

Abstract
Sepsis-associated encephalopathy (SAE) is characterized by acute and diffuse brain dysfunction and correlates with long-term cognitive impairments with no targeted therapy. We used a mouse model of sepsis-related cognitive impairment to examine the role of lncRNA nuclear enriched abundant transcript 1 (Neat1) in SAE. We observed that Neat1 expression was increased in neuronal cells from septic mice and that it directly interacts with hemoglobin subunit beta (Hbb), preventing its degradation. The Neat1/Hbb axis suppressed postsynaptic density protein 95 (PSD-95) levels and decreased dendritic spine density. Neat1 knockout mice exhibited decreased Hbb levels, which resulted in increased PSD-95 levels, increased neuronal dendritic spine density, and decreased anxiety and memory impairment. Neat1 silencing via the antisense oligonucleotide GapmeR ameliorated anxiety-like behavior and cognitive impairment post-sepsis. In conclusion, we uncovered a previously unknown mechanism of the Neat1/Hbb axis in regulating neuronal dysfunction, which may lead to a novel treatment strategy for SAE.
Keywords: Neat1, Hbb, PSD-95, sepsis-associated encephalopathy, cognitive impairment, synaptic dysfunction, antisense oligonucleotide
Graphical abstract
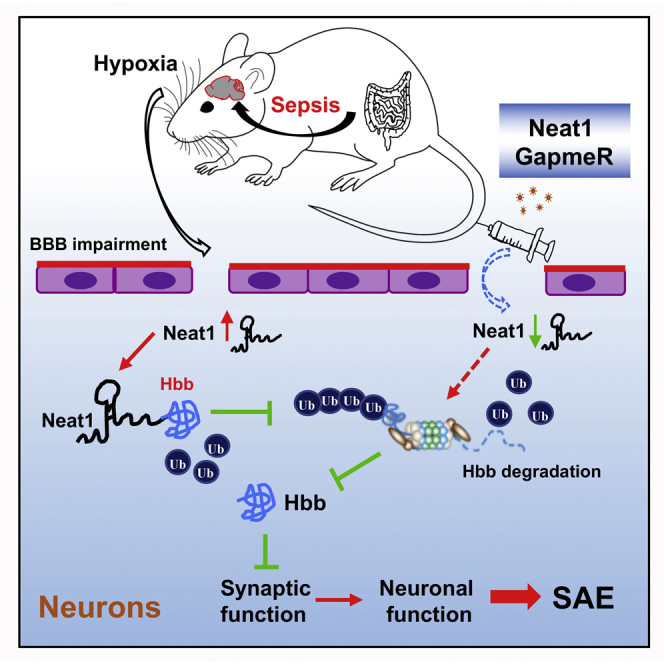
Mice survived sepsis-exhibited cognitive impairment, neuronal dysfunction, and increased expression of lncRNA Neat1. Neat1 associates with hemoglobin subunit beta (Hbb) to regulate PSD-95 expression, dendritic spine density, and cognitive impairment post-sepsis. Neat1 antisense oligonucleotide has the potential to become a novel treatment strategy for sepsis-induced cognitive impairment.
Introduction
Although sepsis incidence has steadily increased in recent years, sepsis morality rates have declined, leading to an expanding population of sepsis survivors.1, 2, 3 This population frequently suffers from sepsis-associated encephalopathy (SAE), which has been associated with long-term functional sequelae including cognitive impairment, anxiety, depression, and post-traumatic stress disorder (PTSD).4,5 This so-called “post-ICU syndrome” contributes to the excess mortality that occurs within five years after sepsis.6 The pathogenesis of sepsis-induced cognitive impairment is poorly understood, although growing evidence indicates that neuroinflammation and related excitotoxicity may play a role.7,8
Neuron synapse loss correlates with the cognitive deficit, and synaptic function is regulated by altering the expression level of synaptic scaffold proteins such as post-synaptic density protein 95 (PSD-95), a member of the membrane-associated guanylate kinase (MAGUK) class of proteins at synapses.9,10 PSD-95 is an essential component involved in synaptic plasticity and dendritic spine morphogenesis during neurodevelopment,11 and several genomic studies link PSD-95 dysfunctions to neuropsychiatric disorders.12, 13, 14 PSD-95 protein expression levels are downregulated in the brain after sepsis15,16 and may represent an underexplored contributor to SAE pathogenesis. Evolving evidence has also demonstrated the potential importance of hemoglobin (Hb) in sepsis-related organ failure via cellular dissociation and resultant inflammatory modulation, nitric oxide scavenging, and free radical injury.17, 18, 19 Hemoglobin expression was previously felt to be specific to erythrocytes; however, it has more recently been observed in several cells, including neurons and glial cells,20 raising the question of its potential role in SAE through local injury. However, little is known about the potential interaction of Hb and PSD-95 and their role in neuronal injury in sepsis.
Long non-coding RNAs (lncRNAs) are a diverse subset of transcripts longer than 200 nucleotides that do not encode for proteins.21 Increasing evidence suggests that lncRNAs regulate almost every cellular function through various molecular mechanisms, including competing with endogenous mRNAs over microRNA binding, scaffolding of RNA-protein structures, and epigenetic regulation.22,23 Neat1, nuclear-enriched abundant transcript 1, belongs to the group of lncRNAs exhibiting highly abundant expression levels in the brain.24 Two isoforms of Neat1 that share the same promoter are recognized but differ in 3′ ends and length (3.7 and 23 kb in humans, and 3.2 and 20 kb in mice).25 Neat1 functions as a core scaffold of nuclear body paraspeckles that regulates transcription,26 which has also been proposed to control stress responses,27 activation of innate immune responses,28 and cellular differentiation29 by sequestering RNA- and DNA-binding proteins,27 thus altering the epigenetic landscape of target gene promoters in favor of transcription.30 Altered Neat1 expression has been reported in many major neurodegenerative and psychiatric diseases, including frontotemporal dementia (FTD), Alzheimer’s, Huntington’s, and Parkinson’s diseases, amyotrophic lateral sclerosis (ALS), epilepsy, traumatic brain injury, and schizophrenia.31, 32, 33 Recently, several studies reported that Neat1 participated in sepsis-induced acute kidney injury,34 myocardial injury,35 and liver injury.36 However, the role of Neat1 in SAE remains to be investigated.
In the present study, we determined neuronal Neat1 expression in sepsis and its impact on neuronal synapse formation through regulation of PSD-95-hemoglobin subunit beta (Hbb) interaction. Utilizing a Neat1 knockout mouse, we demonstrated the critical role of this lncRNA in neuronal dysfunction in sepsis and established a novel therapeutic proof of concept using a Neat1-silencing GapmeR. We determined the impact of Neat1 on post-sepsis cognition, memory, and anxiety-like behavior using behavioral assessments in a murine sepsis model.
Results
Septic mice exhibit anxiety, cognitive impairment and decreased dendritic spine density
To establish a model of SAE, we induced sepsis in C57BL/6 mice via cecal ligation and puncture (CLP). We used a moderate-severity CLP model (one puncture with 22-gauge needle), which results in a mortality rate of 47%, and the surviving mice were subjected to an open-field test for anxiety-like behavior and contextual-fear-conditioning (CFC) test for hippocampus-dependent memory impairment (Figure S1A). We found that septic mice exhibited anxiety-like behavior, as evidenced by visiting the center less frequently, spending less time in the center, and taking more time to first enter the center compared with the sham group (p < 0.05; Figures 1A, 1B, and S1B). In addition, using a CFC test, we observed that mice had no substantial difference in freezing behavior during the training phase. However, when returned to the testing 24 h later, septic mice exhibited significantly (p < 0.05) decreased freezing time compared with sham mice (Figures 1C and S1C). These data demonstrated that mice who survive sepsis exhibit anxiety-like behavior and memory impairment.
Figure 1.

Mice surviving CLP exhibit anxiety-like behavior, memory impairment, and a decrease of dendritic spine density
(A and B) Sham (n = 23) and CLP (n = 27) mice were subjected to open-field test. (A) Representative tracks and heatmap are shown. (B) The frequency, time in the center zone, and the first time for the mice to go to the center zone were analyzed (∗p < 0.05). (C) Sham (n = 12) and CLP (n = 16) mice were subjected to a contextual fear-conditioning test, and freezing behavior was analyzed ( ∗p < 0.05). (D) Differential expression of IEGs in neuronal cells between Sham and CLP mice were analyzed at 24 h after surgery (∗p < 0.05, n = 3 mice/group). (E) The PSD-95 protein levels were determined in the hippocampus of mice at 24 h after Sham or CLP operation (∗p < 0.05, n = 3 mice/group). (F) Neuron dendrite from Sham and CLP mice were stained with Golgi stain (left). Box plots of dendritic spine number in Sham and CLP groups were analyzed at 8 weeks post-surgery (right). Scale bar, 20 μm. ∗p < 0.05, n = 3–4 mice/group.
