

Abstract
In this perspective we focus on recent developments in our understanding of nutrient-induced insulin secretion that challenge a key aspect of the “canonical” model, in which an oxidative phosphorylation-driven rise in ATP production closes KATP channels. We discuss the importance of intrinsic β-cell metabolic oscillations, the phasic alignment of relevant metabolic cycles, shuttles and shunts, and how their temporal and compartmental relationships align with the triggering phase or the secretory phase of pulsatile insulin secretion. Metabolic signalling components are assigned regulatory, effectory and/or homeostatic roles vis-à-vis their contribution to glucose sensing, signal transmission, and resetting the system. Taken together, these functions provide a framework for understanding how allostery, anaplerosis, and oxidative metabolism are integrated into the oscillatory behavior of the secretory pathway. By incorporating these temporal as well as newly-discovered spatial aspects of β-cell metabolism, we propose a much-refined MitoCat-MitoOx model of the signaling process for the field to evaluate.
eTOC BLURB
In this perspective Merrins et al. discuss recent developments in our understanding of nutrient stimulated insulin secretion, with a focus on oscillatory metabolic signaling. By integrating the temporal as well as newly-identified spatial aspects of β-cell metabolism, the authors propose a revised model of pulsatile insulin secretion.
Introduction
The year 2021 celebrated the 100th anniversary of the discovery of insulin and its administration to the first patient with diabetes. The names of Frederick Banting and Charles Best in this landmark discovery in medicine are widely known, and the key roles played by John MacLeod and Cecil Collip are being increasingly recognized. Insulin is considered the most important anabolic hormone in glucose and energy homeostasis, and a rise in blood glucose itself promotes the secretion of insulin by the pancreatic β-cell. Initial in vivo evidence that glucose promotes insulin secretion came from Grafe and Meythaler (Grafe and Meythaler, 1927a, 1927b), who injected glucose in the femoral versus the pancreatic duodenal vein of dogs and found that only the latter caused hypoglycemia. Zunz and La Barre joined two dogs by a pancreatic-jugular anastomosis and observed that pancreatic blood of the donor lowered the blood sugar of the recipient animal (Zunz and La Barre, 1927). In an elegant in vivo experiment, London and Kotschneff placed an angiostomy cannula in the pancreato-duodenal vein and determined the rise in glucose and insulin content of blood from the pancreas after a glucose meal (London and Kotschneff, 1934).
A decade later came key evidence for glucose-stimulated insulin secretion (GSIS) ex vivo. Anderson and Long (Anderson and Long, 1947) established a perfusion system that incorporated the stomach, duodenum and pancreas, and using a bioassay for insulin found that glucose promotes insulin release. Then came the well-known study of Grodsky and colleagues (Grodsky et al., 1963) where a perfused pancreas system was established to demonstrate by immunoassay that glucose dose-dependently promotes insulin secretion from β-cells. Importantly, this study also provided evidence that glucose must be metabolized by the β-cell to cause insulin release.
Much effort and major progress have identified key components of the β-cell metabolic signaling machinery for GSIS. Yet, there is still a fragmented understanding of the relationships among the different metabolic pathways and signaling metabolites of the apparatus. There is a need to better define those signals, traditionally named metabolic coupling factors (MCF), which are either regulatory of key metabolic pathways in the cascade leading to exocytosis, or are effectory as they target key late components of the secretory apparatus, or homeostatic as they bring metabolic signals and secretion back to the ground state. Neither is the temporal assembly that coordinates GSIS mechanistically well-defined. Understanding this metabolic machinery is of importance not only for the islet-cell field but for understanding cellular nutrient sensing at large.
The biochemical components of insulin secretion promoted by the intracellular metabolism of fuel stimuli like glucose, fatty acids, and amino acids have been extensively reviewed (Campbell and Newgard, 2021; Prentki et al., 2013; Rorsman and Braun, 2013; Rutter et al., 2015). To date, they have been interpreted largely vis-à-vis the “canonical” model where oxidative phosphorylation (OxPhos) raises ATP/ADP to trigger KATP channel closure, Ca2+ influx and insulin release. However, more recent discoveries of spatially and temporally compartmentalized metabolism have encouraged a re-examination of this model, which has reached a level of dogma that we believe should be revised.
The canonical model of GSIS should be reconsidered
In 1968 glucokinase (GK) was discovered as a ‘glucose-sensing’ enzyme in the β-cell that regulates glycolytic flux (Matschinsky and Ellerman, 1968). Endowed with a Km for the sugar in its physiological concentration range, around 7–8 mM, GK traps glucose by producing glucose 6-phosphate (G6P). G6P is generated continuously because GK lacks product inhibition unlike the other hexokinases. In 1976, a reconstituted system for insulin secretion from cod islet plasma membrane provided evidence that Ca2+ as well as the metabolites G6P and phosphoenolpyruvate (PEP) play a role in secretion (Davis and Lazarus, 1976; Lazarus et al., 1976), and pyruvate kinase (PK) activity was identified in rat islet lysates soon after (Sugden and Ashcroft, 1977). These early studies were quickly overshadowed by the subsequent discovery of the β-cell ATP-sensitive K+ (KATP) channel (Cook and Hales, 1984), which provided a mechanism to explain how the plasma membrane responds to glucose metabolism and controls the timing of Ca2+ influx and insulin secretion (Ashcroft et al., 1984; Misler et al., 1986; Rorsman and Trube, 1985). Since this discovery, and with the recognition that KATP channels also respond to the corresponding fall in Mg-ADP (Dunne and Petersen, 1986; Kakei et al., 1986), the “canonical” model inferred the intermediary signaling between GK and KATP with a postulate where substrate-driven mitochondrial oxidative phosphorylation (OxPhos) raises cytosolic ATP/ADP (ATP/ADPc) to close KATP channels and activate Ca2+ influx to induce insulin secretion (Figure 1A). It is especially this ‘intermediary signaling’ mechanism that should be reconsidered.
Figure 1. Reconsidering the canonical model of glucose-stimulated insulin secretion.
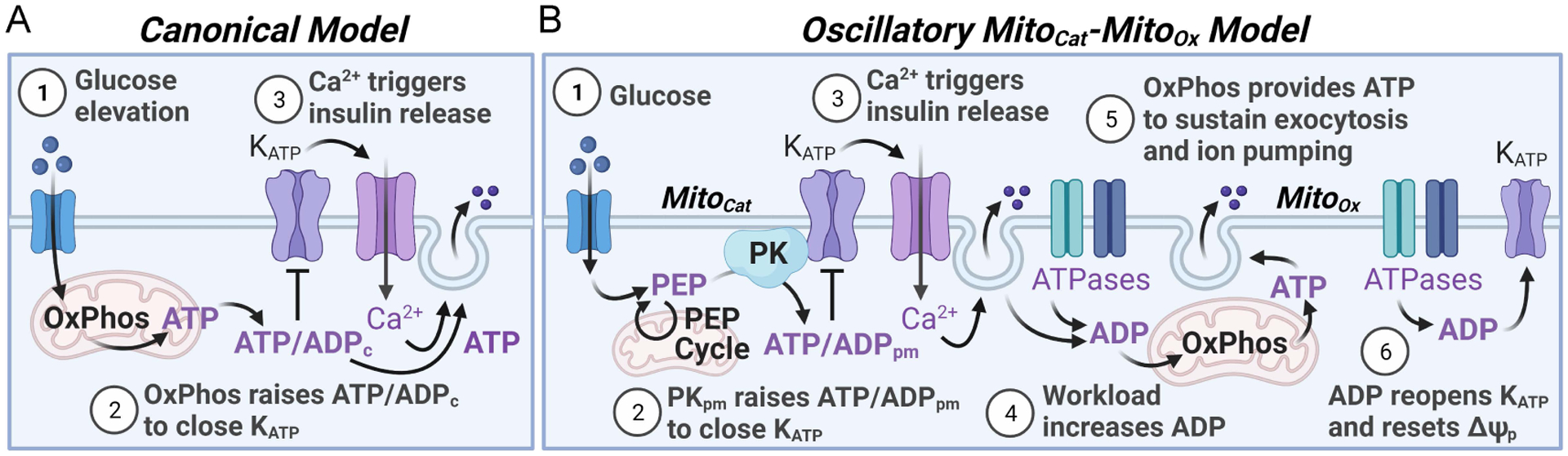
(A) In the canonical model, glucose metabolism drives an initial rise in ATP/ADP ratio via oxidative phosphorylation (OxPhos) that plays the dominant role in both KATP channel closure and ATP production to sustain exocytosis. (B) A new spatially and temporally compartmentalized model of stimulus-secretion coupling, termed the MitoCat-MitoOx model, is motivated by the recent discovery that plasma membrane-associated pyruvate kinase (PKpm), rather than OxPhos, locally generates the rise in ATP/ADP to close KATP channels and initiate insulin secretion. OxPhos has been repositioned after KATP closure, membrane depolarization and the rise in cytosolic Ca2+ to highlight its complimentary role in generating ATP to sustain secretion. While the figure shows that MitoCat and MitoOx are separated by the onset of membrane depolarization, they are not all or none processes. It remains possible that OxPhos may help to sustain KATP channel closure after the channel is initially closed by PK and the PEP cycle. Abbreviations: ATP/ADPc, cytosolic ATP/ADP ratio; ATP/ADPpm, plasma membrane ATP/ADP ratio; ΔΨp, plasma membrane potential.
Pros and cons of the canonical model related to mitochondrial ATP-dependent KATP closure
An intuitive part of the canonical model is that OxPhos, being an efficient ATP generator (up to 31 ATP/glucose vs. 2 ATP from glycolysis alone), should generate the bulk of the ATP/ADP rise to close KATP channels. Below we summarize the key evidence that has been identified in support of the canonical model and discuss the caveats of each argument.
In support of the canonical model, a rise in NAD(P)H fluorescence and substantial O2 consumption occurs prior to Ca2+ influx following a step increase in glucose (Jung et al., 2000; Kennedy et al., 2002; Luciani et al., 2006). Also, ATP/ADPc and total ATP levels correlate with GSIS in many studies. In addition, imaging tools demonstrate that cytosolic and plasma membrane ATP both rise prior to Ca2+ influx (Ainscow and Rutter, 2002; Kennedy et al., 1999; Li et al., 2013) (Figure 1A). However, the common protocol used in these studies was to rescue β-cell ATP/ADPc from a fuel-starved state (2–3 mM glucose) to fuel saturation (11–16 mM glucose, or even higher). This may have little to do with physiological stimulus-secretion coupling, where β-cell oscillations in metabolism and insulin secretion are observed in vitro and in vivo (discussed in detail below). Thus, when metabolic signaling was examined during such steady-state oscillations at ~9–11 mM glucose, multiple groups using different technologies observed that the rise in O2 consumption and the fall in mitochondrial membrane potential (ΔΨm) occur after KATP closure (Jung et al., 2000; Kennedy et al., 2002; Krippeit-Drews et al., 2000; Lewandowski et al., 2020) (Figure 1B).
β-cells have a unique metabolism that allows ATP/ADPc to change in response to substrate availability, a feature that is not present in most tissues (Nicholls, 2016). A rigorous bioenergetic approach to understanding the β-cell electron transport chain (ETC), termed metabolic control analysis (MCA), has identified stronger substrate control of ATP/ADPc in β-cells than in muscle due to a much higher proton leak (Affourtit and Brand, 2006). Furthermore, many β-cell studies show that glucose increases mitochondrial ATP synthesis (Affourtit et al., 2018), including in the presence of other physiological fuels (Mugabo et al., 2017) (Figure 1B). However, MCA measures only O2 consumption to estimate ATP production by mitochondria. Therefore, MCA is blind to anaplerotic cycles, and other sources of local ATP production that may be key for signaling, in particular the anaplerotic PEP cycle (Foster et al., 2022; Stark et al., 2009). In this cycle, mitochondria convert pyruvate to PEP, which leaves the mitochondria and augments the glycolytic PK reaction that produces plasma membrane ATP/ADP (ATP/ADPpm) at the KATP channel (Foster et al., 2022; Lewandowski et al., 2020) (Figure 1B). Noteworthy, the high proton leak in β-cells identified in respirometry experiments (Affourtit and Brand, 2009; Gerencser, 2015; Gerencser et al., 2017, 2016; Taddeo et al., 2020; Wikstrom et al., 2012) facilitates flux through PK and detracts from the ability of mitochondria to generate ATP.
To date, mathematical models have universally started with the assumption that OxPhos drives KATP closure, and have successfully modeled oscillatory glucose metabolism (Bertram et al., 2018; Fridlyand et al., 2005, 2003; Merrins et al., 2016, 2012, 2010). Indeed, the most updated models faithfully reproduce the glycolytic and mitochondrial phase relationships observed in β-cells by incorporating the feedback of Ca2+ on metabolism (Marinelli et al., 2021; McKenna et al., 2016). While these models are compatible with the canonical model, they do not demonstrate causality. Furthermore, like MCA, a significant drawback of these kinetic models is that they lack anaplerosis-dependent cycles known to play key roles in GSIS and lack spatially compartmentalized metabolism that plays a key role in signaling (Foster et al., 2022; Lewandowski et al., 2020) (Figure 1b).
Often-cited evidence for the canonical model is that so-called mitochondrial fuels (e.g., glutamine/leucine, α-ketoisocaproate, methyl-pyruvate, as well as pyruvate in β-cell lines) increase O2 consumption and ATP/ADPc levels, close KATP channels, and stimulate insulin secretion (Prentki et al., 2013). In this review we integrate the roles of anaplerosis, PK and the PEP cycle into a model of GSIS. Suffice is to say here, although these so-called “pure” mitochondrial fuels provide mitochondrial acetyl-CoA, they also induce anaplerosis by activating pyruvate carboxylase (PC) or glutamate dehydrogenase (GDH) and generate mitochondrial PEP. Therefore these non-glucose fuels do not bypass PK and the PEP cycle (Foster et al., 2022; Stark et al., 2009). Insulin stimulation by membrane permeant methyl-pyruvate was taken as another argument for the canonical model (Mertz et al., 1996), but it also directly closes KATP channels independently of metabolism (Düfer et al., 2002).
Mitochondrial poisons (rotenone, oligomycin, etc.) prevent KATP closure, demonstrating that mitochondria are essential to this process (Dukes et al., 1994; Gregg et al., 2019; Kiranadi et al., 1991). However, this does not mean that ETC-derived ATP is responsible for closing KATP channels. An alternative explanation is that because β-cells do not express plasma membrane pyruvate/lactate transporters and consequently lack the Pasteur effect (Pullen et al., 2012; Schuit et al., 1997), mitochondrial poisons will also stall glycolytic flux through PK, which is present on the plasma membrane and generates sufficient ATP/ADPpm to close KATP channels (Foster et al., 2022; Lewandowski et al., 2020).
Although the direct ETC electron donor TMPD/ascorbate closes KATP channels, and this was taken as evidence that mitochondrially-derived ATP is sufficient to close KATP channels (Duchen et al., 1993), this treatment also reduces cytosolic ATP (Prentki, unpublished observations). Notably, the complex II fuel methyl-succinate does not stimulate insulin secretion in mouse islets despite raising NADH and ATP (MacDonald, 2002), arguing that mitochondrial ATP is, in fact, insufficient to close KATP channels. Indeed, mitochondrial fuels (amino acids including glutamine/leucine) that raise ATP/ADPc do not close KATP channels in the absence of PK activity (Foster et al., 2022).
Overall, we believe that the evidence for the canonical model where glucose and other fuels “push” OxPhos to induce mitochondrial ATP production that closes KATP channels is circumstantial, and has significant caveats that are incompatible with recent and historical data showing the importance of spatial and temporal metabolic signaling.
Spatially and temporally compartmentalized glycolysis controls OxPhos and KATP closure
Glucose-induced insulin secretion is associated with a rise in Ca2+, which increases workload (ATP hydrolysis) from ion pumps and elevates ADP. In response, the mitochondrial ETC will “pull” nutrients from the surrounding milieu to be oxidized (Chance and Williams, 1955). It remains possible that the elevated glucose and downstream pyruvate (a metabolic “push”) will benefit mitochondrial ATP generation and insulin secretion after KATP closure, when Ca2+ and ADP are high. But what about before KATP closure, which is dependent upon a rise in ATP/ADP and therefore occurs in the setting of low Ca2+ and ADP levels? As the ATP/ADP ratio rises, and ADP levels fall, OxPhos is less and less able to generate ATP, as evidenced by the fall in O2 consumption and the rise in ΔΨm that precede each Ca2+ pulse during stead-state oscillations (Jung et al., 2000; Kennedy et al., 2002; Krippeit-Drews et al., 2000; Lewandowski et al., 2020). These time-resolved measurements, while not exclusive of the canonical model, indicate that mitochondrial ATP production is at its lowest at the time of KATP closure. This motivated the search for an alternative source of ATP production for KATP channel closure that functions prior to membrane depolarization.
There is now strong evidence that glycolytic, rather mitochondrially-generated ATP, closes KATP channels to initiate insulin secretion, and that this process involves the PEP cycle (Figure 1B). First, the timing of PK activity can explain membrane depolarization. During glucose-estimulated Ca2+ oscillations, allosteric activation of PK by fructose 1,6-bisphosphate (F16BP) increases prior to KATP closure (Lewandowski et al., 2020; McKenna et al., 2016; Merrins et al., 2013), whereas ΔΨm decreases and O2 consumption occurs after KATP closure (Jung et al., 2000; Kennedy et al., 2002; Krippeit-Drews et al., 2000; Lewandowski et al., 2020). In fact, respiration in permeabilized β-cells is actually inhibited by PEP (Lewandowski et al., 2020), and in intact islets by pharmacologic PK activation (Regeenes et al., 2022). Second, PEP is a strong candidate for a signaling molecule. Among all cellular metabolites, PEP has the highest free energy bond, twice that of ATP, and is present at high (~1 mM) levels (Sugden and Ashcroft, 1977). About 40% of PEP derives from the mitochondrial PEP cycle that returns pyruvate from the PK reaction to the cytosol as PEP (Abulizi et al., 2020; Alves et al., 2015; Foster et al., 2022; Lewandowski et al., 2020; Stark et al., 2014, 2009). Third, the substrates for PK, PEP and ADP, are sufficient to stimulate biphasic insulin release in permeabilized β-cells (Pizarro-Delgado et al., 2016), and pharmacologic PK activators stimulate GSIS in mouse, rat, and human islets (Abulizi et al., 2020; Foster et al., 2022; Lewandowski et al., 2020). Fourth, PK is localized to the β-cell plasma membrane, where it is sufficient to raise ATP/ADPpm and close KATP channels (Foster et al., 2022; Lewandowski et al., 2020). These data match prior observations in cardiac myocytes (Dhar-Chowdhury et al., 2007, 2005; Weiss and Lamp, 1989, 1987). Fifth, although the OxPhos-dependent rise in ATP/ADPc may help buffer this plasma membrane-compartmentalized effect, mitochondrial fuels (amino acids) that raise ATP/ADPc were shown to be insufficient to close KATP channels in the absence of PK activity (Foster et al., 2022). Sixth, not only glucose but also amino acids promote PK- and PEP cycle-dependent KATP closure, as shown by whole-body and β-cell deletion of mitochondrial PEPCK (PCK2) (Abulizi et al., 2020; Foster et al., 2022). Hence, the emergent evidence indicates that glucose and other nutrients promote KATP channel closure primarily via PK and the PEP cycle.
In the sections below, we propose an oscillatory model of glucose signaling in the β-cell that we wish to name the MitoCat-MitoOx model (Figure 1B). This novel model surmises that during MitoCat, PK raises ATP/ADPpm to close KATP channels, whereas during the MitoOx phase, OxPhos responds to ADP rise to produce ATP that sustains the energy demands of insulin secretion and ion pumping. Looking at the bigger picture, the MitoCat-MitoOx model recognizes that multiple metabolic and ionic processes accelerate and decelerate with time, coincident with the electrically-silent triggering phase (MitoCat) or the electrically-active secretory phase (MitoOx) of insulin secretory oscillations. Synchronized oscillations (e.g., in ADP and Ca2+) strongly dictate which metabolic and signalling processes are active at any given time. While we do not yet know with precision the relative phase relationship of the many oscillating processes, this model describes the causal relationships among key pathways that collectively influence GSIS.
Metabolic oscillations separate β-cell metabolism into two states: MitoCat and MitoOx
Excess insulin causes hypoglycemia and in extreme cases death; appropriately turning insulin secretion on and off is therefore key to survival. In vivo, insulin secretion is pulsatile even in the basal state (Pørksen, 2002). The primary effect of glucose elevation is to increase the amplitude of the pulses (Goodner et al., 1977; Lang et al., 1979), in part by recruiting more cells to respond (Heart et al., 2006). Insulin oscillations are lost early in the progression to diabetes (Lang et al., 1981; O’Rahilly et al., 1988; Polonsky et al., 1988). Disrupted islet pulsatility is associated with glucose intolerance (Head et al., 2012), and the increased efficacy of insulin pulses at target tissues, relative to continuous delivery, further demonstrates their physiologic relevance (Matthews et al., 1983; Matveyenko et al., 2012).
