit senescence pathways (Kadota et al., 2020). Another SR family gene upregulated in the S1 by SGLT2 inhibition, Srsf5, is involved in the regulation of glucose metabolism and acetyl-CoA production (Chen et al., 2018). These observations suggest the possibility that one mechanism of action of SGLT2 inhibitors may be to regulate alternative splicing to activate a protective metabolic switch.
We confirmed that only SGLT2i, and not ACEi or Rosi, rescued Srsf7 expression in the S1 segment (Figure 7D). We next sought to develop an in vitro cell culture model to study Srsf7 regulation of proximal tubule. We found that human primary renal proximal tubule cells (RPTEC) express Srsf7, and that exposure to glycated albumin down-regulated Srsf7 mRNA in a dose-dependent fashion (Figure S7G). We then knocked down Srsf7 by siRNA in RPTEC, achieving > 80% knockdown of both mRNA and protein (Figure 7E, F). Bulk RNA-seq of scramble vs Srsf7 siRNA in RPTEC revealed dramatic changes in gene expression, with almost 2,000 genes changed (Figure 7G). Pathway analysis of these gene changes revealed upregulation of a variety of pro-inflammatory terms after Srsf7 knockdown, suggesting that Srsf7 normally promotes a healthy and anti-inflammatory phenotype in the S1 segment (Figure 7H, Figure S7H). Importantly, a number of genes that were rescued by SGLT2i were also differentially expressed after Srsf7 knockdown, providing a mechanistic link between our model and the in vivo results (Figure 7I, J).
Discussion
Success in identifying mechanisms of DKD and how different treatments are protective has been complicated by the large number of tissue compartments affected. While murine models of DKD have been useful, they also have been limited by the variable degrees to which they reflect the full spectrum of DKD phenotypes. scRNA-seq offers a powerful and unbiased way to survey kidney responses during the progression of DKD, and how different therapies act to protect against progression. We combined scRNA-seq with a recent murine model of DKD that recapitulates the major clinical and histologic features of DKD, including hyperglycemia, hypertension, proteinuria, progressive loss of kidney function and glomerulosclerosis (Harlan et al., 2018a). To our knowledge, this is the largest mouse kidney single cell atlas to date. The data herein provide a useful resource for understanding the cell-specific kidney transcriptional responses to DKD and to five common treatment regimens. The DEGs identified in this atlas of DKD and its treatment can be used to gain mechanistic insight as well as serving as potential biomarkers for diagnostics.
With the addition of SGLT2 inhibitors for the treatment of DKD, past three years has witnessed a revolution in therapy for this disease. However it should not be ignored that while Dapagliflozin and Canagliflozin reduce ESRD risk by a third, two-thirds of “at risk” trial subjects on both ACEi and SGLT2i continue to progress to ESRD (Perkovic et al., 2019). This residual unmet need requires novel therapies. The present studies identify non-overlapping cell types and transcriptional changes induced by each treatment regimen. Furthermore, combination therapy of ACEi plus rosiglitazone or ACEi plus SGLT2 inhibitor had the strongest normalizing effect and this was not simply additive of either monotherapy alone. These non-overlapping transcriptional effects may reflect residual disease mechanisms not targeted by current therapies which might be exploited through future combination therapies to treat patients unresponsive to current standards of care.
There are a variety of ways that our single cell atlas may be productively mined. As an example, the large size of our study allowed the construction of a detailed intercellular communication network within the glomerulus. This suggested glomerular endothelial cell to podocyte BMP-6 signaling induced by DKD. A member of the transforming growth factor (TGF)-β superfamily, BMP-6 exerts tissue and cell-specific effects. In liver, endothelial-derived BMP-6 acts in a paracrine fashion to activate hepatocyte hepcidin transcription and thereby maintain iron homeostasis (Canali et al., 2017). Global deletion of BMP-6 exacerbated renal fibrosis, suggesting that its induction in DKD may play a protective role (Dendooven et al., 2011).
A major challenge for cell-specific DEG analyses such as this one is distinguishing between gene expression that is causing vs. a consequence of the DKD phenotype (Porcu et al., 2021). We attempted to address this by comparing the S1 and S2 segment responses to SGLT2i, reasoning that SGLT2i targets S1 and not S2, so early transcriptional changes may reflect the most proximate signaling events induced by this drug class. This analysis suggested that SGLT2 inhibitors induce starvation mimicry and hypoxia responses (Packer, 2020). A hypothesis generated from this analysis is that SGLT2i accomplish their protective effects in part by regulating alternative splicing in the S1 segment. This is because SGLT2 inhibition specifically upregulated two spliceosome genes, Srsf5 and Srsf7, that have been previously shown to regulate inflammation and metabolism. Alternative splicing increases transcript diversity by generating multiple variant mRNAs from one gene and ~95% of multiexonic genes undergo alternative splicing (Pan et al., 2008). Our finding that Srsf7 knockdown promotes a pro-inflammatory PT transcriptional state strongly supports a role for alternative splicing in both DKD pathogenesis and in mediating the protective effect of SGLT2 inhibition. Lending support to this hypothesis, insulin itself regulates alternative splicing in some contexts (Chalfant et al., 1995) and both obesity and the ZSF1 rat model of DKD are associated with alternative splicing (Pihlajamäki et al., 2011; Zhang et al., 2018).
This resource should be of value to the DKD research community. We have generated a data visualization tool that can be accessed online (http://humphreyslab.com/SingleCell/). In addition, all codes and the R object can be downloaded from Github and GEO. In conclusion, our single cell atlas of DKD and its treatment should allow detailed exploration of the mechanisms underlying disease progression and yield a deeper understanding of how current therapies act to slow progression of DKD.