To further determine whether these behavioral changes were associated with neuronal dysfunction, we determined the expression of immediate-early genes (IEGs), which regulate anxiety, memory, and neuronal dendritic spine density. Our data showed that the expression of IEGs such as c-fos, Egr1, Arc, Bdnf, Nrn1, and Homer1 were significantly decreased in neuronal cells at 24 h after CLP compared with the sham group (Figure 1D). Additionally, we found that the protein levels of PSD-95 (post-synaptic marker) were decreased after CLP compared with the sham group (Figure 1E). These data suggest that neuronal dysfunction occurs as early as 24 h post-sepsis. We also stained neuronal synapses at 8 weeks after CLP sepsis. Dendritic spine density in the hippocampus was significantly (p < 0.05) lower in CLP mice compared with sham mice (Figure 1F). These data demonstrated that mice surviving sepsis exhibited anxiety-like behavior and memory impairment as well as neuronal dysfunction.
Neat1 expression is increased during sepsis
Since previous studies demonstrated that lncRNAs play important roles in sepsis and neurodegenerative disease,32,37,38 we determined sepsis-associated lncRNAs (Neat1, HOTAIR, Malat1, and PVT1) in mouse brain tissue at 1 day after CLP. The results showed that only Neat1 levels were increased 4-fold (Figure S2A); the other lncRNAs were not singificantly changed (Figures S2B and S2C). The expression levels of PVT1 were not detectable in brain tissue. Therefore, the Neat1 levels were detected in mouse brain tissues and neurons at 1, 7, and 14 days after CLP. Neat1 levels were significantly (p < 0.05) increased in the sepsis group at 1 and 7 days after CLP compared with the sham group and returned to baseline levels at 14 days (Figures 2A and 2B). We also performed RNA fluorescent in situ hybridization (RNA-FISH) with a probe specific to the Neat1 variant in the hippocampus region from sham and septic mice at 24 h after CLP. Neurons were stained with NeuN (neuron marker), and Neat1-positive cells were analyzed in all neurons (Figure 2C). The mean number of Neat1-positive cells was significantly (p < 0.05) increased in the septic mice compared with the sham mice, although nuclear localization of Neat1 was not affected by sepsis (Figure 2D). Since hypoxia and inflammation may occur in brain tissue during sepsis,39,40 we further determined Neat1 expression levels in Neuro-2a (N2a) cells in the condition of hypoxia, or treated with IL-1β, TNF-α, or LPS for 16 h. We found that N2a cells exposed to hypoxia (1% O2 levels) exhibited upregulation of Neat1 compared with cells exposed to normoxia (21% O2 levels) (Figure 2E), whereas IL-1β, TNF-α, or LPS did not upregulate Neat1 levels (Figure S3). RNA-FISH assays further revealed that Neat1 was rarely detectable in the control group (Figure 2F, top), but was significantly (p < 0.01) detectable in the nuclei of N2a cells exposed to hypoxia (Figures 2F, bottom, and 2G). In addition, hypoxia can induce Neat1 through hypoxia-inducible factor (HIF)-2α-mediated transcriptional activation.41,42 We treated N2a cells with siRNA against HIF-2α and incubated the cells in hypoxic conditions for 16 h (Figures S4A and S4B). Treatment with HIF-2α siRNA attenuated hypoxia-induced increases of Neat1 (Figure S4C). These data demonstrated that Neat1 levels were upregulated in sepsis through HIF-2α-mediated signaling pathway.
Figure 2.

Neat1 expression is increased during sepsis
(A and B) The Neat1 levels in brain tissues (A) and neurons (B) at different intervals after CLP were assessed ( ∗p < 0.05, ∗ ∗p < 0.01, n = 3–6 mice/time point/group). (C) Neat1 levels in the hippocampus of Sham and CLP mice were determined with RNA-FISH to visualize Neat1 (red), neuron (green), and nuclei (blue) at 24 h after CLP. Scale bar, 25 μm. (D) Neat1-positive neuronal cells in hippocampus were quantitated ( ∗p < 0.05, n = 4 mice/group). (E) N2a cells were incubated in normoxic or hypoxic conditions (1% O2) for 16 h; Neat1 mRNA levels were detected by qRT-PCR (∗ ∗p < 0.01, n = 4). (F and G) RNA-FISH assays (F) and quantitative data (G) for Neat1-positive N2a cells incubated in normoxic or hypoxic conditions (1% O2) for 16 h. Arrows indicate Neat1. Scale bar, 8 μm. ∗ ∗p < 0.01, n = 4.
Neat1 directly interacts with hemoglobin subunit beta
To determine how Neat1 regulates neuronal cell function, we first wanted to identify proteins binding to Neat1 by use of unbiased methods. Brain neuronal cells were obtained from mice at 24 h after sham or CLP. We performed RNA-protein pull-down assays in lysed neuronal cells followed by LC-MS/MS analysis to identify proteins that bind to Neat1 in neurons. We identified several paraspeckle proteins associated with Neat1. However, those proteins were not significantly altered after sepsis. The proteins that bind to Neat1 and their expression levels were significantly altered (≧2-fold) in the CLP group compared with the sham group, as shown in Figure 3A. Among these identified proteins, we found that Hbb not only bound to Neat1 but also increased most (6.7-fold, p < 0.05) in the CLP group compared with the sham group. The details of proteins that bound to Neat1 and their expression levels that were altered in sepsis are listed in Table S1. We further validated this finding by repeating an RNA protein pull-down assay in neuronal cell lysate by using Neat1 sense and antisense RNA probes and performing Western blot against Hbb on the isolated protein, using c-Fos as a negative control. The sense Neat1 probes clearly pulled down Hbb protein, and the antisense probes pulled down a small amount of Hbb, whereas both of them did not pull down the negative control c-Fos (Figure 3B). These data confirmed the interaction of Neat1 with Hbb. To further confirm the association between Neat1 and Hbb, we also used an RNA immunoprecipitation (RIP) assay to perform a protein-RNA pull-down assay in lysed N2a cells by using Malat1 as a negative control. Cell lysates were precipitated with Hbb antibody coupled to protein G beads, and pulled-down RNA was amplified with Neat1 primers (Figure 3C) but not Malat1 primers (Figure S5A), using RT-qPCR. The amplified products were run on an agarose gel. A 150-bp Neat1 PCR product but not Malat1 was observed in the Hbb pull-down group (Figures 3D and S5B). To further confirm the association of Neat1 and Hbb in neuronal cells, we performed an RNA-FISH/immunofluorescence (IF) assay. RNA-FISH/IF revealed that the colocalization of Neat1 and Hbb in the nucleus of N2a cells (Figure 3E).
Figure 3.

Neat1 directly interacts with Hbb and regulates its expression
(A) Hbb was identified as an interacting target of Neat1 by MS analysis. (B) RNA pull-down assay followed by western blotting directly revealed interaction between Neat1 and Hbb in N2a cells (n = 5). (C and D) RIP assays were performed in N2a cells. Protein-RNA complexes immunoprecipitated by anti-Hbb or IgG were determined by qRT-PCR using primer for Neat1 (C), and the qRT-PCR products were analyzed by electrophoresis (D) (M, marker; ∗p < 0.05 compared with the IgG group, n = 5). (E) RNA-FISH/IF was performed to determine the co-localization of Neat1 (red) and Hbb (green) in N2a cells incubated in hypoxic (1% O2) conditions for 6 h. Arrows indicate the co-localized Neat1 and Hbb nuclei (blue) (n = 6). Scale bar, 5 μm. (F and G) Western blot (F) and immunofluorescence (G) analysis of Hbb expression level in N2a cells incubated in normoxic or hypoxic (1% O2) condition for 16h ( ∗p < 0.05, n = 3, nuclei: blue, Hbb: green. Scale bar = 25 μm). (H) The Hbb protein levels were determined in hippocampus of mice at 24 h after Sham and CLP operation ( ∗p < 0.05, n = 3 mice/group).
Since Neat1 expression levels were increased by hypoxia, we wanted to determine whether Hbb levels were similarly altered. Western blot (Figure 3F) and immunofluorescence staining (Figure 3G) data revealed that Hbb levels were also significantly increased after exposure to hypoxia. Furthermore, we demonstrated that protein levels of Hbb were increased after CLP compared with the sham group (Figure 3H). Collectively, these results demonstrated Neat1 directly interacts with Hbb.