Pulsatility is equally important for the β cell itself – by ensuring the coordination and integration of metabolism. Proportionate only to a rise in glucose, the GK reaction continually pushes carbon into the system (Matschinsky and Ellerman, 1968). Virtually every subsequent step of central carbon metabolism is oscillatory (for reviews see (Bertram et al., 2018; Henquin, 2009; Idevall-Hagren and Tengholm, 2020)). Energetic and redox metabolic pairs, such as ATP/ADP and NAD(P)H/NAD(P)+, are used by multiple reactions across the entire metabolic network. For instance, as the ATP/ADP ratio increases, all ADP-dependent reactions begin to slow and thermodynamically favor ATP hydrolysis.
There is an oscillatory release of insulin not only when pancreas or isolated islets are perifused with a fixed level of glucose, but also in single β-cells, indicating that the oscillator is cell-intrinsic (Ainscow and Rutter, 2002; Civelek et al., 1997, 1996; Detimary et al., 1998; Jung et al., 2000; Juntti-Berggren et al., 2003; Kennedy et al., 2002; Kindmark et al., 2001; Luciani et al., 2006). The reasonably short period (~5 minutes) of oscillatory processes (e.g., insulin secretion, membrane potential, Ca2+) (Gilon et al., 1993; Kindmark et al., 2001), plasma membrane ATP (Li et al., 2013) and cAMP (Dyachok et al., 2006), as well as NAD(P)H fluorescence (Luciani et al., 2006)) largely excludes oscillatory transcriptional and translational control mechanisms. The evidence further indicates that super-connected subpopulations of β-cells within the islet (including hubs, leaders, and wave initiators) drive Ca2+ dynamics and pulsatile insulin secretion (Benninger and Kravets, 2021).
Glucose-driven metabolic and Ca2+ oscillations are complex (Bertram et al., 2007; Dean and Matthews, 1968), with fast electrical oscillations (lasting tens of seconds) superimposed on slower metabolically-driven oscillations (~5 min). The slow oscillations better reflect the timing of Ca2+ and insulin oscillations observed in living animals (Adams et al., 2021; Lang et al., 1979; Nunemaker et al., 2006, 2005). Although Ca2+ can reinforce metabolic oscillations, they can exist independently of oscillations in membrane potential or Ca2+ (Merrins et al., 2010). Substantial evidence implicates activation of glycolytic phosphofructokinase-1 (PFK1) by its product fructose 1,6-bisphosphate (F1,6BP) as the mechanism that initiates slow metabolic oscillations in β-cells (Tornheim, 1997; Merrins et al., 2012, 2013; Bertram et al., 2018). PFK1 is also activated by fructose 2,6-bisphosphate (F2,6BP), generated from fructose 6-phosphate (F6P) by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) (Merrins et al., 2012). While glucose concentration determines GK flux, the PFK1 reaction separates glycolysis into two segments (upper and lower glycolysis) (Figure 2A). As PFK1 accelerates and F1,6BP accumulates, G6P and F6P are consumed. The near-equilibrium enzymes of lower glycolysis from aldolase to enolase propagate the F1,6BP signal to PK (Wilson and Matschinsky, 2021). A second node is found at the PK step, after which pyruvate/lactate levels fall as mitochondria consume pyruvate faster than it is generated (Civelek et al., 1997; Lewandowski et al., 2020; Sdao et al., 2021). Consequently, the lower glycolytic intermediates between PFK1 and PK oscillate in phase (Figure 2A) and are reinforced by the positive allosteric control of PFK1 by F1,6BP and F2,6BP, ultimately creating synchronous oscillations in ATP/ADP produced by phosphoglycerate kinase and PK and NADH/NAD+ produced by glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Due to the PFK1 and PK nodes in glycolysis, the upper glycolytic intermediates and the post PK metabolites oscillate roughly in phase.
Figure 2. Allosteric regulation of glycolysis and oscillations of β-cell metabolism and insulin secretion during MitoCat and MitoOx.
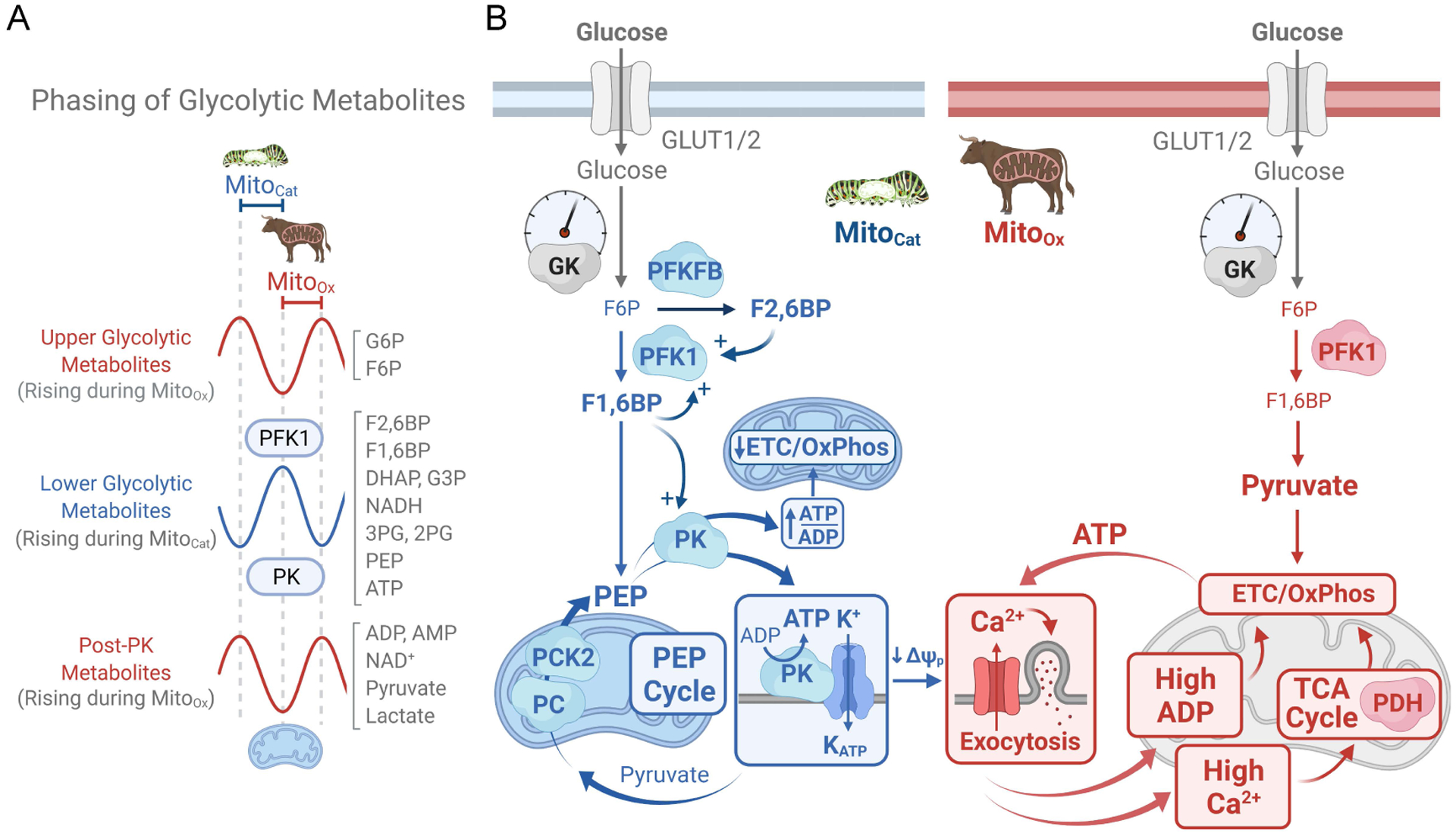
(A) Phosphofructokinase-1 (PFK1), responding to activation by its product fructose 1,6-bisphosphate (F1,6BP), generates glycolytic oscillations that separate the phases of upper glycolysis (red) and lower glycolysis (blue), while pyruvate kinase (PK) controls glycolytic efflux. During MitoCat the lower glycolytic metabolite levels rise, as the flux through PFK1 increases while PK is slowed by rising ATP/ADP; oscillations in post-PK metabolites (red) are out of phase with lower glycolysis due to rapid metabolism of pyruvate by mitochondria. During MitoOx the fall in ATP/ADP stalls PFK1 and maximizes flux through PK, causing a crash in the lower glycolytic metabolite levels. (B) Oscillations in glycolysis and anaplerosis-cataplerosis during MitoCat (blue) are matched by antiphase oscillations in Ca2+, ADP, TCA cycle activity and OxPhos during MitoOx (red). Importantly, PK is localized to both the mitochondrial and plasma membranes. This compartmentation of PK is central to β-cell oscillatory metabolism and insulin secretion. Before plasma membrane depolarization, during MitoCat, PFK1 generates F1,6BP to activate PK, which lowers ADP at the inner mitochondrial membrane, reducing flux through the adenine nucleotide translocator (ANT), slowing the ETC (which becomes ADP-starved and state 4-like) and therefore TCA cycle, while activating anaplerosis/cataplerosis and the phosphoenolpyruvate (PEP) cycle. During the PEP cycle, anaplerosis (filling of TCA cycle intermediates) is due to the PC reaction that carboxylates pyruvate to oxaloacetate, whereas cataplerosis (egress of the TCA cycle intermediates to the cytosol) results from the exit of mitochondrial PEP to the cytosol following the mitochondrial PEP carboxykinase (PCK2) reaction. PEP then exits the mitochondrion to feed mitochondrial and plasma membrane PK. While PK reinforces the PEP cycle, plasma membrane compartmentalized PK drives a rise in ATP/ADP that closes KATP channels. Following membrane depolarization and the rise in Ca2+, during MitoOx, the high workload (i.e. ATP hydrolysis) associated with ion pumping and insulin secretion restores ADP and increases flux through the ETC (which is now ADP replete and state 3-like), the TCA cycle, and lower glycolysis. Abbreviations: DHAP, dihydroxyacetone phosphate; ETC, electron transport chain; F6P, fructose 6-phosphate; F2,6BP, fructose 2,6-bisphosphate; G3P, glyceraldehyde 3-phosphate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate, GK, glucokinase; G6P, glucose 6-phosphate; PC, pyruvate carboxylase; PCK2, phosphoenolpyruvate kinase 2; PDH, pyruvate dehydrogenase; PFKFB, phosphofructo-2-kinase/fructose 2,6-bisphosphatase.
Glycolytic oscillations propagate into the mitochondria driving two opposed “metabolic states” that are separated in time – MitoOx and MitoCat. MitoOx is named for the dominant activity of OxPhos and is predominantly controlled by ADP and supported by Ca2+. MitoCat is named for the matched processes of anaplerosis (i.e., the net expansion of the TCA cycle metabolite pool) and cataplerosis (i.e., the egress of TCA cycle intermediates into other pathways). Figure 2B outlines the MitoCat-MitoOx of oscillatory β-cell metabolism proposed by Lewandowski et al. (Lewandowski et al., 2020) where the progressive closure of KATP channels occurs during the electrically-silent triggering phase of metabolic signaling (during MitoCat), and secretion during the electrically-active phase delineated by membrane depolarization (during MitoOx):
MitoCat. The electrically quiet period leading up to KATP channel closure and β-cell depolarization, MitoCat, is dominated by the progressive lowering of cytosolic ADP by PK thus creating an ADP-starved “state-4-like” mitochondria with low ETC flow but high ΔΨm. In the cytosol, rising NADH/NAD+ due to flux through GAPDH slows glycolysis and the ensuing buildup of F1,6BP activates PFK1 in a positive feedback loop, generating the upstroke of an oscillation. F1,6BP also feeds forward to allosterically activate PK, lowering its Km for PEP and also raising its Vmax. Pyruvate-mediated elevations in mitochondrial NADH/NAD+ and acetyl-CoA then activate pyruvate carboxylase (PC) to generate OAA that is converted to PEP in the mitochondria via PEP carboxykinase (PEPCK, or PCK2). Mitochondrial PEP then exits the mitochondrion, thus feeding cataplerosis to further augment the ADP-lowering power of PK which produces ATP. MitoCat concludes after plasma membrane-associated PK raises ATP/ADP sufficiently to close KATP channels, depolarizing the cell. Although the PEP cycle is ‘futile’, it spatially and temporally redistributes ATP, and is therefore essential for β-cell signaling. This occurs when the PEP produced from pyruvate within the mitochondria, having burned 1 ATP (at PC) and 1 GTP (at PCK2), is delivered following its cataplerotic output to a plasma membrane-associated PK that locally lowers ADP and produces 1 ATP in the close vicinity of KATP channels.
MitoOx. Following KATP channel closure and β-cell depolarization, Ca2+ influx triggers exocytosis. The resulting increase in workload (i.e. ATP hydrolysis) – due to the energy requirement for exocytosis, ion pumps, etc. – elevates ADP to stimulate OxPhos in this “state-3-like” phase with active glycolytic and TCA cycle fluxes. Eventually ADP rises sufficiently to reopen KATP channels and repolarize the plasma membrane.
The MitoCat-MitoOx model describes β-cell metabolism at intermediate levels of glucose that support oscillatory behavior. Unlike the plasma membrane potential, which has a square wave pattern, MitoCat and MitoOx are not binary, all-or-none processes. Indeed the ETC also functions during MitoCat although at a lower pace than MitoOx. Of note, an additional step not included in the model is needed to describe a step elevation in glucose from starvation (2–3 mM). In this case, MitoCat is preceded by massive O2 consumption that raises ATP/ADPc to restore the cellular energy state; then, as described above, PK raises ATP/ADPpm to close KATP channels. As this situation never occurs in vivo, we will not discuss it further. At very high glucose (16–20 mM) when Ca2+ reaches a plateau, both MitoCat and MitoOx metabolic fluxes are maximal, oscillations in metabolism and insulin secretion do not occur (Kjems et al., 2002).
“Metabolic coupling factors” propel the β-cell through MitoCat and MitoOx
The temporal separation inherent to oscillatory behavior is conducive to specific metabolic cycles and signals that sequentially propel the β-cell through the electrically-silent (MitoCat) and electrically-active energy consuming (MitoOx) phases of GSIS. These signals, termed “metabolic coupling factors” (MCFs), link the intracellular metabolism of fuel stimuli in the β-cell to insulin exocytosis (Prentki et al., 2013). Figure 3 presents a reorganized model of β-cell metabolic signaling by aligning MCFs into one of three categories: ‘regulatory’, ‘effectory,’ and ‘homeostatic.’ The latter category is a new designation and includes the important need of a strong “off switch” of metabolic signaling and insulin secretion to reset the system after depolarization. Thus, “regulatory MCFs” (e.g., F1,6BP, ATP/ADP, mitochondrial GTP, citrate, malonyl-CoA, etc.) facilitate switching between metabolic pathways and cycles to modulate the effectory networks. “Effectory MCFs” (e.g. ATP/ADP, Ca2+, reactive oxygen species (ROS), monoacylglycerol (MAG)) directly trigger and/or amplify GSIS via late effectors (e.g., KATP channels, Ca2+ channels, exocytotic proteins). Note that effectory MCFs are sufficient signals for insulin secretion, and are necessarily transient. They must be counterbalanced by even stronger homeostatic signal removers that prevent hyperinsulinemia by resetting the system between oscillations (e.g., ATP, by Ca2+-ATPases; cAMP, by phosphodiesterases; MAG, by ABHD6; ROS and S-S proteins, by NADPH, GSH/GSSG, catalase, and superoxide dismutase).
Figure 3. Metabolic coupling factors and homeostatic signal removers are temporally compartmentalized.
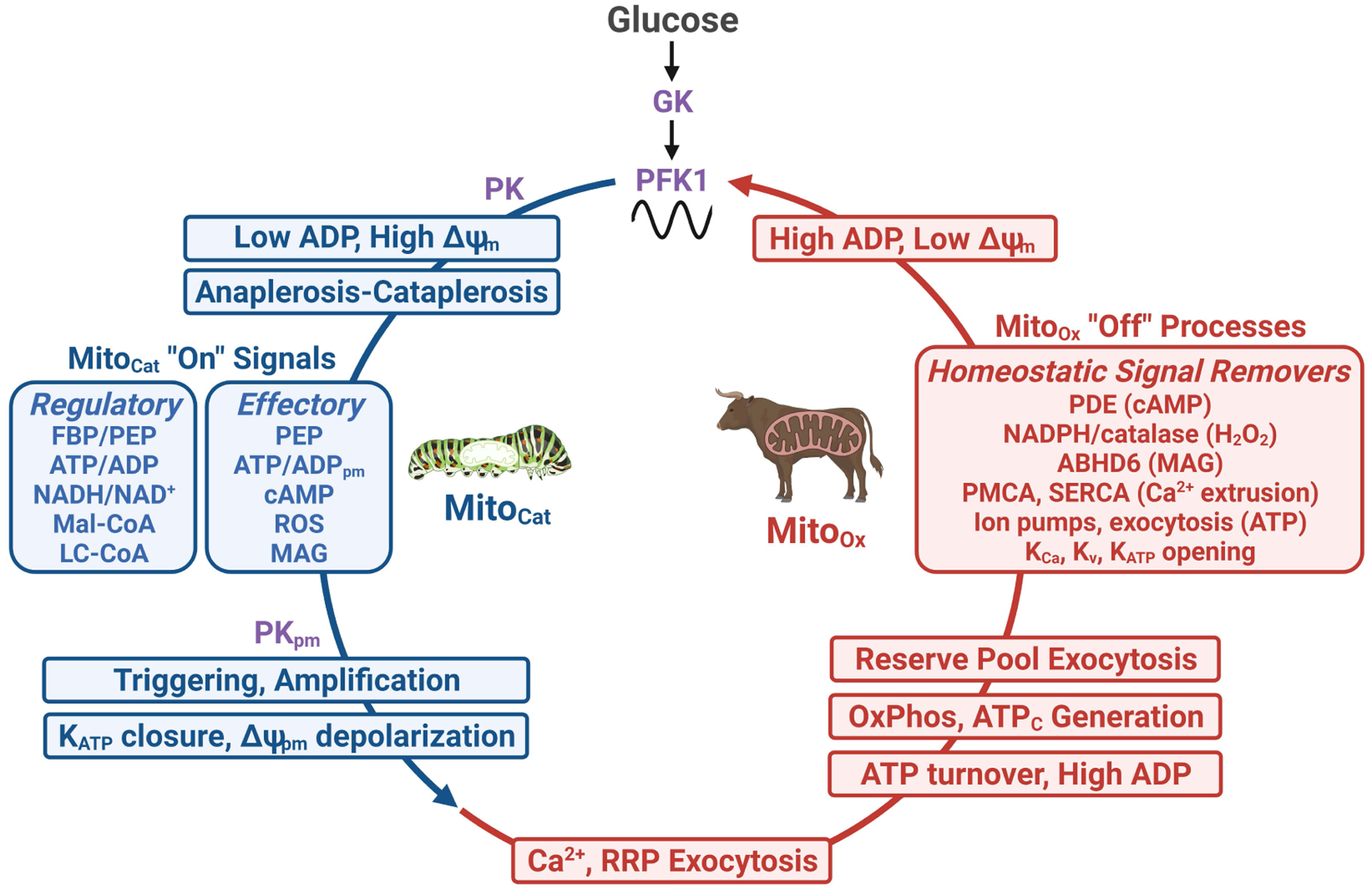
Glucose metabolism via phosphofructokinase-1 (PFK1) creates two metabolic states separated in time: an initial increase in mitochondrial anaplerotic/cataplerotic fluxes (termed MitoCat), followed by enhanced OxPhos (MitoOx) (see Fig. 2). Viewed sequentially, the net result of glycolysis during MitoCat is a pyruvate kinase (PK)-driven reduction in ADP that is sensed by the mitochondrial adenine nucleotide translocase and therefore ATP synthase, causing increased voltage across the mitochondrial inner membrane (ΔΨm), slowing NADH consumption by the electron transport chain, and increasing signals for secretion. These MitoCat “on” signals (blue boxes) include both regulatory and effectory metabolic coupling factors. Several MitoCat signals, such as PEP, plasma membrane ATP/ADP (ATP/ADPpm) and reactive oxygen species (ROS), participate in both the triggering and amplification arms of glucose-stimulated insulin secretion. Following KATP closure by plasma membrane PK (PKpm)-driven ATP/ADPpm, membrane depolarization and Ca2+ influx terminates the MitoCat phase by initiating a cascade of MitoOx processes that consume ATP, including ion pumping and exocytosis. The ensuing rise in ADP stimulates respiration, increasing cytosolic ATP (ATPc) that provides energy to sustain secretion until homeostatic signal removers (inset red box) reset the membrane potential. A rise in cytosolic ADP plays key role in this resetting as it will activate PFK1 and PK, which control oscillations in glycolysis. Abbreviations: ABHD6, α/β-hydrolase domain containing 6; KATP, ATP-sensitive K+ channels; KCa, Ca2+-activated K+ channels; Kv, voltage-dependent K+ channels; LC-CoA long chain acyl-CoA; MAG, monoacylglycerol; Mal-CoA, malonyl-CoA; PDE, phosphodiesterase; PMCA, plasma membrane Ca2+ ATPase; RRP, readily-releasable pool; SERCA, sarco/endoplasmic reticulum Ca2+-ATPase; ΔΨpm, plasma membrane potential.
