

Abstract
Objective
Gilteritinib received approval for the treatment of FLT3-mutated relapsed or refractory acute myeloid leukemia in Japan in 2018. In accordance with regulatory requirements, we conducted a multicenter, observational surveillance of gilteritinib use in Japan.
Methods
Patients were followed for 6 months from gilteritinib treatment initiation. The primary endpoint of the surveillance was incidence of adverse drug reactions related to each element of the safety specification defined in the Japanese Risk Management Plan. This interim analysis presents data collected from 3 December 2018 to 20 September 2020.
Results
Among 204 patients with case report forms, 107 consented to data publication. Of these 107 patients, 59.8% (n = 64) were male and 58.9% (n = 63) were aged ≥65 years; most received a 120-mg/day initial (80.4%; 86/107) and maximum (74.8%; 80/107) daily dose. The discontinuation rate was 61.7% (66/107); the most common reasons for discontinuation were disease progression (18.7%), transplantation (16.8%) and adverse events (15.0%). The adverse drug reaction rate was 77.6%. The incidences of adverse drug reactions (grade ≥ 3) related to each element of the safety specification were myelosuppression, 44.9% (38.3%); liver function disorder, 24.3% (6.5%); infections, 24.3% (21.5%); prolonged QT interval, 10.3% (2.8%); hemorrhage, 9.3% (6.5%); renal dysfunction, 6.5% (0); hypersensitivity, 5.6% (1.9%); interstitial lung disease, 4.7% (3.7%); cardiac failure/pericarditis/pericardial effusion, 1.9% (0.9%); pancreatitis, 0.9% (0); posterior reversible encephalopathy syndrome, 0.9% (0.9%). The composite complete remission rate was 62.7%; the 6-month overall survival rate was 77.7%.
Conclusion
Gilteritinib treatment for 6 months in Japan was associated with acceptable efficacy and no new safety concerns were observed.
Keywords: acute myeloid leukemia, fms-like tyrosine kinase 3, adverse drug reaction
Interim results from this Japan postmarketing surveillance study show that gilteritinib appears safe and efficacious in patients with relapsed/refractory FLT3-mutated AML. These data support the real-world benefit of gilteritinib.
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous hematologic malignancy characterized by the clonal expansion of undifferentiated myeloid precursors in the bone marrow leading to failed hematopoiesis (1). The gene encoding fms-like tyrosine kinase 3 (FLT3), a transmembrane cytokine receptor expressed on hematopoietic progenitor cells, is the most commonly mutated gene identified in patients with AML (2,3). The wild-type FLT3 protein functions to promote differentiation and maturation of hematopoietic progenitor cells. In contrast, activating mutations in FLT3, which include internal tandem duplication (ITD) mutations and tyrosine kinase domain (TKD) mutations, are associated with poor prognosis (4).
The availability of FLT3-targeted therapies has improved the treatment of AML. First-generation multikinase inhibitors such as midostaurin and sorafenib target FLT3 as well as other kinases. As such, these agents require administration in combination with chemotherapy to maximize their clinical benefit (5–8). More potent and FLT3-specific second-generation FLT3 inhibitors, such as gilteritinib and quizartinib, were subsequently developed and demonstrated clinical benefits in patients with FLT3 mutation-positive (FLT3mut+) relapsed or refractory (R/R) AML (9–11). However, despite its benefit in patients with FLT3-ITD+ R/R AML, quizartinib is not clinically active against FLT3-TKD mutations, which develop as a consequence of FLT3 inhibitor therapy (12). Gilteritinib is a highly specific, oral, small molecule FLT3 inhibitor that inhibits the signaling of FLT3 derived from both FLT3-ITD and FLT3-TKD mutations, resulting in apoptosis (13).
In September 2018, gilteritinib received approval from the Japan Ministry of Health, Labour and Wealth for patients with FLT3mut+ R/R AML based on interim results from the Phase 3 ADMIRAL trial, which evaluated gilteritinib versus conventional salvage chemotherapy in this patient population (14). Subsequently, gilteritinib also received approval for this indication from the United States Food and Drug Administration (November 2018) and European Medicines Agency (October 2019) (15,16). In the ADMIRAL trial, the median age of patients in the gilteritinib arm was 62 years (range, 20–84), 39.7% of patients had primary refractory AML and 60.3% had relapsed AML. Final results from the ADMIRAL trial showed that gilteritinib-treated patients showed significantly longer overall survival (OS) than chemotherapy-treated patients (9.3 vs. 5.6 months, respectively) and achieved a 1-year OS rate of 37.1% versus 16.7% with chemotherapy (10). Rates of complete remission (CR) or CR with partial hematologic recovery were 34.0% with gilteritinib and 15.3% with salvage chemotherapy, and the median duration of gilteritinib response was 11.0 months (10). Gilteritinib treatment was also associated with a lower incidence of exposure-adjusted grade ≥ 3 adverse events (AEs) versus chemotherapy (19.34 vs. 42.44 events per patient-year, respectively) (10).