Neat1 stabilizes Hbb via inhibiting Hbb ubiquitination
We analyzed the effects of Neat1 on Hbb expression levels using custom-designed antisense Neat1 GapmeR based on locked nucleic acids (LNA) technology. N2a cells were transfected with control or Neat1 GapmeR, resulting in a significant (p < 0.01) decrease in Neat1 levels (Figures 4A and S6A). Knockdown of Neat1 in N2a cells did not significantly change the mRNA levels of Hbb; however, the protein levels of Hbb were significantly reduced (Figures 4B, 4C, and S6B). These data suggested that Neat1 does not regulate the transcriptional activity of Hbb, but it participates in the regulation of Hbb at the posttranscriptional level. We verified this observation in primary neuron cultures (Figures 4D–4F, S6C, and S6D).
Figure 4.

Neat1 stabilizes Hbb via inhibiting Hbb ubiquitination
(A) The Neat1 levels were measured in N2a cells transfected with Neat1 GapmeR (∗ ∗p < 0.01, n = 3). (B and C) The Hbb mRNA (B) and protein levels (C) in N2a cells after transfection with Neat1 GapmeR ( ∗p < 0.05, n = 3–6). (D) The Neat1 levels were determined in primary neuronal cells transfected with Neat1 GapmeR for 24 h ( ∗p < 0.05, n = 5). (E and F) The Hbb mRNA (E) and protein levels (F) in primary neuronal cells after transfection with Neat1 GapmeR for 24 h ( ∗p < 0.05, n = 3–4). (G) N2a cells transfected with Neat1 GapmeR were treated with cycloheximide (CHX, 200 μg/mL) for the indicated times, and Hbb protein levels were determined. (H) The protein levels of Hbb in N2a cells transfected with control GapmeR or Neat1 GapmeR for 24 h and treated with MG-132 (5 μM) or DMSO for 16 h were determined by western blot assay. (I) N2a cells transfected with control GapmeR or Neat1 GapmeR were treated with MG-132 (5 μM) for 16 h. Cell lysates were immunoprecipitated with antibodies against Hbb or IgG. The levels of ubiquitination were analyzed by western blot. Bottom: input from cell lysates. IB, immunoblot.
To further determine how Neat1 regulates Hbb expression at the post-transcriptional level, we used the protein synthesis inhibitor cycloheximide (CHX) to block new protein synthesis in N2a cells, which were transfected with control or Neat1 GapmeR. Knockdown of Neat1 significantly (p < 0.05) shortened the half-life of Hbb, suggesting that Neat1 stabilizes the Hbb protein (Figure 4G). Furthermore, treatment of control or Neat1 GapmeR-transfected N2a cells with the proteasome inhibitor MG-132 reversed Neat1 GapmeR-induced suppression of Hbb levels (Figure 4H). To further validate that the ubiquitin-proteasome pathway was responsible for the Neat1 knockdown-mediated degradation of Hbb, we performed co-immunoprecipitation (Co-IP) assays to detect the ubiquitination of Hbb. The lysate of control or Neat1 GapmeR-transfected N2a cells were immunoprecipitated with Hbb antibody and immune-blotted with ubiquitin antibody. The ubiquitination of Hbb in N2a cells was significantly (p < 0.05) increased by Neat1 knockdown (Figures 4I and S6E). Taken together, these results demonstrated that Neat1 directly binds to Hbb and prevents proteasome-dependent ubiquitination and degradation of Hbb.
Neat1/Hbb axis suppresses PSD-95 expression and dendritic spine density
To assess the functional role of the Neat1/Hbb axis in cognitive dysfunction in SAE, we focused on PSD-95 expression levels as it plays a critical role in synaptic plasticity and memory.43 Transfection of Neat1 GapmeR into N2a cells significantly (p < 0.05) increased PSD-95 levels (Figures 5A, 5B, and S7A). We extended this finding in primary neurons isolated from the neonatal C57BL/6 mice. These cells were transfected with control or Neat1 GapmeR for 24 h. Neat1 knockdown increased PSD-95 levels (Figures 5C and S7B). We determined whether Neat1/Hbb regulates neuronal dendritic spine density by measuring the number of post-synaptic PSD-95 clusters. The cultured primary neurons were transfected with control or Neat1 GapmeR and stained with βIII-tubulin for axons and PSD-95 for dendritic spines. Knockdown of Neat1 significantly (p < 0.05) increased dendritic spine density, as evidenced by an increased number of PSD-95-positive clusters per 20 μm of dendritic section (Figures 5D and S7C). To further determine the effect of Hbb on PSD-95 expression and dendritic spine density, we transfected Hbb GapmeR into N2a cells and primary neuronal cells. Hbb GapmeR significantly (p < 0.05) decreased Hbb levels and increased PSD-95 levels in N2a cells (Figure 5E) and primary neuronal cells (Figure 5F). In addition, knockdown of Hbb also increased dendritic spine density similarly to Neat1 knockdown (Figure 5G). Taken together, these data demonstrated that the Neat1/Hbb axis suppresses PSD-95 expression and dendritic spine density in neuronal cells.
Figure 5.

Neat1/Hbb axis suppresses PSD-95 expression and dendritic spine density
(A) The Neat1 levels were measured in N2a cells after transfection with Neat1 GapmeR for 48 h ( ∗p < 0.05, n = 4). (B) The PSD-95 protein levels were determined after transfection of N2a cells with Neat1 GapmeR for 48h ( ∗p < 0.05, n = 6). (C) The protein levels of PSD-95 were detected after transfection of the primary neuronal cells with Neat1 GapmeR for 24 h (∗p < 0.05, n = 4). (D) Primary neurons were transfected with control or Neat1 GapmeR for 24 h. The dendritic spine numbers were analyzed by immunostaining to label PSD-95 puncta and axons ( ∗∗p < 0.01, n = 6; PSD-95, green; βⅢ-tubulin, red. Scale bar, 5 μm. (E and F) The protein levels of Hbb and PSD-95 were determined after transfection of the N2a cells (E) and primary neuron cells (F) with control or Hbb GapmeR ( ∗p < 0.05, ∗ ∗p < 0.01, n = 5–6). (G) Control or Hbb GapmeR was transfected into primary neuronal cells for 24 h. The dendritic spine numbers were analyzed by immunostaining to label PSD-95 puncta and axons (∗ ∗p < 0.01, n = 6; PSD-95, green; βⅢ-tubulin, red. Scale bar, 5 μm.
Neat1 deficiency attenuates anxiety and cognitive dysfunction post-sepsis
In vitro loss-of-function assays suggested a unique role of the lncRNA Neat1 in promoting post-synaptic-specific gene expression while suppressing synaptic function (Figure 5). Therefore, we next determined the functional role of Neat1 in synaptic formation in vivo in CLP mice by using Neat1−/− mice. Neat1−/− mice do not exhibit apparent gross abnormalities at baseline except for dysfunction of the corpus luteum and mammary gland development in female knockout (KO) mice.44,45 No significant difference in mortality was observed between the wild-type (WT) and the Neat1−/− mice (WT mice: 42%, Neat1−/− mice: 55%; Figure S8). We confirmed that Neat1 expression is completely depleted in the hippocampus of Neat1−/− mice after CLP (Figure 6A). Neat1−/− mice suppressed post-sepsis anxiety-like behavior, as evidenced by visiting of the center more frequently, spending more time in the center zone, and taking less time to first enter the center compared with the WT group (Figures 6B and 6C). In addition, using CFC testing, we observed that Neat1−/− mice exhibited significantly (p < 0.05) increased freezing time compared with WT mice (Figure 6D). The protein levels of Hbb were significantly (p < 0.05) decreased, whereas the levels of PSD-95 were significantly (p < 0.05) increased in the hippocampus of Neat1−/− mice compared with the WT mice at 24 h after CLP (Figure 6E). Dendritic spine density in the hippocampus was significantly (p < 0.05) increased in Neat1−/− mice compared with WT mice at 8 weeks after CLP (Figure 6F). Taken together, these data demonstrate that the lncRNA Neat1 plays a critical role in neuronal synaptic function in vitro and in vivo and in post-sepsis neuropsychiatric sequelae.
Figure 6.

Neat1 deficiency attenuates anxiety-like behavior and cognitive dysfunction post-sepsis
(A) qRT-PCR was performed to measure Neat1 expression in hippocampus from WT and Neat1−/− mice (∗ ∗p < 0.01, n = 3–4 mice/group). (B and C) WT (n = 13) and Neat1−/− (n = 12) mice were subjected to open-field test at 2 weeks after CLP. (B) Representative tracks and heatmap are shown. (C) The frequency, time in the center zone, and the first time for the mice go to the center zone were analyzed ( ∗p < 0.05). (D) WT (n = 12) and Neat1−/− (n = 11) mice were subjected to contextual fear-conditioning test at 6 weeks after CLP, and freezing behavior as percent freezing time was analyzed ( ∗p < 0.05). (E) The protein level of Hbb and PSD-95 were analyzed in the hippocampus of WT and Neat1−/− mice at 24 h after CLP ( ∗p < 0.05, n = 3–4 mice/group). (F) Neuron dendrite from WT and Neat1−/− mice at 8 weeks after CLP were stained with Golgi stain, and dendritic spine numbers were analyzed (Scale bar, 20 μm. ∗p < 0.05, n = 3 mice/group).
