

Summary
Understanding which biological pathways are specific versus general across diagnostic categories and levels of symptom severity is critical to improving nosology and treatment of psychopathology. Here, we combine transdiagnostic and dimensional approaches to genetic discovery for the first time, conducting a novel multivariate genome-wide association study of eight psychiatric symptoms and disorders broadly related to mood disturbance and psychosis. We identify two transdiagnostic genetic liabilities that distinguish between common forms of psychopathology versus rarer forms of serious mental illness. Biological annotation revealed divergent genetic architectures that differentially implicated prenatal neurodevelopment and neuronal function and regulation. These findings inform psychiatric nosology and biological models of psychopathology, as they suggest that the severity of mood and psychotic symptoms present in serious mental illness may reflect a difference in kind rather than merely in degree.
Keywords: psychiatric genetics, psychiatric disorders, transdiagnostic, genome-wide association study, genetic correlation, pleiotropy, genomics, neurodevelopment
Graphical abstract
Highlights
-
•
Identification of two dimensions of genetic risk in mood and psychotic psychopathology
-
•
Pleiotropic genes broadly implicate neuronal pathways in psychopathology
-
•
Dimensions of genetic risk differ in their associations with health and disease
Mallard et al. identified two transdiagnostic dimensions of genetic risk in a large genome-wide association study of psychiatric phenotypes related to mood disturbance and psychosis. While these genetic risk factors were modestly correlated, they were largely unique from one another and differed in their relationships with health and wellbeing.
Introduction
Psychiatric disorders are one of the leading causes of global disease burden, affecting more than 25% of the world’s population at some point during their lifetime.1 Twin- and family-based studies have established that a substantial portion of individual differences in liability to psychiatric disorders is attributable to genetic variation.2 Genome-wide association studies (GWASs) have identified numerous genetic loci that have replicable associations with severe and debilitating psychiatric disorders, including schizophrenia,3 bipolar disorder,4 and major depressive disorder.5
GWASs have also identified a substantial degree of genetic overlap across psychiatric disorders, finding high genetic covariances and many pleiotropic loci.6,7 This genetic overlap complicates efforts to identify causes, consequences, and treatments that are specific to any individual psychiatric disorder.8 In response to these challenges, transdiagnostic approaches in psychiatry aim to identify biological systems that are perturbed across many forms of illness.9,10 Transdiagnostic research may yield new therapeutic targets with broad utility as well as inform nosological classification and stratification of at-risk populations.
Concurrent with the emergence of transdiagnostic research, efforts to identify disorder-specific genetic loci have turned toward studying self-report measures in population-based cohorts,11, 12, 13 as case-control study designs require diagnostic schedules that can be slow and costly. If valid, this dimensional approach in non-clinical samples has the potential to accelerate genetic discovery via dramatic increases in sample size, as self-report survey measures of psychiatric symptoms can be administered at scale to large, genotyped population-based samples, such as UK Biobank.14,15 However, while this approach may be valid for some common forms of psychopathology,16 it is unknown whether the biology that influences normative variation in subthreshold symptoms also underlies rarer psychiatric conditions, such as those characterized by mania and/or psychosis.
Here, we combine transdiagnostic and dimensional research approaches to genetic discovery in two important ways. First, we use a novel combination of Bayesian item response theory and linear mixed models to perform GWASs of depressive (DEP), manic (MAN), and psychotic (PSY) symptoms on more than 250,000 individuals in the UK Biobank. This approach has been shown to yield higher heritability estimates than single-item measures or simple composite measures.17,18 Second, we used genomic structural equation modeling (Genomic SEM)19 to characterize the shared genetic architecture among these three symptom dimensions and five additional psychiatric disorders: major depressive disorder (MDD), bipolar II disorder (BD2), bipolar I disorder (BD1), schizoaffective disorder (SZA), and schizophrenia (SCZ).
These analyses yield insight into three conceptual questions in the biological study of psychopathology. First, how do the genetic bases of mood and psychotic symptoms compare and contrast with the genetic bases of psychiatric diagnoses that are characterized by those symptoms? Second, to what extent can the shared genetic architecture of these symptoms and disorders be summarized with a single dimension of liability, a “p factor,” as has been previously proposed on the basis of phenotypic analyses?20 Third, how are dimensions of transdiagnostic liability similar and dissimilar in their genetic architecture, underlying biology, and associations with other aspects of human wellbeing and disease?
Results
Novel loci associated with lifetime endorsement of mood and psychotic symptoms
We used a combination of Bayesian item response theory and linear mixed models to conduct univariate GWASs for self-reported measures of lifetime depression, mania, and psychosis from 252,252 individuals in the UK Biobank. We observed substantial inflation of the median test statistic for all three phenotypes, and the linkage disequilibrium (LD) score regression intercepts and attenuation ratios suggest that test-statistic inflation is primarily due to polygenic signal rather than bias (Figure 1; Table 1). After applying a standard clumping algorithm via FUMA (r2 = 0.1, 250 kb merge window), we identified 23 independent loci associated with lifetime depressive, manic, and/or psychotic symptoms (Tables S1A–S1C). Nine of these loci were significantly associated with two or more phenotypes, and six loci were associated with all three psychiatric phenotypes.
Figure 1.

Univariate association results for lifetime measures of mood disturbance and psychosis
(A–C) Manhattan plots and a quantile-quantile plots for (A) depressive, (B) manic, and (C) psychotic symptoms. In the Manhattan plots, the x axis refers to chromosomal position, the y axis refers to the significance on a -log10 scale, the horizontal dashed line denotes genome-wide significance (p = 5 × 10−8), and the horizontal dotted line marks suggestive significance (p = 5 × 10−5). In the quantile-quantile plots, the x axis refers to expected p value, while the y axis refers to the observed p value. For each plot, the nearest gene for the lead SNP in the top five genome-wide significant loci is labeled.
Table 1.
Summary of study phenotypes
| GWAS (abbr.) | Source | N | h2 | λGC | Mean χ2 | Intercept | Ratio |
|---|---|---|---|---|---|---|---|
| Depressive symptoms (DEP) | present study | 252,252 | 0.08 | 1.31 | 1.38 | 1.01 | 0.02 |
| Manic symptoms (MAN) | present study | 252,252 | 0.08 | 1.31 | 1.39 | 1.00 | 0.00 |
| Psychotic symptoms (PSY) | present study | 252,252 | 0.07 | 1.31 | 1.33 | 1.00 | 0.01 |
| Major depressive D/O (MDD) | Wray et al., 20185 | 138,884 | 0.10 | 1.19 | 1.20 | 1.00 | <0 |
| Bipolar II D/O (BD2) | Stahl et al., 20194 | 25,576 | 0.10 | 1.07 | 1.08 | 1.03 | 0.42 |
| Bipolar I D/O (BD1) | Stahl et al., 20194 | 45,871 | 0.22 | 1.31 | 1.37 | 1.04 | 0.09 |
| Schizoaffective D/O (SZA) | Stahl et al., 20194 | 9,667 | 0.27 | 1.06 | 1.06 | 1.02 | 0.35 |
| Schizophrenia (SCZ) | Ruderfer et al., 201821 | 65,967 | 0.23 | 1.49 | 1.63 | 1.05 | 0.08 |
| F1 (mood disturbance) | present study | 377,518 | N/A | 1.44 | 1.53 | 1.05 | 0.10 |
| F2 (serious mental illness) | present study | 51,276 | N/A | 1.46 | 1.61 | 1.02 | 0.04 |
Heritability (h2) was estimated using LD score regression. λGC refers to the median χ2 statistic of the GWAS divided by the expected median of the χ2 distribution with 1 degree of freedom. Mean χ2 refers to the average χ2 statistic of the GWAS. Intercept refers to the estimated intercept from univariate LD score regression. Ratio refers to a measure of stratification bias that is defined as (Intercept – 1)/(Mean χ2 − 1). To harmonize measurement approaches among psychiatric disorders, summary statistics for MDD were obtained for the clinically ascertained cohorts, excluding 23andMe and UK Biobank. D/O, disorder; The effective sample size is reported for F1 and F2 (STAR Methods).
The identified risk loci span 12 chromosomes and include variants tagging the major histocompatibility complex region on chromosome 6 as well a well-known inversion polymorphism on chromosome 17 previously associated with several psychiatric phenotypes.22 Many of these risk loci replicated previous findings from GWASs of psychopathology or were in high LD with previous hits for phenotypes including neuroticism23 (e.g., rs7111031, rs10503002, rs4245154), broadly defined depression11 (e.g., rs9586, rs191800971, rs7111031), and schizophrenia24 (e.g., rs4245154, rs4702). However, several loci contained lead SNPs that were new GWAS signals altogether, identifying new regions of the genome that confer risk for psychopathology, such as rs4722389, rs7324564, and rs570217967.
Moreover, our gene-based association analyses performed via MAGMA identified 124 genes associated with at least one of the psychiatric symptoms (depression, mania, or psychosis), 31 of which were associated with all three. For all phenotypes, we observed enriched expression in brain tissue as well as an enriched signal for brain-related gene sets. We report detailed biological annotation (e.g., gene mapping, gene set enrichment, tissue enrichment) for each of these GWASs in Tables S1D–S1L.
Two transdiagnostic genetic liabilities underlie mood and psychotic psychopathology
To describe the genomic relationships among psychiatric symptoms and disorders commonly characterized by depression, mania, and/or psychosis4,5,21 (see Table 1 for overview of study phenotypes), we first used bivariate LD score regression to estimate genetic correlations between all pairs of psychiatric phenotypes. While we observed very large positive genetic correlations among the three psychiatric symptoms (mean rg = 0.95, SEM = 0.02), we observed more modest genetic correlations for the five psychiatric disorders (mean rg = 0.55, SEM = 0.09). We found that schizophrenia, schizoaffective disorder, and bipolar I were highly correlated with one another (rgSCZ-SZA = 0.87 [SE = 0.13], rgSCZ-BD1 = 0.72 [SE = 0.03], rgSZA-BD1 = 0.81 [SE = 0.12]), but these disorders generally had markedly smaller genetic correlations with bipolar II and major depressive disorder (rgSCZ-BD2 = 0.53 [SE = 0.03], rgSCZ-MDD = 0.39 [SE = 0.04], rgSZA-BD2 = 0.28 [SE = 0.21], rgSZA-MDD = 0.06 [SE = 0.12], rgBD1-MDD = 0.33 [SE = 0.04]). Bipolar I and bipolar II were highly correlated, though (rgBD1-BD2 = 0.88 [SE = 0.11]). Interestingly, we found that bipolar II and major depressive disorder were also highly correlated with each other (rgBD2-MDD = 0.69 [SE = 0.13]) as well as with all psychiatric symptoms (rgBD2-DEP = 0.75 [SE = 0.11], rgBD2-MAN = 0.71 [SE = 0.11], rgBD2-PSY = 0.70 [SE = 0.11], rgMDD-DEP = 0.85 [SE = 0.03], rgMDD-MAN = 0.77 [SE = 0.03], rgMDD-PSY = 0.80 [SE = 0.04]). Notably, many of these genetic correlations differed from the phenotypic correlations observed in UK Biobank (Figure S1).
