

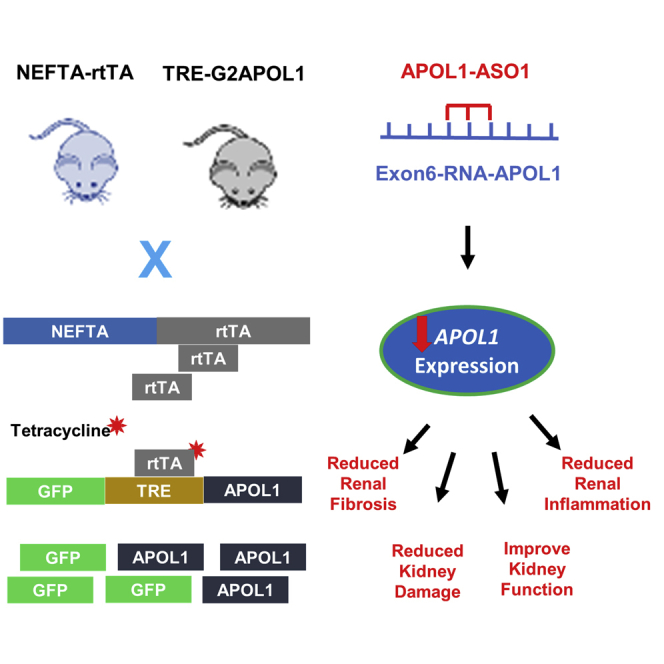
Abstract
Coding variants (named G1 and G2) in Apolipoprotein L1 (APOL1) can explain most excess risk of kidney disease observed in African American individuals. It has been proposed that risk variant APOL1 dose, such as increased risk variant APOL1 level serves as a trigger (second hit) for disease development. The goal of this study was to determine whether lowering risk variant APOL1 levels protects from disease development in a podocyte-specific transgenic mouse disease model. We administered antisense oligonucleotides (ASO) targeting APOL1 to podocyte-specific G2APOL1 mice and observed efficient reduction of APOL1 levels. APOL1 ASO1, which more efficiently lowered APOL1 transcript levels, protected mice from albuminuria, glomerulosclerosis, tubulointerstitial fibrosis, and renal failure. Administration of APOL1 ASO1 was effective even for established disease in the NEFTA-rtTA/TRE-G2APOL1 (NEFTA/G2APOL1) mice. We observed a strong correlation between APOL1 transcript level and disease severity. We concluded that APOL1 ASO1 may be an effective therapeutic approach for APOL1-associated glomerular disease.
Keywords: genetic diseases, nephrology, drug therapy, APOL1, CKD, ASO
Graphical abstract
Introduction
Two coding region variants (G1 and G2) in the Apolipoprotein L1, APOL1 gene have been shown to increase the risk of chronic kidney disease and end-stage renal disease (ESRD) in people of African ancestry.1, 2, 3, 4 About 45% of African American individuals carry one APOL1 risk variant (named G1 and G2), while 13% carry two copies (high-risk genotype).1, 2, 3, 4 High-risk APOL1 genotype explains much of the increased kidney disease risk observed in African American individuals.5,6 Unlike other disease-associated high-penetrant variants, G1 and G2 are present in the general population at a high frequency.7,8
For a long time, it was difficult to establish the causal role of APOL1 risk variants in disease development. The APOL1 gene is only present in some primates and humans, and the gene is missing from the genomes of other species. Several APOL1 transgenic mouse models have been reported, two of them driven by a podocyte-specific (Nephrin) promotor, resulting in APOL1 expression only in podocytes.3,9, 10, 11 In the first model, APOL1 is constitutively expressed in podocytes (albeit at a lower level), which resulted in mild podocyte dysfunction, but no overt glomerulosclerosis or renal failure. Fosmid APOL1 G1 transgenic mice developed significant proteinuria following interferon injection, but no glomerulosclerosis of kidney function declined at the analyzed short timescale.11 Our group generated mice with podocyte-specific inducible expression of the APOL1 alleles using the nephrin rtTA and tet responsive APOL1. Animals expressing higher level of G1 or G2 variants developed functional, structural, and molecular changes that resembled human kidney disease, while mice expressing the reference allele (G0) showed no observable abnormalities.12 Recently, a BAC transgenic model was developed by the Pollak group. These animals developed severe disease following interferon induction.13
People with only one risk allele (G1 or G2) have either absence or minimally elevated kidney disease risk, whereas individuals with two risk alleles have 7- to 10-fold increased risk of developing focal segmented glomerular sclerosis.5,6,8,14 People who carry two copies of G1 or G2 APOL1 allele (so-called high-risk genotype) have an increased risk of developing chronic kidney disease and ESRD. Therefore, kidney disease risk associated with APOL1 variants follow an autosomal recessive inheritance.
The high risk genotype APOL1 associated lifetime CKD risk is estimated to be around 20%, indicating that additional environmental triggers are also critical for nephropathy development.15 For example, interferon injection has been shown to increase albuminuria in subjects with high-risk genotypes and in fosmid APOL1 G1 transgenic mice.11,16,17 Consistently, APOL1 expression in human kidney glomeruli, cultured cell systems, and transgenic mouse models correlated with kidney disease severity. The dose-dependent association between APOL1 and disease severity could provide an important therapeutic opportunity for drugs that reduce APOL1 levels.12,18
Antisense oligonucleotides (ASOs) are powerful therapeutic approaches that reduce RNA transcripts with an RNase H1-mediated mechanism of degradation, inhibiting splicing or translation.19 To date, the Food and Drug Administration has approved ASO therapeutics for systemic and neurological disorders.17,20, 21, 22, 23, 24 Systemically administered ASOs exhibit broad activity, including liver and kidney proximal convoluted tubule epithelial cells, and demonstrate tissue elimination half-lives of around 2 to 4 weeks.25,26 Recently, using the more potent Generation 2.5 ASO chemistry,25 APOL1 ASO treatment reduced APOL1 expression in glomerular cells of APOL1 fosmid transgenic mice and ameliorated kidney disease.11 These animals, however, only developed low-grade and transient albuminuria following interferon injection, preventing the evaluation of the impact of APOL1 ASO treatment on clinically meaningful outcomes, such as kidney function and histopathology, fibrosis, and glomerulosclerosis.