Neat1 GapmeR ameliorates anxiety and cognitive impairment post-sepsis
Our data suggested that the lncRNA Neat1/Hbb axis could be a therapeutic target for SAE. To explore the therapeutic potential of inhibiting the Neat1/Hbb axis, we designed a novel antisense oligonucleotide, LNA GapmeR, to target Neat1. Previous studies have suggested that under normal physiological conditions LNA GapmeRs cannot pass across the blood-brain barrier (BBB) with systemic administration.46 Since it is known that the BBB is damaged during sepsis,47 we injected septic mice intravenously with control or Neat1 GapmeR #1 at 4 h after CLP surgery to determine whether Neat1 GapmeR entered the brain after sepsis. Neat1 GapmeR decreased Neat1 levels in brain tissue from septic mice and increased the expression of IEGs (c-Fos and Bdnf), suggesting biologic activity (Figure 7A). These unexpected data suggest that Neat1 GapmeRs take advantage of BBB breakdown during sepsis and enter the brain tissue to exert their biological function. The Neat1 GapmeR injection did not affect mouse survival (control GapmeR: 57%, Neat1 GapmeR: 55%; Figure S9A). Survivors were subjected to open-field and CFC tests at 2 weeks and 6 weeks after CLP (Figure S9B). Neat1 GapmeR treatment ameliorated CLP sepsis-induced anxiety-like behavior, as evidenced by visiting the center more frequently and taking less time to first enter the center. The time spent in the center zone was not different between control GapmeR- and Neat1 GapmeR-treated mice (Figures 7B and 7C). Neat1 GapmeR-treated mice displayed significantly increased freezing time compared with control GapmeR-treated mice (Figure 7D). At 24 h after CLP, the protein levels of PSD-95 were significantly (p < 0.05) increased in brain tissue of mice treated with Neat1 GapmeR compared with the control group (Figure 7E). Dendritic spine density was determined at 8 weeks after CLP. Treatment with the Neat1 GapmeR ameliorated sepsis-induced dendritic spine loss (Figure 7F). Taken together, these data demonstrated that systemic administration of Neat1 GapmeR ameliorates CLP sepsis-induced anxiety-like behavior, memory impairment, and decrease of dendritic spine density.
Figure 7.

Neat1 GapmeR ameliorated anxiety and cognitive impairment post-sepsis
(A) Mice were subjected to cecal ligation and puncture (CLP)-induced sepsis and treated with control and Neat1-GapmeR, and the Neat1 levels and mRNA levels of c-fos and Bdnf in brain tissue were determined at 24 h after CLP ( ∗p < 0.05, ∗ ∗p < 0.01, n = 4). (B and C) Septic mice were treated with control (n = 12) or Neat1 (n = 13) GapmeR and subjected to open-field test at 2 weeks after CLP. (B) Representative tracks and heatmap are shown. (C) The frequency and time for mice to enter the center zones as well as the first time for the mice to enter the center zone were analyzed ( ∗p < 0.05). (D) Septic mice were treated with control (n = 18) or Neat1 (n = 14) GapmeR and subjected to a contextual fear-conditioning test at 6 weeks after CLP. Freezing behavior as percent freezing time was analyzed ( ∗p < 0.05). (E) PSD-95 protein levels were analyzed in brain tissues of control or Neat1 GapmeR-treated septic mice at 24 h after CLP ( ∗p < 0.05, n = 3 mice/group). (F) Neuron dendrite from Sham mice, and septic mice treated with control or Neat1 GapmeR were stained with Golgi stain, and dendritic spine numbers were analyzed at 8 weeks post-surgery (Scale bar, 20 μm.∗p < 0.05, n = 3 mice/group).
Discussion
Despite their frequency and profound impact on the health and well-being of sepsis survivors, little is known about the mechanisms underlying post-septic cognitive and psychiatric sequelae. This knowledge gap has significantly impacted the development of effective preventative and therapeutic strategies to address this major health concern. We identified an important role of the lncRNA Neat1 in the modulation of neuronal synaptic density and neuropsychiatric dysfunction among murine survivors of experimental sepsis. Specifically, we showed that CLP-induced sepsis replicated clinical cognitive impairments, including anxiety-like behavior and long-term cognitive deficits, in the mouse model and increased the Neat1 expression in brain tissue, especially in neuronal cells. Furthermore, Neat1 mediated PSD-95 expression by interacting with Hbb protein and regulated dendritic spine density, which is associated with cognition and learning. Deficiency of Neat1 was sufficient to protect post-sepsis cognitive function in Neat1−/− mice. We further found that inhibition of Neat1 by use of systemic Neat1 GapmeR ameliorated sepsis-related dendritic spine loss and reduced cognitive dysfunction.
Sepsis survivors develop psychiatric sequelae and long-term cognitive impairment following discharge from intensive care units (ICUs), including anxiety, depression, alterations in memory, attention, concentration, and/or global cognitive decline, which has a profound impact on their quality of life.48 The use of behavioral testing in animal models of sepsis is a potentially valuable strategy to understand the onset and the time course of the mental health problems and cognitive dysfunction associated with sepsis. Open-field tests have been frequently used for anxiety-like behavior, and contextual fear-conditioning tests along with other inhibitory avoidance tests have been widely used for assessing hippocampus-dependent memory.49 Sepsis-induced by LPS and CLP are the most frequently used models, and both have been effective in inducing short- and long-term behavioral impairment.49 Compared with the LPS model, the CLP model more closely resembles the progression and characteristics of human sepsis,50 and it could be a useful tool to study cognitive impairment, anxiety, and depression after sepsis, as well as the mechanisms associated with the recovery from such disorders.49 In agreement with previous studies,51 we demonstrated that sepsis-surviving mice show persistent impairment, since anxiety-like behavior persists for at least 2 weeks after CLP, and memory impairment can be observed at least 6 weeks after sepsis, and dendritic spin density remains decreased at 8 weeks after sepsis. These data suggest that the CLP sepsis model may recapitulate human SAE and reinforce the necessity of developing SAE-targeted therapy to improve the quality of life for sepsis survivors. In addition, synapses are particularly vulnerable in neurodegenerative diseases and neurological disorders, and LPS decreases synaptic proteins and contributes to the memory deficit,16 which is consistent with our data obtained from the CLP model. PSD-95, a major component responsible for synaptic maturation that regulates dendritic spines and developing synapses in the hippocampus, has been recently associated with neuropsychiatric disorders, and reduction of PSD-95 was observed in septic mice.52 Our data uncovered a novel mechanism, that the Neat1/Hbb axis regulated PSD-95 levels and dendritic spine density in SAE.
Neat1 is an essential component of nuclear paraspeckles,26 which consist of ribonucleoprotein complexes formed around Neat1.53 Previous studies investigating Neat1 in the context of epilepsy have reported that, in the excitotoxic conditions of this neurodegenerative disorder, activity-dependent downregulation of NEAT1 expression is impaired.54 However, observations from studies on other neurodegenerative diseases would suggest that NEAT1 upregulation is deleterious to neuronal survival.55 Moreover, another study found that increased NEAT1 expression might play a notable role in the age-related decline of hippocampus-dependent memory formation.32 Therefore, abnormal Neat1 expression and/or regulation might underlie at least some of the symptoms and phenotypes in common neurological diseases. Interestingly, ample evidence also indicates that Neat1 participates in sepsis-induced organ injury in mice.34, 35, 36 However, until now, the role of Neat1 in sepsis-associated neuronal dysfunction has been unknown. Since hypoxia may occur in brain tissue during sepsis, a key regulator of the transcriptional responses to hypoxia is HIF.56 Recently, HIF has been shown to play a critical role in regulating the non-coding transcriptional response.57 It is reported that hypoxia can induce Neat1 through HIF-2α-mediated transcriptional activation,41,42 we found that inhibition of HIF-2α attenuated hypoxia-induced increases of Neat1 in N2a cells. Historically, Neat1 was thought to be exclusive to the nucleus and to mainly function as a transcriptional regulator. In our study, we found that Neat1 was present both in the nucleus and in the cytoplasm and exerted post-translational regulation of Hbb via direct binding and inhibition of ubiquitination. Given a report suggesting that Hbb may be a part of a mechanism linking neuronal energetics with epigenetic changes and may function by supporting neuronal metabolism,58 Hbb was further analyzed. Silencing of Neat1 or Hbb both led to increased PSD-95 and dendritic spines as well as reductions of post-sepsis neuropsychiatric sequelae. The exact role of hemoglobin in neurons is debated, although changes in its expression and subcellular localization have been associated with a few neurodegenerative disorders, including multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease.58, 59, 60 This study’s observations, combined with emerging data suggesting the importance of cell-free hemoglobin in sepsis-related organ failure, provide a strong rationale for ongoing investigation into the role of Hbb in SAE. Previous studies demonstrated that septic mice exhibited decreases of β2-adrenoceptor (β2-AR), increases of neuroinflammation, and decreases of brain-derived neurotrophic factor(BDNF) and PSD-95 expression,61 which are consistent with our findings. The authors demonstrated that treatment with β2-AR agonist clenbuterol ameliorated sepsis-induced cognitive impairment, reduced proinflammatory cytokines, and upregulated BDNF and PSD-95 levels.61 Whether activation of β2-AR could alter Neat1 expression remains to be further investigated.
