

Abstract
A major advance in the study of Huntington’s disease (HD) has been the development of human disease models employing induced pluripotent stem cells (iPSCs) derived from patients with HD. Because iPSCs provide an unlimited source of cells and can be obtained from large numbers of HD patients, they are a uniquely valuable tool for investigating disease mechanisms and for discovering potential disease-modifying therapeutics. Here, we summarize some of the important findings in HD pathophysiology that have emerged from studies of patient-derived iPSC lines. Because they retain the genome and actual disease mutations of the patient, they provide a cell source to investigate genetic contributions to the disease. iPSCs provide advantages over other disease models. While iPSC-based technology erases some epigenetic marks, newly developed transdifferentiation methods now let us investigate epigenetic factors that control expression of mutant huntingtin (mHTT). Human HD iPSC lines allow us to investigate how endogenous levels of mHTT affect cell health, in contrast to other models that often rely on overexpressing the protein. iPSCs can be differentiated into neurons and other disease-related cells such as astrocytes from different brain regions to study brain regional differences in the disease process, as well as the cell-cell dependencies involved in HD-associated neurodegeneration. They also serve as a tissue source to investigate factors that impact CAG repeat instability, which is involved in regional differences in neurodegeneration in the HD brain. Human iPSC models can serve as a powerful model system to identify genetic modifiers that may impact disease onset, progression, and symptomatology, providing novel molecular targets for drug discovery.
Keywords: induced pluripotent stem cells (iPSCs), Huntington’s disease, neurodegeneration, neurodegenerative disease
Introduction
Huntington’s disease (HD) is caused by the expansion of a CAG repeat in the Huntingtin gene (HTT), leading to the expression of a mutant protein (mHTT) with an expanded polyglutamine domain near its N-terminus. Progress towards a cure has been slow, in part because the function of the normal HTT protein is unclear and because the expanded gene and protein appear to impinge on multiple biological processes. The disease manifests as progressive degeneration of striatal neurons and has been modelled in a variety of systems, including animal models and animal or human cell in vitro systems.
A major advance for in vitro cell modeling of HD was the development of induced pluripotent stem cell (iPSC) technology by groups led by Yamanaka1 and Thomson2. This technology allows somatic cells to be reprogrammed into a pluripotent state and subsequently differentiated into cell types of interest1,2. Unlike other cell models to study HD, human iPSCs retain the genome of their patient source, making them a valuable tool to investigate the genetic contributions to disease. Because iPSCs can be propagated indefinitely, they provide a nearly inexhaustible source of patient-derived material to study disease mechanisms and for use in drug discovery.
The methods to efficiently generate iPSCs have evolved over time and have been extensively reviewed3,4. The initial methods used viruses expressing reprogramming factors1,2,5. This approach has drawbacks in that the virus would randomly integrate across the genome, thereby causing genomic insults to the DNA that could alter gene expression in unwanted and unknown ways. More advanced methods have been developed that reprogram cells without leaving a genomic fingerprint4,6. These include using non-integrating episomes7, mRNA8, or small molecules9,10. Most of the iPSC models with expanded CAG repeats reviewed here were made with non-integrating episomal vectors (Table 1).
Table 1. Induced pluripotent stem cell lines to study Huntington’s disease pathogenesis.
| iPSC line* | CAG repeat size** |
Summary of cellular phenotypes in HD i-tissues compared with controls*** |
|---|---|---|
| CS97iHD-180nX (GM09197)18 |
180 | • Gene expression changes: ↑&↓ in ECM signaling, synapse assembly, axon guidance, neurotrophic signaling, and the BDNF pathway in cortical i-neurons31, ↑ in TGF-β pathway and other developmental pathways in i-NSCs32, ↑&↓ in neuronal development pathways, cell cycle, proliferation, axon guidance, and nervous system function pathways in i-NSCs18 Functional phenotypes: • ↑&↓ in fatty acid metabolism and PPAR signaling in i-NSCs32 • ↑ H3K9 methylation in iPSCs33 • ↑ ATM and P53 protein levels in iPSCs34 • ↑ P53 phosphorylation in iPSCs34 • ↑ γH2AX phosphorylation in iPSCs34 • ↑ cell death18 and ↑ susceptibility to cell death after growth factor withdrawal32,35 as well as ↑ susceptibility to cell death after exposure to stressors such as glutamate, 3-MA, H2O2 in i-neurons18 • Proteasome inhibition by MG132 or autophagy inhibition by bafilomycin induces mHTT aggregates in iPSCs14,36 • Neuronal patterning and rosette formation32 is delayed • OCT4 remains high after neuronal induction in i-NPCs37 • ↓ intracellular ATP levels in i-NSCs18,19 • ↑ levels of caspase-3/7 activity in i-NSCs18 • ↑ number of TUNEL-positive staining iPSCs18 |
| CS109iHD-109nX /HD109i.1(ND39258)18,38 | 109 | • Gene expression changes: ↓ P53 expression in i-NSCs39,40, ↑&↓ in neurodevelopmental pathways in i-neurons17, ↑ in TGF-β pathway i-neurons17,38,41, ↑&↓ in ECM signaling in i-neurons17 and i-NPCs18, ↑&↓ in synapse assembly, axon guidance, neurotrophic signaling, and BDNF pathway in i-neurons17, ↑&↓ in ECM organization, synapse assembly, axon guidance, neurotrophic signaling, and the BDNF pathway in cortical i-neurons31, ↑&↓ in neurodevelopmental pathways, DNA damage, and apoptosis in iPSCs38 and others. Functional phenotypes: • ↑ nuclear envelope abnormalities in i-NPCs42 • Mis-localization of nuclear pore proteins and alterations of nucleocytoplasmic transport in i-neurons43 • ↑ susceptibility to cell death after growth factor withdrawal in i-neurons35 • ↑&↓ epigenetic alterations in i-neurons17 • Alterations in neuronal patterning and rosette formation is delayed and OCT4 remains high after neuronal induction in i-NPCs32,37 • Delay of mature electrophysiological currents in i-neurons31 • ↑&↓ neurite length in i-neurons17,31 • ↓ intracellular ATP levels iPSCs and i-neurons18,19,44 • ↓ bioenergetics and ↓ glycolytic capacity in i-neurons and i-NSCs18,19,44 • CAG repeat instability in iPSCs and i-NPCs18 over passage as well as over time during differentiation45. |
| CS77iHD-77nX31 | 77 | • Gene expression changes: ↑&↓ in ECM organization, synapse assembly, axon guidance, neurotrophic signaling, and the BDNF pathway in cortical i-neurons31. Functional phenotypes: • Delay of mature electrophysiological currents in i-neurons31 • ↓ neurite length in cortical i-neurons31 |
| HD7646 | • ↑ SOCE calcium currents i-neurons46 • ↑ mHTT protein expression in i-neurons46 • ↑ VGCC currents in i-neurons46 |
|
| HD-iPS412, HD and HD215 HD-iPSC(Q71)12 |
72 (Some studies33 report this line as having 71 CAG repeats) |
• Gene expression changes: ↑ in DNA damage pathways15, ↑ in oxidative stress15, ↓ in cytoskeletal genes in i-neurons15, ↑ ATM gene expression15, ↑ in TGF-β pathway in i-NSCs16,47, ↓ in cadherin pathway in i-NSCs16, ↑&↓ in ECM functions, cell adhesion and cell surface signaling, transmembrane support, and axon guidance in i-NSCs16, ↑&↓ in ECM organization, synapse assembly, axon guidance, neurotrophic signaling, and the BDNF pathway48, ↑&↓ in developmental pathways and cell adhesion in NPCs33 and ↓ in ECM organization, development, and differentiation and neurodevelopmental pathways in NPCs33 and others Functional phenotypes: • ↑ H3K9 methylation in iPCS33 • Proteasome inhibition by MG132 or autophagy inhibition by bafilomycin induces mHTT aggregates i-tissues14,36 • Neural rosette formation and neural patterning is delayed14 • ↓ neurite length in cortical i-neurons31 • ↑ levels of caspase-3/7 activity in i-NPCs13 • ↑ number of TUNEL-positive staining iPSCs15 i-NSCs16 and in differentiated i-neurons32 • ↑ P53 and ATM expression and phosphorylation in iPSCs16 • ↓ mitochondrial respiration and ATP levels as well as other differences in mitochondrial dynamics i-NSCs49 |
| CS77iHD-71nX38 | 71 | • Gene expression changes: ↑&↓ in neurodevelopmental pathways, TGF-β pathway, Wnt signaling, DNA damage and apoptosis in iPSCs38 |
| HD70 (GM21756)34 | 70 | • ↑ P53 phosphorylation i-tissues?15,16,34 • ↑ ATM and P53 protein levels iPSCs34 • ↑ γH2AX phosphorylation34 in iPSCs. |
| CS21iHD-60nX18 | 60 | • Gene expression changes: ↑ in TGF-β pathway in i-neurons17, ↑&↓ in ECM functions in i-neurons17 and i-NPCs17,18, ↑&↓ in synapse assembly, axon guidance, neurotrophic signaling, and BDNF pathway, and key developmental pathways in i-neurons17 and cortical i-neurons31 Functional phenotypes: • ↑&↓ epigenetic alterations in i-neruons17 • ↑ levels of TUNEL-positive staining in iPSCs18 • ↑ cell death in i-neruons18 • Alterations in neuronal patterning and OCT4 remains high after neuronal induction in i-NPCs37 • ↓ intracellular ATP levels in i-NSCs17, iPSCs11 and i-neurons18,19,44 • ↓ bioenergetics and ↓ glycolytic capacity in i-tissues18,19,44 • ↑ glutamate sensitivity in i-neurons18 • ↑ caspase-3/7 activity in i-neurons18 • ↑ P53 phosphorylation in i-tissues15,16,34 |
| CS03iHD-53nX17,43 | 53 |
Gene expression changes: ↑ in cell cycle and Wnt/β-Catenin signaling in MSN i-neurons50 Functional phenotypes: • Mis-localization of nuclear pore proteins and alterations of the nucleocytoplasmic transport in i-neurons43 • ↑ cell death in MSN i-neurons17 • ↑ neurite length in MSN i-neurons17 • ↑ neurite length in cortical i-neurons51 • ↓ population of enduring mitotically active and resistant-to-differentiation subset of proliferating cells amongst MSN i-tissues50 |
| iPSHD11, iPSHD22, and iPSHD3452 |
40, 47, and 42, respectively |
• ↑ nuclear envelope abnormalities in i-neurons52 • ↑ lysosomes and autophagosomes in i-neurons52 • ↑ susceptibility to cell death by proteasome inhibition in i-neurons52 • ↓ mitochondrial density (only in iPSHD11 iPSHD22)53 • ↑ SOCE calcium currents in HD i-neurons52,54 |
| CS04iHD-46nX17 | 46 | • Gene expression changes: ↑ in cell cycle and Wnt/β-Catenin signaling in MSN i-neurons50 Functional phenotypes: • ↑ cell death in i-neurons17 • ↑ neurite length in MSN i-neurons17 • ↑ population of enduring mitotically active and resistant-to-differentiation subset of proliferating cells amongst MSN i-tissues50 |
| HD1 (GM04022) and HD2 (GM02191)55 |
44 and 42 repeats |
• ↑ 5-hydroxymethylation in i-NSCs • ↑ DNMT family gene expression in i-NPCs55 • ↑ DNA repair gene expression in NSCs and iPSCs55 |
| ChiPS31-HD-hiPS56 | 42/44 homozygous |
• DNA hypermethylation in iPSCs57 • ↑ lysosomes and LC3 BII expression in iPSCs and i-NSCs • ↑ lysosomes and LC3 BII expression56 |
Summary of phenotypes associated with HD i-tissues. ↑ = increased compared with controls, ↓ = decreased compared with controls, ↑&↓ = components or steps of this pathway may be either increased or decreased relative to controls. Gene expression changes are italicized. Underlined font indicates phenotypes found across at least two lines with different CAG repeat lengths.
