

Abstract
Despite being the most common childhood bone tumor, the genomic characterization of osteosarcoma remains incomplete. In particular, very few osteosarcoma metastases have been sequenced to date, critical to better understand mechanisms of progression and evolution in this tumor. We performed an integrated whole genome and exome sequencing analysis of paired primary and metastatic pediatric osteosarcoma specimens to identify recurrent genomic alterations. Sequencing of 13 osteosarcoma patients including 13 primary, 10 metastatic, and 3 locally recurring tumors revealed a highly heterogeneous mutational landscape, including cases of hypermutation and microsatellite instability positivity, but with virtually no recurrent alterations except for mutations involving the tumor suppressor genes RB1 and TP53. At the germline level, we detected alterations in multiple cancer related genes in the majority of the cohort, including those potentially disrupting DNA damage response pathways. Metastases retained only a minimal number of short variants from their corresponding primary tumors, while copy number alterations showed higher conservation. One recurrently amplified gene, KDR, was highly expressed in advanced cases and associated with poor prognosis.
Keywords: childhood bone sarcomas, osteosarcoma, metastasis, genome sequencing, KDR, VEGF
Introduction
Osteosarcoma is the most common bone malignancy among childhood cancers, accounting for ∼5% of all tumors in this age group. Importantly, 5-year survival rates have not seen substantial improvement in the last 35 years [1] and overall event free survival drops significantly (to 20–30%) when metastases are present at diagnosis [2]. As has been observed in other tumors, most deaths (75%) are associated with metastasis, with the lungs being the most common distant site [3]. While the origin of osteosarcoma remains unclear, certain environmental factors such as irradiation [4] and hereditary conditions such as Li–Fraumeni syndrome, familial retinoblastoma, and Rothmund–Thomson syndrome, associated with germline mutations in TP53, RB1, and RECQL4 genes, respectively, predispose to osteosarcoma [5,6]. Moreover, several mouse models implicate TP53 alterations as a major initiating event for osteosarcoma, with RB1 inactivation reducing the latency of tumor formation in TP53 inactivated models [7–9].
A number of osteosarcoma next-generation sequencing studies have been published in recent years [10–14], focusing mainly on primary tumors. In these studies, TP53 was the most recurrently altered gene, with its inactivation appearing to derive mostly from structural variations (SVs), particularly translocations, except in a single study where point mutations were the most common alteration [14]. Somatic single nucleotide variants (SNVs) and SVs were reported for RB1, ATRX, and DLG2 in 29–53% of patients [10,13]. In one study, 80% of tumors demonstrated a copy number signature previously associated with ovarian and breast cancer with underlying pathogenic germline mutations in BRCA1 or BRCA2 [11]. Together, these studies suggest a complex mutational background and high chromosomal instability across different cohorts, highlighting the challenge in determining which component(s) of this convoluted landscape play a primary role in tumor progression and metastasis.
By performing whole genome sequencing (WGS) and whole exome sequencing (WES), we hereby describe the genomic characterization of a cohort of matched primary, local recurrence and metastatic pediatric osteosarcomas, providing further insights into the heterogeneity of osteosarcoma relapse and metastatic progression in this disease.
Materials and methods
Samples
Normal, primary, locally recurrent and metastatic samples were provided by Drs David Malkin (The Hospital for Sick Children, Toronto, Ontario, Canada), Timothy J. Triche (Children Hospital Los Angeles, Los Angeles, CA, USA), Ching C. Lau (Texas Children’s Hospital, Houston, TX, USA) and the B.C. Children’s Hospital (Vancouver, BC, Canada).
Whole-genome and whole-exome sequencing
WGS for the primary, locally recurrent, metastatic and matched normal samples was performed using the HiSeq 2000 platform (Illumina, San Diego, CA, USA). WGS samples for three of the patients (D11, D12, and D13) were processed with Illumina TruSeq kit v2 (Illumina, San Diego, CA, USA) and sequencing was performed on HiSeq 2500 instrument (Illumina, San Diego, CA, USA). WES samples were prepared with Agilent Sure-Select whole exome v4 kit (Agilent Technologies, Santa Clara, CA, USA) and the sequencing was done on a HiSeq 2500 instrument.
Read alignment and variant calling
Read sequences were aligned to the human reference genome (GRCh37) using the BWA-SW algorithm [15] to generate binary alignment/map (BAM) files. SNVs and indels were called using Strelka (version 1.0.14) [16] from BAM files that were pre-processed using the clipOverlap tool from the bamUtils package (version 1.0.13) [17]. Strelka depth filters were disabled for exome data (isSkipDepthFilters). SNVs and indels were annotated using the Ensembl Variant Effect Predictor (release 79) [18] and transcript prioritization was performed using vcf2maf (version 1.5.2) [19]. SNVs and indels called from genome and exome data were merged using the vcftools vcf-merge tool (version 0.1.12b) [20].
Germline variants
SNVs and indels were called on 13 normal samples with varscan (2.4.9) [21] with min coverage 10 and -min reads2 5. Variants were annotated with variant effect predictor ensemble release 79 [18]. Manually curated alterations predicted to be deleterious (SIFT [22]) or damaging (PolyPhen [23]), with a CADD-PHRED [24] score >10 and present in less than 1% of the population (ExAC) [25], were retained. We included cancer associated genes from OncoKB [26] and genes previously identified as recurrently mutated in other tumor cohorts [27,28].
Structural variations
SVs were jointly called using DELLY (version 0.6.7) [29] on all tumor and normal samples. This allowed for the removal of SVs or artifacts that are recurrent in normal samples. Somatic events were identified using a modified version of the somaticFilter.py script.
Copy number alterations
Copy number alterations (CNAs) were called by applying the TITAN workflow [30]. MutationSeq (version 4.3.3) [31] was used to call germline heterozygous SNVs and quantify their allele counts in the matched tumor sample. Read counts were quantified for 1000-bp windows using the readCounter tool from the HMMcopy utilities [32]. GC content and mappability were calculated for 1000 bp bins using gcCounter and map-Counter tools from the HMMcopy utilities. Read counts generated by readCounter were corrected for GC content and mappability. The TitanCNA R package (version 1.8.0) [30] was used to detect somatic CNAs using the binned read counts and allele counts for germline heterozygous positions. One to five clusters were attempted, and the optimal parameter value was selected by maximizing ‘S Dbw validity index (Both)’.
Phylogenetic trees
For construction of phylogenetic trees, only genes that were previously found altered in four previous osteosarcoma genomic studies [10,11,13,14] were included in the analysis. For CNAs, altered genes situated in the same cytoband were counted as a single alteration.