A key limitation in the field is that the oscillatory behaviors of many metabolites and MCFs are not fully characterized in the intact β-cell. Furthermore, even if a metabolite is oscillatory, its assignment to MitoCat or MitoOx is often lacking. Aside from ATP/ADP (Ainscow and Rutter, 2002; Lewandowski et al., 2020; Li et al., 2013; Merrins et al., 2016), F1,6BP (Merrins et al., 2013), glutamate (Lewandowski et al., 2020), lactate (Sdao et al., 2021), citrate (Gregg et al., 2019), mitochondrial pH and ΔΨm (Krippeit-Drews et al., 2000; Lewandowski et al., 2020; Li et al., 2013), there is limited clarity on whether other metabolites and MCFs oscillate or remain static (e.g. cytosolic GSH/GSSG (Santos et al., 2017)) as part of their regulatory contribution. Cytosolic ATP/ADP, F1,6BP-dependent PK activation, citrate, and mitochondrial matrix alkalization all positively associate with the MitoCat phase (Gregg et al., 2019; Lewandowski et al., 2020). Mitochondrial and cytosolic Ca2+, lactate, and O2 consumption are positively associated with the MitoOx phase (Jung et al., 2000; Kennedy et al., 2002; Lewandowski et al., 2020; Sdao et al., 2021). Citrate, in particular, is a long-established regulator of multiple key enzymes involved in GSIS, such as PFK1 and acetyl-CoA carboxylase, and together with Ca2+ it may reinforce oscillatory metabolism (MacDonald et al., 2003). These lists are certainly incomplete and some components may be found to belong to multiple classes. Finally, it should be emphasized that ADP plays a critical role in GSIS regulation. ADP acts as both as key effectory and regulatory signal (Figure 3), since in the cytosol it controls, together with ATP, KATP channel closure and is the main regulator of PFK1 that controls oscillations in glycolytic flux. High ADP in cytosol resets the balance between MitoCat and MitoOx and elevated mitochondrial ADP is central to the regulation of the TCA cycle, ETC activity, and ATP production. In the next section, we provide a rationale for assigning mitochondrial cycles and signals to either MitoCat or MitoOx.
What regulates different states of mitochondria?
Oscillations of metabolic networks are largely coordinated by allosteric regulations that are evolutionarily hard-wired into the tertiary and quaternary structures of enzymes. These prevent metabolic traffic jams that would otherwise render signaling sluggish and unresponsive. Oscillations dynamically encode nutrient signaling into frequency and amplitude domains, rather than just relying on static variations in metabolite concentrations (Henquin, 2009). An equally important attribute of allosteric regulation and oscillatory signaling is that they provides a fast mechanism to turn off insulin secretion to prevent hypoglycemia. It is important to point out that the functional contribution to allosteric regulations in intact β-cells has not been formally evaluated in most cases. Therefore, a collective analysis of allosteric control is used inferentially here to disentangle phasic metabolism.
Allosteric regulation during MitoCat
Allosteric regulation of mitochondrial metabolism during MitoCat is more complex than that during MitoOx since the TCA cycle during this phase is fragmented (Figure 4A). When ATP demand is low because insulin secretion is also low, the energy level of the β-cell increases. This is indicated by the elevated cytosolic and mitochondrial ATP/ADP and NADH/NAD+ ratios and ΔΨm all moving toward their apices (Krippeit-Drews et al., 2000; Lewandowski et al., 2020). At the same time cytosolic Ca2+ is lowered (to ~50 nM relative to ~250 nM during MitoOx), with mitochondrial Ca2+ following suit. ETC activity and O2 consumption rates during MitoCat approach their nadir (Kennedy et al., 2002). The slowing of the ETC and consistent “pressure” from the glucose-driven NADH/NAD+ rise increases ΔΨm and ROS generation. During this time, inhibitory allostery remodels mitochondrial metabolism. The enzymatic targets are the proximal, NAD+-dependent dehydrogenases. For instance, ATP inhibits PDH (via PDH kinase), IDH1, IDH3, and αKGDH in addition to citrate synthase. NADH, in turn, inhibits IDH3 and αKGDH (Rafalowska et al., 1974; Seelig and Colman, 1978). Succinyl-CoA inhibits αKGDH. This inhibitory allostery tends to be Km raising for several enzymes and as such, raises substrate concentrations and redirects metabolites out of the TCA cycle. Thus, the inhibition of the matrix dehydrogenases during MitoCat disconnects the sequential enzymatic activities of the TCA cycle downstream of IDH2/3, and this favors anaplerosis/cataplerosis. Indeed, this fragmented TCA cycle accumulates matrix oxaloacetate and citrate whose carbons can then flow out of the matrix into the cytosol as PEP and citrate. Both of these provide additional routes, besides oxidation to CO2, to clear pyruvate. Accumulation of acetyl-CoA also promotes anaplerosis/cataplerosis as it is a potent allosteric activator of PC. Thus, activated PC anaplerosis expands the TCA cycle with OAA. The increased OAA can be balanced by cataplerotic export of PEP as part of the GTP-dependent mitochondrial PEPCK reaction (PCK2) and the PEP cycle (discussed below). Once in the cytosol, citrate can form malonyl-CoA, an important regulatory MCF (Prentki et al., 1992).
Figure 4. Allosteric and Ca2+ regulation of mitochondrial enzymes dictates the mitochondrial cycles, shuttles, and shunts during MitoCat and MitoOx.
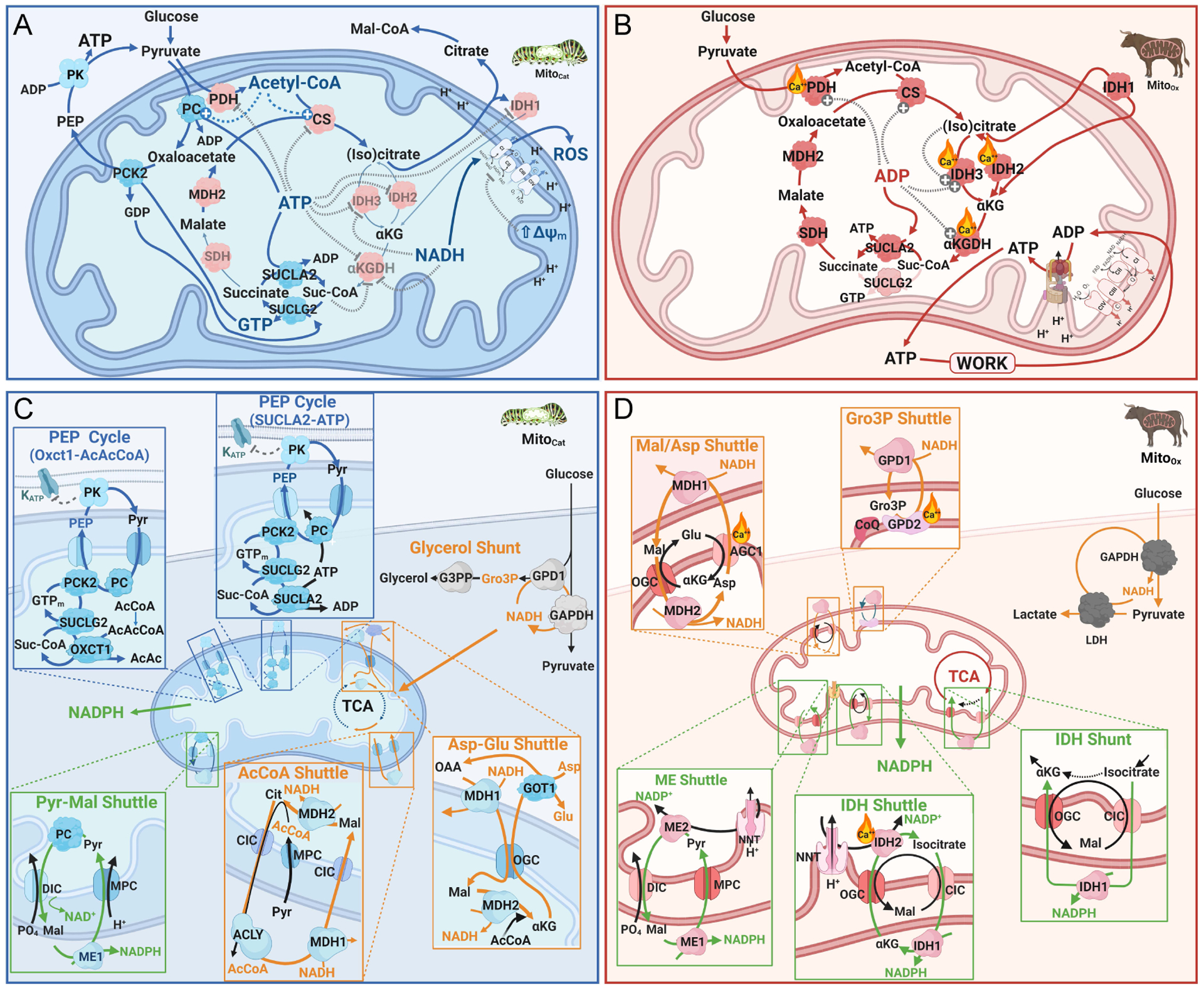
(A) MitoCat is a high-energy mitochondrial state-4-like condition that exerts strong allosteric control over the PEP cycle, the TCA cycle, and the ETC through increased ATP/ADP, NADH/NAD+, Ac-CoA/CoA and Suc-CoA/CoA ratios. Key to MitoCat is the blockade of isocitrate dehydrogenase (IDH2 and IDH3), and α-ketoglutarate dehydrogenase (αKGDH), an energy level sensor in the mitochondrial matrix that supports the following events through partial TCA cycle blockade: anaplerosis and increased matrix oxaloacetate levels via allosteric activation of PC stimulated by a rise in acetyl-CoA; cataplerotic output of citrate and the formation of cytosolic acetyl-CoA and malonyl-CoA (Mal-CoA), which is also favored by cytosolic IDH1 inhibition; enhanced PCK2 flux and PEP cycle activity promoted via oxaloacetate production. The accompanying rise in NADH drives reactivate oxygen species (ROS) production by the ETC in this low ADP state. (B) During MitoOx, ADP activates OxPhos, reduces ΔΨm, and fully engages the TCA cycle. Ca2+-activated dehydrogenases are critical to support an NADH burst for OxPhos, and ADP activation of IDH2 and IDH3 reinforces citrate commitment to the TCA cycle for ATP production to cope with the workload of insulin secretion. (C, D) Putative assignments of metabolic cycles (blue boxes), as well as NADH and NADPH shuttles and shunts (orange and green boxes, respectively), and are shown for MitoCat (C) and MitoOx (D). GAPDH during glycolysis reduces NAD+ in the cytosol that has to be reoxidized during MitoCat to maintain glycolytic flux. This occurs through the acetyl-CoA shuttle, the aspartate-glutamate exchange, as well as the glycerol shunt and the glycerolipid/fatty acid cycle (not shown). During MitoOx, NAD+ is reoxidized via the malate-aspartate shuttle, the Gro3P shuttle, or pyruvate lactate exchange performed by LDH. During MitoCat, succinyl-CoA (Suc-CoA) that supports mitochondrial GTP synthesis for the PCK2 reaction during the PEP cycle is generated from succinate by SUCLA2 in reverse mode that uses ATP. The increased Suc-CoA can then drive GTP synthesis by SUCLG2 (PEP cycle SUCLA2-ATP). Alternatively, in a variant PEP cycle (OXCT1-AcAcCoA), Suc-CoA and mitochondrial GTP are generated from AcAc-CoA via the action of OXCT1. However, during MitoOx, Suc-CoA is directly generated by αKGDH in the TCA cycle (not shown). NADPH can be generated in the cytosol by the pentose phosphate pathway (not shown) or the pyruvate-malate shuttle during MitoCat or from electron transfer from the mitochondria to the cytosol through the malic enzyme (ME) shuttle, the isocitrate dehydrogenase (IDH) shuttle, or the IDH shunt during MitoOx. Abbreviations: AcCoA, acetyl-CoA; AcAcCoA, aceto-acetyl-CoA; DIC, dicarboxylate carrier; GPD1, cytosolic glycerol 3-phosphate dehydrogenase; GPD2, mitochondrial membrane associated glycerol 3-phosphate dehydrogenase; GOT1, glutamic-oxaloacetic transaminase; Gro3P, glycerol 3-phosphate; GTPm, mitochondrial GTP; OGC, oxoglutarate carrier; ME, malic enzyme; MDH, malate dehydrogenase; MPC, mitochondrial pyruvate carrier; OXCT1, 3-oxoacid CoA transferase, also called SCOT1; Pyr-Mal shuttle, pyruvate-malate shuttle; SUCLA2, ATP-specific succinyl-CoA synthase; SUCLG2, GTP specific succinyl-CoA synthase; ACLY, ATP citrate lyase; NNT, nicotinic nucleotide transhydrogenase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase, PC, pyruvate carboxylase; PCK2, mitochondrial PEP carboxykinase.
Allosteric regulation during MitoOx
The more familiar allosteric regulation of metabolism during MitoOx favors OxPhos capacity (Figure 4B). With the exception of uncoupling, the main control point for OxPhos is via ADP as substrate for complex V (ATP synthase), which through the consumption of the proton motive force promotes ETC activity and O2 consumption. ADP itself is also a substrate for the ATP-specific isoform of succinyl-CoA synthase (SUCLA2) in the TCA cycle for OxPhos-independent synthesis of ATP in the mitochondrial matrix. In contrast to its substrate role, through allostery ADP lowers the substrate Km of many TCA cycle enzymes, especially the NADH dehydrogenases. ADP is an allosteric activator of multiple mitochondrial enzymes, including pyruvate dehydrogenase (PDH) (via PDH kinase), citrate synthase, isocitrate dehydrogenase 3 (IDH3), and α-ketoglutarate dehydrogenase (αKGDH) (Sankaran et al., 1996). Citrate also can feed forward to allosterically activate IDH3 to promote isocitrate lowering. Likewise, Ca2+ favors substrate lowering for the dehydrogenases of the proximal TCA cycle including PDH (via PDH phosphatase), all three IDH isoforms, and αKGDH (Glancy and Balaban, 2012; Strumiło, 1995). Transport across membranes is also regulated by allostery during MitoOx (Figure 4D). Thus, a rise in cytosolic Ca2+ activates components of redox shuttles such as the aspartate-glutamate carrier (AGC1, ARALAR/SLC25A12) and mitochondrial Gro3P dehydrogenase (GPD2) (addressed later). These reactions favor the activity of both the malate/asparate and glycerol-3-phosphate (Gro3P) shuttles that transfer cytosolic NADH electrons into the ETC, and regenerate NAD+ to facilitate glycolysis. Importantly, cytosolic rather than mitochondrial Ca2+ controls the pyruvate supply for OxPhos through these shuttles and transporters (Mármol et al., 2009; Szibor et al., 2020). Worth emphasis, the allosteric roles of ADP and Ca2+ to promote fuel oxidation and ATP synthesis are likely secondary in control strength to the regulation of ATP synthase by ADP (Glancy and Balaban, 2012).
Mitochondrial shuttles and shunts in nutrient signaling
The chemical potential stored in the covalent bonds of glucose and other nutrients are distributed via metabolism into energy generation, signaling, and/or homeostatic processes in both the cytosol and mitochondria. This distribution occurs through the transfer of electrons from fuel stimuli into redox intermediates such as NAD+, NADP+, and FAD. Biochemical products of these electron exchanges are diverse and include molecules such as ATP, ROS, CoA esters, and GSH. Electron transfer across membranous cellular compartments does not occur directly but is coupled mostly via shuttles and shunts. Shuttles are defined as mass-balanced cycles that transfer reducing equivalents or metabolites in and out of the mitochondria. A shunt, in contrast, is an offshoot of the main pathways (e.g., glycolysis or the TCA cycle) that can either divert metabolites to another pathway, or allow them to rejoin the main pathway at a later point. During β-cell glucose stimulation the pathways and destinations of the electrons are not constant and change temporally with the oscillations of MitoOx and MitoCat. Mitochondrial shuttles and shunts are key components of GSIS in the β-cell and we will discuss below those which are implicated during the oscillatory MitoCat and MitoOx states.
Re-oxidation of cytosolic NADH and acetyl-CoA export during MitoCat
Sustaining glycolysis during MitoCat requires reoxidation of cytosolic NADH. Four electron sinks, allowing glycolytic flux through GAPDH, are considered during MitoCat (Figure 4C).
Glycerol shunt.
The recently characterized enzyme in the β-cell, glycerol 3-phosphate phosphatase (G3PP) (Mugabo et al., 2016), provides a pathway for electron disposal/NAD+ regeneration during MitoCat. In this shunt NADH is oxidized to NAD+ by cytosolic glycerol-3-phosphate dehydrogenase (GPD1) generating Gro3P from dihydroxyacetone-phosphate (DHAP). But instead of regenerating DHAP as in the Gro3P shuttle discussed below, the glycerol shunt utilizes G3PP to directly generate glycerol from Gro3P, bypassing lipogenesis and lipolysis. In the absence of β-cell glycerol kinase (Prentki et al., 2013), glycerol is lost from the cell via aquaporins, thus lowering the maximal yield of ATP/glucose to about 12.5. The high Km of G3PP for Gro3P (Mugabo et al., 2016) make this pathway ideal for the β-cell to manage metabolic excess via carbon spillage to glycerol at the higher end of the glucose range (Possik et al., 2021). That said, peaks of glycolytic oscillations may be sufficiently close to the Km to spill over into this pathway during physiologic glucose. β-cell specific G3PP KO mice show enhanced GSIS in association with increased anaplerosis and mitochondrial metabolism, and are more susceptible to glucotoxicity. Interestingly, activation of this pathway protects from glucotoxicity and fat deposition and promotes healthspan in C.elegans (Possik et al., 2022).
Glycerolipid shunt.
This shunt is part of the glycerolipid/free fatty acid (FFA) cycle (not pictured in Figure 4C) and is implicated in the amplification arm of GSIS and fuel excess detoxification (Poursharifi et al., 2020; Prentki et al., 2020a). An additional NAD+ regeneration pathway occurs via the incorporation of Gro3P into complex lipids via Gro3P acyltransferase. Stimulatory glucose increases incorporation of Gro3P into triglyceride 2-fold and into phospholipids by 5-fold (Berne, 1975) and impacts signaling by long chain acyl-CoA (LC-CoA) and complex lipids as well as supporting cycles of triglyceride synthesis and breakdown (Prentki et al., 2013). The initial steps of the glycerolipid/FFA cycle (Prentki and Madiraju, 2012) are to esterify FFA into glycerolipids followed by lipolysis into FFA and glycerol. The energy requirement is significant and includes phosphorylation of glucose for the Gro3P and the activation of three FFA to acyl-CoAs with a net cost of 7 ATP per turn. Because this cycle is energetically costly and removes the glucose carbons available for ATP production, it may therefore be more important for signaling or fuel excess than the reoxidation of NADH.
Aspartate-Glutamate shuttle.
Reversed glutamate dehydrogenase (GDH) flux from αKG to glutamate was proposed as a regulatory/effectory MCF (Maechler and Wollheim, 1999). However, this is much debated (MacDonald and Fahien, 2000; Prentki et al., 2013), and the genetic evidence goes against this view (Stanley et al., 1998). Recent evidence suggests that glutamate may be more important for incretin/cAMP signaling but not for GSIS per se (Gheni et al., 2014). In the absence of stimulatory cytosolic Ca2+, AGC1 a is not activated and limits the full malate-aspartate shuttle described below. Thus, we refer to the AGC1-independent half of the malate-aspartate shuttle as the aspartate-glutamate shuttle (Figure 4C). In this mechanism, a high cytosolic NADH/NAD+ ratio pulls aspartate into malate via glutamate/oxaloacetate transaminase (GOT1) and MDH1, and malate enters the mitochondria in exchange for αKG. Without net cellular uptake of aspartate, the aspartate-glutamate exchange is a time-limited electron sink into glutamate. Notably, its relevance aligns well with the long known marked drop in aspartate levels and the small increase in cellular glutamate that occurs with glucose stimulation (Alves et al., 2015; Lamontagne et al., 2017; Li et al., 2003; Mugabo et al., 2017), and the rise in cytosolic glutamate that occurs during the MitoCat phase of oscillations (Lewandowski et al., 2020).
Acetyl-CoA shuttle.
In this pathway, mitochondrial acetyl-CoA and oxaloacetate are incorporated into citrate using citrate synthase. Citrate is then transported via the CIC to the cytosol where acetyl-CoA plus oxaloacetate is re-generated by ATP citrate lyase (ACLY). A high cytosolic NADH/NAD+ favors the transfer of NADH electrons into oxaloacetate to form malate, regenerating NAD+ (Palmieri, 2004). This shuttle functions to short circuit the otherwise inhibited TCA cycle during MitoCat. Importantly, malate returns to the matrix in coupled exchange for citrate via the CIC and stoichiometrically balances the shuttle. Therefore, we distinguish this from the so-called “pyruvate-citrate cycle” (Farfari et al., 2000; Jensen et al., 2008), because of a required exchange of citrate for malate as well as independence from PC anaplerosis. Importantly, the malate-citrate exchange via CIC rules out the possibility of malate decarboxylation by cytosolic malic enzyme 1 (ME1). In addition to regeneration of cytosolic NAD+, important signaling benefits of this shuttle are supporting glycolytic flux during MitoCat, and malonyl-CoA generation. In other words, transformation of pyruvate into cytosolic acetyl-CoA allows glycolysis to continue despite a stalled TCA cycle. As such, the acetyl-CoA shuttle supports continued PEP metabolism by PK to support ATP production for KATP closure (El Azzouny et al., 2016; Guay et al., 2007; Joseph et al., 2007). Malonyl-CoA formed from cytosolic acetyl-CoA also blocks lipid oxidation via carnitine palmitoyl-transferase 1 inhibition to favor pyruvate metabolism and the PEP cycle, and to support the generation of other lipid MCF signals such as MAG (Prentki et al., 2020a).