The conditions of the approval of gilteritinib in Japan based on the strategy of SAKIGAKE, the practical application of innovative medical products and devices, required an assessment of the safety of its real-world use in the postmarketing setting. The objective of this surveillance study was to assess the safety and tolerability of gilteritinib therapy in patients with R/R FLT3mut+ AML in actual clinical settings in Japan.
Patients and methods
Surveillance design
An all-case postmarketing drug-use surveillance of patients with R/R FLT3mut+ AML treated with commercially available gilteritinib was initiated on 3 December 2018, the start date of gilteritinib marketing in Japan. Case report forms (CRFs) were collected for all patients who had started gilteritinib therapy by 31 October 2019. This multicenter, uncontrolled, observational surveillance of gilteritinib is being conducted across 362 medical institutions in Japan. The surveillance was conducted in accordance with Good Postmarketing Study Practice in Japan to comply with data integrity standards for regulatory submission and documentation. In the international Phase 3 ADMIRAL trial, most AEs related to the safety specifications and most grade ≥ 3 AEs occurred within the first 6 months after initiation of gilteritinib therapy. Based on this finding, a 6-month observation period from the start of treatment was considered appropriate for monitoring the occurrence of AEs related to the safety specifications. As such, the 6-month observation period for each patient was initiated from the start date of treatment. For patients who died or were transferred to another hospital before the end of the 6-month surveillance, the observation period spanned from the start of treatment to the date of death or hospital transfer.
Data collection and analysis
Patient data were collected by investigators at individual sites using CRFs for the following survey items: demographic and baseline characteristics, medical history related to AML and AML type, dosing parameters for gilteritinib, other AML therapies, AEs and adverse drug reactions (ADRs), response to treatment and transplant status. The primary endpoint was the incidence of ADRs related to each element of the safety specification defined in the Japanese Risk Management Plan, which is based on clinical trial results. The ADRs that were related to elements of the safety specification included myelosuppression, infection, hemorrhage, prolonged QT interval, cardiac failure/pericarditis/pericardial effusion, liver function disorder, renal dysfunction, gastrointestinal perforation, interstitial lung disease (ILD), hypersensitivity, pancreatitis and posterior reversible encephalopathy syndrome (PRES). Secondary endpoints were the incidence of all ADRs and serious AEs. The ADRs and AEs were categorized by preferred term and system organ class (Medical Dictionary for Regulatory Activities v23.0) and graded using National Cancer Institute Common Terminology Criteria for Adverse Events. An ADR was defined as an AE for which a causal relationship to gilteritinib could not be ruled out. Treatment response assessed using modified International Working Group criteria (17) and OS were evaluated as exploratory endpoints. The composite CR (CRc) rate was defined as the sum of patients who achieved CR, CR with incomplete hematologic recovery (CRi) and CR with incomplete platelet recovery (CRp). The cumulative continuation rate and OS rate at 1, 2, 3, 4, 5 and 6 months after initiation of gilteritinib were estimated using the Kaplan–Meier method. For patients without an event resulting in treatment discontinuation, cumulative continuation rates were calculated using the date of completion of the observation period or the date of the last visit as the end date; OS was determined using the date of completion of the observation period or the date of the last gilteritinib dose as the end date.
Statistical analyses
Continuous variables were summarized using descriptive statistics, including mean (standard deviation), median (interquartile range) and minimum and maximum values. Discrete variables were summarized as frequencies and proportions. Data were analyzed using SAS® v9.4 (SAS Institute Inc., Cary, NC).
Results
Patient disposition and baseline characteristics
A total of 328 patients have been enrolled between the date of initiation of surveillance on 3 December 2018 and 20 September 2020 (end of surveillance for the fourth safety update report). Of the 204 patients with complete CRFs, 107 patients across 81 sites provided consent to publish survey results and were included in the safety and efficacy analysis sets.