A comprehensive analysis of the molecular perturbations produced by Neat1 in neuronal cells provides the first evidence of its potential as a therapeutic target in neurons of SAE. To efficiently target Neat1, we used a novel LNA GapmeR antisense oligonucleotide, which serves as a unique tool to efficiently knock down lncRNAs.62 LNA GapmeRs have a central DNA gap that binds the RNA target and triggers its RNase H-dependent degradation; the presence of phosphorothioate confers nuclease resistance in bodily fluids,63 while LNA increases affinity to the target.64 LNA GapmeRs are becoming an attractive therapeutic modality to target undruggable pathways in vivo. Prior studies suggest that, in physiological conditions, LNA GapmeRs cannot pass through the BBB to reach the brain by systemic administration.46 However, our investigation capitalized on the known BBB interruption associated with sepsis, which allowed intravenously injected Neat1 GapmeRs to enter the brain and impart beneficial effects on both synaptic density and post-sepsis neuropsychiatric symptoms. These findings support the therapeutic potential of LNA GapmeRs in human illness and serve as the basis of ongoing translational studies in SAE.
There are limitations in our studies. Notably, we do not have access to human brain samples from septic patients to determine Neat1 levels in the human brain post-sepsis. Neat1 expression and function in neuronal cells were studied, but we cannot exclude the possibility for Neat1 to also modulate the function of other brain cells post-sepsis. Although we clearly showed the relationship between Neat1 and Hbb, it is possible that Neat1 impacts SAE via other mechanisms that were not investigated. Finally, the potential off-target effects of Neat1 GapmeR were not investigated here and clearly deserve further investigation.
In summary, we identified the interaction between Neat1 and Hbb, which together regulate dendritic spine density and impact cognitive dysfunctions after sepsis (Figure 8). As a result, Neat1 and Hbb may become a potential target for diagnosis and a treatment strategy for sepsis-associated acute brain dysfunction. Furthermore, these roles may extend to other sources of acute brain dysfunction including COVID-19. Finally, we report the first evidence of mitigating SAE through a novel antisense oligonucleotide, GapmeR-based reduction of Neat1, which could lead to a novel therapeutic strategy to combat SAE.
Figure 8.

Schematic model for Neat1/Hbb axis regulating post-sepsis cognitive impairment
During sepsis, neurons are exposed to hypoxic environments, which results in upregulation of Neat1 levels. Neat1 directly interacts with Hbb and stabilizes its protein levels via inhibiting Hbb ubiquitination. Furthermore, Hbb suppression of PSD-95 expression results in decreased dendritic spine density, which is critical for cognition and memory. In addition, suppression of Neat1 via GapmeR promotes degradation of Hbb, resulting in increases of PSD-95 levels, reduction of synaptic dysfunction, and mitigation of cognitive impairment post-sepsis.
Materials and methods
Animals
Male and female C57BL/6 Mice (8–12 weeks old) were purchased from the Jackson Laboratory and housed in a pathogen-free environment. Neat1 knockout mice were generated by Dr. Shinichi Nakagawa’s group44 and provided by Dr. Jiliang Zhou (Augusta University). Male and female Neat 1 knockout mice (8–12 weeks) and littermate WT mice in C57BL/6 background were used. Genotyping of these mice was performed as previously described in detail.65 The primer sequences for Neat1 knockout mouse genotyping were as follows: forward primer (5′–3′), GCCATTCAGGCTGCG-CAACTG; reverse primer (5′–3′), AGCAGGGATAGCCTGGTCTT; reverse primer (5′–3′), CTAGTGGTGGGGAGGCAGT. The use of experimental animals was approved by the IACUC at Medical University of South Carolina in accordance with NIH guidelines. All procedures complied with the standards for care and use of animal subjects as stated in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Resources, National Academies of Sciences, Washington, DC).
Cecal ligation and puncture-induced sepsis
Cecal ligation and puncture were performed as described previously.66 Mice were anesthetized with isoflurane, and a midline incision was made below the diaphragm to expose the cecum. The cecum was ligated at the cecal junction with a 5-0 silk suture without interrupting intestinal continuity and punctured once with a 22-gauge needle. A small amount of stool was extruded through the puncture site. Sham operation was performed in the same way as CLP but without ligation and puncture of the cecum. Mice received the antibiotic imipenem (25 mg/kg, subcutaneously) at 6, 24, and 48 h after CLP.
Behavioral tests
All behavioral tests were performed between 2:00 pm and 5:00 pm during the light cycle in a sound-isolated room. To avoid interference of feces and urine from other mice, the trial site was thoroughly cleaned with 75% ethanol after each test. Mice were subjected to behavioral tests at least 2 weeks after surgery to allow mice to physically recover from sepsis so that physical weakness would not be considered as a confounder of the measurements. Mice were subjected to an open-field test at 2 weeks after CLP and a contextual fear-conditioning test at 6 weeks after CLP. Behavioral tests were rigorously conducted by an experimenter blinded to the genotype and treatment of the animals.
Open-field test
The open-field test is a widely used model of anxiety-like behavior developed to evaluate emotionality in animals. It involves subjecting an animal to an unknown environment whose escape is prevented by surrounding walls.49 Mice were placed in an open-field arena (40 × 40 cm square Plexiglas box with walls ∼30 cm tall) in a brightly lit room and allowed to explore freely for 5 min. Time spent in center (s), and time spent in the periphery (s) were recorded by live video tracking and EthoVision XT software.
Contextual fear-conditioning test
Sepsis-induced cognitive impairment was evaluated by assessing the fear memory processes through re-experiencing symptoms. The measured variable was the time the animal spent freezing, taken as an index of fear in rodents.49 An animal was considered to be freezing when crouching, without any visible body movement of the body and head except for breathing. To evaluate freezing behavior in response to contextual cues, a paradigm using fear-conditioning chambers was employed. For this test, mice were singly placed into a Plexiglas arena (17 × 17 cm with foot shock grid floor) within a sound-attenuated chamber (Ugo Basile). Before testing began, mice were acclimated to the testing room for 30 min. On day 1, mice were singly placed into arenas for 119 s before delivery of a 1-s, 1-mA foot shock. On day 2 (24 h later), mice were singly placed into the same context and allowed to explore freely for 10 min. Freezing behavior at the last 5 min was recorded using a high-speed digital IR camera and analyzed using EthoVision XT software (Noldus Information Technology). Memory was expressed as the percentage of time the animals spent exhibiting this defensive behavior, and this measure could be used as a retention score: the better the memory, the more time the animal spent in freezing behavior.
RNA extraction, reverse transcription, and real-time qPCR
Total RNA was extracted with Trizol (Invitrogen, Waltham, MA USA) according to the manufacture’s instruction. Then, 500 ng of RNA was transcripted into cDNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA USA). Quantitative real-time PCR was completed with SYBR PCR Master Mix (Qiagen, Germantown, MD, USA) according to manufacturer’s protocol. GAPDH was used as the internal control to normalize the mRNA level. Relative mRNA level was calculated by the 2−ΔΔCt comparative method. Related primer sequences are listed in Table S2.
Protein extraction and western blotting
Protein lysates were extracted from N2a cells, primary neuronal cells, or brain hippocampus tissue. Briefly, after being rinsed with PBS, cells were harvested using RIPA buffer (Fisher) with protease/phosphatase inhibitor cocktail (CST, 5872S) to extract proteins. Following sonication and centrifugation, protein lysate was quantified by BCA assay and loaded onto 10–15% SDS-PAGE gel at 20–40 μg per lane. Antibodies used in this study were: β-actin (CST, A5316, rabbit, 1:1,000), Hbb (NOVUS, H00003043-M02, mouse, 1:500), PSD-95 (Invitrogen, 51-6900, rabbit, 1:500), ubiquitin (CST, 43124S, rabbit, 1:1,000). Images were acquired by ChemiDoc Touch Imaging System (Bio-Rad), and band densities were quantified using ImageJ software.
