

Abstract
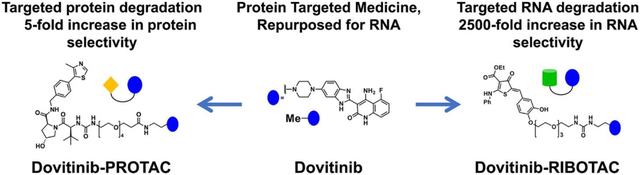
Reprogramming known medicines for a novel target with activity and selectivity over the canonical target is challenging. By studying the binding interactions between RNA folds and known small-molecule medicines and mining the resultant dataset across human RNAs, we identified that Dovitinib, a receptor tyrosine kinase (RTK) inhibitor, binds the precursor to microRNA-21 (pre-miR-21). Dovitinib was rationally reprogrammed for pre-miR-21 by using it as an RNA recognition element in a chimeric compound that also recruits RNase L to induce the RNA’s catalytic degradation. By enhancing the inherent RNA-targeting activity and decreasing potency against canonical RTK protein targets in cells, the chimera shifted selectivity for pre-miR-21 by 2500-fold, alleviating disease progression in mouse models of triple-negative breast cancer and Alport Syndrome, both caused by miR-21 overexpression. Thus, targeted degradation can dramatically improve selectivity even across different biomolecules, i.e., protein versus RNA.
Graphical Abstract
Introduction
Historically, RNA has been perceived as recalcitrant to small-molecule medicines, and as such, the inherent RNA-binding capacity of approved and clinically used drugs has to date been largely ignored. Yet, the reprogramming of such drugs, shown to be safe in humans, (1) for RNA targets could significantly decrease the time necessary to bring medicines to the clinic. Here, we have evaluated the inherent RNA-binding properties of the most comprehensive collection of approved and clinically used small molecules,(2) investigated the physiological relevance of the identified interactions, and used the results to reprogram the activity of a known receptor tyrosine kinase (RTK) inhibitor.
To devise a strategy to reprogram medicines rationally, we selected the optimal RNA folds that bind small-molecule medicines and then defined the biological relevance of these interactions by comparison to the folds present in all folded human RNAs. This approach, i.e., applying a fundamental understanding of molecular recognition to biological systems, has previously afforded a cohort of small molecules that modulate RNA function in patient-derived cells and in vivo. Indeed, a privileged interaction between the RTK inhibitor Dovitinib and a processing site in an oncogenic microRNA (miRNA), miR-21, was identified through this selection process. Although Dovitinib is not selective for the RNA target, we hypothesized that its conversion into a ribonuclease targeting chimera (RIBOTAC) may shift its selectivity. This reprogramming shifted the inhibitor’s selectivity by 2500-fold toward the disease-causing RNA, alleviating disease progression in vivo.
Results and Discussion
Identifying Medicines with Avidity for RNA
Here, we studied the inherent RNA-binding capacity of known medicines in the state-of-the-art small-molecule collection, ReFRAME (Repurposing, Focused Rescue, and Accelerated Medchem) (2,3) (Figures 1A and S1). Binding assays between the ReFRAME library and an RNA library that displays 3×2 nucleotide internal loops (3×2 ILL; Figures 1A, S1, and S2), containing 1024 unique three-dimensional (3D) folds found in human RNAs, (4) revealed that 68 of the 9300 members of ReFRAME (0.7%) bound members of the 3×2 ILL. Collectively, the screen probed >9.5 million potential interactions (Figure 1A). (5)
Figure 1.
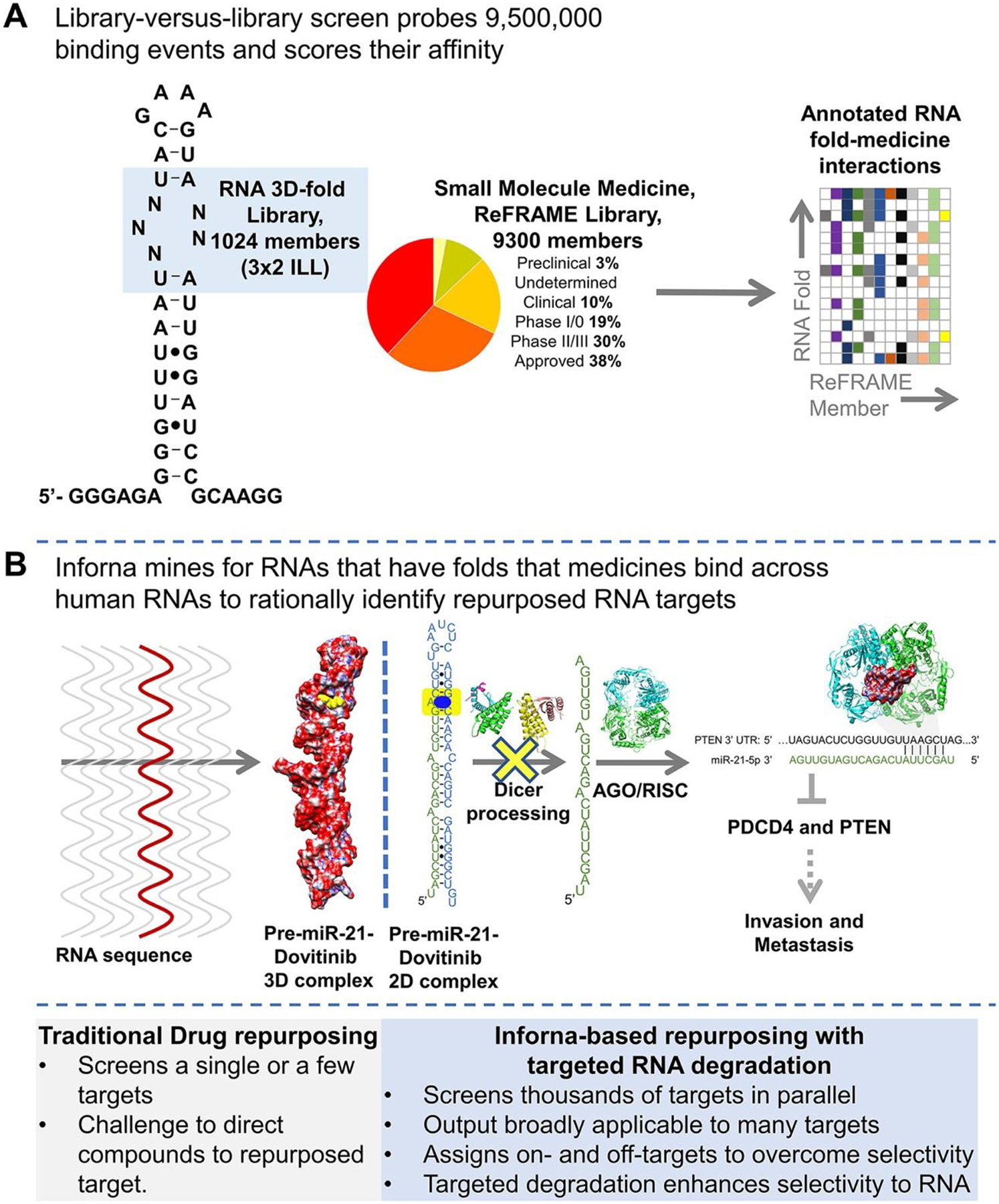
Inforna-based reprogramming of the ReFRAME small-molecule library by identifying RNA binders. (A) Left, secondary structure of the 1024-member 3×2 RNA internal loop library (3×2 ILL) and summary of the 9300-member ReFRAME library. Right, the library-versus-library screen probes over 9,500,000 combinations of RNA motif–small molecule interactions to identify the privileged RNA structures that bind small-molecule medicines with high affinity. (B) Inforna-based identification of Dovitinib (1) targeting pre-miR-21’s Dicer site, which inhibits its processing and hence its oncogenicity.
The precise RNA 3D folds that avidly bound each medicine were identified using a profiling strategy named AbsorbArray. (6) In this experiment, small molecules were non-covalently absorbed to a microarray surface, which was incubated with the 3×2 ILL under highly stringent conditions (Figure S1). Four medicines, Dovitinib, Delparantag, Piroxantrone, and Metiazinic Acid, currently used to treat multiple diseases, bound RNA targets avidly (Figures S3 and S4). RNAs that bound each small-molecule medicine were excised from the surface of the microarray, purified, amplified, and sequenced by RNA-sequencing (RNA-seq) to determine the identities and structures of the selected RNAs (Figure S1). (6) Rigorous statistical analyses of the RNA-seq data, as we have previously described, (7) defined the affinity landscape of the RNA 3D folds for each medicine.
