

Abstract
Microbial degradation of synthetic chelating agents, such as EDTA and nitrilotriacetate (NTA), may help immobilizing radionuclides and heavy metals in the environment. The EDTA- and NTA-degrading bacterium BNC1 uses EDTA monooxygenase to oxidize NTA to iminodiacetate (IDA) and EDTA to ethylenediaminediacetate (EDDA). IDA- and EDDA-degrading enzymes have not been purified and characterized to date. In this report, an IDA oxidase was purified to apparent homogeneity from strain BNC1 by using a combination of eight purification steps. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis revealed a single protein band of 40 kDa, and by using size exclusion chromatography, we estimated the native enzyme to be a homodimer. Flavin adenine dinucleotide was determined as its prosthetic group. The purified enzyme oxidized IDA to glycine and glyoxylate with the consumption of O2. The temperature and pH optima for IDA oxidation were 35°C and 8, respectively. The apparent Km for IDA was 4.0 mM with a kcat of 5.3 s−1. When the N-terminal amino acid sequence was determined, it matched exactly with that encoded by a previously sequenced hypothetical oxidase gene of BNC1. The gene was expressed in Escherichia coli, and the gene product as a C-terminal fusion with a His tag was purified by a one-step nickel affinity chromatography. The purified fusion protein had essentially the same enzymatic activity and properties as the native IDA oxidase. IDA oxidase also oxidized EDDA to ethylenediamine and glyoxylate. Thus, IDA oxidase is likely the second enzyme in both NTA and EDTA degradation pathways in strain BNC1.
Synthetic chelating agents are used in large quantities for a variety of applications in nuclear waste processing, household detergents, water treatments, descaling boilers, and removing the precipitation of sparingly soluble salts (4, 23, 27, 33, 38). Aminopolycarboxylic acids and their salts, primarily EDTA, diethylenetriaminepentaacetate, and nitrilotriacetate (NTA), are by far the most dominant group of substances used as chelating agents worldwide. The annual sales of EDTA, NTA, and diethylenetriaminepentaacetate in Europe were 32,550, 18,600, and 14,000 tons in 1997 (27). The environmental disposal of EDTA and NTA can have undesirable consequences. Chelating agents form soluble complexes with radionuclides or heavy metals, increasing their mobility in subsurface environments (9). The mobilized radionuclides and toxic heavy metals can be directly consumed by humans or accumulated by plants and transferred to humans through the food chain, causing health problems.
Microbial degradation of chelating agents may decrease the mobilization of radionuclides and heavy metals in the environment. Several enrichment cultures have been reported to mineralize EDTA under strictly aerobic conditions (28, 29, 40). Three EDTA-degrading microorganisms have been isolated: an Agrobacterium sp. (19), the bacterial strain DSM 9103 (47), and the bacterial strain BNC1 (28). There are also several microorganisms that can use NTA as a sole source of nitrogen, carbon, and energy (3, 10, 12, 41). Although the NTA-degrading bacteria cannot degrade EDTA, the EDTA-degrading bacterium BNC1 can grow on both EDTA and NTA (16). The biochemistry of NTA and EDTA degradation has been studied. NTA is degraded to iminodiacetate (IDA) by either the NTA monooxygenase of Chelatobacter heintzii (45) or EDTA monooxygenases of the EDTA-degrading bacteria BNC1 and DSM 9103 (7, 32, 47). Then, IDA is transformed to glycine and glyoxylate by a membrane-bound IDA dehydrogenase in C. heintzii (44). For EDTA degradation, EDTA monooxygenases of strains BNC1 and DSM 9103 oxidize EDTA to ethylenediaminetriacetate and then to ethylenediaminediacetate (EDDA) (7, 32, 47). However, the enzymes for IDA and EDDA degradation have never been purified. The accumulation of IDA in the natural environment would be undesirable because of the possible formation of putatively carcinogenic N-nitroso-IDA from IDA and nitrite (13, 34).
We report here the identification, purification, and characterization of an IDA oxidase from the bacterium BNC1. The corresponding gene was identified from a previously sequenced hypothetical oxidase gene (7, 31) with the determined N-terminal amino acid sequence. When the gene was overexpressed in Escherichia coli, the gene product was purified and had IDA oxidase activity. IDA oxidase used both IDA and EDDA as its substrates.
(A preliminary account of this work was presented previously [Y. Liu, T. M. Louie, and L. Xun, Abstr. 99th Gen. Meet. Am. Soc. Microbiol., abstr. Q-395, 1999].)
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The EDTA-degrading bacterium BNC1 was kindly provided by Bernd Nörtemann (Technical University of Braunschweig, Braunschweig, Germany). BNC1 was cultured with disodium EDTA (0.3 g/liter) and glycerol (2 ml/liter) in a mineral medium (28). Large quantities of cells were obtained by culturing BNC1 cells in a 20-liter carboy containing 15 liters of the medium bubbled with sterile air for 3 days at 29°C. Toward the end of log phase, cells were harvested by concentrating the culture volume down to 2 liters in a hollow-fiber filtration unit (model DC10L system; Amicon, Beverly, Mass.) and were then centrifuged at 17,000 × g for 15 min at 4°C. The cells were stored at −20°C for a maximum of 3 days. E. coli strain NovaBlue (Novagen, Madison, Wis.) was used for pET-30 LIC cloning, and strain BL21(DE3) (Novagen) was used for gene expression. E. coli strains were routinely grown at 37°C in Luria-Bertani medium with shaking or on Luria-Bertani agar (36). Kanamycin (Sigma, St. Louis, Mo.) was used at 30 μg per ml in culture media when required.
Chemicals.