Limitations of study
The cell-specific transcriptional responses to this model of DKD and its treatment may differ between different strains and ages of mice. Furthermore, our single cell atlas of DEGs cannot distinguish between gene transcription that is causal vs. a consequence of DKD. A recent report suggests that SGLT2 inhibition may attenuate mitochondrial dysfunction in PT (Wu et al., 2021), but we cannot directly assess this because our nuclear preparations lack mitochondrial RNA. Finally, through single nucleus dissociation, contextual information from neighboring cells and tissue microenvironment is lost. This complicates our ability to infer intercellular communication networks. Future spatially resolved transcriptomic studies will be required to complement our single cell atlas of DKD and its treatment.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Benjamin D. Humphreys (humphreysbd@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The accession number for the RNA sequencing data reported in this paper is NCBI GEO: GSE184652 for snRNA-seq and GSE199437 for bulk RNA-seq. A searchable database, including gene expression in all kidney cell types, glomerular cell types, and PCT cell types is available at our Kidney Interactive Transcriptomics (K.I.T.) website: http://humphreyslab.com/SingleCell/. ).DEG lists were deposited in Zenodo (https://zenodo.org/record/6344528).
Original codes for data analysis were deposited on GitHub at https://github.com/TheHumphreysLab/Mouse_DKD_Rx_Atlas. An R package to visualize the single cell RNA-seq data with complex group design is available on GitHub (https://github.com/TheHumphreysLab/plot1cell.
All values used to generate the graphs of the paper and original western blot images can be found in the file Data S1 - Source Data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Mouse model of diabetic nephropathy and drug treatments
The in vivo experiments (conducted at Plato Biopharma, Inc. Fort Collings, CO) were performed in the ReninAAV db/db mouse after uninephrectomy (Unx), a murine model of severe progressive DKD. As in the human condition, mice subject to this model display severe renal dysfunction characterized by increased urinary albumin excretion, reduced GFR, increased sCr, BUN, and hypertension. Further, the kidneys of these mice display prominent features of DKD including mesangial expansion, glomerular sclerosis, tubular degeneration as well as tubulointerstitial inflammation and fibrosis (Harlan et al., 2015, 2018b).
Mice were maintained and treated in accordance with the guidelines approved by the Association for Assessment and Accreditation of Laboratory Animal Care. Mice were given ad libitum access to Formulab Diet 5008 (LabDiet, Cat. #: 0001325) and water, housed under standard conditions, and allowed to acclimate for at least one week prior to any procedures. To establish the model, female db/db mice (BKS.Cg-Dock7m +/+ Leprdb/J from Jackson, BKS background, Lot 642) were subject to left nephrectomy at 5 weeks of age and, at 12 weeks, infected with a renin expressing adeno-associated virus (ReninAAV, 109 genomic copies) to induce hypertension and accelerate disease progression. For weeks later, a battery of tests (UACR, sCR, BUN, blood glucose, body weight, SBP, and HR) was conducted to randomize the mice into several treatment as follows:
Vehicle (drinking water)
Lisinopril (An ACE inhibitor, 10 mg/kg/d in the drinking water),
Rosiglitazone (A PPARꙋ activator, 0.007% w/w via admix diet, p.o. ~10 mg/kg/d)
JNJ-39933673 (A SGLT2 inhibitor, 0.0075% w/w via admix diet, p.o. 1 ~ mg/kg/d)
ACEi plus SGLT2i (dosed as before)
ACEi plus rosiglitazone (dosed as before)
In addition to the ReninAAV Unx db/db mice, db/m and Unx db/db mice infected with an empty viral vector (LacZ AAV) were included as healthy and diabetic non-hypertensive controls, respectively.
Terminal samples were collected 48 hours and 2 weeks after treatment inception (n=5/group). For this, mice were placed in metabolic cages to collect urines and later, under general anesthesia with inhalational isoflurane, mice were exsanguinated via cardiac puncture and the kidneys harvested. The right kidneys were cut transversally in roughly three equal size sections. The top section was fixed in 10% neutral buffered formalin and further processed for histopathology analysis. The mid-section was cut in half trough the papilla, one half was snapped frozen, stored at −80C and transferred to Washington University in St. Louis for nuclear transcriptomic analysis. The remaining mid- and bottom sections were stored in RNA later.
The SBP and HR were measured at baseline and after 2 weeks of treatment via tail-cuff plethysmography using a CODA® High Throughput Noninvasive Blood Pressure System (Kent Scientific; Cat. #: CODA-HT8). Plasma and urine samples underwent standard clinical chemistry analysis on an Olympus AU400e Clinical Chemistry Analyzer (Beckman Coulter, Inc.) for the determination BUN and creatine. Creatinine concentration was measured using an enzymatic creatinine assay and other reagents from Sekisui Diagnostics (Sekisui Diagnostics, LLC. Lexington, MA, Cat No. 265–30, SE-035, SM-052 and SM-056). Albumin concentration was analyzed using a using a sandwich-type ELISA developed and optimized at Plato Biopharma, Inc that is based on a monoclonal antibody raised against murine albumin (Bethyl Laboratories, Cat. #: A90-134A, Lot #: 17).
For pathology analysis, the formalin-fixed kidneys were transversely trimmed, routinely processed, paraffin embedded, microtome sectioned at a thickness of 5 μm, and stained with hematoxylin and eosin (H&E), Masson’s Trichrome and Periodic acid-shift staining (PAS). Tissue sections were examined by light microscopy and graded for glomerular injury (mesangial matrix expansion, glomerulosclerosis), tubular degeneration, interstitial inflammation, and fibrosis by a veterinary pathologist.