Using our lead identification strategy, Inforna, this annotated dataset of RNA fold–small molecule recognition events was mined in a target agnostic fashion against the folded RNA structures in the human genome, with overlap affording lead medicines and lead cellular RNA targets (Figure 1A). That is, Inforna folds cellular RNA primary sequences and then mines the structures therein against a database of privileged RNA–small molecule interactions, thereby identifying physiologically relevant RNA–small molecule binding interactions. (8)
Dovitinib, a Receptor Tyrosine Kinase (RTK) Inhibitor, Binds to a Functional Site in the Oncogenic MicroRNA Precursor Pre-miR-21 and Inhibits Its Processing in Cells
Indeed, Inforna identified Dovitinib (1), a clinically used receptor tyrosine kinase (RTK) inhibitor, (9) might be amenable to reprogramming for the precursor of miR-21 (pre-miR-21) (Figures 1B, 2A, and S5). This miRNA is associated with many diseases, including cancer (10–13) and the genetically defined kidney disease Alport Syndrome (AS). (14) MiRNAs suppress translation of messenger RNAs (mRNAs) by binding to those with complementary sequences in their 3′ untranslated regions (UTRs) and sterically blocking ribosomal assembly or inducing cleavage via the RNA-induced silencing complex (RISC) (Figure 1B). (15,16) After transcription, primary miRNAs (pri-miRNAs) are processed into pre-miRNAs by the nuclear ribonuclease Drosha. After cytoplasmic export, they are further processed into mature miRNAs by the nuclease Dicer (Figure 1B). Dovitinib (1) binds specifically to the Dicer processing site in pre-miR-21, which harbors an A bulge, with a Kd of 3 μM; no saturable binding was observed to a control RNA in which the target Dicer site was mutated to a base pair (Figure S6A). Compound binding was further confirmed by NMR spectroscopic studies. Here, we studied a derivative of Dovitinib, 1a, which was used for synthesis of the reprogrammed RIBOTAC, as it has superior solubility in buffer conditions required for NMR studies. WaterLOGSY experiments showed that 1a bound to model of pre-miR-21 but not a deletion mutant in which the putative A bulge binding site was removed (Figure S7). Additionally, Dovitinib inhibited in vitro Dicer processing of pre-miR-21 with an IC50 of 5 μM, with no effect on a mutant pre-miR-21 in which Dovitinib’s binding site was ablated (Figure S6B), indicating specificity for the A bulge and that 1 did not inhibit Dicer itself.
Figure 2.
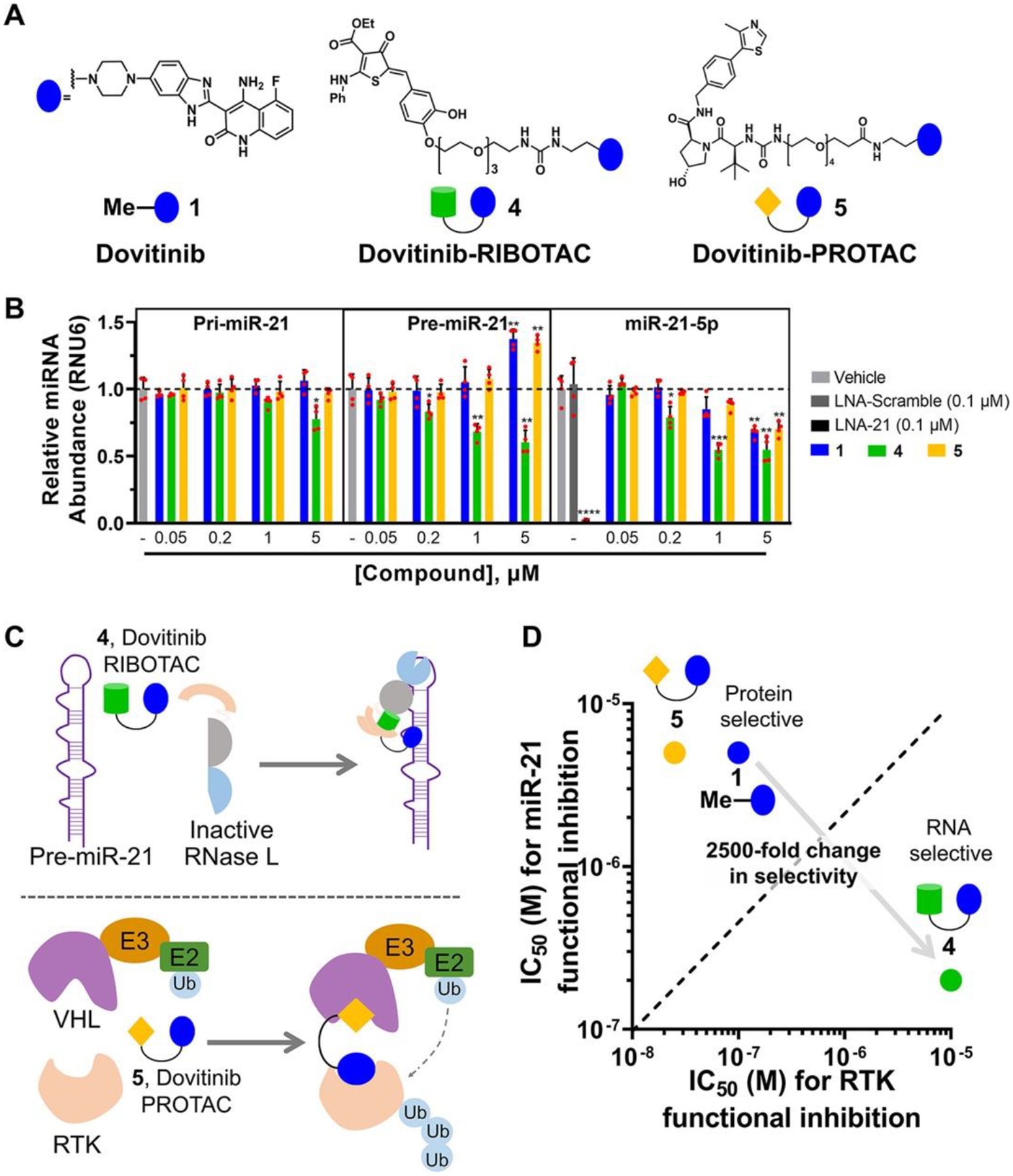
Dovitinib-based rational design of a RIBOTAC for RNA and a PROTAC for protein. (A) Structures of parent compound 1, RIBOTAC 4, and PROTAC 5. (B) Compounds 1, 4, and 5 reduced mature miR-21 levels in the TNBC cell line MDA-MB-231, as determined by RT-qPCR analysis (n = 4). RIBOTAC 4 decreased pre-miR-21 levels via RNase L cleavage, while 1 and 5 increased pre-miR-21 levels, consistent with their mode of action, binding and inhibiting Dicer cleavage. Compounds 1, 4, and 5 (up to 1 μM) had no significant effect on pri-miR-21 levels in MDA-MB-231 cells, as determined by RT-qPCR (n = 4). Compound 4 had a modest effect on pri-miR-21 levels at 5 μM (n = 4). (C) Scheme of RIBOTAC 4 dimerizing RNase L onto pre-miR-21 to cleave it enzymatically (left) and PROTAC 5 recruiting VHL onto an RTK to induce its ubiquitination, leading to subsequent proteasome-mediated down-regulation (right). (D) RIBOTAC 4 exhibited 25-fold increased miR-21 inhibitory activity and 100-fold decreased RTK inhibition, both as compared with 1, while 5 exhibited similar miR-21 inhibitory activity and 5-fold increased RTK inhibition. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as determined by a two-tailed Student’s t test.
To study cellular engagement of pre-miR-21 by 1, we synthesized a diazirine-containing derivative that can react with both cellular RNAs and proteins upon exposure to UV light. (17,18) Chemical synthesis, guided by the structure of Dovitinib bound to an RTK (19) (see the chemical structure in Figure S5), provided a derivative of Dovitinib (2) that cross-links to its cellular targets, via a method dubbed Chemical Cross-linking and Isolation by Pull-down (Chem-CLIP). (20) That is, probe 2 enabled us to study occupancy of both the canonical RTK target and the reprogrammed target, pre-miR-21, in cells (Figures S8A and S9A). Notably, 1 and Chem-CLIP probe 2 have similar in vitro binding affinities for and cellular activity against pre-miR-21 (Figures S6A and S8B); thus, the latter is suitable to study cellular occupancy.
The Chem-CLIP probe (2) was added to the triple-negative breast cancer (TNBC) cell line MDA-MB-231 to study cellular occupancy of RNA and protein targets (Figures S8A and S9A). Both Vascular Endothelial Growth Factor Receptor 1 (FLT3), an RTK avidly bound by Dovitinib, (21) and pre-miR-21 were significantly enriched in the pulled-down fraction, consistent with the hypothesis that the medicine binds both targets (Figures S8 and S9). No enrichment was observed with a control Chem-CLIP probe (3) that lacks the 1 RNA-binding module (Figures S8 and S9). Competitive Chem-CLIP (C-Chem-CLIP) was used to assess the relative binding of 1 itself to the RNA and protein targets (Figures S8 and S9). In MDA-MB-231 cells, 1 reduced the binding and cross-linking of Chem-CLIP probe 2 to FLT3 at 0.1 μM (1 μM of 2) and to pre-miR-21 at 5 μM (5 μM of 2) (Figures S8E and S9B). [Note: the concentrations of 2 for the two targets were chosen based on their IC50s in cells for cross-linking.] These studies demonstrated direct binding of 1 to both FLT3 and pre-miR-21, indicating that the small molecule has potential for RNA reprogramming, provided its selectivity can be improved.
Given the direct engagement of pre-miR-21 by Dovitinib in TNBC cells, we studied if 1 inhibited the RNA’s biogenesis and its downstream biological functions. Indeed, 1 inhibited pre-miR-21 processing in TNBC cells, significantly reducing mature miR-21 levels at a 5 μM dose (p < 0.01; Figure 2B). Consistent with its presumed mode of action, inhibition of Dicer processing, 1 increased pre-miR-21 levels but left pri-miR-21 levels unchanged (Figure 2B). Profiling the effect of 1 on all expressed miRNAs showed that 1 was modestly selective for the reprogrammed target (Figure S10A). [As an aside, Inforna was able to predict binding of 1 to other cellular miRNA precursors, as eight of the ten detectable targets were also pulled down in Chem-CLIP studies (Figure S11A). Of these 10 miRNAs, none were affected by 1-treatment as determined by a false discovery rate (FDR) < 1%, as expected since the binding site within the precursors is not harbored within Drosha or Dicer processing sites (Figures S3 and S10A).
