

Abstract
Chimeric antigen receptor (CAR) T cells are a cancer immunotherapy of extremes. Unprecedentedly effective, but complex and costly to manufacture, they are not yet a therapeutic option for all who would benefit. This disparity has motivated worldwide efforts to simplify treatment. Among the proposed solutions, the generation of CAR T cells directly in the patient, i.e., in vivo, is arguably simultaneously the most technically challenging and clinically useful approach to convert CAR therapy from a cell-based autologous medicinal product into a universally applicable off-the-shelf treatment. Here, we review the current state of the art of in vivo CAR therapy, focusing especially on the vector technologies used. These cover lentiviral vectors and adenovirus-associated vectors as well as synthetic polymer nanocarriers and lipid nanoparticles. Proof of concept, i.e., the generation of CAR cells directly in mouse models, has been demonstrated for all vector platforms. Receptor targeting of vector particles is crucial, as it can prevent CAR gene delivery into off-target cells, thus reducing toxicities. We discuss the properties of the vector platforms, such as their immunogenicity, potency, and modes of CAR delivery (permanent versus transient). Finally, we outline the work required to advance in vivo CAR therapy from proof of concept to a robust, scalable technology for clinical testing.
Keywords: nanoparticle, AAV, lentiviral vector, off the shelf, T lymphocyte, LNP
Graphical abstract
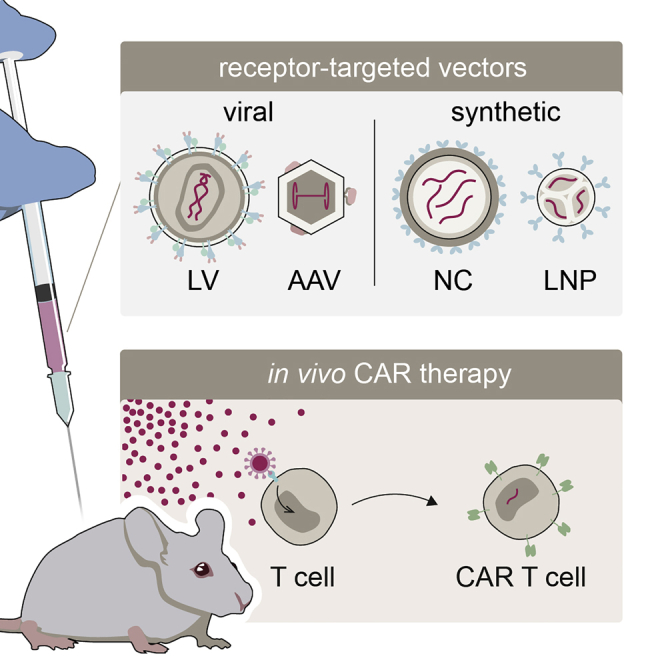
Buchholz and colleagues review recent publications providing preclinical proof of concept for the generation of CAR T cells directly in vivo. They find that in this advance toward a new generation of immunotherapies, receptor-targeted viral and synthetic vector systems mediating selective gene transfer into T lymphocytes have played a critical role.
Introduction
Gene therapy is looking back at over 30 years of history and an ever-increasing number of therapeutic concepts and indications.1 Toward the end of the 2000s, initial success stories were soon followed by reports of severe side effects and deaths.2,3 The field overcame these setbacks through technological innovation in vector design and has since then entered a booming phase with the present worldwide momentum involving academic laboratories, agile biotech companies, and big pharmaceutical players. The main driver for this development was an extension in indications from monogenetic diseases to complex acquired illnesses like cancer, which was facilitated by the use of genes encoding artificial proteins. The most prominent example is chimeric antigen receptors (CARs), which have revolutionized cancer immunotherapy with four medicinal products on the European market.4, 5, 6, 7 Still, major technological challenges for gene therapy remain: Besides the implementation of minimally invasive gene editing in treatment of monogenetic diseases and improving therapies’ safety profiles, cell-type-selective in vivo delivery of therapeutic genes is currently regarded as one of the problems with the highest priority.8 The conversion of therapy-relevant cells into corrected or therapeutic protein-producing cells directly in the patient would make complex ex vivo manufacturing of genetically modified cells unnecessary and enable previously unviable therapeutic approaches.
In vivo gene delivery has already reached the market. Based on adenovirus-associated vectors (AAVs) or oncolytic viruses, they rely on local application or the non-toxicity of their genetic cargo to offset the vectors’ broad cellular tropism. For many indications, however, the therapy-relevant cells form a small, precisely defined fraction, with off-target delivery to the majority of other cells being potentially deleterious. This is also true for CAR T cell therapy, which requires genetic modification of the patient’s T lymphocytes.
In vivo CAR T cell therapy
CAR T cell therapy is a form of adoptive T cell therapy, in which cancer patients receive tumor-specific T cells that were genetically altered and expanded ex vivo. Chimeric antigen receptors (CARs) are composed of an extracellular antigen-binding domain, connected via a hinge region and a transmembrane domain to one or more intracellular signalling domains. Upon binding to their targets, CARs induce intracellular signaling that results in antigen-specific killing of the target cell and simultaneous proliferation of the CAR T cell.9,10 Thus, CAR T cells can be regarded as living drugs that amplify in patients when they encounter target cancer cells. In recent years, this unique therapeutic concept has boosted research worldwide, with four products having been granted marketing authorization in Europe. Initial authorizations were granted to Yescarta and Kymriah, both targeting lymphoma cells via the B cell marker CD19. Besides lymphoma cells, healthy B lymphocytes are eliminated by these products, resulting in B cell depletion as a prominent side effect in treated patients.11 Recently, two additional products, Tecartus and Abecma, have received regulatory approval, the latter extending indications for CAR therapy to multiple myeloma via B cell maturation antigen (BCMA).7 The approval of the first CAR cell product not directed to CD19 marks an important step in this new field in cancer immunotherapy. Indeed, several hundred CAR T cell trials are ongoing worldwide, many of which aim at facilitating manufacturing and addressing additional malignancies.12
Use of CAR T cells in patients has started a public debate about cost explosions in health systems due to innovative therapeutics.13 CAR T cells are especially expensive, since they are individualized cell therapy products requiring time-consuming manufacturing procedures that rely on ex vivo gene transfer protocols. Following the isolation of lymphocytes from patients, the cells are activated and subsequently transduced, often using lentiviral or γ-retroviral vectors. The modified lymphocytes are then expanded and finally re-infused into the patient (Figure 1A). This complex manufacturing process is expensive and, through the necessary manipulations of patients’ T lymphocytes, can alter their phenotype and activity.14 Moreover, T cells have to be isolated from the patient’s blood with utmost stringency, as the presence of residual tumor cells in the preparation during transduction may lead to accidental transfer of the CD19-CAR into leukemic cells during manufacturing. This resulted in a CAR T-cell-resistant clone, in which CD19 was masked by the single-chain variable antibody fragment (scFv) of the CAR, thus preventing its recognition. Cancer relapse and death of the patient followed.15 Although only a single such case has been reported thus far, it highlights the complexities of the manufacturing process for CAR T cells that prevent their broad application in standard medical care as well as the risk associated with off-target transfer of CAR.
Figure 1.

Ex vivo versus in vivo CAR therapy
The two strategies for converting T cells into CAR T cells are compared on the cellular level (A) and regarding their implications for clinical use (B). (A) Ex vivo generation of CAR T cells usually entails isolation of T cells from patient blood (1), followed by activation, transduction, and ex vivo expansion. After conditioning treatment (2), CAR T cells are infused into the patient (3). In the in vivo approach, vector particles (depicted as red dots) are infused directly into the patient, where they encounter T cells and selectively deliver genetic material encoding the CAR (red). (B) Due to their autologous nature, ex vivo-generated CAR cell products currently have to be prepared individually for each patient (left). The vector preparations currently under evaluation for in vivo CAR therapy constitute universally applicable off-the-shelf medicinal products (right).
Consequently, various paths are currently being pursued in preclinical and clinical research to improve CAR technology. Strategies aiming at facilitating the manufacturing process reach from automated systems16 to allogeneic CAR T cells.17 Although automation combined with the possibility to generate CAR T cells close to the patient’s bedside will greatly facilitate the logistics of manufacturing, this will not change the autologous, highly individualized nature of the product. In the allogeneic approach, CAR T cells prepared from a healthy donor are genetically manipulated to decrease their alloreactivity in the recipients. This is a step toward off-the-shelf CAR T cells, although the resulting products will most likely not be completely universal, owing to human leukocyte antigen barriers that necessitate adaptation to patient subgroups.18 Graft-versus-host reactions as well as manufacturing complexity are circumvented when CAR T cells are generated directly in vivo. Here, a single, universally applicable medicinal product in the form of systemically administered vectors encoding the CAR would be used to transduce the patient’s T cells directly in their body. The resulting CAR T cells would be truly autologous (Figure 1). Although theoretically ideal for meeting the growing demand for CAR therapy, the in vivo approach has not yet made it to the clinic, mainly because suitable vector platforms were lacking, until recently. In the last five years, several groups have reported the successful in vivo generation of CAR T cells in mouse models (Table 1). Following advances in vector design, especially in vector targeting, these reports should be appreciated as breakthroughs in cancer immunotherapy and as showcases for the relevance of vector engineering in gene therapy in general.
Table 1.
