

Abstract
Matrix stiffness promotes hepatocellular carcinoma (HCC) metastasis. This study examined the contribution of lipid metabolic reprogramming to matrix stiffness-induced HCC metastasis. HCC cells were cultured on mechanically tunable polyacrylamide gels and subjected to lipidomic analysis. The key enzyme that responded to matrix stiffness and regulated lipid metabolism was identified. The comparative lipidomic screening revealed that stearoyl-CoA desaturase 1 (SCD1) is a mechanoresponsive enzyme that reprogrammed HCC cell lipid metabolism. The genetic and pharmacological inhibition of SCD1 expression/activity altered the cellular lipid composition, which in turn impaired plasma membrane fluidity and inhibited in vitro invasive motility of HCC cells in response to high matrix stiffness. Knockdown of SCD1 suppressed HCC invasion and metastasis in vivo. Conversely, the overexpression of SCD1 or exogenous administration of its product oleic acid augmented plasma membrane fluidity and rescued in vitro invasive migration in HCC cells cultured on soft substrates, mimicking the effects imposed by high matrix stiffness. In human HCC tissues, collagen content, a marker of increasing matrix stiffness, and increased expression of SCD1 together predicted poor survival of HCC patients. An SCD1-dependent mechanoresponsive pathway that responds to increasing matrix stiffness in the tumor microenvironment promotes HCC invasion and metastasis through lipid metabolic reprogramming.
Keywords: extracellular matrix stiffness, hepatocellular carcinoma, membrane fluidity, lipid metabolism
Graphical abstract
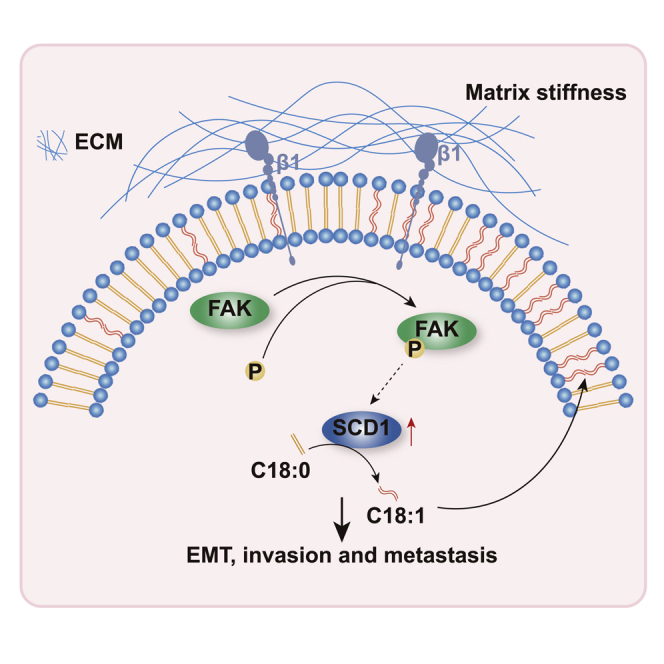
Liu et al. provide evidence that SCD1, the key enzyme for lipid metabolism, is a mechanoresponsive enzyme. Upregulated by stiffness, SCD1 increases the proportion of unsaturated acyl chains in phospholipids, promotes membrane fluidity of HCC cells, and plays a driving role in the invasion and metastasis of HCC.
Introduction
Hepatocellular carcinoma (HCC) is the third most common cause of cancer-related deaths worldwide.1 This dismal outcome has been attributed to patients being diagnosed at late stages and the high frequency of metastasis in HCC. Even among patients with resectable HCC (most of them belonging to early stage), up to 70% will experience intra- or extra-hepatic recurrent metastasis after resection.2 Metastasis remains the major concern that causes death in HCC patients. Elucidating the mechanism underlying HCC metastasis will provide a basis for the prevention of metastasis.3,4
The majority (80%) of HCC develop in liver fibrosis or cirrhosis,5 a condition in which altered biochemical and biophysical microenvironment initiates cancer onset and progression. Matrix stiffness is a characteristic of biophysical signals in the microenvironment and potently regulates cellular behaviors in various physio-pathological processes. Abnormal matrix stiffness plays a significant role during tumor progression. Increasing matrix stiffness is a strong predictor of HCC incidence, regulates HCC cell proliferation, chemotherapeutic response,6 invasion and metastasis,7 and correlates with poor survival in HCC patients.8 The signal from physical matrix stiffness is converted into various biochemical responses to drive malignant behaviors; among these, metabolic input that occurs during matrix stiffness-induced tumor progression is poorly understood.
While glycolysis accounts for rapid proliferation in cancer cells,9 lipid metabolism not only produces more energy but also provides the raw materials for cellular components and signaling molecules for oncogenesis and metastasis.10 Reportedly, matrix stiffness rewires metabolism in the tumors, including HCC. Matrix stiffening activates a mechano-transduction pathway of MAPK-YAP to promote aerobic glycolysis in HCC cells.11 In addition, a matrix stiffness-dependent amino acids aspartate/glutamate exchange between stromal fibroblast and cancer cells initiates tumor progression and metastasis in squamous cell carcinoma.12 These studies highlight the crosstalk between matrix stiffening and tumor metabolic rewiring. However, whether matrix stiffness controls lipid metabolic reprogramming to accelerate tumor progression is yet to be elucidated.
Previously, we found that high matrix stiffness promotes HCC metastasis.13 Herein, we examined the specific contribution of lipid metabolic reprogramming to HCC metastasis triggered by matrix stiffness. Stearoyl-CoA desaturase 1 (SCD1) is an essential mechano-mediator that reprograms HCC cell lipid metabolism. The genetic and pharmacological modulation of SCD1 expression/activity altered the cellular lipid composition, which in turn impaired plasma membrane fluidity and inhibited invasive migration in vitro in HCC cells in response to matrix stiffness and reduced metastasis in vivo. Collagen deposit indicates increased matrix stiffness in the tumor microenvironment. In human HCC tissues, collagen content, a marker of increasing matrix stiffness, and increased expression of SCD1 together predicted poor survival in HCC patients. These findings reveal that an SCD1-dependent mechanoresponsive pathway responds to increasing matrix stiffness (the biomechanical signal in the tumor microenvironment) and promotes HCC invasion and metastasis through lipid metabolic reprogramming, thereby providing a new therapeutic target to attenuate HCC metastasis.