In agreement with the reduction of mature miR-21 levels, 1 (5 μM) de-repressed the expression of miR-21’s downstream protein targets, particularly Programmed Cell Death 4 (PDCD4) and Phosphatase and Tensin Homolog (PTEN) (Figure S10B,C). Further, 1 (5 μM) inhibited the invasiveness of MDA-MB-231 TNBC cells, a phenotype associated with miR-21 overexpression (Figure S12). (10) MCF-10A cells, a model of healthy breast epithelium, do not appreciably express miR-21 and thus are not invasive. Forced expression of pre-miR-21 in MCF-10A cells, however, triggered an invasive phenotype that was reduced by 1-treatment (Figure S12). Interestingly, overexpression of the pre-miR-21 mutant in which the 1 binding site was ablated also increased the invasiveness of MCF-10A cells, but the cells were insensitive to 1 treatment, further supporting miR-21’s Dicer processing site as 1’s binding site (Figure S12). Altogether, these data show 1 selectively rescues a miR-21-mediated phenotype in cells.
Dovitinib RIBOTAC Cleaves Pre-miR-21 with Enhanced Potency and Selectivity
Given that Dovitinib modulates the canonical protein target more potently than the reprogrammed RNA target, we sought a strategy to direct 1’s activity and selectivity toward pre-miR-21. One such method is targeted RNA degradation, which has not been previously applied to tune biomolecule selectivity, i.e., RNA versus protein selectivity. Thus, we converted 1 into both an RNA degrader, or ribonuclease targeting chimera (4, RIBOTAC), (22) and a protein degrader, or proteolysis targeting chimera (5, PROTAC) (23) (Figures 2A and S5). The latter was synthesized as a tool to enable target occupancy studies of both pre-miR-21 and an RTK by 1 and 4.
The RIBOTAC 4 was designed to bind pre-miR-21 and recruit and activate ribonuclease L (RNase L) locally to cleave the target (Figure 2C). (24) RNase L is ubiquitously expressed in cells in minute quantities as an inactive monomer. In response to viral infection, the cell synthesizes 2′–5′ polyadenylate, which binds RNase L, both dimerizing and activating the enzyme. (25) A small-molecule activator of RNase L was recently described, (24) which was appended to 1 to create the chimeric RIBOTAC. As expected, 4 recruited and dimerized inactive monomeric RNase L into the active dimer in vitro, cleaving pre-miR-21 proximal to the Dicer site (Figure S13).
In MDA-MB-231 cells, 4 significantly enhanced selectivity and potency against pre-miR-21 as compared to 1. In particular, 1 decreased mature miR-21 levels by ~30% at a 5 μM dose, while a similar reduction was observed with only 0.2 μM of 4, a 25-fold increase in potency (Figure 2B). [Although the exocyclic methylene of the RNase L recruiter might appear to be a promiscuous electrophile, analysis by mass spectrometry showed that 4 did not react with the common nucleophile glutathione when added to cell culture medium (Figure S14), and the molecule is stable in mice, vide infra.] In addition, pre-miR-21 levels were decreased by 4-treatment while 1 enhanced these levels, as expected for compounds that degrade the pre-miRNA or inhibit Dicer processing, respectively (Figure 2B). Additionally, 4 preferentially cleaved cytoplasmic pre-miR-21 over nuclear pri-miR-21, in accordance with the predominant subcellular localization of RNase L (cytoplasmic). (26) Co-addition of increasing amounts of 1 and a constant concentration of 4 competed away cleavage of pre-miR-21, indicating 1 and 4 bind the same site in pre-miR-21 (Figure S15B).
Three negative control compounds were also studied to investigate potential off-target effects of RIBOTAC 4. A negative control compound (6), which lacks the RNA-binding module, had no effect on miR-21 levels in cells (Figures S5 and S15C). A regioisomer of RIBOTAC 4 with an ablated RNase L-recruiting module (7; binds but does not induce significant degradation) inhibited miR-21 biogenesis, reduced mature miR-21 levels and increased pre-miR-21 levels, similar to 1, as expected (Figures S5 and S15C). Additionally, a Dovitinib-derived less active analog (8), which is 10-fold less avid for binding to pre-miR-21, was synthesized along with its RIBOTAC analog (9) (Figures S5 and S16A,B). Neither compound had an effect on pre- or mature miR-21 levels at the same concentration of 4, demonstrating that cleavage of pre-miR-21 by RIBOTAC 4 is not a general effect of RNase L activation but a specific effect of RNase L cleavage driven by the binding of Dovitinib (1) (Figure S16C).
Two lines of experimental evidence confirmed that cleavage by 4 was indeed RNase L-dependent: (i) an RNase L antibody immunoprecipitated pre-miR-21 in cells treated with 4, showing direct formation of the ternary complex between 4, RNase L, and pre-miR-21 (Figure S15D); and (ii) the cleaving capacity of 4 was reduced when RNase L was knocked down by two different RNase L-targeting siRNAs and in MDA-MB-231 cells in which RNase L was knocked out by CRISPR (Figures S15F–H and S17A–C). Ternary complex formation was specific to pre-miR-21, as complex formation was not observed with pre-miR-210 (Figure S15D). Notably, neither control compound, 6 nor 7, formed a ternary complex with pre-miR-21 and RNase L, as determined by immunoprecipitation studies (Figure S15D). Of the six other pre-miRNAs validated by Chem-CLIP to bind 1 with detectable mature miRNA levels (Figure S11A,C), five formed a ternary complex with 4 and RNase L (Figure S15E). Notably, no statistically significant effect was observed on their mature miRNA levels (Figure S10A), a function of the location of the 1-binding site outside a Drosha or Dicer processing site and the lack of a nearby preferred RNase L cleavage site (Figures 3 and S3).
Figure 3.
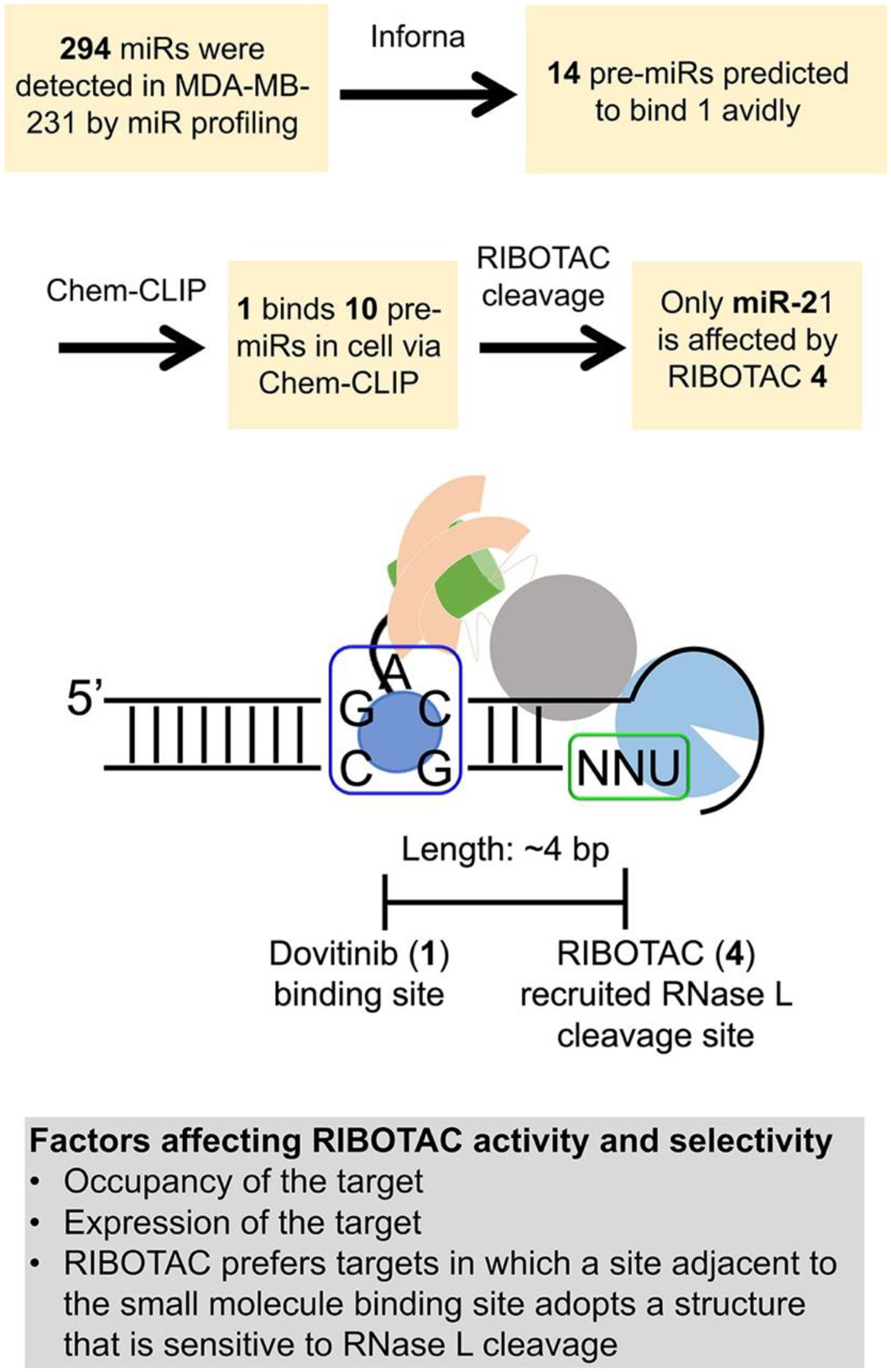
Rationale of selective inhibition of miR-21 by RIBOTAC 4. Top, experimental flowchart of how selective inhibition of miR-21 by RIBOTAC 4 was assessed. Bottom, scheme of factors that contribute to RIBOTAC activity and selectivity, including: occupancy of the target by the RNA-binding module (e.g., 1); expression level of the target where more abundant targets are occupied to a greater extent than less abundant ones; and the juxtaposition of the site bound by the RNA-binding module and a site sensitive to RNase L cleavage (unpaired UNN) and the distance between them.