Preclinical data providing evidence for in vivo CAR T cell generation
| Vector |
Mouse model |
CAR |
Reference | ||||
|---|---|---|---|---|---|---|---|
| Platform | Targeted receptor | Dosing and routeb | Straind | Immune transplantse | Configurationf | Target cellsg | |
| LV with NiV glycoproteins | Human CD8 | 2 × 106 TU (i.p.) | NSG | Activated human PBMCs (i.p.) | hCD19-28ζ-CAR | Raji (i.p.) | Pfeiffer et al.19 |
| 2 × 106 TU (i.v.) | NSG | Human CD34+ CB cells | hCD19-28ζ-CAR | Endogenous B cells | |||
| LV with NiV glycoproteins | Human CD8 | 2.5 × 1011 particles (i.v.) | NSG | Activated human PBMCs (i.v.) | hCD19-28ζ-CAR | Nalm-6 luc, (i.v.) | Agarwal et al.20 |
| LV with MV glycoproteins | Human CD4 | 4 × 1010 particles (i.p.) | NSG | Activated human PBMCs (i.p.) | hCD19-28ζ-CAR | Endogenous B cells | Agarwal et al.21 |
| 1 × 1011 particles (i.v.) | NSG | Activated human PBMCs (i.v.) | hCD19-28ζ-CAR | Nalm-6 luc, (i.v.) | |||
| 4 × 1010 particles (i.v.) | NSG | Human CD34+ CB cells | hCD19-28ζ-CAR | Endogenous B cells | |||
| LV with NiV glycoproteins | Human CD3 | 2 × 1011 particles (i.v.) | NSG | Human CD34+ CB cells | hCD19-28ζ-CAR | Endogenous B cells | Frank et al.22 |
| LV with SINV glycoproteins | Human CD3 | 5 × 1010 particles (i.v.) | NSG | Activated human PBMCs (i.v.) | hCD19-28ζ-CAR | BV-173 luc (i.v.) | Huckaby et al.23 |
| AAV-DJ | N/Aa | 1 × 1011 vg (i.p.) | NCG | Human PBMCs (i.p.) | hCD4-28-4-1BBζ-CAR | Endogenous CD4 T cells | Nawaz et al.24 |
| 2 × 1011 vg (i.p.) | NCG | Human PBMCs (i.p.) | hCD4-28-4-1BBζ-CAR | MT-2 ATL (i.p.) | |||
| NC with DNA | Mouse CD3 | 3 × 1011 particles/day on five consecutive days (i.v.c) | Albino B6 | N/A | mCD19-4-1BBζ-CAR | N/A | Smith et al.25 |
| 3 × 1011 particles/day on five consecutive days (i.v.c) | Albino B6 | N/A | mCD19-4-1BBζ-CAR | Eμ-ALL(i.v.) | |||
| NC with mRNA | Human CD8 | 6 weekly doses; 50 μg mRNA/dose (i.v.) | NSG | Human T cells (i.v.) | hCD19-28ζ-CAR | Raji-luc (i.v.) | Parayath et al.26 |
| 6 weekly doses; 50 μg mRNA/dose (i.v.) | NSG | Human T cells (i.v.) | hROR1-4-1BBζ-CAR | LNCaP C42 prostate cancer cells (orthotopic) | |||
| Human CD3 | 4 weekly bursts of 3 daily doses; 15 μg mRNA/dose (i.v.) | B6 | N/A | mCD19-28ζ-CAR | Eμ-ALL (i.v.) | ||
| LNP with mRNA | Mouse CD5 | Single dose of 10 μg LNPs (i.v.) | B6 | N/A | mFAP-28ζ-CAR | Endogenous fibrotic cardiac fibroblast | Rurik et al.27 |
N/A, not applicable.
TU, transducing unit; i.p., intraperitoneal injection; i.v., intravenous injection; vg, vector genome.
over 20 min with infusion pump.
NSG, NOD.Cg.PrkdcscidIL2rgtmWjl/SzJ; NCG, NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt; Albino B6, C57BL/6J-Tyrc−2J/J; B6, C57BL/6J.
PBMCs, human peripheral blood mononuclear cells; CB, cord blood-derived.
h, anti-human; m, anti-mouse.
luc, luciferase expressing.
Surface engineering of vector particles for receptor-targeted gene delivery
Despite approved medicines and the breakthroughs in clinical research outlined above, gene therapies are far from being widely administered. Approved products address severe diseases where the risks associated with treatment are acceptable. As gene therapies begin to be considered for more indications, vectors need to be improved. This is especially relevant for systemic administration, which resulted in potentially dose-limiting toxicities, including but not limited to liver toxicities, as described for lipid nanoparticles (LNPs)28, 29, 30, 31 and AAVs.32 In a trial investigating treatment of X-linked myotubular myopathy with AAV8, all three patients receiving the higher dose experienced severe hepatotoxicity and two died.33 When organs other than liver are the intended target, high vector doses are required, since only a small fraction of the administered particles reaches the therapy-relevant cell type, as was impressively shown by human biodistribution data from two deceased children treated with Zolgensma, an AAV9-based gene therapy for the treatment of spinal muscular atrophy.34
Vectors that preferentially (or even exclusively) transduce the therapy-relevant cell type upon systemic administration are expected to reduce the required dose and toxicities.35 For in vivo CAR therapy, use of such T-cell-targeted vectors is expected to prevent delivery of the CAR to non-T cells, especially cancer cells, avoiding fatal consequences for the patient. The degree of T cell-selectivity will have to be experimentally determined for any new type of T-cell-targeted vector. Receptor targeting is an approach of rational vector design that renders vectors selective for a defined cell surface receptor, e.g., for immune cell markers like CD3, CD4, CD5, or CD8. Importantly, selectivity is achieved at the level of cell entry. Accordingly, such vectors preferentially transduce cells displaying the targeted receptor on their surface. While some selectivity for organs of interest can be achieved by library-based empirical permutation of the chemical composition of nanoparticles36, 37, 38, 39 or residues in the AAV capsid,40,41 the incorporation of high-affinity binders, such as scFvs,42 or designed ankyrin repeat proteins43 (DARPins) into the particle structure in the course of receptor targeting has resulted in near-absolute cell type selectivity (>95%), especially for lentiviral vectors (LVs).
Lentiviral vectors
LVs (Figure 2A) are usually pseudotyped with the glycoprotein G of vesicular stomatitis virus (VSV-G), resulting in particles 120–150 nm in diameter. VSV-G mediates cell entry via the low-density lipoprotein receptor (LDLR) and its family members, which are expressed on many cell types.44 Accordingly, VSV-G pseudotyped LVs exhibit a broad tropism, achieving high transduction efficiencies on different human cell types, including activated T lymphocytes. CAR therapy mostly relies on lentiviral or γ-retroviral vectors for ex vivo transduction. In 2018, 54% of clinical studies in the US used LVs for CAR T cell generation.45 Beyond that, over 100 registered clinical trials are ongoing, mostly using LVs in the context of immunological and hematological disorders and cancer.46,47
Figure 2.

Vector platforms used for in vivo CAR delivery
The vector particles’ main features (genetic information shown in red) and their cell entry modes, including nuclear entry and potential genomic integration, are depicted from top to bottom, respectively. (A) LVs are enveloped particles containing one or more viral glycoproteins (blue) and two copies of a ssRNA genome packaged in a nucleocapsid. Depending on the glycoprotein, cell entry occurs directly at the cell membrane or is dependent on endocytosis. The transferred gene is reverse transcribed, shuttled into the nucleus, and integrated into the host genome. (B) AAVs consist of a ssDNA genome packaged into an icosahedral protein capsid. Cellular uptake by endocytosis is followed by release of the transferred gene into the nucleus, where it resides episomally, separate from host chromatin. (C) In synthetic vectors, CAR encoding nucleic acids are complexed with positively charged, biodegradable polymers (NCs) or lipids (LNPs). After endosomal escape, mRNA payloads (1) are available for translation. Packaged DNA (2) may reach the nucleus, where it can integrate into host chromatin when transposase is co-delivered.
Engineering of LV-incorporated envelope proteins for receptor targeting is a two-step approach consisting of (1) destroying natural receptor usage and (2) adding the binder for target recognition. Attempts to alter receptor usage of the VSV-G protein are ongoing48 but challenging, since it mediates both receptor binding and membrane fusion. While the recent identification of the contact residues for LDLR has already facilitated the use of receptor-targeted VSV-LVs in the study of cellular interactions,49,50 targeting approaches for LVs relying on glycoproteins from alpha- and paramyxoviruses, which have separate envelope proteins for binding and fusion, are more advanced (Figure 3A).52 Initially developed for the measles virus (MV) glycoproteins,53 the approach was later extended to the envelope proteins of Nipah virus (NiV). When membrane-proximal receptor domains are targeted, NiV-LVs exhibit higher vector titers than MV-LVs and are resistant to human serum antibodies.54 For both approaches, DARPins may be used instead of scFvs,43,55,56 which can be conveniently selected in vitro from large combinatorial libraries by ribosome display.57 Our laboratory has done pioneering work in LV targeting, describing LVs specific for more than a dozen different cell surface proteins. In addition to surface markers of tumor cells, neuronal subtypes, and endothelial cells,58 these include markers of immune and hematopoietic cells with high relevance for in vivo CAR gene delivery.56 The first T lymphocyte marker successfully targeted with engineered paramyxoviral proteins was CD8.42 Interestingly, that study revealed that receptor targeting with LVs not only mediates high target cell selectivity but can also transfer activating stimuli to target cells. When scFv from agonistic CD3 antibodies are used, this results in activation of resting T lymphocytes, making the use of activating antibodies in cell culture obsolete and enabling gene transfer directly in unprocessed blood.22 Another important aspect, illustrated by targeting human CD4, is target receptor downmodulation, which can be detected over a few days after addition of vector particles but is always transient with full reconstitution of target receptor expression.59 Whether this transient receptor downmodulation has functional consequences for in vivo CAR therapy remains to be investigated.
Figure 3.