Signature analysis
We obtained the signature probabilities matrix from COSMIC (version 2) [33] and applied a decomposition algorithm from the R package deconstrucSigs (version 1.8.0) [34].
Gene expression microarrays
Two separate publicly available gene expression microarray datasets, GSE21257 and GSE42352, were retrieved from Gene Expression Omnibus [35].
Immunohistochemistry
For immunohistochemistry (IHC), KDR antibody (#2479, Cell Signaling, Danvers, MA, USA) was used at 1:300 dilution on an osteosarcoma tissue microarray (TMA) obtained by the Children’s Oncology Group, on a Ventana Discover XT platform. The staining was scored by a trained pathologist using the H-score system. Only pediatric tumors (age < 21) were included. MMR immunostaining was performed in the Vancouver General Hospital Department of Anatomical Pathology, using standardized clinical staining protocols. FFPE sections were immunostained for PMS2 (rabbit clone EP51; Dako, Santa Clara, CA, USA), MLH1 (mouse clone ES05, Dako), MSH2 (mouse clone FE11; Dako) and MSH6 (rabbit clone EP49; Dako) on the Dako Omnis automated IHC instrument, using the Dako EnVision FLEX+ detection system (Dako, Santa Clara, CA, USA).
Western blot analysis
Protein extraction and Western blotting were carried out using standard protocols [36]. Multiple antibodies were used for the detection of FLT1 (#2893, New England Biolabs, Whitby, ON, Canada) and KDR (#2479, Cell Signaling). GAPDH (D16H11, Cell Signaling) and GRB2 (610111, BD Biosciences, San Jose, CA, USA), all antibodies were used at a dilution of 1:1000.
Data visualization
R packages maftools (version 1.8.0) [37], GenVisR (version 1.12.1) [38], Mutationmapper (version 1.0.1) [39] were used to create some of the visualizations.
Ethics approval and consent to participate
This project was approved by UBC BC Cancer Research Ethics Board H10–02722. All clinical samples were analyzed in a deidentified fashion. Informed written consent was provided by patient families.
Results
Patient cohort
We analyzed primary and matched normal samples from 13 children suffering from osteosarcoma using WGS to an average of ×30 coverage and WES to an average of ×90 coverage. Matched metastatic samples for 10 of the patients (77%) and local recurrence biopsies for 3 of the patients were also sequenced (see supplementary material, Table S1).
Single nucleotide variants, indels, and structural variants
To detect SNVs and indels we applied a paired somatic variant detection workflow (Strelka; [16]) to both the WES and WGS datasets. Results from each individual analysis were pooled to create a comprehensive list of somatic variants. The most highly represented was RB1, which was mutated in four different patients (retained in all specimens except for one patient), and ASXL1, MGA, PDGFRB, PREX2, RBM10, and SETBP1, which were each altered in three different patients (see supplementary material, Table S2). The cohort showed a wide range in terms of nonsynonymous mutation burden, from a single variant up to samples exceeding 20 000 alterations. Nevertheless, the breakdown of coding variant classes was dominated by missense mutations (mean 80%) followed by nonsense mutations (mean 7%). Interestingly, one of the metastatic samples, D2M, showed a significant enrichment in frameshift insertions (52%) compared to the rest of the cohort (1% average) (binomial test P value <2.2e−16) (Figure 1A). Since frameshift mutations are typically associated with microsatellite instability (MSI), we computed the MSI status for all tumors using an MSI classifier (MSIseq) [40]. Unlike the rest of the cohort, D2M was predicted to be microsatellite unstable. Furthermore, by screening mutations acquired in the D2M metastasis sample, we found alterations in two DNA mismatch repair (MMR) pathway genes, MSH3 and MSH6, associated with MSI tumors [41,42]. These alterations were absent from the primary tumor.
Figure 1.
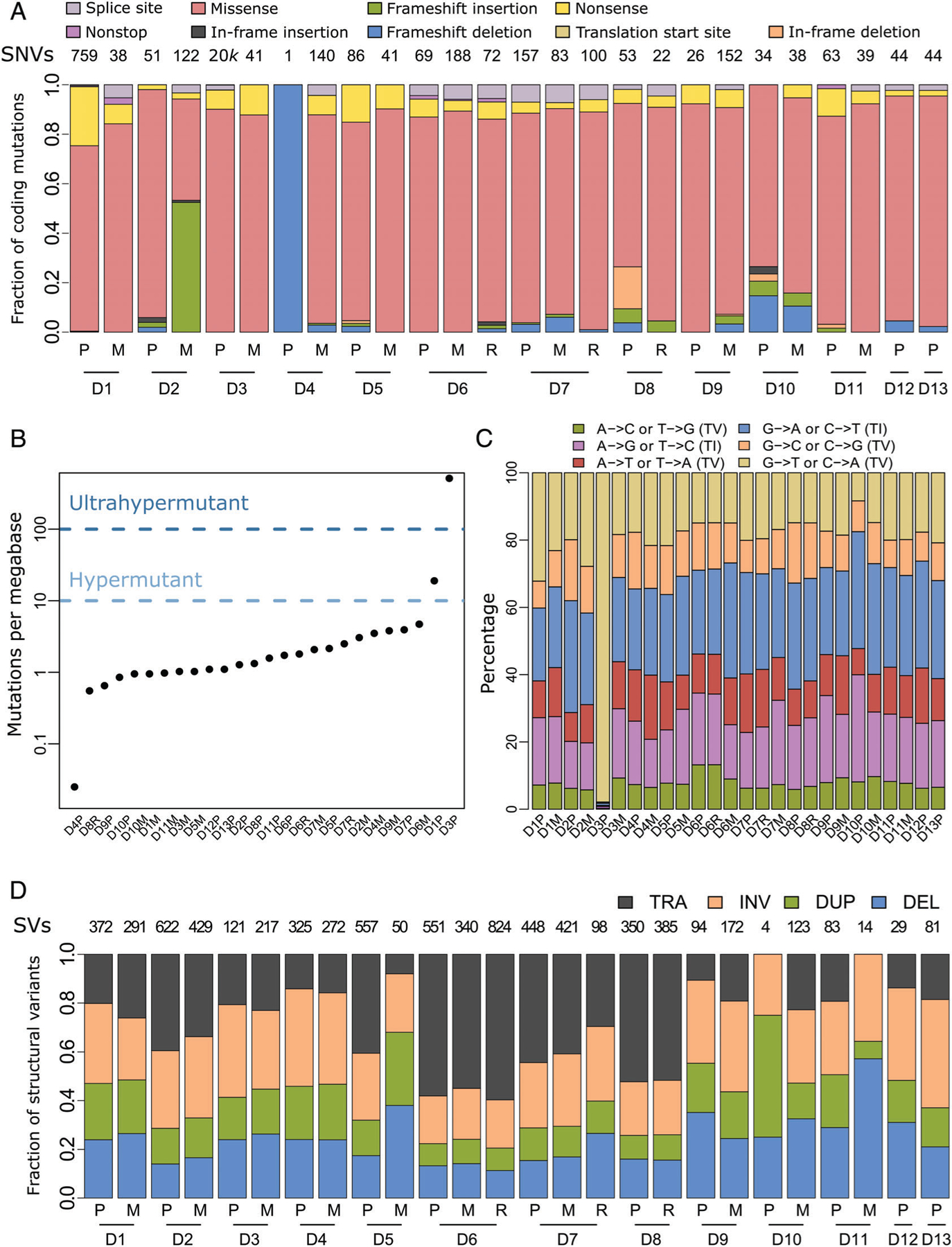
Somatic mutations in the pediatric osteosarcoma cohort. (A) Fractions of the indicated color-designated coding SNVs identified in primary (P), local recurrence (R) and metastatic (M) samples grouped by patient numbers (D1 –13). Total SNVs per patient are indicated above each column. (B) Number of somatic mutations per megabase (Mb) identified in coding regions of each of the 26 tumor samples. One tumor was classified as hypermutant (>10 Mb) and one as ultrahypermutant (>100 Mb), while the remainder were classified as nonhypermutated (<10 Mb). (C) Transition (TI)/transversion (TV) graph showing the percentage of each type of the six possible base pair substitutions. For the hypermutated tumor, D3P, C to A transversions contributed to the vast majority (>90%) of substitutions. (D) Fractions of structural variant (SV) types, translocations (TRA), inversions (INV), duplications (DUP) and deletions (DEL) across the tumor cohort. Total numbers of SVs per patient are reported along the top. Matching tumors are grouped by the indicated patient numbers.