Consistent with its enhanced potency for reduction of mature miR-21 levels, RIBOTAC 4 was 10-fold more potent than 1 in modulating downstream miR-21-associated phenotypes. That is, 4 de-repressed PTEN and PDCD4 and reduced the invasive character of MDA-MB-231 cells at a dose of 1 μM (Figures S10C, S15I, and S18A,B). Importantly, the reduction of invasion by 4 was ablated by overexpression of pre-miR-21 and RNase L knockout, indicating this phenomenon is linked to a miR-21-mediated circuit as well as RNase L-dependent (Figure S17D and S18A,B). Further, 4 rescued the invasive phenotype induced in MCF-10A cells by overexpression of pre-miR-21 but not that induced by the mutated pre-miR-21 that lacks a 1-binding site (Figure S18C,D). Notably, 4 at 1 μM (45% reduction of miR-21) is more selective than 1 at 5 μM (32% reduction of miR-21), as determined by profiling all miRNAs expressed in MDA-MB-231 cells (Figure S10A). Although 1 binds structural elements in other miRNA precursors (Figures S3 and S11A), only the abundance of miR-21 was significantly affected by RIBOTAC treatment (Figure S11C). The observed selectivity is likely a composite of many factors, including expression levels (pre-miR-21 is the most abundant; Figure S11B), the functionality of the site that Dovitinib binds within pre-miR-21 (Dicer processing site), inability to form a ternary complex (Figure S15E), and/or lack of an adjacent preferred RNase L substrate (Figure 3).
The RIBOTAC’s activity (4) was not limited to MDA-MB-231 TNBC cells, as it reduced pre- and mature miR-21 levels in other cancer cell lines, including A549 (lung carcinoma), A375 (melanoma), MCF-7 (breast cancer), and MDA-LM2 (lung metastatic breast cancer) (Figure S15J). RT-qPCR analysis completed by a TaqMan assay further confirmed the RIBOTAC’s activity on miR-21 in these cell lines (Figure S15K).
We next converted 1 to PROTAC 5 by coupling a recruiter of von Hippel–Lindau (VHL) E3 ubiquitin ligase (27) to direct the RTK FLT3 to the proteasome (Figure 2C,D). PROTAC 5 reduced the levels of FLT3 with low nanomolar activity and increased pre-miR-21 levels to a similar extent as 1 (Figures 2B and S19A,B). A 20S proteasome inhibitor, Carfilzomib, ablated degradation of FLT3 by PROTAC 5, which indicates that the PROTAC facilitated target degradation via the proteasome, as expected (Figure S19C). Interestingly, co-treatment with 1 or 4 both reduced the targeted degradation of PROTAC 5. However, a 100-fold greater concentration of RIBOTAC 4 (10 μM) was required than 1 (0.1 μM) (Figure S19D,E), indicating that conversion of a binder into a RIBOTAC decreased its target cellular protein occupancy by 100-fold. These data suggest that Dovitinib’s functional activity may have indeed been reprogrammed away from the canonical RTK and toward the RNA target.
RIBOTAC 4 Shifts Selectivity for Pre-miR-21 over RTK by 2500-Fold
Next, a series of experiments were conducted to study whether 1, RIBOTAC 4, and PROTAC 5 functionally inhibit RTK and downstream phosphorylation of extracellular signal-regulated kinase (28) as well as miR-21-regulated proteins in MDA-MB-231 cells. For the canonical protein RTK target, 1, 4, and 5 all inhibited phosphorylation of ERK (pERK), with IC50’s of about 0.1, 10, and 0.02 μM, respectively (Figure S20). Thus, relative to 1, a 5-fold increase of RTK inhibition is observed with PROTAC 5 while a 100-fold decrease of RTK inhibition is observed with RIBOTAC 4 (Figure 2B). Altogether, the selectivity of the miR-21-targeted medicine is shifted by 2500-fold: 4 is 25-fold more potent for inhibition of miR-21 than 1 and 100-fold less potent than 1 for functional inhibition of RTKs (Figure 2D). In addition to their effects on FLT3 in cells, we also studied the activity of 1 and 4 against a panel of 24 kinases representative of the human kinome in vitro. As expected of a broad-spectrum kinase inhibitor, 1 significantly inhibited eight kinases (p < 0.01) and modestly inhibited one kinase (p < 0.05), while RIBOTAC 4 did not significantly inhibit any kinases (p < 0.01), with modest inhibition of two at 1 μM (p < 0.05) (Figure S21).
To understand the origin of this remarkable enhancement in selectivity for the RNA target, we performed C-Chem-CLIP experiments to study target engagement of 1, 4, and 5 in MDA-MB-231 cells. The IC50s for inhibition of the Chem-CLIP probe (1 μM of 2) for binding to FLT3 were ~0.1 μM for 1 and ~10 μM for 4 (Figure S9; includes control compound 7 and 10). In agreement with competition experiments between PROTAC 5 and 1 or 4 (Figure S19D,E), these studies demonstrate that conversion of a binder to a RIBOTAC decreases cellular protein occupancy by ~100-fold (Figure S9E). Notably, 10, with a urea linker attached to 1, has a similar IC50 as 1 does, indicating the linker has no contribution toward the decrease of protein occupancy by 4 (Figure S9D).
Analogously, C-Chem-CLIP studies were completed for the reprogrammed pre-miR-21 target. Here, control compound 7 was used in C-Chem-CLIP studies rather than RIBOTAC 4 as its cleavage activity would obscure analysis. The IC50s for PROTAC 5 and 7 for inhibiting cross-linking of Chem-CLIP probe 2 (5 μM) to pre-miR-21 were similar to 1, both ~5 μM (Figure S8E). These data indicate these modifications do not affect the compounds’ avidities for pre-miR-21 in cells, consistent with in vitro binding studies (Figure S6A).
Thus, occupancy of 4 accounts for much of the observed change in selectivity for the RNA target while the remaining 25-fold is likely due to the catalytic nature of RIBOTACs, together giving a final 2500-fold shift in selectivity. (24,29)
RIBOTAC 4 Is More Selective for Pre-miR-21 than 1 Proteome- and Transcriptome-wide
We next investigated the global effect of 1 (1 μM, where mature miR-21 levels are not reduced), 4 (1 μM, where mature miR-21 levels are reduced by 45%), and an oligonucleotide that targets miR-21 (LNA-21, 0.1 μM where mature miR-21 levels are reduced by 97%) on the proteome of MDA-MB-231 cells. Overall, modest effects on the proteome were observed for all three compounds (2965 proteins detected, ~1.5% or less were affected), indicative of selectivity (Figure 4A–C). In 1-treated cells, 46 proteins were significantly affected, 14 of which are associated with ERK pathways. For 4 and LNA-21, 28 and 25 proteins were affected, respectively. Principle component analysis (PCA) of all detectable proteins upon 1, 4, or LNA-21 treatment revealed that RIBOTAC 4 was similar to treatment with LNA-21 and dissimilar to 1, as expected since the RIBOTAC and oligonucleotide share a common target. [Note that 1, at the concentration studied in proteomics analysis, did not affect miR-21 (Figure S22).] A context score was calculated to quantify the effects of all three compounds on protein levels encoded by mRNAs regulated by miR-21. With treatment of both 4 and LNA-21, these proteins were significantly upregulated (Figure 4E,F). Treatment with 1 μM of 1, however, affected RTK-associated pathways but had no effect on miR-21 regulated proteins, as expected based on 1’s inability to reduce miR-21 levels at this concentration (Figure 4D). Neither treatment with 1 nor 4 affected proteins associated with let-7, selected as a control as its expression level is similar to that of miR-21 (Figure 4D,E).
Figure 4.
