

Abstract
Although neurologic symptoms occur in two-thirds of lysosomal storage disorders (LSDs), for most we do not understand the mechanisms underlying brain dysfunction. A major unanswered question is if the pathogenic hallmark of LSDs, storage accumulation, induces functional defects directly or is a disease bystander. Also, for most LSDs we do not know the impact of loss of function in individual cell types. Understanding these critical questions are essential to therapy development. Here, we determine the impact of genetic rescue in distinct cell types on neural circuit dysfunction in CLN3 disease, the most common pediatric dementia and a paradigmatic neurodegenerative LSD. We restored Cln3 expression via AAV-mediated gene delivery and conditional genetic rescue in a CLN3 disease mouse model. Surprisingly, we found that low-level rescue of Cln3 expression in neurons alone normalized clinically relevant electrophysiologic markers of network dysfunction, despite the presence of substantial residual histopathology, in contrast to restoring expression in astrocytes. Thus, loss of CLN3 function in neurons, not storage accumulation, underlies neurologic dysfunction in CLN3 disease. This impliesies that storage clearance may be an inappropriate target for therapy development and an ineffectual biomarker.
Keywords: lysosomal storage disease, CLN3 disease, neuronal dysfunction, gene therapy
Graphical abstract
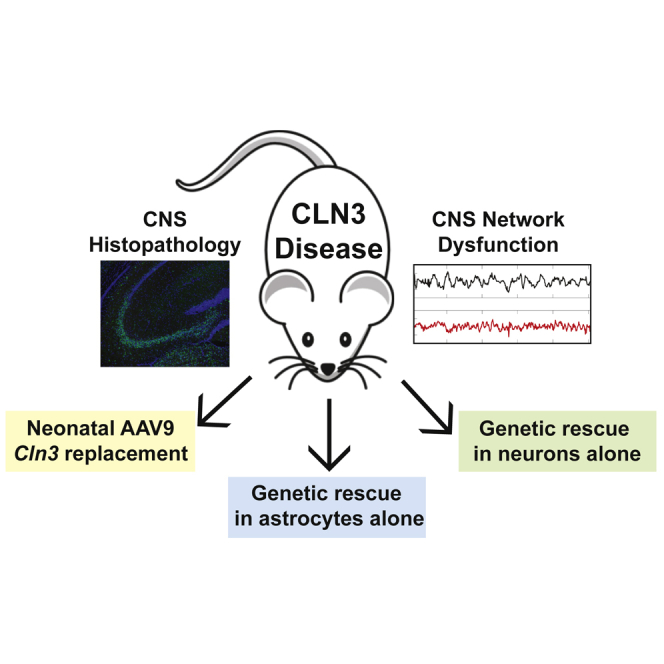
Using AAV-mediated gene therapy and genetic recombination, Ahrens-Nicklas et al. demonstrate that early gene replacement can improve neuronal network function in a mouse model of CLN3 disease, a representative lysosomal storage disorder. Importantly, normalization of function requires correction of neurons but does not depend on prevention of storage accumulation.
Introduction
Lysosomal storage disorders (LSDs) are a group of approximately 50 rare metabolic diseases characterized by progressive accumulation of storage material in lysosomes. Neurological symptoms, including neurocognitive regression, seizures, and psychiatric symptoms, occur in more than two-thirds of LSDs and are generally not ameliorated by current therapies.1 For many LSDs, including CLN3 disease, the most common cause of childhood dementia, the mechanisms underlying neurologic dysfunction are poorly understood.
The function of CLN3 protein remains unknown, hindering mechanism-based therapy development. The brains of CLN3 disease mouse models demonstrate progressive storage accumulation, predominantly comprised of subunit C of the mitochondrial ATP synthase (SCMAS),2,3 mirroring what occurs in CLN3 disease patients.4 Therefore, most drug development efforts rely on histopathologic markers of efficacy, including reduction of storage material, reactive astrocytosis, and microgliosis.5, 6, 7 However, whether these histopathologic changes cause disease, augment the disease process, or are simply harmless epiphenomenon, is unclear.
In addition to lack of clarity on the impact of storage, the role of various central nervous system (CNS) cell types in driving functional deficits in CLN3 disease is unknown. Astrocytes and microglia isolated from CLN3 disease mouse models demonstrate altered cellular properties and physiology8, 9, 10 and, in animal models, localized glial activation occurs in regions where neurons will later die11 supporting a role for glia as a disease driver. On the other hand, neurons from CLN3 animal models exhibit both histopathologic and functional differences.2,12,13 Ultimately, patients’ symptoms, such as seizures and neurologic regression, arise directly from activity changes in key neuronal circuits, i.e., neuronal network dysfunction. Nonetheless, we do not understand what cell types underlie these pathologic network dynamics in the brain.