Having observed a high number of coding mutations in multiple tumors, we computed the mutational burden for each sample. While three recent pan-cancer analyses have reported that most ultrahypermutated pediatric tumors are classified as gliomas [28,43,44], we found a case of an ultrahypermutated (>100 mutations per Mb) primary pediatric osteosarcoma (sample D3P), in addition to a hypermutated (>10 mutations per Mb) primary tumor (D1P) (Figure 1B). Other than these two cases, the overall mutation burdens of primary and metastatic samples were comparable (Wilcoxon rank sum test p = 1).
In the ultrahypermutant case (D3P), we identified multiple alterations in mismatch repair pathway genes, including missense mutations in MMR complex genes (MLH1, MSH3, MLH3, MSH2, and MSH6) and in the DNA polymerase epsilon gene (POLE), already associated with hypermutant cancers [44]. We also observed a high frequency of C to A transversions, a feature of POLE defective tumors [45] (Figure 1C). Additionally, by comparing to the distribution of noncoding mutations across the cohort, we found an enrichment of silent substitutions and a decrease in intergenic alterations in the ultrahypermutant tumor (binomial test, p < 2.2e–16; see supplementary material, Figure S1A). In the hypermutated case (D1P), we failed to detect any alterations that could account for the phenotype. Both hyper- and ultrahypermutated cases showed microsatellite stability, which is perhaps not surprising since an inverse relationship between mutation burden and microsatellite stability has recently been described [44].
IHC for four known MMR markers, MLH1, MSH2, MSH6, and PMS2, was performed on each available specimen from patients with hypermutant or MSI+ tumors (see supplementary material, Figure S2 and Table S3). While all six tumors showed at least some positive staining, we observed reduced percentages of positive cells and a significantly decreased overall staining (one tailed paired Wilcoxon rank sum test; p = 1.2e−03) across all four markers in the hypermutated/MSI+ cases compared to matching tumors.
We next applied the DELLY workflow to detect SVs in our cohort. Overall, we observed a heterogeneous distribution of SV events between patients and a fairly homogenous distribution between patient-matched tumors (Figure 1D). Multiple genes were recurrently affected by SV alterations in the cohort, including TP53 and DLG2, known to reside in a common fragile site, FRA11F [46], both genes having previously been implicated in osteosarcoma [10]. A total of 9 out of 13 patients in the cohort showed SVs in TP53. For most of these patients, SVs in TP53 were found in all available tumor samples (primary, metastasis and recurrent), with the exception of patients 10 and 11, which had TP53 SVs in only the metastasis or primary tumor, respectively. Despite intron 1 representing roughly half of the gene length, all nine SV breakpoints are restricted to intron 1, consistent with previous reports [47] (see supplementary material, Figure S1B). In contrast, SVs in DLG2, a gene spanning over 2 Mbp, were scattered across the entire locus.
Mutation signatures
To evaluate the mutational processes that may underlie osteosarcoma development, we inferred mutation signature profiles based on the fraction of mutations in each possible trinucleotide combination [48]. This was done for each tumor with the exception of D4P, which had too few mutations. By deconvoluting the mutational profiles, we detected 10 out of 30 COSMIC signatures as the most highly represented in our cohort (i.e. representing at least 15% of individual tumors) (Figure 2A). Furthermore, these 10 COSMIC signatures collectively represented more than 40% of each signature profile. Signatures 3, 5, and 8 contribute between 10 and 20% of most tumor profiles, consistent with what has been previously described in osteosarcoma [43]. Although signatures 5 and 8 have not been associated with a specific etiology, signature 3 has been linked to deficiency in DNA double-strand break-repair by homologous recombination. Furthermore, signature 3 is strongly associated with BRCA1/2 alterations and has been linked to ‘BRCAness’, described in osteosarcoma [11]. The ultrahypermutated D3P primary sample showed a unique signature contribution, with signatures 4 and 18 accounting for most of its mutation profile. Signature 4 is commonly associated with tobacco mutagens although its significance in this case remains unknown [48], while signature 18 is of unknown etiology. With the exception of D3P, the mutational profiles were similar between tumors, although we note that metastatic samples tended to have a more homogeneous distribution in signature composition compared to primary samples.
Figure 2.