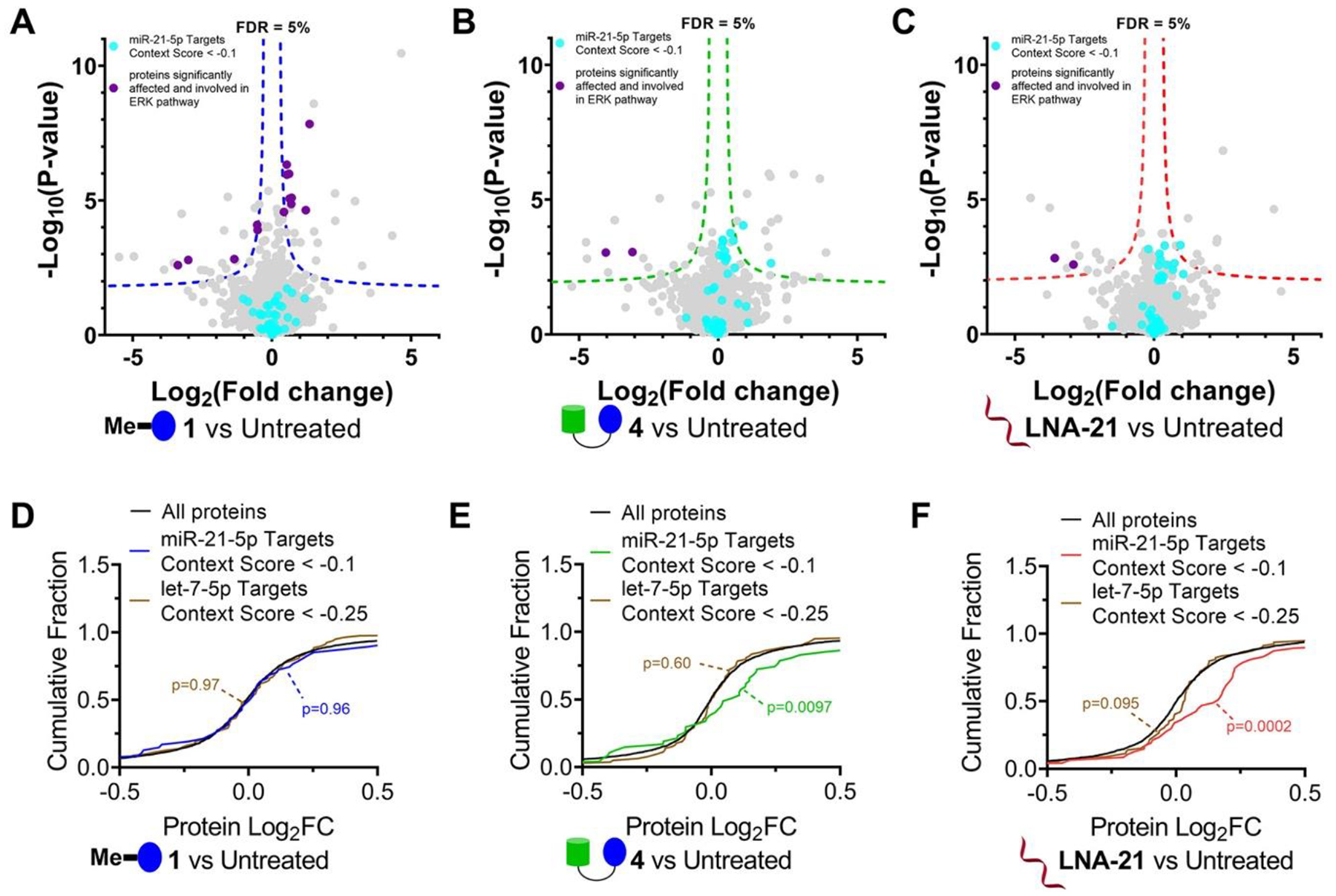
Effects of 1, RIBOTAC 4, and LNA-21 on global protein expression in MDA-MB-231 cells. (A) Volcano plot showing proteome-wide changes induced by 1 (1 μM, a concentration that does not inhibit pre-miR-21 biogenesis but does inhibit RTKs) (n = 3). (B) Volcano plot showing proteome-wide changes triggered by RIBOTAC 4 (1 μM) (n = 3). (C) Volcano plot showing proteome-wide changes caused by LNA-21 (0.1 μM) (n = 3). Dotted lines represent a false discovery rate (FDR) of 5% and an S0 of 0.1. (D) Cumulative distribution plot shows no significant changes for miR-21 regulated proteins upon treatment with 1 μM of 1, a concentration that does not inhibit pre-miR-21 biogenesis. Proteins regulated by miR-21 were predicted by TargetScanHuman v7.2 (n = 390). Approximately 18% of miR-21–5p targets (70/390) were detectable in MDA-MB-231 cells. (E, F) Significant increase in abundance was observed for miR-21 regulated proteins upon treatment with 4 (1 μM; E) or LNA-21 (0.1 μM; F). As a control, we investigated changes in proteins regulated by let-7–5p, which has an abundance similar to that of miR-21 in MDA-MB-231 cells. Downstream protein targets of let-7–5p (n = 1207) were predicted by TargetScanHuman v7.2. Approximately 13% of let-7–5p targets (160/1207) were detectable in the global proteomics analysis. Targets for context + + scores < –0.1 or −0.25 in miR-21–5p or let-7–5p were calculated for cumulation distributions.
In parallel, we studied the transcriptome-wide effects of 1 (1 μM), 4 (1 μM), and LNA-21 (0.1 μM) in MDA-MB-231 cells. Among the 3567 genes commonly detected in all three treatments, 28% (1070/3567) were significantly affected (adjusted P < 0.05) by 1-treatment while only 10.8% (387/3567) and 11.8% (422/3567) were affected by 4 and LNA-21, respectively (Figure 5A–C). Similarly, comparison of the fold change of each detectable transcript showed that treatment with RIBOTAC 4 and LNA-21 were nearly identical transcriptome-wide, which were dissimilar to 1 (Figure 5D–F). Collectively, these studies show that 4 and LNA-21 are more selective for pre-miR-21 than 1 both proteome- and transcriptome-wide and further supports that the RIBOTAC shifts selectivity to RNA.
Figure 5.
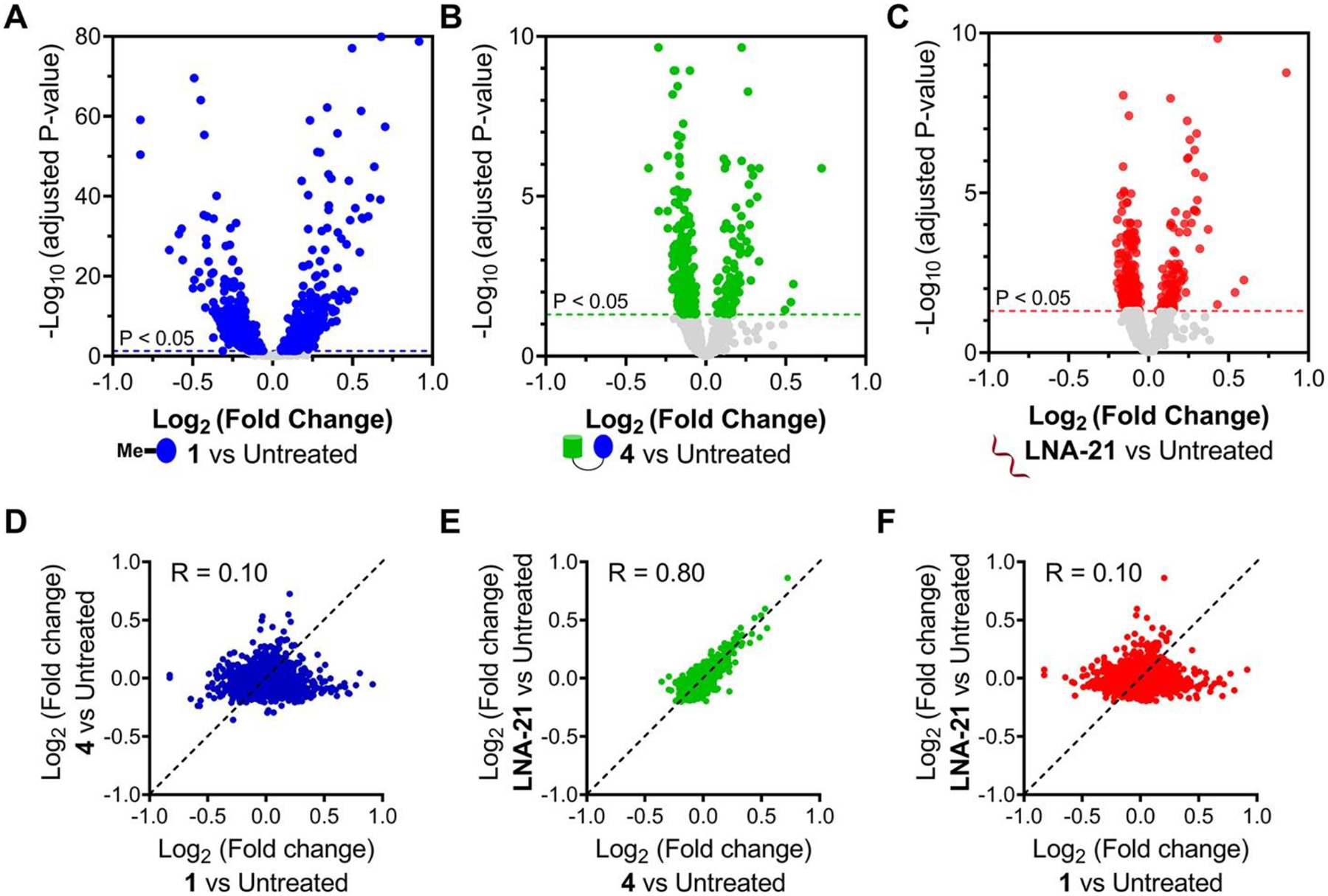
Effects of 1, RIBOTAC 4, and LNA-21 on global gene expression in MDA-MB-231 cells. (A) Volcano plot showing transcriptome-wide changes induced by 1 (1 μM, a concentration that does not inhibit pre-miR-21 biogenesis but does inhibit RTKs; n = 3). (B) Volcano plot showing transcriptome-wide changes triggered by RIBOTAC 4 (1 μM; n = 3). (C) Volcano plot showing transcriptome-wide changes caused by LNA-21 (0.1 μM; n = 3). Dotted lines represent adjusted p-value of <0.05. Fold change comparison of 4 vs 1 (D), LNA-21 vs 4(E), and LNA-21 vs 1 (F) suggests that MDA-MB-231 cells treated with RIBOTAC 4 are similar to samples treated with LNA-21 (green, R = 0.80). (43) Both 4 and LNA-21 are dissimilar to samples treated with 1 (blue and red, R = 0.10), as determined by analysis of RNA-seq data.