Mass spectrometry
Mice were subjected to sham and CLP, and neuronal cells were isolated 24 h after surgery. The protein was precipitated by RNA protein pull-down assay (Pierce magnetic RNA-protein pull-down kit, Thermo Scientific) in lysed neuronal cells and subjected to specific digestion with trypsin. The peptides were subsequently analyzed using LC-MS/MS with an EASY nLC 1200 in-line with the Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Scientific) with instrument control software v.4.2.28.14 (scanning range was 375–1,500 m/z). For protein identification and label-free quantification, the LC-MS/MS data were searched using MaxQuant v.1.6.10 against a mouse SwissProt-reviewed database with 17,034 proteins and a database of common contaminants. Common contaminants, reversed database hits, and proteins identified by one modified peptide were removed, and the LFQ-normalized protein intensities were log2 transformed.
RNA-protein pull-down assay
RNA-protein pull-down assays were carried out using the Pierce Magnetic RNA-Protein pull-down kit essentially following the protocol provided by the manufacturer (Thermo Scientific). A pBLUNT vector containing full-length mouse Neat1 V1 sequence (provided by Dr. Jiliang Zhou, Augusta University) was utilized to synthesize sense or antisense probes in vitro by using T3 or T7 promoter, respectively. Subsequently, the RNA probes were labeled with biotinylated cytidine bisphosphate and captured by streptavidin magnetic beads. Proteins were extracted from N2a cell protein and then incubated with the biotin-labeled sense or antisense Neat1 probe coupled to the streptavidin magnetic beads. The RNA-bound proteins were eluted for MS or western blot analysis as described above.
RNA immunoprecipitation
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation Kit according to the manufacturer’s instructions (Millipore). Briefly, N2a cells were lysed and incubated with Hbb antibody (NOVUS, H00003043-M02, mouse, 10 μg) or control IgG (10 μg) conjugated to magnetic beads 50 μL (Dynabeads Protein G, Invitrogen) overnight. The beads, protein, and mRNA complexes were immunoprecipitated and then magnetically separated. The mRNAs were purified and quantified by RT-qPCR using mouse Neat1 and Malat1 primer, listed in Table S2. The RT-qPCR product was also visualized in an agarose gel.
RNA-FISH/IF
Briefly, seeded cells or sections of brain tissues were fixed with 4% paraformaldehyde and treated with 0.5% Triton for 10 min. The sections were blocked with goat serum (5%, Invitrogen) for 30 min. The sections were incubated with anti-NeuN (Abcam, mouse, 1:500) antibodies and stained with secondary antibody (488-nm anti-mouse secondary antibody, 1:250 dilution, Invitrogen) diluted in blocking buffer for 1 h at room temperature. The sections were fixed with 4% paraformaldehyde for 15 min followed by pre-hybridization. Overnight hybridization was performed with a 10 mM mouse Neat1 probe labeled with Quasar 570 dye (Biosearch Technologies). RNA-FISH/IF was performed using an RNA-FISH kit (Biosearch Technologies) according to the manufacturer’s instructions. Sections were immersed with mounting medium (ProLong Gold anti-fade reagent with DAPI, Invitrogen) to visualize nuclei and imaged using confocal microscopy (TCS SP8, Leica). The numbers of Neat1 focus-positive cells were counted in four random fields from three biological samples in each group. To determine Neat1 co-localization with Hbb in N2a cells, The cells were seeded on chamber slides. Subsequently RNA-FISH/IF was carried out as described above using anti-Hbb (Invitrogen, PA5-60287, rabbit, 1:2,000) antibody and Neat1 probe.
GapmeR delivery
Cells were transfected at 60–80% confluence with 20–50 nmol/L control GapmeR or GapmeR (Qiagen) targeting Neat1 (#1, #2) or Hbb using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions. Forty-eight hours after transfection, the cells were harvested for further analysis.
Mice were subjected to CLP and administered intravenously control or Neat1 GapmeR #1 (10 nmol/kg body wt) at 4 h after CLP.
Cell culture
Mouse neural crest-derived cell line (Neuro-2a, N2a) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were grown at 37°C and in 5% CO2. Cells were incubated for 16 h in an atmosphere of either normoxia (21% oxygen) or hypoxia (1% oxygen). HIF-2α was suppressed by specific siRNA (75,377,553, Invitrogen).
For isolation and culture of primary mouse neuron cells, primary neuronal cultures were prepared from newborn 1- to 3-day-old C57BL/6 mouse brains. Neurons were isolated using a neuron isolation kit (Miltenyi Biotec Inc., Auburn, CA, USA). Briefly, non-neuronal cells in single-cell preparations from whole mouse brain digests were depleted by negative selection. Isolated neurons were plated onto poly-D-lysine-coated dishes and cultured in neuronal medium (catalog no. 1521; ScienCell Research Laboratories, Carlsbad, CA, USA) supplemented with neuronal growth supplement and 1% penicillin-streptomycin (ScienCell Research Laboratories). Cells were grown at 37°C and in 5% CO2.
Golgi staining
We used the FD Rapid Golgi Stain kit (FD NeuroTechnologies) to perform Golgi staining following the vendor’s protocol.67 The brains were then sliced (100 μm/slice) using a cryostat, 5–6 sections/mice. Slides were coded before quantitative analysis, and the person analyzing the slide was blind to the code.
Quantitative differences in the number of dendritic spines on individual dendritic branch orders between conditions were determined as previously described.67 Golgi-stained neurons were examined, and z stack images were captured microscopically via bright-field imaging on a Keyence BZ-X800 microscope and processed using the Keyence software package. Dendritic spines on apical pyramidal neurons were analyzed in the CA1 sub-region of the hippocampus (approximately −1.6 to −2.46 mm from bregma). Spine numbers were counted and estimated by from first- to fifth-order dendrites.67 All protrusions were counted as spines if they were in direct contact with the dendritic shaft and were less than 1 μm in length and 0.5 μm in width. For every mouse, six segments of dendrites were selected per branch order per region of interest.
Statistical analysis
Statistical significance was determined by analysis of variance (ANOVA) or Student’s t test using GraphPad Prism software. A value of p < 0.05 was considered statistically significant.
Acknowledgments
We thank Dr. James A. Cook (Department of Neuroscience, Medical University of South Carolina) for critical reading of the manuscript. This work was supported in part by National Institutes of Health grant 1R01GM113995 (H.F.), National Institutes of Health grant 1R01GM130653 (H.F.), National Institutes of Health grant 1 R41AI157378 (H.F.), National Institutes of Health grant 5R21AG067445 (M.L.), National Institutes of Health grant 5TL1TR001451 (P.V.H.), National Institutes of Health grant UL1TR001450 (P.V.H. and A.G.).
Author contributions
Y.W. and P.L. performed experiments, data collection and analysis, and wrote and edited the manuscript. L.L., A.G., and P.H. helped with study design and manuscript editing. T.H. and S.N. generated the knockout mice. J.L.Z. provided the knockout mice and plasmid. M.L. edited and commented on the manuscript. H.F. was responsible for study conception and design, data analysis and interpretation, study supervision, and manuscript writing and editing. All authors read and approved of this manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.011.