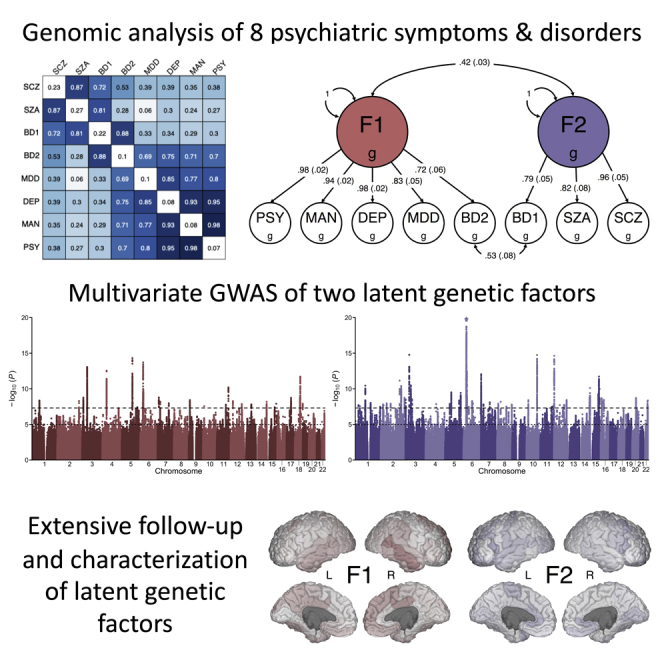
After applying a hierarchical-clustering algorithm to the genetic-correlation matrix, we found two distinct clusters of psychiatric phenotypes (Figure 2A). The first cluster comprised the three psychiatric symptoms, major depressive disorder, and bipolar II, and the second cluster comprised bipolar I, schizoaffective disorder, and schizophrenia. We then conducted an exploratory factor analysis (EFA) of the genetic covariance matrix, which produced results that were consistent with the groupings suggested by the hierarchical clustering algorithm. The correlated two-factor model with approximate simple structure suggested that phenotypes principally loaded onto one of two latent genetic factors with negligible cross-loadings (Figure 2B). Combined, these two correlated latent factors explained 81.3% of the total SNP-based genetic variances across phenotypes.
Figure 2.

Relationships between eight psychiatric symptoms and disorders
(A) Matrix of bivariate genetic-correlation estimates, where the diagonal elements correspond to SNP h2 and the off-diagonal elements correspond to genetic correlations. Estimates that are non-significant are crossed out.
(B) Scatterplot of standardized factor loadings from the exploratory factor analysis.
(C) Path diagram for the final confirmatory factor model with standardized parameter estimates.
Finally, we formally modeled the genetic covariance matrix via confirmatory factor analysis (CFA). We based our model on the EFA results, which consisted of two correlated latent factors, F1 and F2. F1 can be conceptualized as capturing common psychopathology related to mood disturbance (including self-reported depressive, psychotic, and manic symptoms, as well as bipolar II and major depressive disorder), while F2 can be conceptualized as capturing rarer forms of serious mental illness (bipolar I, schizoaffective disorder, and schizophrenia). We did not estimate any cross-loadings. Instead, we estimated correlated residuals between bipolar I and bipolar II, as inspection of the genetic correlation matrix suggested a unique relationship between these disorders. The path diagram for this model is presented in Figure 2C.
We compared the correlated factor model with a common factor model, where all phenotypes were indicators of a single latent factor (i.e., a p factor) (Section 1.1 of supplemental methods; Figure S2). Briefly, we found that the common factor model showed suboptimal fit to the data, while the correlated factors model with correlated residuals for BD1 and BD2 showed excellent fit (Figure S3). Fit indices from the CFA indicated that the correlated factors model closely approximated the observed genetic covariance matrix (χ2(18) = 496.16, Akaike information criterion = 532.16, comparative fit index = 0.99, standardized root mean square residual = 0.06). That is, the patterns of covariance among the eight psychiatric phenotypes were most parsimoniously represented by two transdiagnostic latent factors at the genetic level, which were correlated only modestly (rg = 0.42; SE = 0.03). This is a notable divergence from the factor structure frequently observed at the phenotypic level, including that seen with similar phenotypes in the UK Biobank (Section 1.1 of supplemental methods). However, we note that a direct comparison of phenotypic and genetic factor structure cannot be performed in the UK Biobank due to an insufficient number of clinical cases.
Transdiagnostic factors have markedly divergent genetic architectures
We then conducted a multivariate GWAS of the two latent genetic factors, F1 ( = 377,518) and F2 ( = 51,276). The results of these analyses are summarized in Table 1 and Figure 3. Briefly, we observed substantial inflation of the median test statistic for both F1 (λGC = 1.44, mean χ2 = 1.53) and F2 (λGC = 1.46, mean χ2 = 1.61), which is indicative of a robust polygenic signal for both factors (Figure S4). The LD score regression intercepts and attenuation ratios for F1 (intercept = 1.05, SE = 0.01; ratio = 0.10, SE = 0.02) and F2 (intercept = 1.02, SE = 0.01; ratio = 0.04, SE = 0.02) suggest that test-statistic inflation is primarily due to polygenic signal rather than bias.
Figure 3.

Multivariate association results for the two transdiagnostic latent genetic factors
(A and B) Miami plots for (A) F1 and (B) F2. The top of each Miami plot corresponds to the significance of SNP effects on each latent factor, as traditionally conveyed in a Manhattan plot, while the bottom corresponds to the significance of heterogeneity tests for SNP effects (QSNP; i.e., the degree to which SNP effects are not mediated by F1 or F2). For each plot, the x axis refers to chromosomal position, the y axis refers to the significance on a -log10 scale, the horizontal dashed line denotes genome-wide significance (p = 5 × 10−8), and the horizontal dotted line marks suggestive significance (p = 5 × 10−5). For each plot, the nearest gene for the lead SNP in the top five genome-wide significant loci is labeled.
(C and D) UpSet plots illustrating the intersection of the five gene-mapping methods, ranked by degree of overlap.
After applying a standard clumping algorithm, we identified 26 and 59 independent loci associated with F1 and F2, respectively (Table 2; Tables S2A and S2B). Only five loci were associated with both factors. While many of these genomic regions have been previously identified in either the constituent GWASs or related studies, several contain novel discoveries. For example, several loci associated with F1 contain lead SNPs that have not been previously associated with psychopathology, such as rs13153844 (p = 2.09 × 10−9, nearest gene = PSMC1P5), rs1551765 (p = 3.89 × 10−8, nearest gene = GRIA1), rs147584788 (p = 1.08 × 10−8, nearest gene = AC003088.1), and rs8035987 (p = 3.94 × 10−8, nearest gene = SIN3A). Several loci associated with F2 also contain lead SNPs that were also novel risk variants for psychopathology, including rs2953329 (p = 3.27 × 10−8, nearest gene = AKT3), rs10199182 (p = 1.56 × 10−8, nearest gene = AC068490.2), rs9463650 (p = 3.34 × 10−8, nearest gene = RPS17P5), rs11603014 (p = 2.32 × 10−8, nearest gene = RP11-890B15.2), rs10777957 (p = 1.79 × 10−8, nearest gene = ANKS1B), and rs11064837 (p = 2.43 × 10−8, nearest gene = RP11-768F21.1).
Table 2.
Lead SNPs for the top ten loci per latent factor from multivariate association analyses
| Lead SNP | CHR:BP | A1 | A2 | MAF | Z | p | Nearest gene | Function | CADD | RDB |
|---|---|---|---|---|---|---|---|---|---|---|
| F1 (depressive symptoms, manic symptoms, psychotic symptoms, major depressive disorder, and bipolar II disorder) | ||||||||||
| rs30266 | 5:103972357 | G | A | 0.32 | −7.83 | 4.94 × 10−15 | RP11-6N13.1 | ncRNA intronic | 2.275 | N/A |
| rs148682985 | 6:29288001 | G | A | 0.03 | −7.66 | 1.93 × 10−14 | DDX6P1 | intergenic | 2.643 | 5 |
| rs9586 | 3:49213637 | C | T | 0.02 | 7.46 | 8.80 × 10−14 | KLHDC8B | UTR3 | 11.63 | N/A |
| rs28656217 | 4:42099424 | T | C | 0.16 | 7.30 | 2.85 × 10−13 | SLC30A9 | intergenic | 4.861 | 6 |
| rs67526282 | 18:53471187 | T | C | 0.33 | −7.04 | 1.97 × 10−12 | RP11-397A16.3 | intergenic | 7.656 | 6 |
| rs7934649 | 11:113372671 | C | T | 0.36 | −6.53 | 6.50 × 10−11 | DRD2 | intergenic | 1.426 | 7 |
| rs17410557 | 18:50776391 | T | C | 0.38 | −6.04 | 1.52 × 10−9 | DCC | intronic | 4.502 | 7 |
| rs3807866 | 7:12250378 | G | A | 0.40 | −6.03 | 1.69 × 10−9 | TMEM106B | upstream | 7.544 | N/A |
| rs184262 | 3:12134740 | A | G | 0.15 | 6.02 | 1.77 × 10−9 | SYN2 | ncRNA intronic | 6.559 | 7 |
| rs2696673 | 17:44315803 | A | C | 0.22 | −6.01 | 1.89 × 10−9 | RP11-259G18.2 | intergenic | 3.3 | N/A |
| F2 (bipolar I disorder, schizoaffective disorder, and schizophrenia) | ||||||||||
| rs7746199 | 6:27261324 | C | T | 0.17 | 9.34 | 9.95 × 10−21 | POM121L2 | intronic | 0.879 | 1f |
| rs9834970 | 3:36856030 | T | C | 0.49 | −7.96 | 1.67 × 10−15 | TRANK1 | intergenic | 11.17 | 4 |
| rs12764899 | 10:104635103 | G | A | 0.23 | 7.95 | 1.82 × 10−15 | C10orf32-ASMT:AS3MT | intronic | 1.133 | 7 |
| rs4298967 | 12:2408194 | A | G | 0.34 | 7.92 | 2.37 × 10−15 | CACNA1C | intronic | 10.73 | 5 |
| rs6461049 | 7:2017445 | C | T | 0.44 | −7.15 | 8.80 × 10−13 | MAD1L1 | intronic | 0.914 | 5 |
| rs12902973 | 15:85105982 | G | C | 0.28 | 7.04 | 1.89 × 10−12 | UBE2Q2P1 | ncRNA intronic | 1.808 | 7 |
| rs4380187 | 2:185811940 | A | C | 0.45 | 6.86 | 6.95 × 10−12 | ZNF804A | intergenic | 2.923 | 7 |
| rs2535627 | 3:52845105 | T | C | 0.50 | 6.63 | 3.36 × 10−11 | ITIH4 | intergenic | 5.307 | N/A |
| rs1198588 | 1:98552832 | A | T | 0.23 | −6.62 | 3.65 × 10-11 | NFU1P2 | intergenic | 1.829 | 3a |
| rs11693528 | 2:200736507 | C | G | 0.18 | −6.60 | 4.18 × 10−11 | AC073043.1 | ncRNA intronic | 2.344 | 6 |
Results for all lead SNPs are presented in Tables S2A and S2B. Lead SNPs refer to approximately independent lead SNPs identified via FUMA. CHR:BP refers to genomic location of the lead SNP, specifically the chromosome and base pair location on that chromosome. A1 and A2 refer to the alleles for that SNP. MAF, minor allele frequency; CADD, Combined Annotation Dependent Depletion score; RDB, RegulomeDB score.