Concepts for receptor targeting
(A) LVs are pseudotyped with paramyxoviral (left) or alphavirus (right) glycoproteins. The receptor-binding proteins measles virus H, and Sindbis virus E1 are shown in complex with the membrane fusion proteins F and E2, respectively. Ablation of natural receptor binding by point mutations is indicated by red crosses. Target receptor binding is mediated through DARPins C-terminally fused to H protein by flexible linkers (left) or a tandem Fab serving as adapter between E2 protein and the target receptor (right). (B) An AAV particle displaying DARPins (red) at its 3-fold symmetry axis is shown. Zooming in to a single viral protein (VP), the DARPin is shown inserted into the GH2-GH3 loop. Residues in the GH12-GH13 loop mutated to ablate binding to the attachment receptor heparan-sulfate proteoglycan are labeled by the triangle. The ribbon structure was generated using ColabFold.51 Linker length was adapted manually to improve clarity. (C) Receptor targeting of NCs (left) and LNPs (right). Antibodies and antibody fragments used for targeting are shown in blue, the CAR encoding nucleic acid in red. Electrostatic coupling to the positively charged PBAE in NCs is enabled by conjugating antibodies to polyglutamic acid. In LNPs, antibodies functionalized with sulfhydryl groups are conjugated to the particles via thioether bonds.
LVs targeted through the alphavirus-derived entry machinery, specifically the envelope proteins of Sindbis virus (SINV), differ in the engineering approach as in their cell entry mode from those based on paramyxovirus glycoproteins. In one approach, the SINV E1 glycoprotein is co-displayed on the LV surface together with a selectivity-determining molecule such as a complete CD3-specific antibody.60 In another, more frequently used strategy, the natural tropism of the SINV E2 receptor-binding protein is abrogated by mutation, and particles are redirected to a receptor of choice by means of a molecular adapter that interacts with E2 and the cellular target receptor simultaneously. One such configuration uses E2 with the Fc-binding ZZ domain from protein A to which an antibody of choice, e.g., directed against CD4, can be bound.61 Building on the adapter concept, but improving its stability, especially for in vivo applications, other configurations used SpyTag-SpyCatcher62 or disulfide-bond-forming protein-peptide pairs,63 biotinylated antibodies,64 or tandem Fab23 as adapters (Figure 3A).
One crucial difference between paramyxo- and alphaviral pseudotypes is their pH-dependency. SINV E1 depends on low pH in the endosome for membrane fusion and cell entry.65 In line with this, transduction by a SINV-based LV targeted to CD4 was inhibited by dynamin mutation and acidification inhibitors.61 In sharp contrast, paramyxo-based Her2/neu- or EPCAM-targeted MV- and NiV-LVs fuse with the cell membrane independently of pH, as demonstrated by a strong increase in transduction when endosomal acidification was blocked.54,66 They can therefore also transduce cells via receptors that are not frequently endocytosed. Possibly linked to this, the target cell selectivity of paramyxo-pseudotyped LVs is close to absolute. While many studies found high selectivity on the reporter protein level, a recent study applied single-cell transcriptomics to investigate the target cell selectivity of LVs targeted to human CD8 (CD8-LV) in human peripheral blood mononuclear cells (PBMCs).67 Careful investigation revealed an on-target rate of more than 99%. If the few cells categorized as off-target were in fact target receptor negative at the moment of vector particle entry remains to be investigated.
Adeno-associated vectors
AAVs are in many ways polar opposites to LVs. Consisting of non-enveloped protein capsids containing a single-stranded DNA genome, genetic modification by AAVs is usually transient, especially in proliferating cells such as activated lymphocytes.55 After interaction with their entry receptor, clathrin-mediated endocytosis and intracellular trafficking—the specifics of these processes being determined by the serotype of the capsid used—vectors enter the nucleus and release a single-stranded transgene, which, after synthesis of the second strand, is available for transcription (Figure 2B).68
The lack of a membranous envelope requires that binders have to be incorporated into the rigid capsid for receptor targeting. The different capsid proteins, VP1 through VP3, share the VP3 common region and differ in their flexible N-terminal tails. AAV capsids produced from plasmids assemble stochastically following a random draw principle, with virtually every capsid in a given preparation having a unique stochio-spatial configuration of VPs.69 Similarly, binding and cellular uptake of AAVs is a complex and serotype-dependent process. AAVs often use glycans as attachment factors, such as heparan sulfate proteoglycan (HSPG) in the case of AAV2. Additionally, dozens of cellular proteins are implicated in AAV uptake, among them the transmembrane AAV receptor (AAVR), which is crucial for most serotypes.70,71
Capsid engineering of AAVs is usually based on selecting libraries of vector particles generated by shuffling serotypes or inserting combinatorically diversified peptides.72,73 Although this concept has resulted in many novel capsids with preferential biodistribution to the target tissue, the resulting vector particles usually lack complete cell-type selectivity. Accordingly, AAVs similarly specific for T lymphocytes as the LVs described above have not been obtained with this approach. Proof of principle for the rational approach as established for LVs was provided by displaying a DARPin directed against the tumor marker Her2/neu on the N terminus of VP2. To abrogate binding of the AAV2 capsid to HSPG, two relevant arginine residues were mutated (Figure 3B). Notably, targeting reduced substantially the accumulation of intravenously administered vector to the liver.74 Likewise, AAVs were generated for the specific transduction of GluA4-, CD105-, EpCAM-, and CD4-positive cells; all were used successfully in mouse models and/or human blood.75, 76, 77 In extensions of the N-terminal-targeting approach, covalent coupling of DARPins and also scFvs as proteins to the capsid of a universal AAV has been achieved through protein splicing.78 A light-inducible transduction system was generated by coupling phytochrome-interacting factor 6 to VP2. Interestingly, transduction efficiency improved with incubating vector stocks at 65°C.79 This may indicate that not all of the N-terminally fused binders were located outside of the capsid and that heat-induced conformational changes in the capsids exposed these previously inaccessible binders at the particle surface.
A surface-exposed insertion site in the GH2-GH3 loop of the VP3 common region was identified by screening a library of mCherry insertions for capsid assembly and gene delivery.80 Utilizing this insertion site for nanobodies, Eichhoff et al. targeted vectors to CD38, ARTC2.2, and P2X7.81 AAVs targeted to murine CD8 were generated by inserting a DARPin into the VP1 GH2-GH3 loop. On primary murine splenocytes, the resulting mCD8-AAV achieved 26-fold higher transduction efficiency than the parental AAV2, with >99% of GFP-positive cells being CD8+. The unexpectedly high gene delivery activity makes this new class of vector, termed DART-AAV (DARPin-targeted AAV), highly relevant for in vivo CAR T cell generation (Figure 3B).55
Non-viral vectors
Through the international rollout of mRNA-based SARS-CoV-2 vaccines, non-viral vectors—especially LNPs, for which dozens of clinical trials are ongoing28—have recently gained worldwide attention, and research into their use for gene therapy is booming. Non-viral vector particles are less highly ordered than virus-derived particles; cell entry, trafficking, and transgene delivery are mediated by the (physico)chemical properties of the particle and payload instead of a sophisticated viral machinery.82 Besides LNPs, nanocarriers (NCs) have been used for in vivo CAR delivery. NCs are complexes of negatively charged nucleic acid and positively charged poly(β-amino ester) (PBAE) polymer. They are taken up endocytotically. After uptake, the nucleic acid escapes the endosome, entering the cell. NCs with plasmid DNA25 or in vitro-transcribed (IVT) mRNA26,83 have been described. Plasmid DNA can be directed toward the nucleus by nuclear localization-mictrotubule-associated signal peptides (NLS-MTAS), which are covalently linked to the PBAE carrier material. When an expression plasmid for a transposase such as PiggyBac is co-delivered along with the transgene plasmid, integration of the latter into the host cell chromatin can occur. In LNPs, mRNA is encapsulated at the core of a lipid particle through electrostatic interaction.84 After uptake of the particles by the host cell and endosomal escape, mRNA is available for translation until it is degraded (Figure 2C). To address the issues of limited biostability and immunogenicity of RNA, modifications may be made: These include the addition of a 5′ cap analog and 3′ poly(A) tail, the introduction of stabilizing sequences in 5′ and 3′ untranslated regions (UTRs), the codon optimization of the transcript, and the use of less-immunogenic-modified nucleotides, such as N1-methylpseudouridine.85, 86, 87
Illustrating the fundamental difference between viral and non-viral vectors, decoration of the latter with protein binders, such as antibodies or Fabs, for receptor targeting is an exercise in organic chemistry. Naturally, the specifics of conjugation depend on the composition of the nanoparticle in question, and detailed reviews of the available strategies have been provided.88 Recent successes in the targeting of mRNA-LNPs have relied on N-succinimidyl S-acetylthioacetate (SATA)-maleimide chemistry. Here, micelles containing a polyethylene glycol-lipid derivative functionalized with maleimide (DSPE-PEG-mal) are mixed with already-formed mRNA-LNPs to insert DSPE-PEG-mal into the LNPs.89 The antibodies are then functionalized by introduction of protected sulfhydryl groups on primary amines using SATA. After deprotection, antibodies are conjugated to LNPs by formation of a thioether bond between sulfhydryl and maleimide groups (Figure 3C). By use of this approach, mRNA was delivered specifically to PECAM-1-,90 CD4-91 and CD5-positive cells.27
For NCs, electrostatic interactions are crucial for coupling of the targeting ligand. For this purpose, polyglutamic acid (PGA) is activated with ethyl-N′-(3-dimethylaminopropyl)carbodiimide before conjugation to antibodies or Fabs (Figure 3C). The negatively charged PGA antibody conjugates then binds the positively charged complexes of DNA and PBAE through electrostatic interaction.25,26 It should be noted that when chemical conjugation methods such as the ones described above are used, not all binders may be in the correct orientation on the particle surface.
Generating CAR T cells in vivo—State of the art and perspectives
After the proof-of-concept studies with CD3-targeted NCs for murine T cells25 and CD8-LV for human T cells,19 seven additional studies explored this strategy in preclinical mouse studies, making use of all vector platforms described above. The heterogeneity of reported model systems, application routes, vector doses, cellular targets, time points, and readouts impedes direct quantitative comparisons between platforms (Table 1). Still, the reports shed light on important qualitative differences between the vector platforms that may critically influence each platform’s developmental trajectory.