Of these 107 patients, most were male (59.8%; n = 64), aged ≥65 years (58.9%; n = 63) and had an Eastern Cooperative Oncology Group performance status of 0–1 (76.6%; n = 82) (Table 1). Overall, 57% (n = 61/107) of patients had refractory AML and 78.5% (n = 84/107) had comorbidities, with hypertension (29.9%; n = 32/107), diabetes mellitus (13.1%; n = 14/107), dyslipidemia (12.1%; n = 13/107) and pneumonia (10.3%; n = 11/107) being the most common. Overall, 76.6% (n = 82/107) had FLT3-ITD mutations only, 7.5% (n = 8/107) had FLT3-TKD mutations only, 4.7% (n = 5/107) had both FLT3-ITD and FLT3-TKD mutations and 11.2% (n = 12/107) had other FLT3 mutations. Although gilteritinib is not indicated for use in pediatric patients, six patients aged 11–14 years received gilteritinib off label. All six pediatric patients had FLT3-ITD mutations without FLT3-TKD point mutations, and three had relapsed after prior hematopoietic stem cell transplantation (HSCT). Of the 107 patients evaluated, 19 (male, n = 15; female, n = 4) had AML secondary to myelodysplastic syndrome (MDS); all but one was FLT3-ITD+, 14 had refractory disease and five had relapsed AML. Thirteen of these 19 patients had previously received high-intensity chemotherapy.
Table 1.
Demographic and baseline characteristics of patients with R/R FLT3-mutated AML
| Characteristic | N = 107 |
|---|---|
| Median age, years (range) | 69 (11–85) |
|
Sex
Male Female |
64 (59.8) 43 (40.2) |
| Median body weight, kg (range) | 53.6 (25.7–95.0) |
|
ECOG performance status
0–1 ≥2 |
82 (76.6) 25 (23.4) |
| Median duration of AML, months (range) | 6.0 (0–72.0) |
| AML type New onset AML secondary to MDS Secondary AML |
85 (79.4) 19 (17.8) 3 (2.8) |
|
FAB classification
M0 M1 M2 M3 M4 M5 M6 M7 |
5 (4.7) 20 (18.7) 29 (27.1) 0 21 (19.6) 23 (21.5) 1 (0.9) 0 |
|
Central nervous system leukemia
No Present with symptoms Present without symptoms |
95 (88.8) 2 (1.9) 3 (2.8) |
|
FLT3 mutation type, n (%)
FLT3-ITD only FLT3-TKD only FLT3-ITD and FLT3-TKD Other |
82 (76.6) 8 (7.5) 5 (4.7) 12 (11.2) |
|
Response to AML treatment before initiation of gilteritinib
Relapsed Refractory |
46 (43.0) 61 (57.0) |
|
AML treatment immediately before initiation of gilteritinib
Remission induction Consolidation Salvage Other |
68 (63.6) 6 (5.6) 24 (22.4) 9 (8.4) |
|
Prior HSCT
Yes No |
21 (19.6) 86 (80.4) |
|
Presence of comorbidities
Yes No |
84 (78.5) 23 (21.5) |
Data are reported as n (%) unless otherwise noted.
AML, acute myeloid leukemia; ECOG, Eastern Cooperative Oncology Group; FAB, French–American–British; HSCT, hematopoietic stem cell transplantation; ITD, internal tandem duplication; MDS, myelodysplastic syndrome; R/R, relapsed or refractory; TKD, tyrosine kinase domain.
At the time of treatment initiation with gilteritinib, five patients were diagnosed with central nervous system (CNS) leukemia; two were symptomatic. One case of symptomatic CNS leukemia was observed in a 69-year-old female patient with FLT3-ITD+ AML who displayed symptoms of peripheral neuropathy, insomnia, chronic gastritis, chronic enteritis and constipation. The second case of symptomatic CNS leukemia was observed in a heavily pretreated 70-year-old male patient with FLT3-ITD+ AML secondary to MDS. The patient had symptomatic epilepsy with CNS infiltration.
Treatment with gilteritinib
During the surveillance period, most patients received 120-mg gilteritinib as the initial daily dose (80.4%; n = 86/107) as well as the maximum daily dose (74.8%; n = 80/107) (Table 2). Approximately 52% (n = 56/107) of patients required dose modifications, most frequently within the first 2 months of treatment. Discontinuation of gilteritinib was observed in 61.7% (n = 66/107) of patients; the most common reasons for discontinuation were disease progression (18.7%; n = 20), transplantation (16.8%; n = 18) and AEs (15.0%; n = 16). Initial doses for two pediatric patients weighing <30 kg were 70 mg in an 11-year-old male patient and 40 mg in a 12-year-old female patient; the dose of gilteritinib was subsequently increased to 120 mg in the 12-year-old patient following disease progression.