All chemicals were of analytical grade and were purchased from Fisher (Fair Lawn, N.J.), Sigma, or Merck (Rahway, N.J.).
Enzyme assays.
IDA oxidase activity was assayed either by following the reduction of the artificial electron acceptor potassium ferricyanide [K3Fe(CN)6] or by measuring the production of glyoxylate. A standard K3Fe(CN)6 assay mixture contained 25 mM Tris-HCl buffer (pH 8.0), 25 mM IDA, 1 mM K3Fe(CN)6, and an appropriate amount of IDA oxidase in a total volume of 800 μl. IDA oxidase activity was assayed by measuring the absorbance change of the reaction mixture by a UV/Visible Spectrophotometer (model 4000; Pharmacia, Alameda, Calif.) at 420 nm [ɛ420 = 990 M−1 cm−1 for Fe(CN)63−] at room temperature. One unit of IDA oxidase was defined as the reduction of 2 μmol of K3Fe(CN)6 per min under the defined conditions. The K3Fe(CN)6 assay was also performed under anaerobic conditions by assembling the reaction mixture in a stoppered cuvette in an anaerobic chamber (98% N2 and 2% H2). For the glyoxylate method, a standard assay mixture contained 25 mM Tris-HCl buffer (pH 8.0), 25 mM IDA, and an appropriate amount of IDA oxidase in a total volume of 250 μl. The reaction was initiated by adding IDA oxidase. The assay was stopped by adding 100 μl of 0.1 N HCl. The glyoxylate produced was detected by using phenylhydrazine-K3Fe(CN)6 as previously described (45).
Purification steps for IDA oxidase.
All purification steps were performed at 4°C. All buffers contained 1 mM dithiothreitol. Ammonium sulfate saturation levels were those at 25°C.
(i) Extraction of cells.
About 60 to 75 g (wet weight) of cells was suspended in 20 mM potassium phosphate (KPi) buffer (pH 7.0) containing 2.5 mM EDTA. The protease inhibitor phenylmethylsulfonyl fluoride was freshly prepared in absolute ethanol at a concentration of 30 mM and was added to the cell suspension to a final concentration of 0.5 mM. The cells were then broken by passing through a French pressure cell (model FA-030; Aminco, Urbana, Ill.) three times at 260 MPa. The lysate was centrifuged at 17,000 × g for 25 min to remove debris and unbroken cells. The supernatant was saved as the cell extract.
(ii) Protamine sulfate fractionation.
A 2% (wt/vol) solution of protamine sulfate in 20 mM KPi buffer (pH 7.0) was added to the cell extract to a final concentration of 0.05% with constant stirring. After 5 min, the mixture was centrifuged at 17,000 × g for 15 min. The supernatant was saved.
(iii) Ammonium sulfate fractionation.
Solid ammonium sulfate was added to the supernatant to 30% saturation with constant stirring. The pH of the solution was not adjusted. After being stirred for 10 min, the mixture was centrifuged at 17,000 × g for 15 min. The precipitate was discarded. Additional solid ammonium sulfate was added to the supernatant to 70% saturation with constant stirring. The precipitate was saved.
(iv) Ultracentrifugation.
The precipitated protein was dissolved in an equal volume of 20 mM KPi buffer (pH 7.0). The solution was centrifuged at 230,000 × g for 60 min, and the supernatant was saved.
(v) Phenyl agarose chromatography.
The ultracentrifuged supernatant was loaded onto a phenyl agarose (Sigma) column (12 by 1.5 cm) equilibrated with 20 mM KPi buffer (pH 7.0) containing 25% saturation of ammonium sulfate. The column was first washed with 45 ml of the starting buffer. Then, proteins were eluted with a decreasing gradient of ammonium sulfate (percentages of saturation in the same buffer: 25 to 0%, 200-ml linear gradient; and 0%, 45 ml). The enzyme was eluted at around 5 to 0% saturation of ammonium sulfate. Fractions containing the enzyme activity were concentrated and desalted to 20 mM KPi buffer (pH 7.0) with Centriprep-10 tubes (Millipore, Bedford, Mass.).
(vi) UnoQ chromatography.
The activity-containing fractions from the phenyl agarose column in 20 mM KPi (pH 7.0) were injected onto a 1.3-ml UnoQ column (Bio-Rad, Hercules, Calif.) previously equilibrated with 20 mM KPi buffer (pH 7.0). Proteins were eluted with a step and linear gradient of NaCl (percentages of 1 M NaCl in the same buffer: 0%, 5 ml; 0 to 20%, 20-ml linear gradient; 100%, 5 ml; and 0%, 6 ml) by a Biological Workstation System (Bio-Rad). IDA oxidase was eluted as a small peak around 140 mM NaCl. The fractions containing enzyme activity were pooled and concentrated.
(vii) Hydroxyapatite chromatography.
The buffer for IDA oxidase was changed to 10 mM KPi (pH 6.6) containing 0.3 mM CaCl2. The sample was injected onto a Bio-Scale CHT2-I hydroxyapatite column (7 by 52 mm; Bio-Rad) equilibrated with the same buffer. The proteins were eluted with a step and linear gradient of KPi (pH 6.6) (concentrations of KPi: 10 mM, 5 ml; 10 to 200 mM, 20-ml linear gradient; 500 mM, 5 ml; and 10 mM, 6 ml). IDA oxidase was eluted as a major peak around 80 mM KPi and was concentrated to 1 ml with a Centriprep-10 tube.
(viii) Second UnoQ chromatography.
The buffer for IDA oxidase was changed to 20 mM Tris-HCl (pH 8.0) with Centriprep-10. The sample was injected onto the UnoQ column equilibrated with the same buffer. Proteins were eluted with a step and linear gradient of NaCl (percentages of 1 M NaCl in the same buffer: 0%, 5 ml; 0 to 20%, 20-ml linear gradient; 100%, 5 ml; and 0%, 6 ml). IDA oxidase was eluted as a major peak around 155 mM NaCl.