Supplemental information
References
- 1.Rhee C., Klompas M. Sepsis trends: increasing incidence and decreasing mortality, or changing denominator? J. Thorac. Dis. 2020;12:S89–S100. doi: 10.21037/jtd.2019.12.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaukonen K.M., Bailey M., Suzuki S., Pilcher D., Bellomo R. Mortality related to severe sepsis and septic shock among critically ill patients in Australia and New Zealand, 2000-2012. JAMA. 2014;311:1308–1316. doi: 10.1001/jama.2014.2637. [DOI] [PubMed] [Google Scholar]
- 3.Iwashyna T.J., Cooke C.R., Wunsch H., Kahn J.M. Population burden of long-term survivorship after severe sepsis in older Americans. J. Am. Geriatr. Soc. 2012;60:1070–1077. doi: 10.1111/j.1532-5415.2012.03989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pandharipande P.P., Girard T.D., Ely E.W. Long-term cognitive impairment after critical illness. N. Engl. J. Med. 2014;370:185–186. doi: 10.1056/NEJMc1313886. [DOI] [PubMed] [Google Scholar]
- 5.Rahmel T., Schmitz S., Nowak H., Schepanek K., Bergmann L., Halberstadt P., Horter S., Peters J., Adamzik M. Long-term mortality and outcome in hospital survivors of septic shock, sepsis, and severe infections: the importance of aftercare. PLoS One. 2020;15:e0228952. doi: 10.1371/journal.pone.0228952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Annane D., Sharshar T. Cognitive decline after sepsis. Lancet Respir. Med. 2015;3:61–69. doi: 10.1016/S2213-2600(14)70246-2. [DOI] [PubMed] [Google Scholar]
- 7.Stubbs D.J., Yamamoto A.K., Menon D.K. Imaging in sepsis-associated encephalopathy--insights and opportunities. Nat. Rev. Neurol. 2013;9:551–561. doi: 10.1038/nrneurol.2013.177. [DOI] [PubMed] [Google Scholar]
- 8.Zhou R.X., Li Y.Y., Qu Y., Huang Q., Sun X.M., Mu D.Z., Li X.H. Regulation of hippocampal neuronal apoptosis and autophagy in mice with sepsis-associated encephalopathy by immunity-related GTPase M1. CNS Neurosci. Ther. 2020;26:177–188. doi: 10.1111/cns.13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El-Husseini A.E., Schnell E., Chetkovich D.M., Nicoll R.A., Bredt D.S. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- 10.Diering G.H., Huganir R.L. The AMPA receptor code of synaptic plasticity. Neuron. 2018;100:314–329. doi: 10.1016/j.neuron.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilman S.R., Iossifov I., Levy D., Ronemus M., Wigler M., Vitkup D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron. 2011;70:898–907. doi: 10.1016/j.neuron.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fromer M., Pocklington A.J., Kavanagh D.H., Williams H.J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D.M., et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fossati G., Morini R., Corradini I., Antonucci F., Trepte P., Edry E., Sharma V., Papale A., Pozzi D., Defilippi P., et al. Reduced SNAP-25 increases PSD-95 mobility and impairs spine morphogenesis. Cell Death Differ. 2015;22:1425–1436. doi: 10.1038/cdd.2014.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coley A.A., Gao W.J. PSD-95 deficiency disrupts PFC-associated function and behavior during neurodevelopment. Sci. Rep. 2019;9:9486. doi: 10.1038/s41598-019-45971-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong C.Q., Zhou H.C., Wu J., Guo N.Z. The protective effects and the involved mechanisms of tanshinone IIA on sepsis-induced brain damage in mice. Inflammation. 2019;42:354–364. doi: 10.1007/s10753-018-0899-z. [DOI] [PubMed] [Google Scholar]
- 16.Moraes C.A., Santos G., de Sampaio e Spohr T.C., D'Avila J.C., Lima F.R., Benjamim C.F., Bozza F.A., Gomes F.C. Activated microglia-induced deficits in excitatory synapses through IL-1beta: implications for cognitive impairment in sepsis. Mol. Neurobiol. 2015;52:653–663. doi: 10.1007/s12035-014-8868-5. [DOI] [PubMed] [Google Scholar]
- 17.Shaver C.M., Paul M.G., Putz N.D., Landstreet S.R., Kuck J.L., Scarfe L., Skrypnyk N., Yang H., Harrison F.E., de Caestecker M.P., et al. Cell-free hemoglobin augments acute kidney injury during experimental sepsis. Am. J. Physiol. Renal Physiol. 2019;317:F922–F929. doi: 10.1152/ajprenal.00375.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meegan J.E., Shaver C.M., Putz N.D., Jesse J.J., Landstreet S.R., Lee H.N.R., Sidorova T.N., McNeil J.B., Wynn J.L., Cheung-Flynn J., et al. Cell-free hemoglobin increases inflammation, lung apoptosis, and microvascular permeability in murine polymicrobial sepsis. PLoS One. 2020;15:e0228727. doi: 10.1371/journal.pone.0228727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janz D.R., Bastarache J.A., Peterson J.F., Sills G., Wickersham N., May A.K., Roberts L.J., 2nd, Ware L.B. Association between cell-free hemoglobin, acetaminophen, and mortality in patients with sepsis: an observational study. Crit. Care Med. 2013;41:784–790. doi: 10.1097/CCM.0b013e3182741a54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biagioli M., Pinto M., Cesselli D., Zaninello M., Lazarevic D., Roncaglia P., Simone R., Vlachouli C., Plessy C., Bertin N., et al. Unexpected expression of alpha- and beta-globin in mesencephalic dopaminergic neurons and glial cells. Proc. Natl. Acad. Sci. U S A. 2009;106:15454–15459. doi: 10.1073/pnas.0813216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beermann J., Piccoli M.T., Viereck J., Thum T. Non-coding RNAs in development and disease: background, mechanisms, and therapeutic approaches. Physiol. Rev. 2016;96:1297–1325. doi: 10.1152/physrev.00041.2015. [DOI] [PubMed] [Google Scholar]
- 22.Marchese F.P., Raimondi I., Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206. doi: 10.1186/s13059-017-1348-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devall M., Roubroeks J., Mill J., Weedon M., Lunnon K. Epigenetic regulation of mitochondrial function in neurodegenerative disease: new insights from advances in genomic technologies. Neurosci. Lett. 2016;625:47–55. doi: 10.1016/j.neulet.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakagawa S., Naganuma T., Shioi G., Hirose T. Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J. Cell Biol. 2011;193:31–39. doi: 10.1083/jcb.201011110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamazaki T., Hirose T. The building process of the functional paraspeckle with long non-coding RNAs. Front. Biosci. (Elite Ed.) 2015;7:1–41. doi: 10.2741/715. [DOI] [PubMed] [Google Scholar]
- 26.Anantharaman A., Jadaliha M., Tripathi V., Nakagawa S., Hirose T., Jantsch M.F., Prasanth S.G., Prasanth K.V. Paraspeckles modulate the intranuclear distribution of paraspeckle-associated Ctn RNA. Sci. Rep. 2016;6:34043. doi: 10.1038/srep34043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirose T., Virnicchi G., Tanigawa A., Naganuma T., Li R., Kimura H., Yokoi T., Nakagawa S., Benard M., Fox A.H., Pierron G. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell. 2014;25:169–183. doi: 10.1091/mbc.E13-09-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang P., Cao L., Zhou R., Yang X., Wu M. The lncRNA Neat1 promotes activation of inflammasomes in macrophages. Nat. Commun. 2019;10:1495. doi: 10.1038/s41467-019-09482-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shui X., Chen S., Lin J., Kong J., Zhou C., Wu J. Knockdown of lncRNA NEAT1 inhibits Th17/CD4(+) T cell differentiation through reducing the STAT3 protein level. J. Cell. Physiol. 2019;234:22477–22484. doi: 10.1002/jcp.28811. [DOI] [PubMed] [Google Scholar]
- 30.West J.A., Davis C.P., Sunwoo H., Simon M.D., Sadreyev R.I., Wang P.I., Tolstorukov M.Y., Kingston R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell. 2014;55:791–802. doi: 10.1016/j.molcel.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.An H., Williams N.G., Shelkovnikova T.A. NEAT1 and paraspeckles in neurodegenerative diseases: a missing lnc found? Noncoding RNA Res. 2018;3:243–252. doi: 10.1016/j.ncrna.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butler A.A., Johnston D.R., Kaur S., Lubin F.D. Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aaw9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simchovitz A., Hanan M., Niederhoffer N., Madrer N., Yayon N., Bennett E.R., Greenberg D.S., Kadener S., Soreq H. NEAT1 is overexpressed in Parkinson's disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J. 2019;33:11223–11234. doi: 10.1096/fj.201900830R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y., Qiu J., Chen B., Lin Y., Chen Y., Xie G., Qiu J., Tong H., Jiang D. Long non-coding RNA NEAT1 plays an important role in sepsis-induced acute kidney injury by targeting miR-204 and modulating the NF-kappaB pathway. Int. Immunopharmacol. 2018;59:252–260. doi: 10.1016/j.intimp.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 35.Wang S.M., Liu G.Q., Xian H.B., Si J.L., Qi S.X., Yu Y.P. LncRNA NEAT1 alleviates sepsis-induced myocardial injury by regulating the TLR2/NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019;23:4898–4907. doi: 10.26355/eurrev_201906_18078. [DOI] [PubMed] [Google Scholar]
