

Abstract
A series of microcosm experiments was performed using serial dilutions of a sewage microbial community to inoculate a set of batch cultures in sterile sewage. After inoculation, the dilution-defined communities were allowed to regrow for several days and a number of community attributes were measured in the regrown assemblages. Based upon a set of numerical simulations, community structure was expected to differ along the dilution gradient; the greatest differences in structure were anticipated between the undiluted–low-dilution communities and the communities regrown from the very dilute (more than 10−4) inocula. Furthermore, some differences were expected among the lower-dilution treatments (e.g., between undiluted and 10−1) depending upon the evenness of the original community. In general, each of the procedures used to examine the experimental community structures separated the communities into at least two, often three, distinct groups. The groupings were consistent with the simulated dilution of a mixture of organisms with a very uneven distribution. Significant differences in community structure were detected with genetic (amplified fragment length polymorphism and terminal restriction fragment length polymorphism), physiological (community level physiological profiling), and culture-based (colony morphology on R2A agar) measurements. Along with differences in community structure, differences in community size (acridine orange direct counting), composition (ratio of sewage medium counts to R2A counts, monitoring of each colony morphology across the treatments), and metabolic redundancy (i.e., generalist versus specialist) were also observed, suggesting that the differences in structure and diversity of communities maintained in the same environment can be manifested as differences in community organization and function.
Ecological diversity, the variety and abundance of species in different habitats and communities, is one of the central themes of ecology. Diversity is commonly thought to be a useful indicator of the well-being of an ecological system; however, there is considerable debate over the role diversity plays in ecosystem function (4, 18, 23, 24, 26, 32, 33, 36). Most of this uncertainty arises from the practical limitations of measuring and manipulating diversity for experimental studies. Testing the effects of diversity on any community property or ecosystem function requires knowledge of the diversity of the community under examination; however, there are no methods currently available that allow microbial diversity to be measured. Numerous procedures are available for monitoring changes in community structure (e.g., culture-based analyses, community level physiological profiling (CLPP), analysis of the lipid content of microbial cells, and molecular genetic techniques); these approaches each have biases and limitations that are well documented (for reviews, see references 1, 3, 8, 13, 16, 28, 39, 41, and 46).
Despite the inability to directly measure diversity, Garland et al. (9, 11) and Morales et al. (25) successfully used dilution to manipulate microbial diversity for several applications. The premise behind these studies was that dilution of a relatively diverse community would remove rare organism types, creating mixtures of cells differing in species richness. Regrowth of the diluted mixtures should then produce cultures of roughly the same biomass but differing in overall diversity. In these studies, the various dilution-diversity communities responded differently to invasion attempts (11, 25) and to environmental stress (11), with more diverse (less dilute) communities being more stable and better able to withstand invasion. However, with no good way to place a numerical value on microbial diversity, the magnitude of the differences being evaluated remains unknown.
The present work sought to define the relationship between dilution and resultant changes in diversity and community structure. First, several numerical simulations were performed in order to develop a set of expectations about how overall diversity (expressed as the Shannon-Wiener index), richness (number of species or types of organisms in the community), and evenness (the relative distribution of individuals among these types) change with dilution. Next, a series of microcosm experiments was conducted using batch cultures of sterile sewage inoculated with serial dilutions of fresh sewage. After regrowth of these batch cultures, several methods were used to characterize the communities, including traditional microbiological procedures, CLPP, and molecular genetic techniques. The regrown communities differed along the dilution gradient, and the results followed a pattern similar to that observed in the simulated dilution of a relatively uneven mixture of organisms. The results of this work will be useful in planning future studies, as the ability to create natural communities systematically differing in complexity could allow researchers to manipulate diversity, perhaps in a quantifiable way, while evaluating its relationship to other community level properties (e.g., stability, invasibility, or ecosystem function).
MATERIALS AND METHODS
Numerical simulations.
To examine the theoretical effect of dilution on community inoculum composition, a series of numerical simulations (coded in MATLAB) was performed. Communities were constructed by assigning each of 106 individuals a random species identification based on a normal distribution of integers from 1 to 1,000. The mean of this distribution was set at 500; the variance (var) was adjusted in order to create communities of different initial evenness (Fig. 1A). The var levels used were 100, 250, 1,000, and 20,000, and a perfectly even initial community was also generated (1,000 types, each containing 1,000 individuals). Dilution of each of these five initial communities was simulated by randomly selecting 1/10 of the individuals from the array representing the undiluted community. The species identification of each individual in this subset was copied to a second array, which served as the initial community for the next dilution in the series (dilutions extended through 10−5). For each community, at each dilution level, richness, evenness, and diversity were calculated. Richness (S) was taken to be the number of organism types (species) in the community. Diversity was expressed as the Shannon-Wiener index (H′) as fol- i lows: H′ = Σ pi ln pi, where i indicates each species or category and pi is the 1 proportion of individuals of each species (37). Evenness (E) was calculated as E = H′/H′max, where H′max = In S (31).
FIG. 1.

(A) Distribution of individuals among 1,000 types in the initial communities used in simulations. Note that both total abundance and richness were the same in each of the five communities. The mean of each distribution was set at 500, and the variance was altered to simulate communities with a dominant (var = 100, 250, or 1,000) or relatively even (var = 20,000 or even) distribution. (B to D) Simulation results showing how community structure differed in the various initial communities for each serial dilution. The x axis represents the negative exponent of the dilution factor (e.g., 4 corresponds to a 10−4 dilution), and the y axis represents richness (number of types or species) (B), evenness (C), or the calculated value of the Shannon-Wiener diversity index (D).
Batch culture experiments. (i) Microcosm setup.
Raw sewage was collected from the Cape Canaveral Air Station Wastewater Treatment Facility (Kennedy Space Center, Fla.) and used as the inoculum for the microcosm experiments. A single large sample (2 liters) was collected from the equilibration basin and allowed to settle for approximately 2 h to remove large particles. Tenfold serial dilutions (through 10−6) of the supernatant were prepared in autoclaved sewage; each of these different dilutions then served as an inoculum for a series of batch culture incubations. Before dilution, the concentration of cells in the supernatant, determined by acridine orange direct counting (AODC; 15), was 1.8 × 106 cells/ml.
The batch cultures were established by adding 1 ml of inoculum to 60 ml of autoclaved sewage in a 125-ml Erlenmeyer flask. Seven treatments (100 through 10−6) were maintained in this experiment, with three replica communities at each dilution. All flasks were capped with sterile foam plugs to prevent contamination and kept on a shaker table, operated at 150 rpm, to maintain aerobic conditions. Each day, 20 ml of liquid was removed from each flask and replaced with 20 ml of sterile sewage. After 9 days (three retention times), flasks were harvested and analyzed.
(ii) Cultural counts and diversity of colony morphology.
For each flask, a serial dilution of the regrown community was plated, in duplicate, onto both R2A agar (35) and sterile sewage medium (SM; prepared by mixing 15 g of agar per liter of sewage supernatant). Plates were incubated at room temperature (ca. 23°C), and the number of CFU on R2A agar was determined after 3 days. Growth on SM was evaluated after 6 days.