* Line may have a slightly different name depending on the study or publication. ** The CAG repeat size listed here is an estimate of the number of contiguous CAG triplets and does not include CAA interruptions, which are likely to occur and have been identified in human58,59 and mouse models60. *** To generate this list, we focused on the most prominent phenotypes reported between 2012 and 2022. We acknowledge that this list may not be exhaustive, as we may have missed some reports or new phenotypes may have been described since. Note: Not all HD lines that have been generated and studied are listed here. Please refer to the Coriell database (https://www.coriell.org/) for the complete list from the HD iPSC Consortium and for other lists that are publicly available. BDNF, brain-derived neurotrophic factor; ECM, extracellular matrix; HD, Huntington’s disease; i-NPC, neural precursor cell developed from induced pluripotent stem cells; i-NSC, neural stem cell developed from induced pluripotent stem cells; i-neuron, neuron developed from induced pluripotent stem cells; iPSC, induced pluripotent stem cell; i-tissue, tissue developed from induced pluripotent stem cells; mHTT, mutant huntingtin; PPAR, peroxisome proliferator-activated receptor; SOCE, store-operated calcium entry; TGF-β, transforming growth factor beta; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling; VGCC, voltage-gated Ca2+ channel.
In 2008, the Daley lab was the first to develop human iPSC-derived HD models and to demonstrate that the fibroblasts of patients with HD could be reprogrammed into pluripotent stem cells11. The first HD patient iPSC line harbored 72 CAG repeats12 and was subsequently differentiated into GABAergic, DARPP32-positive neurons13, demonstrating that iPSCs could be patterned towards striatal neurons, a cell type that is vulnerable to degeneration in HD. Others have also generated iPSCs from patients with HD and differentiated them into neurons that can be patterned into striatal cultures or neural progenitor cells to interrogate HD-related phenotypes14–19. The use of HD iPSC models has made it possible to study the links between mHTT, the activity of multiple biological pathways, and pathogenesis20. Work in other HD models has found that mHTT disrupts mitochondrial function21–23. In addition, neurons depend on proteasome activity24–26 and autophagy flux27 to remove misfolded proteins like mHTT. Cells that express mHTT show impairment of these clearance pathways leading to mHTT buildup, which accelerates neurodegeneration28–30. In the next section, we discuss how the use of human HD iPSC models has aided in our understanding of disease mechanisms, thus providing insights into the development of effective therapies for HD.
For simplicity, we refer to neurons, neural stem cells, neural progenitor cells, and tissues developed from iPSCs as i-neurons, i-NSCs, i-NPCs, and i-tissues, respectively. (See Table 1 and Figure 1 for a summary of data on HD iPSC lines.)
Figure 1. Cartoon summarizing the most prominent phenotypes identified in Huntington’s disease tissues developed from induced pluripotent stem cells.
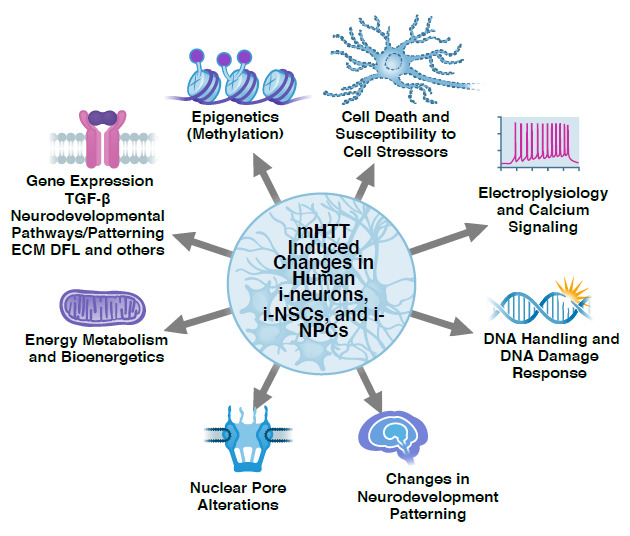
DFL, dermal fibroblast; ECM, extracellular matrix; i-NPC, neural precursor cell developed from induced pluripotent stem cells; i-NSC, neural stem cell developed from induced pluripotent stem cells; i-neuron, neuron developed from induced pluripotent stem cells; TGF-β, transforming growth factor beta.
Somatic instability and DNA handling in HD
It is generally believed that symptomatic HD is found in patients with CAG repeat lengths greater than 40. The number of CAG repeats in HTT is not permanently fixed and instead undergoes somatic expansion in some human tissues, particularly in the striatum and cortex61–64. The cerebellar cortex appears to display the lowest degree of CAG repeat instability61, while other brain tissues display a high degree of CAG repeat mosaicism61,62,65. Somatic instability in the cortex in HD brains is a predictor of age of onset63 and increases with age66. Recently, it has been observed that individuals who lack a CAA interruption in the CAG repeat have a significantly earlier age of onset in HD as well as increased somatic instability58,59,60,67. Somatic instability is often linked to an altered DNA damage response. Indeed, genome-wide association studies (GWAS) have identified numerous variants significantly associated with DNA damage control pathways that modify HD age of onset, including FAN1, LIG1, MLH1, MSH3, PMS1, and PMS268–73. Moreover, reducing expression of a subset of these genes in mice caused changes in somatic instability across different tissues, including iPSCs71,72. Together, these data suggest that the DNA repair machinery is tightly connected with the somatic instability of the CAG repeats in HTT and may play a major role in HD onset and progression.
The number of CAG repeats is generally stable during reprogramming of patient fibroblasts into iPSCs, and, as a general rule, neither iPSC passaging, when some cells in a culture are used to generate a new cell population, nor differentiation into brain cells seems to alter the CAG repeat length significantly5,18,56. There are exceptions such as iPSCs from patients with very long CAG repeats (e.g., the HD109 line), where additional expansion of the CAG repeat length can occur with passaging for both iPSCs45 and i-NPCs18 as well as during differentiation to medium size spiny neurons (MSNs)45.
Interestingly, in lines with large CAG expansions such as the HD109 and HD72 lines, knocking down one of the GWAS modifiers of DNA damage control, FAN1, exacerbated CAG expansions45,72, suggesting that FAN1 is needed to limit somatic expansion. In fact, perturbations in DNA handling genes and the DNA damage response occur in HD74,75. For example, p53 signaling is increased in HD models35,76, and HD iPSCs exhibit elevated p53 levels and phosphorylation15,34, as well as ATM expression15 and phosphorylation15,34 and γH2AX phosphorylation15,34. All of these proteins are widely used as markers of DNA damage15,34,77,78. Surprisingly, p53 expression was found to be decreased in iPSCs and i-NSCs with very long CAG expansions from patients with juvenile-onset HD39,40. The reasons for this perplexing finding are currently unclear. In studies employing a number of HD i-neurons with a range of CAG repeat lengths of 66, 71, and 109, Morozko et al. (2021)79 reported that the SUMO E3 ligase PIAS1 is involved in DNA damage control and that its reduction in HD iPSC neurons normalized HD transcriptional dysregulation associated with DNA damage repair. This served to increase genomic integrity in HD iPSC-derived neurons, supporting a role for SUMOylation in DNA damage repair in HD. Nevertheless, many genes involved in the DNA damage response pathway were found to be altered in HD i-neurons, iPSCs, and i-NSCs compared with controls15,34,38,55, further confirming the importance of the DNA damage pathway in the pathology of HD neurons.
Nuclear pore alterations in HD
The nuclear pore serves as a master regulator for nuclear import and export and thereby controls numerous cellular processes involved in gene transcription and translation80. Nuclear pore dynamics may be impaired in neurons vulnerable to HD since the RanGTP gradient, a critical regulator of nuclear pore dynamics, is compromised in HD, and numerous nuclear pore proteins such as NUP62 and RanGAP1 are mislocalized in HD i-neurons compared with controls43. Altered nuclear pore functions may be responsible for aberrations of nuclear morphology in HD i-neurons52, and alterations in nuclear envelope morphology have been shown in human HD brain tissue as well as i-NSCs42. The shape and size of the nucleus are also altered in HD i-neurons53, although how this affects nuclear dynamics or function is not understood. As mHTT alters chromatin and gene expression, it is perhaps not surprising that gatekeepers of transport in and out of the nucleus are also disrupted in HD.
mHTT-induced gene expression changes
Shortly after iPSC technology was introduced1,2, the effort to create patient-derived lines was of great interest to the HD field. The National Institutes of Health formed a consortium of HD researchers, called the “HD iPSC Consortium”, to investigate both the transcriptomic changes and functional and structural degeneration associated with HD. This team reported gene expression differences between i-NSCs derived from controls and individuals with juvenile- and adult-onset HD, harboring 180 and 60 CAG repeats, respectively. Their microarray studies identified differential expression of genes in numerous pathways involved in development, cell cycle, proliferation, and nervous system function, and they found a subset of gene expression changes that were unique to the juvenile onset cells(CAG180 line)18. Later, the HD iPSC Consortium performed RNA sequencing (RNA-seq) analysis on a subset of the same lines (HD i-neurons harboring 60 and 109 CAG expansions with multiple clones of each) and again found a dysregulation of key developmental pathways and master regulators of neurogenesis17. More than half of the genes altered were involved in nervous system function, cellular and tissue development, and axon guidance17.
One of the mechanisms by which mHTT is suspected to cause neurodegeneration is by altering gene transcription. This is supported by the findings of the Ellerby group16, who used i-NSCs generated from HD iPSCs that harbored 72 CAG repeats and compared their gene expression profiles with gene-corrected control lines11 by using microarray analysis. They found that the transforming growth factor beta (TGF-β) signaling pathway was significantly upregulated as a consequence of CAG repeat expansion16. The TGF-β signaling pathway supports numerous cellular activities involved in development and growth, including proliferation, differentiation, patterning81, apoptosis, and tumor suppression82. This finding has been corroborated: dysregulation of the TGF-β pathway has been observed in i-NSCs harboring 72 CAG repeats16,47,48, 180 CAG repeats32, and 60 and 109 CAG repeats17. In addition, SMAD transcription factors, the downstream effectors of TGF-β signaling, have been implicated as central players in the regulation of HTT itself41.
