

Abstract
Natural metalloproteins perform many functions — ranging from sensing to electron transfer and catalysis — in which the position and property of each ligand and metal, is dictated by protein structure. De novo protein design aims to define an amino acid sequence that encodes a specific structure and function, providing a critical test of the hypothetical inner workings of (metallo)proteins. To date, de novo metalloproteins have used simple, symmetric tertiary structures — uncomplicated by the large size and evolutionary marks of natural proteins — to interrogate structure-function hypotheses. In this Review, we discuss de novo design applications, such as proteins that induce complex, increasingly asymmetric ligand geometries to achieve function, as well as the use of more canonical ligand geometries to achieve stability. De novo design has been used to explore how proteins fine-tune redox potentials and catalyse both oxidative and hydrolytic reactions. With an increased understanding of structure-function relationships, functional proteins including O2-dependent oxidases, fast hydrolases, and multi-proton/multi-electron reductases, have been created. In addition, proteins can now be designed using xeno-biological metals or cofactors and principles from inorganic chemistry to derive new-to-nature functions. These results and the advances in computational protein design suggest a bright future for the de novo design of diverse, functional metalloproteins.
TOC summary
This Review describes the de novo design of metalloproteins, which perform numerous functions essential to life. By understanding, the relationship between the symmetry of the protein structure and the metal active site, we can design novel, functional metalloproteins from scratch.
Introduction
Metalloproteins perform diverse functions that are essential to life, including electron transfer,1 transition metal ion transport/storage,2 gas sensing/transport3 and the catalysis of difficult transformations.4 This impressive range of functions is performed with the limited toolbox of earth-abundant metals and biosynthetically accessible ligands. Given these limitations, the ability of proteins to control not only the primary coordination environment, but also the secondary and tertiary structure at the metal site is critical to success.5 De novo protein design can provide insight on all three levels of structure and allows us to build a knowledge base that should let us reproduce, or even surpass, the achievements of nature.6–11
Successful de novo design of metalloproteins requires an understanding of the relationship between the secondary and tertiary structure of the protein and the desired primary structure around the metal center. There is a push and pull between these factors: the protein superstructure can enforce a coordination geometry on the metal ion12 or the coordination preference of the metal ion can enforce its geometric preference on the protein tertiary or quaternary structure.13 De novo protein design can elucidate this push and pull,14 and recapitulate the structural and functional properties of many metal centers seen in nature.7,15–17 Thereby, designers are now able to generate proteins using metal ions and metallocofactors not found in nature.
Different metalloprotein functions pose distinct challenges to the protein designer. The simplest designs are for structural sites, which serve to increase the thermodynamic stability of a protein. To achieve maximal stabilization, these sites tend to have coordinative saturation and idealized geometries with strong metal–ligand bonds.18 Similarly, highly stable ligand geometries are also often found in allosteric sites that respond to metal ion binding, such as Ca2+. A second challenge is the design of proteins that function in multiple states. These include electron transfer proteins, which tune redox potentials and minimize changes to the coordination geometry between different redox states – thereby lowering the reorganization energy, and tuning the electron transfer rate.19,20 Similarly, ligand binding proteins, such as those involved in O2 transport or small-molecule sensing, facilitate active site access for small molecules, feature vacant or labile ligand sites, and balance the energetics of the bound and unbound state.16 A final level of complexity is observed in catalysts that bind to and act on substrates such as small organic molecules.21 In this case, a cavity must be introduced near the active site to accommodate the substrates, which is energetically destabilizing and requires a highly stable underlying tertiary structure. Stability can be achieved by precise positioning of polar residues within the binding site, which aid catalysis and binding. Moreover, the protein must be protected against undesired modification by strongly reactive species formed during turnover. Nonetheless, natural proteins commonly use these features to achieve a many challenging transformations with high regio- and stereo-selectivity, and de novo design is now beginning to scratch the surface of nature’s skill set.
Metalloprotein design
When beginning metalloprotein design, it is first necessary to have a clear vision of the function you wish to explore. The desired function leads to a proposed metalloprotein active site with appropriate geometric constraints and ligands. These constraints can be sourced informatically from databanks, such as the Protein Data Bank (PDB)22,23 or the Cambridge Crystallographic Data Centre (CCDC ).24 Alternatively, they can be derived from quantum mechanical calculations on the metal site, which include its full ligand geometry and bound substrate(s).25 Here, a protein scaffold is selected that is capable of precisely positioning each ligand in the desired geometry. This entails: 1) identifying a protein tertiary structure capable of positioning sidechains appropriately and 2) stabilizing the fold and active site ligands in the appropriate geometry.
There are generally two distinct methods to achieve the appropriate tertiary structure: selection from a large library of natural proteins or to build the tertiary structure from scratch using mathematical parameterizations or fragment assembly. With respect to the first approach, we refer the reader to appropriate reviews on this subject.26–33 We focus on the second approach, de novo design, which most critically tests our understanding of both structure and function. Furthermore, some cofactor targets might not fit into any natural protein tertiary structure. For example, nature does not provide scaffolds that are appropriate for highly elongated cofactors with covalently linked multi-porphyrin or other porphyrin-cofactor assemblies; however, they can be accommodated in elongated helical bundles.34,35
Once a library of tertiary structures has been selected, a search is performed to identify sites that can precisely position the ligands in the desired geometry. This can be done in a forward direction — all the rotamers of the desired ligating residues at each possible site of the tertiary structure are scored against: a function that incorporates the energy of the rotamer; the agreement with the geometric constraints; and the spatial positioning of the metal ion.36,37 Candidates for convergent binding sites can be further filtered to ensure there are no steric clashes and the desired geometry has been achieved. An alternative approach is to begin with the metal ion geometry and build backwards to find positions where ligating sidechains can attach to backbone atoms in low-energy rotameric configurations (referred to as rotamer interaction fields).38,39 This approach is akin to the “inside-out” design approach, in which an idealized transition state geometry, known as a theozyme, is defined using density functional theory (DFT). An exhaustive search of known backbones (that is, PDB or parameterized coiled coils) is then performed to assess which backbone best accommodates the amino acid side chains necessary to stabilize the theozyme.40
Irrespective of the method employed, it is important to check that the site does not have accessible geometries that are lower in energy than the desired one. This consideration is particularly important for sites that bind metal ions in somewhat distorted or unusual geometries, which represents one aspect of the general approach of negative design. Wherein, not only the desired outcome needs to be stabilized, but also the undesired states destabilized.41–43
Even with the discussed constraints, the number of possible metalloprotein structures is large. This presents exciting opportunities for the designer, particularly as computational power increases. However, it also presents significant challenges to search and score the conformational space for plausible designs. Thus, most designs have emphasized the use of parameterizable protein backbones, which have structures that can be specified with a limited number of adjustable parameters (Box 1). Parametric approaches can identify pools of hyperstability on the conformational landscape and allow for interrogation of the fundamental relationship between the metal ion and protein structure. Coiled coils and helical bundles are particularly easily parameterized, so it is not surprising that most work on the de novo design of metalloproteins has focused on these classes of tertiary structures.44
Box 1 |. Coiled coil fundamentals.