The diversity of colony morphologies on R2A was compared across the different dilution treatments. For each flask, two plates were selected; on each plate, 25 colonies were randomly chosen and colony morphology was described based on size, pigmentation, form, elevation, and surface. Richness (number of distinct colony morphologies), evenness, and diversity (Shannon-Wiener diversity index) were then calculated.
(iii) CLPP.
For each flask, a 10−1 dilution of the microbial community was prepared (in sterile water) and inoculated into Biolog GN microplates, which were then incubated at room temperature (ca. 23°C). Color formation in each of the 96 wells of each plate was monitored by periodically (every 2 to 4 h) measuring the A590 using a Biotek EL 320 microplate reader. Data were normalized using a blank-corrected average well color development of 0.75 absorbance unit and analyzed using a principal-components analysis (PCA) (8, 10).
(iv) Dilution-extinction analysis of CLPP.
Dilution-extinction analysis was performed on a subset of the regrown communities (one replicate flask from each of the 100, 10−2, 10−4, and 10−6 dilution treatments) to determine the relationship between cell density (I) and functional richness (number of positive wells, R) (9). Serial dilutions of the microbial suspensions were inoculated into Biolog GN microplates and incubated at room temperature for 7 days, and A590 was measured. A positive response was defined as any value greater than 0.25 absorbance unit (after correction for the control well), and a hyperbolic model, R = (Rmax × I)/(KI + I), where Rmax equals the maximum (asymptotic level) of R and KI is the value of I when R is one-half of Rmax, was fitted to the data (9).
(v) Molecular analysis of whole-community DNA. (a) DNA extraction and quantification.
At harvest, an approximately 40-ml sample was collected from each flask and the suspended microbial community was concentrated by centrifugation (23,000 × g, 20 min). The cell pellet was resuspended in 200 μl of phosphate-buffered saline and stored at −20°C. Whole-community DNA was extracted using the High Pure PCR Template Preparation Kit (Boehringer Mannheim, Indianapolis, Ind.) and quantified using the PicoGreen double-stranded DNA quantification reagent (Molecular Probes, Eugene, Oreg.).
(b) AFLP.
Amplified fragment length polymorphism (AFLP) was completed using the Perkin-Elmer Microbial Fingerprinting Kit (PE Applied Biosystems, Foster City, Calif.) following the manufacturer's instructions for analysis of individual bacterial strains. Three different pairs of primers, each with a different fluorescent label, were used for selective AFLP amplification: EcoRI-AA (JOE labeled) with MseI-CA, EcoRI-AC (FAM labeled) with MseI-CC, and EcoRI-AT (NED labeled) with MseI-CT. For the complete primer and adapter sequences and an explanation of the primer selection criteria used, see the PE Applied Biosystems AFLP Microbial Fingerprinting Protocol (PE Applied Biosystems, Foster City, Calif.).
Selective amplification products were resolved using an ABI Prism 310 Genetic Analyzer by following the manufacturer's instructions with slight modification; for each sample with each primer pair, 1 μl of PCR product was analyzed using a sample injection time of 10 s. Data were analyzed using the Genotyper software (PE Applied Biosystems), and the presence or absence of each peak in each sample was coded as 1 or 0. Such a data matrix was prepared for each primer pair, and the information from the three primer pairs was pooled into a single large data set. The Jaccard coefficient was used to determine distances between samples (relative similarity), and a cluster analysis (unweighted pair group clustering using arithmetic averages and between-groups linkage) was performed. A bootstrapping analysis was then used to assess the significance of each group and subgroup in the cluster analysis (6).
A PCA was also performed on the original pooled data matrix (SPSS 9.0), and plots of the first two principal components (PCs) were made. As PCA is not mathematically appropriate for use with binary data, its application in this study was solely to aid in visualization of the relationships among the samples and not for statistical evaluation. Such an approach has been used several times to compare samples profiled using a variety of similar genetic techniques (6, 7, 47, 48); PCA generally provides the same information (groupings and relative distances among samples) as the above-outlined cluster analysis.
(c) T-RFLP.
Each T-RFLP (terminal restriction fragment length polymorphism) reaction mixture (50 μl) contained 25 ng of community DNA; 10 mM Tris-Cl (pH 8.3); 50 mM KCl; 1.5 mM MgCl2; 200 μM each dATP, dCTP, dGTP, and dTTP; each primer at a concentration of 0.1 μM; and 1.25 U of Taq DNA polymerase (21). The bacterial 16S rRNA gene was amplified using two primers: 1392 Reverse (5′ ACGGGCGGTG TGTRC) and 27 Forward (5′ AGAGTTTGATCCTGGCTCAG [labeled with the fluorescent tag 6-FAM]). The PCR program was 94°C for 3 min, followed by 30 cycles of 94°C for 30 s, 56°C for 45 s, and 72°C for 2 min, with a final extension at 72°C for 3 min. PCR products were purified using the Wizard PCR Preps DNA Purification System (Promega, Madison, Wis.) and eluted in a final volume of 50 μl. Portions (10 μl) of the purified PCR product were then digested with either the HhaI or MspI restriction enzyme, using the manufacturer's recommended reaction buffer and 20 U of enzyme (New England Biolabs, Beverly, Mass.). Digests were incubated at 37°C for 4 h.
The lengths of the fluorescently labeled terminal restriction fragments were determined for each sample using the ABI 310 Genetic Analyzer. Three-microliter portions of each digested product were mixed with 24 μl of deionized formamide and 1 μl of GeneScan-1000 size standard (PE Applied Biosystems), denatured at 95°C for 5 min, and quickly chilled on ice. Electrophoresis was performed using the same conditions as for AFLP but with a 40-min run time. Data were analyzed using the GeneScan software with a peak height detection of 100. As with the AFLP analysis, the presence or absence of each T-RFLP in each sample was determined and the data from each restriction enzyme were pooled for cluster analysis, bootstrapping analysis, and PCA.
RESULTS
Numerical simulations.
The results of the numerical simulations show that while microbiologists generally consider dilution to be a linear process, the response of various community level parameters (richness, evenness, and diversity) to such a manipulation may produce nonlinear results (Fig. 1B, C, and D). There was no change in the diversity (H′) of the even community with dilution until the number of individuals (103 individuals) equaled the number of species (103 types) at the 10−3 dilution. With further dilution, H′ decreased in response to the rapid loss of species richness (Fig. 1B) although evenness remained 1.0 (Fig. 1C). In each of the other initial communities, a similar trend was observed; in general, H′ remained constant until the 10−3 (var = 250, 1,000, and 20,000) or 10−4 (var = 100) dilution. After this point, diversity decreased, corresponding to the loss of species richness and an increase in community evenness. At the end of the dilution series (10−5), the H′ of all of the communities decreased to the maximum theoretical value of H′ for a perfectly even distribution among 10 organisms. The sole exception to this was in the community created by setting var = 1,000; here, the value of H′ at the 10−5 dilution was 2.16 because only 9 species were recovered in that particular simulation whereas 10 were recovered in all of the others.