Another major cellular pathway dysregulated in HD i-tissues is the extracellular matrix (ECM). The ECM provides structural support to cells and modulates the signaling events and force generation required for cell communication, migration, polarity, and movement83,84. Genes related to ECM functions such as cell adhesion and cell surface signaling, transmembrane support, and axon guidance are altered in HD i-NSCs and i-neurons16–18. In particular, cadherins, calcium-dependent molecules that regulate cell adhesion and connection85, were among the most prominent downregulated genes in HD i-NSCs with 72 CAG repeats16.
RNA-seq of i-neurons derived from this same HD line48 and its gene-corrected counterpart as well as from other HD lines harboring 60 and 109 CAG repeats17 revealed more complex gene dysregulation in numerous pathways, such as synapse assembly, axon guidance, neurotrophic signaling, and the brain-derived neurotrophic factor (BDNF) pathway17,48. RNA-seq on cortical-patterned i-neurons harboring 77 and 109 CAG repeats compared with controls showed similar patterns of transcriptional dysregulation in nervous system development, ECM, and axon guidance molecules31. The changes in synaptic assembly and axon guidance may underlie the synapse degeneration and dysfunction of neuronal circuitry, while loss of supportive neurotropic signaling may contribute to neuronal death over time.
Many of the genes downregulated in HD i-neurons are transcriptional regulators related to neuronal development. This includes REST, which represses genes involved in neuronal fate in non-neuronal cells86 and prevents neuronal differentiation in stem cells87, and NEUROD117, a key regulator of neuronal differentiation. Of interest, activation of NEUROD1 by either its overexpression or stimulation by a small molecule has been shown to ameliorate disease phenotypes in HD neurons. Other studies have reported similar gene expression changes in developmental pathways31,32,38. Together, these findings suggest that cellular activities involved in neuronal maturation are dysregulated in HD, which is consistent with other developmental phenotypes observed in HD i-tissues.
Epigenetic changes in HD
One of the mechanisms controlling gene expression involves epigenetic modulators, and epigenetic changes underlie many of the transcriptional alterations in HD88–90. Studies have shown altered epigenetics in HD iPSCs, including changes in histone methylation and acetylation. Transcriptional changes identified by RNA-seq were highly correlated with alterations in the H3K4me3 and H3K27ac epigenetic marks17. In i-NSCs derived from HD patients harboring 71 CAG repeats, H3K9 methylation was increased compared with controls, and this change was associated with altered expression of genes involved in proliferation, development, differentiation, ECM, and axon guidance33. Changes in histone H3K9 methylation have also been observed in postmortem HD brains91.
ATF7IP, which regulates H3K9 methylation, interacts with wild-type HTT, but this interaction is lost in iPSCs harboring 71 CAG repeats33. mHTT directly alters the deposition of H3K27me3 at active sites of mouse embryonic stem cells (ESCs) and neural precursor cells (NPCs), suggesting that mHTT may directly regulate histone methylation92. Sequence motif analysis revealed that the largest changes in H3K27ac marks occur near transcription factor binding sites at genes involved in neurodevelopmental pathways17.
Another study examining a rare homozygous CAG expanded line harboring 42/44 CAG repeats reported DNA hypermethylation in promoter regions, and the extent of hypermethylation increased as the cells matured and patterned57. Methylation was observed at the promoter region of WDR5, a chromatin remodeler that itself regulates methylation of genes involved in pluripotency57. Finally, there was a global increase of 5-hydroxymethylation in HD i-NSCs that harbor 43 and 44 CAG repeats compared with controls55. These data suggest that CAG expansion in HTT or the resulting glutamine expansion in mHTT (or both) may alter the fate of neuronal development by exerting effects at the epigenome level.
Inclusion bodies and proteostasis
An important use of HD i-neurons is that they allow for the study of the role of endogenous levels of mHTT on cell health rather than requiring overexpression of the protein, as in most other cell models of the disease. mHTT is misfolded and forms inclusion bodies (IBs) in cells of the HD brain. Pathological mHTT inclusions have long been associated with HD and are found in the brains of murine models of HD, as shown primarily by immunohistochemical staining using antibodies against mHTT, such as EM48 or MW893–97. Some investigators have reported that HD i-tissues in culture do not develop EM48- or MW8-positive aggregates or intranuclear inclusions14,18. However, HD i-NSCs grafted into murine brains at postnatal day 2 exhibited EM48-positive aggregates after about 33 weeks14, suggesting that aging and the brain microenvironment may be required for pathological mHTT IB formation. Nekrasov et al.52 also reported the presence of EM48-positive inclusions in 6-month-old i-neuron cultures. However, no quantification was performed, so the extent of aggregation in this context is unclear52.
The increased levels of mHTT, and its aggregation, are due in part to impairment of the proteostasis system30,98,99. Proteostasis entails the breakdown of misfolded proteins. The breakdown of misfolded proteins in cells normally occurs by two main pathways: the ubiquitin-proteasome system and autophagy. HD iPSCs and i-NSCs with moderate-length CAG repeat expansions (42–45 CAG repeats) display increased lysotracker staining, indicating altered lysosome activity compared with controls56. Another study employing cells with moderate-length CAG repeat expansions found increased lysosomes and autophagosomes in HD i-neurons as well as increased mitophagy52. Likewise, early-stage HD i-NSCs exhibit increased markers of autophagy compared with control i-NSCs56. These findings are consistent with observations in HD mouse models100,101 and suggest that proteostasis is activated in these pluripotent cells, possibly reflecting the cells’ attempt to clear toxic mHTT.
However, the consequences of this activation are not understood. Numerous studies report that mHTT causes significant dysfunction of the proteostasis system, leading to neurodegeneration98,102. Treatment with the autophagy inhibitor 3-MA or the proteasome inhibitor MG132 increases cell death in HD i-neurons18,52, confirming that HD i-tissues may be more sensitive to perturbations of protein homeostasis.
Indeed, treatment with the proteasome inhibitor MG132 or the autophagy inhibitor bafilomycin increases the accumulation of mHTT aggregates in HD iPSCs harboring 71 and 180 CAG repeats14,36. Similarly, knocking down one of the most highly expressed E3 ubiquitin ligases, UBR5, in iPSCs results in the generation of mHTT aggregates and increased levels of mHTT36. Oxidative stress induced by menadione also results in EM48-positive aggregates in HD i-neurons harboring 99 CAG repeats103. Furthermore, mHTT may stress proteasomal pathways, as it has been shown that HD i-NSCs harboring 46, 70, and 99 CAG repeats have an increase in ubiquitinated proteins and delayed degradation of proteasome substrates103.
To study the mechanisms by which mHTT may impact proteostasis, our group has used a longitudinal single-cell imaging technology, robotic microscopy (RM)104,105, that can observe i-neurons from HD patients over long periods of time. The high sensitivity of RM allows it to detect disease phenotypes in HD i-neurons without the need for cellular stressors. Using RM, we found that i-neurons from patients with HD harboring 46, 53, 60, or 180 CAGs displayed signs of degeneration such as the retraction of neurites and loss of soma, resulting in a higher cumulative risk of death than controls17,18.
RM analyses also showed that the formation of mHTT IBs in neurons does not cause degeneration but rather serves as a coping mechanism to slow neurodegeneration104,105. To directly study the impact of mHTT on mechanisms of protein clearance, an optical pulse labeling (OPL) technology using photoswitchable probes was developed. OPL allows us to monitor turnover of mHTT and autophagic flux or proteasome activity in single cells30,106. Using OPL and employing Bayesian regression modeling, we found that the mean lifetime of mHTT in single neurons was a greater predictor of neurodegeneration than absolute levels of mHTT30. Autophagy was determined as the prime cellular mechanism of clearance of mHTT, and small-molecule drugs that increase autophagy increased clearance of mHTT and reduced mHTT toxicity and the risk of death in a primary neuron HD model106, supporting the potential utility of small-molecule autophagy inducers to slow the progression of HD.
RM and OPL analyses were used to discover that deubiquitinase Usp12 protects against mHTT-induced neurotoxicity in a primary rodent model of HD and in HD i-neurons from a patient with 109 CAG repeats107. Interestingly, the catalytic activity of Usp12 was not needed for neuroprotection. Usp12 was found to protect against mHTT toxicity by inducing autophagic flux in neurons, and the enzyme was proposed to be an important regulator of neuronal proteostasis in HD107.
Neuronal development
HD i-neurons show a number of aberrations in neuronal patterning17,32,37,108 as well as a persistence of mitotic populations50. Global changes in neurodevelopmental gene expression have been described in HD and are likely linked to the changes in neuronal differentiation and connectivity.
Nestin is an intermediate filament protein and has been widely used as a marker of NSCs109. Genetic deletion of nestin reduces neuronal self-renewal and increases apoptosis110, whereas overexpression increases proliferation and results in larger brain and heart size111. In HD iPSC lines harboring 60, 109, and 180 CAG repeats, there is an increased number of nestin-positive cells as well as overall increased nestin expression compared with controls108,112. The expression of numerous cell cycle-related genes dramatically increases in HD iPSC lines harboring 46 and 53 CAG repeats, which likely explains the observation of a population of mitotically active cells that are resistant to differentiation50. These changes appear to be directly related to increased Wnt/β-Ca signaling as they are rescued by inhibition of this pathway50.
The developmental impairment in HD i-tissues can occur early in differentiation, even before neural patterning begins. Upon exposure to SMAD inhibition, a typical neural induction methodology113, the pluripotency marker OCT4 is normally downregulated114. In iPSCs that harbor large CAG repeat expansions of HTT, the downregulation of OCT4 and conversion to neural markers such as PAX6 are significantly delayed37. Neural rosette formation, a step in i-NSC differentiation, is also deficient in HD-iNSCs14,32,37. Gene correction of the CAG expansion normalizes rosette formation32. The delay in development appears to persist well into later stages, as striatal markers, such as CTIP2 and DARPP32, and pan neuronal markers MAP-2 and β III-tubulin are reduced in HD i-neurons compared with controls37. These developmental aberrations were restored when HTT expression was reduced by a synthetic zinc finger protein repressor37. When HD iPSCs were patterned to a cortical fate, the neurons harboring 77 and 109 CAG repeats exhibited gene expression and electrophysiological phenotypes that were consistent with delayed maturation31.
To gain insight into how mHTT alters neuronal morphology, several groups have examined neurite length in HD i-neurons because neurite length may be an indicator of synapse formation as well as synapse pruning. In i-neurons patterned towards an MSN fate, neurite length is increased in HD lines harboring 46, 53, or 109 CAG repeats, consistent with previous reports of ESCs that harbor CAG expansions and observations in the brains of patients with HD115–117 and with the finding that numerous axon guidance molecules are altered in HD17. Another study reported that when patterned towards cortical neurons, i-neurons with 5351, 77, 109, or 180 CAG repeats had significantly shorter neurites than the controls did31,51. Notably, in this later study, measurements were made much later in differentiation; thus, morphological changes may vary depending on the stage of differentiation and the cell type.