Results
SCD1 is essential for matrix stiffness-induced lipid metabolic programming and cellular invasion
In agreement with previous observations that increasing matrix stiffness induces malignant phenotype,14 HCC cells presented an invasive migration on stiff supports of 25.6 kPa, showing a protrusive appearance with mesenchymal phenotype and well-spread morphology in a three-dimensional (3D) Matrigel overlay culture system (Figure 1A). Since the basement membrane serves as a barrier separating epithelial cells from the surrounding stroma, the decrease in the basement membrane component laminin occurs in an early invasive stage. In Figure 1B, a pronounced decrease in the basement membrane protein laminin β1 was observed in HCC cells at 25.6 kPa, as an indicator of cellular invasion, in conjunction with changes in the epithelial marker E-cadherin and mesenchymal marker N-cadherin (Figure 1C), which highlights that increased matrix stiffness induces mesenchymal shift and cellular invasion.
Figure 1.

Matrix stiffness promotes the invasion of HCC cells
HCC cells were cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) and plastic plates in 3D Matrigel overlay culture. (A) Bright-field images of HCC cells. The in vitro invasion of HCC cells is quantified by the degree of cell scattering using ImageJ software. (B and C) Expression of basement membrane component laminin β1 and EMT-related markers (E-cadherin and N-cadherin) was assessed by immunofluorescence staining. (D) Matrix stiffness modulated the ratio of monounsaturated/saturated fatty acids in all lipid species. (E) Phospholipids levels were determined by lipidomic mass spectrometry. (F) Western blot analyzed stiffness-induced SCD1 protein expression. (G) Hue-saturation-brightness images. The value of the minimum color (blue) was set to generalized polarization (GP) = −1 and the maximum (red) was set to 1. ∗p < 0.05, ∗∗∗p < 0.001.
To determine the contribution of lipid metabolism to matrix stiffness-induced cellular invasion, we performed a mass spectrometry-based lipidomic analysis in HCC cells plated on polyacrylamide gels with tunable matrix stiffness in a 3D Matrigel overlay culture system. A total of 1,060 unique lipids were identified through the integration of positive and negative electrospray ionization mode, including 20 free fatty acids, 502 glycerophospholipids, 505 glycerides, 12 sphingolipids, and 21 cholesteryl esters (Figure S1A). Phosphatidylcholine and phosphatidylserine had increased trends on 25.6 kPa compared with 1.6 kPa by relative quantification (Figure S1B). A total of 125 unique lipids exhibited changes in HCC cells cultured on stiff versus soft supports; among these, 3 phosphatidic acids, 12 phosphatidylcholines, 15 phosphatidylethanolamines, 4 phosphatidylglycerols, 10 phosphatidylinositols, and 12 phosphatidylserines were significantly increased (Figures S1C and S2). First, we noted that the ratio of monounsaturated fatty acid/saturated fatty acid (MUFA/SFA) was increased across most lipid classes (Figure 1D). Among these lipids, membrane phospholipids containing the C18:1 unsaturated fatty acid was predominantly increased in MHCC97H and PLC/PRF/5 (Figure 1E), with a significant increase of C16:1 fatty acyl chain in PLC/PRF/5. Meanwhile, membrane phospholipids containing C18:0 were significantly decreased in MHCC97H and showed a non-significant decreasing trend in PLC/PRF/5. These findings suggest that SCD1 might be responsible for matrix stiffness-induced lipid reprogramming because SCD1 is a rate-limiting enzyme in monounsaturated fatty acid synthesis. The protein levels of SCD1 increased proportionally to stiffness (Figure 1F). The membrane content phospholipids affect the invasion of cancer cells by modulating plasma membrane fluidity; higher plasma membrane fluidity was detected in HCC cells cultured on stiff versus soft supports, which was measured by a biophysical parameter-generalized polarization (GP) value inversely correlated with membrane fluidity (MHCC97H, 0.07 versus 0.41; PLC/PRF/5 0.09 versus 0.36, Huh7 -0.01 versus 0.42; Hep3B 0.13 versus 0.38; all p < 0.05) (Figure 1G). These phenomena indicate that SCD1 might be involved in matrix stiffness-trigged lipid metabolic reprogramming, which in turn influences membrane fluidity and cellular invasion.
Next, we investigated whether SCD1 is required for matrix stiffness-induced lipid metabolic reprogramming and cellular invasion. We generated HCC cells expressing short hairpin RNAs (shRNA) against SCD1 and tested the impact of SCD1 on the mechanoresponsive pathway. The knockdown of SCD1 significantly attenuated the HCC cell-invasive phenotype at high matrix stiffness; these cells were round with decreased N-cadherin and increased E-cadherin on rigid matrix substrates (Figures 2A–2C). These data demonstrate that high matrix stiffness-induced HCC cell invasion is SCD1 dependent. Importantly, the knockdown of SCD1 reversed lipid metabolism reprogramming induced by high matrix stiffness, as displayed by the notably reduced MUFA/SFA ratio and membrane phospholipids containing the C18:1 fatty acyl chain (Figures 2D and 2E). This led to a significant blocking of membrane fluidity of HCC cells on high matrix stiffness at 25.6 kPa (GP value, MHCC97H, 0.38 versus 0.11; PLC/PRF/5, 0.55 versus 0.11; p < 0.05) (Figure 2F). The pharmacological disruption of SCD1 activity using SCD1 inhibitor CAY10566 significantly tempered matrix stiffness-mediated regulation of cellular invasion, lipid metabolism reprogramming, and plasma membrane fluidity in HCC cells cultured on stiff supports (Figures 3A–3D). These data indicate an essential role for SCD1 in mediating the response of HCC cells to a matrix stiffness-driven lipid metabolic reprogramming and pro-invasive cue.
Figure 2.

SCD1 is required for matrix stiffness-induced invasion in HCC cells
HCC cells expressing control (shControl) or SCD1 shRNA (shSCD1) were cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) and plastic plates in 3D Matrigel overlay culture. (A) Efficiency of SCD1 knockdown was probed by western blot. (B) Bright-field images of HCC cells. The in vitro invasion of HCC cells is quantified by the degree of cell scattering using ImageJ software. (C) Expression of EMT-related markers E-cadherin and N-cadherin was assessed by western blot. HCC cells were treated with or without oleic acid (OA) (the product of SCD1). (D) Ratio of monounsaturated/saturated fatty acids in all lipid species. (E) Phospholipid levels were determined by lipidomic mass spectrometry. (F) Hue-saturation-brightness images of HCC cells. The value of the minimum color (blue) was set to generalized polarization (GP) = −1, and the maximum (red) was set to 1. ∗p < 0.05, ∗∗∗p < 0.001.
Figure 3.