The richness of the simulated communities decreased with increasing dilution, but the magnitude of this change varied depending on the initial evenness of the mixture (Fig. 1B). For communities with low initial evenness (e.g., var = 100 or 250), richness decreased rapidly with the first dilution. For initial communities that were more even, the number of species lost in the first dilution was small; in the perfectly even community, no species were lost. Moreover, the relative distribution of organisms in the perfectly even community did not change upon dilution (Fig. 1C). For all of the other communities, evenness increased with dilution, approaching a theoretical maximum of 1.0 at the 10−5 dilution (except for var = 1,000, as discussed above).
Batch culture experiments. (i) Microscopic and cultural counts.
After 9 days of regrowth, each experimental community was sampled and AODC and cultural counts were performed (Table 1). An analysis of variance was used to determine whether each parameter varied significantly across the different dilution-diversity treatments, and a modified least significant difference (Bonferroni) test was used for multiple comparisons. Total cell concentrations were similar across the first several dilution treatments (100 through 10−4), but in the communities regrown from each of the higher-dilution inoculum (10−5 and 10−6), abundance was significantly greater (df = 20, F = 25.7, P < 0.00001). Cultural counts on both SM and R2A agar showed a trend similar to the AODC, with significantly greater concentrations of organisms in the higher-dilution–lower-diversity treatments (R2A, df = 20, F = 4.3, P = 0.0116; SM, df = 19, F = 3.6, P = 0.0259).
TABLE 1.
Results of direct and plate count analysesa
| Dilutionb | AODC (106 cells/ml) | R2A count (106 CFU/ml) | % Culturability on R2A | SM count (106 CFU/ml) | % Culturability on SM | Ratio of SM count to R2A count |
|---|---|---|---|---|---|---|
| 100 | 22.0 ± 10.0* | 0.7 ± 0.4* | 4.5* | 0.6 ± 0.2* | 3.2* | 1.1 |
| 10−1 | 13.0 ± 8.6* | 0.7 ± 0.5* | 5.1* | 1.2 ± 0.7* | 10.0* | 2.1 |
| 10−2 | 24.0 ± 14.0* | 1.4 ± 1.2* | 7.5* | 2.2 ± 1.2* | 13.3* | 2.2 |
| 10−3 | 30.0 ± 15.0* | 4.6 ± 2.7* | 14.3*† | 5.2 ± 1.7*† | 18.3* | 1.3 |
| 10−4 | 39.0 ± 2.8*† | 5.6 ± 1.7* | 14.4*† | 5.9 ± 1.6*† | 15.3* | 1.2 |
| 10−5 | 73.0 ± 19.0† | 23.6 ± 11.0*† | 32.4† | 12.4 ± 3.8*† | 18.0* | 0.6 |
| 10−6 | 110.0 ± 9.0‡ | 204.0 ± 1.9‡c | 100.0†d | 58.1 ± 83.2† | 44.1* | 0.8 |
Each value is the average ± 1 standard deviation (when listed).
Dilution refers to the original dilution used as the inoculum for each regrown community. Each superscript symbol denotes a significantly different group of communities determined as described in the text; significance was not evaluated for the ratio of SM counts to R2A counts.
On R2A plates spread from two of the three 10−6 treatment flasks, colony growth was too extensive and data were recorded as too numerous to count. The value presented here, and used in all further calculations, was established by assuming a count of 300 CFU on the most-dilute plate, for each of those replica flasks, and averaging this with the counts obtained for the remaining flask.
In the communities regrown from the 10−6 dilution, growth on R2A exceeded the AODC and so a culturability of 100% was inferred.
Percent culturability on R2A agar (R2A counts divided by AODC) showed a major increase from the undiluted inocula to the 10−6 dilution (Table 1). The various treatments could be separated into three statistically significant subgroups as follows: 1, undiluted (100) through 10−4; 2, 10−3 through 10−5; 3, 10−6 (df = 20, F = 61.01, P < 0.00001). The average percent culturability for each subgroup was as follows: 1, 9%; 2, 20%; 3, 100%. Percent culturability on SM was also calculated, and although the results were not statistically significant (df = 19, F = 1.97, P = 0.1445), the same general trend was observed. Using the subgroupings defined in the R2A analysis, average percent culturabilty on SM varied as follows: 1, 12%; 2, 17%; 3, 44%. The average ratio of the SM counts to the R2A counts was also calculated from these data (Table 1). For the lower-dilution treatments, growth was greater on SM than on R2A agar although the difference between the two was generally not large. However, this trend was reversed in the high-dilution–low-diversity treatments (10−5 and 10−6) where growth on R2A plates was substantially greater than on SM.
(ii) Diversity of colony morphologies.
The diversity, richness, and evenness of R2A colony morphotypes were calculated for each of the dilution treatments (Fig. 2). Because of the high growth that occurred on all of the plates spread from the regrown 10−6 dilution community, it was not possible to evaluate these characteristics for that treatment. However, only three colony types could be distinguished on these plates and all three were distinctly different from the colony morphologies described in the other treatments. Overall, colony diversity was highest in the communities regrown from the undiluted inoculum and decreased with increasing dilution (Fig. 2A). The greatest change in colony diversity was observed between the undiluted (100) and 10−1 regrown communities. Richness decreased along the dilution-diversity gradient, and the most types were also lost after the first dilution (Fig. 2B). In general, evenness increased along the dilution-diversity gradient (Fig. 2C) although a decrease in evenness was observed between the undiluted (100) and 10−1 dilution treatments. The distribution of each colony type across each treatment was also examined, and 40% of the colony morphologies encountered in the low-dilution–high-diversity treatments (the 100 and 10−1 treatments were pooled for this calculation) were not recovered from any of the other regrown communities (see Table 3). Furthermore, the colony morphologies identified in the highest-dilution–lowest-diversity (10−6) treatment were all unique.
FIG. 2.

Results of R2A colony morphology comparison. All values are reported as the average per R2A plate ± 1 standard error. Each value was calculated by comparing 25 randomly selected colonies on each plate, using two replicate plates per flask and three flasks for each treatment. The sole exception to this was the 10−4 treatment, where only two of the replicate flasks were compared. The x axis in each of these graphs represents the negative exponent of the dilution factor used to create the original inoculum (e.g., 4 corresponds to a 10−4 dilution). The y axis represents diversity of colony morphologies based upon the Shannon-Wiener diversity index (A), richness (the number of distinct colony morphotypes) (B), or evenness (C).
TABLE 3.
Comparison of cultural and genetic procedures
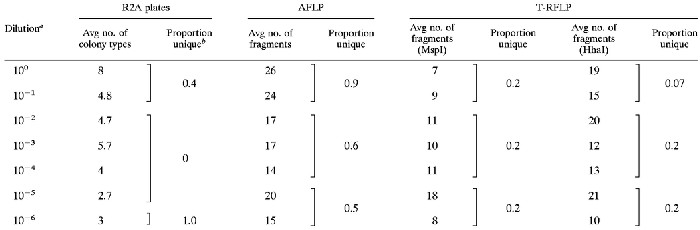 |
Dilution refers to the original dilution used as the inoculum for each regrown community.
Number of unique colony types (or bands) in a group of treatments divided by the total number of colony types (or bands) encountered in each treatment group. Unique refers to a colony type (or band) that was present in a particular group of treatments (e.g., 100 and 10−1) that was not present in either of the other two treatment groups.