Table 2.
Gilteritinib exposure in patients with R/R FLT3-mutated AML
| Parameter | N = 107 |
|---|---|
|
Initial daily dose
40 mg 80 mg 120 mg 160 mg 200 mg Other |
10 (9.3) 10 (9.3) 86 (80.4) 0 0 1 (0.9) |
|
Maximum daily dose
40 mg 80 mg 120 mg 160 mg 200 mg Other |
3 (2.8) 11 (10.3) 80 (74.8) 2 (1.9) 10 (9.3) 1 (0.9) |
| Median dosing duration, days (range) | 105 (3–181) |
|
Drug withdrawal
Yes No |
42 (39.3) 65 (60.7) |
|
Dose modification
Yes No |
56 (52.3) 51 (47.7) |
|
Time from initial dose to dose modification
<1 month ≥1 month–<2 months ≥2 months–<3 months ≥3 months–<4 months ≥4 months–<5 months ≥5 months–<6 months ≥6 months |
21 (19.6) 16 (15.0) 4 (3.7) 5 (4.7) 5 (4.7) 5 (4.7) 0 |
|
Discontinuation
Yes No |
66 (61.7) 41 (38.3) |
|
Reasons for discontinuation
Disease progression Transplantation Adverse event Other Hospital transfer Death Patient request Relapse |
20 (18.7) 18 (16.8) 16 (15.0) 4 (3.7) 4 (3.7) 3 (2.8) 3 (2.8) 1 (0.9) |
Data are reported as n (%) unless otherwise noted.
AML, acute myeloid leukemia; R/R, relapsed or refractory.
The cumulative continuation rate of gilteritinib over the 6-month surveillance period is shown in Fig. 1. The cumulative continuation rate declined to 40.7% over the 6-month period.
Figure 1.

Cumulative continuation rate of gilteritinib during the surveillance period. Abbreviation: CI, confidence interval.
Other treatments for AML
Of the 107 patients receiving gilteritinib, 12 (11.2%) also received other AML therapies, which included antimetabolites (n = 10; 9.3%), antineoplastic antibiotics (n = 4; 3.7%), plant alkaloids (n = 3; 2.8%), other antitumor drugs [mitoxantrone, n = 1 (0.9%); azacitidine, n = 1 (0.9%)] and alkylating agents (n = 1; 0.9%). Six of these 12 patients received concomitant treatment for AML with other agents: five received concomitant treatment with antimetabolites (cytarabine, methotrexate or hydroxycarbamide) and one received concomitant treatment with azacitidine.
Safety outcomes
Overall, 77.6% (n = 83/107) of patients experienced ADRs during the surveillance period. The incidence of ADRs related to each element of the safety specification were as follows: myelosuppression (44.9%; n = 48/107), liver function disorder (24.3%; n = 26/107), infections (24.3%; n = 26/107), prolonged QT interval (10.3%; n = 11/107), hemorrhage (9.3%; n = 10/107), renal dysfunction (6.5%; n = 7/107), hypersensitivity (5.6%; n = 6/107), ILD (4.7%; n = 5/107), cardiac failure/pericarditis/pericardial effusion (1.9%; n = 2/107), pancreatitis (0.9%; n = 1/107) and PRES (0.9%; n = 1/107) (Table 3). There was no incidence of gastrointestinal perforation, which was observed as an important identified risk based on results from clinical studies of gilteritinib (10,18,19) and safety monitoring activities. At the time of this interim analysis, most ADRs related to myelosuppression (n = 32/48), liver function disorder (n = 23/26), infections (n = 21/26) and prolonged QT interval (n = 10/11) had either resolved or were in the process of resolving in most patients. The most common fatal ADRs were related to infections (2.8%; n = 3/107) and hemorrhage (2.8%; n = 3/107).
Table 3.