Pharmacological disruption of SCD1 activity suppresses matrix stiffness-induced invasion in HCC cells
HCC cells were cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) in 3D Matrigel overlay culture and treated with DMSO or SCD1 inhibitor CAY10566. (A) Bright-field images of HCC cells. The in vitro invasion of HCC cells is quantified by the degree of cell scattering using ImageJ software. (B) Ratio of monounsaturated/saturated fatty acid in all lipid species. (C) Phospholipids levels were determined by lipidomic mass spectrometry. (D) Hue-saturation-brightness images of HCC cells. The value of the minimum color (blue) was set to generalized polarization (GP) = −1, and the maximum (red) was set to 1. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
To further characterize the critical function of SCD1 during matrix stiffness-induced cellular invasion, SCD1 was overexpressed in HCC cells. Remarkably, the upregulation of SCD1 induced cell spreading or rescued cellular invasion of HCC cells on the soft supports (Figures 4A and 4B) with increased N-cadherin and reduced E-cadherin levels (Figure 4C). The altered lipid metabolism was observed with respect to increased MUFA/SFA ratio and membrane phospholipids containing the C18:1 fatty acyl chain, and increased plasma membrane fluidity was detected in SCD1-overexpressing HCC cells cultured on soft substrates (MHCC97H, 0.03 versus 0.35; PLC/PRF/5, 0.04 versus 0.39; all p < 0.05) (Figures 4D–4F). However, the overexpression of SCD1 did not show any statistically significant increase in the plasma membrane fluidity of HCC cells at 25.6 kPa (Figure 4F), implying that SCD1 product stimulated by high matrix stiffness is sufficient to maintain membrane fluidity for an invasive phenotype. These results suggested that SCD1 overexpression and its downstream regulators rescue the invasive motility of HCC cells on the soft supports, thereby mimicking the effects imposed by stiff supports.
Figure 4.

Upregulation of SCD1 expression rescues the invasion of HCC cells cultured on soft matrix substrates
HCC cells transfected with mock or SCD1-expressing plasmid (ovSCD1) were cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) and plastic plates in 3D Matrigel overlay culture. (A) SCD1 protein expression was analyzed by western blot. (B) Bright-field images of HCC cells. The in vitro invasion of HCC cells was quantified by the degree of cell scattering using ImageJ software. (C) The expression of EMT-related markers (E-cadherin and N-cadherin) was assessed by western blot. (D) Ratio of monounsaturated/saturated fatty acids in all lipid species. (E) Phospholipids levels were determined by lipidomic mass spectrometry. (F) Hue-saturation-brightness images of HCC cells. The value of the minimum color (blue) was set to generalized polarization (GP) = −1 and the maximum (red) was set to 1. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Taken together, the above data strongly support SCD1 as a mechanoresponsive target mediating matrix stiffness-induced lipid programming and cellular invasion in HCC cells.
SCD1 is upregulated by high matrix stiffness
To determine whether SCD1-mediated protrusive invasion in HCC cells could be reversed if high-stiffness matrices returned to a low level, we first cultured HCC cells on either high- or low-stiffness matrices until the pattern of invasive migration was clear and then transferred the HCC cells to another matrix stiffness environment. The transfer from low- to high-stiffness matrices raised the level of SCD1 and protrusive migration in the new stiffness environment, whereas the transfer from high to low matrices resulted in an SCD1 decrease and a retraction of protrusions and confined growth (Figures 5A and 5B). However, these invasive phenotypic transitions were lost in SCD1 knockdown cells (Figure 5A). These results suggest that the local matrix stiffness environment determines the SCD1-mediated invasive migration strategy in HCC cells.
Figure 5.

Matrix stiffness modulates SCD1 expression
HCC cells expressing control (shControl) or SCD1 shRNA (shSCD1) were grown on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) and then switched from 1.6 to 25.6 kPa (1.6–25.6 kPa) or from 25.6 to 1.6 kPa (25.6–1.6 kPa) in 3D Matrigel overlay culture (A) Representative bright-field images of HCC cells. The in vitro invasion of HCC cells is quantified by the degree of cell scattering using ImageJ software. (B) SCD1 expression was analyzed by western blot. (C) qRT-PCR analysis of SCD1 mRNA expression in HCC cells on polyacrylamide hydrogels with the indicated rigidities (1.6, 3.2, 6.4, and 25.6 kPa) in 3D Matrigel overlay culture. (D) Effect of matrix stiffness on SCD1 protein stability was analyzed by western blot in the presence of cycloheximide (CHX). The half-life (T1/2) of SCD1 was calculated. HCC cells were collected at the indicated time points after exposure to CHX, and cell lysates were subjected to immunoblotting. (E) SCD1 protein expression was increased in HCC cells treated with the proteasome inhibitor MG-132. (F) The level of ubiquitinated SCD1 was reduced in HCC cells cultured on stiff matrix. (G and H) HCC cells were treated with the anti-β1-integrin antibody or p-FAK inhibitor PF562271 (PF). SCD1 expression was analyzed by western blot. ∗∗∗p < 0.001.
As described above, SCD1 protein expression was elevated in HCC cells on stiff versus soft matrix (Figure 1F), whereas its mRNA expression under different matrix rigidities did not differ significantly (Figure 5C). Surprisingly, in the presence of a protein synthesis inhibitor, cycloheximide, SCD1 protein in HCC cells on the soft matrix degraded faster than that on the stiff matrix (Figure 5D). Moreover, treatment with a proteasome inhibitor MG-132 increased the expression of SCD1 protein in HCC cells (Figure 5E). To determine whether stiffness could influence the ubiquitination status of SCD1 and thus promote its degradation, ubiquitination analyses were performed under different matrix rigidities. The ubiquitination level of SCD1 was markedly increased in HCC cells cultured on the soft versus stiff matrix (Figure 5F). Therefore, the SCD1 protein underwent accelerated proteasomal degradation in HCC cells on soft substrates, whereas it was more stabilized in cells cultured on stiff matrices.
To determine the pathway associated with SCD1 upregulation by matrix stiffness, we utilized a function-blocking anti-β1-integrin antibody and pharmacological FAK inhibitor PF562271. The anti-β1-integrin antibody and PF562271 reversed the high matrix stiffness-mediated upregulation of SCD1 (Figures 5G and 5H).
Together, these results indicate that high matrix stiffness-mediated SCD1 increase occurs through integrin β1/FAK signaling pathway activation and downregulation of SCD1 ubiquitin-proteasomal degradation.