Targeting and biodistribution
With the exception of the single report using AAV vectors,24 all publications on in vivo CAR T cell generation have relied on targeted vectors. Overall, targeting of markers on T cell subsets versus pan T cell markers can be distinguished. Among the latter, CD3 as a pan T cell marker is an obvious choice. Although CD5, in contrast to CD3, is not required for T cell effector function, it is also expressed on some B cells. The reduction of potential risks associated with target-receptor downmodulation due to vector particle attachment therefore likely comes with CAR gene delivery to non-T cells.27,92 Further studies will have to investigate this.
CD3 has been targeted by nonviral as well as LV vectors. NCs targeted to the ε-chain of murine CD3 assessed for biodistribution shortly after injection into C57BL/6 mice were mainly associated with T cells (75% of particles), whereas 25% had attached to CD3-negative cells in peripheral blood, mostly B cells, neutrophils, and monocytes. Furthermore, targeting decreased particle burden in the liver by roughly a factor of 3 and improved particle localization into spleen, lymph nodes, and bone marrow.25 The biodistribution of gene expression was well in agreement with particle distribution.26,27 MCD3-NCs exhibited similar reporter activities to NCs decorated with an isotype control in liver, but 3-8-fold higher activities in lymphoid tissues. Although most reporter activity was found in the T cells of these tissues, there was also substantial activity in non-T cells, such as B cells, dendritic cells, and macrophages. These off-target activities being in the range of 20%27 or 30%26 of on-target gene transfer, respectively, demand further investigations, especially with respect to tumor cell transduction.
Vector targeting against human CD3 has also been described for both types (SINV and NiV) of LVs. To not only achieve selectivity for T cells but also facilitate their genetic engineering, LVs pseudotyped with the NiV glycoproteins were targeted by scFvs derived from agonistic CD3 antibodies.22 These CD3-LVs induce T cell activation and proliferation, allowing efficient transduction of primary T lymphocytes in the absence of cytokines or stimulatory CD3/CD28 antibodies. Delivery of the CD19 CAR into NSG mice humanized with CD34+ hematopoietic stem cells (huCD34+ NSG) resulted in functional CAR T cells in the absence of any conditioning with CAR signals restricted to CD3+CD45+ cells.22 Building on this work, Huckaby et al. infused SINV-pseudotyped LVs targeted by a bispecific tandem Fab containing a CD3-binding moiety (CD3-SINV-LV) into NSG mice humanized with PBMCs. At final analysis, approximately 70%–90% of CAR-positive cells were CD3 positive.23
When biodistribution of vector particles targeted against human pan T cell markers is monitored, most humanized mouse models provide only on-target cells among the human cells. Targeting T cell subsets, in contrast, provides the opportunity to monitor target cell selectivity among closely related human on-target and off-target cells when, e.g., addressing CD4 or CD8 as target receptors, since CD4+ and CD8+ T lymphocytes share the vast majority of cell surface proteins. Subset-specific in vivo CAR cell generation has been realized using NiV-pseudotyped CD8-LV. Vectors were administered i.v. into huCD34+ NSG mice resulted in detectable CAR signals only in CD8+ cells in blood, spleen and bone marrow.19 Similarly specific, albeit transient, generation of CD8+ CAR T cells was achieved in T-cell-humanized NSG mice by using mRNA-delivering hCD8-NCs.26 Notably, in a follow-up study with CD8-LV, NK and NKT cells, both positive for the targeted CD8 α-chain, were found to express the CAR besides CD8 T cells.20 The on-target transduction of NK cells by CD8-LVs likely contributed to the observed tumor clearance, as CAR NK cells can be potent antitumoral effectors.93,94
Surprisingly active CAR T cells were generated by a MV-pseudotyped LV targeted to human CD4 by display of a DARPin (CD4-LV). Administration of CD4-LV not only resulted in near-exclusive expression of the CAR on CD4+ cells, but these CAR T cells developed much more rapidly than those generated with CD8-LV and, moreover, completely eliminated their targeted tumor cells. While cytotoxic activities of CD4+ T cells have been described before,95 it is meanwhile well accepted in the CAR T cell field that a 1:1 composition of CD4+ and CD8+ CAR T cells is beneficial for tumor cell clearance.96,97 Further momentum for the relevance of CD4+ CAR T cells originates from the recent observation that CAR T cells persisting in patients for 10 years are mainly CD4+.98 Also, by the in vivo approach, CAR T cells can be generated in a subset-specific manner by combining vectors targeted to CD4 and CD8 in the desired ratios as recently demonstrated for LVs.21
Dosing and expression kinetics
The expression and effector kinetics observed in preclinical in vivo CAR cell generation and the doses administered to achieve them reflect the vector platforms’ different modes of action, resulting in either permanent or transient gene transfer. Although side-by-side comparisons are still missing, the number of vector particles required per animal appears to be higher for nanoparticles and AAVs than for LVs (Table 1). This may be due to the permanent gene transfer mediated by LVs as well as their high target cell specificity. However, other parameters such as the kinetics and efficiency of cell entry and intracellular delivery may play a role as well. Independently of that, ex vivo and in vivo CAR T cell therapies differ substantially in their pharmacokinetic parameters. In the conventional approach, high numbers of CAR T cells are instantly available after administration. Upon vector particle injection, in contrast, orders of magnitude fewer CAR T cells are generated that then have to expand over time in vivo.
LVs and transposase-enabled NCs stably integrate their transgene into host chromatin. Accordingly, single injections of vector were sufficient to induce a persistent presence of CAR T cells. On the low end of the dosing spectrum, doses ranging from 4 × 1010 to 2.5 × 1011 particles per mouse were described for paramyxo-pseudotyped LVs and 5 × 1010 vector genomes for SINV-LV.20, 21, 22, 23 Kinetics depended on the activation status of human immune components: In models reconstituted with activated human PBMCs, CAR T cells and/or target-cell killing became apparent within 1–2 weeks after vector injection and thus earlier than in huCD34-NSG mice. Additionally, peak CAR T levels were higher in PBMC-NSG than in huCD34-NSG mice, reaching up to 40% of the targeted T cell subtype.19, 20, 21, 22 Notably, CAR T cells were detected in some CD34+ huNSG mice over the complete monitoring period of up to 18 weeks after vector administration.19 Permanent CAR expression was also reported using NCs transferring plasmids for CAR and a hyperactive PiggyBac transposase, albeit at particle doses substantially higher than those reported for LVs. A total dose of 1.5 × 1012 particles infused in five daily doses resulted in up to 20% of T cells being CAR positive 12 days after infusion, resulting in elimination of leukemia cells in 7 of 10 treated mice. Although this appeared promising, this strategy was later abandoned due to low transfer efficiencies and concerns about transposase toxicities, instead focusing on transient delivery by mRNA-NCs.26
In transient gene transfer—i.e., when non-integrating vectors are used—transgene expression is eventually lost. This is due to transgene degradation and inactivation as well as its dilution upon cell proliferation. Then, re-dosing may be required to maintain sufficient CAR T cell levels despite antigen-induced proliferation. Indeed, multiple high doses of mRNA-transferring PBAE-NCs were necessary for sustained antitumoral CAR activity. To treat hematologic and solid tumors in T-cell-humanized NSG mice, six doses of 50 μg of mRNA were administered within 6 weeks, for a total of 300 μg of mRNA. In a syngeneic mouse model of acute lymphatic leukemia, 12 doses of 15 μg were administered over 4 weeks, totaling 180 μg of mRNA. A human equivalent dose corresponds to 2.08 μg/mL blood, or about 10 μgof mRNA.26 By comparison, complete vaccination of human adults against SARS-CoV-2 using RNA-based vaccines is achieved with 90–300 μg of mRNA in LNPs.99,100 Notably, both solid and liquid tumors relapsed some weeks after treatment was discontinued, suggesting that sustained CAR activity will be necessary for lasting tumor suppression. The fast kinetics of mRNA transfer likely contributed to these relapses, since CAR levels peaked after 2 days and were almost completely lost 7 days after nanoparticle infusion into NSG mice. Lower doses appear to be sufficient when long-term CAR cell activity is not desired: A single injection of 10 μg of FAP-CAR-mRNA in LNPs resulted in a decreased burden of fibrotic tissue and marked improvements in cardiac function in a syngeneic model of fibrosis, as determined 3 weeks after nanoparticle infusion. The short-term CAR T cell activity in this setting was desired to prevent reactivity of the CAR T cells against healthy fibroblasts.27
Transferring not mRNA but DNA, single doses of 1–2 × 1011 vector genomes of untargeted AAV generated CAR T cells in humanized NCG mouse models of T cell leukemia. Surprisingly, CAR T cells were still detectable 35 days after AAV infusion.24 This unexpected longevity of transgene signal may have been a consequence of the high transduction efficiencies achieved by intraperitoneal injection and/or spontaneous integration of the CAR-coding sequence into the T cell genome. In vitro, transgene signals from mCD8-targeted DART-AAVs were almost completely lost in activated mouse lymphocytes within a week of vector addition.55
Safety and control
For vectors enabling permanent transfer of CAR genes, one imminent safety concern is genotoxicity originating from transcriptional dysregulation through the misplaced insertion of the transferred gene into host chromatin. Children treated for X-linked severe combined immunodeficiency with autologous hematopoietic stem cells (HSCs) transduced ex vivo with first-generation, non-self-inactivating gamma-retroviral vectors developed leukemia after the vector genome’s long terminal repeat (LTR)-intrinsic enhancer caused upregulation of transcription factors like LMO2,3 which led to uncontrolled clonal expansion. Research in the wake of this incident led to a better understanding of retrovirus-mediated insertional oncogenesis and use of self-inactivating retro- and lentiviral vectors. Of note, T lymphocytes are less likely to undergo retrovirus-mediated oncogene transformation than HSCs.101,102 Overall, LVs have been used in numerous clinical applications involving T cell transductions without incidents of insertional oncogenesis.103,104 Yet, a few examples of benign clonal expansion exist.105,106 In contrast to LVs, transposon-mediated gene transfer had its “SCID moment” last year, when two patients receiving CAR T cells generated ex vivo using piggyBac transposase developed T cell lymphoma.107,108 Although the mechanism of oncogenesis is still under investigation, this incident must lead to development of safer transposition protocols in the future. In light of these events, close monitoring in long-term studies is advisable for integrating platforms. Focusing on risk factors such as the vector copy number in CAR T cells will ensure that the benefit of CAR delivery outweighs genotoxic risks.