Mutational signatures and germline variants. (A) Heatmap showing percentages of the mutational profiles of each tumor that can be explained by the corresponding COSMIC signature shown on the right. Only signatures contributing to ≥15% of any individual tumor profile are included. (B) Germline mutations identified in the 13 normal samples (D1N–D13N) and predicted to be potentially deleterious by Polymorphism Phenotyping (PolyPhen) or by sorting intolerant from tolerant (SIFT) and with CADD–PHRED score > 10, in 21 genes previously associated with cancer as part of the OncoKB database. Green scale intensity corresponds to CADD-PHRED score. A score greater than 20 indicates that the substitution is predicted to be more deleterious than 99% of all possible substitutions of the human genome and a score greater than 30 more deleterious than 99.9%. Frameshift mutations (insertion or deletion) are color-coded as indicated. Genes are grouped based on gene ontology categories; gene names colored in a darker shade belong to both adjacent categories.
Germline variants
A previous study found that ∼8% of all pediatric and 18% of osteosarcoma patients carry potential predisposing genetic alterations [27], highlighting the importance of assessing the germline status of cancer-related genes. To identify genes with germline alterations we performed variant calling on the 13 available normal samples. We removed common population variants and focused exclusively on variations predicted to be pathogenic or potentially pathogenic in a set of ∼1000 genes previously implicated in cancer [26]. We found multiple germline alterations in 21 of these genes, with all patients except one showing variations in at least one of these genes (Figure 2B and see supplementary material, Table S4). Four of the altered genes (TSC2 p.A583T, GATA2 p.P250A, NFE2 p.R365W, and PDGFRA p.V544A) showed further loss of heterozygosity in the corresponding tumors. Two DNA polymerases, POLD1 and POLE, which have been associated with early onset cancers [49,50], showed missense alterations in two different patients. This included mutations in residues R19 and D287, all of which have been found substituted in a recent hypermutation screen [44]. Despite this association, we did not detect hypermutated status in any of the tumors derived from these two patients. Underscoring our findings, eight of these genes were found to be altered in germline samples of mixed sarcoma cohorts [27,50]. While a previous study found TP53 germline alterations, a hallmark of Li–Fraumeni syndrome, in 9.5% of pediatric osteosarcoma cases [51], we did not detect any TP53 germline alteration in our cohort.
Copy number alterations
To assess the copy number alteration (CNAs) status of tumor specimens we employed the TITAN [30] probabilistic framework. Overall, we observed a strong correlation between CNAs across matched primary, metastatic and locally recurred tumors (Figure 3), and in particular in those CNAs previously reported in osteosarcoma [11,52]. Multiple regions showed recurrent alterations in more than 25% of both primary and metastatic samples, including amplifications at chromosomes 1p, 1q, 6p21, 8q, 14q, 17p13.1, and 21q and deletions at 3q13, 6q, chr10, chr11, 15, 16p, 17p13.2, and 19q. When we compared the recurrence of CNAs (0.9 quantile threshold), we observed similar profiles at reduced frequency in the metastatic cohort (see supplementary material, Figure S3). A comparison of the CN status of genes in primary and metastatic tumors revealed correlations ranging from moderate (0.46–0.6) to strong (0.73–0.81) (see supplementary material, Figure S4), indicating that CNAs tend to be retained in metastases. Focusing on the genome distribution of relative CN, we found a single patient (D10) with significantly higher prevalence of CNAs in the metastatic sample compared to the primary, while three patients had primary tumors with a significantly higher prevalence of CNAs relative to the matched metastasis (see supplementary material, Figure S5). Therefore, osteosarcoma shows significant heterogeneity in this feature across primary tumor and metastatic lesions, with no clear enrichment for chromosomal instability in either specimen type.
Figure 3.

Copy number alterations in pediatric osteosarcoma. Copy number profiles as computed by TITAN for each tumor in the cohort, showing gains (red) and losses (blue) as indicated in the heat map on the right. Relative genomic locations are represented on the top. Zoomed in areas highlight examples of regions with high (left and center panels) and low degrees of conservation (right panel) across the indicated primary (P)/local recurrence (R)/metastatic (M) tumor trio.
Phylogenetic reconstruction of metastatic osteosarcoma
To determine the fraction of the mutational landscape retained in patient-matched metastases and local recurrences, we performed a phylogenetic reconstruction from all available tumors for each patient. This analysis was restricted to previously reported somatic alterations [10,11,13,14] (Figure 4). The proportion between trunks (showing genes and genomic regions altered in primary or primary-relapse and retained by metastases) and primary branches varied greatly across the cohort, from 3% in the ultrahypermutated patient D3 to more than 80% in some patients. In both cases with a local recurrence, the relapsed tumor shared more similarities with the primary tumor than the metastasis. Amplifications were the most commonly retained event, with seven regions retained in six or more metastases. The genomic region affected by the most trunk mutations was the short arm of chromosome 17, with both deletions, including TP53, and amplifications, including COPS3 [53]. Amplifications on chr6p21, including RUNX2 and CCND3 [54], chr8q24, including MYC [55] and chr4q12, including KDR [14], were also frequently retained by the majority of metastases. Only three genes were recurrently altered by SNVs or SVs in matching primary-metastatic pairs; TP53 and RB1, recognized as potential drivers of osteosarcoma [10,11,13], and SETBP1, which has not been previously associated with osteosarcoma. SETBP1 was also altered in the germline of two additional patients (Figure 2B). When we focused on the private mutations of individual tumors, we failed to identify genes or genomic regions preferentially altered in the primary or metastatic tumors.
Figure 4.