Previously, we described network-level neurologic defects in both the cortex and hippocampus of two CLN3 disease mouse models using in vitro voltage-sensitive dye imaging and in vivo electrophysiology techniques.14 While many brain regions are affected in CLN3 disease, the cortex and hippocampus are especially vulnerable and show early pathologic changes in mouse models and human patients.3,4,13,15
We found that network-level changes begin early in the disease course before substantial storage accumulation in both the hippocampus and cortex. In slice recordings, these changes include hypoexcitability of the dentate gyrus of the hippocampus, a region essential for learning and memory. On in vivo electroencephalogram (EEG), CLN3 disease mice show frequent epileptiform spikes, a feature of many epilepsy models, and a shift toward high-frequency background activity, a change associated with cognitive impairment and present in Alzheimer’s disease models and patients.16,17
To date, CLN3 disease research has been hindered by the fact that animal models have only subtle behavioral and survival phenotypes.18,19 Most manifest histopathologic changes, including storage accumulation and reactive astrocytosis; however, these phenotypes may not correlate with CNS dysfunction. Our network-level electrophysiologic approach provides a robust, clinically meaningful measure of neurologic function that can be used to probe disease mechanisms and evaluate potential therapies.
Here, we developed a new “conditional rescue” mouse model of CLN3 disease. The model is homozygous for the most common human CLN3 disease mutation,2 but also carries one copy of a Cre-inducible wild-type Cln3 transgene, under the control of its endogenous promoter, allowing for cell-type-specific rescue of expression. We employed AAV-mediated gene replacement as a second strategy to restore Cln3 expression and combined these tools with network-level electrophysiology techniques to answer three essential questions: (1) Does gene replacement correct neural circuit dysfunction in a LSD? (2) What cell types must be corrected to improve network dynamics? (3) Does storage accumulation underlie deficits in CNS network dynamics in CLN3 disease?
Results
In previous work, we found that CLN3 disease mouse models demonstrate robust, progressive neuronal network defects.14 We first assessed if neonatal correction of most CNS cell types could prevent circuit-level functional pathology. For this, we employed an adeno-associated virus serotype 9 (AAV9) vector expressing wild-type murine Cln3 under control of 1,665 bp of the endogenous Cln3 promoter, which mimics the low levels of expression seen in the endogenous setting (Figures S1 and S2; supplemental methods). The vector was delivered at p0 via unilateral intracerebroventricular (ICV) injection into CLN3-deficient mice to transduce both astrocytes and neurons20 (Figure S3). The CLN3 disease mouse model harbors the most common human CLN3 disease mutation, a deletion of exons 7 and 8 (Cln3Δex78/Δex78) (2). ICV injection of AAV9.Cln3 at a dose of 1e10 vg increased Cln3 expression (Figure 1A). One year after injection, levels in the Cln3Δex78/Δex78 hemisphere contralateral to the injection site reached 24.5% ± 4.7% of heterozygous controls. When assessed histologically, gene replacement partially prevented storage accumulation in both the hippocampus and motor cortex ipsilateral to the injection site (Figures 1B and S4).
Figure 1.

Intracerebroventricular AAV9.Cln3 gene therapy given at p0 normalizes neuronal network dynamics in vivo at age 12 months
(A) p0 ICV injection of AAV9.Cln3 restored Cln3 expression in Cln3Δex78/Δex78 mouse brain at age 12 months, with levels in the Cln3Δex78/Δex78 hemisphere contralateral to the injection site reaching 24.5% ± 4.7% of heterozygous controls. (B) Subunit C of the mitochondrial ATP synthase (SCMAS) accumulation is partially prevented in the hippocampus ipsilateral to the injection site of Cln3Δex78/Δex78 mice treated with AAV9.Cln3 (n = 3–4 mice, one-way ANOVA, followed by Tukey’s multiple comparisons test). Gene replacement corrects circuit function measured on intracranial EEG, including (C) epileptiform spiking rates throughout the cortex and (D) background frequency band composition in the motor cortex. Groups: Cln3WT/WT (blue); Cln3WT/Δex78 (black); Cln3Δex78/Δex78 (red); Cln3Δex78/Δex78 + AAV9.CLN3 (gray). N = 5–6 animals/condition. Results from 12 h of recordings were averaged for each animal shown as an individual circle. Spikes were analyzed by two-way ANOVA, followed by Tukey’s multiple comparisons test to compare between genotype groups. Frequency bands were analyzed by two-way ANOVA by band and genotype, followed by Tukey’s multiple comparisons test (significant results of multiple comparison testing shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). Data are shown as mean ± SEM.
To determine the impact of p0 AAV9.Cln3 infusion on in vivo network dynamics, we recorded activity throughout the brain via in vivo EEG. Twelve-month-old Cln3Δex78/Δex78 mice have increased spike burden and a shift in background activity toward faster frequencies, with increased power in faster frequency bands compared with Cln3WT/WT and Cln3Δex78/WT controls (Figures 1C and 1D). Specifically, there is decreased power in the slow delta frequency band (0.1–4 Hz) and a trend toward increased power in the faster beta (13–25 Hz) and gamma (25–50 Hz) bands in Cln3Δex78/Δex78 mice, consistent with our previous report in Cln3 knockout mice.14 Remarkably, restoring gene expression at p0 normalized spiking rates (Figure 1C) and corrected background activity (Figure 1D) in 12-month-old Cln3Δex78/Δex78 AAV9.Cln3-treated animals. Correction of EEG phenotypes was equally effective in both the hemispheres. Thus, early gene replacement can normalize network dynamics, and this rescue does not require full inhibition of storage accumulation.