- 36.Zhang C.C., Niu F. LncRNA NEAT1 promotes inflammatory response in sepsis-induced liver injury via the Let-7a/TLR4 axis. Int. Immunopharmacol. 2019;75:105731. doi: 10.1016/j.intimp.2019.105731. [DOI] [PubMed] [Google Scholar]
- 37.Liu W.Q., Wang Y.J., Zheng Y., Chen X. Effects of long non-coding RNA NEAT1 on sepsis-induced brain injury in mice via NF-kappaB. Eur. Rev. Med. Pharmacol. Sci. 2019;23:3933–3939. doi: 10.26355/eurrev_201905_17822. [DOI] [PubMed] [Google Scholar]
- 38.Riva P., Ratti A., Venturin M. The long non-coding RNAs in neurodegenerative diseases: novel mechanisms of pathogenesis. Curr. Alzheimer Res. 2016;13:1219–1231. doi: 10.2174/1567205013666160622112234. [DOI] [PubMed] [Google Scholar]
- 39.Taccone F.S., Su F., De Deyne C., Abdellhai A., Pierrakos C., He X., Donadello K., Dewitte O., Vincent J.L., De Backer D. Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit. Care Med. 2014;42:e114–e122. doi: 10.1097/CCM.0b013e3182a641b8. [DOI] [PubMed] [Google Scholar]
- 40.Meneses G., Cardenas G., Espinosa A., Rassy D., Perez-Osorio I.N., Barcena B., Fleury A., Besedovsky H., Fragoso G., Sciutto E. Sepsis: developing new alternatives to reduce neuroinflammation and attenuate brain injury. Ann. N Y Acad. Sci. 2019;1437:43–56. doi: 10.1111/nyas.13985. [DOI] [PubMed] [Google Scholar]
- 41.Choudhry H., Albukhari A., Morotti M., Haider S., Moralli D., Smythies J., Schodel J., Green C.M., Camps C., Buffa F., et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2alpha dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene. 2015;34:4546. doi: 10.1038/onc.2014.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choudhry H., Mole D.R. Hypoxic regulation of the noncoding genome and NEAT1. Brief. Funct. Genomics. 2016;15:174–185. doi: 10.1093/bfgp/elv050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu W. PSD-95-like membrane associated guanylate kinases (PSD-MAGUKs) and synaptic plasticity. Curr. Opin. Neurobiol. 2011;21:306–312. doi: 10.1016/j.conb.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakagawa S., Shimada M., Yanaka K., Mito M., Arai T., Takahashi E., Fujita Y., Fujimori T., Standaert L., Marine J.C., Hirose T. The lncRNA Neat1 is required for corpus luteum formation and the establishment of pregnancy in a subpopulation of mice. Development. 2014;141:4618–4627. doi: 10.1242/dev.110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Standaert L., Adriaens C., Radaelli E., Van Keymeulen A., Blanpain C., Hirose T., Nakagawa S., Marine J.C. The long noncoding RNA Neat1 is required for mammary gland development and lactation. RNA. 2014;20:1844–1849. doi: 10.1261/rna.047332.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Straarup E.M., Fisker N., Hedtjarn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Orum H., Hansen J.B., Koch T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Erikson K., Tuominen H., Vakkala M., Liisanantti J.H., Karttunen T., Syrjala H., Ala-Kokko T.I. Brain tight junction protein expression in sepsis in an autopsy series. Crit. Care. 2020;24:385. doi: 10.1186/s13054-020-03101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hopkins R.O., Weaver L.K., Collingridge D., Parkinson R.B., Chan K.J., Orme J.F., Jr. Two-year cognitive, emotional, and quality-of-life outcomes in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2005;171:340–347. doi: 10.1164/rccm.200406-763OC. [DOI] [PubMed] [Google Scholar]
- 49.Savi F.F., de Oliveira A., de Medeiros G.F., Bozza F.A., Michels M., Sharshar T., Dal-Pizzol F., Ritter C. What animal models can tell us about long-term cognitive dysfunction following sepsis: a systematic review. Neurosci. Biobehav. Rev. 2021;124:386–404. doi: 10.1016/j.neubiorev.2020.12.005. [DOI] [PubMed] [Google Scholar]
- 50.Dejager L., Pinheiro I., Dejonckheere E., Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 2011;19:198–208. doi: 10.1016/j.tim.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 51.Denstaedt S.J., Spencer-Segal J.L., Newstead M., Laborc K., Zeng X., Standiford T.J., Singer B.H. Persistent neuroinflammation and brain-specific immune priming in a novel survival model of murine pneumosepsis. Shock. 2020;54:78–86. doi: 10.1097/SHK.0000000000001435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang W.Y., Liu K.H., Lin S., Chen T.Y., Tseng C.Y., Chen H.Y., Wu H.M., Hsu K.S. NADPH oxidase 2 as a potential therapeutic target for protection against cognitive deficits following systemic inflammation in mice. Brain Behav. Immun. 2020;84:242–252. doi: 10.1016/j.bbi.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 53.Yamazaki T., Souquere S., Chujo T., Kobelke S., Chong Y.S., Fox A.H., Bond C.S., Nakagawa S., Pierron G., Hirose T. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol. Cell. 2018;70:1038–1053.e7. doi: 10.1016/j.molcel.2018.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Barry G., Briggs J.A., Hwang D.W., Nayler S.P., Fortuna P.R., Jonkhout N., Dachet F., Maag J.L., Mestdagh P., Singh E.M., et al. The long non-coding RNA NEAT1 is responsive to neuronal activity and is associated with hyperexcitability states. Sci. Rep. 2017;7:40127. doi: 10.1038/srep40127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu Y., Lu Z. Long non-coding RNA NEAT1 mediates the toxic of Parkinson's disease induced by MPTP/MPP+ via regulation of gene expression. Clin. Exp. Pharmacol. Physiol. 2018;45:841–848. doi: 10.1111/1440-1681.12932. [DOI] [PubMed] [Google Scholar]
- 56.Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 57.Shih J.W., Kung H.J. Long non-coding RNA and tumor hypoxia: new players ushered toward an old arena. J. Biomed. Sci. 2017;24:53. doi: 10.1186/s12929-017-0358-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown N., Alkhayer K., Clements R., Singhal N., Gregory R., Azzam S., Li S., Freeman E., McDonough J. Neuronal hemoglobin expression and its relevance to multiple sclerosis neuropathology. J. Mol. Neurosci. 2016;59:1–17. doi: 10.1007/s12031-015-0711-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singhal N.K., Alkhayer K., Shelestak J., Clements R., Freeman E., McDonough J. Erythropoietin upregulates brain hemoglobin expression and supports neuronal mitochondrial activity. Mol. Neurobiol. 2018;55:8051–8058. doi: 10.1007/s12035-018-0971-6. [DOI] [PubMed] [Google Scholar]
- 60.Ferrer I., Gomez A., Carmona M., Huesa G., Porta S., Riera-Codina M., Biagioli M., Gustincich S., Aso E. Neuronal hemoglobin is reduced in Alzheimer's disease, argyrophilic grain disease, Parkinson's disease, and dementia with Lewy bodies. J. Alzheimers Dis. 2011;23:537–550. doi: 10.3233/JAD-2010-101485. [DOI] [PubMed] [Google Scholar]
- 61.Zong M.M., Zhou Z.Q., Ji M.H., Jia M., Tang H., Yang J.J. Activation of beta2-adrenoceptor attenuates sepsis-induced hippocampus-dependent cognitive impairments by reversing neuroinflammation and synaptic abnormalities. Front. Cell. Neurosci. 2019;13:293. doi: 10.3389/fncel.2019.00293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lennox K.A., Behlke M.A. Cellular localization of long non-coding RNAs affects silencing by RNAi more than by antisense oligonucleotides. Nucleic Acids Res. 2016;44:863–877. doi: 10.1093/nar/gkv1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stein C.A., Hansen J.B., Lai J., Wu S., Voskresenskiy A., Hog A., Worm J., Hedtjarn M., Souleimanian N., Miller P., et al. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2010;38:e3. doi: 10.1093/nar/gkp841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roux B.T., Lindsay M.A., Heward J.A. Knockdown of nuclear-located enhancer RNAs and long ncRNAs using locked nucleic acid GapmeRs. Methods Mol. Biol. 2017;1468:11–18. doi: 10.1007/978-1-4939-4035-6_2. [DOI] [PubMed] [Google Scholar]
- 65.Ahmed A.S.I., Dong K., Liu J., Wen T., Yu L., Xu F., Kang X., Osman I., Hu G., Bunting K.M., et al. Long noncoding RNA NEAT1 (nuclear paraspeckle assembly transcript 1) is critical for phenotypic switching of vascular smooth muscle cells. Proc. Natl. Acad. Sci. U S A. 2018;115:E8660–E8667. doi: 10.1073/pnas.1803725115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li P., Zhou Y., Goodwin A.J., Cook J.A., Halushka P.V., Zhang X.K., Wilson C.L., Schnapp L.M., Zingarelli B., Fan H. Fli-1 governs pericyte dysfunction in a murine model of sepsis. J. Infect. Dis. 2018;218:1995–2005. doi: 10.1093/infdis/jiy451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kassem M.S., Lagopoulos J., Stait-Gardner T., Price W.S., Chohan T.W., Arnold J.C., Hatton S.N., Bennett M.R. Stress-induced grey matter loss determined by MRI is primarily due to loss of dendrites and their synapses. Mol. Neurobiol. 2013;47:645–661. doi: 10.1007/s12035-012-8365-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.