Mathematical terms for coiled coil parameterization
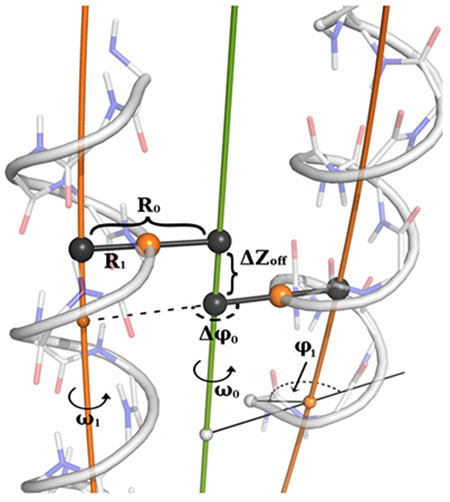
Superhelical radius (R0) is the distance from the central major axis of the coiled coil (green) to the axis helix (orange).
Helical radius (R1) is the distance of closest approach from the axis of the major helix (the superhelix) to a point on the minor helix (here, the α-helix)
Superhelical frequency (ω0) is a measure of the angular rotation of the superhelix, measured about the central axis of the coiled coil (2.9°/residue for an idealized left-handed coiled coil.
Helical frequency (ω1) characterizes the angular rotation of the minor helix around its local axis with each residue. For a canonical coiled coil, this value is approximately 102.8 °/residue.
Chain axial offset (ΔZoff) is the shortest displacement along the central axis between an inward-facing point on one helix and the closest point on an adjacent helix. The sign is set by whether the second helix is shifted in the N-terminus to C-terminus direction (+) or C-terminus to N-terminus direction (−) relative to the first helix.
The superhelical phase offset (Δφ0) is the angular rotation of a minor helix relative to the first helix in a coiled coil. In an idealized, symmetrical, parallel 2-stranded coiled coil it is 180°. In four-helix bundles this parameter can control the shape of a coiled coil (that is, square bundle vs. rectangular bundle).
The starting helical phase (φ1) measures the starting angular register of the α-helix in a coiled coil, controlling the projection of the first residue, relative to the center of the bundle.
To get a more hands-on understanding of these mathematical parameters, the authors suggest that new designers use the CCCP tool (Box 1) to adjust parameters by hand and visualize the changes each parameter endows on a coiled coil.
Reprinted with permission from ref. 1, Elsevier.
Coiled coil fundamentals
The straightforward parameterization (Box 1)45,46 and inherent symmetry47 of coiled coils allows the interplay between the protein and metal center to be explored without the complexity and ambiguity of more complex and irregular tertiary structures.48 The parameterization and symmetry of coiled coils has been reviewed elsewhere,49–51 so will only be described briefly here.
The most commonly observed coiled coils in nature52,53 have left-handed superhelical twists and are defined by a repeating seven-residue geometric repeat, (abcdefg)n (Fig. 1a–b).54 The sidechains of the a- and d-positions project towards the central axis of the bundle – they tend to be apolar and drive the assembly through hydrophobic interactions along the cylindrical core (Fig. 1a–b).41,55–58 However, the a and d positions can also harbor polar residues that serve as metal ligands in both natural and designed proteins. The e- and g-residues are generally partially buried at the helix-helix interfaces, where they can form stabilizing interactions with each other. The more exposed residues at the b-, c-, and f-positions often define the solubility properties.59 In metalloproteins, the e- and g-residues also frequently feature polar residues that form second-shell interactions with primary ligands at the a- and d-positions. The association state and topology (parallel vs. antiparallel) of the bundle is defined by a variety of features that include but are not restricted to: steric packing of a-, d-, e- and g-positions;60,61 buried H-bonding62–64 and metal-binding interactions at a- and d-positions; exposed salt bridges between residues at e-, b-, c- and g-residues;65–67 and the presence or absence of loops connecting the helices.68–70
Fig. 1 |. Three- and four-helix bundles.
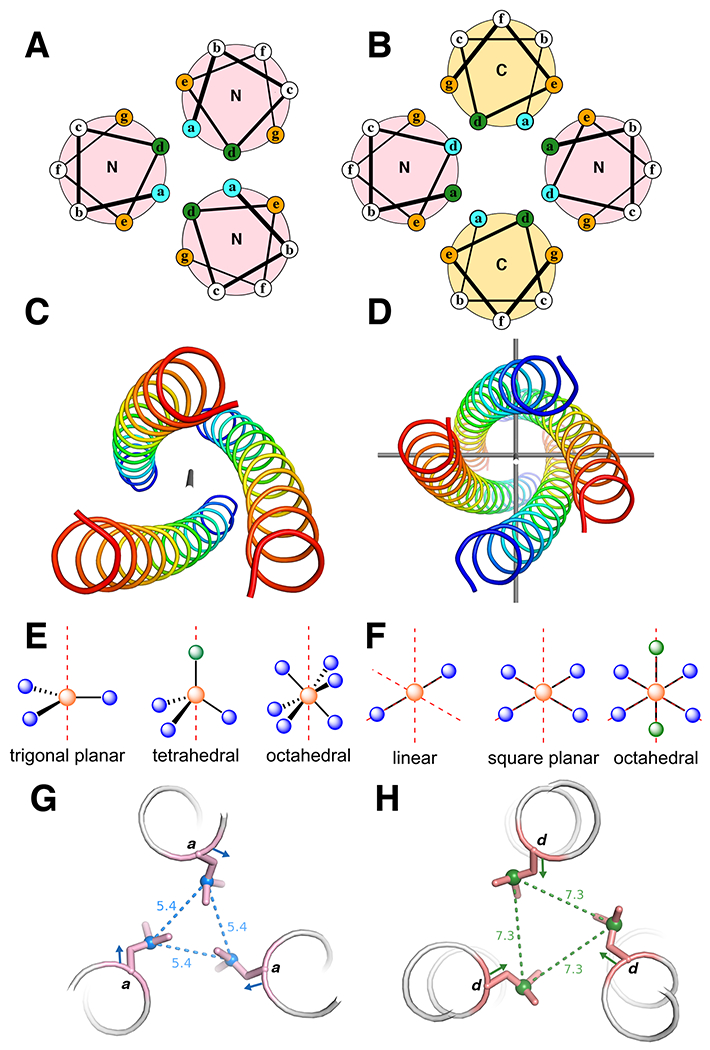
a | Helical wheel diagrams of parallel three-helix bundles. N and C are the terminal ends of the helices. b | Helical wheel diagrams of anti-parallel four-helix bundles. a and b show the heptad arrangements. The buried residues (green and blue) are packed against each other in the core, the orange positions are at the helical interface, and the white positions are on the surface. The N (blue) and C (red) termini of each helix are labelled to show directionality. c |A parallel three-helix bundle with idealized C3-symmetry. The axis of symmetry (in gray) traverses the center of the bundle. d |An antiparallel four-helix bundle with idealized D2-symmetry. The three axes of C2-symmetry are shown in gray. e, f |Examples of idealized coordination geometries accessible in e C3-symmetric three helix bundles and f D2-symmetric four helix bundles. The red axes represent the rotational symmetry axes. Orange spheres are the metal ions, blue spheres are coordinating ligands, and green spheres are empty coordination sites. g, h |Illustration of the difference between an a- g and a d-position h with respect to sidechain orientation. The coloured vector indicates the Cα-Cβ bond direction and the dashed lines and distances (Å) indicate the Cγ-Cγ’ vector.