Because AAV9 delivered to neonates transduces astrocytes and neurons,20 it is unclear whether all cell types must be corrected to improve circuit function, a question critically important to translate gene therapies to children. AAV9 delivered into the CNS of different aged rodents and large animal models transduces different cell populations.21, 22, 23 To address this, we developed a conditional rescue model of CLN3 disease, the FlexCln3 mouse. This model is homozygous for the common Cln3 exon 7–8 deletion and also carries one Cre-inducible FlexCln3 allele, permitting expression of wild-type CLN3 protein in the presence of Cre recombinase (Figures 2 and S5–S7; supplemental methods). The FlexCln3 allele is under control of the same 1,665 bp Cln3 promoter as used for AAV-mediated gene replacement.
Figure 2.

Expression of FlexCln3 allele in all cells reduces storage accumulation in Cln3Δex78/Δex78 brain
(A) In the presence of Cre, the FlexCln3 allele undergoes inversion (step 1) and excision (step 2) and is expressed under control of its endogenous promoter. (B) PCR of genomic DNA from the hippocampus using orientation-specific primers confirms that the FlexCln3 is in the forward orientation only in the presence of Cre (see Figure S7 for uncut gels). (C) In the presence of Zp3-Cre, which drives ubiquitous recombination, the FlexCln3 allele results in expression levels that are 40.0% ± 0.4% of heterozygous controls. (D) Subunit C of the mitochondrial ATP synthase (SCMAS) accumulates in the hippocampus of Cln3Δex78/Δex78 and FlexCln3/Cln3Δex78/Δex78 mice. Storage is reduced in mice with the FlexCln3 allele in the forward confirmation.
We validated the model by crossing FlexCln3 mice with Zp3-Cre mice24 to drive germline level recombination and flip the reverse-oriented wild-type FlexCln3 allele into the forward orientation in all cells. Recombination of the allele was confirmed using orientation-specific PCR primers on genomic DNA extracted from the hippocampus (Figures 2B and S7). In these mice harboring a single FlexCln3 allele, expression was ubiquitously restored in all cell types with expression levels dictated by the transgenic Cln3 1,665 bp promoter. Cln3 expression in the hippocampus was restored to 40% of heterozygous (Cln3Δex78/WT) control levels as determined by qPCR (Figure 2C). Correction (i.e., expression of the FlexCln3 transgene) in all cell types (FlexCln3/Cln3Δex78/Δex78/Zp3-Cre) substantially reduced SCMAS storage in the hippocampus and other brain regions to levels similar to Cln3Δex78/WT or Cln3WT/WT (Figures 2D, 1B, and S8).
For selective rescue of astrocytes or neurons, FlexCln3 mice were crossed to Gfap-Cre25 or Syn1-Cre26 driver lines, respectively. Cre-mediated rearrangement of the FlexCln3 allele was confirmed by PCR of genomic DNA extracted from the hippocampus using orientation-specific primers (Figure 3A). As Cre is only expressed in a subset of cells (either astrocytes or neurons), unlike in the case of Zp3-Cre (Figure 2B), both the forward and reverse alleles were detected in hippocampal tissue.
Figure 3.

Expression of FlexCln3 allele in either neurons or astrocytes alone does not prevent storage accumulation in 12-month-old animals
(A) In the presence of Cre, the FlexCln3 allele undergoes inversion as detected by PCR with orientation-specific primers of genomic DNA from hippocampus. As both the Gfap-Cre and Syn1-Cre only drive recombination in a subset of cells, both the forward and reverse alleles are detected (see Figure S7 for uncut gels). (B) Expression levels as measured by ΔCt values (FlexCln3-βactin) are similar in FlexCln3 brain in the presence of Gfap-Cre and Syn1-Cre (14.1 versus 13.4, p = 0.53 by unpaired t test). (C–G) Expression of FlexCln3 in astrocytes (FlexCln3/Cln3Δex78/Δex78/Gfap-Cre animals) or neurons alone (FlexCln3/Cln3Δex78/Δex78/Syn1-Cre animals) does not prevent accumulation of SCMAS (green). (H–L) Similar findings were seen in the motor cortex. N = 4–5 animals, non-parametric Kruskal-Wallis test followed by Benjamini and Hochberg multiple comparisons correction to detect differences from control. For all panels: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Expression of forward-oriented FlexCln3 transcripts was confirmed in the hippocampus of the astrocyte and neuronal-specific rescue models. As rescue did not occur in all cell types, and Cln3 protomer activity and expression can vary between cells and be induced by disease pathology,3 it is difficult to normalize expression levels to heterozygote controls. However, to get a relative sense of expression levels we compared the level of FlexCln3 transgene expression in the astrocyte-only and neuron-only rescue models. We found no differences in transcript levels (normalized ΔCT values of 14.1 versus 13.4, p = 0.53 by unpaired t test) between the groups (Figure 3B). As there are no available antibodies that can reliably detect endogenous levels of mouse Cln3 protein,27 we could not assess protein expression.
We next determined the impact of cell-type-selective restoration of Cln3 expression on histopathological readouts. Rescue of Cln3 expression in either astrocytes (FlexCln3/Cln3Δex78/Δex78/Gfap-Cre) or neurons (FlexCln3/Cln3Δex78/Δex78/Syn1-Cre) alone did not prevent storage accumulation in the hippocampus or motor cortex in 6- or 12-month-old mice (Figures 3C–3L, S8, and S9). However, there may be region-specific differences as there was a partial reduction of storage in the thalamus after neuronal rescue (Figure S8).