Tests of heterogeneity suggested that the majority of observed SNP effects operate via the latent factors (i.e., associated SNPs primarily had consistent, pleiotropic effects on the constituent phenotypes). Indeed, QSNP tests identified no heterogeneous loci for F1 and only three heterogeneous loci for F2 with lead SNPs rs11696888 on chromosome 20 (QSNP p = 2.04 × 10−8; nearest gene = STAU1), rs1990042 on chromosome 7 (QSNP p = 2.10 × 10−8, nearest gene = AC004854.1), and rs3764002 on chromosome 12 (QSNP p = 2.70 × 10−8; nearest gene = WSCD2). Interestingly, the heterogeneous locus with lead SNP rs11696888 also contains rs200005157, which is a four base pair insertion/deletion that was previously identified as a locus with divergent effects on bipolar disorder and schizophrenia.21 Fine mapping conducted by Ruderfer and colleagues identified CSE1L as a plausible causal gene with divergent effects for bipolar disorder and schizophrenia on chromosome 20.
Transdiagnostic factors are related to different aspects of neurobiology
To characterize the effects of variants associated with the transdiagnostic factors of psychopathology, we used FUMA to conduct a series of gene-mapping analyses. Specifically, we used positional mapping to align lead SNPs to genes based on genomic location, expression quantitative trait loci (eQTL) mapping to match cis-eQTL SNPs to genes whose expression they affect, and chromatin-interaction mapping to link SNPs to genes on the basis of three-dimensional DNA-DNA interactions. These three methods linked the associated SNPs for F1 and F2 to a combined 287 and 570 putative risk genes, respectively (Tables S2C–S2J). We then used MAGMA to conduct gene-based association analyses, which identified 115 and 243 genes associated with F1 and F2 (Tables S2K and S2L). Finally, we used S-Multi-Xcan to identify 50 and 91 genes associated with differential expression levels in brain tissue for F1 and F2, respectively (Tables S2M and S2N).
Collectively, these five approaches mapped a total of 332 genes to F1 and 710 genes to F2 (Figure 3). Only 159 of these putative risk genes were related to both factors. Focusing on genes implicated across all five methods, we found 11 genes robustly linked to F1 and 16 to F2 (Figure 3). Two genes (PGBD1 and XRCC3) were related to both factors across all analyses. This limited overlap in mapped genes underscores the unique genetic architecture of each factor.
The modest genetic correlation between F1 and F2 (rg = 0.42, SE = 0.03) implies that the majority of SNP-based genetic variance in each factor is unique from the other. To further characterize the shared and unique genetic architecture of these factors, we used HESS to estimate the local genetic covariance for 1,698 contiguous, similarly sized partitions across the genome. We found that approximately 27% of the genome explains 80% of the total genetic covariance between F1 and F2 and that only 15 genomic partitions share a significant local genetic correlation after correcting for multiple comparisons (Figure 4A).
Figure 4.

Biological annotation of the two transdiagnostic latent genetic factors
(A) Manhattan plots for local genetic correlation, covariance, and variance for F1 and F2. Black bars indicate significant local genetic correlation.
(B) Scatterplot of gene set enrichment results illustrating convergence and divergence across the latent genetic factors with accompanying histograms for the top 10 gene sets for each factor.
(C) Scatterplot of neuroimaging genetic-correlation results with accompanying figures where the −log10 p values are mapped across the cortex, as parcellated in the Desikan-Killiany-Tourville atlas.
(D) Smoothed line plots of gene set expression across developmental time in the PsychENCODE dataset for prioritized genes with transcriptomic profiles that are spatially similar to the neuroimaging genetic correlation maps for F1 and F2 (as indexed in the Allen Human Brain Atlas). For all plots, the dashed black line corresponds to the Bonferroni-corrected significance threshold when applicable.
Gene set enrichment and gene property (i.e., tissue expression) analyses further suggest that the genetic architectures of F1 and F2 are divergent at more granular levels of analysis, converging only at higher levels. While results from gene set enrichment analyses broadly implicated neurodevelopmental and neurobiological pathways for both factors, the specific molecular functions, cellular components, and biological processes tended to differ (Figure 4B; Tables S3A and S3B). For example, gene sets related to neurons were enriched for F1 and F2, but gene sets for specific parts of neurons were differentially enriched (e.g., the axon for F1 versus the somatodendritic compartment for F2). Similarly, in the tissue-expression analysis, we found that the brain was broadly implicated in the pathogenesis of psychopathology, as nearly all brain-related tissues were enriched for both F1 and F2 (Tables S3C and S3D). At the level of brain tissue, the only regions with divergent effects were the substantia nigra and brainstem, which were not significantly enriched for F1 after correction for multiple comparisons. However, shortcomings of these analyses include the relatively low spatial resolution of brain-related gene expression data and the limited sample size of the underlying data.
Therefore, to gain greater insight into potential etiological relationships between psychopathology and neurobiology, we estimated genetic correlations between the transdiagnostic factors of psychopathology and 101 morphological features of the human brain. Although we generally observed negative genetic correlations with cortical and subcortical features (i.e., greater risk for psychopathology was associated with smaller volumes across the brain) and positive with ventricular features (i.e., greater risk for psychopathology was associated with larger ventricular volumes), specific estimates between morphological features and F1 and F2 showed relatively little concordance (Figure 4C). After correcting for multiple comparisons, only the genetic correlation between F1 and the right middle temporal gyrus remained statistically significant (rg = −0.15, SE = 0.04, p = 3.98 × 10−4) (Tables S3E and S3F).
We then used data from the Allen Human Brain Atlas to identify genes with transcriptomic profiles that were spatially similar to the neuroimaging genetic correlation maps for F1 and F2 (Tables S3G and S3H). Notably, these transcriptomically prioritized gene sets for F1 and F2 were entirely disjointed from one another and differentially expressed in pre-and postnatal cortical tissue from the PsychENCODE dataset (Figure 4D). We found that the developmental expression profile of the F1 gene set most closely resembled that of postnatal inhibitory neuronal genes, while the developmental expression profile of the F2 gene set most closely resembled that of prenatal inhibitory neuronal genes25,26 (Figure S5). We note that the F1 and F2 gene sets also resemble postnatal microglia and prenatal neural progenitor cells, respectively, although to a lesser extent.
Transdiagnostic factors are differentially associated with human health and wellbeing
To better understand how these transdiagnostic genetic liabilities may manifest above and beyond their constituent phenotypes, we conducted a series of genetic correlation and polygenic prediction analyses focused on theoretically relevant phenotypes. In the genetic correlation analyses, we evaluated the relationships between the latent factors of psychopathology and 92 phenotypes broadly related to four broad domains of human health and wellbeing (Figure 5A; Table S4A). We found that genetic correlation estimates for F1 and F2 were moderately correlated across all broad domains (r = 0.60, p = 2.77 × 10−10) as well as within each of the four domains: demography and socioeconomic status (r = 0.55, p = 1.17 × 10−2), health and disease (r = 0.42, p = 1.17 × 10−2), personality and risky behavior (r = 0.60, p = 3.05 × 10−3), and psychopathology and cognition (r = 0.63, p = 1.57 × 10−2). Generally, we found that F1 was more consistently correlated with phenotypes typically related to psychopathology than F2. This pattern was also observed in the partial genetic correlation analyses, where we found strong evidence of divergent genetic correlations after accounting for the overlap between F1 and F2 (Figure 5B; Table S4B). Indeed, partial genetic correlation estimates for F1 and F2 were negatively correlated across all domains (r = −0.43, p = 2.72 × 10−5).
Figure 5.

Genetic-correlation and phenome-wide association results for the two transdiagnostic latent genetic factors
(A) Scatterplot of genetic correlations (rg) with marginal histograms.
(B) Scatterplot of partial genetic correlations (bg) with marginal histograms. For both plots, phenotypes are grouped into one of four broad domains: (1) demography and socioeconomic status, (2) health and disease, (3) personality and risky behavior, and (4) psychopathology and cognition. A line of best fit (with 95% confidence interval) is fit for all 92 data points. Points are colored burgundy if significant only for F1, violet if significant only for F2, black if significant for both, and faded gray if non-significant for both. The standard errors (SEs) for point estimates are plotted for both factors.