(ix) Size exclusion chromatography.
The activity-containing fractions from the second UnoQ column were pooled, concentrated, and then injected onto a Superdex 75 column (10 by 300 mm; Pharmacia) equilibrated with 20 mM Tris-HCl (pH 8.0) containing 150 mM NaCl. The protein was eluted with the same buffer. IDA oxidase was eluted from the column as a single peak with a retention volume of 10 ml.
Analytical methods.
A high-performance liquid chromatography (HPLC) system equipped with a Nova Pak C18 column (3.9 by 150 mm) (Waters, Milford, Mass.) was used to analyze glycine after derivatization (21). The HPLC with a Biosep Sec-S3000 column (7.8 by 300 mm; Phenomenex, Torrance, Calif.) was used to determine the native molecular weight of IDA oxidase. Ethylenediamine (ED) was assayed by HPLC after dansyl chloride derivatization (18). The sample was separated on the Nova Pak C18 column by an isocratic mobile phase of a mixture of methanol-water-acetic acid (60:38.5:1.5) at a flow rate of 1 ml per min. The ED-dansyl derivative was eluted off at 12.5 min, and the peak was collected. The collected sample was further analyzed by a liquid chromatography-mass spectrometry system (Waters) with the same setting, except a mass detector (ZMD4000) was used to detect the compound instead of a UV detector. Both standard ED-dansyl and reaction product-dansyl derivatives were analyzed. The mass spectrometry detector was operated at 50 eV in a positive electrospray mode. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was done according to the method described by Laemmli (17). Gels were stained for proteins with Gel Code Blue stain reagent (Pierce, Rockford, Ill.). Protein concentrations were determined with a protein dye reagent (8) with bovine serum albumin as the standard. The N-terminal amino acid sequence of the pure protein was determined on an Applied Biosystems 476A sequencer (Perkin-Elmer, Foster City, Calif.) at the Biotechnology Laboratory, The University of British Columbia, Vancouver, British Columbia, Canada, as previously described (48). The type of flavin of the pure IDA oxidase was determined by thin-layer chromatography on a silica gel (Eastman Kodak, Rochester, N.Y.) (30).
Oxygen consumption.
Oxygen consumption by IDA oxidase was determined in a closed reaction vessel (0.67 ml) fitted with a Clark-type oxygen electrode (Instech, Plymouth Meeting, Pa.). The electrode was calibrated with a chemical method by using N-methylphynazonium methosulfate and NADH to quantitatively consume O2 (35). The reaction mixture contained 25 mM IDA in 25 mM Tris-HCl buffer (pH 8.0). The reaction was initiated by adding IDA oxidase to the reaction mixture. Catalase (Sigma) was added (450 U) to detect the presence of any H2O2 by O2 production.
pH and temperature optima.
IDA oxidase activity was measured at various pH values within the range of 6.8 to 8.8 by using 25 mM Tris-HCl buffer in a total volume of 800 μl. The reaction mixture was the same as described above for the enzyme assay, which was conducted by using the K3Fe(CN)6 method. The temperature optimum for the enzyme activity was also determined in the standard K3Fe(CN)6 assay mixture at various temperatures.
Determination of kinetic parameters.
The Michaelis-Menten kinetic parameters were determined by measuring the initial rate of glyoxylate produced by pure IDA oxidase. The IDA and EDDA concentrations used in these experiments were from 4 to 25 mM. Three minutes after the pure IDA oxidase was added to the reaction mixture, the amount of glyoxylate produced was determined by the phenylhydrazine-K3Fe(CN)6 method (45). All of the experiments were performed in triplicate, and average values were used in calculation.
Gene cloning and expression.
To overproduce IDA oxidase as a C-terminal terminal His tag fusion protein (IdaA) in E. coli, PCR primers were designed to clone idaA into the pET-30 LIC vector (Novagen). The forward primer (IdaA5, 5′-CAT-TGG-TCG-TGA-AAC-ATA-TGC-GTG-3′) was located at positions 9760 to 9783 of a gene cluster (GenBank accession no. AF176664) (7, 31), with an NdeI site (underlined) introduced by altering two bases. The reverse primer (IdaA3, 5′-AGG-CTT-GGA-TCC-CCG-GCT-TC-3′) was at base positions of 10881 to 10900, in which a BamHI site (underlined) was introduced by altering four bases to fuse the C terminus to a His tag. The idaA gene starts at position 9777 and ends at position 10889 (7, 31). The gene was amplified from strain BNC1 genomic DNA isolated by standard methods (36). The PCR thermal profile was 30 s at 94°C, 30 s at 50°C, and 30 s at 72°C for 30 cycles. The PCR product was cut by NdeI and BamHI and was then ligated into the plasmid pET-30 LIC (Novagen) (49), which was previously digested by NdeI and BamHI, to produce plasmid pEI1. Plasmid pEI1 was electroporated into E. coli NovaBlue (Novagen) for plasmid identification and recovery. The recovered plasmid was transformed into E. coli strain BL21(DE3) for protein production upon induction by isopropyl-β-d-thiogalactoside (Fisher).
Purification of the C-fusion IdaA.
Since the protein was overproduced and had a His tag, a one-step nickel affinity chromatography procedure that was previously reported (11) was used to purify the C-fusion His tag IdaA to homogeneity.
RESULTS
Detection of IDA oxidase activities.
