

Abstract
Genome sequencing of the thermophilic archaeon Pyrococcus horikoshii OT3 revealed a gene which had high sequence similarity to the gene encoding the carboxypeptidase of Sulfolobus solfataricus and also to that encoding the aminoacylase from Bacillus stearothermophilus. The gene from P. horikoshii comprises an open reading frame of 1,164 bp with an ATG initiation codon and a TGA termination codon, encoding a 43,058-Da protein of 387 amino acid residues. However, some of the proposed active-site residues for carboxypeptidase were not found in this gene. The gene was overexpressed in Escherichia coli with the pET vector system, and the expressed enzyme had high hydrolytic activity for both carboxypeptidase and aminoacylase at high temperatures. The enzyme was stable at 90°C, with the highest activity above 95°C. The enzyme contained one bound zinc ion per one molecule that was essential for the activity. The results of site-directed mutagenesis of Glu367, which corresponds to the essential Glu270 in bovine carboxypeptidase A and the essential Glu in other known carboxypeptidases, revealed that Glu367 was not essential for this enzyme. The results of chemical modification of the SH group and site-directed mutagenesis of Cys102 indicated that Cys102 was located at the active site and was related to the activity. From these findings, it was proven that this enzyme is a hyperthermostable, bifunctional, new zinc-dependent metalloenzyme which is structurally similar to carboxypeptidase but whose hydrolytic mechanism is similar to that of aminoacylase. Some characteristics of this enzyme suggested that carboxypeptidase and aminoacylase might have evolved from a common origin.
Pyrococcus horikoshii OT3 is one of the thermophilic archaea collected from a volcanic vent in the Okinawa Trough (16, 20, 21). This archaeon's optimum growth temperature ranges from 90 to 105°C. Most of the proteins from P. horikoshii are expected to be thermostable and active at high temperatures.
Carboxypeptidase (CP) (peptidyl-l-amino-acid hydrolase; EC 3.4.12.1), which hydrolyzes the peptide bond at the C termini of peptides and proteins, is widely distributed in many organisms. Mammalian carboxypeptidase A (CPA) and CPB have been studied in detail (1, 11, 24). A thermostable CP is useful for high-temperature analysis of the C-terminal amino acid sequences of proteins. Recently, several thermostable CPs from the thermophilic bacteria Thermoactinomyces vulgaris (35, 36) and Thermus aquaticus (27, 28, 29) and the thermophilic archaea Sulfolobus solfataricus (12, 13, 38) and Pyrococcus furiosus (8) have been characterized. Using genome sequencing for P. horikoshii (20, 21), we found two kinds of genes encoding CP-homologous proteins. One protein has 40% identity to the CP of T. aquaticus (28). The other has approximately 45% identity to the CP of S. solfataricus (13) and aminoacylase (N-acyl-l-amino acid amidohydrolase; EC 3.5.1.14) (3) of Bacillus stearothermophilus (33). In the latter protein, some of the amino acids which corresponded to the essential active-site residues in CP (11, 13) were not found. The hyperthermostable aminoacylase is useful for the industrial production of stereoisomers from racemates (9, 10). In addition, the aminoacylase activity in CP has biological significance from the viewpoint of enzyme evolution (4, 33). There has, however, been no report of an enzyme which has significant activities of both CP and aminoacylase. In this study, we tried to clone the latter from P. horikoshii and express the protein in Escherichia coli to examine its characteristics.
MATERIALS AND METHODS
Cloning and expression of the gene.
The gene that is homologous to the S. solfataricus CP gene was amplified using PCR with two primers containing unique restriction sites (NdeI and XhoI). The synthesis of DNA primers was performed at the custom service center of Takara Shuzo (Otsu, Shiga, Japan). Amplification by PCR was carried out at 94°C for 1 min, 55°C for 2 min, and 72°C for 3 min for 35 cycles using Pfu DNA polymerase from Takara Shuzo. The amplified gene was hydrolyzed using the restriction enzymes and inserted into a pET11a vector—the BamHI site was changed to a XhoI site with site-directed mutagenesis—cut by the same restriction enzymes. The DNA sequencing was carried out with an ABI model 373 sequencer (Applied Biosystems, Foster City, Calif.). The amplified gene was expressed using the pET11a vector system in the host E. coli BL21(DE3) according to the manufacturer's instructions (Novagen, Madison, Wis.). The transformant cells were grown in 2YT medium (1% yeast extract, 1.6% tryptone, and 0.5% NaCl) containing CaCl2 (0.2 mM), CoCl2 (0.2 mM), MnCl2 (0.2 mM), ZnCl2(0.2 mM), and ampicillin (100 μg/ml) at 37°C. After incubation with shaking at 37°C until the optical density at 600 nm reached 0.6 to 1.0, the induction of the recombinant protein was carried out by adding isopropyl-β-d-thiogalactopyranoside at a final concentration of 1 mM and shaking for 4 h at 37°C.
Purification of the recombinant enzyme from E. coli.