Using the podocyte-specific inducible G2APOL1 transgenic mice, here we evaluated the effect of ASOs on kidney function and histological changes.
Results
In vitro screening of ASOs
Previous screening efforts to identify an active APOL1 ASO resulted in the discovery of IONIS-APOL1Rx11; however, as IONIS-APOL1Rx targets an intronic region of APOL1, and the nephrin rtTA APOL1 transgenic mice express APOL1 cDNA (coding region). Here we used HEK293T cells stably expressing APOL1 cDNA27 to screen for an active exon-targeting APOL1 ASO. This strategy resulted in the identification of two in vitro active APOL1 ASOs (Figure S1), APOL1 ASO1 and APOL1 ASO2. As wild-type mice do not express APOL1, we could not confirm in vivo activity of these ASOs in wild-type mice and therefore, we chose to carry forward both ASOs into APOL1 transgenic mice.
ASOs efficiently reduce APOL1 levels in mice with podocyte-specific conditional inducible expression of risk variant APOL1
We used mice with conditional inducible expression of risk variant APOL1 in podocytes by crossing the reverse tetracycline transactivator (rtTA) expressed under the nephrin promoter (NEFTA) and TRE-G2APOL1, containing the full-length G2APOL1 variant under the control of tetracycline response elements. In the NEFTA-rtTA/TRE-G2APOL1 (NEFTA/G2APOL1) mice, doxycycline administration drives the expression of risk variant (RV) APOL1 in a podocyte-specific manner.12
We have extensively studied the disease severity in the male versus female NEFTA-rtTA/TREG2APOL1-GFP mice; however, our previous study failed to indicate differences between male and female mice. Here, we used both sexes in an equal proportion in this study. We tested the effect of APOL1 ASO1 (n = 8; male = 4 and female = 4) and APOL1 ASO2 (n = 8; male = 4 and female = 4) in NEFTA/G2APOL1 mice and compared it with that of a non-targeting control ASO (n = 6; male = 3 and female = 3) of the same chemistry. Animals were injected weekly with 50 mg/kg ASO intraperitoneally beginning at 4 weeks of age. We induced RV APOL1 expression by doxycycline diet after the fourth weekly dose of ASO. One final ASO dose was administered after the administration of the doxycycline diet for a total of five doses. Our protocol was based on prior experiments of ASO treatment in APOL1-transgenic mice as well as ASO treatment in other mice models.11,17,28, 29, 30, 31 Two days after the last ASO injection, mice were euthanized (Figure 1A).
Figure 1.

APOL1 ASO effectively lowers APOL1 level in NEFTA/G2APOL1 transgenic mice
(A) Experimental design: NEFTA-rtTA/TREG2APOL1-GFP (NEFTA/G2APOL1) mice were generated. Animals were injected with control ASO, APOL1 ASO1, and ASO2 weekly and placed on a doxycycline diet for 10 days. (B) Representative images of APOL1 in situ hybridization in kidneys of NEFTA/G2APOL1 mice treated with control ASO, APOL1 ASO1, and ASO2. Scale bars of upper panels, 20 μm; lower panels, 10 μm. (C) Relative APOL1 mRNA levels measured by qRT-PCR in whole kidney lysates, ∗∗p ≤ 0.01 versus control ASO-treated mice. (D) Protein levels of APOL1 in kidneys of NEFTA/G2APOL1 mice treated with control ASO, APOL1 ASO1, or ASO2. Each lane represents one mouse. GAPDH and actin were used as a loading control. (E) Quantification of relative APOL1 protein levels in groups from (D), ∗∗p ≤ 0.01 versus control ASO-treated mice.
In situ hybridization showed APOL1 expression in podocytes of NEFTA/G2APOL1 transgenic animals (Figure 1B). APOL1 transcript levels were quantified by qRT-PCR. Kidneys of animals injected with APOL1 ASO1 had lower APOL1 expression, but APOL1 transcript levels did not show a detectable change in kidneys of mice injected with APOL1 ASO2 (Figure 1C). Expression of other APOL genes, including Apol2 and Apol6, were not affected by ASO1 (Figure S2). We did not observe liver toxicity when we analyzed liver sections from ASO-injected animals (Figure S3). The protein level of APOL1 was analyzed on western blots in whole kidney lysates using an APOL1-specific antibody. Animals injected with ASO1 had lower APOL1 expression, while mice injected with ASO2 did not show observable differences (Figures 1D and 1E).
In summary, our in vivo studies of podocyte-specific APOL1 transgenic mice indicated that APOL1 ASO1 lowered APOL1 transcript and protein levels while ASO2 did not.
ASOs ameliorate risk variant APOL1-induced kidney function changes
Kidney functional analysis indicated that podocyte-specific risk variant APOL1 mice manifested with severe albuminuria. NEFTA/G2APOL1 mice treated with APOL1 ASO1 showed lower albuminuria, evident by the lower urinary albumin creatinine ratio (ACR) at the time of euthanasia (Figure 2A). Albuminuria change in mice treated with APOL1 ASO2 did not reach significance.
Figure 2.

APOL1 ASO1 improves kidney function parameters in NEFTA/G2APOL1 transgenic mice
(A) Urine albumin/creatinine ratio (ACR mg albumin/mg creatinine) at baseline (start of doxycycline diet) and at the time of euthanasia in control ASO, APOL1 ASO1, and ASO2-treated mice, ∗∗p ≤ 0.01 versus control ASO1-treated mice. (B) Serum BUN (mg/dL) at the time of euthanasia in control ASO, APOL1 ASO1, and ASO2-treated mice, ∗∗p ≤ 0.01 versus control ASO-treated mice. (C) Serum creatinine (mg/dL) at the time of euthanasia in control ASO, APOL1 ASO1, and ASO2-treated mice, ∗∗p ≤ 0.01 versus control ASO-treated mice.
Podocyte-specific APOL1 transgenic mice injected with control ASO showed severe azotemia evidenced by higher blood urea nitrogen (BUN) and higher serum creatinine levels when compared with wild-type mice (Figures 2B and 2C). The BUN and serum creatinine levels of APOL1 ASO1-treated mice were markedly lower than those of control ASO-treated mice. Mice treated with ASO2 showed no observable differences compared with control ASO-treated animals.