In addition to storage accumulation, CLN3 disease mice develop progressive, reactive astrocytosis. At age 12 months, both the hippocampus and motor cortex of Cln3Δex78/Δex78 showed increased astrocytosis compared with wild-type controls. Rescue of low-level Cln3 expression in either neurons or astrocytes alone only partially improved astrocytosis (Figure S10), suggesting that higher level of expression to all cell types may be required to normalize astrocytosis.
To investigate if the FlexCln3 transgene could prevent storage accumulation in the specific cell type where it was expressed, we investigated co-localization of autofluorescent lipofuscin with a neuronal marker, NeuN, in cortical tissue (Figure 4). There is little lipofuscin accumulation in the Cln3WT/WT cortex compared with the Cln3Δex78/Δex78 cortex, where there is storage in both neurons and non-neuronal cells. Ubiquitous FlexCln3 allele expression (in the presence of Zp3-Cre) prevented storage accumulation in all cell types. FlexCln3 allele expression in neurons (in the presence of Syn1-Cre) did not prevent storage in either neurons or non-neuronal cells.
Figure 4.

Expression of the FlexCln3 allele in neurons alone does not prevent autofluorescent lipofuscin accumulation in cortical neurons
(A–C) There is little autofluorescent lipofuscin accumulation (green) in wild-type cortical neurons (red) (blue DAPI) compared with (D–F) Cln3Δ78/Δ78 cortical neurons in 12-month-old animals. (G–I) Zp3-Cre-induced expression of the FlexCln3 allele in all cell types prevents autofluorescent lipofuscin accumulation in both neuronal and non-neuronal cells. (J–L) FlexCln3 expression in neurons (Syn1-Cre) did not prevent storage accumulation in neurons (arrowheads) or non-neuronal cells (arrows). (M) Quantification of autofluorescence in neurons confirms storage accumulation in NeuN-positive cells after neuronal rescue. Three mice per group were analyzed with three images per mouse. Each image was fragmented for NeuN immunostaining, and autofluorescence was quantified in 238 or more NeuN-positive cells per group. Data represent mean gray value for each cell. Statistical analysis was performed with repeated measures one-way ANOVA followed by Sidak’s multiple comparisons test. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Next, we determined if restoring Cln3 expression could improve electrophysiologic measures despite residual storage accumulation. In vitro voltage-sensitive dye imaging of hippocampal slices from 6-month-old animals was completed in FlexCln3/Cln3Δex78/Δex78/Gfap-Cre, FlexCln3/Cln3Δex78/Δex78/Syn1-Cre, and FlexCln3/Cln3Δex78/Δex78/Zp3 mice. In this early stage of disease, the internal blade of the dentate gyrus of the Cln3Δex78/Δex78 hippocampus was hypoexcitable in response to perforant path stimulation compared with heterozygous Cln3Δ78/WT mice (Figures 5A–5C), consistent with our previous studies.14 Specifically, 31.6% of pixels in the internal blade of the Cln3Δex78/Δex78 dentate gyrus were significantly hypoexcitable compared with slices obtained from heterozygous control animals. Interestingly, restoring expression in astrocytes did not correct hypoexcitability of the dentate gyrus (Figures 5D and 5E), with 23.6% of pixels remaining hypoexcitable. However, restoring Cln3 expression in neurons was sufficient to prevent disease-induced hypoexcitability (Figures 5F and 5G), reducing hypoexcitable pixels to 1% compared with heterozygous controls. This rescue of dynamics occurred despite the presence of residual histopathology (Figures 3, 4, and S8–S10).
Figure 5.

Restoring low-level Cln3 expression in neurons alone corrects hypoexcitability of the dentate gyrus
Dentate gyrus excitability measured through in vitro voltage-sensitive dye imaging of hippocampal slices from 6-month-old animals is shown as raster plots of average fluorescence change (ΔF/F warm colors excitation, cool colors inhibition) over time (x axis) and location within the internal blade of the dentate gyrus (y axis). (A) Four-pulse stimulation of the perforant path in heterozygous control (Cln3Δex78/WT) mice reveals robust excitation of the dentate gyrus (DG) compared with (B) Cln3Δex78/Δex78 mice. (C) Cln3Δex78/WT versus Cln3Δex78/Δex78 rasters were compared using a permutation sampling method with 1,000 trials. Pixels with p > 0.05 are masked in gray. For regions of significant difference in excitability with p < 0.05, the difference in fluorescence change (ΔF/FΔex78/WT − ΔF/FΔex78/Δex78) is shown. (D and E) In animals expressing Cln3 in astrocytes (FlexCln3/Cln3Δex78/Δex78/Gfap-Cre), the DG remained hypoexcitable compared with the Cln3Δex78/WT DG. (F and G) Expression of Cln3 in neurons alone (FlexCln3/Cln3Δex78/Δex78/Syn1-Cre) was sufficient to reverse hypoexcitability of the DG compared with Cln3Δex78/WT animals. Group sizes (n = slices, N = mice): Cln3Δex78/WT n = 23 N = 4; Cln3Δex78/Δex78 n = 30, N = 6; FlexCln3/Cln3Δex78/Δex78/Gfap-Cre n = 19 slices, N = 4; FlexCln3/Cln3Δex78/Δex78/Syn1-Cre n = 17; N = 4.