After induction, the transformant cells were harvested by centrifugation and frozen at −20°C. The cells were disrupted by sonication for 10 min in 50 mM sodium phosphate buffer (pH 6.0) containing 0.6 M NaCl at room temperature. After incubation with DNase I for 30 min at room temperature, the crude extract was heated at 85°C for 30 min. The supernatant obtained by centrifugation was dialyzed against 50 mM Tris-HCl buffer (pH 8.0). The dialyzed sample was loaded on a HiTrap Q column (Pharmacia, Uppsala, Sweden). The column was washed with 50 mM Tris-HCl buffer (pH 8.0) and eluted with a linear NaCl gradient (0 to 1.0 M in the same buffer). The fractions which showed a protein with the molecular mass calculated from the sequence of the enzyme were pooled and concentrated using a Centricon 10 filter (Amicon, Beverly, Mass.). The concentrated solution was loaded on a HiLoad Superdex 200 column (Pharmacia) and eluted with 100 mM Tris-HCl buffer (pH 8.0) containing 1.0 M NaCl. The fractions demonstrating only one band, with a molecular mass of 43 kDa determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), were collected and used for the detailed characterization of the enzyme. The concentration of the enzyme was determined using a Coomassie protein assay reagent (Pierce Chemical Company, Rockford, Ill.) with bovine serum albumin as the standard protein. The sequencing of the protein was performed at the custom service center of Takara Shuzo
Molecular mass determination.
The molecular mass of the enzyme was determined using SDS-PAGE performed on a 10-to-15% gradient Phast gel with the Phast System (Pharmacia). The sample solution was mixed with the SDS sample buffer and incubated at 100°C for 15 min before sample application. The molecular mass was also determined using high-performance liquid chromatography (HPLC) performed on a Superdex 200 column of Pharmacia. The elution was carried out using 100 mM Tris-HCl buffer (pH 8.0) containing 1.0 M NaCl at 1.5 ml/min.
Enzyme assay.
Carbobenzoxyl (Cbz) amino acids (Cbz-F, Cbz-G, and Cbz-R) and peptides (Cbz-G-A, Cbz-G-F, Cbz-G-M, Cbz-G-N, Cbz-G-W, and Cbz-G-G-F), and N-acetyl (Ac) amino acids were purchased from Sigma (St. Louis, Mo.) or Bachem (Bubendorf, Switzerland). Other peptides (G-F, G-G-F, G-G-G-F, R-Y-M-G-F, F-R-Y-M-G, Ac-G-F, Ac-G-G-F, Ac-G-G-G-F, Cbz-G-R, Cbz-G-Y, and Cbz-Y) were purchased from Peptide Institute (Minou, Osaka, Japan). The hydrolytic activity of the enzyme was measured using the peptides, Cbz amino acids, and Ac amino acids as substrates. The enzyme (0.1 to 1.0 μg/ml) was incubated at 85°C with the substrates in 50 mM sodium phosphate buffer (pH 7.5) containing 5% N,N-dimethylformamide (DMF; solvent for the substrates). Hydrolytic activities for the above substrates were measured by detecting the exposed α-NH2 group using the cadmium-ninhydrin colorimetric method (14). One unit of enzyme activity is defined as the amount of enzyme which hydrolyzes 1 μmol of substrate per min. The values of Km and kcat were determined by the nonlinear least-squares method with the Taylor expansion (34). The products from the peptides were examined using HPLC on a TSKgel octyldecyl silane (ODS)-80Ts column (4.6 mm [inside diameter] by 25 cm; Tosoh, Tokyo, Japan). The flow rate was 0.7 ml/min with 95% water, 5% acetonitrile, and 0.1% trifluoroacetic acid. The products were detected with a UV monitor (at 230 nm). Analysis of the amino acids released from the peptides was performed using an amino acid analyzer (L8500A; Hitachi, Tokyo, Japan) after the hydrolytic enzyme reaction was stopped by the addition of acetic acid to a final concentration of 1 N.
Chemical modification.
Chemical modification of Cys residues in the enzyme was carried out with Ellman reagent [DTNB; 5,5′-dithiobis(2-nitrobenzoic acid)] (15). The enzyme solution (1 mg/ml) was incubated with DTNB (0.5 mM) in 50 mM sodium phosphate buffer (pH 8.0) at 30°C. The number of modified Cys residues was determined spectrophotometrically at 412 nm. After the reaction, excess Cys was added to the reaction mixture and the modified enzyme was dialyzed against 50 mM Tris-HCl buffer (pH 8.0) at 4°C.
Preparation of the mutants.
Construction of the mutant enzymes with the mutations E367Q (Glu367 →Gln), C102A, and C102S was performed by site-directed mutagenesis using PCR (31). The entire region of the DNA fragment was sequenced to verify that only the expected mutation had occurred. The expression and purification of the mutant enzymes were performed using the same methods as for the wild type.
Analysis for bound metal ions.
The purified enzyme was analyzed for bound metals by inductively coupled plasma atomic emission spectroscopy (ICP-AES) (model IRIS AP; TJASolutions, Franklin, Mass.). The purified enzyme was dialyzed against 50 mM sodium phosphate buffer (pH 7.5). The dialyzed enzyme (0.5 mg/ml) was used for the analysis, and the amount of zinc was calculated using a zinc standard solution.
Nucleotide sequence accession number.
The sequence reported in this paper has been deposited in the DDBJ/GenBank/EMBL DNA databases with accession number AB009503.
RESULTS
Expression of the enzyme.