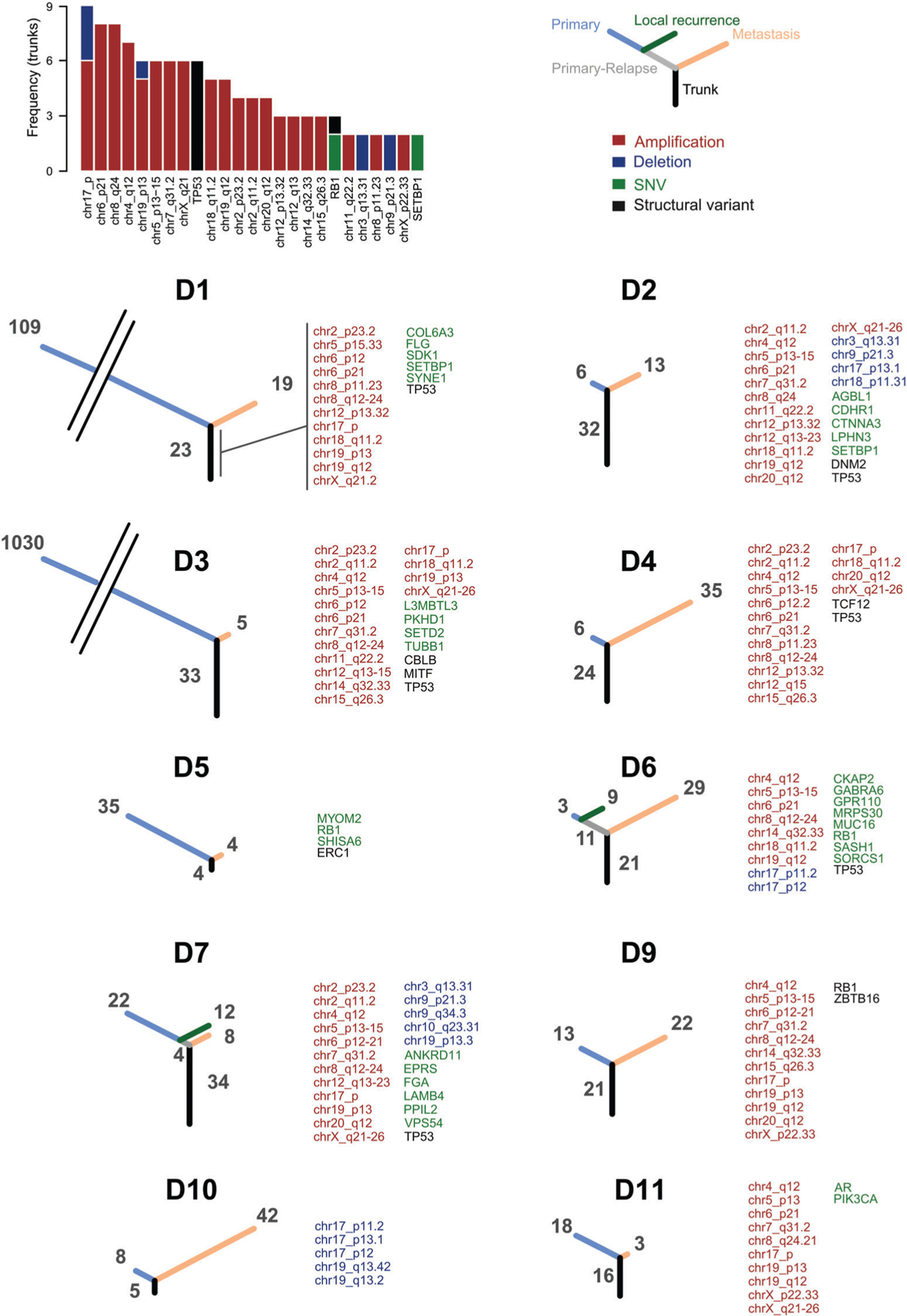
Phylogenetic trees of primary, local relapses and metastatic tumors. The barplot shows the frequency of genes and chromosomal locations recurrently altered in the primary tumors and retained by matching metastasis. The phylogenetic trees show the number of alterations common to primary and metastatic tumors (black), common to primary and local relapse (gray), private to the primary (blue), to the local relapse (green) or to the metastasis (orange). Tree branches are proportional to the number of alterations. Trunk alterations are reported for each matching pair. Adjacent cytoband names are merged into ranges for improved readability.
By examining the overlapping regions between breakpoints identified from DELLY in the primary and metastatic groups, we observed variable degrees of retention (see supplementary material, Figure S6). Some metastases share little (<10%) or no overlap in terms of SVs with the paired primary tumor, with the overlap varying between 0 and 30% of the breakpoints.
Vascular endothelial growth factor receptor alterations in osteosarcoma metastases
While not uniquely mutated in metastatic samples, we found several receptor tyrosine kinases preferentially altered in the metastases. For example, one alteration unique to the metastatic sample was a missense mutation (D964H) in the kinase insert domain receptor (KDR), a VEGF receptor. Additional missense mutations have been found in KDR (S965R and E990G) in separate osteosarcoma cohorts [13,56]. We also found a metastasis-specific mutation (E939A) in the tyrosine kinase domain of another VEGF receptor, Fms Related Tyrosine Kinase 1 (FLT1) in another patient. We detected KDR protein expression in three out of seven osteosarcoma cell lines and variable levels of FLT1 in all tested cell lines (see supplementary material, Figure S7). However, after expressing proteins containing these missense alterations in U2OS cells, we did not detect enhanced activation (phosphorylation) compared to wild type proteins (data not shown). The consequence of these mutations on kinase activity thus remains inconclusive.
KDR is recurrently amplified, highly expressed and associated with poor outcome in osteosarcoma
We next sought to determine whether genes mutated in metastatic tumors were also affected by CNAs across the cohort. We observed that KDR is commonly amplified in chr4q12 (Figure 5A) and that amplifications in this region were recurrently retained in 7 out of 10 metastases (Figure 4). While the size and the position of the amplified regions vary across the cohort, KDR shows CN gains (relative CN status >0.9 quantile) in 50% of the cohort. By comparing gene expression levels between primary osteosarcomas, mesenchymal stem cells and osteoblasts from an independent publicly available dataset (GSE42352) [57], we found KDR mRNA levels were significantly higher in the tumors (see supplementary material, Figure S8A), suggesting that somatic alterations such as amplifications lead to higher KDR expression. Furthermore, when we stratified patients in a separate osteosarcoma dataset (GSE21257) [58], we found KDR mRNA was significantly associated with shorter metastasis-free survival (see supplementary material, Figure S8B). To assess KDR protein levels in osteosarcoma, we performed IHC on a TMA representing 30 pediatric osteosarcoma cases. As controls, six normal cores including muscle, placenta, kidney, testis, lymph node and liver were also included. As expected, vascular endothelial cells in normal organs were strongly immunoreactive for KDR. In addition, osteosarcoma cells exhibited various degrees of positivity. Kaplan–Meier survival analysis revealed that high KDR immunoreactivity was predictive of unfavorable outcome in this cohort (Figure 5B–D), suggesting a potential role for this protein in osteosarcoma progression. Underscoring our findings, a KDR inhibitor, Apatinib, has been shown to repress the growth of osteosarcoma in vivo [59].
Figure 5.