(iii) CLPP.
PCA of the CLPP data showed that the various communities differed in their overall patterns of carbon utilization (Fig. 3). An analysis of variance was performed on the scores from the first two PCs, and two homogeneous subsets were established. The communities regrown from the undiluted (100) through the 10−4 dilution inocula were significantly different from those regrown from the 10−5 and 10−6 dilution treatments. This difference was due primarily to variation in the PC 1 scores (df = 20, F = 18.2, P < 0.00001); PC 2 did not contribute significantly to this separation (df = 20, F = 2.23, P = 0.102).
FIG. 3.

Results of PCA of the CLPP data. Each point represents the average for three replicate flasks maintained at each dilution; error bars represent ± 1 standard error. Each treatment is identified by the negative exponent of the dilution factor used to create the original inoculum (e.g., 4 corresponds to a 10−4 dilution). The percentage of variance explained by each PC is provided.
(iv) Dilution-extinction analysis of CLPP.
The undiluted (100) and 10−2, 10−4, and 10−6 dilution treatments were examined using dilution-extinction analysis of functional characters in the CLPP assays. Plots were made of the number of positive tests obtained in the dilutions made from each regrown community versus the number of cells (AODC) inoculated into each well of the BIOLOG plate (Fig. 4), and the data were fitted with a rectangular hyperbola to estimate the parameters Rmax and KI (Table 2). Rmax decreased along the dilution-diversity gradient; however, considering the confidence intervals about these estimates, it cannot be concluded that this decrease was significant. KI decreased significantly along the dilution-diversity gradient; higher values of KI were found for communities that were predicted to have a higher diversity based upon the extent of dilution.
FIG. 4.

Results of dilution-extinction analysis of CLPP for each regrown community. The x axis represents the inoculum density (as measured by AODC) used in each CLPP assay (presented on a log10 scale). The y axis is the number of positive tests for each incubation. The results are presented as fitted lines generated by modeling the untransformed data with a right rectangular hyperbola; the associated regression statistics for this fit are given in Table 2. The curvature of the regression lines at lower inoculum levels is an artifact of the log scaling of the x axis.
TABLE 2.
Results of dilution-extinction analysis of CLPPa
| Dilutionb | Rmax | KI (104 cells/ml) | Rc |
|---|---|---|---|
| 100 | 163.5 ± 89.9 | 250 | 0.95 |
| 10−2 | 71.6 ± 9.4 | 5.4 ± 3.4 | 0.90 |
| 10−4 | 76.9 ± 8.6 | 3.2 ± 1.5 | 0.92 |
| 10−6 | 65.5 ± 7.1 | 0.31 ± 0.2 | 0.82 |
Error terms are ± 1 standard error.
Dilution refers to the original dilution used as the inoculum for the regrown communities.
R, Multiple correlation coefficient.
(v) AFLP.
Combined, the three AFLP primers generated a total of 106 unique PCR fragments. On average, each sample contained 22 fragments; nearly all (90%) of the bands encountered in the low-dilution treatments (100 and 10−1) were unique, while 50% of the fragments observed in the high-dilution treatments (10−5 and 10−6) were not encountered in any of the other treatments (Table 3).
PCA and cluster analysis of the AFLP data showed that the microbial communities in this experiment could be separated into three distinct groups based on overall genetic composition. The communities regrown from the undiluted inocula (and one of the replicas from the 10−1 dilution) were most unique, a second group was formed from the communities regrown from the middle-dilution inocula (10−1, 10−2, 10−3, 10−4, and one of the 10−5 replicas), and the third cluster included the communities regrown from the very dilute inocula (10−5 and 10−6). This pattern is most easily visualized on the PC plot (Fig. 5), although cluster analysis produced the same separations (results not shown). A bootstrapping procedure (using 100 replications) was performed to assess the significance of the groupings obtained in the cluster analysis (6). The three clusters outlined above were recovered 93% of the time; this high value suggests that the separation was very well supported by the data and represents a significant difference in overall structure among the three sets of communities.
FIG. 5.

Results of PCA of the AFLP profiles. Data are presented for each of three replicate flasks for each treatment, and each value corresponds to the negative exponent of the dilution factor used to create the original inoculum (e.g., 4 corresponds to a 10−4 dilution). The percent variance explained by each PC is provided.
(vi) T-RFLP.
Upon digestion with the HhaI restriction enzyme, 43 different T-RFLP fragments were produced; on average, an individual sample contained 16 of these fragments. When the MspI enzyme was used, 42 different fragments were detected; on average, a sample contained 12 of these. The number of fragments observed across the dilution gradient did not differ for either enzyme (Table 3). The proportion of bands unique to each of the three dilution-diversity groups (100 with 10−1, 10−2 through 10−4, and 10−5 with 10−6) was also compared (Table 3).
Based on the PCA (Fig. 6) and cluster analysis (results not shown) performed on the combined MspI and HhaI data sets, two groups could be distinguished; the first group contained the communities regrown from the undiluted (100) inoculum through the 10−3 inoculum, while the second group contained the lower-diversity–higher-dilution treatments. Again, a bootstrapping procedure (using 100 replications) was used to assess the significance of the results of the cluster analysis. The highest bootstrap value obtained (not considering bootstrap values associated with the subgrouping of the replicate flasks) was associated with the division of the communities into three groups: 100 through 10−3, 10−4, and 10−5 with 10−6. However, the bootstrap value for these groupings was only 47 and it cannot be concluded that these three groups were significantly different. It is possible that the number of fragments compared in the cluster analysis was insufficient and that a higher bootstrap value might have been obtained if additional restriction digests (using different enzymes) had been performed and the data had been pooled prior to statistical analyses.
FIG. 6.

Results of PCA of T-RFLP profiles. Data are presented for each of three replicate flasks for each treatment, and each value corresponds to the negative exponent of the dilution factor used to create the original inoculum (e.g., 4 corresponds to a 10−4 dilution). Due to experimental difficulties with the T-RFLP analysis, only one of the flasks from the 10−4 dilution community and only two of the flasks from the 10−6 dilution community were analyzed. The percent variance explained by each PC is provided.
DISCUSSION
Numerical simulations.
The results (Fig. 1) show that dilution of a complex microbial community does not change the overall diversity of each resultant mixture, regardless of the evenness of the original community, until the size of the community is decreased so much that the number of individuals in the mixture approximates the original number of species. After this point, diversity must decrease with subsequent dilution because each individual that is removed from the system always removes a species from the community. This result was anticipated for relatively even communities, but it was initially surprising to discover that diversity did not change upon dilution of the more dominant mixtures of organisms (e.g., var = 100 or 250 in Fig. 1) for the early stages of the dilution series (through 10−3). It had been expected that dilution of these communities would remove rare organisms from the mixture, causing overall diversity to drop; in fact, the decrease in species richness upon dilution of the highly dominant communities was substantial (Fig. 1B). However, this decrease in species richness was accompanied by a concurrent increase in community evenness, resulting in little change in overall diversity until the 10−4 or 10−5 dilution.