Genome sequencing of P. horikoshii revealed two genes (PH0465 and PH1043) having similarity to the CP gene. In this study, we cloned one (PH1043) of them. The open reading frame of 1,164 bp was preceded by AT-rich regions in which a putative ribosome-binding site, GGCGAT, at position −6 and a putative promoter consensus, TTAAAG, at position −31 from the ATG initiation site were found (DDBJ/GenBank/EMBL DNA databases, accession number AB009503). This consensus resembles the eukaryotic TATA box and has been confirmed to be the archaeal consensus sequence TT(A/T)(T/A)AX, as determined by analysis of over 80 archaeal promoters (32). The amino acid sequence predicted from the gene had approximately 45% identity to CP from S. solfataricus (Fig. 1) (13); however, most of the proposed active-site residues of CP (11, 13) are not found in this amino acid sequence. In addition, the sequence had about 45% identity to aminoacylase from B. stearothermophilus (33) (Fig. 1). In the sequence and in the CP from S. solfataricus (CPS), seven major homology blocks (B1 to B7) in CP, assigned by Colombo et al. (13), were well conserved (Fig. 1). In the sequence and aminoacylase from B. stearothermophilus (33), the last two blocks (B6 and B7) were not conserved (Fig. 1).
FIG. 1.

Sequence comparison of the PP (CP and aminoacylase activities) from P. horikoshii, the CP from S. solfataricus, and the aminoacylase from B. stearothermophilus (top, middle, and bottom, respectively). Dashes indicate gaps. Numbers on the left represent the position of the first residue in the original sequence. Conserved residues between PP and CPS and among the three enzymes are marked with pluses and asterisks, respectively. The seven major homology blocks (B1 through B7) in CPs were assigned by Colombo et al. (13), and the putative residues participating in the activity are in bold.
The gene was amplified with PCR, and the expressed enzyme was purified from 2 liters of culture, yielding 3.1 mg of a 43-kDa protein (as determined by SDS-PAGE [Fig. 2]). The first 15 amino acid residues of the enzyme were determined by sequence analysis of the N terminus. A Met residue at the N terminus of the nascent polypeptide was detected, and the N-terminal sequence was identical to that anticipated from the nucleotide sequence. The molecular mass of the purified enzyme, as determined by SDS-PAGE, was consistent with that calculated from the sequence (43,058 Da). The molecular mass determined by HPLC was 95 kDa.
FIG. 2.

SDS-PAGE (10-to-15% gradient gel) of the purified enzyme (lane 1). The molecular mass standards (lane 2) were phosphorylase b (94 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), carbonic anhydrase (30 kDa), soybean trypsin inhibitor (20 kDa), and α-lactoalbumin (14.4 kDa)
Enzyme activity and substrate specificity.
At 85°C and pH 7.5, crude extracts of E. coli BL21(DE3) not transformed with the cloned gene had no hydrolytic activity for a variety of different peptides and Cbz amino acids, whereas the purified enzyme exhibited hydrolytic activity for them. The rates of these reactions were proportional to the enzyme concentrations examined, and the hydrolyzed substrate was less than 15% of the total substrate. From the peptide substrates Ac-G-F, G-F, Cbz-G-F, Cbz-G-G-F, G-G-F, G-G-G-F, Ac-G-G-F and Ac-G-G-G-F, only the release of F was detected by HPLC analysis, less than 10% of the substrate hydrolyzed. Table 1 shows the substrate specificity of the enzymatic activity, and less than 10% of the substrate hydrolyzed. In Table 1, the specific activity of the hydrolysis at the cleavage sites is shown. The order of amino acids released from the peptides R-Y-M-G-F and F-R-Y-M-G was examined at 85°C and pH 7.5. This enzyme released amino acids sequentially from the C-terminal end of the substrates (data not shown) without a significant multiple-attack mechanism (2). During the reaction, endo-type peptidase activity was not observed by HPLC analysis. From these results, it was concluded that the enzyme has peptidase activity (exo-type peptidase from the C terminus) at high temperatures. This exo-type peptidase (herein referred to as PP) displayed approximately 100-fold greater hydrolytic activity for Cbz-F than Cbz-G-G-F (Table 1). The release of F from Ac-G-F and G-F was faster than that from Cbz-F, G-G-F, and G-G-G-F by PP. The hydrolytic activity for the above substrates is referred to CP activity. In addition to the above substrates, N-acetyl-l-amino acids (Ac amino acids), but not N-acetyl-d-amino acids, were also hydrolyzed by PP at high temperatures. HPLC analysis of Ac peptide products revealed that an Ac residue was not released from Ac peptides by PP. It was concluded that PP also had N-acyl-l-amino acid amidohydrolase activity. This activity is referred to as aminoacylase activity. Table 2 shows the kinetic parameters of the activities. The affinity for Cbz-F was similar to that for Ac-F, whereas the affinity for the peptides was independent of their length. The optimum pH of both the CP and aminoacylase activities at 85°C was around pH 7.5. The temperature-dependent activity of PP was examined for 10 min using Cbz-G-F and Ac-M as substrates in 50 mM phosphate buffer (pH 7.5) containing 0.5 mM ZnCl2 and 5% DMF. The highest CP and aminoacylase activities of PP were observed at temperatures over 95°C, and no decrease of the activities was observed at 90°C for 24 to 48 h.
TABLE 1.
