

Abstract
Bispecific T cell engagers (BiTEs) are bispecific antibodies that redirect T cells to target antigen-expressing tumors. We hypothesized that BiTE-secreting T cells could be a valuable therapy in solid tumors, with distinct properties in mono- or multi-valent strategies incorporating chimeric antigen receptor (CAR) T cells. Glioblastomas represent a good model for solid tumor heterogeneity, representing a significant therapeutic challenge. We detected expression of tumor-associated epidermal growth factor receptor (EGFR), EGFR variant III, and interleukin-13 receptor alpha 2 (IL13Rα2) on glioma tissues and cancer stem cells. These antigens formed the basis of a multivalent approach, using a conformation-specific tumor-related EGFR targeting antibody (806) and Hu08, an IL13Rα2-targeting antibody, as the single chain variable fragments to generate new BiTE molecules. Compared with CAR T cells, BiTE T cells demonstrated prominent activation, cytokine production, and cytotoxicity in response to target-positive gliomas. Superior response activity was also demonstrated in BiTE-secreting bivalent T cells compared with bivalent CAR T cells in a glioma mouse model at early phase, but not in the long term. In summary, BiTEs secreted by mono- or multi-valent T cells have potent anti-tumor activity in vitro and in vivo with significant sensitivity and specificity, demonstrating a promising strategy in solid tumor therapy.
Keywords: chimeric antigen receptor, glioma, glioblastoma, bispecific T cell engager, EGFR, IL13Ra2
Graphical abstract
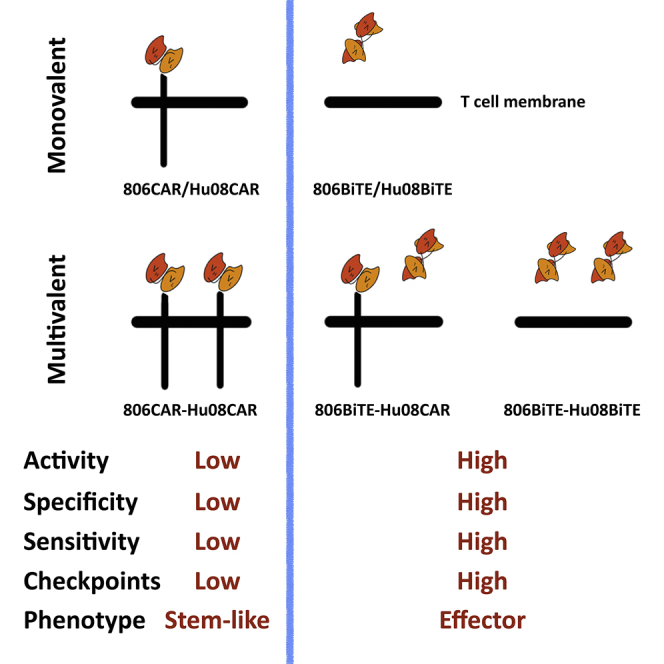
EGFR variant- and IL13Rα2-targeting BiTE secreting T cells are highly responsive, specific, and sensitive anticancer agents in both mono- and multi-valent constructs compared with classic second-generation CAR T cells. BiTE-secreting T cells also demonstrate up-regulated immune checkpoint markers and effector phenotypes.
Introduction
T cells can be redirected to tumors by being genetically modified to express chimeric antigen receptors (CARs).1 Sustained remission was achieved in leukemia and lymphoma patients treated with CD19 targeting CAR T cells.2, 3, 4 Eighty-five percent of patients with relapsed or refractory multiple myeloma responded to B cell maturation antigen targeting.5 In the realm of solid tumors, the clinical application of CAR T cells has had limited success. A patient with recurrent multifocal glioblastoma (GBM) had a 7.5-month clinical response after the treatment of interleukin-13 receptor alpha 2 (IL13Rα2) targeting CAR T cells.6 This patient’s tumor recurred with low levels of the target antigen. This underscores the potential use of CAR T cells against solid tumors, in addition to bringing to light the limitations of monotherapy approaches against dynamic tumors such as GBM.
Another approach to redirect T cells is bispecific T cell engagers (BiTEs). BiTEs combine antigen specificity with the ability to induce cytotoxicity via bystander T cells by linking two single chain variable fragments (scFvs), one recognizing a tumor antigen and the other recognizing the CD3 molecule on T cells.7 Patients with leukemia or lymphoma treated with a CD19-targeting BiTE (blinatumomab) achieved sustained remission.8, 9, 10, 11 However, repeated infusions were needed, as the half-life of BiTEs in circulation was 2–3 h7 In previous studies, EphA2-targeting BiTE secreted by T cells and IL13Rα2 targeting BiTE secreted by neural stem cells were shown to successfully activate bystander T cells and inhibit tumor growth in glioma and lung cancer murine models.12,13 In another report directly comparing the anti-tumor activity of mRNA electroporated blinatumomab-secreting T cells and CD19 CAR T cells, the BiTE T cells demonstrated superior tumor suppression.14
Despite the prominent effect achieved by CAR T cells and BiTEs in the treatment of leukemia and lymphoma, a significant number of treated patients have had limited antitumor activity or tumor relapse.1,7,15 Antigen loss or down-regulation is a common mechanism of treatment resistance, which could be mitigated by combinatorial targeting.16, 17, 18 Antigen loss was also observed in solid tumors after CAR T cell therapy. Significant antigen loss was seen in patients with recurrent GBM after the treatment of IL13Rα2-targeting CAR T cells and epidermal growth factor receptor (EGFR) variant III (EGFRvIII)-targeting CAR T cells.6,19 To address antigen heterogeneity in solid tumors, multivalent CAR T cells have been studied in pre-clinical models, allowing for the expansion of targetable tumors and amplification of treatment effects.20, 21, 22 EGFR-targeting BiTEs secreted from EGFRvIII-targeting CAR T cells have also been developed preclinically. They demonstrated the feasibility of delivering EGFR-targeting BiTEs via CAR T cells,23 which suggests BiTEs can be also used in generating multivalent T cells.
GBMs are the most common primary malignant brain tumor.24,25 With standard-of-care treatment, including surgical resection, radiotherapy, and chemotherapy, the median survival is only 12–17 months.25,26 In our phase I trial of EGFRvIII-targeting CAR T cells, EGFRvIII expression was down-regulated after CAR T infusion, but not amplified wild-type EGFR.19 We hypothesized that the simultaneous targeting of EGFR and IL13Rα2 may result in a meaningful clinical impact for patients with GBM. We have previously reported on the anti-tumor activity of an IL13Rα2-specific CAR.27 Our group and others have also demonstrated the anti-tumor activity of a pan-EGFR alteration-specific CAR based on the 806 monoclonal antibody (mAb).28,29 The 806 mAb recognizes a cryptic epitope on EGFR that is exposed in mutated or amplified conformations found on tumors.30,31 In this study, we generated 806BiTE- and Hu08BiTE-secreting T cells, and compared activity with 806CAR T cells and Hu08CAR T cells in responding to GBM cells. We demonstrate the anti-tumor activity, sensitivity, and specificity in mono- and multi-valent constructs that have application to GBM and other solid tumors.
Results
EGFRvIII is co-expressed with EGFR and IL13Rα2 in GBM
We performed flow cytometry on freshly resected glioma tissue to demonstrate the expression of EGFR and EGFRvIII in glioma cells (Figure 1A). Cases demonstrated significantly more EGFR positivity than EGFRvIII (p = 0.0172). Staining results of four representative cases illustrated that a considerable percentage (10.7%, 10.6%, 22.7%, and 34.1%) of EGFR-positive cells were EGFRvIII negative. This suggested that targeting EGFR would cover a greater percentage of tumor cells than targeting EGFRvIII, highlighting the need for additional targets beyond EGFR.
Figure 1.

EGFR, EGFRvIII, and IL13Rα2 co-express in GBM
(A) Flow cytometry analyses of EGFRvIII and EGFR expression in resected glioma tissues (left). Statistically significant differences were determined using two-tailed paired t test. Flow panels of red dots indicated EGFR expression along the x axis and EGFRvIII expression along the y axis of four representative cases (right), with blue dots as controls of staining. (B) Heat maps showing percentage of IL13Rα2-positive cells in 10 EGFRvIII-positive or -negative (left) and amplified or unamplified EGFR (right) GSC lines. (C) Immunohistochemical stains of IL13Rα2 (brown) and EGFRvIII (red) in a recurrent/residual GBM tissue section. (D) EGFR (upper) and IL13Rα2 (lower) expression segregated by WHO grades (left) and GBM molecular subtypes (right) based on the RNA sequencing data from the TCGA database. The y axis indicates the relative expression levels of each cases. Statistically significant differences were calculated by Kruskal-Wallis test with p < 0.05 being considered statistically significant.