These results suggest that a dilution approach may be used to create communities differing in diversity by comparing undiluted (or barely diluted) mixtures with communities regrown from very dilute inocula. This approach should be successful, regardless of the diversity and dominance relationship of the starting community; however, greater differences are to be expected for more even initial communities. Actual experimental communities regrown from diluted mixtures are not expected to exactly mimic these simulations, which only accounted for the dilution of the inoculum and not for any variance in regrowth. Synergistic and mutualistic interactions among organisms may be disrupted by the dilution procedure, and as a result, not all of the organism types carried through a dilution series to an inoculum may be able to regrow. The dilution procedure also decreases competition among organisms, and this could permit types that were not important in the original community to grow to unanticipated high abundances. Different growth rates among organisms may also impact the diversity of the regrown communities, changing evenness from that of the inoculum.
Batch culture experiments.
The results of the numerical simulations were used to make specific predictions about the behavior of the diluted-regrown communities in the batch culture experiments. If the initial community was evenly distributed, the community structure of the regrown mixtures would not be expected to change along the dilution gradient until the number of cells in the diluted inoculum approximated the total number of types of organisms in the original community. If the initial community was unevenly distributed, the first dilution during inoculum preparation would have removed a large number of types of organisms (e.g., in the var = 100 community, the first dilution removed 827 types out of 1,000). However, this loss of richness would have been offset by a simultaneous increase in community evenness, resulting in no net change in overall inoculum diversity (as calculated using the Shannon-Wiener index). Nevertheless, regrowth of communities so different in richness may have resulted in a measurable change in diversity or community structure between the undiluted and 10−1 dilution treatments. After the initial dilution of an unevenly distributed community, evenness would be greatly increased, and so, differences in subsequent dilutions (10−2, 10−3, and 10−4) were predicted to be small—until the dilution factor exceeds the original number of types of organisms in the community as described above.
(i) Traditional microbiological methods.
The regrown microbial communities were assayed using a number of traditional microbiological methods; each showed that the microbial communities regrown from the very dilute inocula (10−5 and 10−6) were unique. Abundance (as determined by AODC and plates counts on R2A and SM) was always significantly greater in these communities. It is possible that this variation in bacterial concentration was the result of differential grazing pressure along the dilution gradient (dilution of the inoculum would also have changed the amount of predation pressure in each treatment) (17, 29, 38). However, given the low concentration of ciliates and other grazers in the undiluted inoculum (direct microscopic observation, 1.5 × 102 organisms/ml), the impact of these organisms on bacterial abundance should have been small, especially in comparisons of treatments beyond the 10−2 dilution.
Ecological theory predicts that when interspecific competition is decreased (as was done by dilution in this study), populations can increase substantially in abundance (12). In the present work, the inverse relationship observed between final community size and inoculum dilution suggests that interspecific competition is more important than intraspecific competition in controlling total abundance. In the barely diluted communities (100 and 10−1), where diversity, and therefore interspecific competition, was higher, community size was much smaller compared to that of the very dilute communities (10−5 and 10−6), where diversity and interspecific competition were lower.
Percent culturability on each growth medium was much higher in the communities regrown from the high-dilution inocula. Given that microbial growth on culture media recovers a limited number of organisms, due to inappropriate incubation conditions or an inability of certain types of organisms to metabolize the supplied substrates, enhanced growth by the 10−6 dilution community suggests that those organisms are not as limited in their metabolic capabilities as those in the undiluted–low-dilution communities. Furthermore, the ratio of SM to R2A counts changed along the dilution-diversity gradient; communities regrown from the high dilutions (10−5 and 10−6) preferred R2A agar, while the other communities either had no preference or grew to higher abundances on SM; this result provides further evidence that the communities in the various dilution treatments were physiologically distinct.
The comparison of colony morphology on R2A plates showed that the microbial diversity and richness of the recoverable fraction of the communities decreased along the dilution gradient; the evenness of the communities increased. Based upon the dilution simulations, the greatest difference in community structure was expected between the very dilute (10−4 or 10−5) communities and all of the others, regardless of the structure of the original community; each of the other analytical methods employed in this research showed this to be true. However, the greatest difference for the diversity on R2A plates was between the undiluted (100) inoculum and the 10−1 dilution treatment. The fact that any difference in community structure was detected between these two dilution treatments suggests that the original sewage community had high dominance; the fact that there was no discernible change in the overall diversity of colony types for the high-dilution treatments (e.g., 10−4, 10−5, and 10−6) implies that the procedure is not useful for making inferences about microbial community structure in low-diversity situations.
One of the main criticisms of culture-based studies is that the carbon and nutrient sources found in a single culture medium are not as diverse as those found in nature, so only a small fraction of the organisms in a sample actually form colonies on a spread plate; using a large number of different medium types might increase the variety of organisms recovered with a cultural approach. In this study, a sterile SM was also used in an attempt to increase the number of types to compare when calculating the diversity index. Unfortunately, the colonies that grew on the SM were quite small and generally lacked morphological distinctiveness, making a comparison among treatments impossible. Another concern with regard to the use of culture-based procedures is the difficulty in accurately and consistently identifying community members, given the fact that very similar colony morphologies can occur among taxonomically distinct groups of organisms. However, recent studies have shown that colony morphology can, in fact, provide an accurate basis on which to define “recoverable diversity” (14, 19).
(ii) Genetic measures.
The DNA fingerprinting approaches used in this study showed a significant difference in overall microbial community structure along the dilution gradient; in particular, analysis of both the AFLP and T-RFLP data showed that the very dilute (10−5 and 10−6) communities were distinctly different from the communities regrown from less-dilute inocula (Fig. 5 and 6). AFLP also separated the undiluted community from the remaining treatments. The fact that AFLP distinguished a difference in microbial community structure between the undiluted and 10−1 dilution communities provides further evidence that the original microbial community (before dilution) had high dominance.
With T-RFLP, PCR is used to amplify the 16S rRNA genes directly from each community DNA sample using a pair of primers and analysis of a community sample produces a fingerprint wherein each individual band is, theoretically, derived from a different organism type (a different ribotype). However, it is well known that T-RFLP underestimates the species richness of a community because populations that are not numerically dominant are not represented if their template DNA comprises too small a fraction of the total community DNA (5, 21). Moreover, due to the conservation of restriction site positions in the 16S rRNA gene; the resolution of T-RFLP analysis is not at the species level but instead reflects the distribution of higher-order groups. Another limitation to the resolving power of T-RFLP is the actual universalness of primer pairs, as none of the available universal primers can hybridize to all of the known eukaryotic, bacterial, or archaeal 16S rRNA genes (2, 52). Despite these limitations, researchers commonly use T-RFLP to compare microbial community structures and frequently interpret the “number of T-RFLP peaks” to be reflective of (minimum) community richness (21, 22).