Oleic acid, the product of SCD1, reverses SCD1 knockdown-mediated inhibition of cellular invasion
SCD1 catalyzes de novo synthesis of monounsaturated fatty acids from saturated fatty acids, especially stearic acid (18:0) and palmitic acid (16:0) to their Δ9-cis monounsaturated oleic acid (OA) (18:1) and palmitoleic acid (16:1).15 As described above, knockdown of SCD1 inhibited matrix stiffness-induced cellular invasion in HCC cells by reduced membrane phospholipids containing the C18:1 fatty acyl chain (Figures 2B and 2E). Next, we evaluated the impact of the main lipid product of SCD1, OA, on the matrix stiffness-induced lipid metabolic reprogramming, plasma membrane fluidity, and cellular invasion. The HCC cells were incubated with a non-cytotoxic concentration (100 μM) of OA, and then membrane fluidity and invasive cellular motility were analyzed. The supplementation of OA reversed the SCD1 knockdown-mediated E-cadherin increase and N-cadherin decrease in HCC cells on the stiff supports (Figure 2C). The addition of OA significantly increased the basal membrane fluidity and invasive motility in HCC cells on the soft matrices. Notably, OA significantly reversed the SCD1 knockdown-mediated inhibition of membrane fluidity and invasive migration of HCC cells on stiff supports (Figures 6A, 6D, S3A, and S3D). These results indicate that OA is an effector in matrix stiffness-SCD1-mediated membrane fluidity and cellular invasion.
Figure 6.

Membrane fluidity mediates matrix stiffness-induced cellular invasion in HCC cells
(A) MHCC97H cells expressing control (shControl) or SCD1 shRNA (shSCD1) were grown on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) in the presence of oleic acid (OA) in 3D Matrigel overlay culture. (B) MHCC97H cells were transfected with mock or SCD1-expressing plasmid (ovSCD1) and cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) in 3D Matrigel overlay culture and then treated with membrane-stabilizing agent UDCA. (C) MHCC97H cells expressing control (shControl) or SCD1 shRNA (shSCD1) were cultured on polyacrylamide hydrogels with the indicated rigidities (1.6 and 25.6 kPa) in 3D Matrigel overlay culture and treated with membrane-agonist compound BA. The in vitro invasion of HCC cells was quantified by the degree of cell scattering using ImageJ software and GraphPad Prism 8. (D) Hue-saturation-brightness images of MHCC97H cells expressing shControl or shSCD1 in the presence of OA. (E) Hue-saturation-brightness images of MHCC97H cells transfected with mock or ovSCD1 treated with/without UDCA. (F) Hue-saturation-brightness images of MHCC97H cells expressing shControl or shSCD1 treated with/without compound BA. ∗∗∗p < 0.001.
Increased membrane fluidity mediates matrix stiffness-induced cellular invasion
Membrane fluidization promotes the invasion of cancer cells. As described above, increased matrix stiffness promotes plasma membrane fluidity. To determine the role of membrane fluidity in matrix stiffness-induced cellular invasion, HCC cells were pretreated with the membrane-stabilizing agent ursodeoxycholic acid (UDCA) or the compound benzyl alcohol (BA) that fluidifies plasma membranes. Pre-incubation of HCC cells with UDCA significantly attenuated the high matrix stiffness-mediated increase in membrane fluidity and invasive motility (Figures 6B, 6E, S3B, and S3E). Conversely, BA treatment significantly enhanced cell membrane fluidity in HCC cells on soft supports, leading to the cells acquiring invasive migration (Figures 6C, 6F, S3C, and S3F). More importantly, UDCA significantly abrogated the SCD1 overexpressing increased membrane fluidity and invasive motility in HCC cells on soft supports (Figures 6B, 6E, S3B, and S3E). Concurrently, BA treatment reversed the SCD1-knockdown inhibition of membrane fluidity and invasive motility in HCC cells on stiff supports (Figures 6C, 6F, S3C, and S3F). Collectively, these data indicate a pivotal function of membrane fluidity in the matrix stiffness-SCD1 axis-induced cell-invasive motility.
Knockdown of SCD1 suppressed HCC invasion and metastasis in vivo
To investigate the role of SCD1 in HCC progression in vivo, we employed a metastatic MHCC97H HCC xenograft model, which recapitulates the metastasis of human HCC.4 The knockdown of SCD1 significantly attenuated an invasive phenotype in MHCC97H cells cultured on high matrix stiffness in a 3D Matrigel culture system (Figure 2B), indicating that the SCD1 mechanoresponsive pathway is intact in these HCC cells. Mice were subcutaneously injected with a cell suspension of MHCC97HshSCD1or MHCC97HshControl cells. As shown in Figure 7A, the growth rate of tumors derived from MHCC97HshSCD1 cells was significantly slower than that of control tumors. Similarly, the weight of tumors in MHCC97HshSCD1group was significantly reduced compared with the control group (Figure 7B), suggesting that SCD1 promotes tumor growth and progression. In the tumors from the MHCC97HshSCD1 group, SCD1, Ki-67, N-cadherin, and α-SMA expression was significantly decreased, whereas that of E-cadherin was markedly increased (Figure 7C). To further define the role of SCD1 in HCC metastasis, we orthotopically implanted the subcutaneous tumors into the livers of nude mice and allowed tumor development for 7 weeks. Consistent with the results of subcutaneous tumors, the volume and weight of liver tumors in situ in MHCC97HshSCD1 group were reduced than those in the control group (Figure 7D). Importantly, MHCC97HshSCD1 tumors presented a nested architecture characterized by a less-invasive tumor edge and a significant decrease in the number of distant lung metastases compared with the control tumors (Figure 7E). Together, these results support a critical role of SCD1 in modulating HCC invasion and metastasis in vivo.
Figure 7.

Knockdown of SCD1 suppresses HCC invasion and metastasis in vivo
(A) The growth curve of subcutaneous tumors derived from SCD1 knockdown or control MHCC97H cells (shSCD1 versus shControl) in nude mice. (B) Weight of subcutaneous tumors from SCD1 knockdown or control MHCC97H cells (shSCD1 versus shControl). (C) Expression of SCD1, α-SMA, Ki67, and EMT-related markers (E-cadherin and N-cadherin) was analyzed by immunohistochemistry. (D) Subcutaneous tumors were orthotopically implanted into the livers of nude mice and allowed tumor development. (E) In situ liver tumors derived from SCD1 knockdown versus shControl MHCC97H cells presented a less invasive tumor edge and a decrease in the number of distant lung metastases, as indicated by H&E staining. ∗∗p < 0.01, ∗∗∗p < 0.001.