Method Details
Nuclei isolation from mouse kidney
Single nucleus were isolated using a protocol based upon (Habib et al., 2017) with adaptions for adult mouse kidney including: tissue mincing, homogenization strokes, addition of protease inhibitor and RNasin and adjustments to strainer size and sequence. Mouse kidney were cut into <2 mm pieces and homogenized using a Dounce homogenizer (Kimble Chase #885302–0002) in 2ml of ice-cold Nuclei EZ Lysis buffer (Sigma-Aldrich NUC101) supplemented with protease inhibitor (Roche #5892791001) and RNase inhibitor (Promega #N2615, Life Technologies #AM2696). The samples were then incubated on ice for 5 min with an additional 2ml of lysis buffer. The homogenate was filtered through a 40-μm cell strainer (pluriSelect #43–50040-51) and then centrifuged at 500 x for 5 min at 4 °C. The pellet was resuspended and washed with 4 ml of the buffer and incubated on ice for 5 min. After another centrifugation, the pellet was resuspended with Nuclei Suspension Buffer (1x PBS, 0.07% BSA, 0.1% RNase inhibitor), filtered through a 5-μm cell strainer (pluriSelect 43–50005-01) and counted using a disposable hemecytometer (InCYTO, DHC-F01–5). A full step-by-step protocol was submitted to protocols.io (dx.doi.org/10.17504/protocols.io.nahdab6).
Human primary tubular epithelial cell culture and treatment
Primary human renal proximal tubular cells (RPTECs) were purchased from Lonza (CC-2553) and cultured with Renal Epithelium Cell Growth Medium (CC-3190) supplemented with 10 ng/mL EGF, 5% v/v fetal calf serum, provided with the medium kit. Cells were maintained in a humidified 5% CO2 atmosphere at 37°C. All experiments were carried out on passage 3 of the cells. Cells were treated with glycated albumin or siRNA after a 24-h serum starvation.
SRSF7 siRNA transfection
hRPTECs were grown to 50%–60% confluency and transfected with 100 and 200 nmol/L SRSF7 siRNA (siGenome) or negative control siRNA (siGenome) using Lipofectamine RNAiMAX (Life Technologies) following the manufacturer’s protocol. Cells were harvested at day 1 and day 3 after transfection for protein and RNA isolation in order to validate knockdown. Day 3 mRNA samples were submitted for bulk RNA-seq.
Real-time PCR
RNA was extracted using the RNEASY Mini prep kit (Qiagen) following the manufacturer’s instructions. The extracted RNA (600 ng) was reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative PCR (RT-PCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad). Expression levels were normalized to GAPDH and data analyzed using the 2−ΔΔCt method. Cycling conditions were 95°C for 3 minutes then 40 cycles of 95°C for 15 s and 60°C for 1 minute, followed by one cycle of 95°C for 10 s. Primers used in this study were: SRSF7 forward: 5’-GAAGAAGCAGGTCACGGTCT-3’, SRSF7 reverse: 5’-CGACGGGGATTGGAAATACCT-3’; GAPDH forward: 5’-GACAGTCAGCCGCATCTTCT-3’, GAPDH reverse: 5’-GCGCCCAATACGACCAAATC-3’.
Western blot analysis
RPTEC cell lysates were lysed with RIPA lysis buffer containing protease inhibitor cocktails (Roche) on ice. Protein was collected after centrifugation at 12,000g and concentration was determined by the BCA method (Thermo Fisher). Twenty micrograms of total protein was separated by SDS electrophoresis and transferred to an Immobilon PVDF membrane (Millipore). Membrane was blocked with 5% milk in TBST and probed overnight at 4°C with the primary antibody. After washing the membrane with TBST, it was incubated for 1 hour at room temperature with HRP-conjugated secondary antibody (Dako). The primary antibodies used in this assay were: anti-SRSF7 from Abcam and anti-Tubulin from Proteintech. The membrane was incubated by secondary antibodies (Dako) and developed using the ECL Detection System (GE Healthcare). Image was captured by ChemiDoc imaging system (Bio-Rad).
Immunofluorescence
Formalin-fixed kidney blocks were microtome sectioned at a thickness of 5 μm and mounted on Superfrost slides (Thermo Fisher Scientific). Sections were deparaffinized by xylene and rehydrated by series of ethanol in decreasing concentration. Immunofluorescent staining was performed as follows: kidney sections were washed with 1× PBS for 5 minutes and permeabilized with 0.25% Triton X for 15 minutes. Blocking was done with 5%BSA in PBS for 1 hour. Primary antibodies were incubated for overnight at cold room and sections rinsed with 1× PBS for 5 minutes × 3. Secondary antibodies were incubated for 1 hour at room temperature and rinsed with 1× PBS for 5 minutes × 3. DAPI was used for counterstaining. The following primary antibodies were used: anti-SPP1 from Abcam and anti-HAVCR1 from R&D Systems. Images were obtained by confocal microscopy (Nikon C2+ Eclipse).
Single nucleus RNA-seq
Droplet-based single-cell RNA sequencing was performed using the 10x Genomics Chromium Single Cell Kit v3. Single nucleus suspensions at a concentration of 1000 nuclei/ul were loaded into the Chromium Controller as per manufacturer instructions, which target 10,000 nuclei per sample. 12 PCR cycles were used for cDNA synthesis. Each library was sequenced using an Illumina NovaSeq instrument, 8 samples per S4 lane, with one 150 bp read located near the 3′ end of the mRNA. Illumina runs were demultiplexed and the resulting fastq files were processed by the 10X Genomics cellranger pipeline (version 6.0.1). Both exonic and intronic reads were incorporated in the final molecule counts.