If the helices are arranged in a parallel orientation, the idealized bundle symmetry is Cn (in which n is equal to the number of helices, Fig. 1c). The two most important adjustable parameters are the radius (R0), which affects the inter-helix distance, and the α-helical phase (φ1), which controls the twist of the individual helices relative to the super helical axis of the entire bundle. Additional parameters include the superhelical frequency (ω0) and pitch angle (α). In idealized coiled coils, the superhelical frequency is a constant defined by the difference between the alpha-helix geometry (100 degrees per residue) and the repeat of the coiled coil, and the pitch angle, which is a function of the radius and the superhelical frequency. Both of these parameters can be varied in the design of coiled coils that deviate from ideality. In parallel coiled coils, the interior facing a- and d-residues lie in alternating layers (Fig. 1a), in which planar arrays of metal-binding residues form (primarily Cys, His, Asp, and/or Glu, Box 2) and project from either of these two positions. The a- and d-positions differ in the orientation of their side chains and thus, the extent to which they pre-arrange the metal binding site. The Cα-Cβ vector of an a-position points toward the helical interfaces (Fig. 1g), and, thus, the Cβ-Cγ vector in the lowest energy rotamer points towards the center of the coiled coil to fill the cavity. Conversely, the d-position has the opposite characteristics; its Cα-Cβ vector points towards the center of the bundle, while the Cβ-Cγ vector of the preferred rotamer points towards the helical interfaces resulting in significantly longer Cγ-Cγ’ distances (Fig. 1f).53 Thus, the introduction of, for example, a cysteine at an a- vs. a d-position is inequivalent with respect to the preorganized geometry for metal binding (see below).
Box 2 |. Bioinorganic fundamentals.
Common metal ions in biology
| Redox Active | Mn | Fe | Ni | Cu |
|---|---|---|---|---|
| Common oxidation states | 2+, 3+, 4+ | 2+, 3+, 4+ | 2+ | 1+, 2+ |
| Geometric preferences | Octahedral, trigonal bipyramidal | Octahedral, tetrahedral, trigonal bipyramidal | Square planar | Square planar, tetrahedral |
| Redox inactive | Zn | Ca | Mg | |
| Oxidation state | 2+ | 2+ | 2+ | |
| Geometric preferences | Tetrahedral | Octahedral, 7-coordinate | Octahedral, 7-coordinate | |
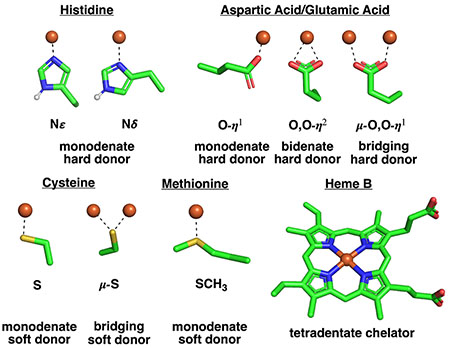
Common ligands and binding modes
The sidechains in antiparallel coiled coils pack into layers composed of residues from both a- and d-positions, which allows the design of more diverse metal coordination sites. For example, antiparallel four-helix bundles tend to place two a- and two d-residues at the corners of a square or rectangle (Fig. 1b). Idealized helical bundles with anti-parallel chains have Dn symmetry (where n is half of the number of helices, Fig. 1d) and two additional degrees of freedom (beyond R0 and φ1): the superhelical phase (Δφ0) and the Z-offset (ΔZoff), which again provides opportunities for diversification of the metal-binding site. The super helical phase controls the placement of the helices relative to one another about the super helical axis, and ΔZoff controls the position of Cα of one heptad position relative to its counterpart on an adjacent helix (that is, a vs a’), allowing helices to be slid up or down the Z-axis to attain better packing or metal-ligand interactions.
We emphasize the bundle symmetry because it defines the set of possible coordination geometries. In homomeric coiled coils, the idealized coordination geometry must contain a common symmetry element that is coincident with the approximate symmetry of the underlying alpha-helical bundle (Fig. 1e–f). For example, a metal lying on the C3-rotation axis running down the bundle of a three-stranded parallel coiled coil could occupy either a coordinatively saturated trigonal planar geometry, a tetrahedral geometry with one vacant site, or an octahedral geometry with ligands from two layers (Fig. 1e). The exact geometry will be determined by the metal ion, ligand choice, and ligand placement. In some cases, the protein fold will enforce a certain geometry on the metal center (entatic state),12 and, in other cases, the metal coordination will enforce a fold on the protein (allostery).13
Finally, it is noteworthy that, while many natural and designed helical bundles are far more asymmetric than a coiled coil, the overall rubric of the heptad repeat is often helpful to analyze helix packing and the local environment around the binding site. Moreover, although the 7-residue repeat of ideal left-handed coiled coils only have two interior-facing residues (a and d), whose projection is restrained by the structure of the coil, the structure can be deliberately varied through insertions and deletions within a single heptad repeat.71–74 Greater diversity could also be achieved by using alternative ideal helical bundle geometries.54,75 Other, largely untapped idealized bundles include a right-handed structure with 11-residues per repeat (that is, three α-helical turns per repeat), which results in 3 geometrically distinct, inwardly focused layers, and straight bundles with 18 residue repeats with five distinct internal layers.49,51,76,77
Examples in Nature
Biology has also exploited coiled coils extensively for the formation of metalloproteins that illustrate many of the above principles. These examples illustrate three tiers of de novo metalloprotein design difficulty: 1) structural stabilization, where the metal ion plays an energetic role in assembly of the secondary, tertiary, and/or quaternary structure of the protein; 2) functional metal sites, in which the metal centre is capable of performing function beyond binding, such as electron transfer or small molecule binding, which require the protein to stabilize a particular, often non-preferred geometry at the expense of structural stability; and 3) catalytic active sites for organic transformations, in which binding sites for both the metal and an organic substrate must be carefully designed to achieve the desired reactivity.