To broaden the range of targetable tumor cells, we also examined IL13Rα2 expression in 10 glioma stem cell (GSC) lines from cases with varying levels EGFR and EGFRvIII expression as determined by next-generation sequencing (Figure 1B).32 The expression of IL13Rα2 was also found to be heterogeneous. Six of 10 cases had more than 40% detectable expression of IL13Rα2. In these six cases, two were positive for EGFRvIII expression and three were positive for EGFR amplification. In the four IL13Rα2-negative cases, only one was negative for both EGFR amplification and EGFRvIII expression. We also detected IL13Rα2 co-expressed with EGFRvIII in a recurrent/residual GBM tissue by immunohistochemistry (Figure 1C) with isotype staining control (Figure S1), further demonstrating the expression of both targets across the tumor. These results suggested that the addition of IL13Rα2 as a target would expand the application of EGFR and EGFRvIII targeting strategies.
To analyze target expression in a larger patient population, we downloaded RNA-seq data of gliomas from the Genomic Data Commons data portal on The Cancer Genome Atlas (TCGA). Glioma cases were grouped by both World Health Organization (WHO) grade and genomic subtype (Figure 1D).33 The expression of EGFR and IL13Rα2 was significantly higher in high-grade gliomas than in low-grade gliomas. EGFR was predominately expressed in the classical subtype, with lower expression levels in the mesenchymal and proneural subtypes. There was no statistical difference in IL13Rα2 expression between glioma subtypes. These results also confirmed the expression of EGFR and IL13Rα2 in glioma tissue and the significance of targeting both antigens with T cell-engaging therapies.
BiTEs secreted from T cells bind targets with a high degree of specificity
Given the heterogeneous antigen expression found in gliomas, we next determined if BiTE-secreting T cells could be used as an antigen-specific targeting strategy. We generated an EGFR-targeting BiTE and an IL13Rα2-targeting BiTE with the 806 and Hu08 scFvs, respectively. Both 806CAR and Hu08CAR constructs were also included to compare the anti-tumor activity of the corresponding BiTE constructs (Figure 2A).
Figure 2.

BiTEs secreted from T cells specifically bind to target antigens and T cells
(A) Schematic vector maps of 806CAR/806BiTE/Hu08CAR/Hu08BiTE constructs. (B) Flow cytometric detection of T cell transduction by mCherry expression. (C) Conditioned media of T cells in each group tested by ELISA to evaluate the binding ability of secreted BiTEs to recombinant EGFR, EGFRvIII, and IL13Rα2 proteins. (D) The GSC line 5077 was lentivirally transduced to overexpress EGFRvIII or IL13Rα2 (red) and assessed via flow cytometry. Staining control was showed (blue). (E) Conditioned media of CAR/BiTE T cells was collected and co-cultured with UTD T cells and target cells. BiTE binding on T cells was detected by biotinylated protein L with secondary streptavidin coupled FITC after 16-h co-culture. The median fluorescence intensity was quantified on CD4- and CD8-positive T cells. (F) Flow based results of representative samples in (E). CD8 was stained to distinguish the CD4-positive and CD8-positive subgroups of T cells along the x axis. Statistically significant differences were calculated by one-way ANOVA with post hoc Tukey test. ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
We normalized CAR expression via mCherry expression detected on T cells by flow cytometry (Figure 2B). Conditioned media from each group was collected and used in a standard direct ELISA to confirm the secretion and binding of 806BiTE and Hu08BiTE from T cells to recombinant EGFR, EGFRvIII, and IL13Rα2 (Figure 2C). 806BiTE secreted from T cells significantly detected plate-bound EGFR and EGFRvIII (p < 0.0001). The optical density value in the EGFRvIII-binding group was higher than the EGFR-binding group, suggesting a high affinity of the 806BiTE for EGFRvIII (p = 0.0006). Hu08BiTE secreted from T cells also significantly bound to IL13Rα2 (p < 0.0001) when compared with the other T cell groups.
To further demonstrate the characteristics of BiTE T cells, we selected GSC line 5077,34 which expressed low levels of endogenous EGFR (7.79%) but not IL13Rα2 or EGFRvIII. We engineered 5077 cells to overexpress EGFRvIII or IL13Rα2 (Figure 2D). Conditioned media from each BiTE T cell group was collected and used in co-culture with untransduced (UTD) T cells and target cells. BiTE binding to T cells was detected with protein L staining by flow cytometry (Figure 2E). 806BiTE or Hu08BiTE binding on T cells was only detected when EGFRvIII or IL13Rα2 was over-expressed in 5077 cells (p < 0.0001). Flow-based results of representative samples are illustrated (Figure 2F), indicating that a BiTE’s ability to bind to T cells was eliminated in the absence of target antigen.
BiTEs and BiTE T cells respond to antigen-positive glioma cells
To determine the ability of the secreted 806BiTEs and Hu08BiTEs to initiate T cell responses, T cell activation marker CD69 was assessed on UTD T cells by flow cytometry in conditioned media from BiTE transduced T cells (Figure 3A). CD69 expression was significantly elevated in co-culture with 5077EGFRvIII+ cells with conditioned media from 806BiTE T cells and co-culture with 5077IL13Rα2+ cells with conditioned media from Hu08BiTE in both CD4+ and CD8+ T cell subgroups (p < 0.0001). Flow panels of representative samples indicating both 806BiTEs and Hu08BiTEs induced UTD T cell activation in the presence of antigen-positive target cells (Figure 3B). Cytokine production (interferon [IFN]γ, IL-2, and tumor necrosis factor [TNF]α) of UTD T cells was also detected when co-cultured with target cells in conditioned media (Figure S2), which was consistent with the result of CD69 expression on these UTD T cells. To determine the ability of 806BiTE and Hu08BiTE to mediate antigen-specific cytotoxicity, we also performed bioluminescence cytotoxicity assays with UTD T cells co-cultured with different target cells (Figure 3C). No cytotoxic activity was observed in co-culture with 5077. Significant killing activity was detected in 5077EGFRvIII+ cells co-cultured with conditioned media from 806BiTE T cells and 5077IL13Rα2+ cells co-cultured with conditioned media from Hu08BiTE T cells. These results demonstrated that T cells transduced to secrete 806BiTEs or Hu08BiTEs can significantly and specifically activate UTD T cells in an antigen-specific manner.
Figure 3.

BiTE T cells respond to target positive glioma cells
(A) UTD T cell activation, as demonstrated by CD69 expression, in CD4+ (top) and CD8+ (bottom) T cells after 16 h co-culture with conditioned media from CAR/BiTE T cells. (B) Flow-based results of representative samples in (A). CD8 was stained to distinguish the CD4-positive and CD8-positive subgroups of T cells along the x axis. (C) Bioluminescence cytotoxicity assay of UTD T cells co-cultured with tumor cell lines and conditioned media of CAR/BiTE T cells. (D) T cell activation, as measured by CD69 expression, in CD4+ (top) and CD8+ (bottom) T cells after 16 h co-culture of CAR/BiTE T cells with target cells. (E) Flow cytometry panels of bar graphs in (D). CD8 was stained to distinguish the CD4-positive and CD8-positive subgroups of T cells along the x axis. (F) Bioluminescence cytotoxicity assays of CAR/BiTE T cells co-cultured with tumor cell lines was analyzed at different effector/target (E:T) ratios (0.625:1, 1.25:1, 2.5:1, 5:1, 10:1, and 20:1) and compared with the UTD T cell group. Statistically significant differences were calculated by one-way ANOVA with post hoc Tukey test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
Next, we considered the ability of 806BiTE T cells and Hu08BiTE T cells to respond to target-expressing tumor cells. Both 806BiTE T cells and Hu08BiTE T cells significantly activated by 5077EGFRvIII+ and 5077IL13Rα2+ cells compared with UTD groups (p < 0.0001). The stimulation level was higher than 806CAR T cells and Hu08CAR T cells in both CD4+ and CD8+ T cell subgroups (p < 0.0001) (Figures 3D and 3E). Bioluminescence cytotoxicity assays also confirmed significant cytotoxicity of 806BiTE T cells and Hu08BiTE T cells by targeting 5077EGFRvIII+ and 5077IL13Rα2+ cells (Figure 3F). To demonstrate the activity of BiTE T cells in vivo, a glioma line, U87MG, which partially expresses IL13Rα2 (Figure S3A), was adopted to generate orthotopically implanted glioma mouse model. Both Hu08BiTE T cells and Hu08CAR T cells significantly inhibited orthotopically implanted tumor growth and prolonged mouse survival compared with UTD T cells (Figure S3B). Bioluminescent tumor signal was significant decreased in these two groups after 11 days of T cell infusion. More strikingly, tumor signal in Hu08BiTE T cells group was more than 30 times lower than Hu08CAR T cells group on that day. Although U87MG cells were only 75% positive for IL13Rα2 (Figure S3A), no tumor recurrence was detected.