Incidence of ADRs for each safety specification
| Safety Specification | Anya,b | Grade ≥ 3a,b | Resolved/ Resolvingc |
Not Resolvedc | Deathc | Median Time to Onset, Days (Range) | Median Time to Resolution, Days (Range) |
|---|---|---|---|---|---|---|---|
| Myelosuppressionc | 48 (44.9) | 41 (38.3) | 32 (66.7) | 14 (29.2) | 1 (2.1) | 9 (1–113) | 21.5 (3–211) |
| Infections | 26 (24.3) | 23 (21.5) | 21 (80.8) | 2 (7.7) | 3 (11.5) | 36 (4–128) | 18 (9–57) |
| Liver function disorder | 26 (24.3) | 7 (6.5) | 23 (88.5) | 3 (11.5) | 0 | 18.5 (3–166) | 34 (6–255) |
| Prolonged QT interval | 11 (10.3) | 3 (2.8) | 10 (90.9) | 1 (9.1) | 0 | 16 (4–122) | 5 (2–129) |
| Hemorrhage | 10 (9.3) | 7 (6.5) | 6 (60.0) | 1 (10.0) | 3 (30.0) | 62.5 (3–109) | 42 (22–157) |
| Renal dysfunction | 7 (6.5) | 0 | 6 (85.7) | 1 (14.3) | 0 | 27 (4–112) | 82.5 (7–239) |
| Hypersensitivity | 6 (5.6) | 2 (1.9) | 6 (100) | 0 | 0 | 33 (1–60) | 12.5 (8–41) |
| Interstitial lung disease | 5 (4.7) | 4 (3.7) | 4 (80.0) | 0 | 1 (20.0) | 58 (20–141) | 18.5 (10–61) |
| Cardiac failure/pericarditis/pericardial effusion | 2 (1.9) | 1 (0.9) | 2 (100) | 0 | 0 | 42.5 (25–59) | 50.5 (43–58) |
| PRES | 1 (0.9) | 1 (0.9) | 1 (100) | 0 | 0 | 25 (25–25) | 3 (3–3) |
| Pancreatitis | 1 (0.9) | 0 | 0 | 1 (100) | 0 | 70 (70–70) | NA |
| Gastrointestinal perforation | 0 | 0 | 0 | 0 | 0 | NA | NA |
Values are reported as n (%) unless otherwise noted.
aPercentage was calculated using a denominator of 107.
b If the same ADR was observed multiple times after gilteritinib administration in the same patient, the earliest occurring ADR is reported here.
cThe outcome was unknown for one patient who experienced myelosuppression.
ADR, adverse drug reaction; NA, not applicable; PRES, posterior reversible encephalopathy syndrome.
Supplementary Table S1 shows a complete list of each safety specification classified by the Medical Dictionary for Regulatory Activities (MedDRA) preferred term and the time-to-onset and time to resolution of ADRs and action taken with respect to gilteritinib therapy. Specific ADRs leading to dose reductions in more than one patient were decreased platelet count (n = 3), bone marrow failure (n = 3), prolonged QT interval (n = 2) and abnormal liver function (n = 2) (Supplementary Table S1). Specific ADRs leading to drug withdrawal in more than one patient were decreased platelet count (n = 6), prolonged QT interval (n = 6), abnormal liver function (n = 4), decreased white blood cell count (n = 4), pneumonia (n = 3), sepsis (n = 2), hemorrhage (n = 2), ILD (n = 2) and renal disorder (n = 2); ADRs leading to discontinuation of gilteritinib in more than one patient were febrile neutropenia (n = 2), neutropenia (n = 2), cellulitis (n = 2) and sepsis (n = 2) (Supplementary Table S1).
Among the 19 patients with AML secondary to MDS, 10 experienced grade ≥ 3 myelosuppression; two cases of myelosuppression were suspected to be related to gilteritinib. In four of the 10 patients, gilteritinib dose was either reduced or treatment was discontinued.
Grade 3 ILD was observed in two patients. One patient was a 35-year-old man who had relapsed after transplantation with chronic graft-versus-host disease (GVHD). Twenty days after initiation of 120-mg gilteritinib, the patient developed Grade 3 ILD. Gilteritinib was withdrawn and the patient was treated with a steroid pulse and recovered over a 10-day period. The patient subsequently underwent a second transplantation. The cause of ILD was thought to be related to both gilteritinib and GVHD. The other patient who developed Grade 3 ILD was a 71-year-old man who had a history of lung cancer and mantle cell lymphoma. He developed Grade 3 ILD 58 days from initiation of 120-mg gilteritinib. Although remaining on gilteritinib therapy, the patient received prednisone and recovered over a 13-day period. The cause of ILD in this patient was thought to be related to gilteritinib and vancomycin.