A simple example involves the modification of bundles resembling classical parallel coiled coils. In the C4-symmetric K+ ion channel KCa3.1 (PDB: 6D42, Fig. 2a), a metal binding site is generated by the introduction of histidines at an a-layer. This placement generates a square planar (locally C4v) metal binding site that can be occupied by Cu2+, thereby inhibiting K+ conduction. In this case, the preferred coordination geometry of the metal center structurally stabilizes this His4 motif.
Fig. 2 |. Crystal structures of natural metalloproteins illustrating symmetry elements in helical bundles.
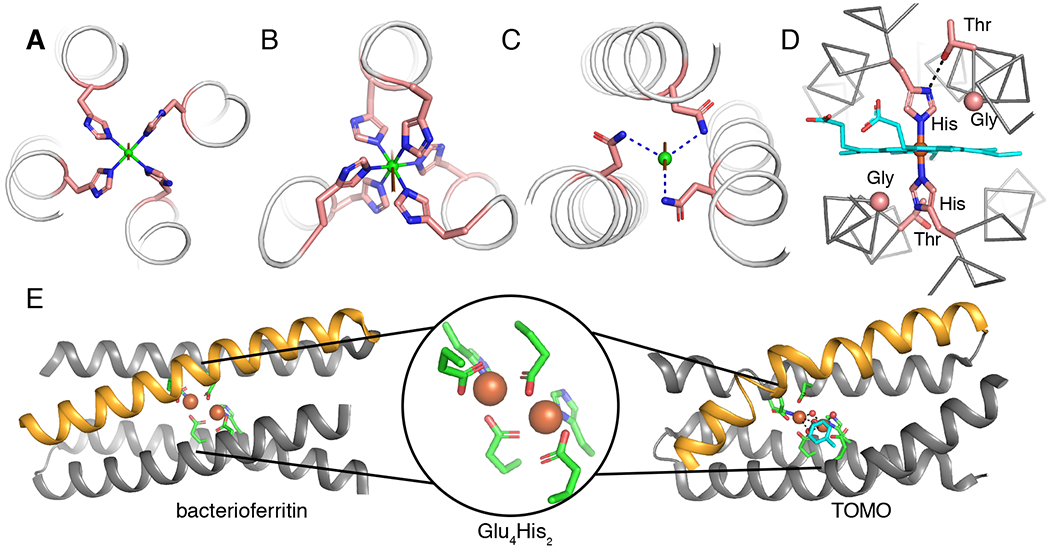
a |A C4-symmetric Cu (green) binding site with a square planar coordination geometry formed by four HIs at a-positions in a parallel helical bundle (PDB: 6D42). b |A C3-symmetry structure with an octahedral Ni (green) coordination geometry generated by His at adjacent a- and d-positions in a parallel helical bundle (PDB: 3NTN).78 c |A C3-symmetry site with a tetrahedral geometry (vacant site on the C3-axis) formed by three Asn residues H-bonded to a chloride (green).78 d |The D2-symmetric heme-binding site from cytochrome bc1 (PDB: 2A06) showing first- and second-shell interactions critical for function. Gly residues are shown as Cα spheres and the heme cofactor is shown in cyan.246 e |D2-symmetric di-Fe binding sites in bacterioferritin (left, PDB: 4AM2)88 and toluene monooxygenase (TOMO) (right, PDB: 5TDT)90 with a representation of the Glu4His2 coordination environment (middle). The orange helices illustrate the ideal helix in bacterioferritin versus the pi-bulge creating a substrate access site in TOMO. The structure of TOMO shows the bound oxidized toluene intermediate (cyan). Dashed lines represent H-bonds and the remainder of the protein structures are hidden for clarity.
Since the symmetry of a bundle is consistent with any coordination geometry that contains the appropriate symmetry element, multiple distinct binding sites can be introduced (Fig. 1). An illustration of this principle can be seen in the structure of the head and neck domain of the UspA1 protein (PDB: 3NTN, Fig 2b–2c).78 The presence of histidine residues at adjacent a- and d-positions of a parallel, three-helix bundle generates an octahedral coordination site for a nickel ion with the C3-axis going through the Ni ion and down the center of the bundle (Fig. 2b). Further down that same axis, a chlorine atom rests above the plane formed by three H-bonded asparagine residues at d-positions (Fig. 2c). One can readily envision a similar approach being used to generate a tetrahedral metal site with the threefold axis (and empty coordination site), again, running down the center of the bundle. Indeed, this approach has been used to great effect in de novo designed metalloenzymes (see below).
To achieve functional metalloproteins, deviations from the ideal symmetry frequently observed in structural metal sites is often necessary. For example, many natural heme-containing helical bundles, such as cytochrome b in the cytochrome bc1 complex, have a specific structural motif.79–81 These electron transfer proteins contain one (or more) heme cofactors that are ligated by two His residues, which are located at d-positions on two pseudo-C2 symmetric (Fig. 2d) helical hairpins that may be the product of gene duplication.81 In each hairpin, one helix contains a ligating His, while the other can contain a Thr or Ser residue to accept a H-bond from the N∂ of the ligating His. This interaction helps lock the imidazole ring in the desired ligation geometry, and presumably also contributes to tuning the redox potential. In addition, Gly residues can be found following the Thr/Ser where the heme ring approaches the helix — these small residues are important in heme packing and overall function.82,83 Taken together, these motifs are responsible for tuning the redox potential of heme cofactor(s) and controlling electron transfer rate. To elicit functions such as gas sensing or C-H bond activation, nature has perturbed the symmetrical ideality of the binding site. Not only must there be an open (or labile) coordination site on the heme iron, but often the helical bundle tertiary structure must be considerably altered. Many catalytically functional 4-helix bundle heme proteins have a more asymmetrically positioned binding site, while others adopt a drastically different tertiary structure (that is, globin-fold or PAS domains) to accommodate both a unique metal ligand sphere and substrate accessibility.84 From a design standpoint, this presents a challenge in building more advanced function in a helical-bundle scaffold.85
Non-heme diiron proteins are also commonly found in four helix bundles and demonstrate helical asymmetry as a strategy to introduce function.86,87 Many non-heme diiron proteins feature a similar, approximately C2V-ligand set of two histidines and four carboxylates. In bacterioferritins, which are used for iron storage, we see near ideal D2 symmetry of the bundle (Fig. 2e, PDB: 4AM2).88,89 However, although the coordination geometry is the same in enzymes such as soluble methane monooxygenase (sMMO) and toluene monooxygenase (TOMO),90 a bulge near the diiron center site lowers the helical symmetry (Fig. 2e, PDB: 6VK5).91,92 This destabilizing pi-bulge is essential to function as it fine-tunes the geometry and dynamics of the protein,93,94 and widens the helix–helix interface to facilitate access to the metal center for an organic substrate. This helical distortion is demonstrated by trapping reactive intermediates in a crystal state, thus elucidating the mechanism of toluene oxidation in TOMO (Fig. 2e, PDB: 5TDT).