Substrate specificity of PP for Cbz peptides, Cbz amino acids, peptides, Ac peptides, and Ac amino acidsa
| Substrate | Activity (μmol/min/mg of enzyme) | Relative activity (%) |
|---|---|---|
| 10 mM | ||
| ↓ | ||
| Cbz-F | 55.9 | 47 |
| ↓ | ||
| Cbz-G-F | 119 | 100 |
| ↓ | ||
| Cbz-G-G-F | 0.571 | 0.48 |
| ↓ | ||
| Cbz-G | 63.0 | 53 |
| ↓ | ||
| Cbz-R | 0.014 | 0.01 |
| ↓ | ||
| G-F | 107b | 90 |
| ↓ | ||
| G-G-F | 1.21b | 1.0 |
| ↓ | ||
| G-G-G-F | 0.551b | 0.46 |
| ↓ | ||
| Ac-G-F | 105a | 88 |
| ↓ | ||
| Ac-G-G-F | 0.773b | 0.65 |
| ↓ | ||
| Ac-G-G-G-F | 0.48b | 0.40 |
| 5 mM | ||
| ↓ | ||
| Ac-M | 38.1 | 43 |
| ↓ | ||
| Ac-Y | 37.3 | 29 |
| ↓ | ||
| Ac-F | 24.9 | 27 |
| ↓ | ||
| Ac-A | 10.5 | 15 |
| ↓ | ||
| Ac-W | 1.99 | 2.7 |
| ↓ | ||
| Ac-G | 7.40 | 2.1 |
The hydrolytic reaction was carried out at 85°C in 50 mM phosphate buffer (pH 7.5) containing 5% DMF. The cleavage site for the substrate is shown by the arrow.
The hydrolytic activity (release of F) was measured by HPLC.
TABLE 2.
Catalytic parameters of wild-type PP, E367Q, C102A, and C102S for Cbz peptides and Ac amino acidsa
| Substrate | Wild-type PP
|
E367Q
|
C102A
|
C102S
|
||||
|---|---|---|---|---|---|---|---|---|
| Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | |
| Cbz-F | 7.35 ± 1.40 | 91.4 ± 11.6 | ||||||
| Cbz-G-F | 4.51 ± 0.90 | 101 ± 6.6 | 5.16 ± 0.99 | 150 ± 9.2 | ND | <0.001 | 7.10 ± 1.10 | 0.0108 ± 0.0030 |
| Gcz-G-G-F | 3.10 ± 0.51 | 0.792 ± 0.19 | ||||||
| Ac-F | 5.20 ± 0.49 | 34.0 ± 4.2 | 6.64 ± 0.90 | 37.3 ± 5.1 | ND | <0.001 | 6.20 ± 1.90 | 0.0208 ± 0.0051 |
| Ac-M | 4.18 ± 0.53 | 45.6 ± 4.9 | ||||||
| G-F | 5.07 ± 0.99 | 103 ± 7.9 | ||||||
The hydrolytic reaction was measured at 85°C in 50 mM sodium phosphate buffer (pH 7.5) containing 5% DMF, and the substrate that hydrolyzed was less than 10% of the total substrate. The parameters for Ac-F and Ac-M were measured using less than 12 mM substrate. Values are means ± standard deviations for five determinations. ND, not determined.
Figure 3 shows the Lineweaver-Burk plot of CP and aminoacylase activities in PP. In the case of aminoacylase activity, the activity for Ac-M decreased remarkably at concentrations of Ac-M higher than 15 mM (Fig. 3B). Even at high concentrations of the substrate, products other than the hydrolytic products from Ac-M were not detected using HPLC analysis. No other condensation reaction was observed with HPLC. Similar phenomena were observed for Ac-Y, Ac-F, Ac-A, Ac-W, Ac-G, and Ac-R. No decrease in activity was observed in the other substrate peptides and Cbz amino acids (Fig. 3A). The dependence of the activities of PP on Ac amino acids and Cbz peptides was examined (Fig. 3). The change in CP activity for Cbz-G-F was measured in the presence of Ac-M (Fig. 3A). CP activity was measured from the release of F, detected by HPLC analysis. CP activity was inhibited competitively by Ac-M acid with a Ki of 4.90 ± 1.12 mM. The change in aminoacylase activity for Ac-M was measured in the presence of Cbz-G-G-F (Fig. 3B). During the assay, the release of F was not detected by HPLC. Aminoacylase activity was inhibited competitively by Cbz-G-G-F, with a Ki of 2.93 ± 0.98 mM.
FIG. 3.

Effect of substrate concentration (Cbz-G-F [A] and Ac-M [B]) on the initial rate (v) of hydrolysis (double-reciprocal plot). The hydrolytic activities were measured in the presence (●) (5 mM) and absence (○) of Ac-M (A) and Cbz-G-G-F (B), at 85°C in 50 mM sodium phosphate buffer (pH 7.5) containing 5% DMF.
When PP was incubated with 5 mM l-benzylsuccinate, a strong competitive inhibitor of CP (6), in the enzyme assay for 10 min at 85°C, neither CP nor aminoacylase activity was detected using the substrates Cbz-F, Cbz-G-F, Ac-M, and Ac-F. l-Benzylsuccinate competitively inhibited both activities at pH 7.5 and 85°C, with the following result: Ki ≓ 1.0 × 10−2 mM.