Besides the risk of genotoxicity, permanent transfer of the CAR gene may necessitate tools for better pharmacokinetic control over CAR activity, especially when high-grade cytokine release syndrome (CRS) and/or immune effector-cell-associated neurotoxicity syndrome (ICANS) occurs.109 Both syndromes often coincide and are characterized by high levels of proinflammatory cytokines.110 Interestingly, CRS-like symptoms were also observed when CAR T cells were generated in CD34+ huNSG mice using CD8-LVs.19 This correlated with infiltrates of CD8+ cells in lung, brain, liver, and splenic B cell zones as well as elevated cytokines such as IL-6, IFNγ, GM-CSF, and TNFα. In the clinic, patients with CRS and/or ICANS are treated with tocilizumab, a monoclonal anti-IL6R antibody, to attenuate systemic inflammation without compromising CAR effector function. Even more granular control can be achieved using the tyrosine kinase inhibitor dasatinib, which has been used preclinically to intermittently switch off CAR T cells in mouse models of CRS, decreasing the inflammatory burden and improving survival.111 Another approach, which has already been evaluated in humans, utilizes inert UniCARs, which are “armed” by infusion of soluble antigen-specific targeting modules (TMs). Control is achieved by choosing TMs with longer or shorter half-life, as CAR T cell activity stops once all TMs are degraded.112,113 Complementing these strategies for short-term control, CAR cassettes that include an inducible caspase-9 (IC9) suicide gene offer true long-term control, potentially allowing physicians to discontinue CAR cell treatment after the tumor is cleared. Through administration of a small molecule, dimerization of IC9 proteins is induced, leading to apoptosis of CAR T cells.114,115 These mechanisms might make CAR cells generated with permanently transferring vectors more attractive for treatment of less severely diseased patients. Importantly, all these strategies can be combined with in vivo CAR T cell generation, potentially facilitating the translation of this approach into the clinic.
When transiently transferring vectors are used, runaway CAR cell activity is not a concern. Conversely, it is unclear at this time whether CAR activity can be maintained long enough to achieve lasting tumor clearance. Currently available safety data from short-term experiments suggest no severe acute toxicities;24, 25, 26 however, increased cytokine levels in response to in vivo administration of both synthetic26 and viral vectors,24 as well as NC-induced activation of complement components and increased mitochondrial oxidative stress in vitro,26 indicate that inflammatory reactions might interfere with repeated vector administration. Long-term experiments in syngeneic models would enable assessment of the risk of inflammatory reactions and other host immune responses upon (repeated) administration of vector.
Host immunity
Immune reactions to the administered vector influence not only the safety profile but also efficacy. Antibody-mediated immune responses against vector particles can interfere with both initial and repeated vector administration, reducing the effective on-target particle dose (Figure 4A) and inducing inflammation. This is of particular concern for LVs and AAVs, which, because of their viral origin, are markedly immunogenic. In fact, single systemic injections into rodents induced the formation of neutralizing antibodies (NAbs) against virion and envelope proteins of VSV-LV.116,117 Additionally, pre-existing antibodies from measles vaccination complicate the use of MV-pseudotyped LVs, a problem that has been addressed by engineering NiV-pseudotyped vectors. Although there is no vaccine against AAV, a considerable percentage of individuals harbor neutralizing antibodies against AAVs.118 Although the immunogenicity profile of LNPs carrying modified mRNA seems to be moderate,28 the introduction of protein components for receptor targeting likely increases the risk of antibody formation following repeated administrations. An alternative to the removal of serum IgG by plasmapheresis—which is a time-consuming process requiring complex instrumentation—is the use of streptococcal IgG-degrading enzymes such as IdeS (Figure 4B). Systemic treatment with IdeS enabled not only liver transduction in macaques with pre-existing anti-AAV8 NAbs but also effective multiple administrations.119
Figure 4.

Mechanisms detracting from the infused vector dose and potential solutions
(A) Several mechanisms can detract from the administered vector dose, reducing the effective on-target dose (shown as darker pink fraction). Among them are the antibody-mediated incapacitation of particles, their uptake by phagocytes and off-target transduction. (B) Antibody-mediated immune responses against vector particles can be reduced by cleaving serum IgG before vector administration using IgG-degrading enzyme from Streptococcus pyogenes (IdeS). (C) LVs can be shielded against phagocytosis by incorporating CD47 into the LV envelope during production (left). CD47 engages signal-regulatory protein alpha (SIRPα) when LV particles (red dots) encounter myeloid cells. Through interaction of CD47 and SIRPα, phagocytosis is inhibited.
Another major contributor to particle clearance is phagocytosis. In mice treated with CD3-targeted NCs, approximately 20% of both macrophages and monocytes were positive for NCs, indicating phagocytic activity against the vectors.25 Of note, phagocytosis seemed unaffected by targeting, as it affected similar particle fractions of CD3-NCs and isotype-NCs.26 In liver transduction experiments with LVs and adenoviral vectors,120,121 non-linear dose responses were observed, as, up to a threshold dose, most administered particles were sequestered by liver-resident Kupffer macrophages. Thanks to their enveloped architecture, however, phagocytosis of LVs can be reduced by incorporation of the phagocytosis inhibitor CD47 into vector particles. Uptake of such particles by macrophages in cell culture was reduced, and phagocytosis-shielded vectors achieved higher hFIX transfer efficiencies in non-human primates than unshielded LVs (Figure 4C).121 Similar effects were observed when CD4- and CD8-targeted LVs were produced in these packaging cells. While the presence of macrophages reduced mean transduction efficiencies of regular CD4- and CD8-LVs on human T cells by approximately 25%–50%, phagocytosis shielding of the vectors improved gene transfer into T cells substantially. Furthermore, in CD34+ HSC humanized NSG-SGM3 mice, which reconstitute physiological levels of human myeloid cells, more efficient in vivo CAR T cell generation and more pronounced target cell clearance were achieved upon shielding.122
Moving on from proof of principle
The reports of in vivo CAR T cell generation reviewed here are an excellent showcase for the state of the art of targeted-vector technology (Table 2). Through the use of cutting-edge vector platforms, CAR T cells can be generated in vivo and clear tumor cells. Although this is a milestone for both CAR therapy and gene therapy in general, important questions, referring to the short-, medium-, and long-term safety and efficacy of both permanent and transient transfer approaches, remain unanswered. Recent and earlier fatalities in gene therapy urge us to thoroughly examine the vectors’ effects on the host in long-term preclinical studies before advancing to first-in-human trials. In addition to ensuring patient safety, rigorous characterization of each vector’s interaction with the host will help us understand how pre-existing and acquired host immunity interferes with vector administration, thereby contributing to both toxicity and particle clearance, and how the host immune system is best modulated to avoid this interference. This is critical, as broad immunosuppression or even lymphodepletion are not an option when targeting lymphocytes. Instead, conditioning regimens must be identified that minimize inflammation related to vector administration and immune-mediated loss of particle dose while retaining lymphocyte viability and activity. For this, fully immunocompetent syngeneic models are instrumental. Mouse T-cell-targeted vectors required for these experiments are available for all four vector platforms with LVs and AAVs targeted to mouse CD8 via DARPins having been described recently.55
Table 2.
Key characteristics of vector platforms for in vivo CAR T cell therapy
| LV | AAV | mRNA-NC | mRNA-LNP | |
|---|---|---|---|---|
| T cell targeting demonstrated | + | + | + | + |
| Synthetic product | – | – | + | + |
| Permanent gene transfer | + | – | – | – |
| Clinical experience | + | + | – | + |
| Immunogenic | + | + | TBD | TBD |
TBD, to be determined.
An additional concern is manufacturing, as the availability of sufficient quantities of high-quality vector stock is essential for further development. Importantly, production processes should be designed with the demands of “good manufacturing practice” (GMP) in mind. Development of GMP-compliant processes for the production of targeted LVs, AAVs, and LNPs will likely benefit from experiences made in the manufacturing of untargeted GMP-grade vectors for approved medicines4,31,123. Although the worldwide rollout of SARS-CoV-2 vaccines99,100 has demonstrated the considerable scalability of synthetic particle production, it remains to be determined whether the incorporation of cell-produced target receptor binders can be integrated into production processes without compromising yield.
Incidentally, progress made toward addressing the problems of host immunity and manufacturing (and other, as yet unidentified problems) in the context of in vivo CAR therapy will likely benefit the entire field of gene therapy, helping to make in vivo therapies for hematologic, cardiac, infectious, and developmental diseases available to many patients in need of therapeutic options. Enabled by targeted vectors and continued exacting work and patience, the gene therapy field may soon enter the “Age of In Vivo.”
Acknowledgments
This work was supported by grants of the LOEWE Center Frankfurt Cancer Institute funded by the Hessen State Ministry for Higher Education, Research and the Arts (III L 5-519/03/03.001-(0015)) and the Deutsche Krebshilfe (70114099) to C.J.B. C.J.B. is listed as inventor on patents covering T-cell-targeted lentiviral vectors.
Author contributions
Literature research: A.M., N.H., and C.J.B.; Writing – original draft: A.M., N.H., and C.J.B.; Writing – revision: A.M., N.H., and C.J.B.; Visualization: A.M.; All authors read and approved the final manuscript.
Declaration of interests
The authors declare no conflict of interest.