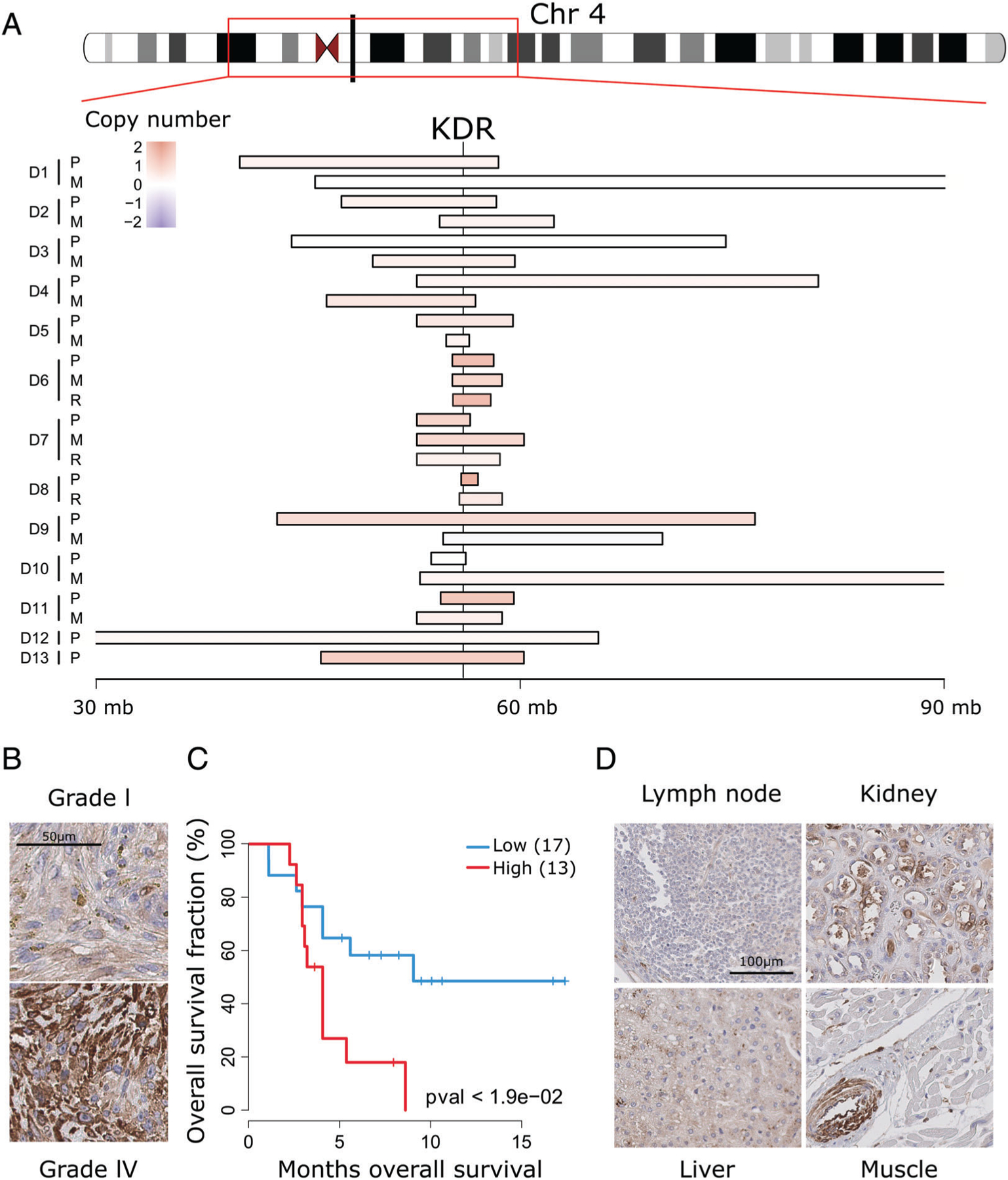
KDR is recurrently amplified in osteosarcoma and associates with poor outcome. (A) Relative copy number status of different chromosome four segments, including the KDR genomic locus, showing recurrent copy number gains (red) across the tumor cohort. (B) Representative examples of KDR IHC of Grade I and IV osteosarcomas. (C) Kaplan–Meier curves of patients with high (H-score > 15) compared to low (H-score ≤ 15) KDR (p < 0.05, log rank test). (D) KDR IHC of the indicated normal organ tissue sections. Only vascular endothelial cells show appreciable immunoreactivity, as shown in the inserts.
Discussion
While multiple studies in recent years have identified high chromosomal instability with numerous SVs as the most characteristic hallmark of primary osteosarcoma, little is known regarding the mutational landscape of metastatic tumors. In this study, we used high-throughput sequencing to shed light on osteosarcoma evolution and metastatic progression. While most cases in our cohort showed low overall mutational burdens, two primary tumors fell into the hyper- and ultrahypermutated tumor category, the latter of which is to our knowledge the first reported example of this process in osteosarcoma. Most SNVs in the two primary tumors were not retained in the matched metastatic clones. This may be due to negative selection during treatment, or an uncharacterized interaction with the metastatic microenvironment. In fact, we failed to identify recurrent mutational events among the metastatic tumors, similar to another study of primary and matched metastatic tumor pairs in osteosarcoma [60]. While several alterations found in metastatic tumors might lead to common pathway activation or disruption, validation with larger cohorts will be necessary to resolve this question.
Heterogeneous losses for multiple MMR markers were detected in the hypermutant/MSI+ tumors and while the interpretation of variable staining for these proteins is still the subject of debate [61], it reflects the heterogeneity observed at the genomic level. Notably, while no genomic alterations of the MMR complex were detected in D1P, loss of MLH1 protein expression is often the result of promoter hypermethylation [62], and PMS2 stability appears to depend directly on the presence of MLH1 [63].
At the germline level, patients showed multiple potentially pathogenic alterations in a set of 21 genes previously associated with malignancy. Germline alterations in 8 of the 21 genes have previously been reported in osteosarcoma and mixed sarcoma cohorts [27,50]. A total of 7 out of 13 patients showed multiple alterations in different members of the DNA damage response pathway, suggesting that loss of DNA repair activity might play a role in osteosarcoma.
Phylogenetic analysis showed a higher overall conservation of CNAs between primary and metastatic lesions from the same patients, compared to SNVs, in line with another matched primary and metastatic osteosarcoma cohort [60]. In the metastatic samples, several recurrent mutations affected genes previously identified as implicated in osteosarcoma formation: TP53, RUNX2, COPS3, MYC, CCND3, KDR, and RB1 [10,11,13,14, 53–55]. This finding underscores the functional role for these alterations in primary tumors as well as in metastatic progression. The greater degree of retention of CNAs compared to SNVs suggests that chromosomal aberrations likely arise earlier in tumor evolution. Due to the central role observed for SVs and CNAs, future studies will need to rely on higher-coverage WGS to determine which of these events play a driver role in osteosarcoma progression. Metastasis specific alterations may have been missed in sequencing of the primary tumors due to tumor heterogeneity or limited coverage, or they may have arisen later, potentially induced by treatment. Sequencing spatially and temporally separated tumor samples at higher coverage will be essential to better understand tumor evolution and clonality.