Chemical modification.
It has been speculated that aminoacylase has an essential SH group at the active site (18, 22, 25). PP has two Cys residues. When PP (1.0 mg/ml) in 50 mM phosphate buffer (pH 8.0) was incubated with DTNB (0.5 mM) for 1 h at 30°C, 1.81 ± 0.41 mol of Cys per mol of monomer enzyme protein were modified by DTNB, and both CP and aminoacylase activities of PP were decreased to less than 0.1% of the those of native PP. Figure 4 shows the rate of loss of the activity for Cbz-G-F by DTNB. The reaction followed pseudo-first-order kinetics during the initial phase of inactivation. The pseudo-first-order rate constant (kapp) for the decrease of activity was obtained by plotting the data on a semilogarithmic scale (Fig. 4A). The pH dependence of the reaction of the essential SH group with DTNB was examined. The pH effect on the kapp value was analyzed to determine the sulfhydryl pK (= −log K) value according to the following equation (7):
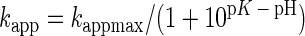 |
1 |
where kappmax and K represent the maximal first-order rate constant and proton dissociation constant for ionizing SH residues, respectively. A plot of these data according to equation 1 yielded a pK of 8.51 ± 0.04 (Fig. 4B).
FIG. 4.

pH dependence of DTNB inactivation. (A) The enzyme solution (1 mg/ml) was incubated with DTNB (0.5 mM) at 30°C in 50 mM sodium phosphate buffer (pH 6 to 8), and 50 mM NaH2PO4-Na2B4O7 buffer (pH 8 to 9.5). The reaction was terminated at the indicated times by transferring an aliquot of the mixture to an equal volume of 50 mM phosphate buffer (pH 7.5) containing 40 mM Cys. The residue activity of the modified enzyme was measured with Cbz-G-F as the substrate at 85°C in 50 mM sodium phosphate buffer (pH 7.5) containing 5% DMF. (B) Replot of pseudo-first-order rate constant versus pH. The solid line represents the fit of the data to equation 1 with a pK of 8.51 and a kappmax of 0.050 s−1.
Metal ions bound to PP.
Analysis by ICP-AES showed that PP contained 1.22 mol of zinc ion and 0.17 mol of calcium ion per mol of monomer enzyme protein. The analysis also showed that PP did not contain cobalt, manganese, molybdenum, nickel, magnesium, or copper ions. The effect of the metal-chelating reagent EDTA on the activity of PP was examined. When PP was dialyzed against 50 mM EDTA (pH 7.5) for 10 h at 4°C and then against 50 mM sodium phosphate buffer (pH 7.5), both CP and aminoacylase activities of PP decreased to less than 1.0%. Both activities were restored by incubation in 50 mM sodium phosphate buffer (pH 7.5) containing ZnCl2, MnCl2, or CoCl2 at 4°C for 1 h. The restorative effect was dependent on the concentration of the metal ions. Eighty-eight, 23, and 20% of activities were restored by adding 1.0 mM ZnCl2, 1.0 mM MnCl2, and 0.1 mM CoCl2, respectively.
Mutants of PP.
According to the alignment of the primary sequence (13) and the structure (11) of CPA, Glu367 in PP is the conserved residue which has been thought to be one of the essential active-site residues for CP. In addition, from the sequence similarity of aminoacylase (22), it was speculated that Cys102 in PP might be important for the activity of aminoacylase. The three mutant PPs (E367Q, C102A, and C102S) were constructed, expressed, and purified. The yields of the expressed mutants were similar to that of wild-type PP. These mutant enzymes contained 1 mol of zinc ions per mol of monomer enzyme protein. Table 2 shows the catalytic parameters of wild-type PP, E367Q, C102A, and C102S. In E367Q, the kcat value increased slightly, whereas the Km value did not change for Cbz-G-F and Ac-F as substrates. Substrate inhibition of aminoacylase activity in E367Q was also observed. The temperature-dependent activity and pH profile of the activity of E367Q were similar to those of wild-type PP. The hydrolytic activity of C102A was not detected. The kcat values of C102S were reduced by a factor of about 104-fold relative to those of wild-type PP with Cbz-G-F and Ac-F as substrates. The Km values of C102S were similar to those of wild-type PP.
For E367G, C102A, and C102S, 1.70, 0.80, and 0.73 mol of Cys per mol of monomer proteins were modified by DTNB, respectively, using the method described above. As a result of this chemical modification, the activity of E367Q was decreased to less than 0.1% of wild-type PP activity but that of C102S was not influenced.
DISCUSSION
Thermostable CPs from microorganisms have been found recently (12, 13, 27–29, 35, 36, 38). Most CPs have the conserved amino acid residues which have been thought to be essential to their activity. The gene from P. horikoshii, which has high sequence similarity to the CPS gene, was expressed using E. coli. From the N-terminal sequence analysis and the activity of the expressed enzyme, it was concluded that efficient translation of the P. horikoshii gene occurred inside the E. coli cells. In spite of the absence of the speculated active-site residues for CP, the expressed enzyme (PP) had both CP and aminoacylase activities at high temperatures. Data from HPLC and SDS-PAGE suggested that PP was unlikely to be a monomer structure. Competitive inhibition by l-benzylsuccinate, Ac amino acids, and Cbz peptides indicates that the active center for CP activity is the same as for aminoacylase activity in PP. Tables 1 and 2 show that the active site of PP is suitable for relatively small substrates, and PP may primarily be a dipeptidase with greater specificity for the C-terminal carboxyl group and lesser specificity for the acyl group. Peptides, amino acids, acyl groups, and Cbz can be accommodated on the α-amino group of the C-terminal residue by PP. The decrease in aminoacylase activity at high substrate concentrations indicates that PP has a strong substrate inhibition for aminoacylase. Ac amino acids seem to participate in nonproductive binding modes in PP.