In this study, the total number of T-RFLP peaks was expected to decrease along the dilution-diversity gradient, corresponding to a loss of species richness; instead, this number remained essentially constant (Table 3). However, the identity of the T-RFLP peaks changed and this shift is illustrated by comparing the number of unique fragments found in each dilution-diversity group (Table 3). The fact that several unique peaks were observed in the low-diversity treatments (10−5 and 10−6) was surprising. Probability suggests that the dominant organisms in the original community are the ones that should persist through the dilution procedure and would therefore be used to inoculate these flasks. Consequently, nearly all of the organisms observed in the very dilute treatments should also have been detected in the less-dilute treatments. The fact that this was not found with T-RFLP suggests either (i) a lack of discriminative power of T-RFLP due to the bias of the procedure toward specific and/or more-abundant community members (a bias that seems to change, depending on the evenness of the community being analyzed) or (ii) a failure of these organisms to survive in the less-dilute treatments, despite their dominance in the original community and their ability to thrive in culture in the more-dilute treatments.
Since the original sewage community contained 1.8 × 106 cells/ml (AODC), the most-dilute community maintained in this experiment (10−6) should have been inoculated with approximately 2 cells. After regrowth, diversity in this community was expected to be very low. On the R2A plates, percent culturability was high (100%) and only three colony morphologies were observed, further suggesting that diversity in these flasks was quite low. Given this information, it was surprising to find that the average number of T-RFLP fragments in the 10−6 treatment was so high (8 for MspI and 10 for HhaI). It is possible that this discrepancy is the result of a technical error with the T-RFLP, e.g., incomplete restriction digestion, which could produce a number of differently sized T-RFLP fragments for each organism type. However, experimental controls in which the T-RFLP analysis was applied to DNA from a pure culture were also performed and a single T-RLFP peak was generated in each case. Another possible explanation is that because an individual organism can contain multiple, heterogeneous copies of the 16S rRNA gene (20, 27, 30, 43, 44), each organism type could actually have been responsible for more than one T-RFLP peak. However, the extent to which such sequence deviations occur has not been well studied and it is unlikely that the detection of multiple, divergent copies of a 16S rRNA gene can account for the results presented here.
Recent work has discovered that related strains of bacteria can have the same 16S rRNA gene but may not have the same physiological profiles or the same ecological strategies in the environment (E. Jaspers and J. Overmann, Abstr. ASM Conf. Microb. Diversity, abstr. B30, 1999). Presumably, this additional variability is coded elsewhere on the bacterial chromosome. Consequently, analysis of a single gene may not provide as much resolution when distinguishing among communities, compared to procedures that can survey the entire genome. With AFLP, a restriction digest is performed on a DNA sample (similar to RFLP) and then a set of primer recognition sequences (adapters) is used to amplify the restriction fragments using PCR (51); the primers and restriction enzymes used are not specific for a given gene or group or genes but can, theoretically, interact in numerous random places throughout a genome. AFLP is very similar in premise and application to randomly amplified polymorphic DNA fingerprinting, which has been used a number of times to compare microbial community structures (6, 7, 47–50).
AFLP is fundamentally different from each of the other procedures applied in this work, and from most other techniques used to compare microbial community structures; in that it is sensitive to overall differences between communities—including taxonomic distances between organisms. Watve and Gangal (45) pointed out that most procedures would not detect a difference in diversity between one community composed of four biotypes of coliforms and another composed of one coliform, one archaebacterium, one myxobacterium, and one actinomycete—although many microbial ecologists would agree that the latter should be treated as more diverse. The ability to differentiate between such mixtures is important, and it has been suggested that one way to incorporate this additional information is to simply calculate the mean taxonomic distance between all pairs of isolates in a community as a diversity index (34); however, the problem of isolation and taxonomic characterization of individuals remains. In this study, AFLP was used to compare overall diversity, considering richness, evenness, and taxonomic relatedness of community members without attempting to evaluate each of these elements separately.
(iii) CLPP.
CLPP compares patterns of carbon substrate utilization among communities by evaluating the extent to which a community metabolizes each of 95 different sole carbon sources (10). When CLPP was applied in this study, the different dilution communities separated into two distinct groups (10−5 and 10−6 were unique; Fig. 3). These results are important, as they demonstrate that there were phenotypic differences among the regrown communities, as well as the genetic differences already described. This means that the dilution process not only changed the identity of the organisms in the communities, as revealed by the genetic analyses, but also changed the communities' overall metabolic capabilities—the most-diverse (undiluted) community did not have the same functional potential as the low-diversity (10−6) community. The fact that the genetic and phenotypic measurement methods gave similar results in this study is also meaningful, as the correlation between the two suggests that genetic differences among communities actually have the potential to be manifested as differences in function.
Dilution-extinction analysis of CLPP was used to compare the relative structural diversities of different regrown communities. This procedure uses the dilution to extinction of a heterotrophic microbial community to evaluate the rate of character loss from the mixture; assuming that the rate of character loss is somehow proportional to the diversity of the original community, the relative diversity of the sample can be estimated (9). In this study, dilution-extinction analysis showed no significant change in the maximum functional richness (Rmax) of each community along the diversity gradient. The other regression parameter, KI, the half-maximum richness, describes the rate at which functional characters can be diluted out of a mixed community and has been used to assess relative structural diversity in a number of different experimental systems; a higher KI correlates with a higher diversity and also with increased niche specialization (9). In this study, KI decreased along the dilution-diversity gradient, confirming that the communities regrown from the less-dilute inocula were more diverse. The community regrown from the undiluted inoculum was able to perform a wide variety of functions but lost this ability rapidly upon dilution; this suggests a community composed primarily of specialists. The low-diversity community, regrown from the 10−6 dilution inocula, had a much lower KI; this increased conservation of function among the individuals in the group may suggest a community of generalists. The results of the percent culturability calculations also showed the high-dilution–low-diversity communities to be more generalized in their metabolic capabilities.
Oftentimes when researchers are advocating the use of molecular techniques over culture-based procedures, the reason presented is that culture-based analyses are too biased toward certain groups of organisms. As such, they underestimate the total richness of a community in an inconsistent and unpredictable manner. In this research, 23 colony types were observed on R2A agar (across all treatments) and only 42 unique T-RFLPs were encountered. Certainly, the actual total number of organism types in the original sample was much greater. Considering the fact that each of the analytical methods employed in this study showed a clear change in overall community structure between the 10−4 and 10−5 dilution treatments (Table 4), the numerical simulations suggest that there were between 1,000 and 10,000 types of organisms in the original sewage community. This value is consistent with results of Torsvik et al., who found that a soil sample can contained between 4,000 and 10,000 different bacterial types (40, 42). A more precise estimate of the number of types in the originial community might have been obtained if a different dilution scheme had been used (e.g., intervals smaller than 10-fold).
TABLE 4.
Summary of the groupings obtained using each techniquea
| Dilutionb | AODC | R2A count | % Culturability on R2A | Diversity on R2A platesc | SM count | Ratio of SM count to R2A countc | CLPP | Dilution-extinction analysis of CLPPc | AFLP | T-RFLP |
|---|---|---|---|---|---|---|---|---|---|---|
| 100 | a | a | a | a | a | a | a | a | a | a |
| 10−1 | a | a | a | b | a | a | a | b | a | |
| 10−2 | a | a | a | b | a | a | a | b | b | a |
| 10−3 | a | a | ab | b | ab | a | ab | b | a | |
| 10−4 | ab | a | ab | b | ab | a | ab | b | b | ab |
| 10−5 | b | ab | b | b | ab | b | b | c | b | |
| 10−6 | c | b | c | b | b | b | c | c | c | b |
Different letters represent significantly different communities, determined as described in the text.