Altered bioenergetics
In mouse models of HD, CAG repeat expansions are associated with mitochondrial pathologies such as altered respiration, energy generation, and metabolic rate118–121. Similar findings have been observed in HD i-tissues. For example, HD i-NSCs with various CAG expansions exhibit decreased ATP levels compared with controls18,19,44,49, suggesting impaired energy production. HD i-NSCs and HD iPSCs from lines harboring 72 CAG repeats display numerous changes associated with mitochondrial dysfunction, biogenesis, dynamics, and morphology, and many of these phenotypes were restored after removal of the CAG repeat49. A quantitative proteomics analysis performed on i-neurons harboring 60 and 109 CAG repeats revealed a unique set of differentially expressed proteins in metabolic pathways and bioenergetic processes that could not be explained by gene expression changes17,19. This suggested that the metabolic defects may be due to post-translational events rather than transcriptional dysregulation. HD i-NSCs have also been shown to have compromised glycolysis19, and HD i-neurons harboring 42–44 repeats have lower mitochondrial density than controls do53. However, in an exhaustive examination of numerous aspects of mitochondrial function, such as mitochondrial membrane potential, ATP levels, respiration, and oxygen consumption rates, Hamilton et al. observed no differences between control and HD i-neurons containing 46, 53, or 72 CAG repeats122.
The reason for the differences in these studies is unclear. They may reflect differences in differentiation methods. Hamilton et al. (2020)122, who observed no apparent bioenergetic changes in HD i-neurons, used a method that generated embryoid bodies before generating neural rosettes, which were then harvested by rosette selection medium before differentiation. By contrast, most of the studies that showed changes in mitochondrial function used different methods for the neuronal differentiation, which may have led to cultures with a very different cellular composition. It may also be that the harvested i-NSCs are sub-stratified to generate a slightly different neuronal population of MSNs. Differentiation of MSN cultures is primarily validated by the staining of the MSN marker, DARPP32. However, in our experience (unpublished results), existing antibodies against DARPP32 are problematic and have exhibited variations in sensitivity and specificity, thereby rendering them unreliable in characterizing the outcomes of a particular differentiation. In other words, without better markers for the different differentiation outcomes, it remains difficult to compare findings from different groups and to ascertain whether and at what stages bioenergetics may differ between HD and control neuron populations differentiated in vitro.
Mechanisms of cell death in HD
Patients with HD exhibit a gradual degeneration of striatal MSNs that contributes to movement abnormalities123–126. Cerebral cortical cells also degenerate over time and likely contribute to cognitive deficits as well as motor abnormalities in HD124,127. Numerous cellular signals may contribute to cell death in HD, including altered calcium signaling128, loss of the supportive trophic factor BDNF129,130, proteolytic cleavage of mHTT131–133, and activation of the cell death pathways such as apoptosis and necrosis134.
HD i-neurons and i-tissues have been used to further refine our understanding of the mechanisms involved in neurodegeneration and cell death in HD. For example, apoptosis is known to activate caspases, which initiate a cascade of downstream signaling events leading to cell death135. Several studies have found activation of caspases in HD i-tissues. The Ellerby group has shown that, after growth factor withdrawal, HD i-NSCs have increased levels of caspase-3/7 activity13, and these findings have been confirmed by other groups and in different lines18,136. Gene correction of the 72 CAG expansion prevented the increase in caspase-3/7 activity, suggesting that activation of the caspase-mediated pathway is related to CAG expansion in HTT16. However, in an independent study of an HD iPSC line with just 42–44 CAG repeats, no elevation of caspase-3 activity was observed, suggesting that activation of this enzyme may be dependent on the length of the CAG repeat56.
Growth factors such as BDNF are critical for the survival of brain neurons, and levels of BDNF are diminished in HD brains. The reduction in BDNF is due to both decreased expression129,130,137,138 and altered BDNF transport in cortical neurons, which reduces BDNF release in the corticostriatal synapse139,140. HD i-tissues have increased sensitivity to loss of trophic factors, and after growth factor withdrawal, increased numbers of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells have been observed in HD i-NSCs16 and in differentiated HD i-neurons32. Specifically, reducing the level of BDNF in the media was shown to increase the number of TUNEL-positive HD i-neurons18,35,44,108 as well as the cell death rate and caspase-3/7 activity18. Overexpression of BDNF prevented the increased cell death rate in HD i-neurons18, as did treatment with an agonist of TrkB, a receptor that mediates the actions of BDNF, suggesting that impaired TrkB signaling due to loss of BDNF makes HD i-neurons susceptible to cell death108. Interestingly, the population of cells most susceptible to BDNF withdrawal appears to be nestin-positive progenitors108, suggesting that underdeveloped neurons are more sensitive to the insults of mHTT than mature cells.
The primary excitatory neurotransmitter in the brain is glutamate, and excess glutamatergic transmission is known to cause neurodegeneration in HD. Like HD brain tissues, HD i-tissues are more sensitive to glutamate, which induces calcium dysregulation and increased numbers of TUNEL-positive cells18. Furthermore, mHTT increases glutamate release from i-neurons, which in turn hyperactivates the glutamate N-methyl-d-aspartate receptors (NMDARs)141, resulting in excess calcium signaling. It also leads to downstream changes such as recruitment of extrasynaptic NMDARs, decreased CREB-mediated transcription, and induction of apoptosis128. Aberrant calcium signaling through store-operated calcium entry (SOCE) has also been observed in HD i-neurons, as inward calcium currents are about two-fold higher in HD i-neurons (notably in numerous patient lines that came from typical and juvenile-onset HD and contained 40–47 and 76 CAG repeats, respectively) compared with controls46,52,54. Calcium dysregulation in HD has long been studied, and aberrant SOCE signaling is likely a major player in this observation142.
CAG expanded lines are also uniquely sensitive to cellular stressors, particularly to oxidative stressors such as H2O218,143. In HD i-neurons, H2O2 was also shown to increase expression of the DNA damage marker γH2AX143. Exposure to the endoplasmic reticulum stress inducers thapsigargin and dithiothreitol increased nuclear indentations in HD i-neurons, with 42 and 44 CAG repeats significantly more than in control i-neurons. However, the molecular nature of this pathology is unclear53. Studies by Machiela et al.144 reported a role for aging in accelerating the sensitivity of HD i-neurons to cellular stressors. In a study of HD iPSCs differentiated towards the astrocytic lineage, both cells with the juvenile-onset 109 CAG repeats and the adult-onset 50 CAG repeats exhibited cytoplasmic vacuoles not present in control cells. These vacuoles were exacerbated by the cellular stressor chloroquine145. This is a curious observation as cytoplasmic vacuolization is thought to occur after cytotoxic stimuli such as bacterial pathogens or exposure to toxic compounds and precedes activation of cell death pathways146.
Isogenic embryonic stem cell lines to study CAG expansions
When iPSCs were first used to study HD, it remained controversial whether there were major differences compared with ESCs and whether they were better or worse for disease modeling147. It has been reported that they are functionally similar in terms of gene expression148 and differentiation potential149,150. Human ESCs have been used in numerous studies to study the effect of CAG expansions. Unlike iPSCs derived from patients, ESCs are unlikely to carry a CAG expansion unless derived from patients with HD. Normal ESCs have been engineered to develop allelic series of isogenic lines with varying numbers of CAG repeats151,152, which is a powerful way to avoid differences in genetic backgrounds when comparing different lines, as described in the next section. One allelic series was generated in the RUES2 line153 using clustered regularly interspaced short palindromic repeats (CRISPR) technology and includes lines with 45, 50, 56–58, 67, and 72–74 CAG repeats151. These lines displayed developmental abnormalities154, such as yielding an increased number of progenitor cells154 and forming aberrant rosettes151,154. Other novel phenotypes such as disruptions in cytokinesis and chromosomal instability151 were detected, and changes in gene expression in developmental pathways previously detected in iPSCs were also found151,154.
Another isogenic CAG allelic series, coined IsoHD lines152, was developed in the long-established H9 ESC background151,155. The CAG repeats were engineered using transcription activator-like effector nuclease (TALEN) technology, and the series consists of lines with 30, 45, 65, and 81 CAG repeats. The authors investigated numerous phenotypes; notably, many of them were sensitive to CAG repeat length, including defects in mitochondrial respiration, increased reactive oxygen species, and increased sensitivity to DNA damage152. Furthermore, the authors took an especially powerful approach to elucidate cell-specific phenotypes by generating different lineages and captured both cell-specific and CAG repeat-specific changes in gene expression at both the RNA and proteomic level152. Another study differentiated a subset of these lines into microglia and also found increased activation of reactive oxygen species as well as susceptibility to stressors and increased cytokine production in the cells with CAG expansions compared with isogenic controls156. Such allelic series of CAG lengths are powerful tools to dissect the contribution of the CAG expansion without the confounding effects of other genomic or epigenomic differences compared with control lines.
Transdifferentiation of HD fibroblasts directly to neurons
Recently, researchers have investigated the potential of directly converting fibroblasts into specific cell types rather than passing through a pluripotent cell stage157,158. This inventive method is believed to retain many of the epigenetic marks embedded within the chromatin, including marks associated with aging, which may be especially important in the context of neurodegenerative disease modeling, as disease onset is typically late in life159. In one study157, the authors expressed the brain-enriched microRNAs miR-9/9* and miR-124 in HD patient fibroblasts and induced expression of MSN markers. Most importantly, these MSN-like cells displayed aggregates of mHTT and IB formation, which is remarkable given that the parental fibroblasts showed no signs of mHTT aggregation157. In addition, the transdifferentiated HD cells exhibited phenotypes associated with neurodegeneration, such as increased oxidative damage, aberrant mitochondrial function, and increased cell death, and these differences correlated with the disease stage of the patients157. Because these cells harbor numerous neurodegenerative marks associated with HD, this approach has great potential. However, there still are several caveats to their use. First, the source material is limited. Unlike iPSCs, fibroblasts tolerate only a finite number of passages. Another is that each experiment requires a fresh batch of fibroblasts, which again becomes limiting. Transdifferentiation is long, complex, and difficult, so batch issues become challenging, which reduces the sensitivity, rigor, and reproducibility of the system. The last caveat is that one cannot make gene-corrected controls, which can confound the interpretation of phenotypes found associated with CAG expanded lines.
The pros and cons of using iPSC technology to study HD
Most of this review has focused on emphasizing the contributions of human iPSC technology to the study of HD and the advantages of this approach. An additional advantage is that iPSCs provide a relatively unlimited source of HD i-neurons that can be employed in high-throughput screens to identify potential therapeutics to treat the disease. Because of the availability of a large number of HD lines from patients with varying numbers of CAG repeats12,15,17,18,31,34,38,52,55,56, one can test potential therapeutics for efficacy in a range of genetic backgrounds and disease severity160. This is important because for most neurodegenerative diseases, the models employed, including human iPSC lines, are from patients with rare, inherited forms of the diseases that may not be reflective of the general patient population, where disease etiology is unknown. This is not the case in HD, which is unquestionably caused by the expansion of CAG repeats in the HTT gene. Nevertheless, it has become clear that variants in genes other than HTT contribute to HD69. Using the human HD iPSC lines, it is possible to understand the role of these other variants in the disease since these variants may not be expressed or may not affect disease in the same way in animal models.