Some CPs belong to metalloproteases (5, 17, 30) containing zinc ions. Analysis by ICP-AES and the observation that activity can be restored by adding metal ions indicate that the binding of one zinc ion to the enzyme has an important role in the activity of PP. Weak activation of PP by cobalt or manganese ions similar to that of some zinc-dependent metallocarboxypeptidases has also been observed (26, 28). The results indicate that PP is one of the zinc-dependent metalloenzymes. It has been reported that the zinc-dependent metallocarboxypeptidases carry a zinc ion at the active site associated with two His side chains, one Glu side chain, and one water molecule (23). His69, Glu72, and His196 are involved in zinc chelation in CPA (11) and conserved well in CP. However, PP has only one conserved His, at position 100 in region B2, corresponding to His69 in CPA (11) and His104 in CPS (13) (Fig. 1). Aminoacylases from S. solfataricus and B. stearothermophilus were also activated by zinc, cobalt, or nickel (33; A. Boyen, C. Legrain, A. Pierard and N. Glansdorff, Thermophiles '96, abstr. 179, 1996). The MHACGHD sequence in B2 (Fig. 1) was not conserved as the zinc chelation motif of CP (29, 37), but was conserved well in PP, CPS, and aminoacylase from B. stearothermophilus. The mechanism of zinc chelation in PP may be a little different from those in the known zinc-dependent metallocarboxypeptidases.
A common origin for CP and aminoacylase has been surmised from their sequence similarity (4, 33) and the similarity of the hydrolytic pattern (hydrolysis of the carboxyl amide of the amino residue at the C-terminal amino acid) in the substrates. In S. solfataricus, aminoacylase (Boyen et al., Thermophiles '96) and CP (12) were present but existed as separate enzymes. To date, no bifunctional (CP and aminoacylase) enzyme has been found. To the best of our knowledge, PP is the only enzyme having significantly high levels of both activities. Glu270 in CPA is one of the most important residues for proton transfer in the hydrolytic mechanism (11). In PP, Glu270 of CPA has been conserved and identified at position 367 (Glu367). The result of site-directed mutagenesis of Glu367 indicates that Glu367 is not required for hydrolytic activity in PP. We have no information about the structure and hydrolytic mechanism of aminoacylase. However, it has been suggested that aminoacylase has an essential Cys residue which is located at the active site (18, 22, 25). This chemical modification study of PP, producing a reasonable value of pK for the Cys residue, suggests that at least one of the Cys residues in PP is important for the activity. The characteristics of C102A and C102S indicate that Cys102 is important for the activity, the substrate binding site of C102S is similar to that of wild-type PP, and the hydroxyl group of Ser102 could serve as the thiol group of Cys102 in PP, as observed in the case of trypsin (19). From the above results and the results of the chemical modification of mutant CPs, it was concluded that Cys102 was located at the active center and was one of the essential amino acids for the activity in PP. It seems that PP has a structure similar to those of other CPs and a hydrolytic mechanism similar to that of aminoacylase. Some characteristics of PP suggest that CP and aminoacylase evolved from a common origin similar to PP. Detailed studies of the hydrolytic mechanism of PP should prove informative with regard to the molecular evolution of these enzymes.
Because of its thermostability, PP is expected to be useful for hydrolysis of some peptides and for the production of l-amino acid derivatives from racemates at temperatures over 90°C.
ACKNOWLEDGMENTS
We thank J. Ishikawa, T. Hashimoto, Y. Kosugi, and S. Ando for their assistance with the experiments in this study.