References
- 1.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359 doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 2.Somia N., Verma I.M. Gene therapy: trials and tribulations. Nat. Rev. Genet. 2000;1:91–99. doi: 10.1038/35038533. [DOI] [PubMed] [Google Scholar]
- 3.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E., et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 4.European Medicines Agency Kymriah: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/kymriah-epar-product-information_en.pdf
- 5.European Medicines Agency Tecartus: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/tecartus-epar-product-information_en.pdf
- 6.European Medicines Agency Yescarta: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/yescarta-epar-product-information_en.pdf
- 7.European Medicines Agency Abecma: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/abecma-epar-product-information_en.pdf
- 8.Eisenstein M. Seven technologies to watch in 2022. Nature. 2022;601:658–661. doi: 10.1038/d41586-022-00163-x. [DOI] [PubMed] [Google Scholar]
- 9.June C.H., O’Connor R.S., Kawalekar O.U., Ghassemi S., Milone M.C. CAR T cell immunotherapy for human cancer. Science. 2018;359:1361–1365. doi: 10.1126/science.aar6711. [DOI] [PubMed] [Google Scholar]
- 10.Sadelain M. CD19 CAR T cells. Cell. 2017;171:1471. doi: 10.1016/j.cell.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 11.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D., et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sterner R.C., Sterner R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gene therapies should be for all. Nat. Med. 2021;27:1311. doi: 10.1038/s41591-021-01481-9. [DOI] [PubMed] [Google Scholar]
- 14.Caruso H.G., Tanaka R., Liang J., Ling X., Sabbagh A., Henry V.K., Collier T.L., Heimberger A.B. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J. Neurooncol. 2019;145:429–439. doi: 10.1007/s11060-019-03311-y. [DOI] [PubMed] [Google Scholar]
- 15.Ruella M., Xu J., Barrett D.M., Fraietta J.A., Reich T.J., Ambrose D.E., Klichinsky M., Shestova O., Patel P.R., Kulikovskaya I., et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018;24:1499–1503. doi: 10.1038/s41591-018-0201-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lock D., Mockel-Tenbrinck N., Drechsel K., Barth C., Mauer D., Schaser T., Kolbe C., Al Rawashdeh W., Brauner J., Hardt O., et al. Automated manufacturing of potent CD20-directed chimeric antigen receptor T cells for clinical use. Hum. Gene Ther. 2017;28:914–925. doi: 10.1089/hum.2017.111. [DOI] [PubMed] [Google Scholar]
- 17.Qasim W. Allogeneic CAR T cell therapies for leukemia. Am. J. Hematol. 2019;94:S50–S54. doi: 10.1002/ajh.25399. [DOI] [PubMed] [Google Scholar]
- 18.Smirnov S., Petukhov A., Levchuk K., Kulemzin S., Staliarova A., Lepik K., Shuvalov O., Zaritskey A., Daks A., Fedorova O. Strategies to circumvent the side-effects of immunotherapy using allogeneic CAR-T cells and boost its efficacy: results of recent clinical trials. Front. Immunol. 2021;12:780145. doi: 10.3389/fimmu.2021.780145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeiffer A., Thalheimer F.B., Hartmann S., Frank A.M., Bender R.R., Danisch S., Costa C., Wels W.S., Modlich U., Stripecke R., et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018;10:e9158. doi: 10.15252/emmm.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agarwal S., Weidner T., Thalheimer F.B., Buchholz C.J. In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology. 2019;8 doi: 10.1080/2162402x.2019.1671761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agarwal S., Hanauer J.D., Frank A.M., Riechert V., Thalheimer F.B., Buchholz C.J. In vivo generation of CAR T cells selectively in human CD4+ lymphocytes. Mol. Ther. 2020;28:1783–1794. doi: 10.1016/j.ymthe.2020.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frank A.M., Braun A.H., Scheib L., Agarwal S., Schneider I.C., Fusil F., Perian S., Sahin U., Thalheimer F.B., Verhoeyen E., Buchholz C.J. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020;4:5702–5715. doi: 10.1182/bloodadvances.2020002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huckaby J.T., Landoni E., Jacobs T.M., Savoldo B., Dotti G., Lai S.K. Bispecific binder redirected lentiviral vector enables in vivo engineering of CAR-T cells. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nawaz W., Huang B., Xu S., Li Y., Zhu L., Yiqiao H., Wu Z., Wu X. AAV-mediated in vivo CAR gene therapy for targeting human T-cell leukemia. Blood Cancer J. 2021;11:119. doi: 10.1038/s41408-021-00508-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith T.T., Stephan S.B., Moffett H.F., McKnight L.E., Ji W., Reiman D., Bonagofski E., Wohlfahrt M.E., Pillai S.P.S., Stephan M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017;12:813–820. doi: 10.1038/nnano.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parayath N.N., Stephan S.B., Koehne A.L., Nelson P.S., Stephan M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020;11:6080. doi: 10.1038/s41467-020-19486-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rurik J.G., Tombácz I., Yadegari A., Méndez Fernández P.O., Shewale S.V., Li L., Kimura T., Soliman O.Y., Papp T.E., Tam Y.K., et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375:91–96. doi: 10.1126/science.abm0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hou X., Zaks T., Langer R., Dong Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021;6:1078–1094. doi: 10.1038/s41578-021-00358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sedic M., Senn J.J., Lynn A., Laska M., Smith M., Platz S.J., Bolen J., Hoge S., Bulychev A., Jacquinet E., et al. Safety evaluation of lipid nanoparticle-formulated modified mRNA in the sprague-dawley rat and cynomolgus monkey. Vet. Pathol. 2018;55:341–354. doi: 10.1177/0300985817738095. [DOI] [PubMed] [Google Scholar]
- 30.Kedmi R., Ben-Arie N., Peer D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials. 2010;31:6867–6875. doi: 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 31.European Medicines Agency Onpattro: EPAR - Product information. 2022. https://www.ema.europa.eu/en/documents/product-information/onpattro-epar-product-information_en.pdf
- 32.Food and Drug Administration Briefing Document Food and Drug Administration (FDA) Cellular, Tissue, and Gene Therapies Advisory Committee (CTGTAC) Meeting #70 Toxicity Risks of Adeno-Associated Virus (AAV) Vectors for Gene Therapy (GT) September 2-3, 2021. 2021. https://www.fda.gov/media/151599/download
- 33.Wilson J.M., Flotte T.R. Moving forward after two deaths in a gene therapy trial of myotubular myopathy. Hum. Gene Ther. 2020;31:695–696. doi: 10.1089/hum.2020.182. [DOI] [PubMed] [Google Scholar]
- 34.Thomsen G., Burghes A.H.M., Hsieh C., Do J., Chu B.T.T., Perry S., Barkho B., Kaufmann P., Sproule D.M., Feltner D.E., et al. Biodistribution of onasemnogene abeparvovec DNA, mRNA and SMN protein in human tissue. Nat. Med. 2021;27:1701–1711. doi: 10.1038/s41591-021-01483-7. [DOI] [PubMed] [Google Scholar]
- 35.Buchholz C.J., Friedel T., Büning H. Surface-engineered viral vectors for selective and cell type-specific gene delivery. Trends. Biotechnol. 2015;33:777–790. doi: 10.1016/j.tibtech.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 36.Dahlman J.E., Kauffman K.J., Xing Y., Shaw T.E., Mir F.F., Dlott C.C., Langer R., Anderson D.G., Wang E.T. Barcoded nanoparticles for high throughput in vivo discovery of targeted therapeutics. Proc. Natl. Acad. Sci. U S A. 2017;114:2060–2065. doi: 10.1073/pnas.1620874114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paunovska K., Da Silva Sanchez A.J., Sago C.D., Gan Z., Lokugamage M.P., Islam F.Z., Kalathoor S., Krupczak B.R., Dahlman J.E. Oxidized cholesterol: nanoparticles containing oxidized cholesterol deliver mRNA to the liver microenvironment at clinically relevant doses. Adv. Mater. 2019;31 doi: 10.1002/adma.201970098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Paunovska K., Gil C.J., Lokugamage M.P., Sago C.D., Sato M., Lando G.N., Gamboa Castro M., Bryksin A.V., Dahlman J.E. Analyzing 2000 in vivo drug delivery data points reveals cholesterol structure impacts nanoparticle delivery. ACS Nano. 2018;12:8341–8349. doi: 10.1021/acsnano.8b03640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu S., Cheng Q., Wei T., Yu X., Johnson L.T., Farbiak L., Siegwart D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 2021;20:701–710. doi: 10.1038/s41563-020-00886-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deverman B.E., Pravdo P.L., Simpson B.P., Kumar S.R., Chan K.Y., Banerjee A., Wu W.-L., Yang B., Huber N., Pasca S.P., Gradinaru V. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat. Biotechnol. 2016;34:204–209. doi: 10.1038/nbt.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Byrne L.C., Day T.P., Visel M., Strazzeri J.A., Fortuny C., Dalkara D., Merigan W.H., Schaffer D.V., Flannery J.G. In vivo-directed evolution of adeno-associated virus in the primate retina. JCI Insight. 2020;5 doi: 10.1172/jci.insight.135112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou Q., Schneider I.C., Edes I., Honegger A., Bach P., Schönfeld K., Schambach A., Wels W.S., Kneissl S., Uckert W., Buchholz C.J. T-cell receptor gene transfer exclusively to human CD8(+) cells enhances tumor cell killing. Blood. 2012;120:4334–4342. doi: 10.1182/blood-2012-02-412973. [DOI] [PubMed] [Google Scholar]
- 43.Frank A.M., Weidner T., Brynza J., Uckert W., Buchholz C.J., Hartmann J. CD8-Specific designed ankyrin repeat proteins improve selective gene delivery into human and primate T lymphocytes. Hum. Gene Ther. 2020;31:679–691. doi: 10.1089/hum.2019.248. [DOI] [PubMed] [Google Scholar]
- 44.Finkelshtein D., Werman A., Novick D., Barak S., Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. U S A. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vormittag P., Gunn R., Ghorashian S., Veraitch F.S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018;53:164–181. doi: 10.1016/j.copbio.2018.01.025. [DOI] [PubMed] [Google Scholar]
- 46.Kohn L.A., Kohn D.B. Gene therapies for primary immune deficiencies. Front. Immunol. 2021;12:648951. doi: 10.3389/fimmu.2021.648951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.clinicaltrials.gov. ClinicalTrials.gov, https://clinicaltrials.gov/, Last accessed: May 2022.