Further research is needed to clarify which of the NADH reoxidation systems are quantitatively important during MitoCat. However, the glycerolipid shunt and the glycerol shunt because of their high energy cost, as discussed above, are unlikely to play prominent roles. The acetyl-CoA shuttle together with the glutamate-aspartate shuttle likely accounts for the majority of the cytosolic NADH reoxidation during MitoCat.
Re-oxidation of cytosolic NADH and mitochondrial transfer of reducing equivalents during MitoOx
During MitoOx, when OxPhos is the dominant activity, the main goal of metabolism is to move the electrons of glucose into the ETC to generate ATP. Once pyruvate, with its 10 disposable electrons, is transported into the mitochondria, the mitochondrial dehydrogenases use NADH and FADH2 to transfer electrons into the ETC. It is the 4 electrons per glucose (2 NADH) accumulated in the cytosol by the GAPDH reaction that have a more complicated pathway to reach the ETC. To prevent a slowing of glycolysis at the GAPDH step from a high cytosolic NADH/NAD+ ratio, several shuttles, shunts and/or exchanges re-oxidize cytosolic NADH with varying efficiency (Figure 4D). Of these, the malate-aspartate shuttle, the Gro3P shuttle, and the pyruvate/lactate exchange are likely to be more active during MitoOx.
Malate-aspartate shuttle.
This is the most efficient shuttle in term of ATP production that transfers electrons to site 1 of the ETC and plays key role in GSIS, although it is not essential due to redundancy with the Gro3P shuttle discussed below (Ravier et al., 2000). It uses the two isozymes of malate dehydrogenase (MDH1 and MDH2) for electron transfer from the cytosol to the matrix, yielding up to 31 ATP per glucose. The two aspartate transaminase isoforms (GOT1 and GOT2) repackage oxaloacetate as aspartate, and glutamate as αKG, to be compatible for mitochondrial transport via inner membrane antiporters. The energetic cost of balancing this shuttle is one co-transported proton used by AGC1. Mitochondrial NADH then transfers its electrons into complex I to eventually translocate 10 protons out of the matrix via the ETC for a net gain of 9 protons available to ATP synthase.
Gro3P shuttle.
This slightly less efficient shuttle (up to 29 ATP/glucose) uses the two isoforms of Gro3P dehydrogenase (GPD1 and GPD2) to move an electron pair first into coenzyme Q and then into complex III for the net translocation of 6 protons out of the matrix. β-cells are endowed with very high levels of mitochondrial bound Gro3P dehydrogenase (GPD2) pointing to an important role of this shuttle in GSIS (MacDonald, 1981). However, the Gro3P shuttle is not essential as β-cell specific deletion of GPD2 does not impair GSIS, due to compensation by the malate-aparate shuttle (Ravier et al., 2000). Worth emphasizing, in the β-cell the AGC1 and mitochondrial GPD2 used by the malate-aspartate and Gro3P shuttles, respectively, require Ca2+ activation (Civelek et al., 1996; Rutter et al., 1992, p.). Ca2+ activation of both shuttles assigns their dominant roles as redox equivalent transfer pathways to the ETC during MitoOx after depolarization when β-cell cytosolic Ca2+ is elevated.
Pyruvate-lactate exchange.
In contrast to the general belief that β-cells lack lactate dehydrogenase (LDH), only one of the four isoforms, LDHA, is a ‘repressed’ gene (Pullen et al., 2010). The LDHA/B/D proteins are expressed at significant levels (Mitok et al., 2018), and in response to glucose, catalyze the reversible accumulation of lactate during MitoOx (Sdao et al., 2021). Lactate synthesis, as an electron sink to regenerate NAD+, is both temporary and limited because the dead-end metabolite is trapped in the cytosol due to the lack of MCT1 plasma membrane monocarboxylate transporters, explaining the low level of glucose-dependent lactate output from β-cells (Pullen et al., 2012; Sekine et al., 1994). Eventually during MitoOx, this transient sink reverses to regenerate pyruvate and NADH and pyruvate enters the mitochondria to be metabolized. However, this exchange can only provide short-term NADH/NAD+ buffering and plays a minor role in NADH reoxidation.
The mitochondrial GTP cycle and spatially compartmentalized PEP cycling
As discussed above, PEP metabolism by PK controls a key regulatory switch from MitoOx to MitoCat. PEP is generated either from glycolysis or in the mitochondria, in the PEP cycle which requires the mitochondrial GTP cycle for its capacity to generate GTP for the mitochondrial PEPCK (PCK2) reaction (Abulizi et al., 2020; Foster et al., 2022; Jesinkey et al., 2019; Lewandowski et al., 2020; Stark et al., 2014). In the PEP cycle (Figure 4C), pyruvate, formed from cytosolic PEP by PK, undergoes ATP-dependent carboxylation to oxaloacetate via PC, followed by the GTP-dependent generation of PEP by PCK2. Mitochondrial PEP3− is then transported out of the mitochondria in exchange for another di- or trivalent anion. Since β-cell CIC deletion has no effect on GSIS (Bauchle et al., 2021), PEP transport may be mediated by the ANT as occurs in the heart (Sul et al., 1976). Once mitochondrial PEP arrives in the cytosol, cytosolic PK regenerates pyruvate to further lower ADP, completing the PEP cycle. It should be pointed out that, upon glucose stimulation, glycolytic PEP is sufficient to close KATP channels without the PEP cycle; however, the PEP cycle accelerates cytosolic PEP production because pyruvate is recycled to PEP itself (Foster et al., 2022). Importantly, the PEP cycle has two different compartmentalized effects. Cytosolic PK reduces ADP to inhibit mitochondrial respiration (Lewandowski et al., 2020; Regeenes et al., 2022), while plasma membrane-associated PK raises ATP/ADP to close KATP channels (Foster et al., 2022; Lewandowski et al., 2020). Spatially compartmentalized PEP cycles use the favorable energetics from hydrolysis of one mitochondrial ATP at the PC step and one mitochondrial GTP to form one cytosolic ATP. This sustains the low ADP of MitoCat until ADP levels are increased by the cellular work during MitoOx that follows depolarization.
Crucial to the PEP cycle is the generation of mitochondrial GTP. Since there is no GTP transport activity in the inner mitochondrial membrane (McKee et al., 2000), the primary source of mitochondrial GTP, then, is the GTP-specific subunit of succinyl-CoA synthase (SUCLG2) (Kibbey et al., 2007). Mitochondrial nucleotide diphosphokinase (NDPK), despite the initial suggestion of its matrix localization (Kowluru et al., 2002), is in the intermembrane space and unavailable to convert ATP to GTP in the matrix (Lacombe et al., 2009). Mitochondrial GTP can be made either during MitoOx or MitoCat. During MitoOx, mitochondrial GTP synthesis is produced in proportion to the fraction of the TCA cycle that flows through SUCLG2 relative to SUCLA2, the ATP-specific subunit of succinyl-CoA synthase. The dynamics of this enzyme pair (SUCLA2 vs SUCLG2) changes dramatically during MitoCat since: 1) the high ATP/ADP ratio reverses the direction of SUCLA2 towards formation of succinyl-CoA and ADP (Ottaway et al., 1981); 2) pyruvate anaplerosis makes OAA to support GTP hydrolysis by PCK2 during the PEP cycle further favoring the generation of succinate by SUCLG2 (Figure 4C, panel named PEP cycle/SUCLA2-ATP); 3) during MitoCat the succinyl-CoA synthetase reaction is bookended by the inhibited αKG dehydrogenase and succinate dehydrogenase (Zeyelmaker and Slater, 1967) (Figure 4A) that together limit net loss or gain of succinyl-CoA or succinate. As such, in the mitochondrial GTP cycle SUCLG2 and SUCLA2 use the high ATP/ADP ratio to favor GTP synthesis with GDP regenerated by PCK2 which is a component of both the PEP cycle and the mitochondrial GTP cycle (Figure 4C). In this manner the two isoforms of succinyl CoA synthase (A2 and G2) work antiparallel akin to NDPK but instead cycling succinyl-CoA and succinate.
In light of mutations of the fat oxidizing enzyme hydroxy-acyl dehydrogenase (HADH, also named SCHAD) causing hyperinsulinemia (see also Figure 6) it is worth considering it as another source of succinyl-CoA to support mitochondrial GTP synthesis (Figure 4C, panel named PEP cycle/OXCT1-AcAcCoA). The mitochondrial enzyme 3-oxoacid CoA transferase (OXCT1, also named succinyl-CoA:3-ketoacid coenzyme A transferase 1, SCOT1) catalyzes the reversible transfer of the CoA moiety from succinate to acetoacetate during mitochondrial acetoacetate metabolism. This aligns with a reported role for ketone bodies in a pathway implicating acetoacetyl-CoA in the regulation of GSIS (Hasan et al., 2010). Thus, it may be speculated that besides a role in the activation of GDH to favor glutamine anaplerosis (Li et al., 2010) and fat oxidation inhibition causing the accumulation of LC-CoA or acyl-carntine compounds (Pepin et al., 2010), HADH/SCHAD mutations causing hyperinsulinemia also implicates the PEP cycle (Figure 6).
Figure 6. Familial hyperinsulinemic hypoglycemia (HHF): human genetic clues to the metabolic mechanisms of insulin secretion.
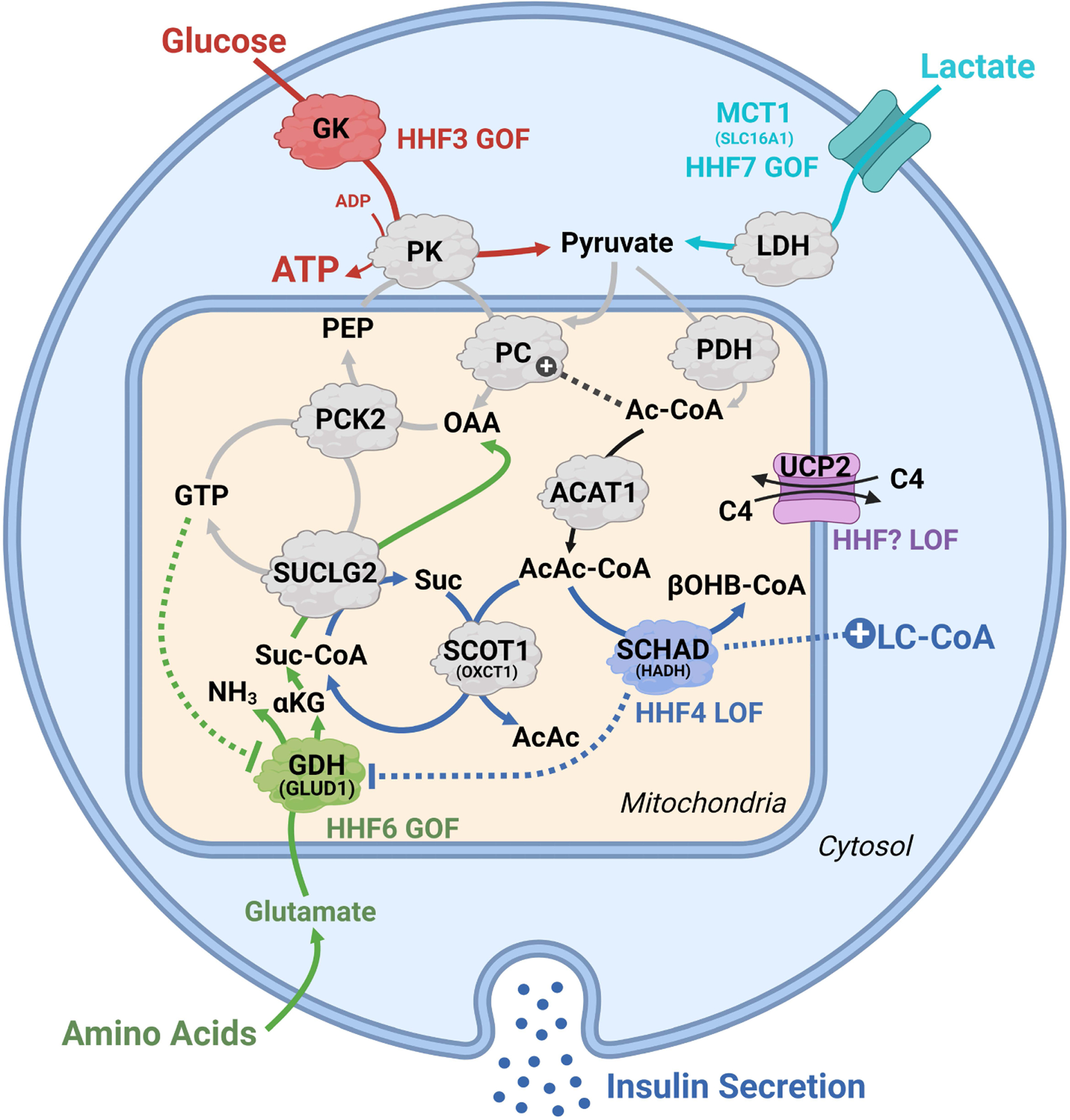
Several inborn errors of metabolism associated with congenital hyperinsulinemia intersect with pathways that inappropriately increase insulin release. Gain-of-function (GOF) mutations in glucokinase (red, HHF3) and ectopic expression of the lactate transporter (teal, MCT1/HHF7) increase pyruvate delivery to the mitochondria. GOF in glutamate dehydrogenase (green, GLUD1/HHF6), from loss of mitochondrial GTP inhibition, increases glutamate anaplerosis by providing α-ketoglutarate (αKG), mitochondrial GTP and mitochondrial PEP in the setting of a protein rich meal. Loss-of-function (LOF) of SCHAD (blue, HADH/HHF4) promotes hyperinsulinemia via three potential mechanisms: restriction of AcAc-CoA clearance to β-hydroxybutryate-CoA (βOHB-CoA) to favor succinyl-CoA generation by SCOT1/OXCT1, promoting activity of the PEP cycle; the clearance of LC-CoAs to AcCoA, thus favoring LC-CoA accumulation; disinhibition of GDH to favor anaplerosis via provision of αKG. A novel HHF has been proposed from LOF of the four carbon (C4) carboxylate exchanger (purple, UCP2), which may promote hyperinsulinemia by altering anaplerosis-cataplerosis and/or ΔΨm. Abbreviations: ACAT1, acetyl-CoA acetyltransferase 1; Ac-CoA, acetyl-CoA; AcAcCoA, acetoacetyl-CoA; GDH, glutamate dehydrogenase; GK, glucokinase; αKG, α-ketoglutarate; LDH, lactate dehydrogenase; LC-CoA, long chain acyl-CoA; MCT1, lactate/pyruvate transporter; OAA, oxaloacetate; PEP, phosphoenolpyruvate; PC, pyruvate carboxylase; PCK2, mitochondrial PEP carboxykinase; PDH, pyruvate dehydrogenase; PK, pyruvate kinase; SUCLG2, GTP specific succinyl-CoA synthase; SCHAD, short-chain acyl-CoA dehydrogenase/HADH; SCOT1, succinyl-CoA:3-ketoacid coenzyme A transferase/OXCT1; UCP2, uncoupling protein 2.
Hence, during MitoCat a stalled and fragmented TCA cycle – but a highly energized mitochondria – transfers its phosphorylation potential to the cytosol where PEP metabolism via PK restricts OxPhos via ADP privation, while plasma membrane PK metabolizes PEP to increase the ATP/ADP ratio for KATP channel closure followed by a rise in cytosolic Ca2+ that promotes insulin secretion (Figure 2B). Recent work indicates that while the PEP cycle is important for KATP channel closure during MitoCat, it is equally important during MitoOx for switching insulin secretion off by mechanisms that remain to be established (Foster et al., 2022).
Turning “off” the signals for secretion: cytosolic NADPH-generating shuttles and shunts
Which NADPH generating pathway is most important for insulin secretion?
When a metabolic process is essential to the function of a cell, nature often provides redundancy. For instance, cytosolic NADH/NAD+ ratio is maintained highly oxidized in the cytosol through the above-mentioned shuttle and exchange pathways to ensure dynamic glycolysis. Likewise, the cytosolic NADPH/NADP+ ratio is maintained highly reduced in the cytosol through supernumerary pathways that utilize combinations of the enzymes glucose-6-phosphate dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH) in the pentose phosphate pathway, as well as IDH1 and ME1 to generate NADPH. In the setting of very powerful “on signals” for secretion and substantial metabolic oxidative toxicities, a highly reduced cytosolic NADPH pool is likely essential as an “off switch” to reset the system during each oscillation. In addition to improving control strength to prevent hyperinsulinemia, NADPH is consumed to restore redox and to modify proteins via reduced thiols (Ferdaoussi et al., 2015; Santos et al., 2017), and for synthetic functions (e.g., cholesterol and fatty acid synthesis) to maintain cellular homeostasis. One exception may be the NADPH oxidase (NOX) family of enzymes that uses NADPH to generate ROS and especially the constitutively active NOX4 (Figure 5) which generates H2O2 from NADPH, albeit independent of variations in the NADPH/NADP+ ratio that do not regulate the enzyme (Ježek et al., 2021; Nisimoto et al., 2010). As such, for an essential homeostatic metabolite, any knockout, drug, or nutrient condition that lowers the NADPH level is predicted to impair the function and health of the β-cell and thus insulin secretion. The relative importance of the different NADPH synthesizing pathways has been the subject of much debate. Some studies have suggested involvement of the pentose phosphate pathway (Spégel et al., 2013), whereas other did not (Schuit et al., 1997). Likewise, metabolic flux through IDH1 and ME1 are of variable impact on GSIS (Guay et al., 2013; MacDonald, 2002, 1995; Pongratz et al., 2007; Ronnebaum et al., 2008, 2006). Much of this variability could be due to species specific differences in isozyme expression. Given such redundancy it is not surprising that it has been difficult finding consensus for the supremacy of one pathway over another related to cytosolic NADPH production by observed gain or loss of function following knockout, overexpression, and/or inhibition (Campbell and Newgard, 2021; Prentki et al., 2013). This is not easily settled as the specific contributions of each individual pathway are likely dependent upon cell line, species (e.g., mouse vs. rat vs. human), and condition (e.g., the presence of fatty acids or not or presence of high pyruvate levels that scavenge ROS) (MacDonald et al., 2009; Prentki et al., 2013).
Figure 5. Model illustrating the redox cycles and the roles of ROS and NADPH in controlling insulin exocytosis and recovery.