Increased collagen content and SCD1 upregulation synergistically predict poor outcome in HCC patients
Next, we investigated whether the SCD1 mechanoresponsive pathway had a significant role in human HCC progression. SCD1 protein was highly expressed in tumor tissues compared with paired non-tumoral tissues in 12 HCC patients (Figure 8A). SCD1 expression and intratumoral collagen content were determined in the tissue microarrays from a cohort of 110 HCC patients. The clinicopathologic characteristics of patients are summarized in Table S1. SCD1 protein expression was restricted to the HCC cells (Figure 8B). As a surrogate marker for tissue rigidity, collagen content determined by Masson trichrome staining was automatically quantified using Image Pro Plus 6.0 software. The collagen content was significantly correlated with the level of SCD1 expression in HCC (Figure 8B). The patients with stiffer tumors (high collagen content ≥11.98%) had a poor 5-year recurrence-free survival (RFS) and overall survival (OS) of 16.90% and 52.73% compared with 54.70% and 74.06% in patients with compliant tumors (low collagen content <11.98%) (p < 0.01; Figure 8C), respectively. The patients with high SCD1 expression had more unfavorable outcome than those with low SCD1 expression (5-year RFS: 18.20% versus 54.32%; 5-year OS: 51.40% versus 76.70%) (p < 0.01; Figure 8D). Importantly, the level of SCD1 expression, together with collagen content, indicated that matrix stiffness stratified HCC patient prognosis. Patients with collagenhigh/SCD1high tumors had markedly poor outcomes with a 5-year RFS and OS of 6.02% and 44.44% compared with 65.00% and 81.50% in patients with collagenlow/SCD1low tumors (p < 0.001, Figure 8E). Furthermore, patients with tumors presented as either collagenhigh or SCD1high had intermediate survival outcomes (5-year RFS: 36.80% and 39.40%; 5-year OS: 68.40% and 62.80%, respectively; Figure 8E). These indicated the cooperative effect of increasing matrix stiffness and SCD1 upregulation on HCC progression. Similar to the importance of tumor size, the association between collagenhigh/SCD1high tumors and poor prognosis was independent of other clinicopathological parameters, including gender, and Child-Pugh and BCLC staging (Tables S2 and S3). In addition, collagenhigh/SCD1high tumors predicted poor survival in The Cancer Genome Atlas-HCC cohort (Figure 8F). Thus, these findings indicate that increasing matrix stiffness in the HCC tumor microenvironment in concert with SCD1 upregulation promotes HCC recurrence and progression, and predicts poor prognosis.
Figure 8.

Increased collagen content and SCD1 upregulation synergistically predict poor prognosis in HCC patients
(A) SCD1 protein expression in the tumor and non-tumoral tissues detected by western blot. T, tumor tissues; N, non-tumoral tissues. (B) Representative images of HCC tissues analyzed for intratumoral collagen content by Masson’s trichrome staining and SCD1 expression by immunohistochemistry staining. Correlation analysis between intratumoral collagen content and SCD1 expression. (C and D) Kaplan-Meier curve of recurrence-free survival and OS for HCC patients stratified by intratumoral collagen content or SCD1 expression. (E) Kaplan-Meier curve of recurrence-free survival and OS for HCC patients stratified by intratumoral collagen content and SCD1 expression (collagenhigh/SCD1high versus collagenhigh/SCD1low versus collagenlow/SCD1high versus collagenlow/SCD1low tumors). (F) Kaplan-Meier curve of OS for HCC patients stratified by collagen content and SCD1 expression in The Cancer Genome Atlas-HCC cohort. (G) Schematic of the proposed mechanism that an SCD1-dependent mechanoresponsive pathway responsive to increasing matrix stiffness promotes HCC invasion and metastasis through lipid metabolic reprogramming. ECM, extracellular matrix; β1, integrin β1; FAK, focal adhesion kinase; EMT, epithelial mesenchymal transformation.
Discussion
Matrix stiffness is a physical cue in the tumor environment that significantly increases carcinogenesis and tumor progression.16,17 To the best of our knowledge, this is the first study that reveals that the SCD1-mediated mechanoresponsive pathway is upregulated by high matrix stiffness. This leads to lipid metabolic reprogramming and increased plasma membrane fluidity, which in turn promotes HCC cell-invasive migration and metastasis. This study provided a new potential approach by decreasing SCD1 expression/activity to reduce matrix stiffness-induced HCC metastatic dissemination.
HCC frequently develops and progresses in the fibrotic or cirrhotic liver with increased matrix rigidity, which has led to detrimental interactions between the altered biomechanical environment and HCC.18 Moreover, most of the HCC lesions are stiffer than their surrounding liver tissues.19 The increase in extra-/intratumoral tissue rigidity or matrix stiffness plays a significant role in HCC progression.20,21 Reportedly, the mechanical forces derived from matrix stiffness underlie the physio-pathological processes, including development,22 inflammation,23 and cancer.24, 25, 26 For example, stem cells differentiate into specific lineages in response to various matrix elasticities;27 mammary epithelial cells grown on the matrix with rigidity similar to the stiffness of breast cancer exhibit EMT-morphological features and invade into the basement membrane.7 The physical signal derived from matrix stiffness is a characteristic feature in the mechanical properties of extracellular matrix and transformed into intracellular biochemical responses to direct cancer cell behavior.28, 29, 30 However, metabolic input during matrix stiffness-induced tumor metastasis is largely unknown. Only a few studies have yet addressed the interconnection between matrix stiffening and tumor metabolic rewiring in cancer. This study explored the direct connection between matrix stiffness and lipid metabolic reprogramming. We used lipidomic analysis and identified changes in the lipid composition in HCC cells in response to matrix stiffness. Moreover, SCD1 was identified as an unanticipated target enzyme modulated by matrix stiffness. Lipid metabolic reprogramming initiated by matrix stiffness was orchestrated in an SCD1-dependent mechanoresponsive manner.
In this study, we showed that the expression level and activity of SCD1 affected the matrix stiffness-induced MUFA/SFA ratio and the lipid metabolism in HCC cells. An increase in matrix stiffness-induced fatty acid desaturation was reversed by SCD1 inhibition. Membrane fluidity alteration has been described in various pathologies, including cancer, which is regulated by modification of MUFA/SFA ratio. The type of fatty acids in the structure of the cell membrane can influence its fluidity, which is closely related to the fatty acyl chain saturation of phospholipid molecules.31 Saturated fatty acids with a straight, rigid chain form an ordered structure and the phospholipids form a rigid bilayer; hence, the fluidity of membranes formed by saturated fatty acids is low, while membranes formed by cis-unsaturated fatty acids are flexible. Among these lipids in membranes, phospholipids play a critical role in the structural components of membranes on cell growth, differentiation, and motility.32 As components in the two-layered sheets of lipid molecules, phospholipids with a C18:1 monounsaturated fatty acyl chain can induce membrane curvature,33 which may account for the increased membrane fluidity in response to elevated matrix stiffness in this study. In addition, SCD1 inhibition antagonized the effects of high matrix stiffness on membrane fluidity, a prominent feature of cancer cell invasion. SCD1 knockdown-mediated inhibition of membrane fluidization and cell invasion is reversed by pre-incubation with exogenous OA, one of the SCD1 product. Also, a perturbation of membrane fluidity by membrane-stabilizing agent ameliorates a direct action of high matrix stiffness on cellular invasion. Furthermore, SCD1 overexpression led to an increase in fatty acid desaturation and rescued the invasive motility of HCC cells cultured on soft supports. SCD1 knockdown inhibited HCC metastasis in vivo. Taken together, we propose a new mechanism in which a matrix stiffness-SCD1 axis regulates lipid metabolism to promote HCC invasion and metastasis, thereby suggesting that SCD1 is a novel therapeutic target to reduce matrix stiffness-induced HCC metastasis.