Ambient RNA and doublets removal, batch effect correction, and cell clustering
We used CellBender to remove the ambient RNA contamination (Fleming et al., 2019). In brief, the unfiltered count matrices sampleID.h5 from cellranger were input into CellBender. CellBender remove-background was run (on cuda mode) with parameters “expected-cells” and “total-droplets-included” were chosen for each dataset based on the total UMI per cell vs. cell barcode curve in accordance with CellBender documentation. Other parameters were left at their default values. The CellBender corrected count matrices were then input to DoubletFinder (McGinnis et al., 2019) to remove the potential doublets. We used default parameters to run DoubletFinder except for the nExp, which is estimated based on the total cell number of the sample (nNuc) and a linear regression equation derived from the Demuxlet study (Kang et al., 2018), i.e. nExp = nNuc * (0.018+8.4e-06* nNuc). The ambient RNA- and doublet- free count matrices were input into Seurat for clustering analysis. Batch effect was corrected by using the integration pipeline from Seurat6. Due to the large size of our dataset, we used a reference-based approach to make the analysis less computationally intensive (https://satijalab.org/seurat/articles/integration_large_datasets.html). One sample from the healthy group (sample A3020) was used as the ‘reference’ with the remainder designated as ‘query’ datasets. After data integration, the combined data proceeded with joint analysis such as principal component analysis (PCA), clustering and 2D projection by UMAP. We annotated the cell clusters based on their marker gene expression. Cell clusters with confused marker gene expression of multiple cell types were manually removed. We also excluded the tAL cell type from downstream analysis because our initial analysis indicated that the number of cells in tAL varies dramatically across the 14 groups ranging from 12 cells in AAV2w to 1,468 cells in db/m, which greatly skewed the differential gene analysis. Alternative ‘reference’ sample (sample E3019) and data integration method (Harmony) were used to validate the cell clustering results.
snRNA-seq differential gene analysis
Cell type specific markers were identified from the whole dataset after clustering using Seurat function FindAllMarkers, with significant cutoff FDR set to 0.05, and log fold change set to 0.25. Cell class were annotated based on the well-known kidney cell markers. To find cell type specific disease genes for DN, we compared the gene expression between db/db and db/m, AAV 2d and db/m, and AAV 2w and db/m using the Seurat function FindMarkers, with significant cutoff FDR set to 0.05, and log fold change set to 0.5. The DE genes were categorized by cell type where they were detected. To identify the disease genes that were rescued by each drug treatment, we compared the gene expression between treatment versus DN and cross the gene list to the disease gene list. The overlapped DE genes in opposite directions are considered the genes that were rescued by treatment.
Pathway and gene ontology analysis
Pathway analysis of the differentially expressed genes were performed using fgsea R package (Korotkevich et al., 2021). Multiple pathway databases were used for this analysis, including MSigDB, Hallmark, and Reactome. The returning enrichment scores and p-values were employed to compare the pathway activity across cell types and drug treatments. For gene ontology analysis, topGO package (Alexa et al., 2006) was used for testing the gene-sets related to biological processes on the DE gene lists generated by various comparisons. GO terms with greater than 500 genes or less than 5 genes were excluded from the analysis.
Bulk RNA-seq and data analysis
Total RNA integrity was determined using Agilent 4200 Tapestation. Library preparation was performed with 500ng to 1ug of total RNA. Ribosomal RNA was removed by an RNase-H method using RiboErase kits (Kapa Biosystems). mRNA was then fragmented in reverse transcriptase buffer and heating to 94 degrees for 8 minutes. mRNA was reverse transcribed to yield cDNA using SuperScript III RT enzyme (Life Technologies, per manufacturer’s instructions) and random hexamers. A second strand reaction was performed to yield ds-cDNA. cDNA was blunt ended, had an A base added to the 3’ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 15 cycles using primers incorporating unique dual index tags. Fragments were sequenced on an Illumina NovaSeq-6000 using paired end reads extending 150 bases. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a maximum of one mismatch in the indexing read was used in demultiplexing. Fastq files were processed using an in-house pipeline (https://github.com/HaojiaWu/Bulk_RNAseq_analysis_pipeline). RNA-seq reads were then aligned to the mm10 (for mouse kidneys) and GRCh38 (for human RPTEC) with STAR version 2.7.3a. Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 2.0.3. The count matrices were then input to edgeR for data normalization and differential gene analysis.
Cell type deconvolution of bulk RNA-seq data
We used BisqueRNA (Jew et al., 2020) to deconvolute the mouse DKD dataset (this study) and publicly available bulk RNA-seq data from human DKD (Fan et al., 2019; Levin et al., 2020). For the microdissection dataset, raw count matrix was directly downloaded from the link that the authors provided (Levin et al., 2020). For the early and advanced DKD dataset (Fan et al., 2019), fastq files were download from GEO: GSE142025, and processed using our inhouse scripts to generate the count matrix (https://github.com/HaojiaWu/Bulk_RNAseq_analysis_pipeline). Data were normalized using the R package edgeR (Robinson et al., 2010). Human genes were converted to mouse orthologues using R package BiomaRt (Durinck et al., 2009). The bulk data were deconvoluted using the cell type specific markers identified from our snRNA-seq data.
Measurement of the drug effect on injured PT
We used three different approaches to assess the drug effect in ameliorating the injured PT. First, we inspected the expression level of the well-known injured markers (such as Havcr1, Cp, and Cxcl1) before and after drug treatments. Second, we used the top 50 upregulated genes in comparing AAV versus healthy PT to score the cells. PT injury after treatment was measured by the scores assigned to each group. Third, we used a discriminant analysis tool, scID (Boufea et al., 2020), to qualify the percent of cells mapping to disease or healthy PT after treatments. We set a cutoff of logFold-change = 0.6 for a gene to be selected as marker genes to input in to scID.