Cell extracts of BNC1 cultured with EDTA and glycerol in a mineral medium were able to degrade IDA to glyoxylate in the presence of O2. The conversion was enzymatic because formation of glyoxylate was not detected in controls without cell extracts or with boiled cell extracts. After ultracentrifugation at 230,000 × g for 60 min, most of the IDA oxidase activity (about 70%) was present in the supernatant. This finding suggests that the enzyme is not an integral membrane protein. The specific enzyme activity of cell extracts of BNC1 cultured with EDTA as the nitrogen source (0.191 ± 0.057 U/mg of protein) (standard deviation of three samples) was about onefold higher than that cultured with NH4Cl as the nitrogen source (0.101 ± 0.036 U/mg of protein). For ease of detection, a simple assay was developed by using K3Fe(CN)6 as an artificial electron acceptor, and the reduction of K3Fe(CN)6 was spectrophotometrically monitored. The reduction of K3Fe(CN)6 was associated with IDA oxidation because there was no reduction without IDA. The development of the K3Fe(CN)6 assay was essential for the purification of IDA oxidase.
Enzyme purification.
IDA oxidase was purified from BNC1 cell extracts (Table 1). The purification scheme, consisting of eight steps, resulted in 309-fold purification of IDA oxidase relative to the cell extracts. Approximately 1.5% of IDA oxidase activity was recovered. After size exclusion chromatography, a single band with an apparent molecular weight of 40,000 was detected by SDS-PAGE (Fig. 1). The molecular mass of the native enzyme was determined to be about 80 kDa by size exclusion chromatography in the presence of 150 mM NaCl, suggesting that the native enzyme is a homodimer. The purified enzyme (25 μg/ml) could be stored without apparent loss of activity at −80°C for a week in the size exclusion chromatography buffer.
TABLE 1.
Purification of IDA oxidase
| Purification step | Vol (ml) | Protein (mg) | Total activity (U) | Sp act (U/mg) | Recovery (%) |
|---|---|---|---|---|---|
| Cell extract | 116 | 764 | 118 | 0.16 | 100 |
| Protamine sulfate treatment | 117 | 763 | 115 | 0.15 | 96.9 |
| Ammonium sulfate treatment | 33.2 | 510 | 112 | 0.22 | 94.3 |
| Ultracentrifugation | 32.6 | 344 | 71.7 | 0.21 | 60.6 |
| Phenyl agarose column | 38.0 | 68.5 | 46.7 | 0.68 | 39.5 |
| UnoQ column | 12.0 | 10.1 | 23.4 | 2.33 | 19.8 |
| Hydroxyapatite column | 3.0 | 0.28 | 5.62 | 20.0 | 4.7 |
| UnoQ column | 3.0 | 0.17 | 4.32 | 25.3 | 3.7 |
| Size exclusion column | 1.8 | 0.04 | 1.82 | 47.9 | 1.5 |
FIG. 1.

SDS-PAGE of purified IDA oxidase. Lanes 1 and 3, low-range molecular mass standards (Bio-Rad); lane 2, 0.5 μg of IDA oxidase purified from BNC1; lane 4, 3 μg of C-fusion His tag IdaA purified from E. coli.
Gene overexpression.
The N-terminal amino acid sequence of the purified enzyme was determined to be MRVLIIGAGILGASAAYHLARLGAQVEIID. It was identical to the deduced N-terminal sequence of an open reading frame of unknown function, adjacent to a gene cluster involved in EDTA degradation from BNC1 (7, 31). The calculated molecular weight of the encoded protein was 39,026, which matched the determined molecular weight of IDA oxidase. The gene was amplified by PCR and was subsequently cloned into the pET-30 LIC vector to obtain plasmid pEI1. E. coli BL21(DE3) carrying pEI1 produced a C-terminal fusion protein with a His tag. The recombinant protein was overproduced in E. coli and purified by a one-step nickel affinity chromatography procedure (Fig. 1). For a typical purification, 4.9 mg of IDA oxidase was obtained from 280 mg of total protein in cell extract with a 28% recovery of activity. The pure C-fusion IdaA had a specific activity of 42 U mg−1 when reducing K3Fe(CN)6. The purified protein oxidized IDA to glyoxylate and glycine in the presence of oxygen, confirming that it is IDA oxidase. Thus, the gene is named idaA. The specific enzyme activity of the purified fusion protein was very similar to that of the native protein (Table 1). The fusion protein was stable at −80°C for 1 week in 20 mM KPi buffer (pH 8.0) with 1 mM dithiothreitol. The nucleotide sequence of the gene encoding recombinant IdaA has also been confirmed. One single-point mutation was found, which caused one mutation in the amino acid sequence (Asn32Ser). However, the single mutation did not change the apparent enzyme activity.
Enzymatic activity.
The optimal temperature for IdaA activity was 35°C, with 60, 82, and 97% of the optimal activity retained at 25, 30, and 40°C. The highest activity was observed at pH 8.0 in 20 mM Tris-HCl buffer, and the activity was 72, 88, and 91% at pH 6.8, 7.5, and 8.8 in 20 mM Tris-HCl buffers. The best activity was obtained in 20 mM Tris-HCl buffer (pH 8.0) at 35°C. The addition of CaCl2 or MgCl2 to the purified enzyme did not influence its activity, suggesting that the enzyme does not require divalent cations for its activity.
Substrate specificity and role of IDA oxidase in EDTA degradation pathway.