In this study, we show that the pro-invasive activity conferred by matrix stiffness is through SCD1-mediated fatty acid desaturation, providing a basis for evaluating an SCD1 target for inhibiting HCC metastasis. SCD1 inhibition will reduce fatty acid desaturation, modify a pathological interaction between matrix stiffness and lipid metabolism, and decrease membrane fluidity, thus alleviating matrix stiffness-induced cellular invasion. An important feature of cancer cells is the enrichment of unsaturated fatty acids in lipid composition to form various cell membranes, supporting tumorigenesis and malignant progression.34 SCD family converting saturated fatty acids to Δ9-monounsaturated fatty acids is correlated with cancer aggressiveness and poor outcomes of patients.35 Studies have shown that SCD1 is involved in the biological behaviors of cancer cell proliferation, migration, metastasis, and maintaining cancer stem cell characteristics of cancer stem cells,36 implicating that it is a promising target for anticancer therapy. SCD1 inhibitors for the treatment of cancer have been developed and preclinically tested. Currently, there are two SCD isoforms in humans, SCD1 and SCD5,37 that contribute to fatty acid desaturation and exert a high activity on C16 or C18 substrates. Since SCD1 is ubiquitously expressed in various tissues, including the liver, there are potential off-target side effects of SCD inhibition. Surprisingly, SCD1 inhibitors have shown a favorable tolerance in clinical studies.38
Collagen deposition is a surrogate marker for increasing matrix stiffness in the tumor microenvironment and is associated with tumor progression.39 In this study, collagen content, together with increased expression of SCD1, predicted the elevated risk of metastatic relapse in a relatively large cohort of HCC patients, thereby indicating that increased matrix stiffness in the tumor microenvironment leads to SCD1 upregulation that promotes HCC recurrence and metastasis.
Nevertheless, this study has some limitations. Firstly, the direct molecular mechanism by which matrix stiffness fine-tuned SCD1 expression is not determined. Although our data indicate that high matrix stiffness promotes SCD1 expression through activated integrin β1/FAK signaling and decreased SCD1 proteasomal degradation, how the FAK pathway affects the ubiquitination of SCD1 has not been dissected in this study. Also, the ubiquitination pathway that mediates SCD1 ubiquitination and then affects its protein expression level is not clarified. Moreover, how OA (the main product of SCD1) modulates the downstream targets has not been resolved. Secondly, we used pharmacological and genetic approaches and showed that the level of SCD1 expression affected matrix stiffness-triggered EMT. How SCD1 triggered EMT is not clarified in this study. One study reported that a metabolic acyl-CoA synthetase (ACSL)/SCD pathway causes EMT, which promotes migration and invasion of colon cancer cells.40 Another study showed that hyperglycemia induces EMT in colorectal cancer cells via the SCD1-MUFA axis and by suppressing PTEN.41 Thus, it will be interesting to explore whether the matrix stiffness-SCD1 axis induces EMT in HCC cells through the same or different mechanism.
In conclusion (Figure 8G), this study provides a new mechanistic linking matrix stiffness-SCD1 axis to lipid metabolic reprogramming and HCC metastasis, indicating that the SCD1-dependent mechanoresponsive pathway is a potential therapeutic target for anti-HCC metastasis.
Materials and methods
Quantification of membrane fluidity
Plasma membrane fluidity was quantified by the laser scanning platform as described previously.41,42 Living cells were labeled with polarity-sensitive membrane probe laurdan (6-dodecanoyl-2-dimethylamino-napthalene) (Aladdin, China), which provides information about membrane fluidity by the shift in the emission spectrum. The laurdan intensity images were obtained under excitation at 405 nm, and fluorescence emission was recorded simultaneously through two channels of 400–460 and 470–530 nm. Cell membrane fluidity was expressed in the form of pseudo-colored GP values by ImageJ software (National Institutes of Health, Bethesda, MD, USA). The GP values were calculated using the formula: GP = (I400-460 − GI470-530)/(I400-460 + GI 470-530) where G (the calibration factor) was obtained for laurdan in dimethyl sulfoxide (DMSO) (Sigma) and I indicated the intensity of pixels in the images acquired in specified channels. The GP value was calculated in multiple regions of the cell membrane (n = 10) per group, which is inversely correlated with membrane fluidity.
HCC tissues
This study was approved by the Zhongshan Hospital of Fudan University Research Ethics Committee, and written informed consent was obtained from each patient. HCC and matched non-tumoral liver tissues from 12 patients who underwent curative liver resection and did not receive preoperative therapy were collected from Zhongshan Hospital of Fudan University from July 2015 to August 2017. A total of 110 HCC specimens collected between August 2010 and June 2011 were used for tumor tissue microarrays (TMAs). Duplicated tissue cores of 1-mm diameter were obtained from the same tissue block. The TMAs were constructed as described previously43 and stained for SCD1 for retrospective analysis. The TMAs were also stained with Masson’s trichrome to evaluate the degree of collagen deposition. SCD1 staining was scored blindly by two investigators according to the following criteria: 0 for no expression, 1+ for very weak expression, 2+ for moderate expression in >75% of tumor cells, and 3+ for strong expression in >75% of tumor cells. The collagen content in tumors was quantified in the images with five random fields per tumor using the ImageJ method as described previously.44
In vivo study
All animal experiments were approved by the Committee on Animal Research of Zhongshan Hospital (Shanghai, China) and performed in accordance with the guidelines formulated by Shanghai Medical Experimental Animal Care Commission.
A metastatic model of human HCC in nude mice using MHCC97H cells was established as described previously.45 In brief, 5 × 106 HCC cells (MHCC97HshSCD1or MHCC97HshControl) suspended in PBS were injected into the upper left flank region of BALB/c nude mice (male, 4–6 weeks old, weighting 18–20 g) (Shanghai SLAC Laboratory Animal, China) (n = 7/group). Tumor growth was monitored by measuring the length and width of the tumor at the inoculation site. The tumor volume was calculated using the following formula: V = a × b2/2, where a is the largest diameter and b is the smallest diameter. When the tumor reached 10 mm in length, one mouse in each group was sacrificed, and the subcutaneous tumor was minced into small pieces of equal volume (1 × 1 × 1 mm3), which were orthotopically transplanted into the livers in 14 nude mice (n = 7/group). Subsequently, the mice were euthanized, and lung metastasis was assessed at 7 weeks post-tumor implantation. The tumors were fixed in 4% formaldehyde or snap-frozen in liquid nitrogen for further analysis.