In this review, we focus on de novo designed metalloproteins that provide insight into important structure–function relationships. The simplicity of their folds should help the reader appreciate the logic of de novo metalloproteins and provide a springboard for new investigators to participate in the nascent field of functional metalloenzyme design.
Three-helix de novo metalloproteins
One of the earliest and most illustrative examples of de novo metalloprotein design produced a model of the heavy metal binding protein MerR. This natural metalloprotein features an unusual trigonal planar ligation to Hg95 that could be reproduced in a parallel three helix bundle. This work illustrated the delicate interplay between protein fold, which can impose a geometry on a protein (entatic state), and metal preference, which can impose a conformation on the protein (allosteric assembly). The initial designs were based on the previously constructed CoilSer protein,96,97 which features four heptad repeats (Leua-Lysb-Alac-Leud-Glue-Gluf-Lysg). Charge complementary Glu and Lys residues at the interfacial e- and g-positions encourage trimerization at neutral pH; a low pH disrupts these interactions, which leads to dimerization. The Leu residues at a- and d-positions form the hydrophobic core, while other residues were selected to improve solubility and helicity. The introduction of a coordinating ligand at either the a- or d-position facilitates the formation of buried metal coordination sites in peptides termed Tri-peptides.15 These designed scaffolds exemplify the first tier of design, that is structural metal binding sites where the metal controls assembly and stability.
Mercury has a very strong intrinsic preference to form a linear, two-coordinate geometry with soft thiolate ligands; the addition of a third ligand is generally unfavourable in aqueous solution unless enforced by a preorganized protein scaffold.95,98 In theory, protein folding could generate a high local concentration of thiolate ligands that are spatially pre-arranged in a trigonal planar fashion (Fig. 1 and Fig. 3a–c). Computational modeling of C3-symmetric coiled coils demonstrated that cysteines placed at a-positions form an appropriate binding site, while those in d-positions diverged.98 Subsequent crystal structures of the a- and d-substituted apo-peptides confirmed these conformational predictions (Fig. 3a–b).99 Consistent with the computational predictions and the crystal structures, “Tri”-peptides with cysteines at the a-position bind Hg2+ in a trigonal planar geometry when there is a 3:1 peptide:Hg ratio. However, at lower peptide:Hg ratios or in conditions that favour dimer formation (that is, low pH), two-coordinate Hg species were observed.98 Similar species also occur using the peptide Tyr-Gly-Gly-(Iled-Glue-Lysf-Lysg-Ilea-Glub-Alac)4 (termed IZ-peptide), which contains a cysteine substitution for isoleucine at the a-position of the third heptad, to affirm the generality of this result.100
Fig. 3 |. Tri-peptide scaffolds for metalloproteins design.
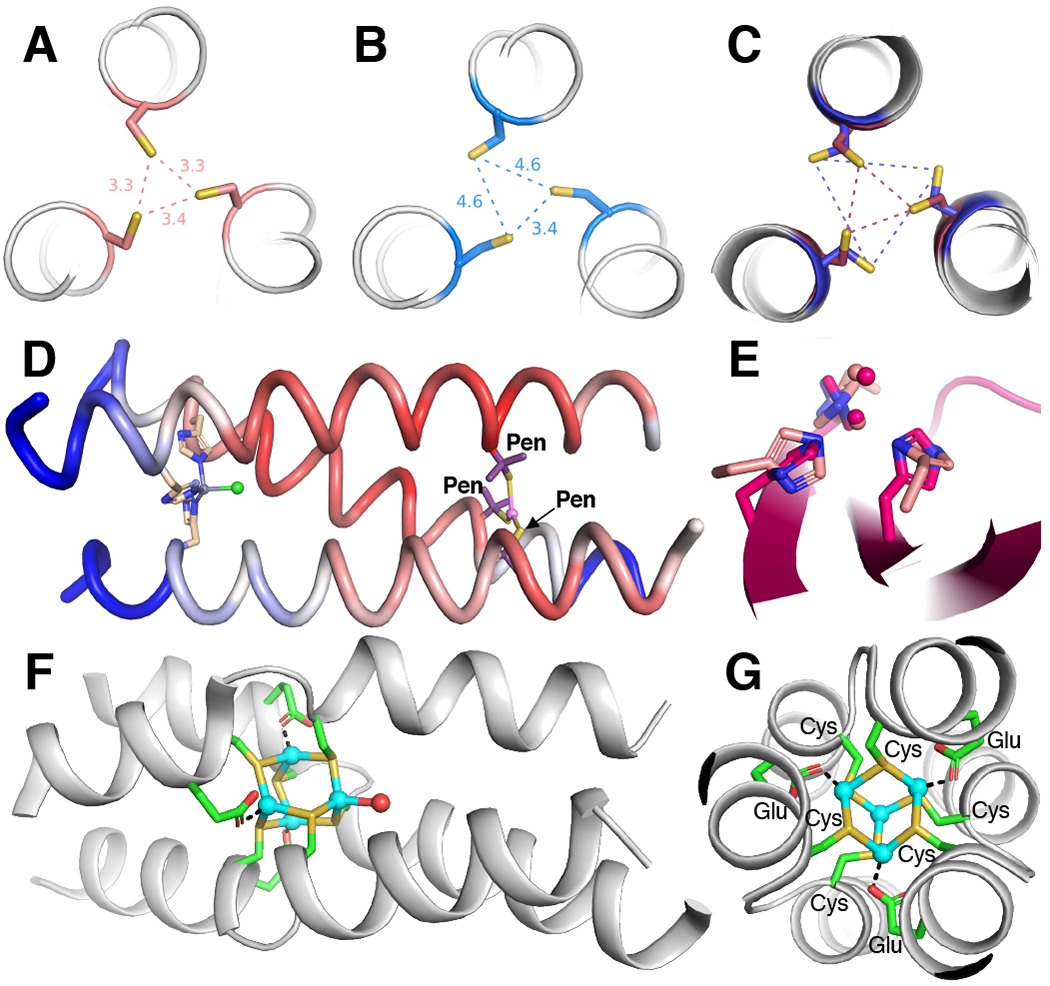
a |Crystal structure of the apo-Tri-peptide substituted with cysteine at an a-position (PDB: 3LJM).99 b |Crystal structure of the apo-Tri-peptide substituted with cysteine at a d-position (PDB: 2X6P).99 One Cys adopts a non-preferred rotamer to facilitate hydrogen bonding. c |Overlay of two crystal structures (PDBs: 3H5F and 3H5G) featuring penicillamine (labelled Pen, β-mercaptovaline, methyl carbons hidden) substituted at an a-position.102 The L-amino acids (maroon) form a small equilateral triangle (maroon dashes) and the D-amino acids (dark blue) form a large equilateral triangle (dark blue dashes). d |Crystal structure (PDB: 3PBJ) of a Zn-carbonic anhydrase mimic with a tetrahedral, catalytic site (left) and a trigonal planar, structural site (right).120 The backbone cartoon is coloured by B-factor with more mobile sites in blue and less mobile sites in red, showing that the catalytic site is more dynamic than the structural metal site. e |Overlay of the tris-histidine active site from a de novo designed (light pink)120 and a natural carbonic anhydrase (dark pink, PDB: 2CBA).247 f |Side-on view of a tetra-Cd2+ cluster showing the disruption of the alpha-helix at the binding site (PDB: 4G1A).248 g |View down the C3-symmetry axis of the tetra-Cd2+ cluster and the three, parallel alpha-helices (PDB: 4G1A).248 Cd2+ ions shown in cyan.