Histological analysis performed by light microscopy indicated that podocyte-specific risk variant APOL1 transgenic mice developed marked global and segmental sclerosis, as well as mesangial expansion. We also observed pseudo-crescent formation in some samples (Figure 3A). Tubules showed atrophy, interstitial fibrosis, and proteaceous casts. The degree of glomerular damage, such as global sclerosis and tubule injury, was lower in APOL1 ASO1-injected animals, while animals showed some heterogeneity, we did not observe marked differences in ASO2-treated animals (Figure 3B).
Figure 3.

APOL1 ASO improves glomerulosclerosis and fibrosis in NEFTA/G2APOL1 transgenic mice
(A) Representative PAS-stained kidney images of NEFTA/G2APOL1 mice treated with control ASO, APOL1 ASO1, or ASO2. Scale bars in upper panels, 20μm; lower panels, 10μm. (B) Semi-quantitative analysis of percent of tubular injury with cast, global glomerulosclerosis, and Sirius red positive area in each group; ∗p ≤ 0.05 versus control ASO-treated mice, ∗∗p ≤ 0.01 versus control ASO-treated mice. (C) Representative Sirius red-stained kidney images of NEFTA/G2APOL1 mice treated with control ASO, ASO1, or ASO2. Scale bars in upper panels, 20μm; lower panels, 10μm.
To further understand changes in kidney fibrosis, we performed Sirius red staining in NEFTA/G2APOL1 transgenic mice. Podocyte RV APOL1 transgenic mice injected with control ASO and APOL1 ASO2 showed noticeable collagen deposition. We observed a marked reduction in Sirius red positive area in APOL1 ASO1-treated animals (Figure 3C).
We next analyzed profibrotic gene expression changes by qRT-PCR. We found that transcript levels of Collagen 1 (Col1a1) (Figure 4A), Collagen 3 (Col3a1) (Figure 4B), and fibronectin (Fn1) (Figure 4C) were lower in APOL1 ASO1-treated NEFTA/G2APOL1 transgenic mice. Furthermore, their levels positively correlated with APOL1 expression measured in the kidney (Figures 4D–4F).
Figure 4.

Treatment with APOL1 ASO1 lowers profibrotic gene expression and protects from podocyte loss
(A–F) Relative transcript level of Collagen 1 (Col1a1) (A), Collagen 3 (Col3a1) (C) and fibronectin (Fn1) (E) in whole kidney lysates of NEFTA/G2APOL1 mice treated with control ASO, APOL1 ASO1 or ASO2; ∗p ≤ 0.05 versus control ASO-treated mice, ∗∗p ≤ 0.01 versus control ASO-treated mice. The correlation of relative transcript levels of APOL1 and Col1a1 (B), Col3a1 (D), and Fn1 (F) ASO-treated NEFTA/G2APOL1 mice. Podocyte number in control and APOL1 ASO-treated animals. (G) Representative images of Wilms’ tumor (WT-1) Immunohistochemistry staining, an indicator of podocyte marker in the glomerulus in wild-type NEFTA/G2APOL1 transgenic mice treated with control, APOL1 ASO1, and ASO2.
APOL1 ASO1 improves podocyte pathology
Podocyte loss plays a critical role in glomerulosclerosis development.32,33 To estimate podocyte number, we performed immunohistochemical staining for Wilms’ tumor protein 1 (WT-1), a key podocyte marker.34 Quantification WT-1 positive cells showed a marked reduction in WT-1 positive cells in NEFTA/G2APOL1 transgenic mice (12 cells/glomeruli) compared with wild-type mice (28 cells/glomeruli) (Figure 4G). WT-1-positive cells were preserved in mice treated with APOL1 ASO1 (24 cells/glomeruli) but not in those treated with APOL1 ASO2 (11 cells/glomeruli) (Figure 4G). In summary, treatment with APOL1 ASO1 protected from loss of WT-1-positive cells in the NEFTA/G2APOL1 mice (Figure 4H).
APOL1 expression correlates with proinflammatory gene expression
Previous studies indicated an important role of inflammatory cell death pathways leading to kidney disease in NEFTA/G2APOL1 transgenic mice.35,36 Therefore, we examined changes in inflammatory markers.
We found that expression of inflammasome components, for example, Nlrp3, was higher in kidneys of NEFTA/G2APOL1 transgenic mice. Furthermore, kidney expression of Nlrp3 was found to be positively correlated with APOL1 transcript level (Figures 5A and 5C). Il1b level was lower both in APOL1 ASO1 and ASO2-treated G2APOL1 transgenic animals (Figure 5G).
Figure 5.

Effects of APOL1 ASO treatment on renal inflammation in NEFTA/G2APOL1 mice
(A–H) The relative transcript level of NLR family pyrin domain containing 3 (Nlrp3) (A), stimulator of interferon genes (Sting or Tmem173) (B), interferon regulatory factor 3 (Irf3) (E), signal transducer and activator of transcription 3 (Stat3) (F), Interleukin 1 beta (Il1b) (G), signal transducer and activator of transcription 1 (Stat1) (H) in whole kidney lysates of NEFTA/G2APOL1 mice treated with control ASO, APOL1 ASO1, or ASO2, ∗p ≤ 0.05 versus control ASO-treated mice. The correlation of relative transcript levels of APOL1 and Nlrp3 (C) and Sting (D) in ASO-treated NEFTA/G2APOL1 mice.
Transcript levels of genes in the nucleotide sensing pathway, such as Sting, was also lower in APOL1 ASO1-treated animals (Figure 5B). Sting level correlated with APOL1 expression (Figure 5D). Downstream targets of the nucleotide sensors such as Irf3 and Stat1 and Stat3 were lower in APOL1 ASO1-treated G2APOL1 animals (Figures 5E, 5F, and 5H).
Expression of cytokines, such as Ccl2 and Cxcl10, was higher in podocytes of NEFTA/G2APOL1 transgenic mice. Their expression was lower in APOL1 ASO1- but not in ASO2-treated animals (Figures 6A–6D).
Figure 6.