To evaluate how cell-type-specific correction modulates CNS functional defects in vivo, we measured network activity through long-term EEG recordings. While restoration of Cln3 expression in either astrocytes or neurons prevented epileptiform spikes throughout the brain (Figure 6A), only rescue in neurons corrected EEG background frequency composition (i.e., frequency band power) and thereby fully normalized network activity (Figures 6B–6F). Intriguingly, this correction of physiology occurred despite the presence of substantial storage burden, revealing for the first time that loss of CLN3 protein function in neurons, not storage burden, underlies functional defects in CLN3 disease.
Figure 6.

Cln3 rescue in either astrocytes or neurons reduces epileptiform spikes, but only neuronal rescue fully normalizes network dynamics as measured on EEG
(A) Correction of either astroctyes (FlexCln3/Cln3Δex78/Δex78/Gfap-Cre, blue) or neurons (FlexCln3/Cln3Δex78/Δex78/Syn1-Cre, green) reduced spike burden on EEG in 12-month-old mice. (B–E) Example EEG traces from Cln3Δex78/Δex78 (red) and FlexCln3/Cln3Δex78/Δex78/Gfap-Cre motor cortex (blue) show decreased slow delta activity and increased fast activity compared with control motor cortex (black). EEG trace from FlexCln3/Cln3Δex78/Δex78/Syn1-Cre motor cortex (green) shows that rescue of Cln3 expression in neurons normalizes EEG background frequency composition. (F) Power spectral analysis of EEG data recorded in the motor cortex confirm that rescue of Cln3 expression in astrocytes (blue) does not correct disease-associated loss of delta activity, and actually exacerbates increased fast gamma activity. However, correction in neurons (green) normalizes EEG frequency composition. N = 4–5 animals/genotype, after passing a Shapiro-Wilk test of normality, data were analyzed by two-way ANOVA, followed by Sidak’s multiple comparisons test. Significant differences from multiple comparisons testing are shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. Frequency data in (F) in Cln3WT/Δex78 and Cln3Δex78/Δex78 are also shown in Figure 1D.
Discussion
Previous cellular-level studies of CLN3 disease have demonstrated disease-associated abnormalities in both neurons and astrocytes.4,9, 10, 11,13,28, 29, 30 Prior to this work, it was unknown how these cellular defects translate to the circuit-level dysfunction that underlies neurological symptoms, such as dementia and seizures, that CLN3 disease patients experience. This work highlights the fact that neurons underlie network-level functional defects in CLN3 disease and must be targeted in future drug development efforts.
A number of novel therapies, including small molecule,5,6,31, 32, 33, 34 antisense oligonucleotides,35 and gene replacement7,36 strategies, are under development for CLN3 disease. These approaches will not target all cell types equally. For example, studies in non-human primates have demonstrated that intravenous administration of AAV9 in juvenile non-human primates predominately transduces glia,23,37 while direct administration into the cerebral spinal fluid improves delivery to neurons.34,38 Our results suggest that maximizing neuronal correction will be essential to develop a treatment that improves functional outcomes in CLN3 disease.
Future in vitro and in vivo studies evaluating different levels of rescue in a variety of cell types could provide further insights into how outcomes vary with level of gene expression rescue. In addition to neurons and astrocytes, Cln3 is expressed at low levels in other CNS cell types including endothelial cells and microglia.39 Abnormalities in these cell types have been reported in CLN3 disease models.6,10 Future investigations of Cln3 rescue in these populations could further our understanding of disease mechanisms.
In previous studies, Cln3Δex78/Δex78 mice have demonstrated abnormal astrocytic-neuronal coupling.9 Monogenic mouse models of isolated astrocyte dysfunction have epileptiform spikes and seizures on EEG because of pathologic crosstalk between astrocytes and neurons.40 In the FlexCln3 mouse, rescue of Cln3 expression in astrocytes alone actually exacerbated EEG background abnormalities, including increasing fast gamma activity. This suggests that balanced Cln3 expression in both neurons and astrocytes may be required to maintain a normal EEG background. However, rescue in astrocytes alone was sufficient to prevent epileptiform spikes. The reduction in spiking likely reflects improved astrocytic-neuronal interactions when Cln3 expression is restored in astrocytes. Collectively, our data suggest that diseased astrocytes may modulate network-level dysfunction in CLN3 disease, but diseased neurons are a primary driver of functional deficits.
Using our cell-type-specific rescue model, we found that storage accumulation was prevented when Cln3 was reintroduced to all cell types embryonically, but not when that correction was restricted to astrocytes or neurons alone. Interestingly, prevention of storage accumulation is not required for correcting neuronal network physiology, suggesting that storage material may not be the primary driver of the disease process. This finding is consistent with a previous study of CLN3 gene therapy in mice, which demonstrated improvement in some disease markers despite residual storage accumulation.7
These studies highlight the importance of using clinically relevant functional measures, such as in vivo electrophysiology studies, to guide treatment development. Histologic measures, such as storage burden, may be incomplete surrogate biomarkers of therapeutic efficacy in CLN3 disease and possibly other neuronopathic LSDs. Future studies are needed to fully elucidate how loss of CLN3 protein in neurons leads to neurologic dysfunction. One emerging theory is that CLN3 may play a role in maintaining synaptic structure and function.13,41,42 The circuit-level abnormalities demonstrated here could arise directly from these synaptic alterations, rather than in response to storage accumulation.