On the other hand, Tri-peptides with cysteines at the d-position generate solely two-coordinate Hg2+ ligation under all conditions.101 Furthermore, the introduction of a noncanonical D-cysteine at the a-position prevents trigonal coordination of Hg2+ (Fig. 3c), which is consistent with our previous discussion on the importance of the Cα-Cβ vector directionality; the D-amino acid essentially converts an a-site into a d-site (Fig. 1g–h).102 These results all suggest that the free energy of protein folding (derived primarily from the burial of hydrophobic residues in the interior of the coiled coil) is sufficient to enforce this unfavourable geometry on the metal center. Consistent with this expectation, the rate and extent of trimerization is altered by peptide length, with longer peptides showing faster and more extensive trimerization. This finding is consistent with a greater expected free energy of folding deriving from the increased number of salt bridges and extent of hydrophobic burial in the longer peptide.103
These studies illustrate important fundamental features of metalloproteins; the structure is determined by a balance between the free energy of protein folding and metal ion ligation (determined by the favourability of the coordination number and geometry). These energies can be similar in magnitude and, depending on the context, can result in either protein-enforced metal geometry (entatic state) or metal-enforced protein fold (structural stabilization and allosteric assembly).104 Thus, As3+, unlike Hg2+, strongly prefers three soft, anionic ligands, which enables Tri peptides with three cysteines at either the a- or d-position to form trimers even at low pH. In this case, the free energy of metal-ligand binding is sufficiently large to compensate for: 1) an energetically unfavourable Cys rotamer required to adopt a trigonal geometry at the d-position, and 2) unfavourable electrostatic interactions within the trimer at low pH.
When the geometry of the metal binding site is not fully consistent with the protein packing, the free energy of metal ligation can play a dominant role to force an unexpected geometry on the bundle. One striking example is a homotrimeric peptide with a Cysa-X-X-Cysd-Glue in which the Glu at the e position, which would ordinarily be at an exterior-facing location, has moved into the interior of the protein to bind a Cd2+ ion, inducing a several-residue break in helical conformation (Fig. 3f–g). A similar Hisa-X-X-Hisd-Glue motif was shown to assemble a tri-Cu+ site in a parallel three-helix bundle with the preferred tetrahedral geometry of Cu+ accommodated by recruitment of a second Glu ligand from another bundle.105 These findings show how peptides can be used as multivalent ligands that assemble to create binding sites that are not always fully anticipated, just as coordination chemists have long used designed ligands to assemble multimetallic complexes in either an empirical or programmatic manner.106–108
The balance between folding free energy and metal-ligand binding free energy can also be used to drive large predetermined conformational changes in protein folding upon metal addition. Negatively charged Asp residues can be introduced into a canonically hydrophobic layer (d-position) of a parallel three helix bundle, to break the helix and form a loop stabilized by an N-capping interaction with the Asp residues. However, this shorter bundle can be switched to a more extended bundle motif by the addition of Ca2+, which stabilizes the interior-facing Asp residues by metal coordination.109 These results demonstrate that designers can use structural metal sites to select a desired fold through the choice of metal ion, as biology does in proteins such as calmodulin and related EF-Hand proteins.110
As noted in Figure 1e, a particular fold symmetry (for example, antiparallel bundle in D2) is consistent with any coordination geometry (for example, square planar) that contains a common symmetry element (for example, a C2 axis down the center of the bundle). This principle is well-illustrated by Cd2+ binding studies.111–114 Cd2+ prefers a tetrahedral geometry and, as a consequence, both trigonal planar (CdS3) and tetrahedral (CdS3(OH2)) coordination modes, which both contain a C3-axis, are observed in the parent Tri-peptide.111–114 By lessening the steric bulk in the hydrophobic layers adjacent to the metal binding site or even in remote layers, a void is created which results in increased occupancy of water in the protein interior and a complete conversion to the intrinsically preferred tetrahedral geometry, at the expense of backbone stability.115 In this way, we can start to see that both the ligands and the hydrophobic core are critical to facilitate the entry of potentially reactive substrates, a prerequisite to catalysis.
Using the full pallet of protein ligands, it is possible to engineer a wider variety of functions. Early work demonstrated that the introduction of a single histidine layer into the hydrophobic core of a protein could also generate trigonal binding sites akin to those in native metalloproteins.116,117 Binding of an apical water at these C3-symmetric sites can also generate a pseudo-tetrahedral metal center similar to the active sites of Zn2+-containing carbonic anhydrase118 and Cu nitrite reductase.119 Therefore, de novo metalloenzymes in non-native folds can recapitulate the activity of native enzymes, if the primary coordination sphere is appropriately recreated (Fig. 3d–e).19
Histidine has a longer side chain than cysteine, therefore a larger superhelical radius (R0) is necessary to accommodate this ligand. The expanded radius worsens the packing of the hydrophobic core resulting in weak metal-ligand binding.116,117 An additional tris-thiolate Pb site can be introduced as a structural metal site to stabilize the overall fold to allow crystallographic characterization.120 Nonetheless, in the crystal structure, increased B-factors (a measure of atom mobility, Fig. 3d) are observed for one helix in the tris-histidine binding site, which suggests local dynamics. The increased flexibility near the Zn2+ site may be critical to enable catalysis given the lack of an obvious substrate pocket in the hydrophobic core. These de novo metalloenzymes are capable of both carbonic anhydrase and ester hydrolase reactivity with observed rates within an order of magnitude of the natural protein. Although, an elevated pH is required (optimal at 9.5), a potential consequence of an absent secondary coordination sphere. Thereby, as the enzymatic activity is only minimally perturbed by the relative or absolute placement of these sites within the coiled coil, it suggests that the activity is largely dictated by the primary coordination sphere.121
Related peptides with a histidine layer, but without a structural metal site, were used to model Cu nitrite reductase.122 Unlike the native enzyme — in which the primary coordination sphere is generated by histidines that are on loop regions — the predictable secondary structure of the coiled coil motif allows for systematic variation of local residues.123 Thorough studies were performed on the effect of exterior charged residues,124 of proximate steric bulk,125 of histidine methylation,126 and of helical distortions on catalytic activity.74 Although, only relatively minimal improvements in catalytic activity were observed, these studies demonstrate the potential of high symmetry scaffolds for interrogation of fundamental structure-function relationships. Moreover, they illustrate key design principles to control the function of designed metal binding sites.