(C) Rotated Miami plot for (left) F1 and (right) F2, where the y axis refers to the ICD-10 code category, the x axis refers to the significance on a −log10 scale, the vertical light red line denotes phenome-wide significance (p = 3.27 × 10−5) following Bonferroni correction, and the vertical light blue line marks nominal significance (p = 0.05). The direction of the triangle refers to the direction of effect. Phecodes closely resembling Genomic SEM model phenotypes are bolded and italicized for emphasis.
In the polygenic prediction analyses, we used electronic health records from the Vanderbilt University Medical Center biobank (BioVU) to evaluate the penetrance and pleiotropy of genetic risk for the transdiagnostic factors of psychopathology across 1,335 disease phenotypes, hereby referred to as “phecodes” (Figure 5C; Tables S4C and S4D). We found that polygenic scores for F1 and F2 were generally associated with all of the constituent phenotypes for both factors, but F1 was more strongly associated with mood-related phecodes, while F2 was more strongly associated with psychosis-related phecodes. Both polygenic scores for F1 and F2 shared associations with some forms of psychopathology (e.g., suicidality, posttraumatic stress disorder, substance-use disorders, and anxiety disorders) but diverged in their associations with others (e.g., personality disorders, paranoid disorders). Beyond psychopathology, F1 was more consistently associated a variety of medical phecodes, including those related to infectious diseases (e.g., viral hepatitis, HIV disease) and pervasive developmental disorders as well as diseases of the circulatory, digestive, endocrine, genitourinary, musculoskeletal, and respiratory systems.
Discussion
By jointly analyzing genome-wide data for eight psychiatric disorders and symptoms in a novel multivariate framework, we identified two distinct transdiagnostic factors that distinguished common forms of psychopathology related to mood disturbance versus rare forms of serious mental illness. Together, these factors explained approximately 80% of the SNP-based genetic variance in mood and psychotic psychopathology but were themselves only moderately correlated. Extensive biological annotation of these two transdiagnostic factors revealed clear differences between their factors in their underlying genetic architecture and biology. Further follow-up analyses highlighted additional differences between the factors in their associations with human wellbeing and disease. Our results provide four critical insights into the genetic architecture of forms of psychopathology characterized by mood disturbance and psychosis.
First, we built on genomic investigations of the dimensional structure of certain forms of psychopathology, such as a large-scale study of the mood-disorder spectrum,27 and identified two transdiagnostic factors that explain the vast majority of SNP-based genetic variance in their constituent phenotypes. Perhaps surprisingly, variation in self-reported manic and psychotic symptoms is much more closely related to common forms of mood psychopathology (self-reported depressive symptoms, major depressive disorder, bipolar II disorder) than to psychiatric disorders characterized by severe mood disturbance and/or psychosis. These findings extend those of an earlier GWAS of psychotic experiences,28 which also reported stronger genetic overlap with major depressive disorder than bipolar disorder or schizophrenia. Here, we also find that the factor structure at the genetic level is different than the factor structure that we observe at the phenotypic level in the UK Biobank with similar indicators. This finding contrasts with what has been called the “phenotypic null hypothesis,” which states that genetic and phenotypic factor structures are expected to converge.29 Overall, these results illustrate how diagnostic boundaries, which are known to be problematic based on widespread phenotypic comorbidity, become even fuzzier at the genetic level of analysis.
Second, our multivariate association analyses identified 80 approximately independent loci associated with one of the transdiagnostic factors. Many of these genome-wide significant loci contain novel lead SNPs and map to genes that have not been previously associated with mood or psychotic psychopathology, such as SIN3A, which has been reported to be a key transcriptional regulator of cortical neurodevelopment, involved in neurogenesis and corticocortical projections in the developing mammalian brain.30 Moreover, by employing multiple gene-mapping techniques, we were also able to triangulate on novel genes associated with psychopathology, including WDR73, the causal gene in a rare recessive autosomal disorder characterized by severe encephalopathy, developmental delay, and neurocognitive impairment.31 Associations such as these are particularly interesting in light of results suggesting that genes disrupted in Mendelian disorders are also dysregulated by non-coding variants in phenotypically similar traits and disorders.32 Furthermore, we build on the results of a large GWAS of eight psychiatric disorders33 by providing novel evidence of factor-specific pleiotropy (i.e., consistent effects across a factor’s constituent indicators) via QSNP results, which also identified several novel loci with significantly heterogeneous effects for bipolar I disorder, schizoaffective disorder, and schizophrenia.
Third, our extensive biological annotation revealed a marked divergence in the biology associated with the two transdiagnostic factors. While we find that the CNS is dually implicated at a broad systems-based level (e.g., non-specific enrichment of brain tissues), the biology associated with the two factors quickly diverges at more molecular levels of investigation. Via our novel approach to gene prioritization based on spatial transcriptomics, we identified two sets of factor-specific genes with contrasting developmental expression profiles. Specifically, we found that transcriptomically prioritized genes associated with the factor broadly characterized by common mood disturbance (F1) exhibited lower expression levels during early prenatal periods, while transcriptomically prioritized genes for the factor broadly characterized by rarer forms of serious mental illness (F2) exhibited higher expression levels during early prenatal periods. Notably, both of these trajectories identify the prenatal epoch as a critical developmental period related to psychopathology, albeit in different ways. These findings coalesce with and build upon previous studies that have begun to characterize developmental expression patterns of transdiagnostic genetic liabilities.34 Here, we found that the two observed trajectories strongly resembled those of postnatal and prenatal inhibitory neuronal genes,25 which have been implicated in the development of mood and psychotic disorders.35, 36, 37
Fourth, we found that the two factors differ substantially in their associations with human wellbeing and disease. Our results expand upon recent phenome-wide association studies of genetic risk for major depressive disorder38 and schizophrenia,39 expanding the list of complex traits and medical phenotypes associated with mood and psychotic psychopathology. We also identified an interesting pattern of results in our genetic correlation and phenome-wide association analyses, where the factor comprising more common forms of mood disturbance (F1) had broader and often stronger negative associations with socioeconomic and health-related outcomes than the factor comprising rarer forms of serious mental illness (F2). This runs counter to associations often observed at the phenotypic level, where individuals diagnosed with more serious mental illnesses tend to face more severe impairments and consequences in these domains.40,41 These results raise questions about the potential ascertainment biases that affect GWASs. For example, clinically ascertained samples of people with diagnosed psychiatric disorders (particularly when those disorders are rare and seriously impairing) are subject to different sources of selection, attrition, and non-response than population-based studies that utilize self-report surveys. Consider, for instance, that individuals who are homeless and incarcerated in Western countries are drastically more likely than the general population to meet diagnostic criteria for a serious mental illness,42,43 but these socially marginalized groups are less likely to have access to adequate mental health care or be included in medical research. This selective representation of psychopathology may induce collider bias and lead to misleading estimates of genetic association.44 Indeed, cohort-level studies have already found that educational attainment polygenic scores are positively associated with research participation, while psychopathology polygenic scores are negatively associated.45,46
Limitations of the study
While we have taken many steps to address potential confounds, these major findings should be interpreted in light of several limitations. First, SEM does not reveal a “ground truth” about the nature of the phenotypes included in the analysis. Instead, it is a useful statistical framework for representing complex data structures, and latent factors are most appropriately considered as convenient statistical entities that explain the (co)variances of their indicators. As such, latent genetic factors are most useful as explanatory devices when accompanied by extensive biological annotation and follow up, as done in the present study. Second, the univariate GWASs are comprised of different samples with different measurement approaches and varying levels of power. However, we have made efforts to harmonize each of the GWASs used in Genomic SEM analyses (e.g., excluding self-rated measures from diagnostic phenotypes, and vice versa), and previous examination of these concerns suggest that the genetic factor structure is not biased by sample overlap or sample-size differences.19,33
Third, the univariate GWASs are comprised of different cohorts that may be subject to different sources of bias that cannot be fully quantified. For example, we note that the UK Biobank is subject to a volunteer selection bias, where study participants are generally healthier than individuals who are not study participants.47 In ideal circumstances, the phenotypic and genetic factor analyses reported here would be performed within the same participants in order to minimize confounding by differences in sample selection. However, such deep phenotypic data are not currently available at the scale required for GWASs. Fourth, the current study focuses on forms of psychopathology that involve a wide variety of disturbances in mood and reality testing but does not comprehensively sample the full range of psychiatric disorders. These results thus complement other transdiagnostic research studies that have illuminated how schizophrenia and bipolar I disorder diverge genetically from other clinically defined disorders, such as compulsive disorders and disorders of childhood.33
Conclusions
In summary, we have conducted a novel multivariate GWAS of multiple symptoms and disorders spanning mood and psychotic psychopathology. This analysis identified two transdiagnostic genetic liabilities operating quite distinctly from one another. Extensive biological annotation revealed contrasting genetic architectures that implicated prenatal neurodevelopment and neuronal function and regulation in markedly different ways. Given the degree of divergence between these two factors, future research is warranted to investigate the utility and appropriateness of even broader spectra of psychopathology (e.g., the p factor20) as explanatory devices at the level of molecular genetics. Collectively, our results suggest that the severity of mood and psychotic symptoms evident in severe psychiatric disorders might actually reflect a difference in kind rather than merely in degree.
STAR★Methods
Key resources table
Resource availability
Lead contact
Any inquiries about analytical results or other information should be directed to lead contact, Travis T. Mallard (tmallard@mgh.harvard.edu).
Materials availability
No new materials were generated as part of this study.
Method details
Phenotype construction in UK biobank
Mplus48 v8 was used to estimate person-specific thetas (i.e., factor scores) for three symptom domains: depression, mania, and psychosis. As each psychiatric phenotypes was assessed by four items, thetas were estimated via a multidimensional two-parameter probit model,61 which allowed item-level responses across measurement occasions to be combined for correlated latent variables simultaneously. Furthermore, a combination of multiple imputation and Bayesian estimation with non-informative priors was used to maximally leverage all available responses for participants to minimize the impact of missing data. See Section 1.1 of Methods S1 for further description of the phenotypic modeling.