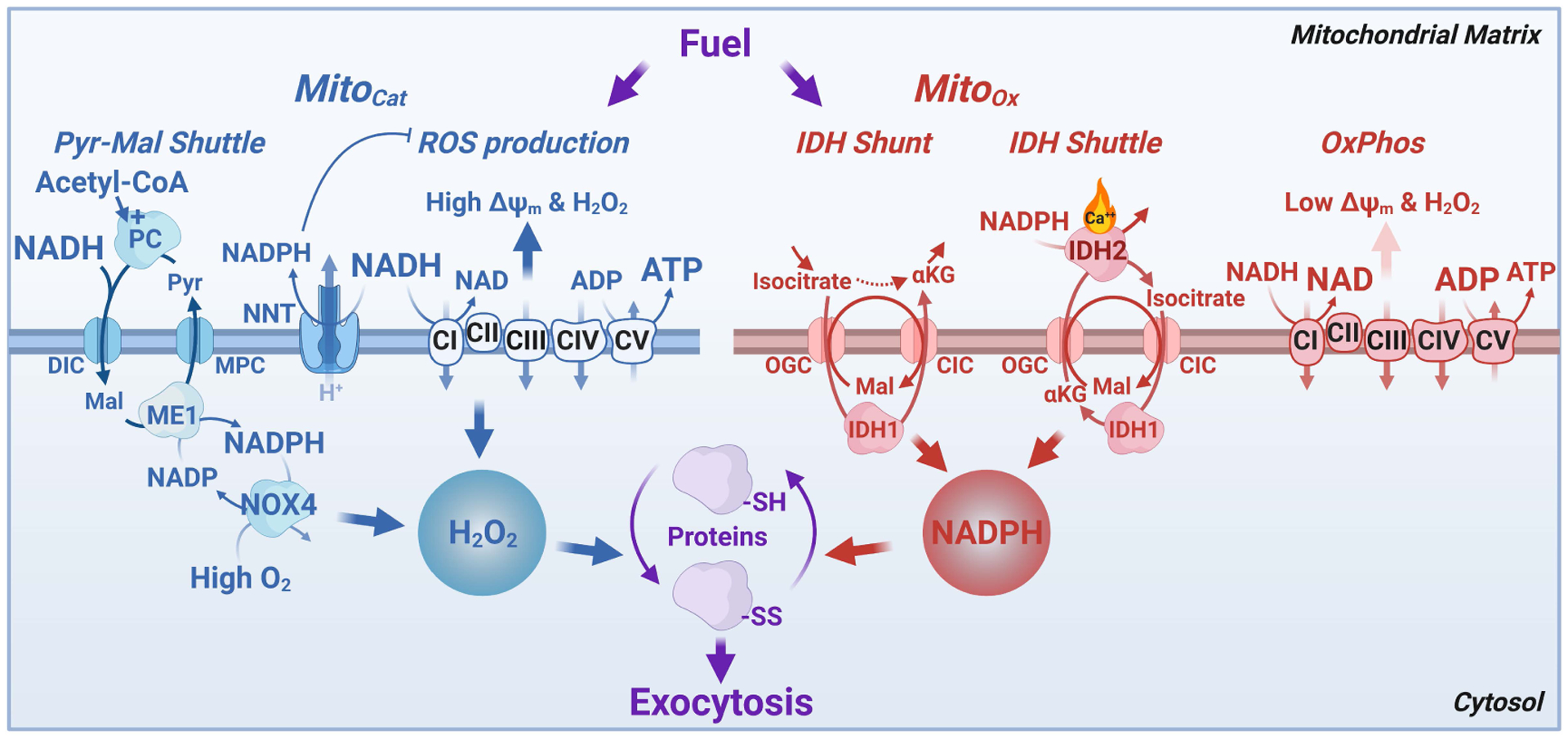
Cellular redox is communicated differently between mitochondria and cytosol during MitoCat and MitoOx. During MitoCat (left) mitochondrial NADH is elevated leading to ROS production via the ETC, NNT activation, and efflux of malate to the cytosol via the pyruvate-malate shuttle. This leads to cytosolic NADPH formation by malic enzyme (ME1) allowing ROS production in part via NOX4. ROS efflux from the mitochondria and generation via NOX4 modifies reactive cysteines in susceptible proteins involved in exocytosis that primarily favor insulin secretion. During MitoOx (right), NADH is consumed to support respiration and cytosolic NADPH is formed as a result of isocitrate transport to the cytosol as part of the IDH shunt and IDH shuttle, the latter associated with Ca2+-dependent IDH2 activity in reverse mode, in the direction of α-ketoglutarate to isocitrate. During metabolic oscillations both mitochondrial and cytosolic NADPH help convert H2O2 to H2O and restore ROS to basal levels. Cytosolic NADPH has dual effects on insulin exocytosis. During MitoCat NADPH-dependent NOX4 generates H2O2 which promotes exocytosis, whereas NADPH produced during MitoOx inhibits exocytosis, thus preventing excessive secretion by reducing the activity of thiol redox sensitive exocytosis effector(s). Abbreviations: IDH, isocitrate dehydrogenase; αKG, α-ketoglutarate; NOX4, NADPH oxidase 4; NNT, nicotinic nucleotide transhydrogenase; Px, peroxidase.
In addition to the pentose phosphate pathway, malic enzyme and IDH1, cytosolic NADPH is generated by moving carbons and/or electrons out of the mitochondrial matrix. Multiple potential mitochondrial pathways to generate cytosolic NADPH have been proposed for the β-cell (Prentki et al., 2013), but plausible mechanisms require balanced translocation of charged metabolites across the inner mitochondrial membrane. Here we critically re-evaluate the generation of cytosolic NADPH with attention to energetics, membrane transport and stoichiometry. Per our nomenclature, NADPH shuttles are defined as mass-balanced cycles that are independent of the TCA cycle activity per se. A NADPH shunt, in contrast, is a balanced extramitochondrial excursion that depends on TCA cycle flux. NADPH shuttles involve carboxylation-decarboxylation cycles of either pyruvate/malate or αKG/isocitrate. An essential player is nicotinamide nucleotide transhydrogenase (NNT) that generates mitochondrial NADPH and NAD+ at the expense of a proton translocated to the mitochondria matrix. We focus on the three mitochondrial shuttles and one shunt where balanced transport across the mitochondria inner membrane occurs (Figure 4, green boxes) and provide a rationale to assign them to MitoOx or MitoCat. While we strongly agree that anaplerotic/cataplerotic metabolism is important for β-cell function, we are unable to firmly identify any direct pathway linking PC anaplerosis and cytosolic NADPH synthesis via IDH1 despite suggestions otherwise (Ferdaoussi et al., 2015; MacDonald et al., 2005; Prentki et al., 2013). The reason for this is explained below.
Is cytosolic NADPH a regulatory or effectory on-switch, or a homeostatic off-switch?
There is abundant evidence that NADPH is absolutely required for numerous cellular functions and signaling cascades but essentiality does not distinguish the regulatory, effectory, or homeostatic nature of their function. For instance, complete washout of essential molecules during a patch clamp experiment (including GSH and NADPH) reduces vesicle fusion, and replacing them correspondingly improves vesicular fusion (Ferdaoussi et al., 2015). As this example shows, it is challenging to distinguish the essentiality from effectory, regulatory, or homeostatic control especially without assessing the dose dependency within the physiologic realm. Even at low glucose concentration, the cytosolic NADPH/NADP+ ratio is highly reduced and in response to 20 mM glucose the free cytosolic NADPH only increases by ~2% (Nicholls, 2016). Thus, a regulatory or effectory processes would have to discriminate between a very high NADPH/NADP+ ratio and a slightly higher ratio upon glucose stimulation. At the time of this writing, such a molecular target of NADPH with these biochemical features remains to be identified. At the very least, NADPH is an homeostatic cofactor supporting multiple reactions in the mitochondria and cytosol. As such, we do not dispute that NADPH-dependent processes (e.g., sumoylation of SENP-1) are essential for GSIS (Ferdaoussi et al., 2015; Lin et al., 2021), nevertheless, additional studies are required to determine if these and others are also regulatory or effectory.
Does reductive carboxylation of mitochondrial αKG to isocitrate and the IDH shuttle regulate insulin secretion?
Mitochondrial enzymes are generally considered in terms of their activity in the oxidative direction. One exception are the IDH isozymes, which can catalyze antiparallel reactions within and across the inner mitochondrial membrane (reductive carboxylation and oxidative decarboxylation). In the late eighties the Boquist lab first provided evidence in islet lysates that reductive carboxylation of cytosolic αKG to isocitrate via IDH1 (Boquist, 1987; Boquist and Alehagen, 1986; Boquist and Lorentzon, 1986). To distinguish directionality of flux through IDH isozymes in an intact system, there are many caveats due to the multiple isoforms (3 altogether) variably working in antiparallel fashion in multiple compartments. Especially challenging to tracer methods is that the IDH reaction lies close to its biochemical equilibrium, so the same isoform can reversibly transfer isotopic label in the retrograde and anterograde direction (i.e., an exchange flux) even without a net flux in either direction. Taking this reversibility into account, Alves et al. (Alves et al., 2015) used quantitative isotopomeric tracer studies, and documented that neither the IDH reversible exchange flux nor the net reductive IDH flux (αKG to isocitrate) correlated with β-cell GSIS. In this study, reductive fluxes were at most about 15–20% of the net forward IDH reaction. Later knockout and inhibitor studies suggested that targeting individual isoforms can change stable isotope labeling patterns, but unfortunately the directionality, magnitude, and isoform specificity of these changes were not determined (G.-F. Zhang et al., 2021). Thus, quantitative measurements show that reductive carboxylation of αKG to isocitrate is small relative to the TCA cycle flux and likely has only limited importance to generate either an NADPH “on”- or “off”-switch for GSIS.
Most cytosolic NADPH generation likely occurs during MitoOx
The pentose phosphate pathway that occurs in the cytosol is inhibited by ATP (MitoCat phase), suggesting a role of this pathway in sustaining the NADPH/NADP+ during MitoOx. Likewise, cytosolic NADPH-producing ME1 and IDH1 are inhibited by ATP (Boquist, 1987; Chang et al., 1991). Cytosolic acetyl-CoA, likely high during MitoCat, also inhibits ME1 (Bartholomé et al., 1972). Thus, the bulk of cytosolic NADPH synthesis likely occurs during MitoOx. This may serve to minimize mitochondrial ROS toxicity during this active phase of secretion and refill the cytosolic NADPH pool.
Cytosolic NADPH synthesis during MitoCat
Pyruvate-malate shuttle.
In the pyruvate-malate shuttle (Figure 4C), anaplerotic PC makes oxaloacetate from pyruvate. A high mitochondrial matrix NADH/NAD+ is required to reverse ME2 to generate malate that enters the cytosol via the DIC. There ME1 generates NADPH and pyruvate from malate in the cytosol. Shuttle completion follows pyruvate return to the matrix via the mitochondrial pyruvate carrier (MPC). This shuttle, also referred in the literature as pyruvate/malate cycling, is likely the only mitochondrial NADPH shuttle to carboxylate pyruvate that is using PC. Driven by high matrix acetyl-CoA/CoA, NADH/NAD+, and ATP/ADP ratios paired with ATP hydrolysis via PC makes it the most likely of the cytosolic NADPH-generating processes coincident with MitoCat. Some studies have proposed a role of this shuttle in GSIS and NADPH reduction (Huypens et al., 2011; Pongratz et al., 2007). However, the role of this shuttle is in mice debated (Prentki et al., 2013) as ME1 is poorly expressed in mouse islets (MacDonald, 2002) and some studies did not support a role of this shuttle in GSIS in normal mouse islet (Heart et al., 2009). Also, it competes with the PEP cycle for PC flux (Jesinkey et al., 2019). However, in this study a decrease in the pyruvate-malate shuttle relative to the PEP cycle was associated with increased GSIS, suggesting that the pyruvate-malate shuttle is less important for GSIS than the PEP cycle.
Cytosolic NADPH synthesis during MitoOx
Malic enzyme shuttle.
An alternative version of the pyruvate-malate shuttle is the PC-independent ME shuttle (Figure 4D) where proton coupled pyruvate uptake in the mitochondrion via the transporter MPC is followed by conversion via ME2 to malate coupled to the high mitochondrial NAD(P)H/NAD(P)+ ratios. Malate is then exported into the cytosol in electroneutral exchange for phosphate via the DIC (Huypens et al., 2011). Once in the cytosol, ME1 decarboxylates malate back into pyruvate making cytosolic NADPH. The role of ME1 in GSIS as discussed above is debated (MacDonald et al., 2009; Prentki et al., 2013) and therefore the significance of this shuttle in GSIS is uncertain.
Isocitrate dehydrogenase shuttle.
Like the ME shuttle, the IDH shuttle (Figure 4D) uses antiparallel operation of cytosolic IDH1 and mitochondrial IDH2 that is driven by the NNT to reduce cytosolic NADP+ (Palmieri, 2004). Balanced translocation of isocitrate out of and αKG into the matrix is achieved via two carriers, the CIC and the OGC, by cycling malate as counterion for both. There is no anaplerotic PC involvement. Consistent with the view that this shuttle is related to an off-signal for secretion via NADPH production, knockdown of IDH1 in the β-cell resulted in enhanced GSIS in association with increased isocitrate, NADP+, and ATP levels (Guay et al., 2013).
Isocitrate dehydrogenase shunt.
Unlike the IDH shuttle, the IDH shunt (Figure 4D) is essentially a transient extramitochondrial extension of the TCA cycle, which assigns its activity to MitoOx. Here cytosolic NADPH is generated (like in the IDH shuttle) after isocitrate exits the matrix to be converted into α-ketoglutarate by IDH1 in the cytosol. Once returned to the matrix, αKG re-enters the oxidative TCA cycle. It is important to emphasize that the IDH shunt is simply a redirected loop of the TCA cycle out of then into mitochondria that has complete independence from anaplerotic pyruvate metabolism via PC, and like the IDH shuttle, for the reasons discussed above, is unlikely to contribute to GSIS but may be an off-pathway that reduces ROS and perhaps also acts as a signal remover for insulin secretion (Prentki et al., 2013).
Unlikely NADPH shuttles and pyruvate cycles.
Given the importance of both PC and NADPH to β-cell function, it has been compelling to try to link pyruvate anaplerosis to cytosolic NADPH generation through a pathway utilizing citrate synthase. As described above, the acetyl-CoA shuttle, the IDH shuttle, and the IDH shunt all are completely independent of pyruvate anaplerosis. Since citrate efflux needs malate as a counter ion, we are unaware of a balanced stoichiometric pathway that can link pyruvate anaplerosis to NADPH from citrate or isocitrate. In the absence of such a connection, we recommend that proposed constructs such as the “pyruvate-citrate cycle” and the “pyruvate-isocitrate cycle” no longer be used. Thus, we now favor the view that inhibition of GSIS following knockdown or pharmacological inhibition of ACLY (Guay et al., 2007) may not be due to an altered putative pyruvate/citrate cycle, but rather to reduced activity of the acetyl-CoA shuttle that provides malonyl-CoA and reoxidizes NAD+ in the cytosol.
Phasic metabolism and exocytosis also implicate oscillatory ROS, lipid signals, and cAMP
Reactive oxygen species
β-cell ROS is a critical participant in insulin secretion and likely oscillate during phasic glucose metabolism and insulin secretion (Leloup et al., 2009). Glucose dose-dependently increases H2O2 in rat islets that plateaus between 10–16 mM (about 7-fold rise above 4 mM glucose) (Mugabo et al., 2017). A consequence of elevated ΔΨm and mitochondrial NADH/NAD+ during MitoCat is increased ROS production by the ETC (Figure 5). During this time, ROS sensitizes KATP channels to closure by ATP/ADP (Plecitá-Hlavatá et al., 2020b). In contrast, when the proton motive force is fully engaged with the production of ATP, then ΔΨm and relative ROS production fall during MitoOx (Plecitá-Hlavatá et al., 2020a). It has been proposed that the NADPH oxidase NOX4 plays a significant role in ROS production, KATP closure and GSIS (Plecitá-Hlavatá et al., 2020b). This potentially important signaling role of NOX4 requires clarification as to how this enzyme is activated upon glucose stimulation. Although H2O2 is made by NOX4 which uses NADPH as a substrate (Plecitá-Hlavatá et al., 2020b), this enzyme may not be limited by the cellular levels of NADPH (Nisimoto et al., 2010), and thus variations in NADPH levels upon metabolic oscillations (if they even occur) should not modulate its activity. The potency of ROS signaling in the β-cell has been attributed to low antioxidant enzyme expression (Lenzen et al., 1996). However, like any strong “on-switch”, ROS needs a powerful “off-switch”, given its potential toxicity. Phasic production of ROS during MitoCat (when ΔΨm production is high) and scavenging of ROS during MitoOx (when NADPH production is high) provide a mechanism to generate and remove pulses of ROS that could otherwise be toxic if sustained (Figure 5).
Overall, we favor the view that ROS levels oscillate during GSIS, and that H2O2 produced by the ETC and possibly by NADPH-dependent (but not NADPH regulated) NOX4 is an important signal for GSIS during MitoCat. How NOX4 is activated, and the ROS targets related to exocytosis remain to be identified. Furthermore, we believe that NADPH production by the IDH shuttle/shunt during MitoOx negatively impacts secretion both by promoting a reduced state of proteins linked to exocytosis and also via ROS removal (Figure 5).
Glycerolipid/FFA cycle signals
There is very strong evidence that a GL/FFA cycle, driven by both glucose and FFA, plays an essential regulatory role in GSIS, and that β-cell lipolysis-derived MAG is the mediator of this pathway via its binding to the exocytosis coordinator Munc13–1. As this has been extensively reviewed by us and others (Campbell and Newgard, 2021; Prentki et al., 2020a) we will not discuss it further. Whether oscillations in β-cell MAG and other lipid signals occur in response to fuels remains to be shown.
cAMP
Glucose induces oscillatory cAMP near the β-cell plasma membrane (Dyachok et al., 2008, 2006). However, the spatial bias of cAMP signaling, well-known to other fields, has not yet been thoroughly examined in β-cells. One striking example of such compartmentation is that cAMP and Ca2+ oscillations in INS-1 cells are in-phase in the microdomain of voltage-dependent Ca2+ channels tethered by A-kinase anchoring protein 79 (AKAP79) scaffolds to the plasma membrane, and out-of-phase in the general plasma membrane domain (Tenner et al., 2020). Also, cAMP may regulate KATP channel trafficking (Cochrane et al., 2021) and KATP activity via the cAMP effector EPAC2 (Kang et al., 2008; Shuai et al., 2021) in response to glucagon-like peptide-1 receptor activation (Holz et al., 1993). New optical tools for compartmentalized cAMP signaling (J.-F. Zhang et al., 2021), may be helpful to assess signaling on the timescale of MitoCat and MitoOx.
Inborn errors of metabolism and insulin secretion
Inborn errors of metabolism that alter islet function provide profoundly informative insights into key mechanistic components of the β-cell glucose sensing in humans. These include those causing familial hyperinsulinemic hypoglycemia (HHF) and those associated with maturity onset diabetes of the young (MODY).
Glucokinase (HHF3) and SUR1/KATP (HHF1/2).
Mutations in the earliest steps of glucose signaling (GK/HHF3/MODY2) and late steps (SUR/KATP channels/MODY12/13/HHF1/HHF2) offer clear human evidence mechanistically connecting the beginning steps of glucose metabolism to plasma membrane depolarization needed for insulin secretion (Snider et al., 2013).
Monocarboxylate transporter 1 (MCT1/HHF7).
Exercise-induced hyperinsulinemic hypoglycemia (HHF7) is a consequence of inappropriate expression of the monocarboxylate lactate transporter, MCT1/SLC16A1, in the plasma membrane of β-cells (Otonkoski et al., 2007; Pullen et al., 2012). Normally, β-cells are impermeable to lactate (Pullen et al., 2012; Sdao et al., 2021; Sekine et al., 1994). Hypoglycemia follows excess insulin secretion caused by plasma lactate generated during exercise in individuals via ectopic β-cell expression of SLC16A1. While LDH activity in primary β-cell extracts is low relative to most tissues, it is sufficient to enable HHF in vivo when the cell membrane is permeable to lactate. This fits well with the view that mitochondria-dependent signals are important for GSIS.
Glutamate dehydrogenase (GDH/HHF6).
Gain-of-function mutations in glutamate dehydrogenase (GLUD1/HHF3) increase anaplerosis due to loss of mitochondrial GTP inhibition of glutamate deamination (Figure 6). In humans, this causes protein-meal-induced HHF as well as asymptomatic hyperammonemia (Kelly et al., 2002). Unopposed oxidation of the anaplerotic amino acid glutamate by this mutation expands the entry of αKG to TCA metabolism. This pathway generates NAD(P)H and FADH2, but also will make mitochondrial GTP and oxaloacetate to support cataplerotic PEP generation via the mitochondrial GTP cycle (Kibbey et al., 2014, 2007). The glutamate-derived PEP then leaves the mitochondria to potently stimulate insulin release by augmenting the PEP cycle (Abulizi et al., 2020; Jesinkey et al., 2019; Lewandowski et al., 2020; Stark et al., 2009). This form of HHF supports the view that anaplerosis and the PEP cycle are central to β-cell activation.
Short-chain acyl-CoA dehydrogenase (SCHAD/HADH/HHF4).
Loss of function of hydroxyacyl-CoA dehydrogenase short chain (HADH, a.k.a., short-chain acyl-CoA dehydrogenase; SCHAD) causes fasting HHF and hyperammonemia (HHF4) possibly via multiple mechanisms. First, via interaction with GDH, HADH inhibits GDH (GLUD1) through a protein-protein complex (Li et al., 2010) and thus, loss of HADH augments glutamine-derived anaplerosis to stimulate the TCA cycle, cataplerosis, and fasting insulin release. In light of the phenotypic differences with HHF3 (GK) and HHF6 (GDH) , it is worth considering alternative explanations (Figure 6). Second, during MitoCat, when acetyl-CoA levels peak, acetoacetyl-CoA (AcAc-CoA) generation by mitochondrial ACAT1 offloads pyruvate oxidation. Since NADH/NAD+ is high during MitoCat, AcAc-CoA gets converted by HADH/SCHAD to β-hydroxybutyrate-CoA. With the loss of HADH, AcAc-CoA is pushed towards acetoacetate by SCOT1 (a.k.a., OXCT1) with succinyl-CoA as a byproduct. Thus loss of HADH augments the ability of acetyl-CoA to drive the production of mitochondrial GTP to support the PEP cycle. SCOT1 deficiency also impairs insulin secretion (Hasan et al., 2010), supporting the functional involvement of this enzymatic step. Third, HADH loss could reduce β-oxidation to potentiate MAG synthesis and insulin secretion via the GL/FFA cycle (Prentki et al., 2020a). Altogether, HHF3, HHF4 and HHF6 point towards a role for anaplerosis and the PEP cycle in the regulation of GSIS.
Disallowed/repressed metabolic genes in the β-cell point to key pathways of insulin secretion
Several metabolic enzymes or transporters are under-represented in the β-cell relative to other tissues and are referred to as “disallowed” or “repressed” genes (Lemaire et al., 2016; Rutter et al., 2020) (Figure 6). This likely prevents metabolic interference with GSIS.
Low Km hexokinases.
β-cells preferentially express the high Km and high Vmax hexokinase 4 isozyme GK, but low levels of the other low Km isoforms. This permits GK to act as a glucose sensor to control glycolytic flux and GSIS according to blood glucose levels.
Lactate/pyruvate transporter (MCT1) and lactate dehydrogenase A (LDHA).