Effects of APOL1 ASO treatment on the expression of chemokines and cytokines in NEFTA/G2APOL1 mice
(A–F) The relative transcript level of C-C motif chemokine ligand 2 (Ccl2 or MCP1) (A), C-X-C Motif Chemokine Ligand 10 (Cxcl10) (C), and soluble urokinase receptor (suPar) (E) in whole kidney lysates of control ASO, APOL1 ASO1, and ASO2-treated NEFTA/G2APOL1 mice, ∗p ≤ 0.05 versus control ASO-treated mice. The correlation of relative transcript levels of APOL1 and Ccl2 (B), Cxcl10 (D), and suPar (F) in ASO-treated NEFTA/G2APOL1 mice.
Recent in vitro and in vivo studies indicate that an increase in the level of soluble urokinase-type plasminogen activator (suPA)/uPA-specific receptor (uPAR) plays role in APOL1 nephropathy. In our study, kidney expression of suPAR was higher in G2APOL1 transgenic mice and it was markedly lowered following APOL1 ASO1 injection (Figure 6E). Further more, the expression of suPAR was found to be correlated with the expression of the APOL1 (Figure 6F). Overall, we observed a correlation between immune and inflammatory gene expression and APOL1 expression, indicating the key role of APOL1 dose in disease severity.
ASOs improve renal functions in mice with established renal disease
To understand the impact of APOL1 ASO1 treatment on established renal disease in the NEFTA/G2APOL1 transgenic mice we changed our experimental strategy (Figure 7A). Both male and female NEFTA-rtTA/TREG2APOL1-GFP (NEFTA/G2APOL1) mice in equal proportions were placed on a doxycycline diet, and ASO injection was initiated 3 days later when proteinuria was already detectable in this model. ASO was administered intraperitoneally 3, 6, and 9 days after the initiation of the doxycycline diet (Figure 7A). Mice were distributed into two groups: 1) treated with control ASO, and 2) treated with APOL1-ASO1 (n = 4/group). The experiment was terminated on day 10 after initiation of the doxycycline diet (Figure 7A).
Figure 7.

Effects of APOL1 ASO treatment in NEFTA/G2APOL1 mice with established disease
(A–F) Experimental design (A). NEFTA-rtTA/TREG2APOL1-GFP (NEFTA/G2APOL1) mice were placed on a doxycycline diet and ASO treatment (ASO1 or control) was initiated on day 3 and repeated on day 6 and day 9. Temporal changes of urinary ACR (mg/mg) in control ASO and APOL1 ASO1-treated mice (B), ∗p ≤ 0.05 versus control ASO-treated mice and ∗∗p < 0.01 versus control ASO-treated mice. Serum BUN (mg/dL) at the end of the experiment in control ASO and APOL1 ASO1-treated mice (C), ∗p ≤ 0.05 versus control ASO-treated mice. Semi-quantitative analysis of Sirius red positive area in each group (D); ∗p ≤ 0.05 versus control ASO-treated mice. Representative images of renal histopathology of NEFTA/G2APOL1 mice treated with control ASO and APOL1 ASO1: Sirius red staining (E) and H&E staining (F). Scale bars in upper panels, 20μm; lower panels, 10μm.
Kidney APOL1 transcript levels were lower (∼50%) in APOL1 ASO1-treated mice compared with control ASO treatment group in NEFTA/G2APOL1 mice (Figure S2A).
The APOL1 ASO1 treatment observably improved renal function compared with control ASO treatment in NEFTA/G2APOL1 mice. Mice treated with ASO1 had lower albuminuria (Figure 7B) and lower BUN levels (Figure 7C).
Histological changes were evaluated by H&E (Figure 7F) and Sirius red stainings (Figures 7D and 7E). APOL1 ASO1-treated mice had less glomerular damage, tubular injury, protein casts, and renal fibrosis (Figures 7E and 7F).
Molecular analysis indicated lower transcript levels of αSMA (Figure 8A) and Col1a1 (Figure 8B), while a difference was not observed in Fn1 (Figure 8C). The expression of Acta2 and Fn1 positively correlated with kidney APOL1 levels (Figures 8D and 8F).
Figure 8.

Effects of APOL1 ASO treatment on renal inflammation and fibrosis in NEFTA/G2APOL1 mice with established disease
(A–L) The relative expression of the transcript of alpha-smooth muscle actin (α-SMA) (A), Collagen 1 (Col1a1) (B), and fibronectin (Fn1) (C) in whole kidney lysates of NEFTA/G2APOL1 mice treated with control ASO and APOL1 ASO1; ∗p ≤ 0.05 versus control ASO-treated mice and ∗∗p < 0.01 versus control ASO-treated mice. The correlation of relative transcript levels of APOL1 with α-SMA (D), Col1a1 (E), and Fn1 (F) in ASO-treated NEFTA/G2APOL1 mice. The relative expression of the transcript of Nlrp3 (G), Sting or Tmem173 (H), and Irf3 (I) in whole kidney lysates of NEFTA/G2APOL1 mice treated with control ASO and APOL1 ASO1; ∗p ≤ 0.05 versus control ASO-treated mice. The correlation of relative transcript levels of APOL1 and Nlrp3 (J), Sting (K), and Sting or Tmem173 (L) in ASO-treated NEFTA/G2APOL1 mice. Genetic risk variants in Apolipoprotein L1 can explain most excess kidney disease risk in African American individuals. Increased APOL1 level likely serves as the second hit for disease development. Antisense oligonucleotide-based lowering of APOL1 administered before or even after disease development improved kidney function in podocyte-specific APOL1 risk variant mice.
Similarly, treatment with APOL1 ASO1 was associated with lower expression of proinflammatory genes, including Sting (Figure 8H) and Irf3 (Figure 8I). The expression of Nlrp3, Sting, and Irf3 positively correlated with kidney APOL1 expression (Figures 8J–8L).
APOL1 ASO1 treatment of mice with established disease was associated with lower APOL1 expression and improved kidney function and less kidney structural damage.