That said, there are limitations to the iPSC technology. iPSC lines can come from individuals with diverse genetic backgrounds, and this heterogeneity can confound the interpretation of findings161–165. This problem has been addressed with great success thanks to the use of gene-editing strategies such as CRISPR166 or TALENs167. These technologies allow researchers to either correct or introduce mutations of interest in a given line or genetic background (or both)168–170. Using gene correction to make isogenic control lines is a powerful strategy to confirm that disease-related phenotypes are due to the mutation of interest and not to clonal variation, cellular heterogeneity, or another mutation or variant in the genetic background. However, generating gene-corrected lines may not entirely solve these problems as genetic background can still influence cell phenotypes165. Nevertheless, CAG expanded lines have proven to display robust cellular changes compared with their respective isogenic control lines13,16,32,49. Additional heterogeneity and confounding factors come from differences in methodologies between experiments and labs, which can produce different subsets of cell types and complicate comparisons between lines6,171. Methods of cell differentiation can be inefficient and complex and result in cultures with heterogeneous cell populations. The longer and more complex the protocol, the more opportunity for batch variation from experiment to experiment, which can confound interpretation of phenotypes. Using linear mixed models or more sophisticated statistical measures to account for all of the batch variation is much needed in the field.
Another challenge with iPSCs is that the reprogramming process changes the epigenetic marks, many of which may influence disease-related phenotypes172. While studies have been performed to investigate epigenetic differences in HD using iPSC lines, the process of generation of iPSC lines can vary173–176, which can confound the interpretation of the effect of patient-specific epigenetic marks. While epigenetic studies can be done directly on the patient fibroblasts, the use of these cells provides limited insight into the role of epigenetics in creating vulnerability to selective neuronal populations in HD. The use of transdifferentiation technology, as described in the previous section, to study HD157 may overcome these limitations as neurons can be generated directly. However, transdifferentiation is still in its early stages, and as fibroblasts have only limited self-renewal potential in culture, transdifferentiation from fibroblasts may never generate the number of cells necessary for high-throughput assays.
Critical view of the topic
Most of the iPSC models of HD are derived from patient cells, and as such, they harbor the actual human disease mutations and genetic background contributing to disease. While most of our discussion so far has emphasized MSNs as the main target of HD, iPSCs can be differentiated into other types of i-neurons as well as into other cell types, including astrocytes and microglia, the immune cells of the brain. Scientists can therefore use human iPSC models to reproduce some of the cell-cell dependencies that underlie neuronal dysfunction, degeneration, and death and to investigate the role of inflammation in neurodegeneration. This can provide insights into potential molecular targets in humans that might be useful for developing disease-modifying therapeutics. Furthermore, it is possible to generate the human i-neurons most vulnerable to HD (MSNs and cortical neurons)177 from large numbers of different patients with HD to identify common disease mechanisms. Since not all aspects of pathogenesis are dependent on CAG repeat length, the in vitro human cell models also provide an approach to screen for potential genetic modifiers that may impact disease onset, progression, and symptomology, which also could be molecular targets for drug discovery.
Some have suggested that findings from the human HD i-neurons and i-tissues may be more reflective of disease processes in patients than animal models since these cell populations express the same genetic mutations and, in the case of transdifferentiated cells, may also maintain the epigenetic status of the patients. However, we can learn only so much from in vitro studies, and findings from in vitro systems may or may not correlate with the disease pathology and behaviors of patients with HD. Both types of models may be necessary to better understand the pathogenesis of HD and to develop novel approaches for disease treatment.
Future perspectives
New artificial intelligence technology and especially deep learning (DL) methods are providing unique ways to use imaging to make predictions on cell fate without invasive procedures of biosensors178,179. This is important because neurodegeneration is heterogeneous in HD, as it is in all neurodegenerative diseases. This heterogeneity is visible at the level of cell models as well, as cultures of human i-neurons carrying the same HD mutation can harbor cells that resist degeneration adjacent to cells that die rapidly. DL technology, with its ability to record and analyze large numbers of individual cells and correlate cell biology to cell fate, can help us unravel the basis of this heterogeneity.
Furthermore, the development of HD i-neurons grown in three-dimensional organoids can provide new approaches to study the degeneration of HD i-neurons under conditions that better maintain their cytoarchitecture than two-dimensional monolayers can. These technologies can also begin to provide the basis for studying dysfunction of neuronal circuitry across distances in the brain, such as cortical-striatal-thalamic circuits, that may contribute to the degeneration of specific cell populations in the brain.
Acknowledgments
We would like to thank Katy Claiborn (Gladstone Institutes), Giovanni Maki (Gladstone Institutes), Gayane Abramova (Gladstone Institutes), Kelley Nelson (Gladstone Institutes), Francoise Chanut (Gladstone Institutes), and all Finkbeiner lab members.
The peer reviewers who approve this article are:
Lesley Jones, MRC Centre for Neuropsychiatric Genetics and Genomics, School of Medicine, Cardiff University, Cardiff, UK
Marta Olejniczak, Department of Genome Engineering, Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan, Poland
Funding Statement
The funding that supported this work was from National Institutes of Health grants R37NS101996, P01 AG054407, and RF1 AG058476.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Julia Kaye, Email: Julia.kaye@gladstone.ucsf.edu.
Steven Finkbeiner, Email: steve.finkbeiner@gladstone.ucsf.edu.
References
- 1. Takahashi K, Tanabe K, Ohnuki M, et al. : Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131(5): 861–72. 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 2. Yu J, Vodyanik MA, Smuga-Otto K, et al. : Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007; 318(5858): 1917–20. 10.1126/science.1151526 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 3. Watson J: Epidemiological situation and surveillance of tuberculosis in England and Wales. Bull Int Union Tuberc Lung Dis. 1990; 65(2–3): 42–3. [PubMed] [Google Scholar]
- 4. Schlaeger TM, Daheron L, Brickler TR, et al. : A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015; 33(1): 58–63. 10.1038/nbt.3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park I-H, Zhao R, West JA, et al. : Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008; 451(7175): 141–6. 10.1038/nature06534 [DOI] [PubMed] [Google Scholar]
- 6. Rivetti di Val Cervo P, Besusso D, Conforti P, et al. : hiPSCs for predictive modelling of neurodegenerative diseases: Dreaming the possible. Nat Rev Neurol. 2021; 17(6): 381–92. 10.1038/s41582-021-00465-0 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 7. Fusaki N, Ban H, Nishiyama A, et al. : Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009; 85(8): 348–62. 10.2183/pjab.85.348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Warren L, Manos PD, Ahfeldt T, et al. : Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010; 7(5): 618–30. 10.1016/j.stem.2010.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 9. Qin H, Zhao A, Fu X: Small molecules for reprogramming and transdifferentiation. Cell Mol Life Sci. 2017; 74(19): 3553–75. 10.1007/s00018-017-2586-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bruck RM, Williams TH: Neurological mouse mutants: New tools in neuroanatomical research. J Anat. 1970; 106(Pt 1): 187. [PubMed] [Google Scholar]
- 11. Park I-H, Lerou PH, Zhao R, et al. : Generation of human-induced pluripotent stem cells. Nat Protoc. 2008; 3(7): 1180–6. 10.1038/nprot.2008.92 [DOI] [PubMed] [Google Scholar]
- 12. Park I-H, Arora N, Huo H, et al. : Disease-specific induced pluripotent stem cells. Cell. 2008; 134(5): 877–86. 10.1016/j.cell.2008.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 13. Zhang N, An MC, Montoro D, et al. : Characterization of Human Huntington's Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010; 2: RRN1193. 10.1371/currents.RRN1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jeon I, Lee N, Li JY, et al. : Neuronal properties, in vivo effects, and pathology of a Huntington's disease patient-derived induced pluripotent stem cells. Stem Cells. 2012; 30(9): 2054–62. 10.1002/stem.1135 [DOI] [PubMed] [Google Scholar]
- 15. Chae J-I, Kim D-W, Lee N, et al. : Quantitative proteomic analysis of induced pluripotent stem cells derived from a human Huntington's disease patient. Biochem J. 2012; 446(3): 359–71. 10.1042/BJ20111495 [DOI] [PubMed] [Google Scholar]
- 16. An MC, Zhang N, Scott G, et al. : Genetic correction of Huntington's disease phenotypes in induced pluripotent stem cells. Cell Stem Cell. 2012; 11(2): 253–63. 10.1016/j.stem.2012.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. HD iPSC Consortium: Developmental alterations in Huntington's disease neural cells and pharmacological rescue in cells and mice. Nat Neurosci. 2017; 20(5): 648–60. 10.1038/nn.4532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. HD iPSC Consortium: Induced pluripotent stem cells from patients with Huntington's disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell. 2012; 11(2): 264–78. 10.1016/j.stem.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. HD iPSC Consortium: Bioenergetic deficits in Huntington's disease iPSC-derived neural cells and rescue with glycolytic metabolites. Hum Mol Genet. 2020; 29: 1757–71. 10.1093/hmg/ddy430 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 20. Cattaneo E, Zuccato C, Tartari M: Normal huntingtin function: An alternative approach to Huntington's disease. Nat Rev Neurosci. 2005; 6(12): 919–30. 10.1038/nrn1806 [DOI] [PubMed] [Google Scholar]
- 21. Shirendeb U, Reddy AP, Manczak M, et al. : Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington's disease: Implications for selective neuronal damage. Hum Mol Genet. 2011; 20(7): 1438–55. 10.1093/hmg/ddr024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shirendeb UP, Calkins MJ, Manczak M, et al. : Mutant huntingtin's interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington's disease. Hum Mol Genet. 2012; 21(2): 406–20. 10.1093/hmg/ddr475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song W, Chen J, Petrilli A, et al. : Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. 2011; 17(3): 377–82. 10.1038/nm.2313 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 24. Jana NR, Zemskov EA, Wang G, et al. : Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001; 10(10): 1049–59. 10.1093/hmg/10.10.1049 [DOI] [PubMed] [Google Scholar]