Two metastatic samples in the cohort showed unique mutations in two different VEGF receptors, KDR and FLT1. When we expressed full-length KDR and FLT1 receptors with the above SNVs in U2OS cells, we did not observe increased kinase activity. These amino acid substitutions may therefore bestow other functions, such as altering the capacity of RTKs to bind critical interaction partners involved in downstream signaling [64–66], such as Src [67].
Recurrent gains of KDR were observed in 7 (54%) of the 13 primary tumors and were retained in patient-matched metastases, suggesting a central role of this kinase in the proliferation of both primary and metastatic tumors. Moreover, IHC revealed a strong association between high KDR protein levels and poor outcome in osteosarcoma, further highlighting the potential importance of this pathway in metastatic osteosarcoma.
Supplementary Material
Table S3. Immunohistochemistry for MMR proteins
Table S2. Cancer genes recurrently altered in the cohort
Table S4. Germline mutations in the osteosarcoma cohort identified in 21 genes previously associated with cancer
Table S1. Data on patient samples analyzed in the study
Figure S1. Somatic noncoding mutations and TP53 SV hotspot
Figure S2. Immunohistochemistry for MMR proteins
Figure S3. Frequency of copy number gains and losses in the primary and metastatic cohorts
Figure S4. Gene copy number correlation between matched primary and metastatic tumors differs across patients
Figure S5. Distribution of copy number alterations differs in primary versus metastatic tumors of individual patients
Figure S6. Limited overlap of structural variant (SV) breakpoints
Figure S7. KDR and FLT1 missense mutations in tumors and protein expression levels in osteosarcoma cell lines
Figure S8. Osteosarcoma shows high expression of KDR which associates with reduced metastasis free survival
Acknowledgements
This project was supported by a grant to PHS from the Canadian Pediatric Cancer Genome Consortium (CPCGC), with funds from Genome Canada, Genome British Columbia, Genome Quebec, C17 Council for Childhood Cancer and Blood Disorders, and the Canadian Institutes of Health Research ‘Advancing Technology Innovation Through Discovery’ program. This research was also supported by the Stand Up To Cancer – St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113). AMEAEN is funded through a Michael Smith Foundation for Health Research (MSFHR) trainee award (Grant ID # 17159).
Footnotes
Availability of data and materials
WGS and WES data have been deposited at the European Genome-Phenome Archive (EGA) (EGAS00001003342).
No conflicts of interest were declared.
References
- 1.Isakoff MS, Bielack SS, Meltzer P, et al. Osteosarcoma: current treatment and a collaborative pathway to success. J Clin Oncol 2015; 33: 3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bacci G, Rocca M, Salone M, et al. High grade osteosarcoma of the extremities with lung metastases at presentation: treatment with neoadjuvant chemotherapy and simultaneous resection of primary and metastatic lesions. J Surg Oncol 2008; 98: 415–420. [DOI] [PubMed] [Google Scholar]
- 3.Huang YM, Hou CH, Hou SM, et al. The metastasectomy and timing of pulmonary metastases on the outcome of osteosarcoma patients. Clin Med Oncol 2009; 3: 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei-wei L, Qiu-liang W, Guo-hao W, et al. Clinicopathologic features, treatment, and prognosis of postirradiation osteosarcoma in patients with nasopharyngeal cancer. Laryngoscope 2005; 115: 1574–1579. [DOI] [PubMed] [Google Scholar]
- 5.Mirabello L, Troisi RJ, Savage SA. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int J Cancer 2009; 125: 229–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang LL, Gannavarapu A, Kozinetz CA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund– Thomson syndrome. J Natl Cancer Inst 2003; 95: 669–674. [DOI] [PubMed] [Google Scholar]
- 7.Berman SD, Calo E, Landman AS, et al. Metastatic osteosarcoma induced by inactivation of Rb and p53 in the osteoblast lineage. Proc Natl Acad Sci U S A 2008; 105: 11851–11856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin PP, Pandey MK, Jin F, et al. Targeted mutation of p53 and Rb in mesenchymal cells of the limb bud produces sarcomas in mice. Carcinogenesis 2009; 30: 1789–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walkley CR, Qudsi R, Sankaran VG, et al. Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 2008; 22: 1662–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen X, Bahrami A, Pappo A, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 2014; 7: 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kovac M, Blattmann C, Ribi S, et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat Commun 2015; 6: 8940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiappetta C, Mancini M, Lessi F, et al. Whole-exome analysis in osteosarcoma to identify a personalized therapy. Oncotarget 2017; 8: 80416–80428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry JA, Kiezun A, Tonzi P, et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci U S A 2014; 111: E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Behjati S, Tarpey PS, Haase K, et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat Commun 2017; 8: 15936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010; 26: 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saunders CT, Wong WSW, Swamy S, et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012; 28: 1811–1817. [DOI] [PubMed] [Google Scholar]
- 17. Available from: https://genome.sph.umich.edu/wiki/BamUtil (version 1.0.13).
- 18.McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol 2016; 17: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Available from: https://github.com/mskcc/vcf2maf (version 1.5.2).
- 20.Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics 2011; 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012; 22: 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vaser R, Adusumalli S, Leng SN, et al. SIFT missense predictions for genomes. Nat Protoc 2016; 11: 1–9. [DOI] [PubMed] [Google Scholar]
- 23.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res 2002; 30: 3894–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019; 47: D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536: 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol 2017; 1: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 2015; 373: 2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature 2018; 555: 321–327. [DOI] [PubMed] [Google Scholar]
- 29.Rausch T, Zichner T, Schlattl A, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012; 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ha G, Roth A, Khattra J, et al. TITAN: inference of copy number architectures in clonal cell populations from tumor whole-genome sequence data. Genome Res 2014; 24: 1881–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ding J, Bashashati A, Roth A, et al. Feature-based classifiers for somatic mutation detection in tumour-normal paired sequencing data. Bioinformatics 2012; 28: 167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Available from: https://github.com/shahcompbio/hmmcopy_utils [Accessed 11 June 2015].