Three-stranded parallel coiled coils are also amenable to the generation of octahedral binding geometry. Substitution of the Ile with His at adjacent a- and d-positions in the third heptad of their IZ-peptide allows the coordination of divalent first row transition metals.127 Peptides can also be designed with similar symmetries to bind xeno-biological metals with biomedical applications.44 The introduction of Asp and Asn mutations at adjacent a- and d-positions on the five-heptad repeat (Ilea-Alab-Alac-Iled-Glue-Asnf-Lysg) produce a pseudo-octahedral, C3-symmetric, tri-anionic binding site (Fig. 4a) suitable for selective coordination to oxophilic trivalent lanthanide ions (for example, Tb3+, Gd3+).128 A helical position and steric bulk in the hydrophobic core has a significant effect on hydration and, in turn, on the physical properties of the lanthanide ions.129–131 These lanthanide-bound coiled coils have significant potential for application in imaging technologies and translate the discussed principles of symmetry and coiled coil formation for the preparation of new-to-nature metalloproteins with novel applications.
Fig. 4 |. Asymmetric active sites in three-helix bundles.
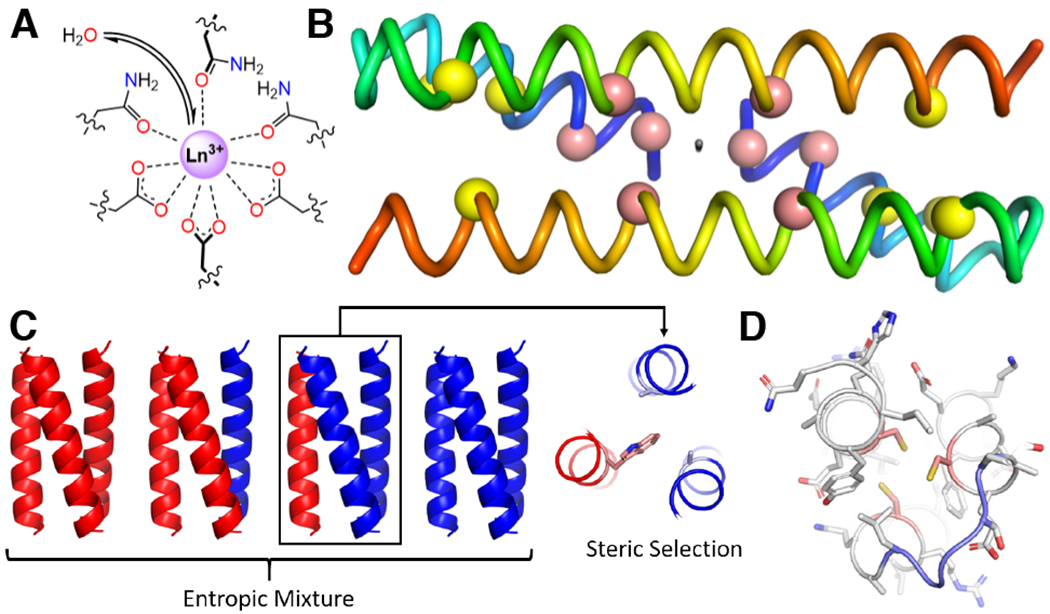
a |Model of the pseudo-C3 symmetric active site in a three-helix bundle. Steric bulk in adjacent layers controls access of water to the lanthanide ion, and thereby the photophysical properties.130 b |Crystal structure of a domain swapped-dimer (PDB: 1G6U)132 with the pseudo-C2 axis shown intersecting the N-termini and the other two helices. The locations of leucine mutations to cysteine to build the two 4Fe-4S binding sites are shown for the first generation (yellow spheres)133 and second generation (pink spheres)134 designs. N-termini are shown in blue and C-termini are shown in red. c |On the left, are the possible parallel, three-helix bundles entropically favoured to assemble upon 1:1 mixing of two different peptides. A model of the knobs-into-holes packing approach shows preferential 2:1 heterooligomer formation by packing a large residue such as tryptophan (pink) against two small residues such as alanine (light blue). d |A single-stranded protein can also be used to generate asymmetry. In this case, secondary structure elements (that is, helices) are connected by loops (light blue) to form a fully asymmetric active site (cysteines in pink).144
An alternative geometry, the domain-swapped dimer (DSD), allows for the self-assembly of three-stranded coiled coils and has been adapted for the de novo design of metalloproteins. The original designed protein (PDB: 1G6U) features two domains each consisting of a long straight helix and a short straight helix connected by a loop. Dimerization results in the two short helices arranged such that their N-termini come together at an interface and pack against the two longer helices, creating two abutting three-helix bundles related by a C2-axis orthogonal to the helical bundle axis.132 This two-fold symmetry can generate metalloproteins with two metal sites at well-controlled distances (Fig. 4b). In particular, the introduction of four Cys residues on each peptide allows for the coordination of two 4Fe-4S clusters whereby the Cys placement controls the inter-cluster distance, which, in turn, effects the electronic coupling and the redox properties.133,134 The replacement of one of the Cys with Leu or Ser causes selective formation of a 3Fe-4S cluster135 that mimics the inactive state of aconitase.136
All of the systems discussed thus far have been homooligomers; however, the ability to fine tune such designs are inherently limited, because any change in sequence is necessarily propagated on each element. While such high symmetry is useful for the construction of metalloproteins, particularly for generating idealized binding sites, the introduction of asymmetry is often highly beneficial to function. One solution is to develop heterooligomers. However, careful design is necessary to avoid the entropically favourable formation of statistical mixtures. Work on non-metal containing scaffolds suggest that electrostatics could be used to encourage heterotrimerization.137,138 In addition, a complementary packing arrangement in which a large tryptophan would preferentially pack against two small residues (that is, Ala) can overcome the entropic penalty of selectively forming A2B heterooligomers (Fig. 4c).139 An approach that was exploited in trimeric coiled coils featured the non-canonical amino acid, γ-carboxy-glutamate, bound to Eu3+.140
Recently, specific heterooligomeric Tri-peptides have been developed that form by packing Leu against Ala in the layer above or below the tris-Cys binding site. Quantum mechanical/molecular mechanical calculations suggest that hetero-oligomerization is not only stabilized by the energetics of knobs-into-holes packing (Fig. 4d), but also by the formation of a cavity that allows the penetration of water molecules into the interior that can then H-bond with the Cys ligands. Thereby, a metalloenzymatic site was introduced into these heteroligomers to further study Zn carbonic anhydrase mimics. The asymmetry now allows comparisons of catalytic performance between systems that are mono-, di-, and tri-substituted in the secondary coordination spheres. Consistent with the native enzyme, the best catalytic performance is observed when a single Thr is introduced.141 This work shows the power of asymmetry to achieve function in de novo metalloenzymes.