GRN inference and metabolic activity analysis
We inferred GRNs independently on the injured PT subsets from different groups of the scRNA-seq data using SCENIC (Aibar et al., 2017; Van de Sande et al., 2020). In brief, we log2-transformed the CPM-normalized counts of each subset with a prior of 1 and used those normalized counts for pySCENIC in a docker container. We create cell GRN networks from injured PT single nuclei data, with the mouse mm10 genome for cis-regulatory analysis and the gene-motif rankings 10 kilobases around the transcription start site (TSS). AUCell values of the regulons from the SCENIC output file were used for differential test and to generate the dot plot. For metabolic analysis, we ran COMPASS (Wagner et al., 2021) on the normalized count data from the injured PT of different groups. Reaction scores for each single cell were processed using the R script provided by the authors. Reactions from fatty acid oxidation were selected and visualized.
Association of cell type with GWAS traits by integrative analysis
MAGMA (de Leeuw et al., 2015) was used to test for a positive association between the binned fractions in each cell type and the gene-level associations. For a given kidney cell type, this tested whether increasing tissue specificity of gene expression was associated with increasing common-variant genetic findings for eGFR using information from all of the genes. This analysis was composed of the following steps. First, GWAS summary statistics of the four kidney function traits eGFR, UACR, BUN, and serum urate were downloaded from the CKDGen Consortium Data website (https://ckdgen.imbi.uni-freiburg.de). To match the European linkage disequilibrium (LD) score, we used the European ancestry–specific GWAS summary statistics only. Second, the summary stats were preprocessed following the public tutorial (https://github.com/NathanSkene/MAGMA_Celltyping). MAGMA v1.0 was used to calculate gene-level association statistics using a window of 10 kb upstream and 1.5 kb downstream of each gene, accounting for LD, gene length, gene density, sample size, and minor allele count. Significant heritability enrichment for kidney GWAS traits and the kidney cell types was reported based on the p value with adjusted by multiple tests (i.e. FDR <0.05).
Cell–cell interaction network analysis
We used CellChat (Version 1.1.3) to infer the cell-cell interactions across all kidney cell types and among the glomerular cell types using the expression of known ligand-receptor pairs. To identify the changes of the cell–cell communication networks induced by diabetic nephropathy and assess how drug treatments can rescue the perturbations, we followed the tutorial for comparison analysis of multiple datasets in the CellChat github repository (https://htmlpreview.github.io/?https://github.com/sqjin/CellChat/blob/master/tutorial/Comparison_analysis_of_multiple_datasets.html) and computed the major signaling changes between DKD and healthy as well as the treated and untreated groups by joint manifold learning and quantitative contrasts of multiple cell-cell communication networks. For the analysis on the whole kidney dataset, we selected the “Secreted Signaling pathways” and used the precompiled mouse protein–protein-Interactions as a priori network information. For the analysis on the glomerular dataset, we selected all databases curated in CellChat including the “Secreted Signaling”, “Cell-Cell contact” and “ECM-Receptor”. We then compared the outgoing and incoming interaction strength of each pair of cell types to identify significant changes in sending or receiving signals between groups, and used the interaction strength to qualify the drug treatment effect. We then visualized the most significant ligand-receptor pairs that mediate the cell-cell interaction changes across comparisons by using the netVisual_bubble function in CellChat.
Supplementary Material
Table S1. DEGs for S1 and S2 segments in DKD and after SGLT2i treatment. Related to Figure 7.
Data S1. Unprocessed source data underlying all blots and graphs. Related to Figures 1, 5 and 7 and Supplemental Figure 7.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SRSF7 Polyclonal Antibody | Thermo Fisher Scientific | PA5-39482; RRID:AB_2556056 |
| HAVCR1 Antibody | R & D Systems | AF1817; RRID: AB_2116446 |
| Streptavidin, Alexa Fluor 647 conjugate | Invitrogen | S21374 |
| SPP1 antibody | Abcam | ab218237; RRID: AB_2732079 |
| Tubulin antibody | Proteintech | 66240; RRID: AB_2881629 |
| Donkey Anti Rabbit, Alexa Fluor 488 conjugate | Invitrogen | A21206 |
| Donkey Anti Goat, Alexa Fluor 568 conjugate | Invitrogen | A11057 |
| Goat anti-rabbit HRP | Dako | P0448 |
| Goat anti-mouse HRP | Dako | P0447 |
| Chemicals, peptides, and recombinant proteins | ||
| 1× DPBS | Gibco | 14190144 |
| Nuclei EZ Lysis Buffer | Sigma | NUC101 |
| Complete ULTRA Tablets, Mini, EDTAfree, EASYpack | Roche | 05 892 791 001 |
| RNasin Plus Ribonuclease Inhibitors | Promega | N2615 |
| SUPERaseIN RNase Inhibitor | Thermo Fisher Scientific | AM2696 |
| RNaseZap | Ambion | AM9780 |
| RNase free H2O | Thermo Scientific | AM9938 |
| Primary RPTEC | Lonza | CC-2553 |
| REGM BulletKit | Lonza | CC-3190 |
| Glycated albumin | Sigma | A8301 |
| Periodic Acid-Schiff (PAS) Kit | Sigma | 395B |
| siGENOME Human SRSF7 | Horizon Discovery | M-015909-01-0020 |
| siGENOME Non-Targeting siRNA | Horizon Discovery | D-001210-05-20 |
| Lipofectamine™ RNAiMAX | Thermo Fisher Scientific | 13778075 |
| Biotinylated Lotus Tetragonolobus Lectin (LTL) | Vector Laboratories | B-1325 |