Discussion
Here, using mice with podocyte-specific APOL1 risk variant expression, we show that administration of an in vivo active APOL1 ASO markedly reduced podocyte APOL1 transcript and protein expression. The reduction in APOL1 expression strongly correlated with improved kidney function, albuminuria, structural damage of the glomerulus and tubules, fibrosis, and inflammation, even when the treatment was initiated after disease onset.6,11,37
APOL1 is a recently evolved gene that is only present in humans and a few primates. APOL1 is likely to be a non-essential gene. As of yet, only a single person has been identified with a biallelic loss of function APOL1 variant.38 He was identified as he presented with recurrent trypanosomiasis, but no kidney disease. In cell systems, animal models, and patient samples, disease development showed a strong correlation with total APOL1 amount, indicating that reducing APOL1 expression could be a potential therapeutic approach3,12; however, other disease triggers such as suPAR have also been proposed.39
Recently, antisense therapy has emerged as a promising strategy for the treatment of various genetic disorders, especially for the gain-of-function mutations and variants. Risk variant APOL1s are such gain-of-function variants, making them excellent ASO targets. One challenge, however, has been the delivery of ASOs to ASO-refractory target cell types. One previous study demonstrated that ASO could be successfully delivered to glomerular cells using a more advanced ASO chemistry; however, kidney-specific delivery of ASOs has not been developed.11 The follow-up time in the initial study was short and animals did not develop severe renal disease, only mild albuminuria without significant glomerulosclerosis.11
Here we developed an ASO that targets the coding region of APOL1 and found that ASO1 successfully targeted podocytes and reduced APOL1 expression. We found that reduced APOL1 levels resulted in a marked reduction in proteinuria and glomerulosclerosis. In addition, it ameliorated tubulointerstitial fibrosis and improved kidney function such as serum BUN and creatinine levels. Furthermore, it has decreased the level of proinflammatory cytokines and inflammation.
It is important to note that treating mice with ASO prior to disease development was highly effective, while differences in vivo effectiveness of ASO1 and ASO2 were observed. We also observed a reduction in APOL1 expression when mice were treated after they developed proteinuria, but the effect of treating animals with the established disease was less pronounced than those treated before the disease, nevertheless, this may indicate an important strategy for patients.
Risk variant APOL1-induced glomerular disease and podocyte damage are poorly understood. Recent studies indicate that APOL1 functions as a channel, not only in Trypanosomes but also in mammalian cells.40 It is interesting to note that risk variant and reference allele APOL1 showed similar channel activities.10 Key differences have been, identified in their activation and membrane insertion. APOL1 activation is poorly understood, complex intracellular trafficking and acidification play important roles in the process. Risk variant APOL1 showed a defect in intracellular trafficking and autophagy leading to the activation of the inflammasome and cytosolic nucleotide sensing pathways, which is consistent with our current data.12,41 Here we observed a significant positive correlation of APOL1 expression with the inflammasome (NlLRP3) and downstream Il1b expression and disease severity. In addition, we also observed a correlation between APOL1 level, disease severity, and Sting expression. Though we cannot conclude whether these NLRP3 and STING pathways are indeed in the causal disease mechanism in this current study, our recently published data indicate the key role of inflammasome and STING in APOL1-associated kidney disease.35 Expression of APOL1 risk variant was associated with impaired mitochondrial function42 cytosolic release of mitochondrial DNA. Cytosolic mtDNA acts as a strong activator of cytosolic nucleotide sensors such as STING.43 downstream of STING is Irf3, Stat1, Stat3, and proinflammatory cytokine release. In our study, we observed a strong correlation between APOL1 transcript level, inflammation, and disease severity.
It is important to note that we observed differences between APOL1 ASO1 and ASO2 in terms of their effectiveness in reducing APOL1 transcript and protein levels. Similarly, we observed differences in their effect in reducing G2APOL1 induced nephropathy. It is important to note that APOL1 transcript and protein levels showed correlation with disease severity parameters including proteinuria, kidney function, glomerulosclerosis, and interstitial fibrosis.27,44 We do not fully understand the in vivo and in vitro differences between APOL1 ASO1 and ASO2. In addition, we also noted significant baseline variation in APOL1 expression and phenotype development in our animal model, which was minimized here by working only with a single transgenic line, which shows consistent results.12
Together, these data suggest that reduction of APOL1 expression may be sufficient to ameliorate disease progression, including clinically meaningful outcomes such as kidney function preservation and fibrosis amelioration. We were able to evaluate APOL1 ASO treatment therapeutically in this study in NEFTA/G2APOL1 transgenic mice following the development of kidney disease. Treatment with APOL1 ASO1 prior to initiation of the doxycycline diet was found to be more effective and ameliorated the associated albuminuria. Therefore, APOL1 ASO1 could be a potential therapeutic intervention in the clinical setting against the development and progression of renal disease associated with APOL1 gene expression.
Materials and methods
Transgenic mice
TRE-APOL1 mice were generated as described before.12 NEFTA-rtTA transgenic mice were generated by Jeffrey Miner.45 We mated the TRE-APOL1 mice with NEFTA-rtTA animals. Transgene expression was induced by doxycycline-containing food (200 mg/kg, BioServ). Animal studies were approved by the Animal Care Committee of the University of Pennsylvania.
ASOs
Control and APOL1 Generation 2.5 ASOs were 16 nucleotides in length and chemically modified with phosphorothioates in the backbone and 2′–4′ constrained ethyl (cEt) residues in the wings, with a central deoxy gap (3-10-3 gapmer design). Synthesis of Gen 2.5 ASOs was previously described.46 ASOs were dissolved in PBS (without Mg2+ or Ca2+; Life Technologies) for in vivo administration. ASO1 binds to exon 6 in the membrane addressing domain region and ASO2 binds to exon 4 prior to the pore-forming domain (which contains the BH3 site) region. Neither ASO targets an exon junction nor binds close to the sites of the G1/G2 risk variants. As the ASOs bind sequences that do not overlap with the G1 risk variants, they are predicted to behave similarly in a G1-specific transgenic mouse model.
Cell culture experiments
HEK293-APOL1 G0 cells27 were cultured in a growth medium (DMEM + 10% fetal bovine serum; Thermo Fisher) at 37°C and 5% CO2. Cells were electroporated in the presence of ASOs at the indicated concentrations and plated. Electroporation of ASOs was carried out at 160V with 20,000 cells/well using the HT-200 BTX Electroporator with the ElectroSquare Porator (ECM 830) voltage source in 96-well electroporation plates (BTX, 2 mm; Harvard Apparatus). Cells were harvested 24 h after electroporation and total cellular RNA was isolated using glass fiber filter plates (Pall) and chaotropic salts. APOL1 mRNA expression levels were quantitated with qRT-PCR on the 7900HT and QuantStudio 7 instruments (Applied Biosystems) using the following primer-probe set: human APOL1, forward primer 5′-GCTACTCCTGCTGACTGATAATG-3′, reverse primer 5′-AAGGTTGTCCAGAGCTTTACG-3′, probe 5′-FAM-TGCCCAGGAATGAGGCAGATGAG-IOWA-BLACK-3′). Relative gene expression was normalized to total RNA measured by Quant-iT RiboGreen RNA assay (Molecular Probes). Data are presented as percentage untreated control.