In summary, partial restoration of Cln3 expression by AAV vectors or through Cre-based systems shows that neuronal expression is critical for preventing deficits in CNS functional circuits in CLN3 disease mice. This work also highlights that improvement in storage burden alone may not be an adequate readout of therapeutic efficacy in CLN3 disease, a phenomenon that should be considered by the broader LSD community.
Materials and methods
For additional information, see supplemental methods.
Study approval
All animal protocols were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia.
Statistical analysis
All statistical analysis of VSDI rasters was completed using a permutation sampling method and the VSDI toolbox in MATLAB (The MathWorks) as described previously.14,43 For summary statistics of regions or groups, GraphPad Prism was also used. In graphs, data are expressed as mean ± SEM. The statistical significance of the observed differences between two groups was assessed by two-tailed t test (for normally distributed data) or Mann-Whitney U test (for non-normally distributed data). For grouped datasets one- or two-way ANOVA followed by multiple comparisons test was completed as detailed in each figure legend. Results were considered significant when p < 0.05. Male and female mice were included in all groups.
Acknowledgments
NINDSR21NS084424 and NICHD HD33532 to B.L.D., NIH K08NS105865 to R.C.A.-N., NCL Stiftung Research Award to R.C.A.-N., Chan Zuckerberg Initiative Neurodegeneration Challenge Network Award to R.C.A.-N., Children’s Hospital of Philadelphia Research Institute support to R.C.A.-N. and B.L.D.
Author contributions
Design of study, R.C.A.-N., L.T., A.F.H., O.K., R.J.C., E.L., E.D.M., C.S.S., and B.L.D.; data acquisition and analysis of data, R.C.A.-N., L.T., A.F.H., O.K., R.J.C., E.L., and C.S.S.; drafting of the manuscript, R.C.A.-N., L.T., A.F.H., O.K., and C.S.S.; critical review of the data and manuscript, R.C.A.-N., L.T., E.D.M., C.S.S., and B.L.D.
Declaration of interests
B.L.D. is a founder of Spark Therapeutics and Spirovant and is on the scientific advisory boards of Intellia Therapeutics, Homology Medicines, Prevail Therapeutics, Resilience, Moment Bio, Spirovant, Saliogen and Panorama Medicines. The other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.03.025.
Contributor Information
Rebecca C. Ahrens-Nicklas, Email: ahrensnicklasr@email.chop.edu.
Beverly L. Davidson, Email: davidsonbl@chop.edu.
Supplemental information
References
- 1.Platt F.M. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018;17:133–150. doi: 10.1038/nrd.2017.214. [DOI] [PubMed] [Google Scholar]
- 2.Cotman S.L., Vrbanac V., Lebel L.A., Lee R.L., Johnson K.A., Donahue L.R., Teed A.M., Antonellis K., Bronson R.T., Lerner T.J., MacDonald M.E. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 2002;11:2709–2721. doi: 10.1093/hmg/11.22.2709. [DOI] [PubMed] [Google Scholar]
- 3.Eliason S.L., Stein C.S., Mao Q., Tecedor L., Ding S.L., Gaines D.M., Davidson B.L. A knock-in reporter model of Batten disease. J. Neurosci. 2007;27:9826–9834. doi: 10.1523/JNEUROSCI.1710-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tyynela J., Cooper J.D., Khan M.N., Shemilts S.J., Haltia M. Hippocampal pathology in the human neuronal ceroid-lipofuscinoses: distinct patterns of storage deposition, neurodegeneration and glial activation. Brain Pathol. 2004;14:349–357. doi: 10.1111/j.1750-3639.2004.tb00077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarczyluk-Wells M.A., Salzlechner C., Najafi A.R., Lim M.J., Smith D., Platt F.M., Williams B.P., Cooper J.D. Combined anti-inflammatory and neuroprotective treatments have the potential to impact disease phenotypes in Cln3 (-/-) mice. Front. Neurol. 2019;10:963. doi: 10.3389/fneur.2019.00963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz M.L., Tecedor L., Lysenko E., Ramachandran S., Stein C.S., Davidson B.L. Modulating membrane fluidity corrects Batten disease phenotypes in vitro and in vivo. Neurobiol. Dis. 2018;115:182–193. doi: 10.1016/j.nbd.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosch M.E., Aldrich A., Fallet R., Odvody J., Burkovetskaya M., Schuberth K., Fitzgerald J.A., Foust K.D., Kielian T. Self-complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis (CLN3) J. Neurosci. 2016;36:9669–9682. doi: 10.1523/JNEUROSCI.1635-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiong J., Kielian T. Microglia in juvenile neuronal ceroid lipofuscinosis are primed toward a pro-inflammatory phenotype. J. Neurochem. 2013;127:245–258. doi: 10.1111/jnc.12385. [DOI] [PubMed] [Google Scholar]