| BCA Protein Assay Kit | Thermo Fisher Scientific | 23252 |
| Critical commercial assays | ||
| Chromium Single Cell 3’ GEM, Library & Gel Bead Kit v3 | 10× Genomics | PN-1000075 |
| Chromium Single Cell B Chip Kit | 10× Genomics | PN-1000153 |
| Deposited data | ||
| Raw and analyzed snRNA-seq data | This study | GSE184652 |
| Raw and analyzed bulk RNA-seq data | This study | GSE199437 |
| Data S1 – Source Data | This study | |
| Zenodo data | This study | https://zenodo.org/record/6344528 |
| Nuclei isolation protocol | Protocols.io | https://dx.doi.org/10.17504/protocols.io.nahdab6 |
| Kidney Interactive Transcriptomics | The Humphreys Lab | http://humphreyslab.com/SingleCell/ |
| Experimental models: Organisms/strains | ||
| BKS.Cg-Dock7m +/− Leprdb/J Leprdb- heterozygous | The Jackson Lab | Lot 000642 |
| BKS.Cg-Dock7m +/+ Leprdb/J Leprdb- homozygous | The Jackson Lab | Lot 000642 |
| Software and algorithms | ||
| Cell Ranger | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| Seurat V4.0 | Satija Lab | https://github.com/satijalab/seurat |
| STAR V2.7.3a | Dubin et al., Bioinformatics 2013 | |
| CellBender | Broad Institute | https://github.com/broadinstitute/CellBender |
| DoubletFinder V2.0 | McGinnis et al., Cell Systems 2019 | https://github.com/chris-mcginnis-ucsf/DoubletFinder |
| BisqueRNA | Jew and Alvarez et al et al., Nature Communications 2020 | https://github.com/cozygene/bisque |
| scID | Boufea et al., iScience 2020 | https://github.com/BatadaLab/scID |
| MAGMA_Celltyping | Skene et al., Nature Genetics 2018 | https://github.com/neurogenomics/MAGMA_Celltyping |
| CellChat | Jin et al., Nature Communications 2021 | https://github.com/sqjin/CellChat |
| circlize | Gu et al, Bioinformatics 2014 | https://jokergoo.github.io/circlize_book/book/ |
| ComplexHeatmap | Gu et al, Bioinformatcis 2016 | https://jokergoo.github.io/ComplexHeatmap-reference/book/ |
| COMPASS | Wagner et al., Cell 2021 | https://github.com/YosefLab/Compass |
| fgsea | Korotkevich et al., bioRxiv 2019 | https://github.com/ctlab/fgsea |
| edgeR | Bioconductor | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| topGO | Bioconductor | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| plot1cell | This study | https://github.com/TheHumphreysLab/plot1cell |
Highlights.
A million cells atlas shows heterogeneity in kidney cell responses to DKD and treatments.
Mono- and combination therapies target different cells and induce non-overlapping changes.
Combination therapies are more effective in rescuing DKD-associated transcriptional changes.
SGLT2i signature reveals a role for modulation of alternative splicing by this drug class.
Acknowledgements
This study was supported by a sponsored research agreement from Janssen Pharmaceuticals, by the Washington University Diabetes Research Center NIH DK20579 and by NIH DK103740.
Footnotes
Declaration of interests
R.G.V., X.Y., D.R., T.C., M.R., C.M., E.M., and M.D.B. are employees of Janssen Research & Development, LLC. M.D.B. holds Johnson and Johnson stock. B.D.H. is a consultant for Janssen Research & Development, LLC, Pfizer and Chinook Therapeutics. B.D.H. holds equity in Chinook Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine J-C, Geurts P, Aerts J, et al. (2017). SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexa A, Rahnenführer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. [DOI] [PubMed] [Google Scholar]
- Anekal PV, Yong J, and Manser E (2015). Arg Kinase-binding Protein 2 (ArgBP2) Interaction with α-Actinin and Actin Stress Fibers Inhibits Cell Migration. J Biol Chem 290, 2112–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal A, Balasubramanian S, Dhawan S, Leung A, Chen Z, and Natarajan R (2020). Integrative Omics Analyses Reveal Epigenetic Memory in Diabetic Renal Cells Regulating Genes Associated With Kidney Dysfunction. Diabetes 69, 2490–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boufea K, Seth S, and Batada NN (2020). scID Uses Discriminant Analysis to Identify Transcriptionally Equivalent Cell Types across Single-Cell RNA-Seq Data with Batch Effect. IScience 23, 100914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, et al. (2001). Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345, 861–869. [DOI] [PubMed] [Google Scholar]
- Bukosza EN, Kratochwill K, Kornauth C, Schachner H, Aufricht C, and Gebeshuber CA (2020). Podocyte RNA sequencing reveals Wnt- and ECM-associated genes as central in FSGS. PLoS One 15, e0231898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canali S, Zumbrennen-Bullough KB, Core AB, Wang C-Y, Nairz M, Bouley R, Swirski FK, and Babitt JL (2017). Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 129, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfant CE, Mischak H, Watson JE, Winkler BC, Goodnight J, Farese RV, and Cooper DR (1995). Regulation of alternative splicing of protein kinase C beta by insulin. J Biol Chem 270, 13326–13332. [DOI] [PubMed] [Google Scholar]
- Chen Y, Huang Q, Liu W, Zhu Q, Cui C-P, Xu L, Guo X, Wang P, Liu J, Dong G, et al. (2018). Mutually exclusive acetylation and ubiquitylation of the splicing factor SRSF5 control tumor growth. Nat Commun 9, 2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, Reeves WB, and Awad AS (2021). Pathophysiology of diabetic kidney disease: impact of SGLT2 inhibitors. Nat Rev Nephrol 17, 319–334. [DOI] [PubMed] [Google Scholar]