Univariate genome-wide association analyses
BOLT-LMM49 v2.3.2 was used to conduct GWASs in the UK Biobank for three lifetime measures of psychiatric symptoms: depression, mania, and psychosis. This approach used a linear mixed model that included a genetic relationship matrix to estimate SNP effects, which offered improved control for population stratification and maximized power by accounting for relatedness among individuals. The first 40 principal components of ancestry computed with flashPCA2 (Section 1.2 of Methods S1), sex, birth year, sex-by-birth year interactions, and batch were included as covariates. EasyQC51 was used to perform extensive quality control on the GWAS summary statistics. The main objective of the quality control was to filter out rare and low-frequency SNPs, as well as SNPs that were not imputed well. Three main filters were imposed: (i) MAF <0.005; (ii) imputation quality score <0.9; (iii) unavailable in reference panel. Additional quality control procedures and filters are further described in Section 1.3 of Methods S1. The reference panel was a combination of the 1000 Genomes phase 3 v5 and UK10K, which has been described in a previous study.14
Multivariate genome-wide association analyses
Genomic SEM19 v0.0.2 was used to conduct multivariate GWAS based on eight phenotypes: depressive symptoms, manic symptoms, psychotic symptoms, major depressive disorder, bipolar II disorder, bipolar I disorder, schizoaffective disorder, and schizophrenia (see Table 1 for overview). Following identification of the confirmatory factor model that best explained the observed genetic covariances among the phenotypes, Genomic SEM was used to estimate the individual SNP effects on each latent factor in the model. Note that Genomic SEM is unbiased in the presence of varying and unknown sample overlap across the contributing GWAS samples, as the cross-trait intercepts estimated via multivariable LD score regression are used to estimate (and account for) sample overlap and phenotypic correlation.
Effective sample size () for each latent factor was estimated as , where is the number of SNPs in the GWAS, is the lower MAF threshold for inclusion in the calculation (here, 10%), the upper limit (here, 40%), and is the effective sample size for SNP j, which is calculated as . QSNP tests were used to evaluate whether SNP effects on the latent factors were driven by heterogeneous effects across constituent phenotypes. Further description of multivariate association analyses and QSNP tests is provided in Sections 2.4 and 2.5 of Methods S1, respectively.
Genetic correlations among study phenotypes
LD score regression52 v1.0.1 was used to estimate genetic correlations between all pairwise combinations of the eight study phenotypes. Standard procedures and best practices for LD score regression were followed (e.g., restricting to HapMap362 SNPs with a minor allele frequency ≥0.01). Default parameters were used for the three new GWASs of psychiatric symptoms. For the existing GWASs of psychiatric disorders, parameters (e.g., sample prevalence, population prevalence) were defined as outlined in the original studies. A hierarchical clustering algorithm was applied to the final genetic correlation matrix to guide factor selection in the exploratory factor analysis. Although the original LD score regression software was used for this preliminary analysis, the multivariable version of LD score regression employed by Genomic SEM was used for all subsequent analyses. Please note that these software produce estimates that are effectively identical.
Exploratory factor analysis
The stats R package was used to conduct an EFA of the genetic correlations among the eight study phenotypes. Specifically, the factanal function was used to conduct an EFA with promax rotation on the standardized S matrix derived from the multivariable version of LD score regression employed by Genomic SEM. This enabled an empirical assessment of (i) the number of latent factors that best explained the multivariate genetic architecture observed among the set of study phenotypes (i.e., the number of transdiagnostic liabilities present), and (ii) how constituent phenotypes load onto separable latent factors. As suggested by the hierarchical clustering algorithm, two factors were extracted that optimally accounted for shared variation among sets of the observed variables. Results from this analysis were subsequently used to guide construction of the confirmatory factor models. A brief overview of factor analysis is provided in Section 2.2 of Methods S1.
Confirmatory factor analysis
Genomic SEM was used to test whether a common factor model or a correlated factors model best fit the data via CFA, where fit reflects the degree to which the specified latent variable structure adequately explains the observed covariances among the set of observed variables. Parameter estimates were derived using weighted least squares estimation. Model fit was assessed using conventional indices in structural equation modeling: the model χ2 statistic, the Akaike information criterion (AIC), the comparative fit index (CFI), and the standardized root mean square residual (SRMR). All fit indices retain their standard interpretations within a Genomic SEM framework. However, the model χ2 statistic is best used as a comparative measure of fit to evaluate competing models rather than a measure of statistical significance given the sensitivity of model χ2 to sample size, which is comparatively extremely large for GWAS samples. For CFI and SRMR, values greater than .90 and less than .08, respectively, were considered reflective of good model fit.63 Further description of structural equation modeling and confirmatory factor analysis are provided in Section 2.3 of Methods S1.
Heritability for observed and latent phenotypes
LD score regression was used to estimate the heritability of the three psychiatric symptom phenotypes, as well as the two latent genetic factors. Standard procedures and best practices for LD score regression were followed. As there is no phenotypic variance for latent genetic factors modeled in Genomic SEM, heritability is more accurately referred to as genetic variance for F1 and F2. Furthermore, as genetic variance estimates are influenced by the heritability estimates of constituent phenotypes and the metric of the latent genetic factor, we note that estimates for F1 and F2 should only be interpreted in the context of the present study.
Local heritability and genetic correlations
HESS53 and its bivariate extension, ρ-HESS,54 were used to estimate local genetic variance, local genetic covariance, and the proportion of the genome that contributes to the total genetic covariance for F1 and F2. For each factor, HESS was first used to estimate local genetic variance and covariance across 1,698 approximately LD-independent contiguous genomic partitions, averaging 1.5 Mb per partition. The European samples from the 1000 Genomes Project Phase 3v564 (n = 503) were used as a reference panel for these analyses. Independent genomic partitions were then ranked by their absolute genetic covariance, and the percentage that accounted for 80% of the total genetic covariance between F1 and F2 was used to further quantify genetic overlap between F1 and F2.65
Gene mapping and identification
The FUMA55 SNP2GENE pipeline was used to apply a standard clumping algorithm that identified associated genomic loci, lead SNPs within loci, and all independent significant SNPs within loci. The European samples from the 1000 Genomes Project Phase 3v5 (n = 503) were used as a reference panel for LD. FUMA was also used to employ an ensemble of methods to identify putative risk genes for the univariate and multivariate GWAS phenotypes. Specifically, FUMA v1.3.6c was used to conduct positional, eQTL, and chromatin interaction mapping to identify risk-conferring genes that map to genome-wide significant loci. Default parameters were used for each of these analyses. ANNOVAR annotations66 were used for positional mapping, the Geno-type-Tissue Expression (GTEx) v8 brain dataset67 was used as the reference tissue data for eQTL mapping, and Hi-C data from adult and fetal human brain samples68 was used to examine enhancer-promoter and promoter-promoter chromatin interactions.
Two additional methods were employed to identify putative risk genes based on genome-wide summary statistics: and MAGMA56 and S-PrediXcan.57 The former was used to calculate gene-based association statistics, and the latter was used to identify functionally expressed genes via joint analysis of SNP effects and eQTL expression effects. Both methods are described in the following section.
Gene-based association and enrichment analyses
MAGMA v1.08, a bioinformatics software for gene-based biological annotation, was used to conduct gene association, gene set enrichment, and gene property analyses for all novel study phenotypes. Default MAGMA parameters were employed and standard procedures were followed for gene-based association analyses based on summary statistics. MAGMA was also used to conduct competitive gene-set enrichment and gene property analyses based on the gene-level p values produced in the association analyses. These analyses tested whether genes within an annotated set are more strongly associated with the phenotype of interest than other genes. For gene set enrichment analyses, up to 9,987 gene sets cataloged in MolSigDB v7.0 were tested, which corresponded to 7,343 biological processes, 1,001 cellular components, and 1,643 molecular functions. For the gene property analyses, 54 tissues from the GTEx v8 dataset were tested. Bonferroni-corrected thresholds of p ≤ 5.01e-6 and p ≤ 9.26e-4 were used to determine significance for gene sets and tissues, respectively.
S-PrediXcan v0.6.2 was used (i) to predict gene expression levels in brain tissues, and (ii) to test whether predicted gene expression correlated with either transdiagnostic factor. Tissue weights were computed using reference data from the GTEx v8 dataset. GWAS summary statistics for F1 and F2, the reference transcriptomic data, and covariance matrices for the SNPs within each gene model were included as input data. Thirteen brain tissues were tested: anterior cingulate cortex, amygdala, caudate basal ganglia, cerebellar hemisphere, cerebellum, cortex, frontal cortex, hippocampus, hypothalamus, nucleus accumbens basal ganglia, putamen basal ganglia, spinal cord and substantia nigra. A Bonferonni-corrected threshold of p ≤ 8.97e-7 was established for transcriptome-wide significance, which corrected for 55,753 gene-based tests.
Genetic correlation analyses between latent factors and other complex traits
Genomic SEM was used to estimate genetic correlations and partial genetic correlations between latent factors of psychopathology and other phenotypes of interest. Specifically, genetic correlations were estimated for two broad sets of phenotypes: (i) morphological features of the human brain, and (ii) complex traits related to human health and well-being. Summary statistics for 101 neuroimaging phenotypes69 (cortical and subcortical gray matter volumes, ventricular volumes, and global measures of brain volume) were downloaded from https://github.com/BIG-S2/GWAS. Summary statistics for 92 phenotypes broadly related to various domains of human health and well-being were downloaded from various online sources, using download links from GWAS Atlas70 whenever possible. All summary statistics were cleaned and processed using the munge function of Genomic SEM, retaining all HapMap3 SNPs outside of the major histocompatibility complex regions with an allele frequency ≥ .01. A Bonferroni correction was applied within each family of tests to adjust p values for multiple comparisons (p ≤ 4.95e-4 for neuroimaging phenotypes; p ≤ 5.43e-4 for complex traits).