- 48.Anastasov N., Höfig I., Mall S., Krackhardt A.M., Thirion C. Optimized lentiviral transduction protocols by use of a poloxamer enhancer, spinoculation, and scFv-antibody fusions to VSV-G. Methods Mol. Biol. 2016;1448:49–61. doi: 10.1007/978-1-4939-3753-0_4. [DOI] [PubMed] [Google Scholar]
- 49.Nikolic J., Belot L., Raux H., Legrand P., Gaudin Y., A Albertini A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018;9:1029. doi: 10.1038/s41467-018-03432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu B., Shi Q., Belk J.A., Yost K.E., Parker K.R., Huang H., Lingwood D., Davis M.M., Satpathy A.T., Chang H.Y. Systematic discovery of receptor-ligand biology by engineered cell entry and single-cell genomics. 2021. https://www.biorxiv.org/content/10.1101/2021.12.13.472464v1
- 51.Mirdita M., Schuetze K., Moriwaki Y., Heo L., Ovchinnikov S., Steinegger M. ColabFold - making protein folding accessible to all. bioRxiv. 2021 doi: 10.1038/s41592-022-01488-1. https://www.biorxiv.org/content/10.1101/2021.08.15.456425v1 Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buchholz C.J., Mühlebach M.D., Cichutek K. Lentiviral vectors with measles virus glycoproteins - dream team for gene transfer? Trends. Biotechnol. 2009;27:259–265. doi: 10.1016/j.tibtech.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 53.Nakamura T., Peng K.-W., Harvey M., Greiner S., Lorimer I.A.J., James C.D., Russell S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005;23:209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 54.Bender R.R., Muth A., Schneider I.C., Friedel T., Hartmann J., Pluckthun A., Maisner A., Buchholz C.J. Receptor-targeted Nipah virus glycoproteins improve cell-type selective gene delivery and reveal a preference for membrane-proximal cell attachment. PLoS Pathog. 2016;12 doi: 10.1371/journal.ppat.1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michels A., Frank A.M., Günther D.M., Mataei M., Börner K., Grimm D., Hartmann J., Buchholz C.J. Lentiviral and adeno-associated vectors efficiently transduce mouse T lymphocytes when targeted to murine CD8. Mol. Ther. Methods Clin. Dev. 2021;23:334–347. doi: 10.1016/j.omtm.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frank A.M., Buchholz C.J. Surface-engineered lentiviral vectors for selective gene transfer into subtypes of lymphocytes. Mol. Ther. Methods Clin. Dev. 2019;12:19–31. doi: 10.1016/j.omtm.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015;55:489–511. doi: 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- 58.Anliker B., Abel T., Kneissl S., Hlavaty J., Caputi A., Brynza J., Schneider I.C., Münch R.C., Petznek H., Kontermann R.E., et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat. Methods. 2010;7:929–935. doi: 10.1038/nmeth.1514. [DOI] [PubMed] [Google Scholar]
- 59.Zhou Q., Uhlig K.M., Muth A., Kimpel J., Lévy C., Münch R.C., Seifried J., Pfeiffer A., Trkola A., Coulibaly C., et al. Exclusive transduction of human CD4+ T cells upon systemic delivery of CD4-targeted lentiviral vectors. J. Immunol. 2015;195:2493–2501. doi: 10.4049/jimmunol.1500956. [DOI] [PubMed] [Google Scholar]
- 60.Yang H., Joo K.-I., Ziegler L., Wang P. Cell type-specific targeting with surface-engineered lentiviral vectors co-displaying OKT3 antibody and fusogenic molecule. Pharm. Res. 2009;26:1432–1445. doi: 10.1007/s11095-009-9853-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liang M., Morizono K., Pariente N., Kamata M., Lee B., Chen I.S.Y. Targeted transduction via CD4 by a lentiviral vector uses a clathrin-mediated entry pathway. J. Virol. 2009;83:13026–13031. doi: 10.1128/jvi.01530-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kasaraneni N., Chamoun-Emanuelli A.M., Wright G., Chen Z. Retargeting lentiviruses via SpyCatcher-SpyTag chemistry for gene delivery into specific cell types. mBio. 2017;8 doi: 10.1128/mbio.01860-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kasaraneni N., Chamoun-Emanuelli A.M., Wright G.A., Chen Z. A simple strategy for retargeting lentiviral vectors to desired cell types via a disulfide-bond-forming protein-peptide pair. Sci. Rep. 2018;8:10990. doi: 10.1038/s41598-018-29253-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Situ K., Chua B.A., Bae S.Y., Meyer A.S., Morizono K. Versatile targeting system for lentiviral vectors involving biotinylated targeting molecules. Virology. 2018;525:170–181. doi: 10.1016/j.virol.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li L., Jose J., Xiang Y., Kuhn R.J., Rossmann M.G. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468:705–708. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rasbach A., Abel T., Münch R.C., Boller K., Schneider-Schaulies J., Buchholz C.J. The receptor attachment function of measles virus hemagglutinin can be replaced with an autonomous protein that binds Her2/neu while maintaining its fusion-helper function. J. Virol. 2013;87:6246–6256. doi: 10.1128/jvi.03298-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Charitidis F.T., Adabi E., Thalheimer F.B., Clarke C., Buchholz C.J. Monitoring CAR T cell generation with a CD8-targeted lentiviral vector by single-cell transcriptomics. Mol. Ther. Methods Clin. Dev. 2021;23:359–369. doi: 10.1016/j.omtm.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C., Samulski R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020;21:255–272. doi: 10.1038/s41576-019-0205-4. [DOI] [PubMed] [Google Scholar]
- 69.Wörner T.P., Bennett A., Habka S., Snijder J., Friese O., Powers T., Agbandje-McKenna M., Heck A.J.R. Adeno-associated virus capsid assembly is divergent and stochastic. Nat. Commun. 2021;12:1642. doi: 10.1038/s41467-021-21935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer N.L., Chapman M.S. Adeno-associated virus (AAV) cell entry: structural insights. Trends Microbiol. 2021;30:432–451. doi: 10.1016/j.tim.2021.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Riyad J.M., Weber T. Intracellular trafficking of adeno-associated virus (AAV) vectors: challenges and future directions. Gene Ther. 2021;28:683–696. doi: 10.1038/s41434-021-00243-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kotterman M.A., Schaffer D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014;15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Büning H., Srivastava A. Capsid modifications for targeting and improving the efficacy of AAV vectors. Mol. Ther. Methods Clin. Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Münch R.C., Janicki H., Völker I., Rasbach A., Hallek M., Büning H., Buchholz C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013;21:109–118. doi: 10.1038/mt.2012.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Münch R.C., Muth A., Muik A., Friedel T., Schmatz J., Dreier B., Trkola A., Plückthun A., Büning H., Buchholz C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015;6:6246. doi: 10.1038/ncomms7246. [DOI] [PubMed] [Google Scholar]
- 76.Hartmann J., Thalheimer F.B., Höpfner F., Kerzel T., Khodosevich K., García-González D., Monyer H., Diester I., Büning H., Carette J.E., et al. GluA4-Targeted AAV vectors deliver genes selectively to interneurons while relying on the AAV receptor for entry. Mol. Ther. Methods Clin. Dev. 2019;14:252–260. doi: 10.1016/j.omtm.2019.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hartmann J., Münch R.C., Freiling R.-T., Schneider I.C., Dreier B., Samukange W., Koch J., Seeger M.A., Plückthun A., Buchholz C.J. A library-based screening strategy for the identification of DARPins as ligands for receptor-targeted AAV and lentiviral vectors. Mol. Ther. Methods Clin. Dev. 2018;10:128–143. doi: 10.1016/j.omtm.2018.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Muik A., Reul J., Friedel T., Muth A., Hartmann K.P., Schneider I.C., Münch R.C., Buchholz C.J. Covalent coupling of high-affinity ligands to the surface of viral vector particles by protein trans-splicing mediates cell type-specific gene transfer. Biomaterials. 2017;144:84–94. doi: 10.1016/j.biomaterials.2017.07.032. [DOI] [PubMed] [Google Scholar]
- 79.Hörner M., Jerez-Longres C., Hudek A., Hook S., Yousefi O.S., Schamel W.W.A., Hörner C., Zurbriggen M.D., Ye H., Wagner H.J., Weber W. Spatiotemporally confined red light-controlled gene delivery at single-cell resolution using adeno-associated viral vectors. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abf0797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Judd J., Wei F., Nguyen P.Q., Tartaglia L.J., Agbandje-McKenna M., Silberg J.J., Suh J. Random insertion of mCherry into VP3 domain of adeno-associated virus yields fluorescent capsids with no loss of infectivity. Mol. Ther. Nucleic Acids. 2012;1:e54. doi: 10.1038/mtna.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eichhoff A.M., Börner K., Albrecht B., Schäfer W., Baum N., Haag F., Körbelin J., Trepel M., Braren I., Grimm D., et al. Nanobody-Enhanced targeting of AAV gene therapy vectors. Mol. Ther. Methods Clin. Dev. 2019;15:211–220. doi: 10.1016/j.omtm.2019.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheng Z., Li M., Dey R., Chen Y. Nanomaterials for cancer therapy: current progress and perspectives. J. Hematol. Oncol. 2021;14:85. doi: 10.1186/s13045-021-01096-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang F., Parayath N.N., Ene C.I., Stephan S.B., Koehne A.L., Coon M.E., Holland E.C., Stephan M.T. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat. Commun. 2019;10:3974. doi: 10.1038/s41467-019-11911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leung A.K.K., Tam Y.Y.C., Chen S., Hafez I.M., Cullis P.R. Microfluidic mixing: a general method for encapsulating macromolecules in lipid nanoparticle systems. J. Phys. Chem. B. 2015;119:8698–8706. doi: 10.1021/acs.jpcb.5b02891. [DOI] [PubMed] [Google Scholar]