RIBOTAC 4 Mitigates Disease-Associated Pathologies in Two Mouse Models of Disease Caused by miR-21 Overexpression
Based on these encouraging results, we studied 4’s ability to inhibit the miR-21-mediated metastasis of breast cancer to the lung (30) in a xenograft mouse model (injection of MDA-MB-231-Luc cells). RIBOTAC 4 was administered by intraperitoneal (i.p.) injection at a dose to achieve 1 μM drug exposure in the lung, as determined by drug metabolism and pharmacokinetics (DMPK) analyses, which also indicated the compound is stable in vivo (Figures 6A and S23A,B). Indeed, 4-treatment inhibited breast cancer metastasis, as evidenced by the decreased number of lung nodules compared to vehicle-treated mice (Figures 6A and S24A). Lung histological studies showed that treatment also reduced hematoxylin and eosin (H&E) staining. Fluorescence in situ hybridization (FISH) showed miR-21 and pre-miR-21 expression were significantly diminished by 4 (Figures 6B and S24B), while immunohistochemistry (IHC) showed 4 de-repressed PDCD4 expression (Figure S24B) while having no effect on ERK or pERK levels (Figure 6B), thereby demonstrating the capacity of the reprogrammed Dovitinib-based RIBOTAC to selectively modulate miR-21 levels over the canonical RTK protein targets in vivo. Of note, we also treated mice with 1 to study its inhibitory effect on metastasis. Dosing was only maintained for 12 days due to 1’s toxicity (Figure S23C), at which point lung nodules have not yet formed in mice. At this time point, 1 did not affect mature miR-21, pre-miR-21, PDCD4, or ERK levels (Figures 6B and S24B). It did, however, decrease pERK levels, as expected (Figure 6B). Altogether, these studies demonstrate that 1 can indeed be reprogrammed by conversion into RIBOTAC 4 to a selective miR-21 inhibitor in vivo.
Figure 6.
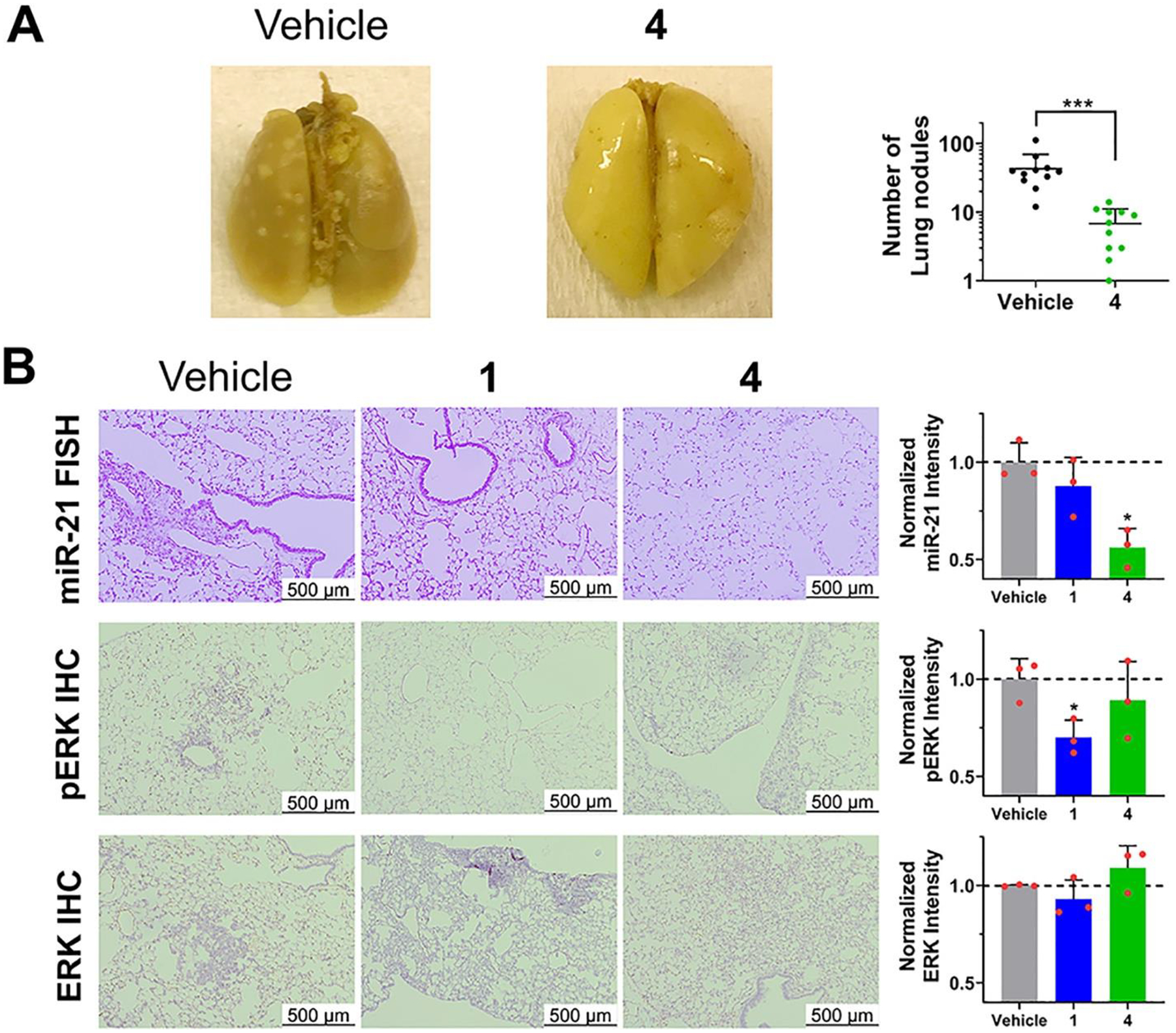
RIBOTAC 4 inhibits invasion of TNBC cells with reprogrammed selectivity in vivo. (A) In vivo treatment of 4 (56 mg/kg, q.o.d., 30 days) decreased the number of lung nodules (white nodules) stained with Bouin’s solution (n = 11 for vehicle or 4). (B) Lung tissue treated with RIBOTAC 4 exhibited decreased miR-21 levels and unchanged pERK levels, while 1 exhibited decreased pERK levels and unchanged miR-21 levels (n = 3). *p < 0.05, as determined by a two-tailed Student’s t test. All images are from biological replicates.
Alport Syndrome (AS) is a genetically defined disease of the kidney where miR-21 inhibition has therapeutic potential. (14) AS patients have a mutation in COL genes, (31) and a mouse model for AS with a knockout of the COL4A3 gene (Col4a3–/–) has been developed. (32) It displays various aspects of AS disease pathology including loss of kidney function, histological changes found in patients, and increased miR-21 levels. (14) Further, treatment of the mouse model with an antagomiR directed at miR-21 improves renal function. (14) Unlike cancers, RTKs have no known function in AS disease.
We delivered 4 to an AS mouse model, which was well tolerated (Figure S23D,E), and measured its effect on kidney function by monitoring urine albumin normalized to urine creatinine levels (Figure 7A). After 1 week of treatment, albumin levels were stabilized indicating improved kidney function (Figure 7A). Kidneys from 4-treated and vehicle-treated AS mice were harvested and analyzed, showing reduced levels of mature and pre-miR-21, as determined by RT-qPCR, and enhanced levels of peroxisome proliferator-activated receptor alpha (PPARα) protein, a target repressed by miR-21 (14) (Figures 7B,C, S25, and S26). In contrast, no effect on pERK levels was observed (Figure S27). Histological studies showed that disease-associated phenotypes were ameliorated by 4; tubulointerstitial pathology and glomerulosclerosis were significantly reduced (Figures 7D and S25). Thus, in a non-cancerous and currently untreatable genetically defined kidney disease, we have reprogrammed 1 into a pre-miR-21 degrader that rescues AS molecular defects and phenotypes.
Figure 7.