- Dendooven A, van Oostrom O, van der Giezen DM, Leeuwis JW, Snijckers C, Joles JA, Robertson EJ, Verhaar MC, Nguyen TQ, and Goldschmeding R (2011). Loss of endogenous bone morphogenetic protein-6 aggravates renal fibrosis. Am J Pathol 178, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Yi Z, D’Agati VD, Sun Z, Zhong F, Zhang W, Wen J, Zhou T, Li Z, He L, et al. (2019). Comparison of Kidney Transcriptomic Profiles of Early and Advanced Diabetic Nephropathy Reveals Potential New Mechanisms for Disease Progression. Diabetes 68, 2301–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. BioRxiv 791699. [Google Scholar]
- Ghezzi C, Yu AS, Hirayama BA, Kepe V, Liu J, Scafoglio C, Powell DR, Huang S-C, Satyamurthy N, Barrio JR, et al. (2017). Dapagliflozin Binds Specifically to Sodium-Glucose Cotransporter 2 in the Proximal Renal Tubule. J Am Soc Nephrol 28, 802–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordin D, Forsblom C, Panduru NM, Thomas MC, Bjerre M, Soro-Paavonen A, Tolonen N, Sandholm N, Flyvbjerg A, Harjutsalo V, et al. (2014). Osteopontin is a strong predictor of incipient diabetic nephropathy, cardiovascular disease, and all-cause mortality in patients with type 1 diabetes. Diabetes Care 37, 2593–2600. [DOI] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan SM, Ostroski RA, Coskun T, Yantis LD, Breyer MD, and Heuer JG (2015). Viral transduction of renin rapidly establishes persistent hypertension in diverse murine strains. Am J Physiol Regul Integr Comp Physiol 309, R467–474. [DOI] [PubMed] [Google Scholar]
- Harlan SM, Heinz-Taheny KM, Overstreet JM, Breyer MD, Harris RC, and Heuer JG (2018a). Pathological and Transcriptome Changes in the ReninAAV db/ db uNx Model of Advanced Diabetic Kidney Disease Exhibit Features of Human Disease. Toxicol Pathol 46, 991–998. [DOI] [PubMed] [Google Scholar]
- Harlan SM, Heinz-Taheny KM, Sullivan JM, Wei T, Baker HE, Jaqua DL, Qi Z, Cramer MS, Shiyanova TL, Breyer MD, et al. (2018b). Progressive Renal Disease Established by Renin-Coding Adeno-Associated Virus-Driven Hypertension in Diverse Diabetic Models. J Am Soc Nephrol 29, 477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Chen P, Zambrano S, Dabaghie D, Hu Y, Möller-Hackbarth K, Unnersjö-Jess D, Korkut GG, Charrin E, Jeansson M, et al. (2021). Single-cell RNA sequencing reveals the mesangial identity and species diversity of glomerular cell transcriptomes. Nat Commun 12, 2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou F-F, Mann JFE, McMurray JJV, Lindberg M, Rossing P, et al. (2020). Dapagliflozin in Patients with Chronic Kidney Disease. N Engl J Med 383, 1436–1446. [DOI] [PubMed] [Google Scholar]
- Jew B, Alvarez M, Rahmani E, Miao Z, Ko A, Garske KM, Sul JH, Pietiläinen KH, Pajukanta P, and Halperin E (2020). Accurate estimation of cell composition in bulk expression through robust integration of single-cell information. Nat Commun 11, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan C-H, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat Commun 12, 1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadota Y, Jam FA, Yukiue H, Terakado I, Morimune T, Tano A, Tanaka Y, Akahane S, Fukumura M, Tooyama I, et al. (2020). Srsf7 Establishes the Juvenile Transcriptome through Age-Dependent Alternative Splicing in Mice. IScience 23, 100929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen L-A, Veys K, et al. (2020). Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 180, 764–779.e20. [DOI] [PubMed] [Google Scholar]
- Kang HM, Subramaniam M, Targ S, Nguyen M, Maliskova L, McCarthy E, Wan E, Wong S, Byrnes L, Lanata CM, et al. (2018). Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat Biotechnol 36, 89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komers R, Xu B, Fu Y, McClelland A, Kantharidis P, Mittal A, Cohen HT, and Cohen DM (2014). Transcriptome-based analysis of kidney gene expression changes associated with diabetes in OVE26 mice, in the presence and absence of losartan treatment. PLoS One 9, e96987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. [Google Scholar]
- de Leeuw CA, Mooij JM, Heskes T, and Posthuma D (2015). MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol 11, e1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin A, Reznichenko A, Witasp A, Liu P, Greasley PJ, Sorrentino A, Blondal T, Zambrano S, Nordström J, Bruchfeld A, et al. (2020). Novel insights into the disease transcriptome of human diabetic glomeruli and tubulointerstitium. Nephrol Dial Transplant 35, 2059–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, et al. (2001). Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345, 851–860. [DOI] [PubMed] [Google Scholar]
- Li Y, Haug S, Schlosser P, Teumer A, Tin A, Pattaro C, Köttgen A, and Wuttke M (2020). Integration of GWAS Summary Statistics and Gene Expression Reveals Target Cell Types Underlying Kidney Function Traits. J Am Soc Nephrol 31, 2326–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzen J, Shah R, Biser A, Staicu SA, Niranjan T, Garcia AM, Gruenwald A, Thomas DB, Shatat IF, Supe K, et al. (2008). The role of osteopontin in the development of albuminuria. J Am Soc Nephrol 19, 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis CS, Murrow LM, and Gartner ZJ (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Systems 8, 329–337.