REFERENCES
- 1.Aviles F X, Vendrell J, Guasch A, Coll M, Huber R. Advances in metallo-procarboxypeptidases. Emerging details on the inhibition mechanism and on the activation process. Eur J Biochem. 1993;211:381–389. doi: 10.1111/j.1432-1033.1993.tb17561.x. [DOI] [PubMed] [Google Scholar]
- 2.Bailey J M, French D. The significance of multiple reactions in enzyme-polymer systems J. Biol Chem. 1957;226:1–14. [PubMed] [Google Scholar]
- 3.Birnbaum S M, Levintow L, Kingsley R B, Greenstein J P. Specificity of amino acid acylases. J Biol Chem. 1952;194:455–470. [PubMed] [Google Scholar]
- 4.Boyen A, Charlier D, Charlier J, Sakanyan V, Mett I, Glansdorff N. Acetylornithine deacetylase, succinyldiaminopimelate desuccinylase and carboxypeptidase G2 are evolutionarily related. Gene. 1992;116:1–6. doi: 10.1016/0378-1119(92)90621-u. [DOI] [PubMed] [Google Scholar]
- 5.Bradshaw R A, Ericsson L H, Walsh K A, Neurath H. The amino acid sequence of bovine carboxypeptidase A. Proc Natl Acad Sci USA. 1969;63:1389–1394. doi: 10.1073/pnas.63.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byers L D, Wolfenden R. Binding of the by-product analog benzylsuccinic acid by carboxypeptidase A. Biochemistry. 1973;12:2070–2078. doi: 10.1021/bi00735a008. [DOI] [PubMed] [Google Scholar]
- 7.Chen C-Y, Emig F A, Schramm V L, Ash D E. Inactivation of chicken mitochondrial phosphoenolpyruvate carboxykinase by o-phthalaldehyde. J Biol Chem. 1991;266:16645–16652. [PubMed] [Google Scholar]
- 8.Cheng T C, Ramakrishnan V, Chan S I. Purification and characterization of a cobalt-activated carboxypeptidase from the hyperthermophilic archaeon Pyrococcus furiosus. Protein Sci. 1999;8:2474–2486. doi: 10.1110/ps.8.11.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chibata I, Tosa T, Sato T, Mori T. Production of L-amino acids by aminoacylase adsorbed on DEAE-Sephadex. Methods Enzymol. 1976;44:746–759. doi: 10.1016/s0076-6879(76)44053-3. [DOI] [PubMed] [Google Scholar]
- 10.Cho H Y, Tanizawa K, Tanaka H, Soda K. Thermostable aminoacylase from Bacillus thermoglucosidius: purification and characterization. Agric Biol Chem. 1987;51:2793–2800. [Google Scholar]
- 11.Christianson D W, Lipscomb W N. Carboxypeptidase A. Acc Chem Res. 1989;22:62–69. [Google Scholar]
- 12.Colombo S, D'Auria S, Fusi P, Zecca L, Raia C A, Tortora P. Purification and characterization of a thermostable carboxypeptidase from the extreme thermophilic archaebacterium Sulfolobus solfataricus. Eur J Biochem. 1992;206:349–357. doi: 10.1111/j.1432-1033.1992.tb16934.x. [DOI] [PubMed] [Google Scholar]
- 13.Colombo S, Toietta G, Zecca L, Vanoni M, Tortora P. Molecular cloning, nucleotide sequence, and expression of a carboxypeptidase-encoding gene from the archaebacterium Sulfolobus solfataricus. J Bacteriol. 1995;177:5561–5566. doi: 10.1128/jb.177.19.5561-5566.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doi E, Shibata D, Matoba T. Modified colorimetric ninhydrin methods for peptidase assay. Anal Biochem. 1981;118:173–184. doi: 10.1016/0003-2697(81)90175-5. [DOI] [PubMed] [Google Scholar]
- 15.Ellman G L. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalez J M, Masuchi Y, Robb F T, Ammerman J W, Maeder D L, Yanagibayashi M, Tamaoka J, Kato C. Pyrococcus horikoshii sp. nov., a hyperthermophilic archaeon isolated from a hydrothermal vent at the Okinawa Trough. Extremophiles. 1998;2:123–130. doi: 10.1007/s007920050051. [DOI] [PubMed] [Google Scholar]
- 17.Hendriks D H, Wang W, Scharpe S, Lommaert M P, van Sande M. Purification and characterization of a new arginine carboxypeptidase in human serum. Biochim Biophys Acta. 1990;1034:86–92. doi: 10.1016/0304-4165(90)90157-r. [DOI] [PubMed] [Google Scholar]
- 18.Henseling J, Rohm K-H. Aminoacylase I from hog kidney: anion effects and the pH dependence of kinetic parameters. Biochim Biophys Acta. 1988;959:370–377. doi: 10.1016/0005-2760(88)90211-1. [DOI] [PubMed] [Google Scholar]
- 19.Higaki J N, Evnin L B, Craik C S. Introduction of a cysteine protease active site into trypsin. Biochemistry. 1989;28:9256–9263. doi: 10.1021/bi00450a004. [DOI] [PubMed] [Google Scholar]
- 20.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, Yamamoto S, Sekine M, Baba S, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Yoshizawa T, Nakamura Y, Robb F T, Horikoshi K, Masuchi Y, Shizuya H, Kikuchi H. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3. DNA Res. 1998;5:55–76. doi: 10.1093/dnares/5.2.55. [DOI] [PubMed] [Google Scholar]