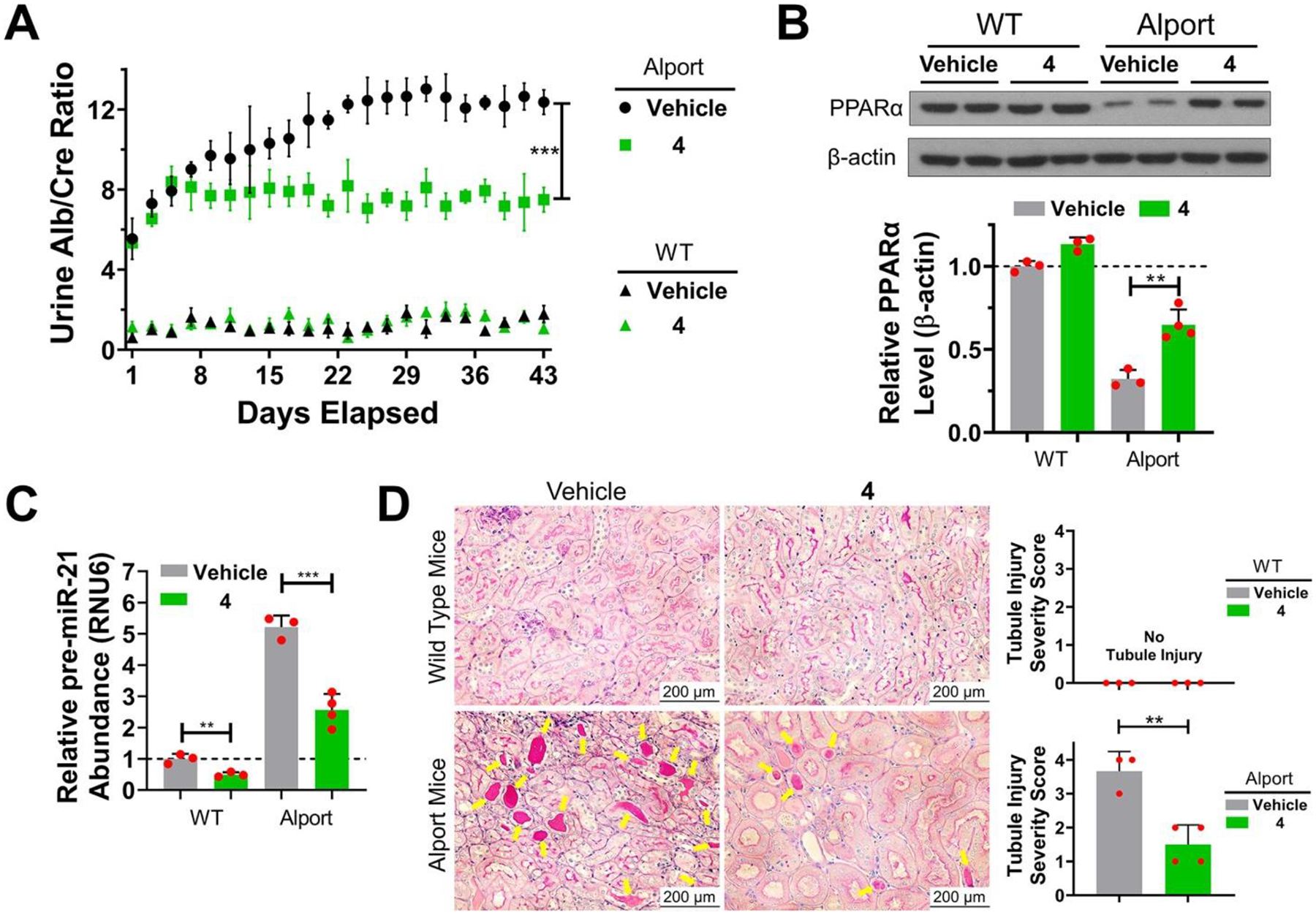
RIBOTAC 4 prevents Alport mice from progression of disease-associated nephropathy by inhibiting miR-21 biogenesis. (A) In vivo treatment of 4 (56 mg/kg, q.o.d., 42 days) decreased urine albumin concentration, as normalized to urine creatinine in Alport mice. In contrast, 4 had no effect on urine from wild-type (WT) mice. (B) RIBOTAC 4 de-repressed PPARα expression in kidneys of Alport mice, with no significant effect on PPARα expression in WT mice, although abundance was slightly boosted. (C) RIBOTAC 4 decreased pre-miR-21 levels in kidneys of both Alport and WT mice. [Note: Genetic ablation of miR-21 is well tolerated in mice. (12)] (D) Periodic acid–Schiff (PAS) staining shows that RIBOTAC 4 prevented tubule injury observed in Alport mice. Tubule injury severity was scored from 0 to 4 by extent of injury as previously described, (14) where 0 is no tubule injury (top left and right) and 4 is severe injury (bottom left). *p < 0.05, **p < 0.01, ***p < 0.001, as determined by a two-tailed Student’s t test. For vehicle- and 4-treated WT mice, n = 3; for vehicle-treated Alport mice, n = 3; for 4-treated Alport mice, n = 4. All images shown are from biological replicates.
With the concern of hyper-activation of the immune response and potential autoimmune disease due to activation of RNase L, (33) we studied changes in the innate immune response to RIBOTAC 4 or 2′–5′A4 treatment. In contrast to 2′–5′A4, no significant upregulation of various mRNAs associated with the antiviral innate immune response were observed upon treatment of MDA-MB-231 cells (Figure S28A) or Alport mice (Figure S28B) with 4. Although these in vivo studies do not unequivocally demonstrate RNase L-dependent cleavage of pre-miR-21 despite the reduction of its abundance (Figures 7C and S24B), these data, in addition to ablation of RIBOTAC 4 activity in MDA-MB-231 cells in which RNase L was knocked out by CRISPR (Figure S17), suggest that the mechanism of action of RIBOTAC 4 is not general, global activation of RNase L but the precise, local recruitment and activation of the enzyme to pre-miR-21.
Outlook
Oligonucleotide antagomiRs have been used to reduce miR-21 levels in cells (34–36) and in vivo. (14,37) However, oligonucleotides suffer from poor cell permeability and accumulation, inherent off-target effects, and problematic pharmacokinetics. (38) Small molecules offer an important alternative that can overcome these liabilities while targeting RNA structure. Indeed, various small molecules have been developed to inhibit miR-21 in cells, including peptides, (39–41) antibiotics, (6) and conjugated chemical probes, (42) although none has reported activity in vivo.
Here, for the first time, we have shown that a protein-targeted medicine, known to be safe in humans, can be rationally reprogrammed to target pre-miR-21, a miRNA that causes cancer, kidney disease, and cardiovascular disease, among other conditions. (10) Conversion of the simple binding compound into a RIBOTAC degrader shifted the selectivity from canonical RTK targets to the reprogrammed RNA target by 2500-fold with decreased toxicity, as evidenced in two mouse models of disease, metastatic breast cancer and Alport Syndrome. We are likely only seeing the tip of the iceberg of small molecules targeting RNA, and these studies may establish a foundation for reprogramming other known drugs for disease-associated RNAs.
Supplementary Material
Acknowledgments
We thank Dr. Jonathan Chen for docking Dovitinib into pre-miR-21 to provide a model of the three-dimensional complex. We thank Tanya Khan for help with mice studies. We thank Prof. Jeffrey H. Miner for providing Alport mice breeding pairs and for experimental advice.
Funding
This work was supported by the National Institutes of Health grants R01 CA249180 and R35 NS116846 (to M.D.D.), the Myotonic US Fellowship Research Grant (to R.I.B.), and the National Ataxia Foundation Fellowship Research Grant (to R.I.B.).
Footnotes
Supporting Information
- Methods (biochemical, cellular, in vivo), Supplementary Figures S1–S29 and Table S1, synthetic experimental methods and schemes, and compound characterization (PDF)
- Data S1: Results of global proteomic analysis of MDA-MB-231 cells treated with 1 (1 μM), 4 (1 μM), or LNA-21 (0.1 μM) and untreated cells (XLSX)
- Data S2: Results of RNA-seq analysis of MDA-MB-231 cells treated with 1 (1 μM), 4 (1 μM), or LNA-21 (0.1 μM) and untreated cells (XLSX)
The authors declare the following competing financial interest(s): M.D.D. is a founder of Expansion Therapeutics.
Proteomics and RNA-seq data files supporting this publication are available from Mendeley Data at DOI: 10.17632/msh9kv79yz.1.
Contributor Information
Peiyuan Zhang, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Xiaohui Liu, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Daniel Abegg, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Toru Tanaka, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Yuquan Tong, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Raphael I. Benhamou, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States
Jared Baisden, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Gogce Crynen, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Samantha M. Meyer, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States
Michael D. Cameron, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States
Arnab K. Chatterjee, California Institute for Biomedical Research (CALIBR), Scripps Research, La Jolla, California 92037, United States
Alexander Adibekian, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
Jessica L. Childs-Disney, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States
Matthew D. Disney, Department of Chemistry, Scripps Research, Jupiter, Florida 33458, United States.
REFERENCES
- 1.Pushpakom S; Iorio F; Eyers PA; Escott KJ; Hopper S; Wells A; Doig A; Guilliams T; Latimer J; McNamee C; Norris A; Sanseau P; Cavalla D; Pirmohamed M Drug repurposing: progress, challenges and recommendations. Nat. Rev. Drug Discovery 2019, 18 (1), 41–58, DOI: 10.1038/nrd.2018.168 [DOI] [PubMed] [Google Scholar]
- 2.Janes J; Young ME; Chen E; Rogers NH; Burgstaller-Muehlbacher S; Hughes LD; Love MS; Hull MV; Kuhen KL; Woods AK; Joseph SB; Petrassi HM; McNamara CW; Tremblay MS; Su AI; Schultz PG; Chatterjee AK The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (42), 10750–10755, DOI: 10.1073/pnas.1810137115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riva L; Yuan S; Yin X; Martin-Sancho L; Matsunaga N; Pache L; Burgstaller-Muehlbacher S; De Jesus PD; Teriete P; Hull MV; Chang MW; Chan JF; Cao J; Poon VK; Herbert KM; Cheng K; Nguyen TH; Rubanov A; Pu Y; Nguyen C; Choi A; Rathnasinghe R; Schotsaert M; Miorin L; Dejosez M; Zwaka TP; Sit KY; Martinez-Sobrido L; Liu WC; White KM; Chapman ME; Lendy EK; Glynne RJ; Albrecht R; Ruppin E; Mesecar AD; Johnson JR; Benner C; Sun R; Schultz PG; Su AI; Garcia-Sastre A; Chatterjee AK; Yuen KY; Chanda SK Discovery of SARS-CoV-2 antiviral drugs through large-scale compound repurposing. Nature 2020, 586 (7827), 113–119, DOI: 10.1038/s41586-020-2577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu B; Childs-Disney JL; Znosko BM; Wang D; Fallahi M; Gallo SM; Disney MD Analysis of secondary structural elements in human microRNA hairpin precursors. BMC Bioinformatics 2016, 17, 112, DOI: 10.1186/s12859-016-0960-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tran T; Disney MD Identifying the preferred RNA motifs and chemotypes that interact by probing millions of combinations. Nat. Commun 2012, 3, 1125, DOI: 10.1038/ncomms2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Velagapudi SP; Costales MG; Vummidi BR; Nakai Y; Angelbello AJ; Tran T; Haniff HS; Matsumoto Y; Wang ZF; Chatterjee AK; Childs-Disney JL; Disney MD Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol 2018, 25 (9), 1086–1094, DOI: 10.1016/j.chembiol.2018.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Velagapudi SP; Luo Y; Tran T; Haniff HS; Nakai Y; Fallahi M; Martinez GJ; Childs-Disney JL; Disney MD Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci 2017, 3 (3), 205–216, DOI: 10.1021/acscentsci.7b00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Velagapudi SP; Gallo SM; Disney MD Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 2014, 10 (4), 291–297, DOI: 10.1038/nchembio.1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi YJ; Kim HS; Park SH; Kim BS; Kim KH; Lee HJ; Song HS; Shin DY; Lee HY; Kim HG; Lee KH; Lee JL; Park KH Phase II study of Dovitinib in patients with castration-resistant prostate cancer (KCSG-GU11–05). Cancer Res. Treat 2018, 50 (4), 1252–1259, DOI: 10.4143/crt.2017.438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krichevsky AM; Gabriely G miR-21: a small multi-faceted RNA. J. Cell Mol. Med 2009, 13 (1), 39–53, DOI: 10.1111/j.1582-4934.2008.00556.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esquela-Kerscher A; Slack FJ Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6 (4), 259–269, DOI: 10.1038/nrc1840 [DOI] [PubMed] [Google Scholar]