Phenotype analysis
Urine albumin and creatinine were determined using mouse albumin-specific ELISA (Bethyl Laboratories) and creatinine reagent (Sciteck Diagnostics), per the manufacturer’s protocol. Serum creatinine and BUN were determined by Mouse Creatinine Kit (Crystal Chem #80350) and TRACE DMA Urea kit (Thermo Electron Corporation #TR12003), respectively, according to the manufacturer’s instructions.
Histopathology assay
Kidneys were fixed in 10% neutral formalin and paraffin-embedded sections. Periodic acid-Schiff staining (PAS) or H&E staining were performed as described previously.47,48 Picrosirius red (Polyscience #24901) was performed to determine the degree of fibrosis.
Immunohistochemical analyses were performed to measure the expression of proteins in the kidney. Briefly, paraffin-embedded sections were incubated in indicated primary antibodies (WT-1, Santa Cruz, 1:50) and visualized using diaminobenzidine substrates. Sections were further counterstained with hematoxylin. For each section, five random fields were quantified in an unbiased manner using ImageJ.
In situ hybridization
In situ hybridization was performed using formalin-fixed paraffin-embedded tissue samples and the RNAscope 2.5 HD Duplex Detection Kit (bio-techne, 322436). We followed the manufacturer’s original protocol. The Hs Apol1 cat# 459791-C1 probes were used for the RNAscope assay.
Western blotting
Whole kidneys were removed from NEFTA-rtTA/TRE-APOL1 transgenic mice and subjected to western blot analysis. Briefly, kidney lysates were homogenized in 1% SDS lysis buffer (1% SDS, 1% Triton X-100, 50 mM Tris pH 7.4, 150 mM NaCl, 1 mM EDTA) containing protease inhibitor cocktail (Complete Mini, Roche). Equal amounts of total protein (40 μg) were resolved on 10% gels, transferred to polyvinylidene difluoride membranes, blocked with 5% non-fat milk in TBS-tween and probed with primary antibodies (Proteintech 66124-1-Ig, 1:1000), at 4°C overnight, and proper secondary antibodies: anti-rabbit (Cell Signaling, 1:3000) and anti-mouse (Cell Signaling, 1:3000) immunoglobulin (Ig)G horseradish peroxidase (HRP) and donkey anti785 goat IgG HRP (Santa Cruz, 1:3000). Blots were detected by enhanced chemiluminescence (Western Lightning-ECL, Thermo Scientific) or Pierce ECL (BioRad).
qRT-PCR
RNA was isolated from kidney tissues and cells using Trizol (Invitrogen). RNA (1 μg) was reverse transcribed using the cDNA Archival Kit (Life Technologies). APOL1 transgene expression was identified by genomic PCR analysis using the TaqMan RT Kit, expression was normalized to the housekeeping gene HPRT (Applied Life Technologies). The following TaqMan probes were used: APOL1 (Hs0106628), HPRT (Mm03024075). qRT-PCR was run in the ViiA 7 System (Life Technologies) instrument using SYBR Green Master Mix and gene-specific primers. The data were normalized and analyzed using the ΔΔCt method. Apol2, Apol7a, and Apol6 were measured by qRT-PCR using the SYBR green kit. The expression of these genes was normalized to various housekeeping genes, including β-actin, GAPDH, and HPRT, and the relative expression was calculated using the ΔΔCt method.
Statistical analyses
Differences between two groups were analyzed using Student’s t test, and one-way or two-way ANOVA was used to analyze intergroup differences (post hoc Tukey tests assuming unequal variance). A p value less than 0.05 was considered to be significant. The analysis was performed using GraphPad Prism 5 (GraphPad software). All data are presented as the means ± SEM and other details such as the level of significance are mentioned in figure legends unless otherwise indicated. Densitometry results of western blots were quantified using ImageJ software. Sample size estimation was not performed, and the sample size was determined based on the number of available age- and gender-matched animals in the colony. No animals or samples were excluded from the analysis.
Acknowledgments
Work in the Susztak lab is supported by the NIH DK076077, DK087635 and DK105821. We thank the University of Pennsylvania Diabetes Research Center (DRC) for the use of the histology core (P30-DK19525).
Author contributions
Y.W.Y., B.P., J.F., P.D., R.S., and Z.M. performed the animal experiments and analyzed the data. S.B., D.G., A.T.W., and M.A. performed the in vitro experiments and analyzed the data. M.P. performed the pathological review. Y.W.Y., B.P., J.F., and P.D. wrote the manuscript. K.O. and J.B.K. reviewed the manuscript. K.S. conceptualized and supervised the experiments, K.S. B.P., Y.W.Y., and J.F. wrote and reviewed the manuscript. All authors have read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.04.007.