Spatiotemporal transcriptomic analyses
Microarray gene expression data from the Allen Human Brain Atlas (AHBA)71 were downloaded from https://human.brain-map.org/static/download, and subsequently aligned to the Desikan-Killiany-Tourville atlas (N = 62 cortical brain regions)72 for spatial compatibility with the cortical neuroimaging phenotypes.73 Spatial correlation coefficients (Spearman’s ρ) were computed for each of 20,647 genes compared against the -log10 p values from F1 and F2. To examine the developmental trajectories of the F1 and F2 gene sets (positive Z-scores of AHBA correlation coefficients, p < .05), weighted gene correlation network analysis58 was used to estimate eigengene values (i.e., gene set expression) for these gene sets in the PsychENCODE dataset, treating each factor-specific gene set as a module. These expression values were then plotted as function of time, using a non-parametric LOESS curve line-of-best-fit to characterize developmental expression trajectories for F1 and F2, which indicated that the prioritized gene sets for each transdiagnostic factor are differentially expressed in pre- and postnatal cortical tissue. Evaluation of cell-type-specific gene sets was performed as above, using available data from a recent cell-specific sequencing study in adult human brain tissue.25
Phenome-wide polygenic prediction
PRS-CS59 and PLINK60 v1.9 were used to calculate polygenic scores for the transdiagnostic latent genetic factors, F1 and F2. PRS-CS, a Bayesian polygenic prediction method, was used to apply a continuous shrinkage prior to SNP effect estimates and infer posterior SNP weights using GWAS summary statistics for F1 and F2 and an external reference panel to model LD. In the present study, PRS-CS was used to adjust weights for 1,027,871 SNPs typed on both the 1000 Genomes Project Phase 3v5 and the HapMap3 reference panels with a minor allele frequency ≥ .01. The European samples from the 1000 Genomes Project Phase 3v5 (n = 503) were used as a reference panel for LD. PLINK was then used to calculate polygenic scores for each individual by summing all included variants weighted by the inferred posterior effect size for the effect allele, and converting that value to a Z-score for each participant within the prediction sample.
The genotyped BioVU sample (n = 66,915) was used to test for associations between polygenic scores for F1 and F2 and a wide array of medical phenotypes. Genotyping and quality control for this sample have been described elsewhere. Case-control medical phenotypes, also referred to as “phecodes,” were constructed from International Classification of Disease (ICD) diagnostic codes in participant electronic health record data. Two instances of an ICD diagnostic code were required to be present to be classified as a case for a given phecode, and 50 cases were required for a phecode to be analyzed. A total of 1,335 phecodes were included in the phenome-wide association analyses. The PheWAS R package was used to conduct phenome-wide association analyses. A logistic regression model was fit to each of 1,335 case/control phenotypes to estimate the odds of each diagnosis given the polygenic scores for F1 and F2. Sex, median age of the longitudinal electronic health record measurements, and the top 10 principal components of ancestry were included as covariates. A Bonferroni-corrected threshold of at p ≤ 3.74e-5 was established for phenome-wide significance.
Consortia
The members of the Bipolar Disorder Working Group of the Psychiatric Genomics Consortium are Eli A. Stahl, Gerome Breen, Andreas J. Forstner, Andrew McQuillin, Stephan Ripke, Vassily Trubetskoy, Manuel Mattheisen, Yunpeng Wang, Jonathan R.I. Coleman, Héléna A. Gaspar, Christiaan A. de Leeuw, Stacy Steinberg, Jennifer M. Whitehead Pavlides, Maciej Trzaskowski, Enda M. Byrne, Tune H. Pers, Peter A. Holmans, Alexander L. Richards, Liam Abbott, Esben Agerbo, Huda Akil, Diego Albani, Ney Alliey-Rodriguez, Thomas D. Als, Adebayo Anjorin, Verneri Antilla, Swapnil Awasthi, Judith A. Badner, Marie Bækvad-Hansen, Jack D. Barchas, Nicholas Bass, Michael Bauer, Richard Belliveau, Sarah E. Bergen, Carsten Bøcker Pedersen, Erlend Bøen, Marco P. Boks, James Boocock, Monika Budde, William Bunney, Margit Burmeister, Jonas Bybjerg-Grauholm, William Byerley, Miquel Casas, Felecia Cerrato, Pablo Cervantes, Kimberly Chambert, Alexander W. Charney, Danfeng Chen, Claire Churchhouse, Toni-Kim Clarke, William Coryell, David W. Craig, Cristiana Cruceanu, David Curtis, Piotr M. Czerski, Anders M. Dale, Simone de Jong, Franziska Degenhardt, Jurgen Del-Favero, J. Raymond De-Paulo, Srdjan Djurovic, Amanda L. Dobbyn, Ashley Dumont, Torbjørn Elvsåshagen, Valentina Escott-Price, Chun Chieh Fan, Sascha B. Fischer, Matthew Flickinger, Tatiana M. Foroud, Liz Forty, Josef Frank, Christine Fraser, Nelson B. Freimer, Louise Frisén, Katrin Gade, Diane Gage, Julie Garnham, Claudia Giambartolomei, Marianne Giørtz Pedersen, Jaqueline Goldstein, Scott D. Gordon, Katherine Gordon-Smith, Elaine K. Green, Melissa J. Green, Tiffany A. Greenwood, Jakob Grove, Weihua Guan, José Guzman-Parra, Marian L. Hamshere, Martin Hautzinger, Urs Heilbronner, Stefan Herms, Maria Hipolito, Per Hoffmann, Dominic Holland, Laura Huckins, Stéphane Jamain, Jessica S. Johnson, Radhika Kandaswamy, Robert Karlsson, James L. Kennedy, Sarah Kittel-Schneider, James A. Knowles, Manolis Kogevinas, Anna C. Koller, Ralph Kupka, Catharina Lavebratt, Jacob Lawrence, William B. Lawson, Markus Leber, Phil H. Lee, Shawn E. Levy, Jun Z. Li, Chunyu Liu, Susanne Lucae, Anna Maaser, Donald J. MacIntyre, Pamela B. Mahon, Wolfgang Maier, Lina Martinsson, Steve McCarroll, Peter McGuffin, Melvin G. McInnis, James D. McKay, Helena Medeiros, Sarah E. Medland, Fan Meng, Lili Milani, Grant W. Montgomery, Derek W. Morris, Thomas W. Mühleisen, Niamh Mullins, Hoang Nguyen, Caroline M. Nievergelt, Annelie Nordin Adolfsson, Evaristus A. Nwulia, Claire O'Donovan, Loes M. Olde Loohuis, Anil P.S. Ori, Lilijana Oruc, Urban Ösby, Roy H. Perlis, Amy Perry, Andrea Pfennig, James B. Potash, Shaun M. Purcell, Eline J. Regeer, Andreas Reif, Céline S. Reinbold, John P. Rice, Fabio Rivas, Margarita Rivera, Panos Roussos, Douglas M. Ruderfer, Euijung Ryu, Cristina Sánchez-Mora, Alan F. Schatzberg, William A. Scheftner, Nicholas J. Schork, Cynthia Shannon Weickert, Tatyana Shehktman, Paul D. Shilling, Engilbert Sigurdsson, Claire Slaney, Olav B. Smeland, Janet L. Sobell, Christine Søholm Hansen, Anne T. Spijker, David St Clair, Michael Steffens, John S. Strauss, Fabian Streit, Jana Strohmaier, Szabolcs Szelinger, Robert C. Thompson, Thorgeir E. Thorgeirsson, Jens Treutlein, Helmut Vedder, Weiqing Wang, Stanley J. Watson, Thomas W. Weickert, Stephanie H. Witt, Simon Xi, Wei Xu, Allan H. Young, Peter Zandi, Peng Zhang, Sebastian Zöllner, Rolf Adolfsson, Ingrid Agartz, Martin Alda, Lena Backlund, Bernhard T. Baune, Frank Bellivier, Wade H. Berrettini, Joanna M. Biernacka, Douglas H.R. Blackwood, Michael Boehnke, Anders D. Børglum, Aiden Corvin, Nicholas Craddock, Mark J. Daly, Udo Dannlowski, Tõnu Esko, Bruno Etain, Mark Frye, Janice M. Fullerton, Elliot S. Gershon, Michael Gill, Fernando Goes, Maria Grigoroiu-Serbanescu, Joanna Hauser, David M. Hougaard, Christina M. Hultman, Ian Jones, Lisa A. Jones, René S. Kahn, George Kirov, Mikael Landén, Marion Leboyer, Cathryn M. Lewis, Qingqin S. Li, Jolanta Lissowska, Nicholas G. Martin, Fermin Mayoral, Susan L. McElroy, Andrew M. McIntosh, Francis J. McMahon, Ingrid Melle, Andres Metspalu, Philip B. Mitchell, Gunnar Morken, Ole Mors, Preben Bo Mortensen, Bertram Müller-Myhsok, Richard M. Myers, Benjamin M. Neale, Vishwajit Nimgaonkar, Merete Nordentoft, Markus M. Nöthen, Michael C. O'Donovan, Ketil J. Oedegaard, Michael J. Owen, Sara A. Paciga, Carlos Pato, Michele T. Pato, Danielle Posthuma, Josep Antoni Ramos-Quiroga, Marta Ribasés, Marcella Rietschel, Guy A. Rouleau, Martin Schalling, Peter R. Schofield, Thomas G. Schulze, Alessandro Serretti, Jordan W. Smoller, Hreinn Stefansson, Kari Stefansson, Eystein Stordal, Patrick F. Sullivan, Gustavo Turecki, Arne E. Vaaler, Eduard Vieta, John B. Vincent, Thomas Werge, John I. Nurnberger, Naomi R. Wray, Arianna Di Florio, Howard J. Edenberg, Sven Cichon, Roel A. Ophoff, Laura J. Scott, Ole A. Andreassen, John Kelsoe, and Pamela Sklar. Their affiliations are provided in the supplemental information.
Acknowledgments
T.T.M. is supported by funds from NIH T32HG010464. S.S.-R. was supported by funds from the California Tobacco-Related Disease Research Program (TRDRP; grant number T29KT0526) and NIDA DP1DA054394. J.S. was supported by NIH T32MH019112. A.O. was supported by the Netherlands Organisation for Scientific Research VENI (016.Veni.198.058). A.A.P. was supported by a grant from the TRDRP (28IR-0070) and by NIH P50DA037844. K.P.H. and E.M.T.-D. were supported in part by Jacobs Foundation Research Fellowships and are faculty research associates of the Population Research Center at the University of Texas at Austin, which is supported by NIH grant P2CHD04284. K.P.H. and E.M.T.-D. were also supported by NIH grants R01-HD083613 and R01-HD092548. E.M.T.-D. was also supported by NIH grant R01-MH120219and is a member of the Center for Aging and Population Studies at the University of Texas at Austin, which is supported by NIH grant P30AG066614. M.C.K. was supported by funds from NIH grants MH100141 and DA044283. P.D.K. was supported by an ERC consolidator grant (647648 EdGe). The dataset(s) used for the PheWAS/LabWAS analyses described were obtained from Vanderbilt University Medical Center’s BioVU, which is supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH-funded Shared Instrumentation Grant S10RR025141 and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, and R01HD074711, and additional funding sources are listed at https://victr.vumc.org/biovu-funding/. L.K.D. obtained support from 1R01MH113362, 1R01MH118233, and 1R56MH120736. The project was approved by the VUMC Institutional Review Board (IRB #160302, #172020, and #190418). Its contents are solely the responsibility of the authors and do not necessarily represent official views of the National Center for Advancing Translational Sciences or the National Institutes of Health. This research was conducted with the UK Biobank Resource under application number 11425 and with the support and collaboration from all investigators who make up the Bipolar Disorder Working Group of the PGC. We would like to thank the many studies that made these consortia possible, the researchers involved, and the participants in those studies, without whom this effort would not be possible. We would also like to thank the research participants and employees of 23andMe for making this work possible.