A 79-year-old male patient with refractory AML secondary to MDS, who had a history of gastric cancer and prostate cancer, developed Grade 2 pancreatitis during treatment with gilteritinib. The patient had initiated gilteritinib therapy at a dose of 120 mg and the dose was subsequently increased to 200 mg due to AML progression. Seventy days after initiation of gilteritinib treatment, the patient developed Grade 2 pancreatitis that was not considered to be serious. The patient continued gilteritinib therapy. A 76-year-old female patient developed a rash that was diagnosed as Grade 3 Sweet syndrome 41 days after initiation of 120-mg gilteritinib. Treatment with gilteritinib was stopped and the patient was treated with prednisone. The patient resumed gilteritinib at an initial dose of 80 mg that was subsequently increased to 120 mg. The rash resolved and did not recur after resumption of gilteritinib.
The incidences of all ADRs as secondary endpoints are shown in Supplementary Table S2. There was no marked difference in the incidence of ADRs between patients aged <65 years and those aged ≥65 years (Fig. 2). Serious AEs occurred in 55.1% of patients (n = 59/107); the most frequent were bone marrow failure (12.1%), febrile neutropenia (10.3%), decreased platelet count (7.5%) and pneumonia (5.6%) (Supplementary Table S3).
Figure 2.

Adverse drug reactions occurring in ≥2% of patients <65 years and ≥65 years.
Response outcomes
Among patients who were assessed for treatment response (n = 67), 34.3% achieved CR and 62.7% achieved CRc (Table 4); the median time to reach CR or CRc was similar (1.8 and 1.6 months, respectively). Four of the six pediatric patients responded to treatment with gilteritinib, with three achieving CR and one achieving CRp. A total of 17 adult patients received starting gilteritinib doses of 40 mg (n = 9) or 80 mg (n = 8); the dose was subsequently increased to 120 mg in four of these patients. Response outcomes were reported for seven of the 17 patients and included CR (n = 1), CRi (n = 3), partial remission (PR; n = 2) and no response (NR; n = 1).
Table 4.
Response outcomes in patients receiving gilteritinib during the surveillance period
| Response Parameter | N = 67a |
|---|---|
| Best response, n (%) CR |
23 (34.3) |
| CRi | 16 (23.9) |
| CRp | 3 (4.5) |
| PR | 14 (20.9) |
| NR | 8 (11.9) |
| NA | 3 (4.5) |
| CRc b | 42 (62.7) |
| ORR c | 56 (83.6) |
| Median time to CR, months (IQR) | 1.8 (0.9–3.6) |
| Median time to CRc, months (IQR) | 1.6 (0.9–3.0) |
| Median time to response, months (IQR) | 1.3 (0.9–2.4) |
Bold italicized font indicates aggregate response rates.
aPatients in the efficacy analysis set who had an assessment of treatment response (CR, CRi, CRp, PR, NR or NA).
bDefined as the sum of patients who achieved CR, CRi or CRp.
cDefined as the sum of patients who achieved CRc and PR.
CR, complete remission; CRc, composite complete remission; CRi, complete remission with incomplete hematologic recovery; CRp, complete remission with incomplete platelet recovery; IQR, interquartile range; NA, not assessable; NR, no response; ORR, overall response rate; PR, partial remission.
Response outcomes were assessed in seven of the 12 patients who received both gilteritinib and chemotherapy during the gilteritinib treatment period. Three of these seven patients had received chemotherapy (azacitidine, cytarabine or hydroxycarbamide) concomitantly with gilteritinib, with responses of PR (n = 1) or NR (n = 2). In the remaining four patients, chemotherapy [daunorubicin (n = 1), aclarubicin plus cytarabine once and an A-triple V (cytarabine, etoposide, vincristine and vindesine) regimen twice (n = 1), cytarabine plus idarubicin and mitoxantrone (n = 1) or cytarabine (n = 1)] was initiated after treatment with gilteritinib was temporarily stopped; gilteritinib was restarted after chemotherapy was stopped. Responses in these four patients were CR (n = 2), PR (n = 1) and NR (n = 1).
After an initial decline during the first 3 months of surveillance, OS rates remained relatively stable until the end of the surveillance period (Fig. 3).
Figure 3.

Overall survival in patients receiving gilteritinib during the surveillance period. Abbreviation: CI, confidence interval.
Patients who underwent transplantation
A total of 14 patients underwent HSCT during the surveillance period (Supplementary Table S4). Six of the 14 patients who underwent HSCT had received prior HSCT and two had GVHD at the start of gilteritinib therapy. Sources of hematopoietic stem cells were peripheral blood (42.9%), umbilical cord blood (28.6%) and bone marrow (28.6%). Engraftment was observed in all but one patient (Supplementary Table S4). Of the 11 patients who had been assessed for response before HSCT, five achieved CR, five achieved CRi and one achieved CRp.