Interestingly, we also detected elevated CD69 expression on 806BiTE T cells when co-cultured with parental 5077 and 5077IL13Rα2+ cells in CD4+ and CD8+ T cells subgroups (Figures 3D and 3E). Significant cytotoxicity was also observed when 806BiTE-secreting T cells were co-cultured with 5077 cells (Figure 3F). The binding of 806BiTE on T cells was also detected when co-cultured with 5077 cells with protein L staining by flow cytometry (Figure S4). This was not observed in UTD T cells co-cultured with 5077 in the presence of 806BiTE-conditioned media or 806CAR T cells co-cultured with 5077, although the initial concentration of 806BiTE in the conditioned media of UTD cells was higher than that in the conditioned media of 806BiTE T cells. These results indicated that autocrine binding of 806BiTE to T cells resulted in a superior response to low antigen-expressing tumor cells.
BiTE T cells respond to low target antigen expression on glioma cancer stem cells
To determine the sensitivity of 806BiTE to detect low levels of EGFR expression on 5077 cells, we generated 806BiTE T cells with high (H), medium (M), and low (L) levels of transduction efficacy. 806CAR T cells with comparable transduction efficacy were included as controls (Figure 4A). CD69 expression on T cells was up-regulated in both 806BiTE and 806CAR T cell groups when co-cultured with 5077 cells in a transduction-dependent manner (Figure 4B). CD69 expression was significantly higher in 806BiTE-transduced T cells than 806CAR-transduced T cells. Conditioned media from 806BiTE T cells and 806CAR T cells at different transduction levels were collected and added to UTD T cells co-cultured with 5077. No significant difference of cytotoxicity was detected as compared with conditioned media added from the UTD T cell group (Figure 4C, left panel). When 806BiTE T cells at different transduction levels were co-cultured with 5077 cells in fresh media, cytotoxicity was observed in every group in a dose-dependent manner (Figure 4C, right panel). No cytotoxicity was observed in 806CAR T cells co-cultured with 5077 at any transduction level. These results demonstrated a dose-dependent activation and killing activity of 806BiTE T cells co-cultured with 5077, expressing low levels of endogenous EGFR, which was not observed in 806CAR T cells or 806BiTE recruited UTD T cells.
Figure 4.

BiTE T cells respond to low antigen expression on glioma cancer stem cells
(A) Flow cytometry showing the high (H), medium (M), and low (L) levels of transduction of 806CARs (top row) and 806BiTEs (bottom row) by mCherry expression. (B) T cell activation, as measured by CD69 expression, in CD4+ and CD8+ T cells after 16 h co-culture of 806CAR/BiTE T cells with 5077. (C) Bioluminescence cytotoxicity assays of UTD T cells co-cultured with 5077 target cells in the presence of conditioned media of 806CAR/BiTE T cells. The volume of conditioned media used in each co-culture was indicated along the x axis (left panel). Bioluminescence cytotoxicity assay of 806CAR/BiTE T cells co-cultured with 5077 at different effector/target (E:T) ratios (2.5:1, 5:1, 10:1, and 20:1) and compared with the UTD T cell group (right panel). (D) Vector maps of the 806 (top) and C225 (bottom) BiTE designs, Hu08CAR lacking signaling motifs (Hu08TM) was used as a cell surface tag. (E) Flow panels showing the transduction of the BiTE constructs, as determined by Hu08TM staining. (F) Conditioned media of T cells in each group tested with a standard direct ELISA to evaluate the binding ability of secreted BiTEs to recombinant EGFR and EGFRvIII protein. (G) 806/C225BiTE T cells and UTD T cells were co-cultured with 5077 or 5077EGFRvIII+ cells. Cytokine secretion was detected with ELISA. (H) Bioluminescence cytotoxicity assays of 806/C225BiTE T cells co-cultured with 5077 cells (left panel) and 5077EGFRvIII+ cells (right panel), analyzed at different effector/target (E:T) ratios (1:1, 3:1, 8:1, and 20:1) and compared with the UTD T cell group. Statistically significant differences were calculated by one-way ANOVA with post hoc Tukey test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
To further demonstrate that the low level of endogenous EGFR was the cause for 806BiTE activity against 5077 cells, another EGFR-targeting BiTE (C225BiTE) was generated (Figure 4D). C225’s epitope is found on the L2 domain of EGFR, which is exposed in both active and inactive states.35 Comparable transduction efficacy of 806BiTE T cells and C225BiTE T cells was achieved, as determined by staining of Hu08CAR lacking signaling motifs (Figure 4E). Conditioned media from 806BiTE and C225BiTE-secreting T cells was collected after overnight culture and used in an ELISA to confirm the secretion and binding of 806BiTE and C225BiTE to recombinant EGFR and EGFRvIII (Figure 4F). Secreted 806BiTEs and C225BiTEs significantly bound to both targets. EGFR binding was more prominent for C225BiTE-secreting T cells than for 806BiTE-secreting T cells (p = 0.0273). Additionally, binding to EGFRvIII was more prominent for 806BiTE-secreting T cells than the C225BiTE-secreting T cells (p = 0.0039). Cytokine secretion was detected in 806BiTE and C225BiTE T cells when co-cultured with either 5077 or 5077EGFRvIII+ cells (Figure 4G). Significant cytokine secretion was detected in C225BiTE T cells co-cultured with 5077 cells when compared with UTD or 806BiTE T cell co-culture groups, indicating that C225BiTE-secreting T cells could be stimulated by the low levels of EGFR expressed in 5077. In co-cultures with 5077EGFRvIII+ cells, significant cytokine secretion was detected in both 806BiTE and C225BiTE T cell groups when compared with UTD T cells. Cytotoxicity was observed in C225BiTE T cells co-cultured with 5077 cells, but not in UTD or 806BiTE T cell co-culture groups. In co-culture with 5077EGFRvIII+ cells, both 806BiTE and C225BiTE T cells showed potent cytotoxicity (Figure 4H). Taken together, the low levels of EGFR found on 5077 glioma cells can be targeted by both 806BiTE-secreting T cells and C225BiTE-secreting T cells. However, this was not observed in 806CAR T cells or 806BiTE-recruited UTD T cells.
806BiTE showed low levels of activity against physiologically expressed EGFR, consistent with the binding property of 806 antibody
After determining the ability of 806BiTE T cells to respond to low levels of EGFR in GBM cells, we next examined the safety of 806BiTE T cells in responding to physiologic EGFR expressed on normal astrocytes (Figure 5A). To determine the response of 806BiTE T cells to astrocytes, we used variable levels of transduction efficacy and compared them with 806CAR T cells at comparable transduction levels as a control. We found T cell activation to be positively correlated with transduction levels after 806BiTE/CAR T cells were co-cultured with astrocytes overnight (Figure 5B). However, in contrast with the response to 5077 or 5077EGFRvIII+ glioma cells, the activation levels in 806BiTE T cell groups were significantly lower than that of 806CAR T cell groups. Cytokine production was also detected in 806BiTE and 806CAR T cells after overnight co-culturing with astrocytes (Figure 5C). In 806CAR T cells, cytokine production was positively correlated with the transduction efficacy in both CD4+ and CD8+ T cells. Consistent with the results of T cell activation, cytokine production in 806BiTE T cell groups was significantly lower than 806CAR T cell groups in corresponding transduction efficacy in both CD4+ and CD8+ T cell subgroups. More strikingly, with the exception of TNFα production in CD4+ 806BiTE T cells in the high transduction efficacy subgroup, no statistical differences of cytokine production in any other 806BiTE T cell co-culture groups were observed when compared with the UTD T cell co-culture group.
Figure 5.

806BiTE was reluctant in responding to physiologically expressed EGFR
(A) Flow profile showing the expression of EGFR in astrocytes (red), with staining control (blue). (B) 806CAR/BiTE T cells with high (H), medium (M), and low (L) levels of transduction were co-cultured with astrocytes. T cell activation, as measured by CD69 expression, in CD4+ and CD8+ T cells after 16 h co-culture. (C) Flow based intracellular cytokine (IFNγ, IL2, and TNFα) staining of 806CAR/BiTE T cells co-cultured with astrocytes. CD4- and CD8-positive subgroups of T cells were distinguished by human CD8 staining. (D) T cell activation, as demonstrated by CD69 expression, in CD4+ (top) and CD8+ (bottom) T cells after 16 h co-culture with target cells and conditioned media of CAR/BiTE T cells. (E) Flow based intracellular cytokine (IFNγ, IL2, and TNFα) staining of UTD T cells co-cultured with astrocytes in conditioned media of CAR/BiTE T cells. Statistically significant differences were determined using log rank test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
To determine the ability of 806BiTE to engage UTD T cells and astrocytes, conditioned media of 806BiTE and 806CAR T cells at different levels of transduction efficacy was collected. CD69 expression and cytokine production was detected in UTD T cells co-cultured with astrocytes in the conditioned media from 806BiTE/806CAR T cells (Figures 5D and 5E). No significant upregulation of CD69 expression or cytokine production was detected in conditioned media from either 806BiTE or 806CAR T cell groups, when compared with conditioned media of UTD T cells.