- 9.Burkovetskaya M., Karpuk N., Xiong J., Bosch M., Boska M.D., Takeuchi H., Suzumura A., Kielian T. Evidence for aberrant astrocyte hemichannel activity in juvenile neuronal ceroid lipofuscinosis (JNCL) PLoS One. 2014;9:e95023. doi: 10.1371/journal.pone.0095023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parviainen L., Dihanich S., Anderson G.W., Wong A.M., Brooks H.R., Abeti R., Rezaie P., Lalli G., Pope S., Heales S.J., et al. Glial cells are functionally impaired in juvenile neuronal ceroid lipofuscinosis and detrimental to neurons. Acta Neuropathol. Commun. 2017;5:74. doi: 10.1186/s40478-017-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pontikis C.C., Cella C.V., Parihar N., Lim M.J., Chakrabarti S., Mitchison H.M., Mobley W.C., Rezaie P., Pearce D.A., Cooper J.D. Late onset neurodegeneration in the Cln3-/- mouse model of juvenile neuronal ceroid lipofuscinosis is preceded by low level glial activation. Brain Res. 2004;1023:231–242. doi: 10.1016/j.brainres.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 12.Brenneman D.E., Pearce D.A., Kovacs A., DeFrees S. Pharmacological effects on ceroid lipofuscin and neuronal structure in Cln3 (ex7/8) mouse brain cultures. J. Mol. Neurosci. 2017;63:100–114. doi: 10.1007/s12031-017-0962-5. [DOI] [PubMed] [Google Scholar]
- 13.Grunewald B., Lange M.D., Werner C., O'Leary A., Weishaupt A., Popp S., Pearce D.A., Wiendl H., Reif A., Pape H.C., et al. Defective synaptic transmission causes disease signs in a mouse model of juvenile neuronal ceroid lipofuscinosis. Elife. 2017;6:e28685. doi: 10.7554/eLife.28685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ahrens-Nicklas R.C., Tecedor L., Hall A.F., Lysenko E., Cohen A.S., Davidson B.L., Marsh E.D. Neuronal network dysfunction precedes storage and neurodegeneration in a lysosomal storage disorder. JCI Insight. 2019;4:e131961. doi: 10.1172/jci.insight.131961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchison H.M., Lim M.J., Cooper J.D. Selectivity and types of cell death in the neuronal ceroid lipofuscinoses. Brain Pathol. 2004;14:86–96. doi: 10.1111/j.1750-3639.2004.tb00502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwak Y.T. Quantitative EEG findings in different stages of Alzheimer's disease. J. Clin. Neurophysiol. 2006;23:456–461. doi: 10.1097/01.wnp.0000223453.47663.63. [DOI] [PubMed] [Google Scholar]
- 17.Kent B.A., Strittmatter S.M., Nygaard H.B. Sleep and EEG power spectral analysis in three transgenic mouse models of Alzheimer's disease: APP/PS1, 3xTgAD, and Tg2576. J. Alzheimers Dis. 2018;64:1325–1336. doi: 10.3233/JAD-180260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faller K.M., Gutierrez-Quintana R., Mohammed A., Rahim A.A., Tuxworth R.I., Wager K., Bond M. The neuronal ceroid lipofuscinoses: opportunities from model systems. Biochim. Biophys. Acta. 2015;1852:2267–2278. doi: 10.1016/j.bbadis.2015.04.022. [DOI] [PubMed] [Google Scholar]
- 19.Johnson T.B., Langin L.M., Zhao J., Weimer J.M., Pearce D.A., Kovacs A.D. Changes in motor behavior, neuropathology, and gut microbiota of a Batten disease mouse model following administration of acidified drinking water. Sci. Rep. 2019;9:14962. doi: 10.1038/s41598-019-51488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammond S.L., Leek A.N., Richman E.H., Tjalkens R.B. Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS One. 2017;12:e0188830. doi: 10.1371/journal.pone.0188830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinderer C., Bell P., Vite C.H., Louboutin J.P., Grant R., Bote E., Yu H., Pukenas B., Hurst R., Wilson J.M. Widespread gene transfer in the central nervous system of cynomolgus macaques following delivery of AAV9 into the cisterna magna. Mol. Ther. Methods Clin. Dev. 2014;1:14051. doi: 10.1038/mtm.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gray S.J., Nagabhushan Kalburgi S., McCown T.J., Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samaranch L., Salegio E.A., San Sebastian W., Kells A.P., Foust K.D., Bringas J.R., Lamarre C., Forsayeth J., Kaspar B.K., Bankiewicz K.S. Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum. Gene Ther. 2012;23:382–389. doi: 10.1089/hum.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shafi R., Iyer S.P., Ellies L.G., O'Donnell N., Marek K.W., Chui D., Hart G.W., Marth J.D. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc. Natl. Acad. Sci. U S A. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gregorian C., Nakashima J., Le Belle J., Ohab J., Kim R., Liu A., Smith K.B., Groszer M., Garcia A.D., Sofroniew M.V., et al. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J. Neurosci. 2009;29:1874–1886. doi: 10.1523/JNEUROSCI.3095-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y., Romero M.I., Ghosh P., Ye Z., Charnay P., Rushing E.J., Marth J.D., Parada L.F. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nelson T., Pearce D.A., Kovacs A.D. Lack of specificity of antibodies raised against CLN3, the lysosomal/endosomal transmembrane protein mutated in juvenile Batten disease. Biosci. Rep. 2017;37 doi: 10.1042/BSR20171229. BSR20171229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pontikis C.C., Cotman S.L., MacDonald M.E., Cooper J.D. Thalamocortical neuron loss and localized astrocytosis in the Cln3Deltaex7/8 knock-in mouse model of Batten disease. Neurobiol. Dis. 2005;20:823–836. doi: 10.1016/j.nbd.