IDA oxidase also degraded EDDA and sarcosine with reduced rates, about 26 and 4% of that for IDA oxidation. No activity was detected when EDTA, NTA, succinate, fumarate, and glycine were used as substrates. When EDDA was oxidized, glyoxylate was also produced. A reaction mixture containing 500 nmol of EDDA produced 794.4 ± 37.3 (standard deviation of three samples) nmol of glyoxylate. The molar ratio of glyoxylate to EDDA was 1.59, indicating that both acetyl groups are removed. ED was shown to be the end product from EDDA. ED was derivatized with dansyl chloride and was analyzed by HPLC. The final reaction product-dansyl derivative had the same retention time as ED-dansyl. Both peaks were collected and analyzed by mass spectrometry. The spectra of the two compounds were almost identical (Fig. 2). When ED (C2H8N2) reacted with two dansyl chlorides (C12H12ClNO2S), the derivative (C26H30N4O4S2) contained two dansyl groups attached to the two amino groups of the ED with the loss of two HCl molecules. The compound should have a mass of 526.4 Da. The mass spectra in a positive mode confirmed this compound. The peaks at 527.44 and 549.02 m/z were the parent compound with cation (H+ or Na+). The peaks at 234.95 and 293.08 m/z were breakdown products with the loss of a dansyl group. The peak at 170 m/z was a fragment of a dansyl group without the −SO2 group. The data showed that the final end product from EDDA oxidation by IDA oxidase is ED.
FIG. 2.

Mass spectra of the ED-dansyl adduct (A) and the end product-dansyl adduct (B).
Oxygen consumption and stoichiometric analysis of the enzymatic reaction.
When IDA oxidase was added to a reaction mixture without the artificial electron acceptor, oxygen consumption was observed (Fig. 3). With 5.0 μg of IDA oxidase in the reaction mixture, 127 nmol of O2 was consumed after approximately 6 min. About 60 nmol of oxygen was produced immediately after 450 U of catalase was added to the reaction mixture, indicating that O2 was quantitatively converted to hydrogen peroxide (H2O2) from IDA oxidation. In several separate experiments, the reactions were stopped at the point of 130 nmol of O2 consumption for glyoxylate analysis. The data showed that about 132 ± 6 (standard deviation of three samples) nmol of glyoxylate was produced. The determined molar ratio of oxygen consumption to H2O2 and glyoxylate production was close to 1. In a reaction mixture containing 250 nmol of IDA, 233.5 ± 60.9 (standard deviation of three samples) nmol of glycine was detected after the completion of the reaction. The molar ratio of glycine produced to IDA consumed was 0.93. Therefore, the complete reaction of IDA oxidation is a typical oxidase reaction as follows:
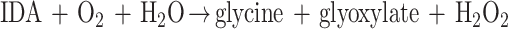 |
FIG. 3.

Oxygen consumption by IDA oxidase. After 6 min of the reaction, catalase was added as indicated by the arrow.
K3Fe(CN)6 was the preferred electron acceptor.
The rate of glyoxylate produced with O2 as electron acceptor was 4.3 ± 0.2 μmol min−1 mg of protein−1. The rate of glyoxylate production in reaction mixtures with K3Fe(CN)6 as artificial electron acceptor was determined to be 29.0 ± 1.0 μmol min−1 mg of protein−1. Thus, the enzymatic reaction was about seven times faster with K3Fe(CN)6 as the electron acceptor than with O2 as the electron acceptor. The preferential use of K3Fe(CN)6 as the electron acceptor was also shown in oxygen consumption experiments. Oxygen consumption was not observed in the presence of K3Fe(CN)6. As soon as K3Fe(CN)6 was completely reduced as evidenced by the disappearance of yellow, O2 consumption started. The ferricyanide reduction rates were identical under both aerobic and anaerobic conditions.
Kinetic analysis.
With IDA or EDDA as the substrate, the kinetic parameters of the enzyme were determined with Lineweaver-Burk plots of initial reaction rates of glyoxylate production in 25 mM Tris-HCl buffer (pH 8.0) at 3 min after the start of the reaction at different concentrations of IDA or EDDA. The apparent Km and kcat values for IDA were 3.97 ± 0.06 mM and 5.23 ± 0.08 s−1 (subunit molecular weight of 40,000), respectively. For EDDA, the apparent Km was 7.35 ± 0.21 mM and the kcat was 1.68 ± 0.01 s−1. The kcat/Km value, a measure of the enzyme's specificity, was 5.8 times higher for IDA than for EDDA, suggesting that IDA is the preferred substrate. The specific activity and kinetic parameters of the native IDA oxidase and C-fusion IDA oxidase were practically identical, indicating that the recombinant enzyme has retained essentially the same catalytic properties.
Determination of prosthetic group of the enzyme.
The purified IDA oxidase had a light yellow color with an absorption peak at 446 nm, which is characteristic for flavoproteins (25). The flavin prosthetic group in the protein was released by boiling, indicating that it is not covalently bound. The flavin extracted from IDA oxidase gave a single fluorescent spot with an Rf of 0.055, which is the same as the Rf of authentic flavin adenine dinucleotide (FAD), identifing the flavin as FAD. A solution containing 15.3 μM IdaA had an A446 of 0.17. The flavin content was calculated to be 15.0 μM, assuming that the associated flavin has the same molar extinction coefficient as free FAD (39). The molar ratio of flavin to protein was 0.98, indicating that each IDA oxidase subunit contains one FAD.
Sequence analysis.