Supplemental information
References
- 1.Bajaj A., Ihegword A., Qiu C., Small A.M., Wei W.Q., Bastarache L., Feng Q., Kember R.L., Risman M., Bloom R.D., et al. Phenome-wide association analysis suggests the APOL1 linked disease spectrum primarily drives kidney-specific pathways. Kidney Int. 2020;97:1032–1041. doi: 10.1016/j.kint.2020.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bajaj A., Susztak K., Damrauer S.M. APOL1 and cardiovascular disease: a story in evolution. Arterioscler. Thromb. Vasc. Biol. 2017;37:1587–1589. doi: 10.1161/atvbaha.117.309756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beckerman P., Susztak K. APOL1: the balance imposed by infection, selection, and kidney disease. Trends Mol. Med. 2018;24:682–695. doi: 10.1016/j.molmed.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiner A.P., Susztak K. APOL1 variants: from parasites to kidney function to cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2016;36:219–220. doi: 10.1161/atvbaha.115.306794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tzur S., Rosset S., Shemer R., Yudkovsky G., Selig S., Tarekegn A., Bekele E., Bradman N., Wasser W.G., Behar D.M., Skorecki K. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum. Genet. 2010;128:345–350. doi: 10.1007/s00439-010-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genovese G., Friedman D.J., Ross M.D., Lecordier L., Uzureau P., Freedman B.I., Bowden D.W., Langefeld C.D., Oleksyk T.K., Uscinski Knob A.L., et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329:841–845. doi: 10.1126/science.1193032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Foster M.C., Coresh J., Fornage M., Astor B.C., Grams M., Franceschini N., Boerwinkle E., Parekh R.S., Kao W.L. APOL1 variants associate with increased risk of CKD among African Americans. J. Am. Soc. Nephrol. 2013;24:1484–1491. doi: 10.1681/asn.2013010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Friedman D.J., Pollak M.R. Genetics of kidney failure and the evolving story of APOL1. J. Clin. Invest. 2011;121:3367–3374. doi: 10.1172/jci46263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruggeman L.A., Wu Z., Luo L., Madhavan S.M., Konieczkowski M., Drawz P.E., Thomas D.B., Barisoni L., Sedor J.R., O'Toole J.F. APOL1-G0 or APOL1-G2 transgenic models develop preeclampsia but not kidney disease. J. Am. Soc. Nephrol. 2016;27:3600–3610. doi: 10.1681/asn.2015111220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Toole J.F., Schilling W., Kunze D., Madhavan S.M., Konieczkowski M., Gu Y., Luo L., Wu Z., Bruggeman L.A., Sedor J.R. ApoL1 overexpression drives variant-independent cytotoxicity. J. Am. Soc. Nephrol. 2018;29:869–879. doi: 10.1681/asn.2016121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aghajan M., Booten S.L., Althage M., Hart C.E., Ericsson A., Maxvall I., Ochaba J., Menschik-Lundin A., Hartleib J., Kuntz S., et al. Antisense oligonucleotide treatment ameliorates IFN-gamma-induced proteinuria in APOL1-transgenic mice. JCI Insight. 2019;4:e126124. doi: 10.1172/jci.insight.126124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beckerman P., Bi-Karchin J., Park A.S.D., Qiu C., Dummer P.D., Soomro I., Boustany-Kari C.M., Pullen S.S., Miner J.H., Hu C.A.A., et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017;23:429–438. doi: 10.1038/nm.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCarthy G.M., Blasio A., Donovan O.G., Schaller L.B., Bock-Hughes A., Magraner J.M., Suh J.H., Tattersfield C.F., Stillman I.E., Shah S.S., et al. Recessive, gain-of-function toxicity in an APOL1 BAC transgenic mouse model mirrors human APOL1 kidney disease. Dis. Model. Mech. 2021;14:dmm048952. doi: 10.1242/dmm.048952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Limou S., Nelson G.W., Kopp J.B., Winkler C.A. APOL1 kidney risk alleles: population genetics and disease associations. Adv. Chronic Kidney Dis. 2014;21:426–433. doi: 10.1053/j.ackd.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kopp J.B., Anders H.J., Susztak K., Podesta M.A., Remuzzi G., Hildebrandt F., Romagnani P. Podocytopathies. Nat. Rev. Dis. Primers. 2020;6:68. doi: 10.1038/s41572-020-0196-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nichols B., Jog P., Lee J.H., Blackler D., Wilmot M., D'Agati V., Markowitz G., Kopp J.B., Alper S.L., Pollak M.R., Friedman D.J. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015;87:332–342. doi: 10.1038/ki.2014.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crooke S.T., Liang X.H., Baker B.F., Crooke R.M. Antisense technology: a review. J. Biol. Chem. 2021;296:100416. doi: 10.1016/j.jbc.2021.100416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freedman B.I., Kopp J.B., Sampson M.G., Susztak K. APOL1 at 10 years: progress and next steps. Kidney Int. 2021;99:1296–1302. doi: 10.1016/j.kint.2021.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crooke S.T., Seth P.P., Vickers T.A., Liang X.H. The interaction of phosphorothioate-containing RNA targeted drugs with proteins is a critical determinant of the therapeutic effects of these agents. J. Am. Chem. Soc. 2020;142:14754–14771. doi: 10.1021/jacs.0c04928. [DOI] [PubMed] [Google Scholar]
- 20.Raal F.J., Santos R.D., Blom D.J., Marais A.D., Charng M.J., Cromwell W.C., Lachmann R.H., Gaudet D., Tan J.L., Chasan-Taber S., et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006. doi: 10.1016/s0140-6736(10)60284-x. [DOI] [PubMed] [Google Scholar]
- 21.Finkel R.S., Chiriboga C.A., Vajsar J., Day J.W., Montes J., De Vivo D.C., Yamashita M., Rigo F., Hung G., Schneider E., et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet. 2016;388:3017–3026. doi: 10.1016/s0140-6736(16)31408-8. [DOI] [PubMed] [Google Scholar]
- 22.Mercuri E., Darras B.T., Chiriboga C.A., Day J.W., Campbell C., Connolly A.M., Iannaccone S.T., Kirschner J., Kuntz N.L., Saito K., et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. N. Engl. J. Med. 2018;378:625–635. doi: 10.1056/nejmoa1710504. [DOI] [PubMed] [Google Scholar]
- 23.Ackermann E.J., Guo S., Benson M.D., Booten S., Freier S., Hughes S.G., Kim T.W., Jesse Kwoh T., Matson J., Norris D., et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-Generation antisense oligonucleotides. Amyloid. 2016;23:148–157. doi: 10.1080/13506129.2016.1191458. [DOI] [PubMed] [Google Scholar]