To determine the cytotoxicity of 806BiTE T cells in co-culturing with astrocyte, impedance cytotoxicity assays were performed with 806CAR T cells as controls (Figure S5A). No cytotoxicity of 806BiTE T cell groups and 806CAR T cell with low transduction efficacy group was detected when co-cultured with astrocyte in 24 h. There was 31% cytotoxicity of astrocytes in 806CAR T cells with high transduction efficacy co-culturing group (p < 0.0001). Impedance cytotoxicity assay was also performed to compare the cytotoxicity of 806BiTE T cells with 806CAR T cells in co-culturing with 5077 cells (Figure S5B) and 5077EGFRvIII cells (Figure S5C). Cytotoxicity can be detected in the 806BiTE T cell groups and 806CAR T cell with high transduction group, but not for 806CAR T cell with the low transduction group in co-culturing with 5077 cells. In co-culturing with 5077EGFRvIII cells, cytotoxicity was detected in both the BiTE T cell groups and the CAR T cell groups (p < 0.001). Cytotoxicity in BiTE T cell groups with high and low transduction efficacy was significantly higher than that in corresponding CAR T cell groups (p < 0.0001). These results demonstrated that, although BiTE T cells lead to superior responding to low-level EGFR in 5077 cells and EGFRvIII, soluble 806BiTE and 806BiTEs secreted by T cells lead to weak activation of effector T cells in response to the EGFR expressed on astrocytes.
BiTEs’ activation significantly up-regulated checkpoints and T cell effector phenotypes
To detect the persistence of BiTE T cells in responding to target cells, the human glioma line, D270, endogenously expresses both EGFR and IL13Rα2 was used in addition to 5077 cells (Figure 6A). After 4 days and 7 days co-culturing with D270 cells, both 806BiTE T cells and Hu08BiTE T cells had higher proliferation compared with UTD T cells (Figure 6B). On day 4, the proliferation of 806BiTE T cells was more evident than 806CAR T cells in both CD4+ and CD8+ T cell subgroups. And the proliferation of Hu08BiTE T cells was less evident than Hu08CAR T cells in CD4+ T cell subgroup. No significant difference between BiTE T cells and CAR T cells in proliferation on day 7. Immune checkpoints (programmed cell death 1 [PD-1], CTLA-4, and TIM-3) were considered to be markers of T cell exhaustion. Comparing with corresponding CAR T cells, PD-1, CTLA-4, and TIM-3 were significantly up-regulated in both CD4+ and CD8+ subgroups of 806BiTE T cells and Hu08BiTE T cells after co-culturing with D270 cells (p < 0.0001) (Figure 6C). We also analyzed T cell phenotypes after 4 days co-culturing with D270 cells (Figure 6D). Comparing with CAR T cells, naive T cell marker, CD45RA, was significantly down-regulated in BiTE T cells. CCR7 was also down-regulated, indicating the tendency of transforming into effector T cell phenotypes rather than memory T cell phenotypes. No significant difference of CD62L expression between CAR T cells and BiTE T cells.
Figure 6.

BiTEs’ activation significantly up-regulated checkpoints and T cell effector phenotypes
(A) Flow panel of EGFR (left) and IL13Rα2 (right) expression (red) on glioma line D270 with staining control (blue). (B) Flow cytometry determined T cell proliferation assay with CFSE staining was performed on UTD T cells, 806CAR T cells, 806BiTE T cells, Hu08CAR T cells, and Hu08BiTE T cells on day 4 (upper panel) and day 7 (lower panel) coculturing with D270 cell line. The median fluorescence intensity was quantified on CD4-positive and CD8-positive subgroups of T cells. (C) The expression of checkpoints (PD-1, CTLA-4, and TIM-3) on the T cells was determined by flow cytometry after overnight co-culturing of CAR/BiTE T cells with D270 cell line. Flow based results of representative samples were illustrated, CD8 was stained to distinguish the CD4-positive and CD8-positive subgroups of T cells along the x axis. The median fluorescence intensity was quantified and compared between CAR T cells and BiTE T cells. (D) The expression of CD45RA, CCR7, and CD62L on the T cells was determined by flow cytometry after 4 days co-culturing of CAR/BiTE T cells with D270 cell line. Flow-based results of representative samples were illustrated. The median fluorescence intensity was quantified and compared between CAR T cells and BiTE T cells on CD4-positive and CD8-positive subgroups. Statistically significant differences were calculated by one-way ANOVA with post hoc Tukey test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
Checkpoints (PD-1, CTLA-4, and TIM-3) expression was also detected in 806BiTE T cells and Hu08BiTE T cells after co-culturing with 5077EGFRvIII+, IL13Rα2+ cells. The expression of PD-1, CTLA-4, and TIM-3 was also significantly higher in both CD4+ and CD8+ subgroups than corresponding CAR T cells (Figure S6A). After 4 days co-culturing with 5077EGFRvIII+, IL13Rα2+ cells, not only were CD45RA and CCR7 significantly down-regulated comparing with corresponding CAR T cells, the expression of CD62L in CD4+ T cell subgroup was also downregulated, which also indicating effector T cell phenotypes transformation (Figure S6B). These results indicated BiTE T cells demonstrated similar proliferation as CAR T cells in 7 days target stimulation in vitro, superior activation of BiTE T cells also accompanied with superior expression of T cell exhaustion markers and effector T cells transformation.
BiTEs responded to target-positive cells in bivalent constructs
To counter the heterogeneous antigen expression found on tumors, we generated bivalent T cells simultaneously targeting tumor-expressed EGFR and IL13Rα2. To further assess the ability of BiTEs to be used in bivalent targeting, we generated 806BiTE-Hu08CAR, Hu08BiTE-806CAR T cells, 806BiTE-Hu08BiTE T cells, and 806CAR-Hu08CAR T cells (Figures 7A and 7B). All bivalent T cells demonstrated activation when co-cultured with modified 5077 or D270 cells (Figure 7C). Strikingly, there were differences in the activation of BiTE-producing T cells and CAR T cells in response to antigen-positive target cells. Both 806BiTE-Hu08CAR and 806BiTE-Hu08BiTE T cells showed superior response to 5077EGFRvIII+ cells and D270 cells when compared with 806CAR-Hu08CAR T cells. Hu08BiTE-806CAR and 806BiTE-Hu08BiTE T cells had a higher response to 5077IL13Rα2+ cells compared with 806CAR-Hu08CAR and 806BiTE-Hu08CAR T cells. Superior response was also observed in Hu08BiTE-806CAR T cells responding to D270 cells compared with 806CAR-Hu08CAR T cells. Consistent results were also observed in cytokine production (Figure 7D). We did not observe enhanced activation in the 806BiTE-Hu08BiTE population responding to D270 cells when compared with 806BiTE-Hu08CAR T cells. In cytotoxicity assays, all bivalent T cells showed dose-dependent cytotoxicity in modified 5077 and D270 cells (Figure 7E). Concordant with the above results, the cytotoxicity of BiTE+ T cells responding to antigen-positive cells was significantly higher than cytotoxicity observed in T cells expressing the equivalent CAR.
Figure 7.

BiTE mediates anti-tumor activity in antigen positive cells in bivalent targeting constructs
(A) Vector maps of 806CAR-Hu08CAR, 806BiTE-Hu08CAR, Hu08BiTE-806CAR, and 806BiTE-Hu08BiTE constructs. (B) Flow cytometric detection of T cell transduction by mCherry expression. (C) Bivalent T cell activation, as measured by CD69 expression, in CD4+ (left) and CD8+ (right) T cells after 16 h co-culture with target cells. (D) Flow-based intracellular cytokine (IFNγ, IL2, and TNFα) staining of bivalent targeting T cells co-cultured with target cells. CD4+ (top) and CD8+ (bottom) subgroups of T cells were distinguished by human CD8 staining. (E) Bioluminescence cytotoxicity assays of bivalent targeting T cells co-cultured with 5077EGFRvIII+ cells (left panel), 5077IL13Rα2+ cells (middle panel) and D270 cells (right panel), analyzed at different effector/target (E:T) ratios (0.625:1, 1.25:1, 2.5:1, 5:1, 10:1, and 20:1) and compared with the UTD T cell group. Statistically significant differences were calculated by one-way ANOVA with post hoc Tukey test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
To demonstrate the anti-tumor effects of bivalent BiTE targeting T cells in vivo, we first treated mice with 806BiTE-Hu08CAR T cells 8 days after D270 subcutaneous tumor cell implantation in NSG mice, using Hu08CAR T cells as a positive control (Figure 8A). Both 806BiTE-Hu08CAR- and Hu08CAR T cell-treated groups significantly controlled tumor growth and prolonged survival compared with mice treated with UTD T cells (p < 0.0001). Notably, we observed earlier control of tumor growth in the 806BiTE-Hu08CAR cohort when compared with the Hu08CAR cohort at day 6 and day 9 after T cell infusion. Next, we infused 806BiTE-Hu08CAR, Hu08BiTE-806CAR, 806BiTE-Hu08BiTE, and 806CAR-Hu08CAR T cells in D270 tumor-bearing mice 8 days after subcutaneous tumor implantation (Figure 8B). All bivalent T cells significantly controlled tumor growth (p < 0.0001) and prolonged survival (p < 0.01) as compared with the UTD T cell cohort. Tumor sizes in 806BiTE-Hu08CAR, Hu08BiTE-806CAR, and 806BiTE-Hu08BiTE T cell groups significantly decreased on day 9 after T cell infusion, which was earlier than the 806CAR-Hu08CAR T cell-infused group. 806CAR-Hu08CAR T cells showed better control of tumor growth and mouse survival than 806BiTE-Hu08CAR, Hu08BiTE-806CAR, and 806BiTE-Hu08BiTE T cell groups in the long run. Taken together, BiTE-secreting bivalent T cells demonstrated significant anti-tumor activity both in vitro and early phage in vivo.