Statistical analysis
Statistical analysis was performed using SPSS 25.0 (SPSS, Chicago, IL, USA) and GraphPad Prism8.0 (GraphPad Software, San Diego, CA, USA). Continuous variables are expressed as mean ± standard deviation and analyzed by Student’s t test between two groups or one-way analysis of variance for multiple groups. Categorical variables are expressed as absolute number (n) and compared using χ2 test or Fisher’s exact test. OS and RFS between different groups were generated using the Kaplan-Meier method and the log rank test. Cox’s proportional hazards regression model was used to analyze independent prognostic factors. A p value of < 0.05 (two-tailed) was considered statistically significant.
Other materials and methods
For more information on other materials and methods, please refer to the supplemental information.
Acknowledgments
We would like to express our sincere gratitude to Prof. Jia Fan and Prof. Jian Zhou for advice and help in the analysis of tissue samples. This work was supported by the National Natural Science Foundation of China (81972715).
Author contributions
R.-X.C., Z.-G.R., and X.-R.Y. participated in the conception and design. R.-X.C. and J.-F.C. designed the experiments and supervised the project. H.-H.L., Y.X., C.-J.L., S.-J.H., X.-H.L., R.Z., Jie Chen, Jun Chen, and D.-M.G. performed the experiments and analyzed the data. R.-X.C. and H.-H.L. wrote the manuscript. All authors contributed in the interpretation of results, manuscript preparation, and revisions. All authors approved the final version to be published.
Conflicts of interest
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.015.
Contributor Information
Xin-Rong Yang, Email: yang.xinrong@zs-hospital.sh.cn.
Zheng-Gang Ren, Email: ren.zhenggang@zs-hospital.sh.cn.
Rong-Xin Chen, Email: chen.rongxin@zs-hospital.sh.cn.
Supplemental information
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Ishizawa T., Hasegawa K., Aoki T., Takahashi M., Inoue Y., Sano K., et al. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology. 2008;134:1908–1916. doi: 10.1053/j.gastro.2008.02.091. [DOI] [PubMed] [Google Scholar]
- 3.Ye Q.H., Qin L.X., Forgues M., He P., Kim J.W., Peng A.C., et al. Predicting hepatitis B virus-positive metastatic hepatocellular carcinomas using gene expression profiling and supervised machine learning. Nat. Med. 2003;9:416–423. doi: 10.1038/nm843. [DOI] [PubMed] [Google Scholar]
- 4.Tang Z.Y., Ye S.L., Liu Y.K., Qin L.X., Sun H.C., Ye Q.H., et al. A decade's studies on metastasis of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2004;130:187–196. doi: 10.1007/s00432-003-0511-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Affo S., Yu L.X., Schwabe R.F. The role of cancer-associated fibroblasts and fibrosis in liver cancer. Annu. Rev. Pathol. 2017;12:153–186. doi: 10.1146/annurev-pathol-052016-100322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrader J., Gordon-Walker T.T., Aucott R.L., van Deemter M., Quaas A., Walsh S., et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192–1205. doi: 10.1002/hep.24108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fattet L., Jung H.Y., Matsumoto M.W., Aubol B.E., Kumar A., Adams J.A., et al. Matrix rigidity controls epithelial-mesenchymal plasticity and tumor metastasis via a mechanoresponsive EPHA2/LYN complex. Dev. Cell. 2020;54:302–316.e307. doi: 10.1016/j.devcel.2020.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marasco G., Colecchia A., Colli A., Ravaioli F., Casazza G., Bacchi Reggiani M.L., et al. Role of liver and spleen stiffness in predicting the recurrence of hepatocellular carcinoma after resection. J. Hepatol. 2019;70:440–448. doi: 10.1016/j.jhep.2018.10.022. [DOI] [PubMed] [Google Scholar]
- 9.Akins N.S., Nielson T.C., Le H.V. Inhibition of glycolysis and glutaminolysis: an emerging drug discovery approach to combat cancer. Curr. Top. Med. Chem. 2018;18:494–504. doi: 10.2174/1568026618666180523111351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo X., Cheng C., Tan Z., Li N., Tang M., Yang L., et al. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer. 2017;16:76. doi: 10.1186/s12943-017-0646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q.P., Luo Q., Deng B., Ju Y., Song G.B. Stiffer matrix accelerates migration of hepatocellular carcinoma cells through enhanced aerobic glycolysis via the MAPK-YAP signaling. Cancers (Basel) 2020;12:490. doi: 10.3390/cancers12020490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bertero T., Oldham W.M., Grasset E.M., Bourget I., Boulter E., Pisano S., et al. Tumor-stroma mechanics coordinate amino acid availability to sustain tumor growth and malignancy. Cell Metab. 2019;29:124–140.e110. doi: 10.1016/j.cmet.2018.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong Y., Zheng Q., Wang Z., Lin X., You Y., Wu S., et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J. Hematol. Oncol. 2019;12:112. doi: 10.1186/s13045-019-0795-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stowers R.S., Shcherbina A., Israeli J., Gruber J.J., Chang J., Nam S., et al. Matrix stiffness induces a tumorigenic phenotype in mammary epithelium through changes in chromatin accessibility. Nat. Biomed. Eng. 2019;3:1009–1019. doi: 10.1038/s41551-019-0420-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hulver M.W., Berggren J.R., Carper M.J., Miyazaki M., Ntambi J.M., Hoffman E.P., et al. Elevated stearoyl-CoA desaturase-1 expression in skeletal muscle contributes to abnormal fatty acid partitioning in obese humans. Cell Metab. 2005;2:251–261. doi: 10.1016/j.cmet.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levental K.R., Yu H., Kass L., Lakins J.N., Egeblad M., Erler J.T., et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei S.C., Fattet L., Tsai J.H., Guo Y., Pai V.H., Majeski H.E., et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015;17:678–688. doi: 10.1038/ncb3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baglieri J., Brenner D.A., Kisseleva T. The role of fibrosis and liver-associated fibroblasts in the pathogenesis of hepatocellular carcinoma. Int. J. Mol. Sci. 2019;20:1723. doi: 10.3390/ijms20071723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie L.T., Gu J.H., Chai W.L., Chen R.D., Zhao Q.Y., Kong D.X., et al. Pre-operative detection of liver fibrosis in hepatocellular carcinoma patients using 2D shear wave elastography: where to measure? Ultrasound Med. Biol. 2020;46:1412–1423. doi: 10.1016/j.ultrasmedbio.2020.02.012. [DOI] [PubMed] [Google Scholar]