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, et al. (2021). A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhl L, Genové G, Leptidis S, Liu J, He L, Mocci G, Sun Y, Gustafsson S, Buyandelger B, Chivukula IV, et al. (2020). Single-cell analysis uncovers fibroblast heterogeneity and criteria for fibroblast and mural cell identification and discrimination. Nat Commun 11, 3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani S, Kakehashi A, Ishimura E, Yamano S, Mori K, Wei M, Inaba M, and Wanibuchi H (2011). Targeted Proteomics of Isolated Glomeruli from the Kidneys of Diabetic Rats: Sorbin and SH3 Domain Containing 2 Is a Novel Protein Associated with Diabetic Nephropathy. Exp Diabetes Res 2011, 979354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura S, Yamamoto Y, Matsumura Y, Ohba K, Sakamaki S, Kimata H, Nakayama K, Kuriyama C, Matsushita Y, Ueta K, et al. (2014). Novel Indole-N-glucoside, TA-1887 As a Sodium Glucose Cotransporter 2 Inhibitor for Treatment of Type 2 Diabetes. ACS Med Chem Lett 5, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Shimizu M, Yamanouchi M, Toyama T, Hara A, Furuichi K, and Wada T (2021). Trajectories of kidney function in diabetes: a clinicopathological update. Nat Rev Nephrol. [DOI] [PubMed] [Google Scholar]
- Packer M (2020). SGLT2 Inhibitors Produce Cardiorenal Benefits by Promoting Adaptive Cellular Reprogramming to Induce a State of Fasting Mimicry: A Paradigm Shift in Understanding Their Mechanism of Action. Diabetes Care 43, 508–511. [DOI] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, and Blencowe BJ (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40, 1413–1415. [DOI] [PubMed] [Google Scholar]
- Patrakka J, Xiao Z, Nukui M, Takemoto M, He L, Oddsson A, Perisic L, Kaukinen A, Szigyarto CA, Uhlen M, et al. (2007). Expression and subcellular distribution of novel glomerulus-associated proteins dendrin, ehd3, sh2d4a, plekhh2, and 2310066E14Rik. [DOI] [PubMed] [Google Scholar]
- Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, Edwards R, Agarwal R, Bakris G, Bull S, et al. (2019). Canagliflozin and Renal Outcomes in Type 2 Diabetes and Nephropathy. N Engl J Med 380, 2295–2306. [DOI] [PubMed] [Google Scholar]
- Pihlajamäki J, Lerin C, Itkonen P, Boes T, Floss T, Schroeder J, Dearie F, Crunkhorn S, Burak F, Jimenez-Chillaron JC, et al. (2011). Expression of the splicing factor gene SFRS10 is reduced in human obesity and contributes to enhanced lipogenesis. Cell Metab 14, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcu E, Sadler MC, Lepik K, Auwerx C, Wood AR, Weihs A, Sleiman MSB, Ribeiro DM, Bandinelli S, Tanaka T, et al. (2021). Differentially expressed genes reflect disease-induced rather than disease-causing changes in the transcriptome. Nat Commun 12, 5647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen T, and Heerspink HJL (2021). A kidney perspective on the mechanism of action of sodium glucose co-transporter 2 inhibitors. Cell Metab 33, 732–739. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande B, Flerin C, Davie K, De Waegeneer M, Hulselmans G, Aibar S, Seurinck R, Saelens W, Cannoodt R, Rouchon Q, et al. (2020). A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc 15, 2247–2276. [DOI] [PubMed] [Google Scholar]
- Vasilopoulou E, Kolatsi-Joannou M, Lindenmeyer MT, White KE, Robson MG, Cohen CD, Sebire NJ, Riley PR, Winyard PJ, and Long DA (2016). Loss of endogenous thymosin β4 accelerates glomerular disease. Kidney Int 90, 1056–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Wang C, Fessler J, DeTomaso D, Avila-Pacheco J, Kaminski J, Zaghouani S, Christian E, Thakore P, Schellhaass B, et al. (2021). Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell 184, 4168–4185.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B, et al. (2016). Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med 375, 323–334. [DOI] [PubMed] [Google Scholar]
- Wilson PC, Wu H, Kirita Y, Uchimura K, Ledru N, Rennke HG, Welling PA, Waikar SS, and Humphreys BD (2019). The single-cell transcriptomic landscape of early human diabetic nephropathy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woroniecka KI, Park AS, Mohtat D, Thomas DB, Pullman JM, and Susztak K (2011). Transcriptome analysis of human diabetic kidney disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun Z, Yang S, Fu J, Fan Y, Wang N, Hu J, Ma L, Peng C, Wang Z, et al. (2021). Kidney single-cell transcriptome profile reveals distinct response of proximal tubule cells to SGLT2i and ARB treatment in diabetic mice. Mol Ther S1525–0016(21)00520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Dower K, Zhang B, Martinez RV, Lin L-L, and Zhao S (2018). Computational identification and validation of alternative splicing in ZSF1 rat RNA-seq data, a preclinical model for type 2 diabetic nephropathy. Sci Rep 8, 7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. DEGs for S1 and S2 segments in DKD and after SGLT2i treatment. Related to Figure 7.
Data S1. Unprocessed source data underlying all blots and graphs. Related to Figures 1, 5 and 7 and Supplemental Figure 7.
Data Availability Statement
The accession number for the RNA sequencing data reported in this paper is NCBI GEO: GSE184652 for snRNA-seq and GSE199437 for bulk RNA-seq. A searchable database, including gene expression in all kidney cell types, glomerular cell types, and PCT cell types is available at our Kidney Interactive Transcriptomics (K.I.T.) website: http://humphreyslab.com/SingleCell/. ).DEG lists were deposited in Zenodo (https://zenodo.org/record/6344528).
Original codes for data analysis were deposited on GitHub at https://github.com/TheHumphreysLab/Mouse_DKD_Rx_Atlas. An R package to visualize the single cell RNA-seq data with complex group design is available on GitHub (https://github.com/TheHumphreysLab/plot1cell.
All values used to generate the graphs of the paper and original western blot images can be found in the file Data S1 - Source Data. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.