Dilution refers to the original dilution used as the inoculum for the regrown communities.
For these data, statistical analyses were not performed to assess significance levels; instead, the different grouping outlined here were determined by visual interpretation of Fig. 2 (diversity on R2A plates) and Fig. 4 (dilution-extinction analysis of CLPP). For the ratio of SM to R2A counts, different groups were defined as having a ratio greater than 1 (a) or less than 1 (b).
In this study, significant differences in community structure were detected using genetic (AFLP and T-RFLP), physiological (CLPP), and culture-based (colony morphology on R2A agar) measurements. Along with this difference in community structure, differences in community size (AODC), composition (ratio of SM counts to R2A counts, monitoring of each colony morphology across the treatments), and metabolic redundancy (generalist versus specialist) were observed, suggesting that structure-diversity differences between communities maintained in the same environment can manifest differences in community organization and function. Although differences in microbial community structure were detected with every measurement method employed, each procedure has different methodological limitations that should be recognized when that technique is applied. The results of the experimental incubation demonstrated that the dilution-regrowth approach might be a useful way of generating communities differing in diversity (richness and evenness) and varying in overall community structure. Moreover, the procedure may be a useful way of analyzing communities—in this study, a great deal of information was gained about the richness and distribution of the original sewage community from analysis of the regrown communities.
ACKNOWLEDGMENTS
This work was supported by a NASA/ASEE Summer Faculty Fellowship (to Aaron Mills), a NASA Planetary Biology Internship (to Rima Franklin), and a NASA GSRP (grant NGT10-52620).
We gratefully acknowledge the assistance of Jennifer Adams, Ravi Bashyal, and Michael Roberts.
REFERENCES
- 1.Bej A K, Mahbubani M H. Applications of the polymerase chain reaction (PCR) in vitro DNA-amplification method in environmental microbiology. In: Griffin H G, Griffin A M, editors. PCR technology: current innovations. Boca Raton, Fla: CRC Press, Inc.; 1994. pp. 327–339. [Google Scholar]
- 2.Brunk C F, Avaniss-Aghajani E, Brunk C A. A computer analysis of primer and probe hybridization potential with bacterial small-subunit rRNA sequences. Appl Environ Microbiol. 1996;62:872–879. doi: 10.1128/aem.62.3.872-879.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse H-J, Denner E B M, Lubitz W. Classification and identification of bacteria: current approaches to an old problem. Overview of methods used in bacterial systematics. J Biotechnol. 1996;47:3–38. doi: 10.1016/0168-1656(96)01379-x. [DOI] [PubMed] [Google Scholar]
- 4.Chapin F S, Walker B H, Hobbs R J, Hooper D U, Lawton J H, Sala O E, Tilman D. Biotic control over the functioning of ecosystems. Science. 1997;277:500–503. [Google Scholar]
- 5.Dunbar J, Ticknor L O, Kuske C R. Assessment of microbial diversity in four southwestern United States soils by 16S rRNA gene terminal restriction fragment analysis. Appl Environ Microbiol. 2000;66:2943–2950. doi: 10.1128/aem.66.7.2943-2950.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Franklin R B, Taylor D R, Mills A L. Chacterization of microbial community structure using randomly amplified polymorphic DNA (RAPD) J Microbiol Methods. 1999;35:225–235. doi: 10.1016/s0167-7012(99)00003-2. [DOI] [PubMed] [Google Scholar]
- 7.Franklin R B, Taylor D R, Mills A L. The distribution of microbial communities in anaerobic and aerobic zones of a shallow coastal plain aquifer. Microb Ecol. 1999;38:377–386. doi: 10.1007/s002489900179. [DOI] [PubMed] [Google Scholar]
- 8.Garland J L. Analytical approaches to the characterization of samples of microbial communities using patterns of potential C source utilization. Soil Biol Biochem. 1996;28:213–221. [Google Scholar]
- 9.Garland J L, Lehman R M. Dilution/extinction of community phenotypic characters to estimate relative structural diversity in mixed communities. FEMS Microbiol Ecol. 1999;30:333–343. doi: 10.1111/j.1574-6941.1999.tb00661.x. [DOI] [PubMed] [Google Scholar]
- 10.Garland J L, Mills A L. Classification and characterization of heterotrophic microbial communities on the basis of patterns of community-level sole-carbon-source utilization. Appl Environ Microbiol. 1991;57:2351–2359. doi: 10.1128/aem.57.8.2351-2359.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garland J L, Mills A L, Morales A, Cook K. Survival of human-associated bacteria in prototype advanced life support systems. SAE tech paper ser. no. 1999-01-2601. Denver, Colo: American Society of Automotive Engineers; 1999. [Google Scholar]
- 12.Giller P S. Community structure and the niche. London, England: Chapman & Hall; 1984. [Google Scholar]
- 13.Griffiths B S, Ritz K, Wheatley R E. Relationship between functional diversity and genetic diversity in complex microbial communities. In: Insam H, Rangger A, editors. Microbial communities. Functional versus structural approaches. Berlin, Germany: Springer-Verlag KG; 1997. pp. 1–9. [Google Scholar]
- 14.Haldeman D L, Amy P S. Diversity within a colony morphotype: implications for ecological research. Appl Environ Microbiol. 1993;59:933–935. doi: 10.1128/aem.59.3.933-935.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hobbie J E, Daley R J, Jasper S. Use of Nuclepore filters for counting bacteria by fluorescence microscopy. Appl Environ Microbiol. 1977;33:1225–1228. doi: 10.1128/aem.33.5.1225-1228.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holben W E. Isolation and purification of bacterial community DNA from environmental samples. In: Hurst C J, Knudsen G R, McInerney M J, Stetzenback L D, Walter M V, editors. Manual of environmental microbiology. Washington, D.C.: American Society for Microbiology; 1997. pp. 431–444. [Google Scholar]
- 17.Juergens K, Arndt H, Zimmermann H. Impact of metazoan and protozoan grazers on bacterial biomass distribution in microcosm experiments. Aquat Microb Ecol. 1997;12:131–138. [Google Scholar]
- 18.Lawton J H. What do species do in ecosystems? Oikos. 1994;71:367–374. [Google Scholar]
- 19.Lebaron P, Ghiglione J F, Fajon C, Batailler N, Normand P. Phenotypic and genetic diversity within a colony morphotype. FEMS Microbiol Lett. 1998;160:137–143. doi: 10.1111/j.1574-6968.1998.tb12903.x. [DOI] [PubMed] [Google Scholar]
- 20.Linton D, Dewhirst F E, Clewley J P, Owen R J, Burnens A P, Stanley J. Two types of 16S rRNA genes are found in Campylobacter helveticus: analysis, applications and characterization of the intervening sequence found in some strains. Microbiology. 1994;140:847–855. doi: 10.1099/00221287-140-4-847. [DOI] [PubMed] [Google Scholar]