A BLAST search (1) with the IdaA amino acid sequence revealed that IdaA had significant sequence similarities to several known oxidases that cleave C N bonds. When entire amino acid sequences were aligned by a GCG Gap program (14), IDA oxidase was 23% identical to glycine oxidase (GloX) in Bacillus subtilis (GenBank accession no. O31616) (26), 25% identical to d-nopaline oxidase (NoxB) in Agrobacterium tumefaciens (50), 27% identical to d-amino acid oxidase (DAAO) in porcine kidneys (GenBank accession no. P00371) (22), and 30% identical to hydrogen cyanide synthase (HcnC) in Pseudomonas fluorescens (GenBank accession no. AAC38596) (20). Sequence alignments of the N terminus of IDA oxidase with the N termini of related enzymes identified the conservative adenine dinucleotide binding domain (46) involved in FAD binding (Fig. 4). Results obtained by using the conserved domain database search available online at GenBank showed that the C-terminal amino acid sequence (residues 196 to 341) of IDA oxidase was 25% identical to the partial amino acid sequence (residues 175 to 320) of DAAO, which is the prototype of the FAD-dependent oxidase (22, 24, 42). Precisely, the C-terminal region of IDA oxidase was homologous to the substrate-binding domain of DAAO. The amino acid residues Arg-283 and Tyr-228 of DAAO are for substrate binding and are also conserved in IDA oxidase (Arg-307 and Tyr-250), NoxB (Arg-306), HcnC (Arg-344 and Tyr-288), and GloX (Arg-302 and Tyr-246) (20, 22, 26, 50). These sequence similarities further support our finding that the IDA-degrading enzyme is an oxidase.
FIG. 4.

The conserved FAD-binding domains (boxed) of IDA oxidase and related enzymes DAAO, HcnC, NoxB, and GloX.
DISCUSSION
IDA oxidase has been identified, purified, and characterized from the EDTA-degrading bacterium BNC1. The enzyme catalyzed the cleavage of C-N bonds in IDA and EDDA. It oxidized IDA to glyoxylate and glycine and EDDA to glyoxylate and ED. Uetz and Egli (44) have previously reported the detection and characterization of a membrane-bound IDA dehydrogenase from C. heintzii ATCC 29600. The IDA dehydrogenase that converts IDA to glycine and glyoxylate is coupled to the respiratory electron transport system with O2 as the terminal electron acceptor, in which O2 is reduced to H2O. Because of the requirement of the electron transport system for the function of the enzyme, IDA dehydrogenase has not been purified and has been characterized with membrane fractions. On the other hand, IDA oxidase is a soluble oxidase that reduces O2 directly to H2O2.
The cleavage of C-N bonds by flavin-containing oxidases has been extensively studied with the mitochondrial monoamine oxidases (2, 37), DAAO (22, 24, 42), monomeric sarcosine oxidase (43), and PAO (6). The oxidases remove two electrons from their substrates to form the corresponding iminium ions, and the FAD prosthetic group is reduced to FADH2. The iminium ions are spontaneously hydrolyzed with the elimination of the nitrogen moiety, and the FADH2 is oxidized by O2 to complete the reaction cycle. IDA oxidase is likely using a similar reaction mechanism for catalysis. Oxygen is used for FADH2 oxidation, and it can be replaced by K3Fe(CN)6.
IDA oxidase showed marginal activity for sarcosine and no detectable activity for NTA, EDTA, glycine, succinate, and fumarate. Its apparent high Km values for IDA and EDDA suggest that IDA and EDDA may not be the natural substrates for the enzyme or that the enzyme has been evolved from an existing protein. However, the relatively high kcat value of IDA oxidase may compensate the enzyme's catalytic efficiency. IDA oxidase specific activity was about onefold higher in the cell extracts of BNC1 cells grown with EDTA as the nitrogen source than with NH4Cl as the nitrogen source. This partial regulation indicates that either the regulation has not been completely evolved or that IDA oxidase has other cellular functions.
The bacterium BNC1 degrades NTA to IDA and glyoxylate by EDTA monooxygenase (32), and then IDA is converted to glycine and glyoxylate by IDA oxidase (Fig. 5). Both glycine and glyoxylate are normal metabolic intermediates and can be completely mineralized by common metabolic pathways. For EDTA degradation, EDTA monooxygenase oxidizes EDTA to EDDA in BNC1 (5, 7, 32), and IDA oxidase oxidizes EDDA to ED (Fig. 5). We assume that IDA oxidase first oxidizes EDDA to ethylenediaminemonoacetate (EDMA) and then to ED. However, EDMA is not commercially available and is not detected in this study. Thus, EDTA monooxygenase and IDA oxidase together can channel NTA to common metabolic intermediates but convert EDTA only to ED (Fig. 5). ED is structurally similar to putrescine, a common biological diamine present in bacterial cell membranes (15). The bacterium BNC1 must have enzymes to further metabolize ED to gain the nitrogen source from it.
FIG. 5.

NTA (A) and EDTA (B) degradation in strain BNC1. EmoA is EDTA monooxygenase.
ACKNOWLEDGMENTS
This research was supported by the Natural and Accelerated Bioremediation Research Program, Office of Biological and Environmental Research, U.S. Department of Energy (DOE). Pacific Northwest National Laboratory is operated for the DOE by Battelle Memorial Institute under contract DE-AC06-76RLO 1830.