Six patients resumed gilteritinib after HSCT (Table 5); the median time from HSCT to the restart of gilteritinib therapy was 37 days (range, 24–102 days). Three of the six patients restarted gilteritinib at the 40-mg dose, one resumed gilteritinib at the 80-mg dose and the remaining two patients resumed gilteritinib at the 120-mg dose (Table 5). In two patients who each resumed gilteritinib at 40 and 80 mg, the dose was increased to 120 mg over a 2-week period. AEs occurred in three of the six patients who resumed gilteritinib after HSCT. Two of these patients had restarted gilteritinib at doses of 40 mg and 120 mg, respectively, and both patients developed liver dysfunction that was suspected to be related to gilteritinib (low possibility of causal relationship; n = 1) or liver GVHD (relationship to gilteritinib was not evaluable; n = 1). The third patient developed sepsis (unrelated to gilteritinib) before restarting gilteritinib therapy.
Table 5.
Gilteritinib dosing parameters before and after transplantation
| Dosing Before HSCT | N = 14 |
|---|---|
| Median duration of dosing before HSCT, days (range) | 93 (24–176) |
| Dosing After HSCT | N = 6 |
| Daily dose immediately before HSCT, n (%) 40 mg 80 mg 120 mg |
0 0 6 (100) |
| Daily dose immediately after HSCT, n (%) 40 mg 80 mg 120 mg |
3 (50) 1 (16.7) 2 (33.3) |
| Median time to restart of gilteritinib after HSCT, days (range) | 37 (24–102) |
HSCT, hematopoietic stem cell transplantation.
Discussion
A limited number of patients with R/R FLT3mut+ AML participated in gilteritinib clinical trials in Japan. Thus, the regulatory authority required a postmarketing use-results surveillance covering all patients treated with gilteritinib to obtain patient characteristics, gilteritinib safety and efficacy and measures taken to ensure proper use of gilteritinib. The results of this interim analysis show that real-world use of gilteritinib in Japan was not associated with any new safety concerns and was effective in a variety of patients with R/R FLT3mut+ AML. At enrollment, >50% of patients had refractory AML and >75% had comorbidities, indicating that this study included patients with multiple comorbidities and those who were ineligible for participation in a clinical trial. The 120-mg/day dose, which was identified in dose-escalation studies as the preferred starting dose (18,19), was used by the majority of patients in this study as the initial dose (80.4%; 86/107) and the maximum dose (74.8%; 80/107). Although gilteritinib is not indicated for pediatric patients with R/R FLT3mut+ AML, it was administered off-label in six pediatric patients at ages ranging from 11 to 14 years.
Overall, the safety profile of gilteritinib observed during the surveillance period did not show any new clinically significant safety signals. Due to the impact of persistent disease, cytopenias and myelosuppression are commonly observed in patients with AML. In general, most ADRs related to myelosuppression, liver function disorder, infections and prolonged QT interval were manageable and had either resolved or were in the process of resolving in a majority of patients at the time of this interim analysis. The proportion of patients who discontinued gilteritinib due to an AE was low (15.0%). Overall, common ADRs were reported less frequently in this survey than in the gilteritinib clinical trials (10,18,19). It should be noted that mild or moderate liver dysfunction does not have a significant effect on the pharmacokinetic parameters of gilteritinib (16). In pharmacokinetic analyses, the ratio of the maximum plasma concentration (Cmax) of unbound gilteritinib in patients with moderate hepatic impairment (Child-Pugh B) to the Cmax of unbound gilteritinib in healthy volunteers was 117.72% [90% confidence interval (CI): 89.90, 154.15]; the corresponding ratio for area under the plasma concentration–time curve from time 0 to infinity was 88.48% (90% CI: 65.97, 118.69) (20). Elevated AST or ALT was found to be mildly correlated with gilteritinib plasma concentration (21).
The rate of CRc in this surveillance (62.7%) was slightly higher than that observed in the Phase 3 clinical trial of gilteritinib (54.3%) (10). The change in OS rate over the 6-month surveillance period was 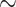 20%, with
20%, with 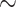 78% of patients remaining alive at the end of surveillance. Of the 14 patients who underwent HSCT, all but one had engraftment, and six resumed gilteritinib treatment posttransplantation. All patients who were assessed for response before undergoing HSCT (n = 11) had achieved CRc before transplantation.
78% of patients remaining alive at the end of surveillance. Of the 14 patients who underwent HSCT, all but one had engraftment, and six resumed gilteritinib treatment posttransplantation. All patients who were assessed for response before undergoing HSCT (n = 11) had achieved CRc before transplantation.