- 21.Liu W, Marsh T L, Cheng H, Forney L J. Characterization of microbial diversity by determining terminal restriction fragment length polymorphisms of genes encoding 16S rRNA. Appl Environ Microbiol. 1997;63:4516–4522. doi: 10.1128/aem.63.11.4516-4522.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu W T, Marsh T L, Forney L J. Determination of the microbial diversity of anaerobic-aerobic activated sludge by a novel molecular biological technique. Water Sci Technol. 1998;37:417–422. [Google Scholar]
- 23.Magurran A E. Ecological diversity and its measurement. Princeton, N.J: Princeton University Press; 1988. [Google Scholar]
- 24.McNaughton S J. Diversity and stability of ecological communities: a comment on the role of empiricism in ecology. Am Nat. 1977;111:515–525. [Google Scholar]
- 25.Morales A, Garland J L, Lim D V. Survival of potentially pathogenic human-associated bacteria in the rhizosphere of hydroponically grown wheat. FEMS Microbiol Ecol. 1996;20:155–162. doi: 10.1016/0168-6496(96)00020-7. [DOI] [PubMed] [Google Scholar]
- 26.Naeem S, Thompson L J, Lawler S P, Lawton J H, Woodfin R M. Declining biodiversity can alter the performance of ecosystems. Nature. 1994;368:734–737. [Google Scholar]
- 27.Ninet B, Monod M, Emler S, Pawlowski J, Metral C, Rohner P, Auckenthaler R, Hirschel B. Two different 16S rRNA genes in a mycobacterial strain. J Clin Microbiol. 1996;34:2531–2536. doi: 10.1128/jcm.34.10.2531-2536.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogram A, Feng X. Methods of soil microbial community analysis. In: Hurst C J, Knudsen G R, McInerney M J, Stetzenback L D, Walter M V, editors. Manual of environmental microbiology. Washington, D.C.: American Society for Microbiology; 1997. pp. 422–430. [Google Scholar]
- 29.Pernthaler J, Posch T, Simek K, Vrba J, Amann R, Psenner R. Contrasting bacterial strategies to coexist with a flagellate predator in an experimental microbial assemblage. Appl Environ Microbiol. 1997;63:596–601. doi: 10.1128/aem.63.2.596-601.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettersson B, Johansson K E, Uhlen M. Sequence analysis of 16S rRNA from mycoplasmas by direct solid-phase DNA sequencing. Appl Environ Microbiol. 1994;60:2456–2461. doi: 10.1128/aem.60.7.2456-2461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pielou E C. An introduction to mathmatical ecology. New York, N.Y: Wiley; 1969. [Google Scholar]
- 32.Pimm S L. The complexity and stability of ecosystems. Nature. 1984;307:321–326. [Google Scholar]
- 33.Putman R J. Community ecology. London, England: Chapman & Hall Publishers; 1994. [Google Scholar]
- 34.Rao C R. Diversity and dissimilarity coefficients: a unified approach. Theor Popul Biol. 1980;21:24–43. [Google Scholar]
- 35.Reasoner D J, Geldreich E E. A new medium for the enumeration and subculture of bacteria from potable water. Appl Environ Microbiol. 1985;49:1–7. doi: 10.1128/aem.49.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenzweig M L. Species diversity in space and time. New York, N.Y: Cambridge University Press; 1995. [Google Scholar]
- 37.Shannon C E, Weaver W. The mathematical theory of communication. Urbana: University of Illinois Press; 1949. [Google Scholar]
- 38.Sinclair J L, Alexander M. Effect of protozoan predation on relative abundance of fast- and slow-growing bacteria. Can J Microbiol. 1989;35:578–582. [Google Scholar]
- 39.Stahl D. Molecular approaches for the measurement of density, diversity, and phylogeny. In: Hurst C J, Knudsen G R, McInerney M J, Stetzenback L D, Walter M V, editors. Manual of environmental microbiology. Washington, D.C.: American Society for Microbiology; 1997. pp. 102–114. [Google Scholar]
- 40.Torsvik V, Goksøyr J, Daae F L. High diversity in DNA of soil bacteria. Appl Environ Microbiol. 1990;56:782–787. doi: 10.1128/aem.56.3.782-787.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torsvik V, Goksøyr J, Daae F L, Sørheim R, Michalsem J, Salte K. Use of DNA analysis to determine the diversity of microbial communities. In: Ritz K, Kighton J, Giller K E, editors. Beyond the biomass. New York, N.Y: British Society of Soil Science, Wiley-Sayce Publishers; 1994. pp. 39–48. [Google Scholar]
- 42.Torsvik V, Sorheim R, Goksoyr J. Total bacterial diversity in soil and sediments: a review. J Ind Microbiol Biotechnol. 1996;17:170–178. [Google Scholar]
- 43.Wang G C-Y, Wang Y. Frequency of formation of chimeric molecules as a consequence of PCR coamplification of 16S rRNA genes from mixed bacterial genomes. Appl Environ Microbiol. 1997;63:4645–4650. doi: 10.1128/aem.63.12.4645-4650.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Zhang Z S, Narendrakumar R. The actinomycete Thermobispora bispora contains two distinct types of transcriptionally active 16S rRNA genes. J Bacteriol. 1997;179:3270–3276. doi: 10.1128/jb.179.10.3270-3276.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watve M G, Gangal R M. Problems in measuring bacterial diveristy and a possible solution. Appl Environ Microbiol. 1996;62:4299–4301. doi: 10.1128/aem.62.11.4299-4301.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.White D C, Pinkart H C, Ringelberg D B. Biomass measurements: biochemical approaches. In: Hurst C J, Knudsen G R, McInerney M J, Stetzenbach L D, Walter M V, editors. Manual of environmental microbiology. Washington, D.C.: American Society for Microbiology; 1997. pp. 91–101. [Google Scholar]
- 47.Wikström P, Andersson A-C, Forsman M. Biomonitoring complex microbial communities using random amplified polymorphic DNA and principal component analysis. FEMS Microbiol Ecol. 1999;28:131–139. [Google Scholar]
- 48.Wikström P, Hägglund L, Forsman M. Structure of a natural microbial community in a nitroaromatic contaminated groundwater is altered during biodegradation of extrinsic, but not intrinsic substrates. Microb Ecol. 2000;39:203–210. doi: 10.1007/s002480000001. [DOI] [PubMed] [Google Scholar]
- 49.Xia X, Bollinger J, Ogram A. Molecular genetic analysis of the response of three soil microbial communities to the application of 2,4-D. Mol Ecol. 1995;4:17–28. doi: 10.1111/j.1365-294x.1995.tb00188.x. [DOI] [PubMed] [Google Scholar]
- 50.Yang Y-H, Yao J, Hu S, Qi Y. Effects of agricultural chemicals on DNA sequence diversity of soil microbial community: a study with RAPD marker. Microb Ecol. 2000;39:72–79. doi: 10.1007/s002489900180. [DOI] [PubMed] [Google Scholar]
- 51.Zabeau, M., and P. Vos. 1993. Selective restriction fragment amplification: a general method for DNA fingerprinting. European Patent Office publication O 534 858 A1.
- 52.Zheng D, Alm E W, Stahl D A, Raskin L. Characterization of universal small-subunit rRNA hybridization probes for quantitative molecular microbial ecology studies. Appl Environ Microbiol. 1996;62:4504–4513. doi: 10.1128/aem.62.12.4504-4513.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]