- 25. Chow WNV, Luk HW, Chan HYE, et al. : Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65. Biochem J. 2012; 443(3): 681–9. 10.1042/BJ20112175 [DOI] [PubMed] [Google Scholar]
- 26. Bhat KP, Yan S, Wang C-E, et al. : Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A. 2014; 111(15): 5706–11. 10.1073/pnas.1402215111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Martin DDO, Ladha S, Ehrnhoefer DE, et al. : Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015; 38(1): 26–35. 10.1016/j.tins.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 28. Ott J, Kamatani Y, Lathrop M: Family-based designs for genome-wide association studies. Nat Rev Genet. 2011; 12(7): 465–74. 10.1038/nrg2989 [DOI] [PubMed] [Google Scholar]
- 29. Mitra S, Tsvetkov AS, Finkbeiner S: Single neuron ubiquitin-proteasome dynamics accompanying inclusion body formation in huntington disease. J Biol Chem. 2009; 284(7): 4398–403. 10.1074/jbc.M806269200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsvetkov AS, Arrasate M, Barmada S, et al. : Proteostasis of polyglutamine varies among neurons and predicts neurodegeneration. Nat Chem Biol. 2013; 9(9): 586–92. 10.1038/nchembio.1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mehta SR, Tom CM, Wang Y, et al. : Human Huntington's Disease iPSC-Derived Cortical Neurons Display Altered Transcriptomics, Morphology, and Maturation. Cell Rep. 2018; 25(4): 1081–1096.e6. 10.1016/j.celrep.2018.09.076 [DOI] [PubMed] [Google Scholar]
- 32. Xu X, Tay Y, Sim B, et al. : Reversal of Phenotypic Abnormalities by CRISPR/Cas9-Mediated Gene Correction in Huntington Disease Patient-Derived Induced Pluripotent Stem Cells. Stem Cell Reports. 2017; 8(3): 619–33. 10.1016/j.stemcr.2017.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Irmak D, Fatima A, Gutiérrez-Garcia R, et al. : Mechanism suppressing H3K9 trimethylation in pluripotent stem cells and its demise by polyQ-expanded huntingtin mutations. Hum Mol Genet. 2018; 27(23): 4117–34. 10.1093/hmg/ddy304 [DOI] [PubMed] [Google Scholar]
- 34. Tidball AM, Neely MD, Chamberlin R, et al. : Genomic Instability Associated with p53 Knockdown in the Generation of Huntington's Disease Human Induced Pluripotent Stem Cells. PLoS One. 2016; 11(3): e0150372. 10.1371/journal.pone.0150372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lu XH, Mattis VB, Wang N, et al. : Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington's disease. Sci Transl Med. 2014; 6(268): 268ra178. 10.1126/scitranslmed.3010523 [DOI] [PubMed] [Google Scholar]
- 36. Koyuncu S, Saez I, Lee HJ, et al. : The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington's disease patients. Nat Commun. 2018; 9(1): 2886. 10.1038/s41467-018-05320-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Conforti P, Besusso D, Bocchi VD, et al. : Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc Natl Acad Sci U S A. 2018; 115(4): E762–E771. 10.1073/pnas.1715865115 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 38. Świtońska K, Szlachcic WJ, Handschuh L, et al. : Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front Cell Neurosci. 2018; 12: 528. 10.3389/fncel.2018.00528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szlachcic WJ, Switonski PM, Krzyzosiak WJ, et al. : Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis Model Mech. 2015; 8(9): 1047–57. 10.1242/dmm.019406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szlachcic WJ, Wiatr K, Trzeciak M, et al. : The Generation of Mouse and Human Huntington Disease iPS Cells Suitable for In vitro. Studies on Huntingtin Function. Front Mol Neurosci. 2017; 10: 253. 10.3389/fnmol.2017.00253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bowles KR, Stone T, Holmans P, et al. : SMAD transcription factors are altered in cell models of HD and regulate HTT expression. Cell Signal. 2017; 31: 1–14. 10.1016/j.cellsig.2016.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gasset-Rosa F, Chillon-Marinas C, Goginashvili A, et al. : Polyglutamine-Expanded Huntingtin Exacerbates Age-Related Disruption of Nuclear Integrity and Nucleocytoplasmic Transport. Neuron. 2017; 94(1): 48–57.e4. 10.1016/j.neuron.2017.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grima JC, Daigle JG, Arbez N, et al. : Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron. 2017; 94(1): 93–107.e6. 10.1016/j.neuron.2017.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rindt H, Tom CM, Lorson CL, et al. : Optimization of trans-Splicing for Huntington's Disease RNA Therapy. Front Neurosci. 2017; 11: 544. 10.3389/fnins.2017.00544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Goold R, Flower M, Moss DH, et al. : FAN1 modifies Huntington's disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. 2019; 28(4): 650–61. 10.1093/hmg/ddy375 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 46. Vigont VA, Grekhnev DA, Lebedeva OS, et al. : STIM2 Mediates Excessive Store-Operated Calcium Entry in Patient-Specific iPSC-Derived Neurons Modeling a Juvenile Form of Huntington's Disease. Front Cell Dev Biol. 2021; 9: 625231. 10.3389/fcell.2021.625231 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 47. Naphade S, Embusch A, Madushani KL, et al. : Altered Expression of Matrix Metalloproteinases and Their Endogenous Inhibitors in a Human Isogenic Stem Cell Model of Huntington's Disease. Front Neurosci. 2017; 11: 736. 10.3389/fnins.2017.00736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ring KL, An MC, Zhang N, et al. : Genomic Analysis Reveals Disruption of Striatal Neuronal Development and Therapeutic Targets in Human Huntington's Disease Neural Stem Cells. Stem Cell Reports. 2015; 5(6): 1023–38. 10.1016/j.stemcr.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopes C, Tang Y, Anjo SI, et al. : Mitochondrial and Redox Modifications in Huntington Disease Induced Pluripotent Stem Cells Rescued by CRISPR/Cas9 CAGs Targeting. Front Cell Dev Biol. 2020; 8: 576592. 10.3389/fcell.2020.576592 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 50. Smith-Geater C, Hernandez SJ, Lim RG, et al. : Aberrant Development Corrected in Adult-Onset Huntington's Disease iPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cell Reports. 2020; 14(3): 406–19. 10.1016/j.stemcr.2020.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 51. Imamura T, Fujita K, Tagawa K, et al. : Identification of hepta-histidine as a candidate drug for Huntington's disease by in silico-in vitro- in vivo-integrated screens of chemical libraries. Sci Rep. 2016; 6: 33861. 10.1038/srep33861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nekrasov ED, Vigont VA, Klyushnikov SA, et al. : Manifestation of Huntington's disease pathology in human induced pluripotent stem cell-derived neurons. Mol Neurodegener. 2016; 11: 27. 10.1186/s13024-016-0092-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nekrasov ED, Kiselev SL: Mitochondrial distribution violation and nuclear indentations in neurons differentiated from iPSCs of Huntington's disease patients. J Stem Cells Regen Med. 2018; 14(2): 80–5. 10.46582/jsrm.1402012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vigont V, Nekrasov E, Shalygin A, et al. : Patient-Specific iPSC-Based Models of Huntington's Disease as a Tool to Study Store-Operated Calcium Entry Drug Targeting. Front Pharmacol. 2018; 9: 696. 10.3389/fphar.2018.00696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mollica PA, Zamponi M, Reid JA, et al. : Epigenetic alterations mediate iPSC-induced normalization of DNA repair gene expression and TNR stability in Huntington's disease cells. J Cell Sci. 2018; 131(13): jcs215343. 10.1242/jcs.215343 [DOI] [PubMed] [Google Scholar]
- 56. Camnasio S, Delli Carri A, Lombardo A, et al. : The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington's disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis. 2012; 46(1): 41–51. 10.1016/j.nbd.2011.12.042 [DOI] [PubMed] [Google Scholar]
- 57. Baronchelli S, La Spada A, Ntai A, et al. : Epigenetic and transcriptional modulation of WDR5, a chromatin remodeling protein, in Huntington's disease human induced pluripotent stem cell (hiPSC) model. Mol Cell Neurosci. 2017; 82: 46–57. 10.1016/j.mcn.2017.04.013 [DOI] [PubMed] [Google Scholar]
- 58. Ciosi M, Maxwell A, Cumming SA, et al. : A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. 2019; 48: 568–80. 10.1016/j.ebiom.2019.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Electronic address: gusella@helix.mgh.harvard.edu, Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium: CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset. Cell. 2019; 178(4): 887–900.e14. 10.1016/j.cell.2019.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 60. Donaldson J, Powell S, Rickards N, et al. : What is the Pathogenic CAG Expansion Length in Huntington's Disease? J Huntingtons Dis. 2021; 10(1): 175–202. 10.3233/JHD-200445 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 61. Telenius H, Kremer B, Goldberg YP, et al. : Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and sperm. Nat Genet. 1994; 6(4): 409–14. 10.1038/ng0494-409 [DOI] [PubMed] [Google Scholar]
- 62. de Rooij KE, de Koning Gans PA, Roos RA, et al. : Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum Genet. 1995; 95(3): 270–4. 10.1007/BF00225192 [DOI] [PubMed] [Google Scholar]
- 63. Swami M, Hendricks AE, Gillis T, et al. : Somatic expansion of the Huntington's disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. 2009; 18(16): 3039–47. 10.1093/hmg/ddp242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kennedy L, Evans E, Chen CM, et al. : Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. 2003; 12(24): 3359–67. 10.1093/hmg/ddg352 [DOI] [PubMed] [Google Scholar]
- 65. Mouro Pinto R, Arning L, Giordano JV, et al. : Patterns of CAG repeat instability in the central nervous system and periphery in Huntington's disease and in spinocerebellar ataxia type 1. Hum Mol Genet. 2020; 29(15): 2551–67. 10.1093/hmg/ddaa139 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 66. Kacher R, Lejeune FX, Noël S, et al. : Propensity for somatic expansion increases over the course of life in Huntington disease. eLife. 2021; 10: e64674. 10.7554/eLife.64674 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 67. Wright GEB, Collins JA, Kay C, et al. : Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased Somatic Instability, Hastening Onset of Huntington Disease. Am J Hum Genet. 2019; 104(6): 1116–26. 10.1016/j.ajhg.2019.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Moss DJH, Pardiñas AF, Langbehn D, et al. : Identification of genetic variants associated with Huntington's disease progression: A genome-wide association study. Lancet Neurol. 2017; 16(9): 701–11. 10.1016/S1474-4422(17)30161-8 [DOI] [PubMed] [Google Scholar]