Figure 8.

BiTE transduced T cells significantly delay early tumor growth in a GBM implanted mouse model
(A) There were 800,000 Hu08CAR- and 806BiTE-Hu08CAR-positive T cells or the same number of UTD T cells that were injected intravenously (n = 8) 8 days after D270 subcutaneous implantation. (B) There were 1,200,000 bivalent targeting construct-transduced T cells (806CAR-Hu08CAR, 806BiTE-Hu08CAR, Hu08BiTE-806CAR, and 806BiTE-Hu08BiTE) or the same number of UTD T cells that were injected intravenously (n = 8) 8 days after D270 subcutaneous implantation. Tumor size was compared between each group. Statistically significant differences of tumor size at each time point were calculated by one-way ANOVA with the post hoc Tukey test. Linear regression was used to test for significant differences between the experimental groups. Survival based on time to end point was plotted using a Kaplan-Meier curve (Prism software). Statistically significant differences were determined using log rank test. ns, not significant; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Data are presented as means ± standard deviation.
Discussion
In this study, we generated T cell-secreting BiTEs directed to tumor-associated EGFR and IL13Rα2, both targets having previously been demonstrated to be expressed on GBM cells.27,28 BiTEs secreted by T cells demonstrated superior anti-tumor activity when compared with their corresponding CAR version. 806BiTE-secreting T cells also demonstrated enhanced sensitivity in responding to low antigen-expressing tumor cells. In bivalent constructs, BiTEs also outperform CARs in killing target-positive tumor cells in vitro and early phase in vivo, but not in the long term.
BiTE therapy and CAR T cell therapy are two main approaches for redirecting T cells against tumors, both of which demonstrated promising effects in pre-clinical and clinical studies.4,7,9,13,36 To integrate the advantages of both strategies, T cells were engineered to constitutively secrete BiTEs to recruit bystander T cells to engage tumor cells.12,37, 38, 39 Consistent with the reported results of mRNA electroporated BiTE T cells,14 lentivirally transduced 806BiTE and Hu08BiTE T cells also demonstrated superior anti-tumor responses when compared with their corresponding 806CAR and Hu08CAR T cells. In our in vivo tumor models, mice treated with BiTE-secreting T cells established tumor control earlier than controls. Remarkably, when co-cultured with EGFR-low 5077, only 806BiTE-secreting T cells significantly attacked target cells, but not 806BiTE-recruited T cells or 806CAR T cells. Combined with a lack of cytotoxic activity against EGFR-expressing astrocytes, this sensitivity and selectivity highlights the safety features of 806BiTEs and 806BiTE-secreting T cells. The rate of response of refractory or relapsed leukemia to blinatumomab in a phase II trial was increased from 43% to 82% when considering only patients with minimal residual disease8, this finding not only shows that BiTEs can "detect" low levels of target antigen, but also that efficacy is better in settings with a low tumor burden.40 In our study, the sensitivity of the T cell-secreted 806BiTE was consistently higher than that of 806CAR T cells, suggesting that BiTE-secreting T cells are a viable strategy in the treatment of antigen-low tumors or with heterogeneous antigen expression in tumors. Our conclusions were based on comparing BiTE secreting T cells with corresponding second-generation CAR T cells. There has been significant work done with third-generation CAR T cells, demonstrating their function compared with second-generation CAR.1 These comparisons are yet to be conducted in our CAR-BiTE system. In addition, scFvs have significant effects on their overall constructs, making broad extrapolation of our findings to unrelated scFvs tenuous.
Compared with monovalent CAR T cells or bivalent CAR T cells, BiTE-CAR T cells and BiTE-BiTE T cells showed superior control of tumor growth in a glioma mouse model 9 days after T cell treatment. At 20 days after T cell administration, BiTE-CAR T cells and BiTE-BiTE T cells demonstrated a loss of tumor control, but this phenomenon was not found in mice treated with bivalent CAR T cells. Although BiTE T cells were shown to have comparable capacity of proliferation as CAR T cells on day 7 after target stimulation in vitro, significant checkpoint expression and loss of memory and naive T cell phenotypes still raised the concern of the persistency of BiTE T cells in the long-term. BiTE-mediated T cell activation does not provide a co-stimulatory signal, presumably resulting in T cell exhaustion and an inability to proliferate.23 Co-stimulatory signals such as 4-1BB and CD28 have significantly enhanced BiTE-mediated T cell activation.41,42 These reports suggest that providing an additional co-stimulatory signal in the BiTEs would be a promising strategy in maintaining the function of BiTE-secreting T cells and integrating the advantages of BiTE and CAR T cell therapies. Additionally, the limited pool of bystander T cells available for a BiTE to recruit in the immunodeficient NSG mouse system could be another reason. Unmodified T cells may not persist in an NSG mouse 20 days after T cell administration, given the lack of stimulation and engraftment.43 Based on our results, repeated infusions of BiTE-secreting T cells could be a potent way to control tumor growth and provide long-term anti-tumor activity.
Antigen loss is a primary mechanism of resistance to redirected T cell therapies in cancer.15 CD19 is homogeneously expressed in mature B cells. Even so, approximately 7%–25% of patients relapsed with CD19-negative disease after treatment with CD19-specific CAR T cells.1 In solid tumors, heterogeneous antigen expression is much more common. In a single-cell RNA-seq study, 430 cells from five GBMs resulted in distinct expression patterns among individual tumor cells derived from the same tumor.44 Clinically, the observed low and heterogeneous antigen expression found on GBM may be contributing factors in limiting the efficacy of CAR T cells targeting IL13Rα2 or EGFRvIII.6 These results call into focus using a multivalent targeting strategy as a valid way in mitigating antigen loss to treat hematological and solid tumors.16,18,20, 21, 22 In this study, we also detected heterogeneously expressed EGFRvIII and EGFR in freshly resected glioma samples. EGFR was found to be more prominent than EGFRvIII expression in individual tumor cells, indicating that a meaningful clinical outcome may be achieved by targeting amplified, oncogenic wild-type EGFR instead of EGFRvIII.
A previous study confirmed that EGFRvIII CAR T cells can traffic to the site of EGFRvIII-positive tumors and secrete EGFR BiTEs into the microenvironment.23 Extending this observation, we adopted the EGFR conformation-specific 806 scFv to generate an 806BiTE. We also found IL13Rα2 to often be co-expressed in the same tumor with EGFR and EGFRvIII. Based on the TCGA data, both IL13Rα2 and EGFR expression positively correlated with WHO glioma grade. Although we cannot quantitively map out overlap and mutual exclusivity between each target, simultaneous targeting of IL13Rα2, EGFR, and EGFRvIII by T cells could result in a prolonged clinical response, as both IL13Rα2 targeting BiTE T cells and CAR T cells persistently controlled 75% target positive U87MG growth in an orthotopically implanted glioma mouse model. In bivalent constructs, BiTEs also mediated superior target specific responses compared to CAR T cells and established earlier control of tumor growth. Besides superior sensitivity to low target antigen expression, bivalent targeting using BiTEs demonstrated potency in mitigating the problem of tumor antigen heterogeneity.