Author contributions
T.T.M., P.D.K., and K.P.H. conceived and designed the study. P.D.K. and K.P.H. oversaw the study. T.T.M. and K.P.H. led the writing of the manuscript with substantive contributions from P.D.K. and M.C.K. A.A.P., E.M.T.-D., and K.S.K. provided valuable feedback on the framing and interpretation of the results. T.T.M. was the lead analyst, responsible for conducting GWASs, quality control, genetic correlations, multivariate analyses with Genomic SEM, and biological annotation with assistance from R.K.L., A.D.G., S.S.-R., and J.S. T.T.M. prepared the data for analysis with assistance from R.K.L., A.O., R.d.V., and S.F.W.M. S.S.-R. performed the phenome-wide association study with assistance from L.K.D. T.T.M. prepared the figures and tables. Investigators from the Bipolar Disorder Working Group of the PGC contributed data for BD1, BD2, and SZA. All authors provided valuable feedback and advice during preparation of the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: June 8, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2022.100140.
Contributor Information
Travis T. Mallard, Email: tmallard@mgh.harvard.edu.
K. Paige Harden, Email: harden@utexas.edu.
Supplemental information
Data and code availability
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. This paper does not report custom code or software. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Vos T., Abajobir A.A., Abate K.H., Abbafati C., Abbas K.M., Abd-Allah F., Abdulkader R.S., Abdulle A.M., Abebo T.A., Abera S.F., et al. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990--2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211–1259. doi: 10.1016/S0140-6736(17)32154-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polderman T.J.C., Benyamin B., de Leeuw C.A., Sullivan P.F., van Bochoven A., Visscher P.M., Posthuma D. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 2015;47:702–709. doi: 10.1038/ng.3285. advance on. [DOI] [PubMed] [Google Scholar]
- 3.Ripke S., Neale B.M., Corvin A., Walters J.T.R., Farh K.-H., Holmans P.A., Lee P., Bulik-Sullivan B., Collier D.A., Huang H., et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahl E.A., Breen G., Forstner A.J., McQuillin A., Ripke S., Trubetskoy V., Mattheisen M., Wang Y., Coleman J.R.I., Gaspar H.A., et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wray N.R., Ripke S., Mattheisen M., Trzaskowski M., Byrne E.M., Abdellaoui A., Adams M.J., Agerbo E., Air T.M., Andlauer T.M.F., et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018;50:668–681. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anttila V., Bulik-Sullivan B., Finucane H.K., Walters R.K., Bras J., Duncan L., Escott-Price V., Falcone G.J., Gormley P., Malik R., et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:eaap8757. doi: 10.1126/science.aap8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J.J., Wedow R., Okbay A., Kong E., Maghzian O., Zacher M., Nguyen-Viet T.A., Bowers P., Sidorenko J., Linnér R.K., et al. Gene discovery and polygenic prediction from a 1.1-million-person GWAS of educational attainment. Nat. Genet. 2018;50:1112. doi: 10.1038/s41588-018-0147-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Donovan M.C., Owen M.J. The implications of the shared genetics of psychiatric disorders. Nat. Med. 2016;22:1214. doi: 10.1038/nm.4196. [DOI] [PubMed] [Google Scholar]
- 9.Insel T., Cuthbert B., Garvey M., Heinssen R., Pine D.S., Quinn K., Sanislow C., Wang P. 2010. Research Domain Criteria (RDoC): Toward a New Classification Framework for Research on Mental Disorders. [DOI] [PubMed] [Google Scholar]
- 10.Cuthbert B.N. The RDoC framework: facilitating transition from ICD/DSM to dimensional approaches that integrate neuroscience and psychopathology. World Psychiatry. 2014;13:28–35. doi: 10.1002/wps.20087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howard D.M., Adams M.J., Clarke T.-K., Hafferty J.D., Gibson J., Shirali M., Coleman J.R.I., Hagenaars S.P., Ward J., Wigmore E.M., et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019;22:343. doi: 10.1038/s41593-018-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanchez-Roige S., Palmer A.A., Fontanillas P., Elson S.L., 23andMe Research Team, the S.U.D.W.G. of the P.G.C. Adams M.J., Howard D.M., Edenberg H.J., Davies G., Crist R.C., et al. Genome-wide association study meta-analysis of the alcohol use disorders identification test (AUDIT) in two population-based cohorts. Am. J. Psychiatry. 2018;176:107–118. doi: 10.1176/appi.ajp.2018.18040369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levey D.F., Gelernter J., Polimanti R., Zhou H., Cheng Z., Aslan M., Quaden R., Concato J., Radhakrishnan K., Bryois J., et al. Reproducible genetic risk loci for anxiety: results From∼ 200,000 participants in the million veteran Program. Am. J. Psychiatry. 2020 doi: 10.1176/appi.ajp.2019.19030256. appi--ajp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bycroft C., Freeman C., Petkova D., Band G., Elliott L.T., Sharp K., Motyer A., Vukcevic D., Delaneau O., O’Connell J., et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez-Roige S., Palmer A.A. Emerging phenotyping strategies will advance our understanding of psychiatric genetics. Nat. Neurosci. 2020;23:475–480. doi: 10.1038/s41593-020-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plomin R., Haworth C.M.A., Davis O.S.P. Common disorders are quantitative traits. Nat. Rev. Genet. 2009;10:872–878. doi: 10.1038/nrg2670. [DOI] [PubMed] [Google Scholar]
- 17.van den Berg S.M., Glas C.A.W., Boomsma D.I. Variance decomposition using an IRT measurement model. Behav. Genet. 2007;37:604–616. doi: 10.1007/s10519-007-9156-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheesman R., Purves K.L., Pingault J.-B., Breen G., Plomin R., Rijsdij K F., Eley T.C., others Extracting stability increases the SNP heritability of emotional problems in young people. Transl. Psychiatry. 2018;8:223–229. doi: 10.1038/s41398-018-0269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grotzinger A.D., Rhemtulla M., de Vlaming R., Ritchie S.J., Mallard T.T., Hill W.D., Ip H.F., Marioni R.E., McIntosh A.M., Deary I.J., et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav. 2019;3:513–525. doi: 10.1038/s41562-019-0566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caspi A., Houts R.M., Belsky D.W., Goldman-Mellor S.J., Harrington H., Israel S., Meier M.H., Ramrakha S., Shalev I., Poulton R., Moffitt T.E. The p factor: one general psychopathology factor in the structure of psychiatric disorders? Clin. Psychol. Sci. 2014;2:119–137. doi: 10.1177/2167702613497473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruderfer D.M., Ripke S., McQuillin A., Boocock J., Stahl E.A., Pavlides J.M.W., Mullins N., Charney A.W., Ori A.P.S., Loohuis L.M.O., et al. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 2018;173:1705–1715. doi: 10.1016/j.cell.2018.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okbay A., Baselmans B.M.L., De Neve J.-E., Turley P., Nivard M.G., Fontana M.A., Meddens S.F.W., Linnér R.K., Rietveld C.A., Derringer J., et al. Genetic variants associated with subjective well-being, depressive symptoms, and neuroticism identified through genome-wide analyses. Nat. Genet. 2016;48:624–633. doi: 10.1038/ng.3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagel M., Jansen P.R., Stringer S., Watanabe K., de Leeuw C.A., Bryois J., Savage J.E., Hammerschlag A.R., Skene N.G., Muñoz-Manchado A.B., et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018;50:920–927. doi: 10.1038/s41588-018-0151-7. [DOI] [PubMed] [Google Scholar]
- 24.Pardiñas A.F., Holmans P., Pocklington A.J., Escott-Price V., Ripke S., Carrera N., Legge S.E., Bishop S., Cameron D., Hamshere M.L., et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018;50:381–389. doi: 10.1038/s41588-018-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lake B.B., Chen S., Sos B.C., Fan J., Kaeser G.E., Yung Y.C., Duong T.E., Gao D., Chun J., Kharchenko P.V., Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018;36:70–80. doi: 10.1038/nbt.4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M., Santpere G., Imamura Kawasawa Y., Evgrafov O.V., Gulden F.O., Pochareddy S., Sunkin S.M., Li Z., Shin Y., Zhu Y., et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615. doi: 10.1126/science.aat7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coleman J.R.I., Gaspar H.A., Bryois J., Byrne E.M., Forstner A.J., Holmans P.A., de Leeuw C.A., Mattheisen M., McQuillin A., Pavlides J.M.W., et al. The genetics of the mood disorder spectrum: genome-wide association analyses of more than 185,000 cases and 439,000 controls. Biol. Psychiatry. 2019;88:169–184. doi: 10.1016/j.biopsych.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Legge S.E., Jones H.J., Kendall K.M., Pardiñas A.F., Menzies G., Bracher-Smith M., Escott-Price V., Rees E., Davis K.A.S., Hotopf M., et al. Association of genetic liability to psychotic experiences with neuropsychotic disorders and traits. JAMA Psychiatry. 2019;76:1256–1265. doi: 10.1001/jamapsychiatry.2019.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turkheimer E., Pettersson E., Horn E.E. A phenotypic null hypothesis for the genetics of personality. Annu. Rev. Psychol. 2014;65:515–540. doi: 10.1146/annurev-psych-113011-143752. [DOI] [PubMed] [Google Scholar]
- 30.Witteveen J.S., Willemsen M.H., Dombroski T.C.D., Van Bakel N.H.M., Nillesen W.M., Van Hulten J.A., Jansen E.J.R., Verkaik D., Veenstra-Knol H.E., van Ravenswaaij-Arts C.M.A., et al. Haploinsufficiency of MeCP2-interacting transcriptional co-repressor SIN3A causes mild intellectual disability by affecting the development of cortical integrity. Nat. Genet. 2016;48:877–887. doi: 10.1038/ng.3619. [DOI] [PubMed] [Google Scholar]