- 21.Kawarabayasi Y, Sawada M, Horikawa H, Haikawa Y, Hino Y, amamoto Y S, Sekine M, Baba S-I, Kosugi H, Hosoyama A, Nagai Y, Sakai M, Ogura K, Otsuka R, Nakazawa H, Takamiya M, Ohfuku Y, Funahashi T, Tanaka T, Kudoh Y, Yamazaki J, Kushida N, Oguchi A, Aoki K, Yoshizawa T, Nakamura Y, Robb F T, Horikoshi K, Masuchi Y, Shizuya H, Kikuchi H. Complete sequence and gene organization of the genome of a hyper-thermophilic archaebacterium, Pyrococcus horikoshii OT3 (supplement) DNA Res. 1998;5:147–155. doi: 10.1093/dnares/5.2.147. [DOI] [PubMed] [Google Scholar]
- 22.Kempf B, Bremer E. A novel amidohydrolase gene from Bacillus subtilis cloning: DNA-sequence analysis and map position of amhX. FEMS Microbiology Lett. 1996;141:129–137. doi: 10.1111/j.1574-6968.1996.tb08374.x. [DOI] [PubMed] [Google Scholar]
- 23.Kester W, Matthews B W. Comparison of the structures of carboxypeptidase A and thermolysin. J Biol Chem. 1977;252:7704–7710. [PubMed] [Google Scholar]
- 24.Kim H, Lipscomb W N. Crystal structure of the complex of carboxypeptidase A with a strongly bound phosphonate in a new crystalline form: comparison with structures of other complexes. Biochemistry. 1990;29:5546–5555. doi: 10.1021/bi00475a019. [DOI] [PubMed] [Google Scholar]
- 25.Kordel W, Schneider F. Chemical investigations on pig kidney aminoacylase. Biochim Biophys Acta. 1976;445:446–457. doi: 10.1016/0005-2744(76)90098-x. [DOI] [PubMed] [Google Scholar]
- 26.Larsen K S, Auld D S. Characterization of an inhibitory metal binding site in carboxypeptidase A. Biochemistry. 1991;30:2613–2618. doi: 10.1021/bi00224a007. [DOI] [PubMed] [Google Scholar]
- 27.Lee S H, Minagawa E, Taguchi H, Matsuzawa H, Ohta T, Kaminogawa S, Yamauchi K. Purification and characterization of a thermostable carboxypeptidase (carboxypeptidase Taq) from Thermus aquaticus YT-1. Biosci Biotechnol Biochem. 1992;56:1839–1844. doi: 10.1271/bbb.56.1839. [DOI] [PubMed] [Google Scholar]
- 28.Lee S H, Taguchi H, Yoshimura E, Minagawa E, Kaminogawa S, Ohta T, Matsuzawa H. Carboxypeptidase Taq, a thermostable zinc enzyme, from Thermus aquaticus YT-1: molecular cloning, sequencing, and expression of the encoding gene in Escherichia coli. Biosci Biotechnol Biochem. 1994;58:1490–1495. doi: 10.1271/bbb.58.1490. [DOI] [PubMed] [Google Scholar]
- 29.Lee S H, Taguchi H, Yoshimura E, Minagawa E, Kaminogawa S, Ohta T, Matsuzawa H. The active site of carboxypeptidase Taq possesses the active-site motif His-Glu-X-X-His of zinc-dependent endopeptidases and aminopeptidases. Protein Eng. 1996;9:467–469. doi: 10.1093/protein/9.6.467. [DOI] [PubMed] [Google Scholar]
- 30.Levin Y, Skidgel R A, Erdos E G. Isolation and characterization of the subunits of human plasma carboxypeptidase N (kininase i) Proc Natl Acad Sci USA. 1982;79:4618–4622. doi: 10.1073/pnas.79.15.4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H A. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp Quant Biol. 1986;51:263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 32.Palmer J R, Daniels C J. In vivo definition of an archaeal promoter. J Bacteriol. 1995;177:1844–1849. doi: 10.1128/jb.177.7.1844-1849.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakanyan V, Desmarez L, Legrain C, Charlier D, Mett I, Kochikyan A, Savchenko A, Boyen A, Falmagne P, Pierard A, Glansdorff N. Gene cloning, sequence analysis, purification, and characterization of a thermostable aminoacylase from Bacillus stearothermophilus. Appl Environ Microbiol. 1993;59:3878–3888. doi: 10.1128/aem.59.11.3878-3888.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sakoda M, Hiromi K. Determination of the best-fit values of kinetic parameters of the Michaelis-Menten equation by the method of least squares with the Taylor expansion. J Biochem (Tokyo) 1976;80:547–555. doi: 10.1093/oxfordjournals.jbchem.a131310. [DOI] [PubMed] [Google Scholar]
- 35.Smulevitch S V, Osterman A L, Galperina O V, Matz M V, Zagnitko O P, Kadyrov R M, Tsaplina I A, Grishin N V, Chestukhina G G, Stepanov V M. Molecular cloning and primary structure of Thermoactinomyces vulgaris carboxypeptidase T. FEBS Lett. 1991;291:75–78. doi: 10.1016/0014-5793(91)81107-j. [DOI] [PubMed] [Google Scholar]
- 36.Teplyakov A, Polyakov K, Obmolova G, Strokopytov B, Kuranova I, Osterman A, Grishin N, Smulevitch S, Zagnitko O, Galperina O, Matz M, Stepanov V. Crystal structure of carboxypeptidase T from Thermoactinomyces vulgaris. Eur J Biochem. 1992;208:281–288. doi: 10.1111/j.1432-1033.1992.tb17184.x. [DOI] [PubMed] [Google Scholar]
- 37.Vallee B L, Auld D S. Zinc coordination, function, and structure of zinc enzymes and other proteins. Biochemistry. 1990;29:5647–5659. doi: 10.1021/bi00476a001. [DOI] [PubMed] [Google Scholar]
- 38.Villa A, Zecca L, Fusi P, Colombo S, Tedeschi G, Tortora P. Structural features responsible for kinetic thermal stability of a carboxypeptidase from the archaebacterium Sulfolobus solfataricus. Biochem J. 1993;295:827–831. doi: 10.1042/bj2950827. [DOI] [PMC free article] [PubMed] [Google Scholar]