These findings are limited in that they are based on the interim analysis prespecified in the study protocol. Thus, the results may change with the collection of additional data. In addition, only data from the 107 patients who provided informed consent were tabulated and analyzed for this report; thus, they may not be representative of the entire population surveyed (N = 328) or of the 204 patients with CRFs. Because only 67 of the 107 patients in this analysis had an assessment of treatment response, a selection bias toward patients with a documented response cannot be ruled out. Furthermore, the reported efficacy data included the effectiveness of gilteritinib in patients who received low, off-label doses and the effectiveness of other drugs in addition to gilteritinib alone. With regard to remission rate, the period from initiation of treatment to the time of response assessment varied among patients. Finally, survival was calculated with the date of discontinuation of gilteritinib as the end date, even in surviving patients.
In conclusion, interim results from this postmarketing surveillance study of gilteritinib suggest that gilteritinib has acceptable safety and efficacy in patients with R/R FLT3mut+ AML in actual use settings in Japan. These findings confirm the clinical benefit provided by gilteritinib in a real-world R/R FLT3mut+ AML patient population.
Supplementary Material
Acknowledgments
We acknowledge physicians across 362 medical institutions in Japan who collected the data for this surveillance. Medical writing/editorial support was provided by Kalpana Vijayan, PhD, Stephanie Leinbach, PhD, Cheryl Casterline, MA and Elizabeth Hermans, PhD, from Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and funded by the study sponsor.
Author Disclosures
Haruko Sugamori, Takumi Lee, Takeshi Mitomi and Chika Yamagishi are employees of Astellas Pharma, Inc.
Funding
This work was supported by Astellas Pharma, Inc.
Data Sharing Statement
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at www.clinicalstudydatarequest.com.
For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
References
- 1. Stanchina M, Soong D, Zheng-Lin B, Watts JM, Taylor J. Advances in acute myeloid leukemia: recently approved therapies and drugs in development. Cancers (Basel) 2020;12:3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cancer Genome Atlas Research Network, Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med 2016;374:2209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Röllig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol 2015;16:1691–9. [DOI] [PubMed] [Google Scholar]
- 6. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med 2017;377:454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Man CH, Fung TK, Ho C, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood 2012;119:5133–43. [DOI] [PubMed] [Google Scholar]
- 8. Fischer T, Stone RM, Deangelo DJ, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol 2010;28:4339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mosquera Orgueira A, Bao Perez L, Mosquera Torre A, et al. FLT3 inhibitors in the treatment of acute myeloid leukemia: current status and future perspectives. Minerva Med 2020;111:427–42. [DOI] [PubMed] [Google Scholar]
- 10. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med 2019;381:1728–40. [DOI] [PubMed] [Google Scholar]
- 11. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3-ITD acute myeloid leukaemia (QuANTUM-R): a multicentre, randomised, controlled, open-label, phase 3 trial. Lancet Oncol 2019;20:984–97. [DOI] [PubMed] [Google Scholar]
- 12. Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015;29:2390–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dhillon S. Gilteritinib: first global approval. Drugs 2019;79:331–9. [DOI] [PubMed] [Google Scholar]
- 14. Quizartinib . Japan PMDA Approval. https://www.pmda.go.jp/files/000240288.pdf. 20 May 2021, date last accessed.
- 15. Tzogani K, Roshol H, Olsen HH, et al. The European Medicines Agency review of gilteritinib (Xospata) for the treatment of adult patients with relapsed or refractory acute myeloid leukemia with an FLT3 mutation. Oncologist 2020;25:e1070–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. XOSPATA [Prescribing Information]. Astellas Pharma US Inc. Northbrook, IL, United States. https://astellas.us/docs/xospata.pdf. 20 May 2021, date last accessed. [Google Scholar]
- 17. Cheson BD, Bennett JM, Kopecky KJ, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003;21:4642–9. [DOI] [PubMed] [Google Scholar]
- 18. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol 2017;18:1061–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Usuki K, Sakura T, Kobayashi Y, et al. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory acute myeloid leukemia: an open-label phase 1 study. Cancer Sci 2018;109:3235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. XOSPATA . [Prescribing Information]. Astellas Pharma Inc. Tokyo, Japan. [Google Scholar]
- 21. Xospata New Drug Application FDA Multisciplinary Review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/211349Orig1s000MultidisciplineR.pdf. 10 June 2021, date last accessed.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.