Specifically, 806 binds to the CR1 domain of EGFR, which is masked in the inactive monomer state. Exposure of this epitope preferentially occurs under tumor-specific conditions, such as constitutive EGFR activation via EGFR amplification and EGFRvIII mutations.31,45 This conformational specificity makes 806BiTE bind to tumor associated oncogenic EGFR and EGFRvIII. Our group has previously confirmed 806CAR T cells successfully target other EGFR oncogenic mutations, including extracellular missense mutants EGFRA289V and EGFRR108K, while demonstrating little cytotoxicity against human astrocytes.28 Similarly, there was no overt response to astrocytes with 806BiTE T cells or 806BiTE-recruited UTD T cells. Given these results, BiTE-redirected T cells could be a tumor-specific approach when using a confirmation-specific scFv. In addition, 806BiTE T cells demonstrated a more potent response to EGFRvIII than C225BiTE T cells. C225BiTE T cells did respond to the EGFR present on 5077 cells, while 806BiTE T cells did not show a response. This result may be explained by the different binding affinity of 806 and C225 scFvs with EGFRvIII and EGFR,35,46 which is also concordant with our results of ELISA-based BiTE binding assays. In our study, BiTE binding on T cells was only detected in the presence of target antigen. That was attributed to the low affinity of the anti-CD3 scFv, in the range of KD = 10−7. In a previous study, BiTE binding on CD3 molecule of T cells was detectable at 10,000 ng/mL, but not at 400 ng/mL.47 The level of BiTE secretion by T cells was in the range of nanograms per milliliter, which was not able to be detected. In the presence of target antigen, BiTE binding on T cells was enhanced by the formation of immune synapses, which could be the reason of detectable by protein L staining. Consistent with a previous study, the property of BiTE binding facilitated BiTE-mediated specific responding.48
In summary, BiTE T cells demonstrated superior activation, sensitivity, and specificity in responding to their cognate antigen than the corresponding CAR construct, but may not be as persistent as CAR T cells. BiTE T cells and BiTE-CAR T cells are promising strategies in addressing the clinical problem of targeting solid tumor antigen heterogeneity and may result in paving the way for targeted T cell therapies.
Materials and methods
Cell lines and culture
Human GSC lines (5041, 5077, 5391, 4701, 4860, 5377, 5560, 4806, 4957, and 4892) were isolated from patient excised tumor tissue obtained under a University of Pennsylvania Institutional Review Board approved protocol and with patient written informed consent (Department of Neurosurgery, Perelman School of Medicine, Philadelphia, PA) and maintained in DMEM/Nutrient Mixture F12 Ham with penicillin/streptomycin, GlutaMAX-1, B27 minus A, EGFR, and basic fibroblast growth factor (Corning, Corning, NY). EGFR copy number amplifications and EGFRvIII detection was carried out as previously described, using the Center for Personalized Diagnostics at the University of Pennsylvania.32 U87MG was purchased from the American Type Culture Collection and maintained in MEM (Richter’s modification) plus GlutaMAX-1, HEPES, pyruvate and penicillin/streptomycin (Thermo Fisher Scientific, Carlsbad, CA), supplemented with 10% fetal bovine serum (FBS). GSC line 5077 was lentivirally transduced to express or co-express EGFRvIII, IL13Rα2, click beetle green (CBG) luciferase, and GFP under control of the EF-1α promoter. D270 glioma cells were grown and passaged in the right flanks of NSG mice. Human astrocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA) and maintained in culture for 3 to 7 passages in astrocyte medium (ScienCell Research Laboratories 1801, Carlsbad, CA), as directed by the vendor.
Vector constructs
The nucleic acid sequences of EGFR targeting scFv (806) or IL13Rα2 targeting scFv (Hu08) in a second-generation CAR construct or a BiTE construct were synthesized and ligated into pTRPE lentiviral vector with a T2A ribosomal skipping sequence and an mCherry gene (Twist Bioscience, San Francisco, CA). We synthesized 806 CAR sequences and 806 BiTE sequences with P2A ribosomal skipping sequences, digested them with NheI and XbaI, and ligated them into pTRPE vector of an Hu08 CAR T2A mCherry construct to generate bivalent constructs. Truncated Hu08 CAR sequences (Hu08TM; Hu08 scFv with leader, hinge, and transmembrane sequences of human CD8α) were digested with XbaI and SalI and ligated into the 806 BiTE and Hu08 CAR bivalent structure in the same enzyme sites to replace Hu08 CAR and mCherry genes. Hu08TM was used as a cell surface tag. C225 BiTE structure was digested with NheI and HpaI to replace the 806 BiTE in the Hu08TM tag construct. Sequences of the vector constructs have been published previously.49
Human T cell transduction and culture in vitro
Human T cells transduction and culture were performed as previously described.50 Briefly, isolated T cells were derived from leukapheresis products obtained from the Human Immunology Core at the University of Pennsylvania, using de-identified healthy donors under an institutional review board-approved protocol. T cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (Life Technologies, Carlsbad, CA) at a bead-to-cell ratio of 3:1. After 24-h stimulation, lentivirus was added into the culture media and thoroughly mixed to produce stably transduced CAR T cells. The concentration of the expanding human T cells was calculated on a Coulter Multisizer (Beckman Coulter, Brea, CA) and maintained at 1.0–2.0 × 106 cells/mL in R10 media (RPMI-1640 plus GlutaMAX-1, HEPES, pyruvate and penicillin/streptomycin (ThermoFisher Scientific, Carlsbad, CA), supplemented with 10% FBS) and 30 IU/mL recombinant human IL-2 (rhIL-2; ThermoFisher Scientific).
Flow cytometry
Fresh human GBM samples were minced and single cell suspensions were washed through a cell strainer (40 μm). Red blood cells were lysed with Ammonium-Chloride-Potassium Lysing Buffer (Lonza, Basel, Switzerland). The size and concentration of cells was measured on a Coulter Multisizer after washing with PBS. GBM cells were distinguished with live/dead viability stain (ThermoFisher Scientific), followed by human CD45 (clone HI30; BioLegend, San Diego, CA) stain. GSC lines were collected and centrifuged from culture in vitro. Cell pellet were re-suspended and digested into single cells in pre-heated Accutase (Sigma-Aldrich, Saint Louis, MO). Other tumor cell lines were detached from flask and digested into single cells with pre-heated Versene solution (ThermoFisher Scientific). Digested single cells were stained with live/dead viability stain. BV711/PE conjugated anti-IL13Rα2 (clone 47, BioLegend), PE-conjugated anti-EGFR (clone AY13, BioLegend), non-conjugated anti-EGFRvIII antibody (clone 3052, Novartis, Basil, Switzerland), and PE-conjugated anti-Rabbit IgG (clone Poly4064, BioLegend) secondary stains were used for detecting the targets. mCherry and Hu08TM were used as tags of T cell transduction. Cells expressing Hu08TM were stained with biotinylated protein L (GenScript, Piscataway, NJ) and streptavidin-coupled PE (BD Biosciences, Franklin Lakes, NJ).
In co-culture experiments, transduced or UTD T cells (2×105 cells per well in 100μL R10 media) were co-cultured with target cells (2×105 cells per well in 100μL R10 media) in 96-well round bottom tissue culture plates, at 37°C with 5% CO2 for 16 h. When conditioned media was used to stimulate UTD T cells, transduced or UTD T cells (4×105 cells in 200μL R10 media) were cultured in 96-well round bottom tissue culture plates at 37°C in 5% CO2 for 16 h. We took 160μL supernatant from each well and added it into UTD T cells (2×105 cells) co-cultured with target cells (2×105 cells) to reach 200μL/well. In T cell proliferation and phenotype assays, transduced or UTD T cells (5×105 cells per well in 500μL R10 media) were co-cultured with target cells (2.5×105 cells per well in 500μL R10 media) in 48-well flat bottom tissue culture plates. CFSE (ThermoFisher Scientific) staining was performed as per the manufacturer’s instructions. Target cells were irradiated with 10,000 rad ahead of co-culture with T cells. On day 2 and day 5, 2.5×105 irradiated target cells were added in each well. We replaced 500μL supernatant in each well with fresh R10 media every day from day 2. Human CD4+ and CD8+ T cells were distinguished with live/dead viability stain (ThermoFisher Scientific), followed by human CD3 and CD8 (clone OKT3 and clone SK1; BioLegend) stain. Biotinylated protein L (GenScript) and the addition of streptavidin-coupled PE (BD Biosciences) was used to detect BiTEs’ binding to T cells. APC conjugated anti-human CD69 (clone FN50, BioLegend) was used to detect the T cell stimulation. BV711-conjugated anti-human PD-1 (clone EH12.2H7, BioLegend), PE-conjugated anti-human CTLA-4 (clone L3D10, BioLegend) and APC-conjugated anti-human TIM-3 (clone F38-2E2, BioLegend) were used to detect the expression of checkpoints. BV711-conjugated anti-human CD45RA (clone HI100, BioLegend), APC-conjugated anti-human CCR7 (clone G043H7, BioLegend) and PE-conjugated anti-human CD62L (clone DREG-56, BioLegend) were used to detect T cell phenotypes. Isotype antibodies were used as an additional control. Before and after each staining, cells were washed twice with PBS containing 2% FBS (FACS buffer). Fluorescence was assessed using a BD LSRFortessa flow cytometer and data were analyzed with FlowJo software.