- 31.Colin E., Huynh Cong E., Mollet G., Guichet A., Gribouval O., Arrondel C., Boyer O., Daniel L., Gubler M.-C., Ekinci Z., et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: galloway-Mowat syndrome. Am. J. Hum. Genet. 2014;95:637–648. doi: 10.1016/j.ajhg.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freund M.K., Burch K.S., Shi H., Mancuso N., Kichaev G., Garske K.M., Pan D.Z., Miao Z., Mohlke K.L., Laakso M., et al. Phenotype-specific enrichment of Mendelian disorder genes near GWAS regions across 62 complex traits. Am. J. Hum. Genet. 2018;103:535–552. doi: 10.1016/j.ajhg.2018.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee P.H., Anttila V., Won H., Feng Y.-C.A., Rosenthal J., Zhu Z., Tucker-Drob E.M., Nivard M.G., Grotzinger A.D., Posthuma D., et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–1482. doi: 10.1016/j.cell.2019.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schork A.J., Won H., Appadurai V., Nudel R., Gandal M., Delaneau O., Revsbech Christiansen M., Hougaard D.M., Bækved-Hansen M., Bybjerg-Grauholm J., et al. A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci. 2019;22:353–361. doi: 10.1038/s41593-018-0320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volk D.W., Lewis D.A. Early developmental disturbances of cortical inhibitory neurons: contribution to cognitive deficits in schizophrenia. Schizophr. Bull. 2014;40:952–957. doi: 10.1093/schbul/sbu111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fine R., Zhang J., Stevens H.E. Prenatal stress and inhibitory neuron systems: implications for neuropsychiatric disorders. Mol. Psychiatry. 2014;19:641–651. doi: 10.1038/mp.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duman R.S., Sanacora G., Krystal J.H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron. 2019;102:75–90. doi: 10.1016/j.neuron.2019.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dennis J., Sealock J., Levinson R.T., Farber-Eger E., Franco J., Fong S., Straub P., Hucks D., Linton M.F., Song W.-L., et al. Genetic risk for major depressive disorder and loneliness in gender-specific associations with coronary artery disease. bioRxiv. 2019:512541. doi: 10.1038/s41380-019-0614-y. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheutlin A.B., Dennis J., Karlsson Linnér R., Moscati A., Restrepo N., Straub P., Ruderfer D., Castro V.M., Chen C.-Y., Ge T., et al. Penetrance and pleiotropy of polygenic risk scores for schizophrenia in 106,160 patients across four health care systems. Am. J. Psychiatry. 2019;176:846–855. doi: 10.1176/appi.ajp.2019.18091085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis D.A., Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu. Rev. Neurosci. 2002;25:409–432. doi: 10.1146/annurev.neuro.25.112701.142754. [DOI] [PubMed] [Google Scholar]
- 41.Laursen T.M., Nordentoft M., Mortensen P.B. Excess early mortality in schizophrenia. Annu. Rev. Clin. Psychol. 2014;10:425–448. doi: 10.1146/annurev-clinpsy-032813-153657. [DOI] [PubMed] [Google Scholar]
- 42.Fazel S., Danesh J. Serious mental disorder in 23 000 prisoners: a systematic review of 62 surveys. Lancet. 2002;359:545–550. doi: 10.1016/S0140-6736(02)07740-1. [DOI] [PubMed] [Google Scholar]
- 43.Fazel S., Geddes J.R., Kushel M. The health of homeless people in high-income countries: descriptive epidemiology, health consequences, and clinical and policy recommendations. Lancet. 2014;384:1529–1540. doi: 10.1016/S0140-6736(14)61132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Munafò M.R., Tilling K., Taylor A.E., Evans D.M., Davey Smith G. Collider scope: when selection bias can substantially influence observed associations. Int. J. Epidemiol. 2018;47:226–235. doi: 10.1093/ije/dyx206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martin J., Tilling K., Hubbard L., Stergiakouli E., Thapar A., Davey Smith G., O’Donovan M.C., Zammit S. Association of genetic risk for schizophrenia with nonparticipation over time in a population-based cohort study. Am. J. Epidemiol. 2016;183:1149–1158. doi: 10.1093/aje/kww009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Taylor A.E., Jones H.J., Sallis H., Smith G.D., Lawlor D., Davies N.M., Zammit S., Lawlor D.A., Munafò M.R., Davey Smith G., et al. Exploring the association of genetic factors with participation in the avon longitudinal study of parents and children. Int. J. Epidemiol. 2018;47:1207–1216. doi: 10.1093/ije/dyy060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fry A., Littlejohns T.J., Sudlow C., Doherty N., Adamska L., Sprosen T., Collins R., Allen N.E. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am. J. Epidemiol. 2017;186:1026–1034. doi: 10.1093/aje/kwx246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muthén L.K., Muthén B. 2016. Mplus. The Comprehensive Modelling Program for Applied Researchers: User’s Guide. [Google Scholar]
- 49.Loh P.-R., Tucker G., Bulik-Sullivan B.K., Vilhjálmsson B.J., Finucane H.K., Salem R.M., Chasman D.I., Ridker P.M., Neale B.M., Berger B., et al. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nat. Genet. 2015;47:284–290. doi: 10.1038/ng.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abraham G., Qiu Y., Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics. 2017 doi: 10.1093/bioinformatics/btx299. [DOI] [PubMed] [Google Scholar]
- 51.Winkler T.W., Day F.R., Croteau-Chonka D.C., Wood A.R., Locke A.E., Mägi R., Ferreira T., Fall T., Graff M., Justice A.E., et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 2014;9:1192–1212. doi: 10.1038/nprot.2014.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bulik-Sullivan B.K., Finucane H.K., Anttila V., Gusev A., Day F.R., Loh P.-R., Duncan L., Perry J.R.B., Patterson N., Robinson E.B., et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015;47:1236–1241. doi: 10.1038/ng.3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shi H., Kichaev G., Pasaniuc B. Contrasting the genetic architecture of 30 complex traits from summary association data. Am. J. Hum. Genet. 2016;99:139–153. doi: 10.1016/j.ajhg.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi H., Mancuso N., Spendlove S., Pasaniuc B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am. J. Hum. Genet. 2017;101:737–751. doi: 10.1016/j.ajhg.2017.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe K., Taskesen E., Van Bochoven A., Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017;8:1826–1911. doi: 10.1038/s41467-017-01261-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Leeuw C.A., Mooij J.M., Heskes T., Posthuma D., Visscher P., Brown M., McCarthy M., Yang J., Manolio T., Collins F., et al. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015;11:e1004219. doi: 10.1371/journal.pcbi.1004219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Wheeler H.E., Torres J.M., Torstenson E.S., Shah K.P., Garcia T., Edwards T.L., et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825–1920. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ge T., Chen C.-Y., Ni Y., Feng Y.-C.A., Smoller J.W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang C.C., Chow C.C., Tellier L.C.A., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheng Y., Wikle C.K. Bayesian multidimensional IRT models with a hierarchical structure. Educ. Psychol. Meas. 2008;68:413–430. [Google Scholar]
- 62.Altshuler D.M., Gibbs R.A., Peltonen L., Altshuler D.M., Dermitzakis E., Schaffner S.F., Yu F., Dermitzakis E., Bonnen P.E., de Bakker P.I.W., et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu L., Bentler P.M. Cutoff criteria for fit indexes in covariance structure analysis: conventional criteria versus new alternatives. Struct. Equation Model. a multidisciplinary J. 1999;6:1–55. [Google Scholar]
- 64.Auton A., Abecasis G.R., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A., et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cai N., Revez J.A.A., Adams M.J., Andlauer T.F.M., Breen G., Byrne E.M., Clarke T.-K., Forstner A.J.J., Grabe H.J.J., Hamilton S.P., et al. Minimal phenotyping yields gwas hits of reduced specificity for major depression. BioRxiv. 2019:440735. doi: 10.1101/440735. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aguet F., Barbeira A.N., Bonazzola R., Brown A., Castel S.E., Jo B., Kasela S., Kim-Hellmuth S., Liang Y., Oliva M., et al. The GTEx Consortium Atlas of Genetic Regulatory Effects across Human Tissues. BioRxiv. 2019:787903. doi: 10.1126/2Fscience.aaz1776. Preprint at. [DOI] [Google Scholar]
- 68.Giusti-Rodriguez P.M.D., Sullivan P.F. Using three-dimensional regulatory chromatin interactions from adult and fetal cortex to interpret genetic results for psychiatric disorders and cognitive traits. BioRxiv. 2019:406330. doi: 10.1101/406330. Preprint at. [DOI] [Google Scholar]
- 69.Zhao B., Luo T., Li T., Li Y., Zhang J., Shan Y., Wang X., Yang L., Zhou F., Zhu Z., Zhu H. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat. Genet. 2019;51:1637–1644. doi: 10.1038/s41588-019-0516-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Watanabe K., Stringer S., Frei O., Umićević Mirkov M., de Leeuw C., Polderman T.J.C., van der Sluis S., Andreassen O.A., Neale B.M., Posthuma D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019;51:1339–1348. doi: 10.1038/s41588-019-0481-0. [DOI] [PubMed] [Google Scholar]
- 71.Hawrylycz M.J., Lein E.S., Guillozet-Bongaarts A.L., Shen E.H., Ng L., Miller J.A., Van De Lagemaat L.N., Smith K.A., Ebbert A., Riley Z.L., et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–399. doi: 10.1038/nature11405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Klein A., Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front. Neurosci. 2012;6:171. doi: 10.3389/fnins.2012.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romero-Garcia R., Whitaker K.J., Váša F., Seidlitz J., Shinn M., Fonagy P., Dolan R.J., Jones P.B., Goodyer I.M., Bullmore E.T., Vertes P.E. Structural covariance networks are coupled to expression of genes enriched in supragranular layers of the human cortex. Neuroimage. 2018;171:256–267. doi: 10.1016/j.neuroimage.2017.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table. This paper does not report custom code or software. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.