- 12.Ma X; Kumar M; Choudhury SN; Becker Buscaglia LE; Barker JR; Kanakamedala K; Liu MF; Li Y Loss of the miR-21 allele elevates the expression of its target genes and reduces tumorigenesis. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (25), 10144–10149, DOI: 10.1073/pnas.1103735108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Medina PP; Nolde M; Slack FJ OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467 (7311), 86–90, DOI: 10.1038/nature09284 [DOI] [PubMed] [Google Scholar]
- 14.Gomez IG; MacKenna DA; Johnson BG; Kaimal V; Roach AM; Ren S; Nakagawa N; Xin C; Newitt R; Pandya S; Xia TH; Liu X; Borza DB; Grafals M; Shankland SJ; Himmelfarb J; Portilla D; Liu S; Chau BN; Duffield JS Anti-microRNA-21 oligonucleotides prevent Alport nephropathy progression by stimulating metabolic pathways. J. Clin. Invest 2015, 125 (1), 141–156, DOI: 10.1172/JCI75852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel DP MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116 (2), 281–297, DOI: 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- 16.Bartel DP MicroRNAs: target recognition and regulatory functions. Cell 2009, 136 (2), 215–233, DOI: 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J; Schultz PG; Johnson KA Mechanistic studies of a small-molecule modulator of SMN2 splicing. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (20), E4604–E4612, DOI: 10.1073/pnas.1800260115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mortison JD; Schenone M; Myers JA; Zhang Z; Chen L; Ciarlo C; Comer E; Natchiar SK; Carr SA; Klaholz BP; Myers AG Tetracyclines modify translation by targeting key human rRNA substructures. Cell Chem. Biol 2018, 25 (12), 1506–1518, DOI: 10.1016/j.chembiol.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Renhowe PA; Pecchi S; Shafer CM; Machajewski TD; Jazan EM; Taylor C; Antonios-McCrea W; McBride CM; Frazier K; Wiesmann M; Lapointe GR; Feucht PH; Warne RL; Heise CC; Menezes D; Aardalen K; Ye H; He M; Le V; Vora J; Jansen JM; Wernette-Hammond ME; Harris AL Design, structure-activity relationships and in vivo characterization of 4-amino-3-benzimidazol-2-ylhydroquinolin-2-ones: a novel class of receptor tyrosine kinase inhibitors. J. Med. Chem 2009, 52 (2), 278–292, DOI: 10.1021/jm800790t [DOI] [PubMed] [Google Scholar]
- 20.Velagapudi SP; Li Y; Disney MD A cross-linking approach to map small molecule-RNA binding sites in cells. Bioorg. Med. Chem. Lett 2019, 29 (12), 1532–1536, DOI: 10.1016/j.bmcl.2019.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trudel S; Li ZH; Wei E; Wiesmann M; Chang H; Chen C; Reece D; Heise C; Stewart AK CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood 2005, 105 (7), 2941–2948, DOI: 10.1182/blood-2004-10-3913 [DOI] [PubMed] [Google Scholar]
- 22.Costales MG; Matsumoto Y; Velagapudi SP; Disney MD Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 2018, 140 (22), 6741–6744, DOI: 10.1021/jacs.8b01233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (15), 8554–8559, DOI: 10.1073/pnas.141230798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costales MG; Aikawa H; Li Y; Childs-Disney JL; Abegg D; Hoch DG; Pradeep Velagapudi S; Nakai Y; Khan T; Wang KW; Yildirim I; Adibekian A; Wang ET; Disney MD Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (5), 2406–2411, DOI: 10.1073/pnas.1914286117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han Y; Whitney G; Donovan J; Korennykh A Innate immune messenger 2–5A tethers human RNase L into active high-order complexes. Cell Rep. 2012, 2 (4), 902–913, DOI: 10.1016/j.celrep.2012.09.004 [DOI] [PubMed] [Google Scholar]
- 26.Silverman RH Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol 2007, 81 (23), 12720–12729, DOI: 10.1128/JVI.01471-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Crews CM Feeding the machine: mechanisms of proteasome-catalyzed degradation of ubiquitinated proteins. Curr. Opin. Chem. Biol 2003, 7 (5), 534–539, DOI: 10.1016/j.cbpa.2003.08.002 [DOI] [PubMed] [Google Scholar]
- 28.Ventura A; Young AG; Winslow MM; Lintault L; Meissner A; Erkeland SJ; Newman J; Bronson RT; Crowley D; Stone JR; Jaenisch R; Sharp PA; Jacks T Targeted deletion reveals essential and overlapping functions of the miR-17 ~ 92 family of miRNA clusters. Cell 2008, 132 (5), 875–886, DOI: 10.1016/j.cell.2008.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Costales MG; Suresh B; Vishnu K; Disney MD Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol 2019, 26 (8), 1180–1186, DOI: 10.1016/j.chembiol.2019.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang S; Zhang JJ; Huang XY Mouse models for tumor metastasis. Methods Mol. Biol 2012, 928, 221–228, DOI: 10.1007/978-1-62703-008-3_17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hudson BG; Tryggvason K; Sundaramoorthy M; Neilson EG Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med 2003, 348 (25), 2543–2556, DOI: 10.1056/NEJMra022296 [DOI] [PubMed] [Google Scholar]
- 32.Cosgrove D; Meehan DT; Grunkemeyer JA; Kornak JM; Sayers R; Hunter WJ; Samuelson GC Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996, 10 (23), 2981–2992, DOI: 10.1101/gad.10.23.2981 [DOI] [PubMed] [Google Scholar]
- 33.Chakrabarti A; Jha BK; Silverman RH New insights into the role of RNase L in innate immunity. J. Interferon Cytokine Res 2011, 31 (1), 49–57, DOI: 10.1089/jir.2010.0120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao YJ; Li YJ; Zheng W; Zhao JJ; Guo MM; Zhou Y; Qin NL; Zheng J; Xu L Antisense oligonucleotides against microRNA-21 reduced the proliferation and migration of human colon carcinoma cells. Cancer Cell Int. 2015, 15, 77, DOI: 10.1186/s12935-015-0228-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nedaeinia R; Sharifi M; Avan A; Kazemi M; Rafiee L; Ghayour-Mobarhan M; Salehi R Locked nucleic acid anti-miR-21 inhibits cell growth and invasive behaviors of a colorectal adenocarcinoma cell line: LNA-anti-miR as a novel approach. Cancer Gene Ther. 2016, 23 (8), 246–253, DOI: 10.1038/cgt.2016.25 [DOI] [PubMed] [Google Scholar]
- 36.Ding T; Cui P; Zhou Y; Chen C; Zhao J; Wang H; Guo M; He Z; Xu L Antisense oligonucleotides against miR-21 inhibit the growth and metastasis of colorectal carcinoma via the DUSP8 pathway. Mol. Ther.--Nucleic Acids 2018, 13, 244–255, DOI: 10.1016/j.omtn.2018.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolling M; Kaucsar T; Schauerte C; Hubner A; Dettling A; Park JK; Busch M; Wulff X; Meier M; Scherf K; Bukosza N; Szenasi G; Godo M; Sharma A; Heuser M; Hamar P; Bang C; Haller H; Thum T; Lorenzen JM Therapeutic miR-21 silencing ameliorates diabetic kidney disease in mice. Mol. Ther 2017, 25 (1), 165–180, DOI: 10.1016/j.ymthe.2016.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juliano RL; Ming X; Nakagawa O The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res 2012, 45 (7), 1067–1076, DOI: 10.1021/ar2002123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bose D; Nahar S; Rai MK; Ray A; Chakraborty K; Maiti S Selective inhibition of miR-21 by phage display screened peptide. Nucleic Acids Res. 2015, 43 (8), 4342–4352, DOI: 10.1093/nar/gkv185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakamoto K; Otake K; Umemoto T Discovery of peptidic miR-21 processing inhibitor by mirror image phage display: A novel method to generate RNA binding D-peptides. Bioorg. Med. Chem. Lett 2017, 27 (4), 826–828, DOI: 10.1016/j.bmcl.2017.01.023 [DOI] [PubMed] [Google Scholar]
- 41.Shortridge MD; Walker MJ; Pavelitz T; Chen Y; Yang W; Varani G A macrocyclic peptide ligand binds the oncogenic microRNA-21 precursor and suppresses Dicer processing. ACS Chem. Biol 2017, 12 (6), 1611–1620, DOI: 10.1021/acschembio.7b00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan H; Bhattarai U; Guo ZF; Liang FS Regulating miRNA-21 biogenesis by bifunctional small molecules. J. Am. Chem. Soc 2017, 139 (14), 4987–4990, DOI: 10.1021/jacs.7b00610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu M; Yi M; Kim CH; Deng C; Li Y; Medina D; Stephens RM; Green JE Integrated miRNA and mRNA expression profiling of mouse mammary tumor models identifies miRNA signatures associated with mammary tumor lineage. Genome Biol. 2011, 12 (8), R77, DOI: 10.1186/gb-2011-12-8-r77 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.