- 69. Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium: Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell. 2015; 162(3): 516–26. 10.1016/j.cell.2015.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 70. Lee JM, Chao MJ, Harold D, et al. : A modifier of Huntington's disease onset at the MLH1 locus. Hum Mol Genet. 2017; 26(19): 3859–67. 10.1093/hmg/ddx286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Loupe JM, Pinto RM, Kim KH, et al. : Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out. Hum Mol Genet. 2020; 29(18): 3044–53. 10.1093/hmg/ddaa196 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 72. Kim KH, Hong EP, Shin JW, et al. : Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects. Am J Hum Genet. 2020; 107(1): 96–110. 10.1016/j.ajhg.2020.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 73. Wright GEB, Black HF, Collins JA, et al. : Interrupting sequence variants and age of onset in Huntington's disease: Clinical implications and emerging therapies. Lancet Neurol. 2020; 19(11): 930–9. 10.1016/S1474-4422(20)30343-4 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 74. Menendez D, Inga A, Resnick MA: The biological impact of the human master regulator p53 can be altered by mutations that change the spectrum and expression of its target genes. Mol Cell Biol. 2006; 26(6): 2297–308. 10.1128/MCB.26.6.2297-2308.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Massey TH, Jones L: The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech. 2018; 11(1): dmm031930. 10.1242/dmm.031930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Bae BI, Xu H, Igarashi S, et al. : p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005; 47(1): 29–41. 10.1016/j.neuron.2005.06.005 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 77. Podhorecka M, Skladanowski A, Bozko P: H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J Nucleic Acids. 2010; 2010: 920161. 10.4061/2010/920161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Al Rashid ST, Dellaire G, Cuddihy A, et al. : Evidence for the direct binding of phosphorylated p53 to sites of DNA breaks in vivo. Cancer Res. 2005; 65(23): 10810–21. 10.1158/0008-5472.CAN-05-0729 [DOI] [PubMed] [Google Scholar]
- 79. Morozko EL, Smith-Geater C, Monteys AM, et al. : PIAS1 modulates striatal transcription, DNA damage repair, and SUMOylation with relevance to Huntington's disease. Proc Natl Acad Sci U S A. 2021; 118(4): e2021836118. 10.1073/pnas.2021836118 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 80. Knockenhauer KE, Schwartz TU: The Nuclear Pore Complex as a Flexible and Dynamic Gate. Cell. 2016; 164(6): 1162–71. 10.1016/j.cell.2016.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Derynck R, Budi EH: Specificity, versatility, and control of TGF-β family signaling. Sci Signal. 2019; 12(570): eaav5183. 10.1126/scisignal.aav5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gal A, Sjöblom T, Fedorova L, et al. : Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene. 2008; 27(9): 1218–30. 10.1038/sj.onc.1210741 [DOI] [PubMed] [Google Scholar]
- 83. Parsons JT, Horwitz AR, Schwartz MA: Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010; 11(9): 633–43. 10.1038/nrm2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hynes RO: The extracellular matrix: Not just pretty fibrils. Science. 2009; 326(5957): 1216–9. 10.1126/science.1176009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Maître JL, Heisenberg CP: Three functions of cadherins in cell adhesion. Curr Biol. 2013; 23(14): R626–33. 10.1016/j.cub.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 86. Lunyak VV, Burgess R, Prefontaine GG, et al. : Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002; 298(5599): 1747–52. 10.1126/science.1076469 [DOI] [PubMed] [Google Scholar]
- 87. Huang Z, Wu Q, Guryanova OA, et al. : Deubiquitylase HAUSP stabilizes REST and promotes maintenance of neural progenitor cells. Nat Cell Biol. 2011; 13(2): 142–52. 10.1038/ncb2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lee J, Hwang YJ, Kim KY, et al. : Epigenetic mechanisms of neurodegeneration in Huntington's disease. Neurotherapeutics. 2013; 10(4): 664–76. 10.1007/s13311-013-0206-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Achour M, Le Gras S, Keime C, et al. : Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington's disease mice. Hum Mol Genet. 2015; 24(12): 3481–96. 10.1093/hmg/ddv099 [DOI] [PubMed] [Google Scholar]
- 90. Bai G, Cheung I, Shulha HP, et al. : Epigenetic dysregulation of hairy and enhancer of split 4 (HES4) is associated with striatal degeneration in postmortem Huntington brains. Hum Mol Genet. 2015; 24(5): 1441–56. 10.1093/hmg/ddu561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ryu H, Lee J, Hagerty SW, et al. : ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci U S A. 2006; 103(50): 19176–81. 10.1073/pnas.0606373103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Biagioli M, Ferrari F, Mendenhall EM, et al. : Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum Mol Genet. 2015; 24(9): 2442–57. 10.1093/hmg/ddv006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schilling G, Klevytska A, Tebbenkamp ATN, et al. : Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J Neuropathol Exp Neurol. 2007; 66(4): 313–20. 10.1097/nen.0b013e318040b2c8 [DOI] [PubMed] [Google Scholar]
- 94. Ko J, Ou S, Patterson PH: New anti-huntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res Bull. 2001; 56(3–4): 319–29. 10.1016/s0361-9230(01)00599-8 [DOI] [PubMed] [Google Scholar]
- 95. Wang CE, Tydlacka S, Orr AL, et al. : Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum Mol Genet. 2008; 17(17): 2738–51. 10.1093/hmg/ddn175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Miller J, Arrasate M, Brooks E, et al. : Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011; 7(12): 925–34. 10.1038/nchembio.694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gutekunst CA, Li SH, Yi H, et al. : Nuclear and Neuropil Aggregates in Huntington’s Disease: Relationship to Neuropathology. J Neurosci.. 1999; 19(7): 2522–34. 10.1523/JNEUROSCI.19-07-02522.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Margulis J, Finkbeiner S: Proteostasis in striatal cells and selective neurodegeneration in Huntington's disease. Front Cell Neurosci. 2014; 8: 218. 10.3389/fncel.2014.00218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Skibinski G, Finkbeiner S: Longitudinal measures of proteostasis in live neurons: Features that determine fate in models of neurodegenerative disease. FEBS Lett. 2013; 587(8): 1139–46. 10.1016/j.febslet.2013.02.043 [DOI] [PubMed] [Google Scholar]
- 100. Lee H, Noh JY, Oh Y, et al. : IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet. 2012; 21(1): 101–14. 10.1093/hmg/ddr445 [DOI] [PubMed] [Google Scholar]
- 101. Martinez-Vicente M, Talloczy Z, Wong E, et al. : Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci. 2010; 13(5): 567–76. 10.1038/nn.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 102. Soares TR, Reis SD, Pinho BR, et al. : Targeting the proteostasis network in Huntington's disease. Ageing Res Rev. 2019; 49: 92–103. 10.1016/j.arr.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Liu Y, Qiao F, Leiferman PC, et al. : FOXOs modulate proteasome activity in human-induced pluripotent stem cells of Huntington's disease and their derived neural cells. Hum Mol Genet. 2017; 26(22): 4416–28. 10.1093/hmg/ddx327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Arrasate M, Mitra S, Schweitzer ES, et al. : Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004; 431(7010): 805–10. 10.1038/nature02998 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 105. Arrasate M, Finkbeiner S: Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A. 2005; 102(10): 3840–5. 10.1073/pnas.0409777102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Tsvetkov AS, Miller J, Arrasate M, et al. : A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010; 107(39): 16982–7. 10.1073/pnas.1004498107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Aron R, Pellegrini P, Green EW, et al. : Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington's disease. Nat Commun. 2018; 9(1): 3191. 10.1038/s41467-018-05653-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Mattis VB, Tom C, Akimov S, et al. : HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity. Hum Mol Genet. 2015; 24(11): 3257–71. 10.1093/hmg/ddv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Mignone JL, Kukekov V, Chiang AS, et al. : Neural stem and progenitor cells in nestin-GFP transgenic mice. J Comp Neurol. 2004; 469(3): 311–24. 10.1002/cne.10964 [DOI] [PubMed] [Google Scholar]
- 110. Park D, Xiang AP, Mao FF, et al. : Nestin is required for the proper self-renewal of neural stem cells. Stem Cells. 2010; 28(12): 2162–71. 10.1002/stem.541 [DOI] [PubMed] [Google Scholar]
- 111. Liu J, Ji X, Li Z, et al. : Nestin overexpression promotes the embryonic development of heart and brain through the regulation of cell proliferation. Brain Res. 2015; 1610: 1–11. 10.1016/j.brainres.2015.03.044 [DOI] [PubMed] [Google Scholar]
- 112. Mathkar PP, Suresh D, Dunn J, et al. : Characterization of Neurodevelopmental Abnormalities in iPSC-Derived Striatal Cultures from Patients with Huntington's Disease. J Huntingtons Dis. 2019; 8(3): 257–69. 10.3233/JHD-180333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chambers SM, Fasano CA, Papapetrou EP, et al. : Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009; 27(3): 275–80. 10.1038/nbt.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 114. Zeineddine D, Hammoud AA, Mortada M, et al. : The Oct4 protein: More than a magic stemness marker. Am J Stem Cells. 2014; 3(2): 74–82. [PMC free article] [PubMed] [Google Scholar]
- 115. Curtis MA, Penney EB, Pearson AG, et al. : Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci U S A. 2003; 100(15): 9023–7. 10.1073/pnas.1532244100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Curtis MA, Waldvogel HJ, Synek B, et al. : A histochemical and immunohistochemical analysis of the subependymal layer in the normal and Huntington's disease brain. J Chem Neuroanat. 2005; 30(1): 55–66. 10.1016/j.jchemneu.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 117. Ferrante RJ, Kowall NW, Richardson EP, Jr: Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: A combined study using the section-Golgi method and calbindin D28k immunocytochemistry. J Neurosci. 1991; 11(12): 3877–87. 10.1523/JNEUROSCI.11-12-03877.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Quintanilla RA, Johnson GV: Role of mitochondrial dysfunction in the pathogenesis of Huntington's disease. Brain Res Bull. 2009; 80(4–5): 242–7. 10.1016/j.brainresbull.2009.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Tobore TO: Towards a comprehensive understanding of the contributions of mitochondrial dysfunction and oxidative stress in the pathogenesis and pathophysiology of Huntington's disease. J Neurosci Res. 2019; 97(11): 1455–68. 10.1002/jnr.24492 [DOI] [PubMed] [Google Scholar]
- 120. Oliveira JM: Nature and cause of mitochondrial dysfunction in Huntington's disease: Focusing on huntingtin and the striatum. J Neurochem. 2010; 114(1): 1–12. 10.1111/j.1471-4159.2010.06741.x [DOI] [PubMed] [Google Scholar]
- 121. Carmo C, Naia L, Lopes C, et al. : Mitochondrial Dysfunction in Huntington's Disease. Adv Exp Med Biol. 2018; 1049: 59–83. 10.1007/978-3-319-71779-1_3 [DOI] [PubMed] [Google Scholar]
- 122. Hamilton J, Brustovetsky T, Sridhar A, et al. : Energy Metabolism and Mitochondrial Superoxide Anion Production in Pre-symptomatic Striatal Neurons Derived from Human-Induced Pluripotent Stem Cells Expressing Mutant Huntingtin. Mol Neurobiol. 2020; 57(2): 668–84. 10.1007/s12035-019-01734-2 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 123. Augood SJ, Faull RL, Emson PC: Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann Neurol. 1997; 42(2): 215–21. 10.1002/ana.410420213 [DOI] [PubMed] [Google Scholar]
- 124. Han I, You Y, Kordower JH, et al. : Differential vulnerability of neurons in Huntington's disease: The role of cell type-specific features. J Neurochem. 2010; 113(5): 1073–91. 10.1111/j.1471-4159.2010.06672.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Vonsattel JP, Myers RH, Stevens TJ, et al. : Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985; 44(6): 559–77. 10.1097/00005072-198511000-00003 [DOI] [PubMed] [Google Scholar]
- 126. Reiner A, Albin RL, Anderson KD, et al. : Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988; 85(15): 5733–7. 10.1073/pnas.85.15.5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cudkowicz M, Kowall NW: Degeneration of pyramidal projection neurons in Huntington's disease cortex. Ann Neurol. 1990; 27(2): 200–4. 10.1002/ana.410270217 [DOI] [PubMed] [Google Scholar]