REFERENCES
- 1.Altschul S F, Lipman D J. Protein database searches for multiple alignments. Proc Natl Acad Sci USA. 1990;87:5509–5513. doi: 10.1073/pnas.87.14.5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson A H, Kuttab S, Castagnoli N., Jr Deuterium isotope effect studies on the MAO-B catalyzed oxidation of 4-benzyl-1-cyclopropyl-1, 2,3,6-tetrahydropyridine. Biochemistry. 1996;35:3335–3340. doi: 10.1021/bi9526701. [DOI] [PubMed] [Google Scholar]
- 3.Auling G, Busse H-J, Egli T, El-Banna T, Stackebrandt E. Description of the gram-negative obligately aerobic, nitrilotriacetate (NTA)-utilizing bacteria as Chelatobacter heintzii, gen. nov., sp. nov., and Chelatococcus asaccharovorans, gen. nov., sp. nov. Syst Appl Microbiol. 1993;16:104–112. [Google Scholar]
- 4.Avers J A. Decontamination of nuclear reactors and equipment. New York, N.Y: The Ronald Press Co.; 1970. [Google Scholar]
- 5.Belly R T, Lauff J J, Goodhue C T. Degradation of ethylenediaminetetraacetic acid by microbial populations from an aerated lagoon. Appl Microbiol. 1975;29:787–794. doi: 10.1128/am.29.6.787-794.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binda C, Coda A, Angelini R, Federico R, Ascenzi P, Mattevi A. A 30 Å-long U-shaped catalytic tunnel in the crystal structure of polyamine oxidase. Structure Fold Des. 1999;7:265–276. doi: 10.1016/s0969-2126(99)80037-9. [DOI] [PubMed] [Google Scholar]
- 7.Bohuslavek J, Payne J W, Liu Y, Bolton H J, Xun L. Cloning, sequencing, and characterization of a gene cluster involved in EDTA degradation from the bacterium BNC1. Appl Environ Microbiol. 2001;67:688–695. doi: 10.1128/AEM.67.2.688-695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 9.Cleveland J M, Rees T F. Characterization of plutonium in Maxey Flats radioactive trench leachates. Science. 1981;200:1506–1509. doi: 10.1126/science.212.4502.1506. [DOI] [PubMed] [Google Scholar]
- 10.Cripps R E, Noble A S. The metabolism of nitrilotriacteate by a pseudomonad. Biochem J. 1973;136:1059–1068. doi: 10.1042/bj1361059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daubaras D L, Saido K, Chakrabarty A M. Purification of hydroxyquinol 1,2-dioxygenase and maleylacetate reductase: the lower pathway of 2,4,5-trichlorophenoxyacetic acid metabolism by Burkholderia cepaciaAC1100. Appl Environ Microbiol. 1996;62:4276–4279. doi: 10.1128/aem.62.11.4276-4279.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egli T, Weilenmann H U, El-Banna T, Auling G. Gram-negative, aerobic, nitrilotriacetate-utilizing bacteria from wastewater and soil. Syst Appl Microbiol. 1988;10:297–305. [Google Scholar]
- 13.Epstein S. Toxicological and environmental implications on the use of nitrilotriacetic acid as a detergent builder. Int J Environ Stud. 1972;2:291–311. [Google Scholar]
- 14.Genetics Computer Group. Program manual for GCG package, version 10. Madison, Wis: Genetics Computer Group; 1998. [Google Scholar]
- 15.Ishizuka H, Horinouchi S, Beppu T. Putrescine oxidase of Micrococcus rubens: primary structure and Escherichia coli. J Gen Microbiol. 1993;139:425–432. doi: 10.1099/00221287-139-3-425. [DOI] [PubMed] [Google Scholar]
- 16.Klüner T, Hempel D C, Nörtemann B. Metabolism of EDTA and its metal chelates by whole cells and cell-free extracts of strain BNC1. Appl Microbiol Biotechnol. 1998;49:194–201. [Google Scholar]
- 17.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 18.Lau-Cam C A, Roos R W. Simultaneous high performance liquid chromatographic determination of theophylline and ethylenediamine in aminophylline dosage forms as their dansyl derivatives. J Liq Chromatogr. 1991;14:1939–1956. [Google Scholar]
- 19.Lauff J J, Steele D B, Coogan L A, Breitfeller J M. Degradation of the ferric chelate of EDTA by a pure culture of an Agrobacteriumsp. Appl Environ Microbiol. 1990;56:3346–3353. doi: 10.1128/aem.56.11.3346-3353.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laville J, Blumer C, Von Schroetter C, Gaia V, Defago G, Keel C, Haas D. Characterization of the hcnABC gene cluster encoding hydrogen cyanide synthase and anaerobic regulation by ANR in the strictly aerobic biocontrol agent Pseudomonas fluorescensCHA0. J Bacteriol. 1998;180:3187–3196. doi: 10.1128/jb.180.12.3187-3196.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim C K. HPLC of small molecules—a practical approach. Oxford, United Kingdom: IRL Press; 1986. [Google Scholar]
- 22.Mattevi A, Vanoni M A, Todone F, Rizzi M, Teplyakov A, Coda A, Bolognesi M, Curti B. Crystal structure of d-amino acid oxidase: a case of active site mirror-image convergent evolution with flavocytochrome b2. Proc Natl Acad Sci USA. 1996;93:7496–7501. doi: 10.1073/pnas.93.15.7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McFadden K M. Organic components of nuclear wastes and their potential altering radionuclide distribution when released to soil. Springfield, Va: National Technical Information Service; 1980. [Google Scholar]
- 24.Mizutani H, Miyahara I, Hirotsu K, Nishina Y, Shiga K, Setoyama C, Miura R. Three-dimensional structure of porcine kidney d-amino acid oxidase at 3.0 A resolution. J Biochem. 1996;120:14–17. doi: 10.1093/oxfordjournals.jbchem.a021376. [DOI] [PubMed] [Google Scholar]
- 25.Muller F, Mayhew S G. Temperature-difference spectra of flavins and flavoproteins. Methods Enzymol. 1980;66:350–360. doi: 10.1016/0076-6879(80)66480-5. [DOI] [PubMed] [Google Scholar]