Both the lactate/pyruvate plasma membrane transporter (MCT1/SLC16A1) and the LDHA isoform have low mRNA expression levels (Ghiasi and Rutter, 2021). However, three isoforms of LDH (A, B, and D) are detectable at the protein level (Mitok et al., 2018), and glucose elevation is accompanied by lactate production in intact mouse islet β-cells (Sdao et al., 2021), indicating that LDH is a viable short-term NAD+ generator. Human β-cells must also express sufficient LDH activity for ectopic SLC16A1 to cause hyperinsulinemic hypoglycemia (HHF7). Low MCT1 in the β-cell (Pullen et al., 2012; Sdao et al., 2021) is appropriate as it spares glucose derived carbons for the TCA cycle and cataplerotic processes linked to insulin secretion.
Adenylate kinase 3 and guanine deaminase.
Two other disallowed enzyme, adenylate kinase 3 (AK3, that can interconvert GDP and GTP) and guanine deaminase (GDA, which converts guanine to xanthine to limit the guanine pool) potentially oppose the mitochondrial GTP cycle and their reduced level should favor mitochondrial GTP production and the PEP cycle.
Ornithine transaminase 1.
Similarly, significant expression of ornithine transaminase 1 (OAT1) would restrict the anaplerotic glutamate pool by diversion of the metabolite glutamate-5-semialdehyde into the ornithine/urea cycle. Disallowance of this gene favors amino acid induced secretion by glutamine, glutamate, and leucine (which activates GDH).
Monoacylglycerol lipase.
Disallowed genes in lipid metabolism point towards interference with LC-CoA and MAG signaling. Limited MAG lipase (Mgll gene) expression permits the accumulation of MAG to activate the exocytotic coordinator Munc13–1 (Zhao et al., 2014). Likewise, signaling MAG is specifically degraded by the hydrolase ABHD6 which is located at the plasma membrane (Poursharifi et al., 2017). In a mouse study to identify plasma biomarkers of β-cell deficits on low and high fat diets, the genes (mRNA levels) that best correlated were first ABHD6 and second glucokinase (Sánchez-Archidona et al., 2021).
Acyl-CoA Thioesterase 7.
Another enzyme opposing LC-CoA accumulation and the glycerolipid/FFA cycle is acyl-CoA thioesterase 7 (ACOT7). Its overexpression reduced GSIS in vitro and in vivo (Martinez-Sanchez et al., 2016, p. 7) and lowered ATP due to increased synthesis and breakdown of acyl-CoA which consumes ATP. This provide genetic evidence that LC-CoA or its derived signals are key to GSIS.
Conclusion
Contrary to what is often believed, the glucose-induced signaling process in β-cells has not been largely solved and the current model implicating an initial rise in mitochondrially-derived ATP driving KATP channel closure and Ca2+ influx is incomplete and possibly wrong. The main reasons are: 1) it does not account for mitochondrial thermodynamics, which govern the way by which mitochondrial ATP generation is accelerated following enhanced workload via a rise in ADP, and not via a rise in NADH and FADH2 that would drive ROS as well as ATP production when ADP is limiting prior to membrane depolarization; 2) it does not account for metabolic compartmentation, including the ability of PK to switch off oxidative phosphorylation, and the sufficiency and essentiality of plasma membrane PK to close KATP channels (Foster et al., 2022; Lewandowski et al., 2020); 3) it does not account for temporal compartmentation, which is provided by oscillatory metabolism. However, very significant recent progress in the area of metabolic signaling may allow the field to reconcile potential caveats and contrasting reports. In this review article, we integrated many aspects of β-cell metabolic signal transduction that have not been previously covered together with new knowledge in the field. From this analysis we developed a testable model of glucose, amino acid, and FFA induced secretion (Figure 7). This model with four main metabolic cycles incorporates the following: spatial and temporal aspects of metabolic signaling (i.e. MitoCat and MitoOx); the relationship between cellular compartments, including the mitochondrion, cytosol and the area beneath the plasma membrane; “on-switch” signals for secretion but also “off-signals” that permit oscillations of metabolism and insulin secretion; and the redundancy among some pathways, in particular redox components that may explain diverging views in the literature.
Figure 7. Integrated metabolic cycles of nutrient induced insulin secretion.
Glucose metabolism in the β-cell drives the oscillatory activity of four interconnected metabolic cycles (PEP, TCA, Acetyl-CoA/Redox, and GL/FFA) that produce regulatory metabolic coupling factors (blue text within the cycles) and effectory metabolic coupling factors (white text within ovals) controlling the distal steps of insulin secretion. The relationships of each of these cycles with the highest activities of MitoCat and MitoOx are shown within the cycles. Some amino acids, like alanine and glutamine, feed the Krebs cycle via the production of Ac-CoA, OAA, and αKG. The TCA and PEP cycles are interlinked by OAA and mitochondrial GTP; the TCA and Ac-CoA/redox cycles are interlinked by citrate and other TCA cycle intermediates; the Ac-CoA/redox and GL/FFA cycles are interlinked by citrate-derived malonyl-CoA, which inhibit fat oxidation, thus preventing the removal of GL/FFA cycle intermediates. FFA promotes insulin secretion via lipolysis derived MAG, which activates the exocytotic effector Munc13–1, and also via the plasma membrane receptor FFAR1 (not shown). NADPH prevents excessive ROS accumulation and possibly excessive insulin secretion as well. Abbreviations: ATGL, adipose triglyceride lipase; DHAP, dihydroxyacetone phosphate; GTPm, mitochondrial GTP; Gro3P, glycerol 3-phosphate; GL/FFA, glycerolipd/free fatty acid; HSL, hormone sensitive lipase; LC-CoA, long-chain acyl-CoA; Mal-CoA, malonyl-CoA; MAG, monoacylglycerol; OAA, oxaloacetate; PEP, phosphoenolpyruvate.
This area of research, although not new, addresses a central question in metabolism at large: how do calorigenic nutrients activate a cell? This is of importance for the islet field but also for many other cell types such as the fuel-sensitive cells in the gut, the portal vein and the brain. In fact discoveries in β-cell metabolism often foresee findings in other related fields and cell types. Thus, we believe that research on nutrient signaling in the β-cell has exciting years of research and discoveries ahead.
Some timely key questions can be identified for future research. Do all regulatory, effectory and homeostatic MCF, or post-translational modifications, oscillate upon glucose stimulation? Are there oscillations in metabolism when a β-cell is stimulated with FFA or some amino acids? What are the MCFs at various glucose concentrations as they may not be necessarily the same or of the same importance? What are the late effectory targets of MCFs besides ATP and MAG, in particular ROS? What are the similarities and differences between β-cell signaling in mice, rats, and humans? How is basal secretion regulated? How is metabolic signaling enhanced in obesity (compensation) and how does it fail precisely in diabetes (decompensation)? Does the integrated model with interlinked metabolic cycles apply to other glucose-sensitive cell types? These are only a few questions and undoubtedly as the field evolves others will be formulated to enhance basic knowledge but also to uncover translational aspects related to the prevention of “nutri-stress” (Prentki et al., 2020b) in various cell types and the treatment of metabolic disorders.
ACKNOWLEDGEMENTS
The authors would like to dedicate this review to the late Franz Matschinsky, who contributed immensely to our understanding of glucose-stimulated insulin secretion and who mentored BEC and MP at the University of Pennsylvania. The authors would like to thank Jude Deeney, S.R Murthy Madiraju, Keith Tornheim and Guy Rutter, as well as all members of the Merrins, Corkey, Kibbey and Prentki laboratories for discussions and critical review. MJM gratefully acknowledges support from the NIH/NIDDK (R01DK113103 and R01DK127637) and the Department of Veterans Affairs (I01B005113). RGK gratefully acknowledges support from the NIH/NIDDK (R01DK127637). MP gratefully acknowledges support from the Canadian Institute of Health Research (143308). Figures created with BioRender.com.
ABBREVIATIONS
- αKG
α-ketoglutarate
- αKGDH
α-ketoglutarate dehydrogenase
- ΔΨm
mitochondrial membrane potential
- ABHD6
monoacylglycerol lipase ABHD6
- Ac-CoA
acetyl-CoA
- AcAc-CoA
acetoacetyl-CoA
- ACLY
ATP citrate lyase
- ACOT7
acyl-CoA thioesterase 7
- AGC1
aspartate-glutamate carrier SLC25A12/ARALAR
- AK3
Adenylate kinase 3
- AKAP79
A-kinase anchoring protein 79
- CIC
citrate-isocitrate carrier SLC25A1
- EPAC2
exchange factor directly activated by cAMP/RAPGEF4
- ETC
electron transport chain
- F6P
fructose 6-phosphate
- F1,6BP
fructose 1,6-bisphosphate
- F2,6BP
fructose 2,6-bisphosphate
- FFA
free fatty acid
- G3PP
glycerol 3-phosphate phosphatase
- G6P
glucose 6-phosphate
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GDA
guanine deaminase
- GDH
glutamate dehydrogenase/GLUD1
- GK
glucokinase
- GLP-1
glucagon-like peptide 1
- GPD2
mitochondrial glycerol-3-phosphate dehydrogenase
- Gro3P
glycerol 3-phosphate
- GSIS
glucose-stimulated insulin secretion
- GSH
glutathione, reduced
- GSSG
glutathione, oxidized
- HADH
short-chain acyl-CoA dehydrogenase/SCHAD/HHF4
- HHF
familial hyperinsulinemic hypoglycemia
- IDH
isocitrate dehydrogenase
- KATP
ATP-sensitive K+ channel
- LC-CoA
long chain acyl-CoA
- LDH
lactate dehydrogenase
- MAG
monoacylglycerol
- MCT1
Monocarboxylate transporter 1
- MDH
malate dehydrogenase
- ME1
cytosolic malic enzyme 1
- ME2
mitochondrial malic enzyme 2
- MODY
maturity onset diabetes of the young
- NNT
nicotinic nucleotide transhydrogenase
- NOX4
NADPH oxidase 4
- OAA
oxaloacetate
- OXCT1
3-oxoacid CoA transferase/SCOT1
- OxPhos
oxidative phosphorylation
- PC
pyruvate carboxylase
- PCK2
mitochondrial phosphoenolpyruvate carboxykinase/PEPCK-M
- PDH
pyruvate dehydrogenase
- PEP
phosphoenolpyruvate
- PEPCK
phosphoenolpyruvate carboxykinase
- PFK1
phosphofructokinase-1
- PFKFB
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- PK
pyruvate kinase
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- SUCLA2
ATP-generating succinyl-CoA synthase/SCSATP
- SUCLG2
GTP-generating succinyl-CoA synthase/SCSGTP
- TCA cycle
tricarboxylic acid cycle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abulizi A, Cardone RL, Stark R, Lewandowski SL, Zhao X, Hillion J, Ma L, Sehgal R, Alves TC, Thomas C, Kung C, Wang B, Siebel S, Andrews ZB, Mason GF, Rinehart J, Merrins MJ, Kibbey RG, 2020. Multi-Tissue Acceleration of the Mitochondrial Phosphoenolpyruvate Cycle Improves Whole-Body Metabolic Health. Cell Metab 32, 751–766.e11. 10.1016/j.cmet.2020.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MT, Dwulet JM, Briggs JK, Reissaus CA, Jin E, Szulczewski JM, Lyman MR, Sdao SM, Kravets V, Nimkulrat SD, Ponik SM, Merrins MJ, Mirmira RG, Linnemann AK, Benninger RK, Blum B, 2021. Reduced synchroneity of intra-islet Ca2+ oscillations in vivo in Robo-deficient β cells. Elife 10, e61308. 10.7554/eLife.61308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Affourtit C, Alberts B, Barlow J, Carré JE, Wynne AG, 2018. Control of pancreatic β-cell bioenergetics. Biochem. Soc. Trans 46, 555–564. 10.1042/BST20170505 [DOI] [PubMed] [Google Scholar]
- Affourtit C, Brand MD, 2009. Measuring mitochondrial bioenergetics in INS-1E insulinoma cells. Methods Enzymol 457, 405–424. 10.1016/S0076-6879(09)05023-X [DOI] [PubMed] [Google Scholar]
- Affourtit C, Brand MD, 2006. Stronger control of ATP/ADP by proton leak in pancreatic beta-cells than skeletal muscle mitochondria. Biochem J 393, 151–159. 10.1042/BJ20051280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainscow EK, Rutter GA, 2002. Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet beta-cells: evidence for a Ca2+-dependent mechanism. Diabetes 51 Suppl 1, S162–170. [DOI] [PubMed] [Google Scholar]
- Alves TC, Pongratz RL, Zhao X, Yarborough O, Sereda S, Shirihai O, Cline GW, Mason G, Kibbey RG, 2015. Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab. 22, 936–947. 10.1016/j.cmet.2015.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E, Long JA, 1947. The effect of hyperglycemia on insulin secretion as determined with the isolated rat pancreas in a perfusion apparatus. Endocrinology 40, 92–97. 10.1210/endo-40-2-92 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Harrison DE, Ashcroft SJ, 1984. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312, 446–448. [DOI] [PubMed] [Google Scholar]
- Bartholomé K, Brdiczka DG, Pette D, 1972. Purification and properties of extra- and intramitochondrial malate dehydrogenase (NADP; decarboxylating) from pig heart. Hoppe Seylers Z Physiol Chem 353, 1487–1495. 10.1515/bchm2.1972.353.2.1487 [DOI] [PubMed] [Google Scholar]
- Bauchle CJ, Rohli KE, Boyer CK, Pal V, Rocheleau JV, Liu S, Imai Y, Taylor EB, Stephens SB, 2021. Mitochondrial Efflux of Citrate and Isocitrate Is Fully Dispensable for Glucose-Stimulated Insulin Secretion and Pancreatic Islet β-Cell Function. Diabetes 70, 1717–1728. 10.2337/db21-0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benninger RKP, Kravets V, 2021. The physiological role of β-cell heterogeneity in pancreatic islet function. Nat Rev Endocrinol. 10.1038/s41574-021-00568-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berne C, 1975. The metabolism of lipids in mouse pancreatic islets. The biosynthesis of triacylglycerols and phospholipids. Biochem J 152, 667–673. 10.1042/bj1520667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R, Satin LS, Sherman AS, 2018. Closing in on the Mechanisms of Pulsatile Insulin Secretion. Diabetes 67, 351–359. 10.2337/dbi17-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R, Sherman A, Satin LS, 2007. Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am. J. Physiol. Endocrinol. Metab 293, E890–900. 10.1152/ajpendo.00359.2007 [DOI] [PubMed] [Google Scholar]
- Boquist L, 1987. NADP-linked dismutation and concentrations of citrate, cytosolic free Ca2+ and phosphoenolpyruvate in islet B-cells stimulated with glucose. Biochem Int 14, 531–538. [PubMed] [Google Scholar]
- Boquist L, Alehagen U, 1986. Ca2+ transport in isolated mouse liver mitochondria; role of reductive carboxylation and citrate? Cell Calcium 7, 275–282. 10.1016/0143-4160(86)90006-0 [DOI] [PubMed] [Google Scholar]
- Boquist L, Lorentzon R, 1986. Factors affecting Ca2+ transport in mouse islet and kidney mitochondria. Biochem Int 13, 181–187. [PubMed] [Google Scholar]
- Campbell JE, Newgard CB, 2021. Mechanisms controlling pancreatic islet cell function in insulin secretion. Nat Rev Mol Cell Biol 22, 142–158. 10.1038/s41580-020-00317-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance B, Williams GR, 1955. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem 217, 383–393. [PubMed] [Google Scholar]
- Chang GG, Wang JK, Huang TM, Lee HJ, Chou WY, Meng CL, 1991. Purification and characterization of the cytosolic NADP(+)-dependent malic enzyme from human breast cancer cell line. Eur J Biochem 202, 681–688. 10.1111/j.1432-1033.1991.tb16423.x [DOI] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Fusonie GE, Corkey BE, Tornheim K, 1997. Oscillations in oxygen consumption by permeabilized clonal pancreatic beta-cells (HIT) incubated in an oscillatory glycolyzing muscle extract: roles of free Ca2+, substrates, and the ATP/ADP ratio. Diabetes 46, 51–56. 10.2337/diab.46.1.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelek VN, Deeney JT, Shalosky NJ, Tornheim K, Hansford RG, Prentki M, Corkey BE, 1996. Regulation of pancreatic beta-cell mitochondrial metabolism: influence of Ca2+, substrate and ADP. Biochem. J 318 (Pt 2), 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane VA, Yang Z, Dell’Acqua ML, Shyng S-L, 2021. AKAP79/150 coordinates leptin-induced PKA signaling to regulate KATP channel trafficking in pancreatic β-cells. J Biol Chem 296, 100442. 10.1016/j.jbc.2021.100442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL, Hales CN, 1984. Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311, 271–273. 10.1038/311271a0 [DOI] [PubMed] [Google Scholar]
- Davis B, Lazarus NR, 1976. An in Vitro system for studying insulin release caused by secretory granules-plasma membrane interaction: definition of the system. J Physiol 256, 709–729. 10.1113/jphysiol.1976.sp011347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean PM, Matthews EK, 1968. Electrical activity in pancreatic islet cells. Nature 219, 389–390. 10.1038/219389a0 [DOI] [PubMed] [Google Scholar]
- Detimary P, Gilon P, Henquin JC, 1998. Interplay between cytoplasmic Ca2+ and the ATP/ADP ratio: a feedback control mechanism in mouse pancreatic islets. Biochem. J 333 (Pt 2), 269–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Harrell MD, Han SY, Jankowska D, Parachuru L, Morrissey A, Srivastava S, Liu W, Malester B, Yoshida H, Coetzee WA, 2005. The glycolytic enzymes, glyceraldehyde-3-phosphate dehydrogenase, triose-phosphate isomerase, and pyruvate kinase are components of the K(ATP) channel macromolecular complex and regulate its function. J. Biol. Chem 280, 38464–38470. 10.1074/jbc.M508744200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar-Chowdhury P, Malester B, Rajacic P, Coetzee WA, 2007. The regulation of ion channels and transporters by glycolytically derived ATP. Cell. Mol. Life Sci 64, 3069–3083. 10.1007/s00018-007-7332-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Smith PA, Ashcroft FM, 1993. Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic beta-cells. Biochem J 294 (Pt 1), 35–42. 10.1042/bj2940035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düfer M, Krippeit-Drews P, Buntinas L, Siemen D, Drews G, 2002. Methyl pyruvate stimulates pancreatic beta-cells by a direct effect on KATP channels, and not as a mitochondrial substrate. Biochem. J 368, 817–825. 10.1042/BJ20020657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes ID, McIntyre MS, Mertz RJ, Philipson LH, Roe MW, Spencer B, Worley JF, 1994. Dependence on NADH produced during glycolysis for beta-cell glucose signaling. J Biol Chem 269, 10979–10982. [PubMed] [Google Scholar]
- Dunne MJ, Petersen OH, 1986. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett 208, 59–62. 10.1016/0014-5793(86)81532-0 [DOI] [PubMed] [Google Scholar]
- Dyachok O, Idevall-Hagren O, Sågetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjärvi G, Gylfe E, Tengholm A, 2008. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab. 8, 26–37. 10.1016/j.cmet.2008.06.003 [DOI] [PubMed] [Google Scholar]
- Dyachok O, Isakov Y, Sågetorp J, Tengholm A, 2006. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting beta-cells. Nature 439, 349–352. 10.1038/nature04410 [DOI] [PubMed] [Google Scholar]
- El Azzouny M, Longacre MJ, Ansari I-UH, Kennedy RT, Burant CF, MacDonald MJ, 2016. Knockdown of ATP citrate lyase in pancreatic beta cells does not inhibit insulin secretion or glucose flux and implicates the acetoacetate pathway in insulin secretion. Mol Metab 5, 980–987. 10.1016/j.molmet.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfari S, Schulz V, Corkey B, Prentki M, 2000. Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49, 718–726. [DOI] [PubMed] [Google Scholar]