Intracellular cytokine analysis
To stimulate UTD T cells, supernatants of T cells were collected in 96-well round bottom tissue culture plates as described above and added into UTD T cells (2×105 cells) co-cultured with target cells (2×105 cells) in 96-well round bottom tissue culture plates in R10 media with the presence of Golgi inhibitors monensin and brefeldin A (BD Bioscience) to reach 200μL/well. In transduced T cells stimulation assay, transduced or UTD T cells (2×105 cells in 160μL R10 media) were cultured in 96-well round bottom tissue culture plates at 37°C in 5% CO2 for 16 h. Target cells (2×105 cells in 40μL) with Golgi inhibitors were added into each well. After 16 h, cells were washed, stained with live/dead viability stain, followed by surface staining for human CD3 and CD8 (clone OKT3 and clone SK1, BioLegend), then fixed and permeabilized, and intracellularly stained for human IFNγ (clone B27, BioLegend), IL2 (clone MQ1-17H12, Fisher Scientific) and TNFα (clone Mab11, BioLegend). Cells were analyzed by flow cytometry (BD LSRFortessa) and gated on live, single-cell lymphocytes and CD3-positive lymphocytes.
Bioluminescence cytotoxicity assay
We seeded 1×104 target cells transduced with CBG luciferase in 96-well plates with opaque walls. In the assays of supernatants, 5×104 UTD T cells were added in each well. Supernatants of specific volumes (160μL, 80μL, 40μL, 20μL, and so on) plus R10 media were added into each well to reach 200μL in total. No supernatant was added in control wells. In the assays of transduced T cells, effector cells were added at different E:T ratios, as indicated in each figure, to reach 200μL per well. No T cells were added in control wells. Plates were incubated at 37°C in 5% CO2 in an incubator. At the end of the incubation period, 15μg D-Luciferin (Gold Biotechnology, St. Louis, MO) in 5μL PBS was added in each well and incubated at room temperature for 10 min. Luminescence from the assay plates was measured with BioTek Synergy H4 hybrid multi-model microplate reader. Percentage specific lysis was calculated as: (control counts – sample counts)/control counts × 100%.
Mouse model
All mouse experiments were conducted according to Institutional Animal Care and Use Committee (IACUC)-approved protocols and as previously described.50 For orthotopic model, 5 × 105 U87MG cells were implanted intracranially into 6- to 8-week-old NSG mice. The surgical implants were done using a stereotactic surgical setup with tumor cells implanted 2 mm right and 2 mm anterior to the lambda and 2 mm into the brain. Tumor progression was evaluated by luminescence emission on a Xenogen IVIS spectrum after intraperitoneal D-luciferin (Gold Biotechnology) injection according to the manufacturer’s directions. In subcutaneous model, NSG mice were injected with 5×105 D270 tumor cells subcutaneously in 100 μL PBS on day 0. Tumor was measured by calipers in length and width for the duration of the experiment. Tumor size was calculated as the area of tumor by multiplying the two dimensions. T cells were injected in 100 μL PBS intravenously via the tail vein 8 days after tumor implantation. Survival was followed over time until a predetermined IACUC-approved endpoint was reached.
Statistical analysis
Data are presented as means ± standard deviation. Target detection on fresh tissue was analyzed with two-tail paired student t test. RNA-seq data from TCGA was analyzed with Kruskal-Wallis test. Survival curves were analyzed with Kaplan-Meier (log rank test). The remaining experiments were analyzed with one-way ANOVA with post hoc Tukey test to compare the differences in each group. For the in vivo tumor study, linear regression was used to test for significant differences between the experimental groups. Survival, based on time to experimental endpoint, was plotted using a Kaplan-Meier curve. All statistical analyses were performed with Prism software version 9.0 (GraphPad, La Jolla, CA).
Acknowledgments
The authors thank the Human Immunology Core at the University of Pennsylvania for providing T cells for the described work, the Stem Cell and Xenograft Core at the University of Pennsylvania for assistance with the animal work, the Small Animal Imaging Facility at the University of Pennsylvania for the bioluminescence imaging, and the Pathology Clinical Service Center of Penn Medicine for immunohistochemistry staining of IL13Rα2 and EGFRvIII, as well as Laura A. Johnson, Donald L. Siegel, Avery D. Posey, Jr., and Alina C. Boesteanu for the technical support on molecular biology. This work was supported by funding from GBM Translational Center of Excellence (D.M.O.), The Templeton Family Initiative in Neuro-Oncology (D.M.O.), The Maria and Gabriele Troiano Brain Cancer Immunotherapy Fund (D.M.O.), National Natural Science Foundation of China (U20A20383, 81772678; Z.L.), Heilongjiang Postdoctoral Scientific Research Developmental Fund (LBH-Q20129; Y.Y.), Innovation Grant of Harbin Medical University (2021-KYYWF-0221; Y.Y.), and Innovation Grant of the First Affiliated Hospital of Harbin Medical University (2020L03; Y.Y.).
Author contributions
D.M.O., Z.A.B., and Y.Y. designed the experiments. Y.Y., J.L.R., N.L., R.T., M.P.N., L.H., L.Z., J.V.Z., M.T.J., D.K., L.H., Y.Z., T.W., E.X.J., Y.L., H.L., J.Q., X.Z., Z.L., Z.A.B., and D.M.O. performed the experiments. D.M.O., Z.A.B., Z.L., and Y.Y. provided funding. All the authors contributed to the writing and editing of the manuscript.
Declaration of interests
Y.Y., R.T., Z.A.B., and D.M.O. are on patent filings related to the research presented here. D.M.O. received monetary support from Tmunity Therapeutics for related laboratory work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.05.011.
Contributor Information
Zhiguo Lin, Email: linzhiguo@hotmail.com.
Donald M. O’Rourke, Email: donald.orourke@pennmedicine.upenn.edu.
Supplemental information
References
- 1.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuster S.J., Svoboda J., Chong E.A., Nasta S.D., Mato A.R., Anak O., Brogdon J.L., Pruteanu-Malinici I., Bhoj V., Landsburg D., et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 2017;377:2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chong E.A., Ruella M., Schuster S.J., Lymphoma Program Investigators at the University of, Pennsylvania Five-year outcomes for refractory B-cell lymphomas with CAR T-cell therapy. N. Engl. J. Med. 2021;384:673–674. doi: 10.1056/NEJMc2030164. [DOI] [PubMed] [Google Scholar]
- 5.Raje N., Berdeja J., Lin Y., Siegel D., Jagannath S., Madduri D., Liedtke M., Rosenblatt J., Maus M.V., Turka A., et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N. Engl. J. Med. 2019;380:1726–1737. doi: 10.1056/NEJMoa1817226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goebeler M.E., Bargou R.C. T cell-engaging therapies - BiTEs and beyond. Nat. Rev. Clin. Oncol. 2020;17:418–434. doi: 10.1038/s41571-020-0347-5. [DOI] [PubMed] [Google Scholar]
- 8.Topp M.S., Gokbuget N., Stein A.S., Zugmaier G., O'Brien S., Bargou R.C., Dombret H., Fielding A.K., Heffner L., Larson R.A., et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 9.Kantarjian H., Stein A., Gokbuget N., Fielding A.K., Schuh A.C., Ribera J.M., Wei A., Dombret H., Foa R., Bassan R., et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017;376:836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goebeler M.E., Knop S., Viardot A., Kufer P., Topp M.S., Einsele H., Noppeney R., Hess G., Kallert S., Mackensen A., et al. Bispecific T-cell engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non-Hodgkin lymphoma: final results from a phase I study. J. Clin. Oncol. 2016;34:1104–1111. doi: 10.1200/JCO.2014.59.1586. [DOI] [PubMed] [Google Scholar]
- 11.Dufner V., Sayehli C.M., Chatterjee M., Hummel H.D., Gelbrich G., Bargou R.C., Goebeler M.E. Long-term outcome of patients with relapsed/refractory B-cell non-Hodgkin lymphoma treated with blinatumomab. Blood Adv. 2019;3:2491–2498. doi: 10.1182/bloodadvances.2019000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iwahori K., Kakarla S., Velasquez M.P., Yu F., Yi Z., Gerken C., Song X.T., Gottschalk S. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Mol. Ther. 2015;23:171–178. doi: 10.1038/mt.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pituch K.C., Zannikou M., Ilut L., Xiao T., Chastkofsky M., Sukhanova M., Bertolino N., Procissi D., Amidei C., Horbinski C.M., et al. Neural stem cells secreting bispecific T cell engager to induce selective antiglioma activity. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2015800118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X., Barrett D.M., Jiang S., Fang C., Kalos M., Grupp S.A., June C.H., Zhao Y. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood Cancer J. 2016;6:e430. doi: 10.1038/bcj.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Majzner R.G., Mackall C.L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 16.Fry T.J., Shah N.N., Orentas R.J., Stetler-Stevenson M., Yuan C.M., Ramakrishna S., Wolters P., Martin S., Delbrook C., Yates B., et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamieh M., Dobrin A., Cabriolu A., van der Stegen S.J.C., Giavridis T., Mansilla-Soto J., Eyquem J., Zhao Z., Whitlock B.M., Miele M.M., et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112–116. doi: 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider D., Xiong Y., Wu D., Hu P., Alabanza L., Steimle B., Mahmud H., Anthony-Gonda K., Krueger W., Zhu Z., et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abc6401. [DOI] [PubMed] [Google Scholar]