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Bosch M.E., Kielian T. Astrocytes in juvenile neuronal ceroid lipofuscinosis (CLN3) display metabolic and calcium signaling abnormalities. J. Neurochem. 2019;148:612–624. doi: 10.1111/jnc.14545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhong Y., Mohan K., Liu J., Al-Attar A., Lin P., Flight R.M., Sun Q., Warmoes M.O., Deshpande R.R., Liu H., et al. Loss of CLN3, the gene mutated in juvenile neuronal ceroid lipofuscinosis, leads to metabolic impairment and autophagy induction in retinal pigment epithelium. Biochim. Biophys. Acta Mol. Basis Dis. 2020;1866:165883. doi: 10.1016/j.bbadis.2020.165883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Augustine E.F., Beck C.A., Adams H.R., Defendorf S., Vierhile A., Timm D., Weimer J.M., Mink J.W., Marshall F.J. Short-term administration of mycophenolate is well-tolerated in CLN3 disease (juvenile neuronal ceroid lipofuscinosis) JIMD Rep. 2019;43:117–124. doi: 10.1007/8904_2018_113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Sitt S., Soueid J., Maalouf K., Makhoul N., Al Ali J., Makoukji J., Asser B., Daou D., Harati H., Boustany R.M. Exogenous galactosylceramide as potential treatment for CLN3 disease. Ann. Neurol. 2019;86:729–742. doi: 10.1002/ana.25573. [DOI] [PubMed] [Google Scholar]
- 33.Petcherski A., Chandrachud U., Butz E.S., Klein M.C., Zhao W.N., Reis S.A., Haggarty S.J., Ruonala M.O., Cotman S.L. An autophagy modifier screen identifies small molecules capable of reducing autophagosome accumulation in a model of CLN3-mediated neurodegeneration. Cells. 2019;8:1531. doi: 10.3390/cells8121531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maalouf K., Makoukji J., Saab S., Makhoul N.J., Carmona A.V., Kinarivala N., Ghanem N., Trippier P.C., Boustany R.M. Exogenous flupirtine as potential treatment for CLN3 disease. Cells. 2020;9:1872. doi: 10.3390/cells9081872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Centa J.L., Jodelka F.M., Hinrich A.J., Johnson T.B., Ochaba J., Jackson M., Duelli D.M., Weimer J.M., Rigo F., Hastings M.L. Therapeutic efficacy of antisense oligonucleotides in mouse models of CLN3 Batten disease. Nat. Med. 2020;26:1444–1451. doi: 10.1038/s41591-020-0986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kleine Holthaus S.M., Aristorena M., Maswood R., Semenyuk O., Hoke J., Hare A., Smith A.J., Mole S.E., Ali R.R. Gene therapy targeting the inner retina rescues the retinal phenotype in a mouse model of CLN3 batten disease. Hum. Gene Ther. 2020;31:709–718. doi: 10.1089/hum.2020.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R., Samulski R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bey K., Deniaud J., Dubreil L., Joussemet B., Cristini J., Ciron C., Hordeaux J., Le Boulc'h M., Marche K., Maquigneau M., et al. Intra-CSF AAV9 and AAVrh10 administration in nonhuman primates: promising routes and vectors for which neurological diseases? Mol. Ther. Methods Clin. Dev. 2020;17:771–784. doi: 10.1016/j.omtm.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Z., van Velthoven C.T.J., Nguyen T.N., Goldy J., Sedeno-Cortes A.E., Baftizadeh F., Bertagnolli D., Casper T., Chiang M., Crichton K., et al. A taxonomy of transcriptomic cell types across the isocortex and hippocampal formation. Cell. 2021;184:3222–3241.e26. doi: 10.1016/j.cell.2021.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robel S., Buckingham S.C., Boni J.L., Campbell S.L., Danbolt N.C., Riedemann T., Sutor B., Sontheimer H. Reactive astrogliosis causes the development of spontaneous seizures. J. Neurosci. 2015;35:3330–3345. doi: 10.1523/JNEUROSCI.1574-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gomez-Giro G., Arias-Fuenzalida J., Jarazo J., Zeuschner D., Ali M., Possemis N., Bolognin S., Halder R., Jager C., Kuper W.F.E., et al. Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol. Commun. 2019;7:222. doi: 10.1186/s40478-019-0871-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Llavero Hurtado M., Fuller H.R., Wong A.M.S., Eaton S.L., Gillingwater T.H., Pennetta G., Cooper J.D., Wishart T.M. Proteomic mapping of differentially vulnerable pre-synaptic populations identifies regulators of neuronal stability in vivo. Sci. Rep. 2017;7:12412. doi: 10.1038/s41598-017-12603-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bourgeois E.B., Johnson B.N., McCoy A.J., Trippa L., Cohen A.S., Marsh E.D. A toolbox for spatiotemporal analysis of voltage-sensitive dye imaging data in brain slices. PLoS One. 2014;9:e108686. doi: 10.1371/journal.pone.0108686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.