- Ferdaoussi M, Dai X, Jensen MV, Wang R, Peterson BS, Huang C, Ilkayeva O, Smith N, Miller N, Hajmrle C, Spigelman AF, Wright RC, Plummer G, Suzuki K, Mackay JP, van de Bunt M, Gloyn AL, Ryan TE, Norquay LD, Brosnan MJ, Trimmer JK, Rolph TP, Kibbey RG, Manning Fox JE, Colmers WF, Shirihai OS, Neufer PD, Yeh ETH, Newgard CB, MacDonald PE, 2015. Isocitrate-to-SENP1 signaling amplifies insulin secretion and rescues dysfunctional β cells. J. Clin. Invest 125, 3847–3860. 10.1172/JCI82498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster HR, Ho T, Potapenko E, Sdao SM, Lewandowski SL, VanDeusen HR, Davidson SM, Cardone RL, Kibbey RG, Merrins MJ, 2022. The isoforms of pyruvate kinase act as nutrient sensors for the β-cell KATP channel. 10.1101/2022.02.09.478817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlyand LE, Ma L, Philipson LH, 2005. Adenine nucleotide regulation in pancreatic beta-cells: modeling of ATP/ADP-Ca2+ interactions. Am. J. Physiol. Endocrinol. Metab 289, E839–848. 10.1152/ajpendo.00595.2004 [DOI] [PubMed] [Google Scholar]
- Fridlyand LE, Tamarina N, Philipson LH, 2003. Modeling of Ca2+ flux in pancreatic beta-cells: role of the plasma membrane and intracellular stores. Am. J. Physiol. Endocrinol. Metab 285, E138–154. 10.1152/ajpendo.00194.2002 [DOI] [PubMed] [Google Scholar]
- Gerencser AA, 2015. Bioenergetic Analysis of Single Pancreatic β-Cells Indicates an Impaired Metabolic Signature in Type 2 Diabetic Subjects. Endocrinology 156, 3496–3503. 10.1210/en.2015-1552 [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Mookerjee SA, Jastroch M, Brand MD, 2017. Positive Feedback Amplifies the Response of Mitochondrial Membrane Potential to Glucose Concentration in Clonal Pancreatic Beta Cells. Biochim Biophys Acta Mol Basis Dis 1863, 1054–1065. 10.1016/j.bbadis.2016.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerencser AA, Mookerjee SA, Jastroch M, Brand MD, 2016. Measurement of the Absolute Magnitude and Time Courses of Mitochondrial Membrane Potential in Primary and Clonal Pancreatic Beta-Cells. PLoS ONE 11, e0159199. 10.1371/journal.pone.0159199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheni G, Ogura M, Iwasaki M, Yokoi N, Minami K, Nakayama Y, Harada K, Hastoy B, Wu X, Takahashi H, Kimura K, Matsubara T, Hoshikawa R, Hatano N, Sugawara K, Shibasaki T, Inagaki N, Bamba T, Mizoguchi A, Fukusaki E, Rorsman P, Seino S, 2014. Glutamate acts as a key signal linking glucose metabolism to incretin/cAMP action to amplify insulin secretion. Cell Rep 9, 661–673. 10.1016/j.celrep.2014.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiasi SM, Rutter GA, 2021. Consequences for Pancreatic β-Cell Identity and Function of Unregulated Transcript Processing. Front Endocrinol (Lausanne) 12, 625235. 10.3389/fendo.2021.625235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilon P, Shepherd RM, Henquin JC, 1993. Oscillations of secretion driven by oscillations of cytoplasmic Ca2+ as evidences in single pancreatic islets. J Biol Chem 268, 22265–22268. [PubMed] [Google Scholar]
- Glancy B, Balaban RS, 2012. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry 51, 2959–2973. 10.1021/bi2018909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodner CJ, Walike BC, Koerker DJ, Ensinck JW, Brown AC, Chideckel EW, Palmer J, Kalnasy L, 1977. Insulin, glucagon, and glucose exhibit synchronous, sustained oscillations in fasting monkeys. Science 195, 177–179. 10.1126/science.401543 [DOI] [PubMed] [Google Scholar]
- Grafe E, Meythaler F, 1927a. Beitrag zur Kenntnis der Regulation der Insulin produktion I. Mitteilung: Der Traubenzucker als Hormon fϋr die Insulin abgabe. Naunyn-Schmiedebergs Arch. Exp. Path. Pharmak 125, 181–192. [Google Scholar]
- Grafe E, Meythaler F, 1927b. Űber den Traubensucker als Hormone der Insulinsekretion. Klin. Wochenschr 6, 1240. [Google Scholar]
- Gregg T, Sdao SM, Dhillon RS, Rensvold JW, Lewandowski SL, Pagliarini DJ, Denu JM, Merrins MJ, 2019. Obesity-dependent CDK1 signaling stimulates mitochondrial respiration at complex I in pancreatic β-cells. J. Biol. Chem 294, 4656–4666. 10.1074/jbc.RA118.006085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grodsky GM, Batts AA, Bennett LL, Vcella C, Mcwilliams NB, Smith DF, 1963. EFFECTS OF CARBOHYDRATES ON SECRETION OF INSULIN FROM ISOLATED RAT PANCREAS. Am J Physiol 205, 638–644. 10.1152/ajplegacy.1963.205.4.638 [DOI] [PubMed] [Google Scholar]
- Guay C, Joly E, Pepin E, Barbeau A, Hentsch L, Pineda M, Madiraju SRM, Brunengraber H, Prentki M, 2013. A role for cytosolic isocitrate dehydrogenase as a negative regulator of glucose signaling for insulin secretion in pancreatic ß-cells. PLoS ONE 8, e77097. 10.1371/journal.pone.0077097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guay C, Madiraju SRM, Aumais A, Joly E, Prentki M, 2007. A role for ATP-citrate lyase, malic enzyme, and pyruvate/citrate cycling in glucose-induced insulin secretion. J Biol Chem 282, 35657–35665. 10.1074/jbc.M707294200 [DOI] [PubMed] [Google Scholar]
- Hasan NM, Longacre MJ, Seed Ahmed M, Kendrick MA, Gu H, Ostenson C-G, Fukao T, MacDonald MJ, 2010. Lower succinyl-CoA:3-ketoacid-CoA transferase (SCOT) and ATP citrate lyase in pancreatic islets of a rat model of type 2 diabetes: knockdown of SCOT inhibits insulin release in rat insulinoma cells. Arch Biochem Biophys 499, 62–68. 10.1016/j.abb.2010.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head WS, Orseth ML, Nunemaker CS, Satin LS, Piston DW, Benninger RKP, 2012. Connexin-36 gap junctions regulate in vivo first- and second-phase insulin secretion dynamics and glucose tolerance in the conscious mouse. Diabetes 61, 1700–1707. 10.2337/db11-1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heart E, Cline GW, Collis LP, Pongratz RL, Gray JP, Smith PJS, 2009. Role for malic enzyme, pyruvate carboxylation, and mitochondrial malate import in glucose-stimulated insulin secretion. Am J Physiol Endocrinol Metab 296, E1354–1362. 10.1152/ajpendo.90836.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heart E, Corkey RF, Wikstrom JD, Shirihai OS, Corkey BE, 2006. Glucose-dependent increase in mitochondrial membrane potential, but not cytoplasmic calcium, correlates with insulin secretion in single islet cells. Am J Physiol Endocrinol Metab 290, E143–E148. 10.1152/ajpendo.00216.2005 [DOI] [PubMed] [Google Scholar]
- Henquin JC, 2009. Regulation of insulin secretion: a matter of phase control and amplitude modulation. Diabetologia 52, 739–751. 10.1007/s00125-009-1314-y [DOI] [PubMed] [Google Scholar]
- Holz GG, Kühtreiber WM, Habener JF, 1993. Pancreatic beta-cells are rendered glucose-competent by the insulinotropic hormone glucagon-like peptide-1(7–37). Nature 361, 362–365. 10.1038/361362a0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huypens P, Pillai R, Sheinin T, Schaefer S, Huang M, Odegaard ML, Ronnebaum SM, Wettig SD, Joseph JW, 2011. The dicarboxylate carrier plays a role in mitochondrial malate transport and in the regulation of glucose-stimulated insulin secretion from rat pancreatic beta cells. Diabetologia 54, 135–145. 10.1007/s00125-010-1923-5 [DOI] [PubMed] [Google Scholar]
- Idevall-Hagren O, Tengholm A, 2020. Metabolic regulation of calcium signaling in beta cells. Semin Cell Dev Biol 103, 20–30. 10.1016/j.semcdb.2020.01.008 [DOI] [PubMed] [Google Scholar]
- Jensen MV, Joseph JW, Ronnebaum SM, Burgess SC, Sherry AD, Newgard CB, 2008. Metabolic cycling in control of glucose-stimulated insulin secretion. Am. J. Physiol. Endocrinol. Metab 295, E1287–1297. 10.1152/ajpendo.90604.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesinkey SR, Madiraju AK, Alves TC, Yarborough OH, Cardone RL, Zhao X, Parsaei Y, Nasiri AR, Butrico G, Liu X, Molina AJ, Rountree AM, Neal AS, Wolf DM, Sterpka J, Philbrick WM, Sweet IR, Shirihai OH, Kibbey RG, 2019. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep 28, 759–772.e10. 10.1016/j.celrep.2019.06.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ježek P, Holendová B, Jabůrek M, Tauber J, Dlasková A, Plecitá-Hlavatá L, 2021. The Pancreatic β-Cell: The Perfect Redox System. Antioxidants (Basel) 10, 197. 10.3390/antiox10020197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JW, Odegaard ML, Ronnebaum SM, Burgess SC, Muehlbauer J, Sherry AD, Newgard CB, 2007. Normal flux through ATP-citrate lyase or fatty acid synthase is not required for glucose-stimulated insulin secretion. J Biol Chem 282, 31592–31600. 10.1074/jbc.M706080200 [DOI] [PubMed] [Google Scholar]
- Jung SK, Kauri LM, Qian WJ, Kennedy RT, 2000. Correlated oscillations in glucose consumption, oxygen consumption, and intracellular free Ca(2+) in single islets of Langerhans. J. Biol. Chem 275, 6642–6650. [DOI] [PubMed] [Google Scholar]
- Juntti-Berggren L, Webb D-L, Arkhammar POG, Schultz V, Schweda EKH, Tornheim K, Berggren P-O, 2003. Dihydroxyacetone-induced oscillations in cytoplasmic free Ca2+ and the ATP/ADP ratio in pancreatic beta-cells at substimulatory glucose. J. Biol. Chem 278, 40710–40716. 10.1074/jbc.M308248200 [DOI] [PubMed] [Google Scholar]
- Kakei M, Kelly RP, Ashcroft SJ, Ashcroft FM, 1986. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett 208, 63–66. 10.1016/0014-5793(86)81533-2 [DOI] [PubMed] [Google Scholar]
- Kang G, Leech CA, Chepurny OG, Coetzee WA, Holz GG, 2008. Role of the cAMP sensor Epac as a determinant of KATP channel ATP sensitivity in human pancreatic beta-cells and rat INS-1 cells. J Physiol 586, 1307–1319. 10.1113/jphysiol.2007.143818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Li C, Gao Z, Stanley CA, Matschinsky FM, 2002. Glutaminolysis and insulin secretion: from bedside to bench and back. Diabetes 51 Suppl 3, S421–426. 10.2337/diabetes.51.2007.s421 [DOI] [PubMed] [Google Scholar]
- Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA, 1999. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J. Biol. Chem 274, 13281–13291. [DOI] [PubMed] [Google Scholar]
- Kennedy RT, Kauri LM, Dahlgren GM, Jung S-K, 2002. Metabolic oscillations in beta-cells. Diabetes 51 Suppl 1, S152–161. [DOI] [PubMed] [Google Scholar]
- Kibbey RG, Choi CS, Lee H-Y, Cabrera O, Pongratz RL, Zhao X, Birkenfeld AL, Li C, Berggren P-O, Stanley C, Shulman GI, 2014. Mitochondrial GTP insensitivity contributes to hypoglycemia in hyperinsulinemia hyperammonemia by inhibiting glucagon release. Diabetes 63, 4218–4229. 10.2337/db14-0783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibbey RG, Pongratz RL, Romanelli AJ, Wollheim CB, Cline GW, Shulman GI, 2007. Mitochondrial GTP regulates glucose-stimulated insulin secretion. Cell Metab. 5, 253–264. 10.1016/j.cmet.2007.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindmark H, Köhler M, Brown G, Bränström R, Larsson O, Berggren PO, 2001. Glucose-induced oscillations in cytoplasmic free Ca2+ concentration precede oscillations in mitochondrial membrane potential in the pancreatic beta-cell. J. Biol. Chem 276, 34530–34536. 10.1074/jbc.M102492200 [DOI] [PubMed] [Google Scholar]
- Kiranadi B, Bangham JA, Smith PA, 1991. Inhibition of electrical activity in mouse pancreatic beta-cells by the ATP/ADP translocase inhibitor, bongkrekic acid. FEBS Lett 283, 93–96. 10.1016/0014-5793(91)80561-g [DOI] [PubMed] [Google Scholar]
- Kjems LL, Ravier MA, Jonas J-C, Henquin J-C, 2002. Do oscillations of insulin secretion occur in the absence of cytoplasmic Ca2+ oscillations in beta-cells? Diabetes 51 Suppl 1, S177–182. 10.2337/diabetes.51.2007.s177 [DOI] [PubMed] [Google Scholar]
- Kowluru A, Tannous M, Chen H-Q, 2002. Localization and characterization of the mitochondrial isoform of the nucleoside diphosphate kinase in the pancreatic beta cell: evidence for its complexation with mitochondrial succinyl-CoA synthetase. Arch Biochem Biophys 398, 160–169. 10.1006/abbi.2001.2710 [DOI] [PubMed] [Google Scholar]
- Krippeit-Drews P, Düfer M, Drews G, 2000. Parallel oscillations of intracellular calcium activity and mitochondrial membrane potential in mouse pancreatic B-cells. Biochem. Biophys. Res. Commun 267, 179–183. 10.1006/bbrc.1999.1921 [DOI] [PubMed] [Google Scholar]
- Lacombe M-L, Tokarska-Schlattner M, Epand RF, Boissan M, Epand RM, Schlattner U, 2009. Interaction of NDPK-D with cardiolipin-containing membranes: Structural basis and implications for mitochondrial physiology. Biochimie 91, 779–783. 10.1016/j.biochi.2009.02.006 [DOI] [PubMed] [Google Scholar]
- Lamontagne J, Al-Mass A, Nolan CJ, Corkey BE, Madiraju SRM, Joly E, Prentki M, 2017. Identification of the signals for glucose-induced insulin secretion in INS1 (832/13) β-cells using metformin-induced metabolic deceleration as a model. J Biol Chem 292, 19458–19468. 10.1074/jbc.M117.808105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang DA, Matthews DR, Burnett M, Turner RC, 1981. Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 30, 435–439. 10.2337/diab.30.5.435 [DOI] [PubMed] [Google Scholar]
- Lang DA, Matthews DR, Peto J, Turner RC, 1979. Cyclic oscillations of basal plasma glucose and insulin concentrations in human beings. N. Engl. J. Med 301, 1023–1027. 10.1056/NEJM197911083011903 [DOI] [PubMed] [Google Scholar]
- Lazarus NR, Davis B, O’Connor KJ, 1976. An approach to a molecular understanding of exocytotic insulin release. J Physiol (Paris) 72, 787–794. [PubMed] [Google Scholar]
- Leloup C, Tourrel-Cuzin C, Magnan C, Karaca M, Castel J, Carneiro L, Colombani A-L, Ktorza A, Casteilla L, Pénicaud L, 2009. Mitochondrial reactive oxygen species are obligatory signals for glucose-induced insulin secretion. Diabetes 58, 673–681. 10.2337/db07-1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaire K, Thorrez L, Schuit F, 2016. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu Rev Nutr 36, 45–71. 10.1146/annurev-nutr-071715-050808 [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M, 1996. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med 20, 463–466. 10.1016/0891-5849(96)02051-5 [DOI] [PubMed] [Google Scholar]
- Lewandowski SL, Cardone RL, Foster HR, Ho T, Potapenko E, Poudel C, VanDeusen HR, Sdao SM, Alves TC, Zhao X, Capozzi ME, de Souza AH, Jahan I, Thomas CJ, Nunemaker CS, Davis DB, Campbell JE, Kibbey RG, Merrins MJ, 2020. Pyruvate Kinase Controls Signal Strength in the Insulin Secretory Pathway. Cell Metab 32, 736–750.e5. 10.1016/j.cmet.2020.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chen P, Palladino A, Narayan S, Russell LK, Sayed S, Xiong G, Chen J, Stokes D, Butt YM, Jones PM, Collins HW, Cohen NA, Cohen AS, Nissim I, Smith TJ, Strauss AW, Matschinsky FM, Bennett MJ, Stanley CA, 2010. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency involves activation of glutamate dehydrogenase. J Biol Chem 285, 31806–31818. 10.1074/jbc.M110.123638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Najafi H, Daikhin Y, Nissim IB, Collins HW, Yudkoff M, Matschinsky FM, Stanley CA, 2003. Regulation of leucine-stimulated insulin secretion and glutamine metabolism in isolated rat islets. J Biol Chem 278, 2853–2858. 10.1074/jbc.M210577200 [DOI] [PubMed] [Google Scholar]
- Li J, Shuai HY, Gylfe E, Tengholm A, 2013. Oscillations of sub-membrane ATP in glucose-stimulated beta cells depend on negative feedback from Ca(2+). Diabetologia 56, 1577–1586. 10.1007/s00125-013-2894-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Smith N, Spigelman AF, Suzuki K, Ferdaoussi M, Alghamdi TA, Lewandowski SL, Jin Y, Bautista A, Wang YW, Manning Fox JE, Merrins MJ, Buteau J, MacDonald PE, 2021. β-Cell Knockout of SENP1 Reduces Responses to Incretins and Worsens Oral Glucose Tolerance in High-Fat Diet-Fed Mice. Diabetes 70, 2626–2638. 10.2337/db20-1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ES, Kotschneff NP, 1934. Mechanismus der Insulininkretion. Pflügers Arch. 234, 194–199. 10.1007/BF01766897 [DOI] [Google Scholar]
- Luciani DS, Misler S, Polonsky KS, 2006. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J. Physiol. (Lond.) 572, 379–392. 10.1113/jphysiol.2005.101766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald MJ, 2002. Differences between mouse and rat pancreatic islets: succinate responsiveness, malic enzyme, and anaplerosis. Am. J. Physiol. Endocrinol. Metab 283, E302–310. 10.1152/ajpendo.00041.2002 [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, 1995. Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270, 20051–20058. [PubMed] [Google Scholar]
- MacDonald MJ, 1981. High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. J. Biol. Chem 256, 8287–8290. [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA, 2000. Glutamate is not a messenger in insulin secretion. J Biol Chem 275, 34025–34027. 10.1074/jbc.C000411200 [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA, Brown LJ, Hasan NM, Buss JD, Kendrick MA, 2005. Perspective: emerging evidence for signaling roles of mitochondrial anaplerotic products in insulin secretion. Am. J. Physiol. Endocrinol. Metab 288, E1–15. 10.1152/ajpendo.00218.2004 [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA, Buss JD, Hasan NM, Fallon MJ, Kendrick MA, 2003. Citrate oscillates in liver and pancreatic beta cell mitochondria and in INS-1 insulinoma cells. J. Biol. Chem 278, 51894–51900. 10.1074/jbc.M309038200 [DOI] [PubMed] [Google Scholar]
- MacDonald MJ, Longacre MJ, Kendrick MA, 2009. Mitochondrial malic enzyme (ME2) in pancreatic islets of the human, rat and mouse and clonal insulinoma cells. Arch Biochem Biophys 488, 100–104. 10.1016/j.abb.2009.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P, Wollheim CB, 1999. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402, 685–689. 10.1038/45280 [DOI] [PubMed] [Google Scholar]
- Marinelli I, Fletcher PA, Sherman AS, Satin LS, Bertram R, 2021. Symbiosis of Electrical and Metabolic Oscillations in Pancreatic β-Cells. Front Physiol 12, 781581. 10.3389/fphys.2021.781581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mármol P, Pardo B, Wiederkehr A, Del Arco A, Wollheim CB, Satrústegui J, 2009. Requirement for aralar and its Ca2+-binding sites in Ca2+ signal transduction in mitochondria from INS-1 clonal beta-cells. J Biol Chem 284, 515–524. 10.1074/jbc.M806729200 [DOI] [PubMed] [Google Scholar]
- Martinez-Sanchez A, Pullen TJ, Chabosseau P, Zhang Q, Haythorne E, Cane MC, Nguyen-Tu M-S, Sayers SR, Rutter GA, 2016. Disallowance of Acot7 in β-Cells Is Required for Normal Glucose Tolerance and Insulin Secretion. Diabetes 65, 1268–1282. 10.2337/db15-1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschinsky FM, Ellerman JE, 1968. Metabolism of glucose in the islets of Langerhans. J. Biol. Chem 243, 2730–2736. [PubMed] [Google Scholar]
- Matthews DR, Naylor BA, Jones RG, Ward GM, Turner RC, 1983. Pulsatile insulin has greater hypoglycemic effect than continuous delivery. Diabetes 32, 617–621. 10.2337/diab.32.7.617 [DOI] [PubMed] [Google Scholar]
- Matveyenko AV, Liuwantara D, Gurlo T, Kirakossian D, Dalla Man C, Cobelli C, White MF, Copps KD, Volpi E, Fujita S, Butler PC, 2012. Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 61, 2269–2279. 10.2337/db11-1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee EE, Bentley AT, Smith RM, Kraas JR, Ciaccio CE, 2000. Guanine nucleotide transport by atractyloside-sensitive and -insensitive carriers in isolated heart mitochondria. Am J Physiol Cell Physiol 279, C1870–1879. 10.1152/ajpcell.2000.279.6.C1870 [DOI] [PubMed] [Google Scholar]