- 19.O'Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J.D., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bielamowicz K., Fousek K., Byrd T.T., Samaha H., Mukherjee M., Aware N., Wu M.F., Orange J.S., Sumazin P., Man T.K., et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20:506–518. doi: 10.1093/neuonc/nox182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hegde M., Corder A., Chow K.K., Mukherjee M., Ashoori A., Kew Y., Zhang Y.J., Baskin D.S., Merchant F.A., Brawley V.S., et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 2013;21:2087–2101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hegde M., Mukherjee M., Grada Z., Pignata A., Landi D., Navai S.A., Wakefield A., Fousek K., Bielamowicz K., Chow K.K., et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016;126:3036–3052. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi B.D., Yu X., Castano A.P., Bouffard A.A., Schmidts A., Larson R.C., Bailey S.R., Boroughs A.C., Frigault M.J., Leick M.B., et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019;37:1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 24.Omuro A., DeAngelis L.M. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310:1842–1850. doi: 10.1001/jama.2013.280319. [DOI] [PubMed] [Google Scholar]
- 25.Wen P.Y., Kesari S. Malignant gliomas in adults. N. Engl. J. Med. 2008;359:492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 26.Stupp R., Taillibert S., Kanner A.A., Kesari S., Steinberg D.M., Toms S.A., Taylor L.P., Lieberman F., Silvani A., Fink K.L., et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314:2535–2543. doi: 10.1001/jama.2015.16669. [DOI] [PubMed] [Google Scholar]
- 27.Yin Y., Boesteanu A.C., Binder Z.A., Xu C., Reid R.A., Rodriguez J.L., Cook D.R., Thokala R., Blouch K., McGettigan-Croce B., et al. Checkpoint blockade reverses anergy in IL-13rα2 humanized scFv-based CAR T cells to treat murine and canine gliomas. Mol. Ther. Oncolytics. 2018;11:20–38. doi: 10.1016/j.omto.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thokala R., Binder Z.A., Yin Y., Zhang L., Zhang J.V., Zhang D.Y., Milone M.C., Ming G., Song H., O’Rourke D.M. High-affinity chimeric antigen receptor with cross-reactive scFv to clinically relevant EGFR oncogenic isoforms. Front. Oncol. 2021;11:664236. doi: 10.3389/fonc.2021.664236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ravanpay A.C., Gust J., Johnson A.J., Rolczynski L.S., Cecchini M., Chang C.A., Hoglund V.J., Mukherjee R., Vitanza N.A., Orentas R.J., Jensen M.C. EGFR806-CAR T cells selectively target a tumor-restricted EGFR epitope in glioblastoma. Oncotarget. 2019;10:7080–7095. doi: 10.18632/oncotarget.27389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orellana L., Thorne A.H., Lema R., Gustavsson J., Parisian A.D., Hospital A., Cordeiro T.N., Bernado P., Scott A.M., Brun-Heath I., et al. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. U S A. 2019;116:10009–10018. doi: 10.1073/pnas.1821442116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrett T.P.J., Burgess A.W., Gan H.K., Luwor R.B., Cartwright G., Walker F., Orchard S.G., Clayton A.H.A., Nice E.C., Rothacker J., et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc. Natl. Acad. Sci. U S A. 2009;106:5082–5087. doi: 10.1073/pnas.0811559106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nasrallah M.P., Binder Z.A., Oldridge D.A., Zhao J., Lieberman D.B., Roth J.J., Watt C.D., Sukhadia S., Klinman E., Daber R.D., et al. Molecular neuropathology in practice: clinical profiling and integrative analysis of molecular alterations in glioblastoma. Acad. Pathol. 2019;6 doi: 10.1177/2374289519848353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P., et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roccograndi L., Binder Z.A., Zhang L., Aceto N., Zhang Z., Bentires-Alj M., Nakano I., Dahmane N., O’Rourke D.M. SHP2 regulates proliferation and tumorigenicity of glioma stem cells. J. Neurooncol. 2017;135:487–496. doi: 10.1007/s11060-017-2610-x. [DOI] [PubMed] [Google Scholar]
- 35.Li S., Schmitz K.R., Jeffrey P.D., Wiltzius J.J., Kussie P., Ferguson K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–311. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Sternjak A., Lee F., Thomas O., Balazs M., Wahl J., Lorenczewski G., Ullrich I., Muenz M., Rattel B., Bailis J.M., Friedrich M. Preclinical assessment of AMG 596, a bispecific T-cell engager (BiTE) immunotherapy targeting the tumor-specific antigen EGFRvIII. Mol. Cancer Ther. 2021;20:925–933. doi: 10.1158/1535-7163.MCT-20-0508. [DOI] [PubMed] [Google Scholar]
- 37.Blanco B., Compte M., Lykkemark S., Sanz L., Alvarez-Vallina L. T cell-redirecting strategies to 'STAb' tumors: beyond CARs and bispecific antibodies. Trends Immunol. 2019;40:243–257. doi: 10.1016/j.it.2019.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Bonifant C.L., Szoor A., Torres D., Joseph N., Velasquez M.P., Iwahori K., Gaikwad A., Nguyen P., Arber C., Song X.T., et al. CD123-Engager T cells as a novel immunotherapeutic for acute myeloid leukemia. Mol. Ther. 2016;24:1615–1626. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Velasquez M.P., Torres D., Iwahori K., Kakarla S., Arber C., Rodriguez-Cruz T., Szoor A., Bonifant C.L., Gerken C., Cooper L.J.N., et al. T cells expressing CD19-specific engager molecules for the immunotherapy of CD19-positive malignancies. Sci. Rep. 2016;6:27130. doi: 10.1038/srep27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Offner S., Hofmeister R., Romaniuk A., Kufer P., Baeuerle P.A. Induction of regular cytolytic T cell synapses by bispecific single-chain antibody constructs on MHC class I-negative tumor cells. Mol. Immunol. 2006;43:763–771. doi: 10.1016/j.molimm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 41.Claus C., Ferrara C., Xu W., Sam J., Lang S., Uhlenbrock F., Albrecht R., Herter S., Schlenker R., Husser T., et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019;11:eaav5989. doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Correnti C.E., Laszlo G.S., de van der Schueren W.J., Godwin C.D., Bandaranayake A., Busch M.A., Gudgeon C.J., Bates O.M., Olson J.M., Mehlin C., Walter R.B. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia. 2018;32:1239–1243. doi: 10.1038/s41375-018-0014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hegde P.S., Chen D.S. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52:17–35. doi: 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 44.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gan H.K., Burgess A.W., Clayton A.H.A., Scott A.M. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012;72:2924–2930. doi: 10.1158/0008-5472.CAN-11-3898. [DOI] [PubMed] [Google Scholar]
- 46.Johns T.G., Adams T.E., Cochran J.R., Hall N.E., Hoyne P.A., Olsen M.J., Kim Y.S., Rothacker J., Nice E.C., Walker F., et al. Identification of the epitope for the epidermal growth factor receptor-specific monoclonal antibody 806 reveals that it preferentially recognizes an untethered form of the receptor. J. Biol. Chem. 2004;279:30375–30384. doi: 10.1074/jbc.M401218200. [DOI] [PubMed] [Google Scholar]
- 47.Dreier T., Lorenczewski G., Brandl C., Hoffmann P., Syring U., Hanakam F., Kufer P., Riethmuller G., Bargou R., Baeuerle P.A. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. Int. J. Cancer. 2002;100:690–697. doi: 10.1002/ijc.10557. [DOI] [PubMed] [Google Scholar]
- 48.Brischwein K., Parr L., Pflanz S., Volkland J., Lumsden J., Klinger M., Locher M., Hammond S.A., Kiener P., Kufer P., et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007;30:798–807. doi: 10.1097/CJI.0b013e318156750c. [DOI] [PubMed] [Google Scholar]
- 49.O'Rourke D., Yin Y., Johnson L., Binder Z., Thokala R. U.S.P.a.T. Office; 2021. Synthetic CARs to Treat IL13R-alpha-2 Positive Human and Canine Tumors. 20210128617. [Google Scholar]
- 50.Johnson L.A., Scholler J., Ohkuri T., Kosaka A., Patel P.R., McGettigan S.E., Nace A.K., Dentchev T., Thekkat P., Loew A., et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015;7:275ra222. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.