- 24.Crooke S.T., Baker B.F., Crooke R.M., Liang X.H. Antisense technology: an overview and prospectus. Nat. Rev. Drug Discov. 2021;20:427–453. doi: 10.1038/s41573-021-00162-z. [DOI] [PubMed] [Google Scholar]
- 25.Hung G., Xiao X., Peralta R., Bhattacharjee G., Murray S., Norris D., Guo S., Monia B.P. Characterization of target mRNA reduction through in situ RNA hybridization in multiple organ systems following systemic antisense treatment in animals. Nucleic Acid Ther. 2013;23:369–378. doi: 10.1089/nat.2013.0443. [DOI] [PubMed] [Google Scholar]
- 26.Proceedings of the European Association for the Study of the Liver (EASL) international consensus conference on Hepatitis B. September 14-16, 2002. Geneva, Switzerland. J Hepatol. 2003;39:S1–S235. [PubMed] [Google Scholar]
- 27.Okamoto K., Rausch J.W., Wakashin H., Fu Y., Chung J.Y., Dummer P.D., Shin M.K., Chandra P., Suzuki K., Shrivastav S., et al. APOL1 risk allele RNA contributes to renal toxicity by activating protein kinase R. Commun. Biol. 2018;1:188. doi: 10.1038/s42003-018-0188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rinaldi C., Wood M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018;14:9–21. doi: 10.1038/nrneurol.2017.148. [DOI] [PubMed] [Google Scholar]
- 29.Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019;70:307–321. doi: 10.1146/annurev-med-041217-010829. [DOI] [PubMed] [Google Scholar]
- 30.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F. RNA-targeted therapeutics. Cell Metab. 2018;27:714–739. doi: 10.1016/j.cmet.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 32.Muller-Deile J., Schiffer M. Podocyte directed therapy of nephrotic syndrome-can we bring the inside out? Pediatr. Nephrol. 2016;31:393–405. doi: 10.1007/s00467-015-3116-4. [DOI] [PubMed] [Google Scholar]
- 33.Tao J., Polumbo C., Reidy K., Sweetwyne M., Susztak K. A multicolor podocyte reporter highlights heterogeneous podocyte changes in focal segmental glomerulosclerosis. Kidney Int. 2014;85:972–980. doi: 10.1038/ki.2013.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kann M., Ettou S., Jung Y.L., Lenz M.O., Taglienti M.E., Park P.J., Schermer B., Benzing T., Kreidberg J.A. Genome-Wide analysis of Wilms' tumor 1-controlled gene expression in podocytes reveals key regulatory mechanisms. J. Am. Soc. Nephrol. 2015;26:2097–2104. doi: 10.1681/asn.2014090940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu J., Ma Z., Raman A., Beckerman P., Dhillon P., Mukhi D., Palmer M., Chen H.C., Cohen C.R., Dunn T., et al. APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity. 2021;54:2632–2649.e6. doi: 10.1016/j.immuni.2021.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu J., Raman A., Coffey N.J., Sheng X., Wahba J., Seasock M.J., Ma Z., Beckerman P., Laczko D., Palmer M.B., et al. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J. Clin. Invest. 2021;131:e136329. doi: 10.1172/jci136329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kopp J.B., Nelson G.W., Sampath K., Johnson R.C., Genovese G., An P., Friedman D., Briggs W., Dart R., Korbet S., et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011;22:2129–2137. doi: 10.1681/asn.2011040388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnstone D.B., Shegokar V., Nihalani D., Rathore Y.S., Mallik L., Ashish Z.V., Ikizler H.O., Powar R., Holzman L.B. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS One. 2012;7:e51546. doi: 10.1371/journal.pone.0051546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayek S.S., Koh K.H., Grams M.E., Wei C., Ko Y.A., Li J., Samelko B., Lee H., Dande R.R., Lee H.W., et al. A tripartite complex of suPAR, APOL1 risk variants and αvβ3 integrin on podocytes mediates chronic kidney disease. Nat. Med. 2017;23:945–953. doi: 10.1038/nm.4362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giovinazzo J.A., Thomson R.P., Khalizova N., Zager P.J., Malani N., Rodriguez-Boulan E., Raper J., Schreiner R. Apolipoprotein L-1 renal risk variants form active channels at the plasma membrane driving cytotoxicity. Elife. 2020;9:e51185. doi: 10.7554/elife.51185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Biasizzo M., Kopitar-Jerala N. Interplay between NLRP3 inflammasome and autophagy. Front Immunol. 2020;11:591803. doi: 10.3389/fimmu.2020.591803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma L., Chou J.W., Snipes J.A., Bharadwaj M.S., Craddock A.L., Cheng D., Weckerle A., Petrovic S., Hicks P.J., Hemal A.K., et al. APOL1 renal-risk variants induce mitochondrial dysfunction. J. Am. Soc. Nephrol. 2017;28:1093–1105. doi: 10.1681/asn.2016050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maekawa H., Inoue T., Ouchi H., Jao T.M., Inoue R., Nishi H., Fujii R., Ishidate F., Tanaka T., Tanaka Y., et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29:1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050. [DOI] [PubMed] [Google Scholar]
- 44.Wakashin H., Heymann J., Roshanravan H., Daneshpajouhnejad P., Rosenberg A., Shin M.K., Hoek M., Kopp J.B. APOL1 renal risk variants exacerbate podocyte injury by increasing inflammatory stress. BMC Nephrol. 2020;21:371. doi: 10.1186/s12882-020-01995-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin X., Suh J.H., Go G., Miner J.H. Feasibility of repairing glomerular basement membrane defects in Alport syndrome. J. Am. Soc. Nephrol. 2014;25:687–692. doi: 10.1681/asn.2013070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seth P.P., Siwkowski A., Allerson C.R., Vasquez G., Lee S., Prakash T.P., Wancewicz E.V., Witchell D., Swayze E.E. Short antisense oligonucleotides with novel 2'-4' conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem. 2009;52:10–13. doi: 10.1021/jm801294h. [DOI] [PubMed] [Google Scholar]
- 47.Zeng Y., Wang P.H., Zhang M., Du J.R. Aging-related renal injury and inflammation are associated with downregulation of Klotho and induction of RIG-I/NF-κB signaling pathway in senescence-accelerated mice. Aging Clin. Exp. Res. 2016;28:69–76. doi: 10.1007/s40520-015-0371-y. [DOI] [PubMed] [Google Scholar]
- 48.Fang J., Yao X., Hou M., Duan M., Xing L., Huang J., Wang Y., Zhu B., Chen Q., Wang H. ApoL1 induces kidney inflammation through RIG-I/NF-κB activation. Biochem. Biophys. Res. Commun. 2020;527:466–473. doi: 10.1016/j.bbrc.2020.04.054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.