- 85.Sahin U., Karikó K., Türeci Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014;13:759–780. doi: 10.1038/nrd4278. [DOI] [PubMed] [Google Scholar]
- 86.Andries O., Mc Cafferty S., De Smedt S.C., Weiss R., Sanders N.N., Kitada T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Release. 2015;217:337–344. doi: 10.1016/j.jconrel.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 87.Karikó K., Muramatsu H., Welsh F.A., Ludwig J., Kato H., Akira S., Weissman D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008;16:1833–1840. doi: 10.1038/mt.2008.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moulahoum H., Ghorbanizamani F., Zihnioglu F., Timur S. Surface biomodification of liposomes and polymersomes for efficient targeted drug delivery. Bioconjug. Chem. 2021;32:1491–1502. doi: 10.1021/acs.bioconjchem.1c00285. [DOI] [PubMed] [Google Scholar]
- 89.Ishida T., Iden D.L., Allen T.M. A combinatorial approach to producing sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett. 1999;460:129–133. doi: 10.1016/s0014-5793(99)01320-4. [DOI] [PubMed] [Google Scholar]
- 90.Parhiz H., Shuvaev V.V., Pardi N., Khoshnejad M., Kiseleva R.Y., Brenner J.S., Uhler T., Tuyishime S., Mui B.L., Tam Y.K., et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control Release. 2018;291:106–115. doi: 10.1016/j.jconrel.2018.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tombácz I., Laczkó D., Shahnawaz H., Muramatsu H., Natesan A., Yadegari A., Papp T.E., Alameh M.-G., Shuvaev V., Mui B.L., et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol. Ther. 2021;29:3293–3304. doi: 10.1016/j.ymthe.2021.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Soldevila G., Raman C., Lozano F. The immunomodulatory properties of the CD5 lymphocyte receptor in health and disease. Curr. Opin. Immunol. 2011;23:310–318. doi: 10.1016/j.coi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Burger M.C., Zhang C., Harter P.N., Romanski A., Strassheimer F., Senft C., Tonn T., Steinbach J.P., Wels W.S. CAR-engineered NK cells for the treatment of glioblastoma: turning innate effectors into precision tools for cancer immunotherapy. Front. Immunol. 2019;10:2683. doi: 10.3389/fimmu.2019.02683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takeuchi A., Saito T. CD4 CTL, a cytotoxic subset of CD4+ T cells, their differentiation and function. Front. Immunol. 2017;8:194. doi: 10.3389/fimmu.2017.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sommermeyer D., Hudecek M., Kosasih P.L., Gogishvili T., Maloney D.G., Turtle C.J., Riddell S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Turtle C.J., Hanafi L.-A., Berger C., Gooley T.A., Cherian S., Hudecek M., Sommermeyer D., Melville K., Pender B., Budiarto T.M., et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 2016;126:2123–2138. doi: 10.1172/jci85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Melenhorst J.J., Chen G.M., Wang M., Porter D.L., Chen C., Collins M.A., Gao P., Bandyopadhyay S., Sun H., Zhao Z., et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature. 2022;602:503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.European Medicines Agency Spikevax: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf
- 100.European Medicines Agency Comirnaty: EPAR - Product information. 2022. https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf
- 101.Newrzela S., Al-Ghaili N., Heinrich T., Petkova M., Hartmann S., Rengstl B., Kumar A., Jäck H.-M., Gerdes S., Roeder I., et al. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia. 2012;26:2499–2507. doi: 10.1038/leu.2012.142. [DOI] [PubMed] [Google Scholar]
- 102.Schambach A., Morgan M., Fehse B. Two cases of T cell lymphoma following Piggybac-mediated CAR T cell therapy. Mol. Ther. 2021;29:2631–2633. doi: 10.1016/j.ymthe.2021.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Goyal S., Tisdale J., Schmidt M., Kanter J., Jaroscak J., Whitney D., Bitter H., Gregory P.D., Parsons G., Foos M., et al. Acute myeloid leukemia case after gene therapy for sickle cell disease. N. Engl. J. Med. 2022;386:138–147. doi: 10.1056/NEJMoa2109167. [DOI] [PubMed] [Google Scholar]
- 104.Marcucci K.T., Jadlowsky J.K., Hwang W.-T., Suhoski-Davis M., Gonzalez V.E., Kulikovskaya I., Gupta M., Lacey S.F., Plesa G., Chew A., et al. Retroviral and lentiviral safety analysis of gene-modified T cell products and infused HIV and oncology patients. Mol. Ther. 2018;26:269–279. doi: 10.1016/j.ymthe.2017.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fraietta J.A., Nobles C.L., Sammons M.A., Lundh S., Carty S.A., Reich T.J., Cogdill A.P., Morrissette J.J.D., DeNizio J.E., Reddy S., et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–312. doi: 10.1038/s41586-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shah N.N., Fry T.J. Mechanisms of resistance to CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Micklethwaite K.P., Gowrishankar K., Gloss B.S., Li Z., Street J.A., Moezzi L., Mach M.A., Sutrave G., Clancy L.E., Bishop D.C., et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood. 2021;138:1391–1405. doi: 10.1182/blood.2021010858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bishop D.C., Clancy L.E., Simms R., Burgess J., Mathew G., Moezzi L., Street J.A., Sutrave G., Atkins E., McGuire H.M., et al. Development of CAR T-cell lymphoma in 2 of 10 patients effectively treated with piggyBac-modified CD19 CAR T cells. Blood. 2021;138:1504–1509. doi: 10.1182/blood.2021010813. [DOI] [PubMed] [Google Scholar]
- 109.Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gust J., Ponce R., Liles W.C., Garden G.A., Turtle C.J. Cytokines in CAR T cell-associated neurotoxicity. Front. Immunol. 2020;11:577027. doi: 10.3389/fimmu.2020.577027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mestermann K., Giavridis T., Weber J., Rydzek J., Frenz S., Nerreter T., Mades A., Sadelain M., Einsele H., Hudecek M. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aau5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Loureiro L.R., Feldmann A., Bergmann R., Koristka S., Berndt N., Máthé D., Hegedüs N., Szigeti K., Videira P.A., Bachmann M., Arndt C. Extended half-life target module for sustainable UniCAR T-cell treatment of STn-expressing cancers. J. Exp. Clin. Cancer Res. 2020;39:77. doi: 10.1186/s13046-020-01572-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wermke M., Kraus S., Ehninger A., Bargou R.C., Goebeler M.-E., Middeke J.M., Kreissig C., von Bonin M., Koedam J., Pehl M., et al. Proof of concept for a rapidly switchable universal CAR-T platform with UniCAR-T-CD123 in relapsed/refractory AML. Blood. 2021;137:3145–3148. doi: 10.1182/blood.2020009759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Minagawa K., Al-Obaidi M., Di Stasi A. Generation of suicide gene-modified chimeric antigen receptor-redirected T-cells for cancer immunotherapy. Methods Mol. Biol. 2019;1895:57–73. doi: 10.1007/978-1-4939-8922-5_5. [DOI] [PubMed] [Google Scholar]
- 115.Amatya C., Pegues M.A., Lam N., Vanasse D., Geldres C., Choi S., Hewitt S.M., Feldman S.A., Kochenderfer J.N. Development of CAR T cells expressing a suicide gene plus a chimeric antigen receptor targeting signaling lymphocytic-activation molecule F7. Mol. Ther. 2021;29:702–717. doi: 10.1016/j.ymthe.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abordo-Adesida E., Follenzi A., Barcia C., Sciascia S., Castro M.G., Naldini L., Lowenstein P.R. Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum. Gene Ther. 2005;16:741–751. doi: 10.1089/hum.2005.16.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Munis A.M., Mattiuzzo G., Bentley E.M., Collins M.K., Eyles J.E., Takeuchi Y. Use of heterologous vesiculovirus G proteins circumvents the humoral anti-envelope immunity in lentivector-based in vivo gene delivery. Mol. Ther. Nucleic Acids. 2019;17:126–137. doi: 10.1016/j.omtn.2019.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Weber T. Anti-AAV antibodies in AAV gene therapy: current challenges and possible solutions. Front. Immunol. 2021;12:658399. doi: 10.3389/fimmu.2021.658399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Leborgne C., Barbon E., Alexander J.M., Hanby H., Delignat S., Cohen D.M., Collaud F., Muraleetharan S., Lupo D., Silverberg J., et al. IgG-cleaving endopeptidase enables in vivo gene therapy in the presence of anti-AAV neutralizing antibodies. Nat. Med. 2020;26:1096–1101. doi: 10.1038/s41591-020-0911-7. [DOI] [PubMed] [Google Scholar]
- 120.Tao N., Gao G.P., Parr M., Johnston J., Baradet T., Wilson J.M., Barsoum J., Fawell S.E. Sequestration of adenoviral vector by Kupffer cells leads to a nonlinear dose response of transduction in liver. Mol. Ther. 2001;3:28–35. doi: 10.1006/mthe.2000.0227. [DOI] [PubMed] [Google Scholar]
- 121.Milani M., Annoni A., Moalli F., Liu T., Cesana D., Calabria A., Bartolaccini S., Biffi M., Russo F., Visigalli I., et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ho N., Agarwal S., Milani M., Cantore A., Buchholz C.J., Thalheimer F.B. In vivo generation of CAR T cells in presence of human myeloid cells. Mol. Ther. Methods Clin. Dev. 2022 doi: 10.1016/j.omtm.2022.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.European Medicines Agency Zolgensma: EPAR - Product information. 2021. https://www.ema.europa.eu/en/documents/product-information/zolgensma-epar-product-information_en.pdf