- 128. Tang TS, Slow E, Lupu V, et al. : Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005; 102(7): 2602–7. 10.1073/pnas.0409402102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zuccato C, Cattaneo E: Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007; 81(5–6): 294–330. 10.1016/j.pneurobio.2007.01.003 [DOI] [PubMed] [Google Scholar]
- 130. Zuccato C, Ciammola A, Rigamonti D, et al. : Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001; 293(5529): 493–8. 10.1126/science.1059581 [DOI] [PubMed] [Google Scholar]
- 131. Graham RK, Deng Y, Slow EJ, et al. : Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006; 125(6): 1179–91. 10.1016/j.cell.2006.04.026 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 132. Wellington CL, Ellerby LM, Gutekunst CA, et al. : Caspase Cleavage of Mutant Huntingtin Precedes Neurodegeneration in Huntington's Disease. J Neurosci. 2002; 22(18): 7862–72. 10.1523/JNEUROSCI.22-18-07862.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Juenemann K, Weisse C, Reichmann D, et al. : Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox Res. 2011; 20(2): 120–33. 10.1007/s12640-010-9227-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Hickey MA, Chesselet MF: Apoptosis in Huntington's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003; 27(2): 255–65. 10.1016/S0278-5846(03)00021-6 [DOI] [PubMed] [Google Scholar]
- 135. Kiraz Y, Adan A, Kartal Yandim M, et al. : Major apoptotic mechanisms and genes involved in apoptosis. Tumour Biol. 2016; 37(7): 8471–86. 10.1007/s13277-016-5035-9 [DOI] [PubMed] [Google Scholar]
- 136. Malankhanova T, Suldina L, Grigor'eva E, et al. : A Human Induced Pluripotent Stem Cell-Derived Isogenic Model of Huntington's Disease Based on Neuronal Cells Has Several Relevant Phenotypic Abnormalities. J Pers Med. 2020; 10(4): 215. 10.3390/jpm10040215 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 137. Ferrer I, Goutan E, Marín C, et al. : Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000; 866(1–2): 257–61. 10.1016/s0006-8993(00)02237-x [DOI] [PubMed] [Google Scholar]
- 138. Zuccato C, Liber D, Ramos C, et al. : Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005; 52(2): 133–9. 10.1016/j.phrs.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 139. Gauthier LR, Charrin BC, Borrell-Pagès M, et al. : Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004; 118(1): 127–38. 10.1016/j.cell.2004.06.018 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 140. Her LS, Goldstein LS: Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J Neurosci. 2008; 28(50): 13662–72. 10.1523/JNEUROSCI.4144-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Song C, Zhang Y, Parsons CG, et al. : Expression of polyglutamine-expanded huntingtin induces tyrosine phosphorylation of N-methyl-D-aspartate receptors. J Biol Chem. 2003; 278(35): 33364–9. 10.1074/jbc.M304240200 [DOI] [PubMed] [Google Scholar]
- 142. Czeredys M: Dysregulation of Neuronal Calcium Signaling via Store-Operated Channels in Huntington's Disease. Front Cell Dev Biol. 2020; 8: 611735. 10.3389/fcell.2020.611735 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 143. Chiu FL, Lin JT, Chuang CY, et al. : Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington's disease iPSCs. Hum Mol Genet. 2015; 24(21): 6066–79. 10.1093/hmg/ddv318 [DOI] [PubMed] [Google Scholar]
- 144. Machiela E, Jeloka R, Caron NS, et al. : The Interaction of Aging and Cellular Stress Contributes to Pathogenesis in Mouse and Human Huntington Disease Neurons. Front Aging Neurosci. 2020; 12: 524369. 10.3389/fnagi.2020.524369 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 145. Juopperi TA, Kim WR, Chiang CH, et al. : Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington's disease patient cells. Mol Brain. 2012; 5: 17. 10.1186/1756-6606-5-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Shubin AV, Demidyuk IV, Komissarov AA, et al. : Cytoplasmic vacuolization in cell death and survival. Oncotarget. 2016; 7(34): 55863–89. 10.18632/oncotarget.10150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Halevy T, Urbach A: Comparing ESC and iPSC-Based Models for Human Genetic Disorders. J Clin Med. 2014; 3(4): 1146–62. 10.3390/jcm3041146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Choi J, Lee S, Mallard W, et al. : A comparison of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol. 2015; 33(11): 1173–81. 10.1038/nbt.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Marei HE, Althani A, Lashen S, et al. : Genetically unmatched human iPSC and ESC exhibit equivalent gene expression and neuronal differentiation potential. Sci Rep. 2017; 7(1): 17504. 10.1038/s41598-017-17882-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zhao MT, Chen H, Liu Q, et al. : Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc Natl Acad Sci U S A. 2017; 114(52): E11111–E11120. 10.1073/pnas.1708991114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Ruzo A, Croft GF, Metzger JJ, et al. : Chromosomal instability during neurogenesis in Huntington's disease. Development. 2018; 145(2): dev156844. 10.1242/dev.156844 [DOI] [PubMed] [Google Scholar]
- 152. Ooi J, Langley SR, Xu X, et al. : Unbiased Profiling of Isogenic Huntington Disease hPSC-Derived CNS and Peripheral Cells Reveals Strong Cell-Type Specificity of CAG Length Effects. Cell Rep. 2019; 26(9): 2494–2508.e7. 10.1016/j.celrep.2019.02.008 [DOI] [PubMed] [Google Scholar]
- 153. Lacoste A, Berenshteyn F, Brivanlou AH: An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell. 2009; 5(3): 332–42. 10.1016/j.stem.2009.07.011 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 154. Conforti P, Besusso D, Brocchetti S, et al. : RUES2 hESCs exhibit MGE-biased neuronal differentiation and muHTT-dependent defective specification hinting at SP1. Neurobiol Dis. 2020; 146: 105140. 10.1016/j.nbd.2020.105140 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 155. Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. : Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282(5391): 1145–7. 10.1126/science.282.5391.1145 [DOI] [PubMed] [Google Scholar]
- 156. O'Regan GC, Farag SH, Casey CS, et al. : Human Huntington's disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J Neuroinflammation. 2021; 18(1): 94. 10.1186/s12974-021-02147-6 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 157. Victor MB, Richner M, Olsen HE, et al. : Striatal neurons directly converted from Huntington's disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat Neurosci. 2018; 21(3): 341–52. 10.1038/s41593-018-0075-7 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 158. Vierbuchen T, Ostermeier A, Pang ZP, et al. : Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010; 463(7284): 1035–41. 10.1038/nature08797 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 159. Horvath S: DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14(10): R115. 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 160. Linsley JW, Reisine T, Finkbeiner S: Cell death assays for neurodegenerative disease drug discovery. Expert Opin Drug Discov. 2019; 14(9): 901–13. 10.1080/17460441.2019.1623784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Burrows CK, Banovich NE, Pavlovic BJ, et al. : Genetic Variation, Not Cell Type of Origin, Underlies the Majority of Identifiable Regulatory Differences in iPSCs. PLoS Genet. 2016; 12(1): e1005793. 10.1371/journal.pgen.1005793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. DeBoever C, Li H, Jakubosky D, et al. : Large-Scale Profiling Reveals the Influence of Genetic Variation on Gene Expression in Human Induced Pluripotent Stem Cells. Cell Stem Cell. 2017; 20(4): 533–546.e7. 10.1016/j.stem.2017.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 163. Kilpinen H, Goncalves A, Leha A, et al. : Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017; 546(7658): 370–5. 10.1038/nature22403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Kyttälä A, Moraghebi R, Valensisi C, et al. : Genetic Variability Overrides the Impact of Parental Cell Type and Determines iPSC Differentiation Potential. Stem Cell Reports. 2016; 6(2): 200–12. 10.1016/j.stemcr.2015.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 165. Volpato V, Webber C: Addressing variability in iPSC-derived models of human disease: Guidelines to promote reproducibility. Dis Model Mech. 2020; 13(1): dmm042317. 10.1242/dmm.042317 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 166. Jinek M, Chylinski K, Fonfara I, et al. : A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012; 337(6096): 816–21. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 167. Boch J, Scholze H, Schornack S, et al. : Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009; 326(5959): 1509–12. 10.1126/science.1178811 [DOI] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 168. Adli M: The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018; 9(1): 1911. 10.1038/s41467-018-04252-2 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 169. Hendriks WT, Warren CR, Cowan CA: Genome Editing in Human Pluripotent Stem Cells: Approaches, Pitfalls, and Solutions. Cell Stem Cell. 2016; 18(1): 53–65. 10.1016/j.stem.2015.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Dabrowska M, Ciolak A, Kozlowska E, et al. : Generation of New Isogenic Models of Huntington's Disease Using CRISPR-Cas9 Technology. Int J Mol Sci. 2020; 21(5): 1854. 10.3390/ijms21051854 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 171. Le Cann K, Foerster A, Rösseler C, et al. : The difficulty to model Huntington's disease in vitro using striatal medium spiny neurons differentiated from human induced pluripotent stem cells. Sci Rep. 2021; 11(1): 6934. 10.1038/s41598-021-85656-x [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 172. Noguchi H, Miyagi-Shiohira C, Nakashima Y: Induced Tissue-Specific Stem Cells and Epigenetic Memory in Induced Pluripotent Stem Cells. Int J Mol Sci. 2018; 19(4): 930. 10.3390/ijms19040930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kim K, Zhao R, Doi A, et al. : Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat Biotechnol. 2011; 29(12): 1117–9. 10.1038/nbt.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Scesa G, Adami R, Bottai D: iPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells. 2021; 10(6): 1470. 10.3390/cells10061470 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 175. Bar S, Benvenisty N: Epigenetic aberrations in human pluripotent stem cells. EMBO J. 2019; 38(12): e101033. 10.15252/embj.2018101033 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 176. Churko JM, Lee J, Ameen M, et al. : Transcriptomic and epigenomic differences in human induced pluripotent stem cells generated from six reprogramming methods. Nat Biomed Eng. 2017; 1(10): 826–37. 10.1038/s41551-017-0141-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Kaye J, Reisine T, Finkbeiner S: Huntington's disease mouse models: Unraveling the pathology caused by CAG repeat expansion. Fac Rev. 2021; 10: 77. 10.12703/r/10-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Christiansen EM, Yang SJ, Ando DM, et al. : In Silico Labeling: Predicting Fluorescent Labels in Unlabeled Images. Cell. 2018; 173(3): 792–803.e19. 10.1016/j.cell.2018.03.040 [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Opinions Recommendation
- 179. Linsley JW, Shah K, Castello N, et al. : Genetically encoded cell-death indicators (GEDI) to detect an early irreversible commitment to neurodegeneration. Nat Commun. 2021; 12(1): 5284. 10.1038/s41467-021-25549-9 [DOI] [PMC free article] [PubMed] [Google Scholar]