- McKenna JP, Ha J, Merrins MJ, Satin LS, Sherman A, Bertram R, 2016. Ca(2+) Effects on ATP Production and Consumption Have Regulatory Roles on Oscillatory Islet Activity. Biophys. J 110, 733–742. 10.1016/j.bpj.2015.11.3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Bertram R, Sherman A, Satin LS, 2012. Phosphofructo-2-kinase/fructose-2,6-bisphosphatase modulates oscillations of pancreatic islet metabolism. PLoS ONE 7, e34036. 10.1371/journal.pone.0034036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Fendler B, Zhang M, Sherman A, Bertram R, Satin LS, 2010. Metabolic oscillations in pancreatic islets depend on the intracellular Ca2+ level but not Ca2+ oscillations. Biophys. J 99, 76–84. 10.1016/j.bpj.2010.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Poudel C, McKenna JP, Ha J, Sherman A, Bertram R, Satin LS, 2016. Phase Analysis of Metabolic Oscillations and Membrane Potential in Pancreatic Islet β-Cells. Biophys. J 110, 691–699. 10.1016/j.bpj.2015.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrins MJ, Van Dyke AR, Mapp AK, Rizzo MA, Satin LS, 2013. Direct measurements of oscillatory glycolysis in pancreatic islet β-cells using novel fluorescence resonance energy transfer (FRET) biosensors for pyruvate kinase M2 activity. J. Biol. Chem 288, 33312–33322. 10.1074/jbc.M113.508127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz RJ, Worley JF, Spencer B, Johnson JH, Dukes ID, 1996. Activation of stimulus-secretion coupling in pancreatic beta-cells by specific products of glucose metabolism. Evidence for privileged signaling by glycolysis. J Biol Chem 271, 4838–4845. 10.1074/jbc.271.9.4838 [DOI] [PubMed] [Google Scholar]
- Misler S, Falke LC, Gillis K, McDaniel ML, 1986. A metabolite-regulated potassium channel in rat pancreatic B cells. Proc Natl Acad Sci U S A 83, 7119–7123. 10.1073/pnas.83.18.7119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitok KA, Freiberger EC, Schueler KL, Rabaglia ME, Stapleton DS, Kwiecien NW, Malec PA, Hebert AS, Broman AT, Kennedy RT, Keller MP, Coon JJ, Attie AD, 2018. Islet proteomics reveals genetic variation in dopamine production resulting in altered insulin secretion. J. Biol. Chem 293, 5860–5877. 10.1074/jbc.RA117.001102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugabo Y, Zhao S, Lamontagne J, Al-Mass A, Peyot M-L, Corkey BE, Joly E, Madiraju SRM, Prentki M, 2017. Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic β-cells. J Biol Chem 292, 7407–7422. 10.1074/jbc.M116.763060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugabo Y, Zhao S, Seifried A, Gezzar S, Al-Mass A, Zhang D, Lamontagne J, Attane C, Poursharifi P, Iglesias J, Joly E, Peyot M-L, Gohla A, Madiraju SRM, Prentki M, 2016. Identification of a mammalian glycerol-3-phosphate phosphatase: Role in metabolism and signaling in pancreatic β-cells and hepatocytes. Proc Natl Acad Sci U S A 113, E430–439. 10.1073/pnas.1514375113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, 2016. The Pancreatic β-Cell: A Bioenergetic Perspective. Physiol. Rev 96, 1385–1447. 10.1152/physrev.00009.2016 [DOI] [PubMed] [Google Scholar]
- Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, Lambeth JD, 2010. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 49, 2433–2442. 10.1021/bi9022285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunemaker CS, Wasserman DH, McGuinness OP, Sweet IR, Teague JC, Satin LS, 2006. Insulin secretion in the conscious mouse is biphasic and pulsatile. Am. J. Physiol. Endocrinol. Metab 290, E523–529. 10.1152/ajpendo.00392.2005 [DOI] [PubMed] [Google Scholar]
- Nunemaker CS, Zhang M, Wasserman DH, McGuinness OP, Powers AC, Bertram R, Sherman A, Satin LS, 2005. Individual mice can be distinguished by the period of their islet calcium oscillations: is there an intrinsic islet period that is imprinted in vivo? Diabetes 54, 3517–3522. 10.2337/diabetes.54.12.3517 [DOI] [PubMed] [Google Scholar]
- O’Rahilly S, Turner RC, Matthews DR, 1988. Impaired pulsatile secretion of insulin in relatives of patients with non-insulin-dependent diabetes. N Engl J Med 318, 1225–1230. 10.1056/NEJM198805123181902 [DOI] [PubMed] [Google Scholar]
- Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, Schuit F, Quintens R, Sipilä I, Mayatepek E, Meissner T, Halestrap AP, Rutter GA, Kere J, 2007. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet 81, 467–474. 10.1086/520960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottaway JH, McClellan JA, Saunderson CL, 1981. Succinic thiokinase and metabolic control. Int J Biochem 13, 401–410. 10.1016/0020-711x(81)90111-7 [DOI] [PubMed] [Google Scholar]
- Palmieri F, 2004. The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447, 689–709. 10.1007/s00424-003-1099-7 [DOI] [PubMed] [Google Scholar]
- Pepin E, Guay C, Delghingaro-Augusto V, Joly E, Madiraju SRM, Prentki M, 2010. Short-chain 3-hydroxyacyl-CoA dehydrogenase is a negative regulator of insulin secretion in response to fuel and non-fuel stimuli in INS832/13 β-cells. J Diabetes 2, 157–167. 10.1111/j.1753-0407.2010.00076.x [DOI] [PubMed] [Google Scholar]
- Pizarro-Delgado J, Deeney JT, Corkey BE, Tamarit-Rodriguez J, 2016. Direct Stimulation of Islet Insulin Secretion by Glycolytic and Mitochondrial Metabolites in KCl-Depolarized Islets. PLoS ONE 11, e0166111. 10.1371/journal.pone.0166111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecitá-Hlavatá L, Engstová H, Holendová B, Tauber J, Špaček T, Petrásková L, Křen V, Špačková J, Gotvaldová K, Ježek J, Dlasková A, Smolková K, Ježek P, 2020a. Mitochondrial Superoxide Production Decreases on Glucose-Stimulated Insulin Secretion in Pancreatic β Cells Due to Decreasing Mitochondrial Matrix NADH/NAD+ Ratio. Antioxid Redox Signal 33, 789–815. 10.1089/ars.2019.7800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecitá-Hlavatá L, Jabůrek M, Holendová B, Tauber J, Pavluch V, Berková Z, Cahová M, Schroeder K, Brandes RP, Siemen D, Ježek P, 2020b. Glucose-Stimulated Insulin Secretion Fundamentally Requires H2O2 Signaling by NADPH Oxidase 4. Diabetes. 10.2337/db19-1130 [DOI] [PubMed] [Google Scholar]
- Polonsky KS, Given BD, Hirsch LJ, Tillil H, Shapiro ET, Beebe C, Frank BH, Galloway JA, Van Cauter E, 1988. Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 318, 1231–1239. 10.1056/NEJM198805123181903 [DOI] [PubMed] [Google Scholar]
- Pongratz RL, Kibbey RG, Shulman GI, Cline GW, 2007. Cytosolic and mitochondrial malic enzyme isoforms differentially control insulin secretion. J. Biol. Chem 282, 200–207. 10.1074/jbc.M602954200 [DOI] [PubMed] [Google Scholar]
- Pørksen N, 2002. The in vivo regulation of pulsatile insulin secretion. Diabetologia 45, 3–20. 10.1007/s125-002-8240-x [DOI] [PubMed] [Google Scholar]
- Possik E, Al-Mass A, Peyot M-L, Ahmad R, Al-Mulla F, Madiraju SRM, Prentki M, 2021. New Mammalian Glycerol-3-Phosphate Phosphatase: Role in β-Cell, Liver and Adipocyte Metabolism. Front Endocrinol (Lausanne) 12, 706607. 10.3389/fendo.2021.706607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possik E, Schmitt C, Al-Mass A, Bai Y, Côté L, Morin J, Erb H, Oppong A, Kahloan W, Parker JA, Madiraju SRM, Prentki M, 2022. Phosphoglycolate phosphatase homologs act as glycerol-3-phosphate phosphatase to control stress and healthspan in C. elegans. Nat Commun 13, 177. 10.1038/s41467-021-27803-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursharifi P, Attané C, Mugabo Y, Al-Mass A, Ghosh A, Schmitt C, Zhao S, Guida J, Lussier R, Erb H, Chenier I, Peyot M-L, Joly E, Noll C, Carpentier AC, Madiraju SRM, Prentki M, 2020. Adipose ABHD6 regulates tolerance to cold and thermogenic programs. JCI Insight 5, 140294. 10.1172/jci.insight.140294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poursharifi P, Madiraju SRM, Prentki M, 2017. Monoacylglycerol signalling and ABHD6 in health and disease. Diabetes Obes Metab 19 Suppl 1, 76–89. 10.1111/dom.13008 [DOI] [PubMed] [Google Scholar]
- Prentki M, Corkey BE, Madiraju SRM, 2020a. Lipid-associated metabolic signalling networks in pancreatic beta cell function. Diabetologia 63, 10–20. 10.1007/s00125-019-04976-w [DOI] [PubMed] [Google Scholar]
- Prentki M, Madiraju SRM, 2012. Glycerolipid/free fatty acid cycle and islet β-cell function in health, obesity and diabetes. Mol Cell Endocrinol 353, 88–100. 10.1016/j.mce.2011.11.004 [DOI] [PubMed] [Google Scholar]
- Prentki M, Matschinsky FM, Madiraju SRM, 2013. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185. 10.1016/j.cmet.2013.05.018 [DOI] [PubMed] [Google Scholar]
- Prentki M, Peyot M-L, Masiello P, Madiraju SRM, 2020b. Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic β-Cell. Diabetes 69, 279–290. 10.2337/dbi19-0014 [DOI] [PubMed] [Google Scholar]
- Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE, 1992. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. Journal of Biological Chemistry 267, 5802–5810. 10.1016/S0021-9258(18)42624-5 [DOI] [PubMed] [Google Scholar]
- Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA, 2010. Identification of genes selectively disallowed in the pancreatic islet. Islets 2, 89–95. 10.4161/isl.2.2.11025 [DOI] [PubMed] [Google Scholar]
- Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA, 2012. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes 61, 1719–1725. 10.2337/db11-1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafalowska U, Pastuszko A, Gromek A, 1974. NADP-dependent isocitrate dehydrogenase from rat brain cytosol. Bull Acad Pol Sci Biol 22, 453–459. [PubMed] [Google Scholar]
- Ravier MA, Eto K, Jonkers FC, Nenquin M, Kadowaki T, Henquin JC, 2000. The oscillatory behavior of pancreatic islets from mice with mitochondrial glycerol-3-phosphate dehydrogenase knockout. J Biol Chem 275, 1587–1593. 10.1074/jbc.275.3.1587 [DOI] [PubMed] [Google Scholar]
- Regeenes R, Wang Y, Piro A, Au A, Yip C, Wheeler MB, Rocheleau JV, 2022. Design of an islet-on-a-chip device reveals glucose-stimulated respiration is substrate limited by glycolytic flux through PKM2. 10.1101/2022.03.02.482671 [DOI] [Google Scholar]
- Ronnebaum SM, Ilkayeva O, Burgess SC, Joseph JW, Lu D, Stevens RD, Becker TC, Sherry AD, Newgard CB, Jensen MV, 2006. A pyruvate cycling pathway involving cytosolic NADP-dependent isocitrate dehydrogenase regulates glucose-stimulated insulin secretion. J. Biol. Chem 281, 30593–30602. 10.1074/jbc.M511908200 [DOI] [PubMed] [Google Scholar]
- Ronnebaum SM, Jensen MV, Hohmeier HE, Burgess SC, Zhou Y-P, Qian S, MacNeil D, Howard A, Thornberry N, Ilkayeva O, Lu D, Sherry AD, Newgard CB, 2008. Silencing of cytosolic or mitochondrial isoforms of malic enzyme has no effect on glucose-stimulated insulin secretion from rodent islets. J Biol Chem 283, 28909–28917. 10.1074/jbc.M804665200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Braun M, 2013. Regulation of insulin secretion in human pancreatic islets. Annu Rev Physiol 75, 155–179. 10.1146/annurev-physiol-030212-183754 [DOI] [PubMed] [Google Scholar]
- Rorsman P, Trube G, 1985. Glucose dependent K+-channels in pancreatic beta-cells are regulated by intracellular ATP. Pflugers Arch. 405, 305–309. [DOI] [PubMed] [Google Scholar]
- Rutter GA, Georgiadou E, Martinez-Sanchez A, Pullen TJ, 2020. Metabolic and functional specialisations of the pancreatic beta cell: gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 63, 1990–1998. 10.1007/s00125-020-05205-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Pralong WF, Wollheim CB, 1992. Regulation of mitochondrial glycerol-phosphate dehydrogenase by Ca2+ within electropermeabilized insulin-secreting cells (INS-1). Biochim Biophys Acta 1175, 107–113. 10.1016/0167-4889(92)90016-5 [DOI] [PubMed] [Google Scholar]
- Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A, 2015. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem J 466, 203–218. 10.1042/BJ20141384 [DOI] [PubMed] [Google Scholar]
- Sánchez-Archidona AR, Cruciani-Guglielmacci C, Roujeau C, Wigger L, Lallement J, Denom J, Barovic M, Kassis N, Mehl F, Weitz J, Distler M, Klose C, Simons K, Ibberson M, Solimena M, Magnan C, Thorens B, 2021. Plasma triacylglycerols are biomarkers of β-cell function in mice and humans. Mol Metab 54, 101355. 10.1016/j.molmet.2021.101355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaran B, Chavan AJ, Haley BE, 1996. Identification of adenine binding domain peptides of the NADP+ active site within porcine heart NADP(+)-dependent isocitrate dehydrogenase. Biochemistry 35, 13501–13510. 10.1021/bi9614592 [DOI] [PubMed] [Google Scholar]
- Santos LRB, Muller C, de Souza AH, Takahashi HK, Spégel P, Sweet IR, Chae H, Mulder H, Jonas J-C, 2017. NNT reverse mode of operation mediates glucose control of mitochondrial NADPH and glutathione redox state in mouse pancreatic β-cells. Mol Metab 6, 535–547. 10.1016/j.molmet.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuit F, De Vos A, Farfari S, Moens K, Pipeleers D, Brun T, Prentki M, 1997. Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J. Biol. Chem 272, 18572–18579. [DOI] [PubMed] [Google Scholar]
- Sdao SM, Ho T, Poudel C, Foster HR, De Leon ER, Adams MT, Lee J-H, Blum B, Rane SG, Merrins MJ, 2021. CDK2 limits the highly energetic secretory program of mature β cells by restricting PEP cycle-dependent KATP channel closure. Cell Rep 34, 108690. 10.1016/j.celrep.2021.108690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelig GF, Colman RF, 1978. Characterization of the physicochemical and catalytic properties of human heart NADP-dependent isocitrate dehydrogenase. Arch Biochem Biophys 188, 394–409. 10.1016/s0003-9861(78)80024-1 [DOI] [PubMed] [Google Scholar]
- Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, Girotti M, Marie S, MacDonald MJ, Wollheim CB, 1994. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem 269, 4895–4902. [PubMed] [Google Scholar]
- Shuai H, Xu Y, Ahooghalandari P, Tengholm A, 2021. Glucose-induced cAMP elevation in β-cells involves amplification of constitutive and glucagon-activated GLP-1 receptor signalling. Acta Physiol (Oxf) 231, e13611. 10.1111/apha.13611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider KE, Becker S, Boyajian L, Shyng S-L, MacMullen C, Hughes N, Ganapathy K, Bhatti T, Stanley CA, Ganguly A, 2013. Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab 98, E355–363. 10.1210/jc.2012-2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spégel P, Sharoyko VV, Goehring I, Danielsson APH, Malmgren S, Nagorny CLF, Andersson LE, Koeck T, Sharp GWG, Straub SG, Wollheim CB, Mulder H, 2013. Time-resolved metabolomics analysis of β-cells implicates the pentose phosphate pathway in the control of insulin release. Biochem J 450, 595–605. 10.1042/BJ20121349 [DOI] [PubMed] [Google Scholar]
- Stanley CA, Lieu YK, Hsu BY, Burlina AB, Greenberg CR, Hopwood NJ, Perlman K, Rich BH, Zammarchi E, Poncz M, 1998. Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338, 1352–1357. 10.1056/NEJM199805073381904 [DOI] [PubMed] [Google Scholar]
- Stark R, Guebre-Egziabher F, Zhao X, Feriod C, Dong J, Alves TC, Ioja S, Pongratz RL, Bhanot S, Roden M, Cline GW, Shulman GI, Kibbey RG, 2014. A role for mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) in the regulation of hepatic gluconeogenesis. J. Biol. Chem 289, 7257–7263. 10.1074/jbc.C113.544759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark R, Pasquel F, Turcu A, Pongratz RL, Roden M, Cline GW, Shulman GI, Kibbey RG, 2009. Phosphoenolpyruvate cycling via mitochondrial phosphoenolpyruvate carboxykinase links anaplerosis and mitochondrial GTP with insulin secretion. J. Biol. Chem 284, 26578–26590. 10.1074/jbc.M109.011775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strumiło E, 1995. Effect of Ca2+ on the activity of mitochondrial NADP-specific isocitrate dehydrogenase from rabbit adrenals. Acta Biochim Pol 42, 325–328. [PubMed] [Google Scholar]
- Sugden MC, Ashcroft SJ, 1977. Phosphoenolpyruvate in rat pancreatic islets: a possible intracellular trigger of insulin release? Diabetologia 13, 481–486. 10.1007/BF01234500 [DOI] [PubMed] [Google Scholar]
- Sul HS, Shrago E, Shug AL, 1976. Relationship of phosphoenolpyruvate transport, acyl coenzyme A inhibition of adenine nucleotide translocase and calcium ion efflux in guinea pig heart mitochondria. Arch Biochem Biophys 172, 230–237. 10.1016/0003-9861(76)90071-0 [DOI] [PubMed] [Google Scholar]
- Szibor M, Gizatullina Z, Gainutdinov T, Endres T, Debska-Vielhaber G, Kunz M, Karavasili N, Hallmann K, Schreiber F, Bamberger A, Schwarzer M, Doenst T, Heinze H-J, Lessmann V, Vielhaber S, Kunz WS, Gellerich FN, 2020. Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J Biol Chem 295, 4383–4397. 10.1074/jbc.RA119.011902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddeo EP, Alsabeeh N, Baghdasarian S, Wikstrom JD, Ritou E, Sereda S, Erion K, Li J, Stiles L, Abdulla M, Swanson Z, Wilhelm JJ, Bellin MD, Kibbey RG, Liesa M, Shirihai OS, 2020. Mitochondrial Proton Leak Regulated by Cyclophilin D Elevates Insulin Secretion in Islets at Nonstimulatory Glucose Levels. Diabetes 69, 131–145. 10.2337/db19-0379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenner B, Getz M, Ross B, Ohadi D, Bohrer CH, Greenwald E, Mehta S, Xiao J, Rangamani P, Zhang J, 2020. Spatially compartmentalized phase regulation of a Ca2+-cAMP-PKA oscillatory circuit. Elife 9, e55013. 10.7554/eLife.55013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornheim K, 1997. Are metabolic oscillations responsible for normal oscillatory insulin secretion? Diabetes 46, 1375–1380. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Lamp ST, 1989. Cardiac ATP-sensitive K+ channels. Evidence for preferential regulation by glycolysis. J. Gen. Physiol 94, 911–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Lamp ST, 1987. Glycolysis preferentially inhibits ATP-sensitive K+ channels in isolated guinea pig cardiac myocytes. Science 238, 67–69. [DOI] [PubMed] [Google Scholar]
- Wikstrom JD, Sereda SB, Stiles L, Elorza A, Allister EM, Neilson A, Ferrick DA, Wheeler MB, Shirihai OS, 2012. A novel high-throughput assay for islet respiration reveals uncoupling of rodent and human islets. PLoS ONE 7, e33023. 10.1371/journal.pone.0033023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DF, Matschinsky FM, 2021. Metabolic Homeostasis in Life as We Know It: Its Origin and Thermodynamic Basis. Front Physiol 12, 658997. 10.3389/fphys.2021.658997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeyelmaker WP, Slater EC, 1967. The inhibition of succinate dehydrogenase by oxaloacetate. Biochim Biophys Acta 132, 210–212. 10.1016/0005-2744(67)90214-8 [DOI] [PubMed] [Google Scholar]
- Zhang G-F, Jensen MV, Gray SM, El K, Wang Y, Lu D, Becker TC, Campbell JE, Newgard CB, 2021. Reductive TCA cycle metabolism fuels glutamine- and glucose-stimulated insulin secretion. Cell Metab 33, 804–817.e5. 10.1016/j.cmet.2020.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J-F, Mehta S, Zhang J, 2021. Signaling Microdomains in the Spotlight: Visualizing Compartmentalized Signaling Using Genetically Encoded Fluorescent Biosensors. Annu Rev Pharmacol Toxicol 61, 587–608. 10.1146/annurev-pharmtox-010617-053137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Mugabo Y, Iglesias J, Xie L, Delghingaro-Augusto V, Lussier R, Peyot M-L, Joly E, Taïb B, Davis MA, Brown JM, Abousalham A, Gaisano H, Madiraju SRM, Prentki M, 2014. α/β-Hydrolase domain-6-accessible monoacylglycerol controls glucose-stimulated insulin secretion. Cell Metab. 19, 993–1007. 10.1016/j.cmet.2014.04.003 [DOI] [PubMed] [Google Scholar]
- Zunz E, La Barre J, 1927. De I’hyperinsulinémie consécutive à I’hyperglycémie provoquée par injection de dextrose. Arch. intern XXIX, 265–280. [Google Scholar]