- 33.Alexandrov LB, Jones PH, Wedge DC, et al. Clock-like mutational processes in human somatic cells. Nat Genet 2015; 47: 1402–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosenthal R, McGranahan N, Herrero J, et al. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol 2016; 17: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.El-Naggar AM, Veinotte CJ, Cheng H, et al. Translational activation of HIF1alpha by YB-1 promotes sarcoma metastasis. Cancer Cell 2015; 27: 682–697. [DOI] [PubMed] [Google Scholar]
- 37.Mayakonda AK, Koeffler HP. Maftools: efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv 2016. 10.1101/052662. [DOI] [Google Scholar]
- 38.Skidmore ZL, Wagner AH, Lesurf R, et al. GenVisR: genomic visualizations in R. Bioinformatics 2016; 32: 3012–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vohra S, Biggin PC. Mutationmapper: a tool to aid the mapping of protein mutation data. PLoS One 2013; 8: e71711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang MN, McPherson JR, Cutcutache I, et al. MSIseq: software for assessing microsatellite instability from catalogs of somatic mutations. Sci Rep 2015; 5: 13321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duraturo F, Liccardo R, Cavallo A, et al. Association of low-risk MSH3 and MSH2 variant alleles with Lynch syndrome: probability of synergistic effects. IntJ Cancer 2011; 129: 1643–1650. [DOI] [PubMed] [Google Scholar]
- 42.Cortes-Ciriano I, Lee S, Park W-Y, et al. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 2017; 8: 15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 2018; 555: 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017; 171: 1042–1056 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Church DN, Briggs SEW, Palles C, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet 2013; 22: 2820–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reshmi SC, Huang X, Schoppy DW, et al. Relationship between FRA11F and 11q13 gene amplification in oral cancer. Genes Chromosomes Cancer 2007; 46: 143–154. [DOI] [PubMed] [Google Scholar]
- 47.Ribi S, Baumhoer D, Lee K, et al. TP53 intron 1 hotspot rearrangements are specific to sporadic osteosarcoma and can cause Li–Fraumeni syndrome. Oncotarget 2015; 6: 7727–7740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature 2013; 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palles C, Cazier J-B, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan SH, Lim WK, Ishak NDB, et al. Germline mutations in cancer predisposition genes are frequent in sporadic sarcomas. Sci Rep 2017; 7: 10660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirabello L, Yeager M, Mai PL, et al. Germline TP53 variants and susceptibility to osteosarcoma. J Natl Cancer Inst 2015; 107: djv101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuijjer ML, Rydbeck H, Kresse SH, et al. Identification of osteosarcoma driver genes by integrative analysis of copy number and gene expression data. Genes Chromosomes Cancer 2012; 51: 696–706. [DOI] [PubMed] [Google Scholar]
- 53.Henriksen J, Aagesen TH, Maelandsmo GM, et al. Amplification and overexpression of COPS3 in osteosarcomas potentially target TP53 for proteasome-mediated degradation. Oncogene 2003; 22: 5358–5361. [DOI] [PubMed] [Google Scholar]
- 54.Lu XY, Lu Y, Zhao YJ, et al. Cell cycle regulator gene CDC5L, a potential target for 6p12-p21 amplicon in osteosarcoma. Mol Cancer Res 2008; 6: 937–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ladanyi M, Park CK, Lewis R, et al. Sporadic amplification of the MYC gene in human osteosarcomas. Diagn Mol Pathol 1993; 2: 163–167. [PubMed] [Google Scholar]
- 56.Joseph CG, Hwang H, Jiao Y, et al. Exomic analysis of myxoid liposarcomas, synovial sarcomas, and osteosarcomas. Genes Chromosomes Cancer 2014; 53: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuijjer ML, Peterse EF, van den Akker BE, et al. IR/IGF1R signaling as potential target for treatment of high-grade osteosarcoma. BMC Cancer 2013; 13: 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Buddingh EP, Kuijjer ML, Duim RAJ, et al. Tumor-infiltrating macrophages are associated with metastasis suppression in high-grade osteosarcoma: a rationale for treatment with macrophage activating agents. Clin Cancer Res 2011; 17: 2110–2119. [DOI] [PubMed] [Google Scholar]
- 59.Liu K, Ren T, Huang Y, et al. Apatinib promotes autophagy and apoptosis through VEGFR2/STAT3/BCL-2 signaling in osteosarcoma. Cell Death Dis 2017; 8: e3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu H, Zhu X, Bao H, et al. Genetic and clonal dissection of osteosarcoma progression and lung metastasis. Int J Cancer 2018; 143: 1134–1142. [DOI] [PubMed] [Google Scholar]
- 61.McCarthy AJ, Capo-Chichi JM, Spence T, et al. Heterogenous loss of mismatch repair (MMR) protein expression: a challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J Pathol Clin Res 2019; 5: 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 1998; 95: 8698–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma AH, Xia L, Littman SJ, et al. Somatic mutation of hPMS2 as a possible cause of sporadic human colon cancer with microsatellite instability. Oncogene 2000; 19: 2249–2256. [DOI] [PubMed] [Google Scholar]
- 64.Sun Z, Li X, Massena S, et al. VEGFR2 induces c-Src signaling and vascular permeability in vivo via the adaptor protein TSAd. J Exp Med 2012; 209: 1363–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Warner AJ, Lopez-Dee J, Knight EL, et al. The Shc-related adaptor protein, Sck, forms a complex with the vascular-endothelial-growth-factor receptor KDR in transfected cells. Biochem J 2000; 347: 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Igarashi K, Shigeta K, Isohara T, et al. Sck interacts with KDR and Flt-1 via its SH2 domain. Biochem Biophys Res Commun 1998; 251: 77–82. [DOI] [PubMed] [Google Scholar]
- 67.Mezquita B, Mezquita J, Pau M, et al. A novel intracellular isoform of VEGFR-1 activates Src and promotes cell invasion in MDA-MB-231 breast cancer cells. J Cell Biochem 2010; 110: 732–742. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3. Immunohistochemistry for MMR proteins
Table S2. Cancer genes recurrently altered in the cohort
Table S4. Germline mutations in the osteosarcoma cohort identified in 21 genes previously associated with cancer
Table S1. Data on patient samples analyzed in the study
Figure S1. Somatic noncoding mutations and TP53 SV hotspot
Figure S2. Immunohistochemistry for MMR proteins
Figure S3. Frequency of copy number gains and losses in the primary and metastatic cohorts
Figure S4. Gene copy number correlation between matched primary and metastatic tumors differs across patients
Figure S5. Distribution of copy number alterations differs in primary versus metastatic tumors of individual patients
Figure S6. Limited overlap of structural variant (SV) breakpoints
Figure S7. KDR and FLT1 missense mutations in tumors and protein expression levels in osteosarcoma cell lines
Figure S8. Osteosarcoma shows high expression of KDR which associates with reduced metastasis free survival