- 20.Masuzaki R., Tateishi R., Yoshida H., Goto E., Sato T., Ohki T., et al. Prospective risk assessment for hepatocellular carcinoma development in patients with chronic hepatitis C by transient elastography. Hepatology. 2009;49:1954–1961. doi: 10.1002/hep.22870. [DOI] [PubMed] [Google Scholar]
- 21.Shili-Masmoudi S., Wong G.L., Hiriart J.B., Liu K., Chermak F., Shu S.S., et al. Liver stiffness measurement predicts long-term survival and complications in non-alcoholic fatty liver disease. Liver Int. 2020;40:581–589. doi: 10.1111/liv.14301. [DOI] [PubMed] [Google Scholar]
- 22.Vining K.H., Mooney D.J. Mechanical forces direct stem cell behaviour in development and regeneration. Nat. Rev. Mol. Cell Biol. 2017;18:728–742. doi: 10.1038/nrm.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Solis A.G., Bielecki P., Steach H.R., Sharma L., Harman C.C.D., Yun S., et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature. 2019;573:69–74. doi: 10.1038/s41586-019-1485-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Discher D.E., Janmey P., Wang Y.L. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 25.Elosegui-Artola A., Andreu I., Beedle A.E.M., Lezamiz A., Uroz M., Kosmalska A.J., et al. Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell. 2017;171:1397–1410.e1314. doi: 10.1016/j.cell.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 26.Provenzano P.P., Keely P.J. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 2011;124:1195–1205. doi: 10.1242/jcs.067009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu N., Zhou M., Zhang Q., Zhang T., Tian T., Ma Q., et al. Stiffness regulates the proliferation and osteogenic/odontogenic differentiation of human dental pulp stem cells via the WNT signalling pathway. Cell Prolif. 2018;51:e12435. doi: 10.1111/cpr.12435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lesage F., Maingret F., Lazdunski M. Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K(+) channel. FEBS Lett. 2000;471:137–140. doi: 10.1016/s0014-5793(00)01388-0. [DOI] [PubMed] [Google Scholar]
- 29.Thumkeo D., Watanabe S., Narumiya S. Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 2013;92:303–315. doi: 10.1016/j.ejcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 30.Tzima E., Irani-Tehrani M., Kiosses W.B., Dejana E., Schultz D.A., Engelhardt B., et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 31.Hac-Wydro K., Jedrzejek K., Dynarowicz-Łatka P. Effect of saturation degree on the interactions between fatty acids and phosphatidylcholines in binary and ternary Langmuir monolayers. Colloids Surf. B Biointerfaces. 2009;72:101–111. doi: 10.1016/j.colsurfb.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 32.Wang B., Tontonoz P. Phospholipid remodeling in physiology and disease. Annu. Rev. Physiol. 2019;81:165–188. doi: 10.1146/annurev-physiol-020518-114444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tiberti M.L., Antonny B., Gautier R. The transbilayer distribution of polyunsaturated phospholipids determines their facilitating effect on membrane deformation. Soft Matter. 2020;16:1722–1730. doi: 10.1039/c9sm02107h. [DOI] [PubMed] [Google Scholar]
- 34.Urbanelli L., Buratta S., Logozzi M., Mitro N., Sagini K., Raimo R.D., et al. Lipidomic analysis of cancer cells cultivated at acidic pH reveals phospholipid fatty acids remodelling associated with transcriptional reprogramming. J. Enzyme Inhib. Med. Chem. 2020;35:963–973. doi: 10.1080/14756366.2020.1748025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyazaki M., Bruggink S.M., Ntambi J.M. Identification of mouse palmitoyl-coenzyme A Delta9-desaturase. J. Lipid Res. 2006;47:700–704. doi: 10.1194/jlr.C500025-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Ran H., Zhu Y., Deng R., Zhang Q., Liu X., Feng M., et al. Stearoyl-CoA desaturase-1 promotes colorectal cancer metastasis in response to glucose by suppressing PTEN. J. Exp. Clin. Cancer Res. 2018;37:54. doi: 10.1186/s13046-018-0711-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bai Y., McCoy J.G., Levin E.J., Sobrado P., Rajashankar K.R., Fox B.G., et al. X-ray structure of a mammalian stearoyl-CoA desaturase. Nature. 2015;524:252–256. doi: 10.1038/nature14549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tracz-Gaszewska Z., Dobrzyn P. Stearoyl-CoA desaturase 1 as a therapeutic target for the treatment of cancer. Cancers (Basel) 2019;11:948. doi: 10.3390/cancers11070948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ng K.Y., Shea Q.T., Wong T.L., Luk S.T., Tong M., Lo C.M., et al. Chemotherapy-enriched THBS2-deficient cancer stem cells drive hepatocarcinogenesis through matrix softness induced histone H3 modifications. Adv. Sci. (Weinh) 2021;8:2002483. doi: 10.1002/advs.202002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sánchez-Martínez R., Cruz-Gil S., Gómez de Cedrón M., Álvarez-Fernández M., Vargas T., Molina S., et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget. 2015;6:38719–38736. doi: 10.18632/oncotarget.5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin L., Ding Y., Wang Y., Wang Z., Yin X., Yan G., et al. Functional lipidomics: palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology. 2017;66:432–448. doi: 10.1002/hep.29033. [DOI] [PubMed] [Google Scholar]
- 42.Owen D.M., Rentero C., Magenau A., Abu-Siniyeh A., Gaus K. Quantitative imaging of membrane lipid order in cells and organisms. Nat. Protoc. 2011;7:24–35. doi: 10.1038/nprot.2011.419. [DOI] [PubMed] [Google Scholar]
- 43.Sun Y.F., Wang P.X., Cheng J.W., Gong Z.J., Huang A., Zhou K.Q., et al. Postoperative circulating tumor cells: an early predictor of extrahepatic metastases in patients with hepatocellular carcinoma undergoing curative surgical resection. Cancer Cytopathol. 2020;128:733–745. doi: 10.1002/cncy.22304. [DOI] [PubMed] [Google Scholar]
- 44.Fenner J., Stacer A.C., Winterroth F., Johnson T.D., Luker K.E., Luker G.D. Macroscopic stiffness of breast tumors predicts metastasis. Sci. Rep. 2014;4:5512. doi: 10.1038/srep05512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W., Zhu X.D., Sun H.C., Xiong Y.Q., Zhuang P.Y., Xu H.X., et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin. Cancer Res. 2010;16:3420–3430. doi: 10.1158/1078-0432.CCR-09-2904. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.