- 26.Nishiya Y, Imanaka T. Purification and characterization of a novel glycine oxidase from Bacillus subtilis. FEBS Lett. 1998;438:263–266. doi: 10.1016/s0014-5793(98)01313-1. [DOI] [PubMed] [Google Scholar]
- 27.Nörtemann B. Biodegradation of EDTA. Appl Microbiol Biotechnol. 1999;51:751–759. doi: 10.1007/s002530051458. [DOI] [PubMed] [Google Scholar]
- 28.Nörtemann B. Total degradation of EDTA by mixed cultures and a bacterial isolate. Appl Environ Microbiol. 1992;58:671–676. doi: 10.1128/aem.58.2.671-676.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nörtemann B, Imberg B, Hempel D C. Biodegradation of ethylenediaminetetraacetate (EDTA) In: Verachert H, Verstraete W, editors. International Symposium on Environmental Biotechnology. Antwerp, Belgium: Koninklijke Vlaamse Ingenieursverenigung ZVW; 1991. pp. 259–262. [Google Scholar]
- 30.Parry R J, Li W. An NADPH:FAD oxidoreductase from the valanimycin producer, Streptomyces viridifaciens. Cloning, analysis, and overexpression. J Biol Chem. 1997;272:23303–23311. doi: 10.1074/jbc.272.37.23303. [DOI] [PubMed] [Google Scholar]
- 31.Payne J W. M.S. thesis. Pullman: Washington State University; 1999. [Google Scholar]
- 32.Payne J W, Bolton H J, Campbell J A, Xun L. Purification and characterization of EDTA monooxygenase from the EDTA-degrading bacterium BNC1. J Bacteriol. 1998;180:3823–3827. doi: 10.1128/jb.180.15.3823-3827.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piciulo P L, Adams J W, Davis M S, Milian L W, Anderson C I. Release of organic chelating agents from solidified decontamination wastes. Springfield, Va: National Technical Information Service; 1986. [Google Scholar]
- 34.Pickaver A H. The production of N-nitrosoiminodiacetate from nitrilotriacetate and nitrite by microorganism growing in mixed culture. Soil Biol Biochem. 1976;8:13–17. [Google Scholar]
- 35.Robinson J, Cooper J M. Method of determining oxygen concentrations in biological media, suitable for calibration of the oxygen electrode. Anal Biochem. 1970;33:390–399. doi: 10.1016/0003-2697(70)90310-6. [DOI] [PubMed] [Google Scholar]
- 36.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 37.Silverman R B. Radical ideas about monoamine oxidase. Acc Chem Res. 1995;28:335–342. [Google Scholar]
- 38.Sisto J D, Jackel M, Ishikawa M. Chemical economics handbook. Menlo Park, Calif: SRI Consulting; 1996. p. 515.50000A. [Google Scholar]
- 39.Strickland S, Massey V. The purification and properties of the flavoprotein melilotate hydroxylase. J Biol Chem. 1973;248:2944–2952. [PubMed] [Google Scholar]
- 40.Thomas R A, Lawlor K, Bailey M, Macaskie L E. Biodegradation of metal-EDTA complexes by an enriched microbial population. Appl Environ Microbiol. 1998;64:1319–1322. doi: 10.1128/aem.64.4.1319-1322.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tiedje J M, Mason B B, Warren C B, Malec E J. Metabolism of nitrilotriacetate by cells of Pseudomonasspecies. Appl Microbiol. 1973;25:811–818. doi: 10.1128/am.25.5.811-818.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Todone F, Vanoni M A, Mozzarelli A, Bolognesi M, Coda A, Curti B, Mattevi A. Active site plasticity in d-amino acid oxidase: a crystallographic analysis. Biochemistry. 1997;36:5853–5860. doi: 10.1021/bi9630570. [DOI] [PubMed] [Google Scholar]
- 43.Trickey P, Wagner M, Jorns M S, Mathews F S. Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure Fold Des. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 44.Uetz T, Egli T. Characterization of an inducible, membrane-bound iminodiacetate dehydrogenase from Chelatobacter heintziiATCC 29600. Biodegradation. 1993;3:423–434. [Google Scholar]
- 45.Uetz T, Schneider R, Snozzi M, Egli T. Purification and characterization of a two-component monooxygenase that hydroxylates nitrilotriacetate from “Chelatobacter” strain ATCC 29600. J Bacteriol. 1992;174:1179–1188. doi: 10.1128/jb.174.4.1179-1188.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wierenga R K, Terpstra P, Hol W G J. Prediction of the occurrence of the ADP-binding βαβ-fold in proteins, using an amino acid sequence fingerprint. J Mol Biol. 1986;187:101–107. doi: 10.1016/0022-2836(86)90409-2. [DOI] [PubMed] [Google Scholar]
- 47.Witschel M, Nagel S, Egli T. Identification and characterization of the two-enzyme system catalyzing oxidation of EDTA in the EDTA-degrading bacterial strain DSM 9103. J Bacteriol. 1997;179:6937–6943. doi: 10.1128/jb.179.22.6937-6943.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu Y, Mortimer M W, Fisher T S, Kahn M L, Brockman F J, Xun L. Cloning, sequencing, and analysis of a gene cluster from Chelatobacter heintziiATCC 29600 encoding nitrilotriacetate monooxygenase and NADH:flavin mononucleotide oxidoreductase. J Bacteriol. 1997;179:1112–1116. doi: 10.1128/jb.179.4.1112-1116.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xun L, Sandvik E R. Characterization of 4-hydroxyphenylacetate 3-hydroxylase (HpaB) of Escherichia colias a reduced flavin adenine dinucleotide-utilizing monooxygenase. Appl Environ Microbiol. 2000;66:481–486. doi: 10.1128/aem.66.2.481-486.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zanker H, Lurz G, Langridge U, Langridge P, Kreusch D, Schroder J. Octopine and nopaline oxidases from Ti plasmids of Agrobacterium tumefaciens: molecular analysis, relationship, and functional characterization. J Bacteriol. 1994;176:4511–4517. doi: 10.1128/jb.176.15.4511-4517.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]