A more general approach to achieve asymmetry is to loop secondary structure elements into single-chain proteins. This topology renders the entire sequence independently designable. Moreover, well-chosen loops can enhance the stability of the desired fold, potentially mitigating the destabilizing effects of introducing polar residues or cavities into the hydrophobic core. However, to connect two helices with short loop sequences they must be antiparallel to one another. Therefore, a parallel, C3-symmetric bundle is no longer possible. Indeed, the helix-loop-helix-loop-helix motif are only pseudo-Cs (that is, with a σH mirror plane and no other symmetry elements relating the ligating atoms) if the chirality of each helix and the loops themselves is omitted; thus, the looped systems are truly C1 symmetric, and their sequence design space is consequently far larger, which makes the design more reliant on computation than the symmetric systems discussed previously.
Alpha-3d is a de novo designed, 73 residue protein consisting of three helices and two loop regions and was one of the first structurally characterized de novo proteins (Fig. 4d).142 A tris-Cys heavy metal binding site could also be introduced into this protein through mutations of three hydrophobic residues to Cys.143 Consistent with the lower protein symmetry, the metal centre, Hg, adopted an approximately Cs-symmetric, T-shaped geometry;144 the loops provide additional flexibility to the design of coordination geometries. The introduction of a fourth Cys into alpha-3d, in this case originating from one of the loops, allows a pseudo-tetrahedral site to form that can bind Fe. This protein mimics the active site of rubredoxin and indeed closely matches the spectroscopic parameters for the native protein. However, this non-native fold does not provide the extended H-bonding network observed in the native beta-hairpin loop environment. Thereby, the redox stability of the construct is reduced, again emphasizing the importance of the secondary coordination environment on function.145 As expected, fully asymmetric binding sites can now also be designed. In particular, a partially functional red-Cu site (two His and one Cys ligand) can be recapitulated in a non-native fold.146–148
Functional 4-helix metalloproteins
The de novo design of four-helix bundles has greatly impacted our understanding of natural metalloproteins and has set a strong foundation for the development of novel functions. Four-helix coiled coils are prevalent in nature as dinuclear metal and heme-binding sites. Many native dinuclear metalloproteins, with otherwise low sequence homology and diverse functionality, feature a Glua-Xxxb-Xxxc-Hisd motif in which two copies of this motif are incorporated in a four-helix bundle with approximate D2 symmetry.149,150 The low sequence homology, diverse function, and relatively high symmetry of the fold provided an excellent test of de novo design to produce well-structure, functional metalloproteins from scratch. Proteins can now be designed that model the cofactor environment in dinuclear metallonenzymes, such as ribonucleotide reductase (RNR), TOMO, and MMO, which feature Glu/Asp and His ligands in four-helix coiled coils.17
The first design, DF1 (Due Ferri 1), consisted of the self-assembly of two non-covalently associated helix–turn–helix motifs to form a four-helix bundle.150 On each helix–turn–helix, two Glu residues were placed at a-positions and one His was placed at a d-position (Fig. 5a, although DF1 was not originally designed as a coiled coil, the heptad nomenclature is useful to illustrate approximate positions of sidechains). These residues provide a Glu4His2 binding environment around two metal ions (Fig. 5b) reducing the D2 symmetry of the bundle to C2. Additionally, an Asp residue was placed in an intermediate g position as a second-shell H-bond to the His ligand. Along with these polar residues, the adjoining hydrophobic core was packed as tightly as possible to maximize the stability of the bundle and had polar residues at exterior positions.150 This protein was stable and well-folded; however, it did not allow access for substrates other than the dimetal cofactor. Therefore, a Leu residue — that sterically prevents cofactor access — was mutated to either Ala or Gly to open a channel to allow binding of small substrates to the di-Mn or di-Fe centers. However, these mutations lowered the amount of solvent accessible surface area that was buried upon folding, which, in turn, destabilized the protein.151 These data demonstrated that in both designed and native metalloenzymes other elements of the protein must stabilize the inherently destabilizing elements necessary for function.
Fig. 5 |. Functional metalloproteins with four-helix bundles.
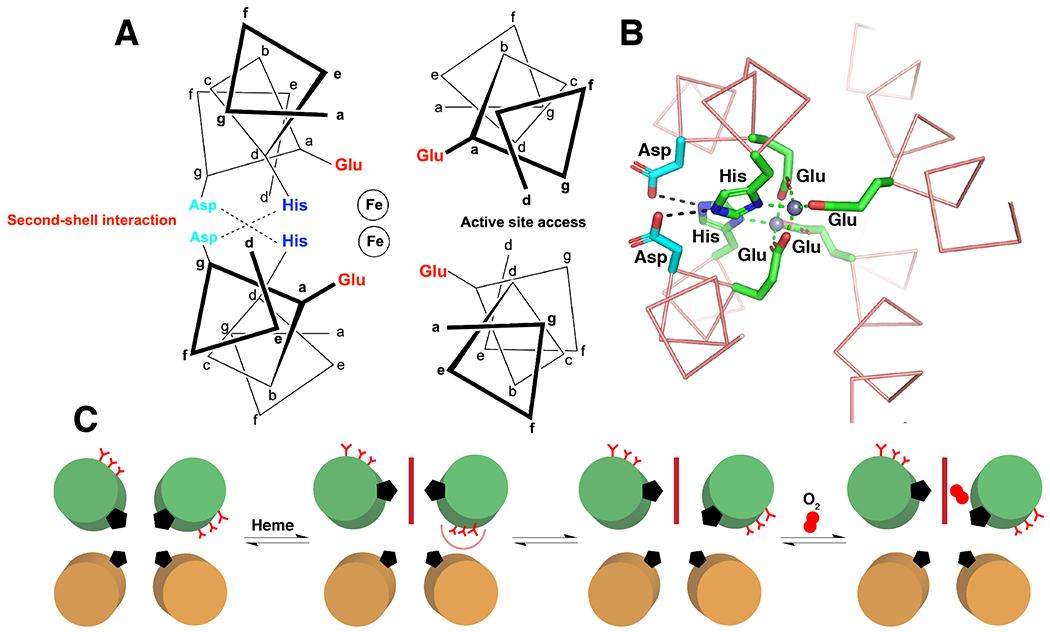
a |Diagram showing the helical backbone and heptad positions chosen for ligating residues and second-shell H-bonding interactions.150 b |Solution structure of di-Zn bound Due Ferri 2 (PDB: 1U7M) showing positions of primary shell ligands (green) and second-shell H-bonding residues (cyan). H-bonds are shown as black dashed lines and metal-ligand interactions are shown in green dashed lines.249 c |Schematic illustrating the entatic state designed to allow reversible O2 binding in a 4-helix maquette. Green and brown helices represent the N- and C-terminal ends of the helices. Addition of heme (red rectangle) forces unfavourable burial of Glu residues (red Y), weakening the His-Fe interaction. Exposure to O2 leads to reversible formation of an Fe-O2 species.199 Part a adapted with permission from ref.150, Elsevier. Part c adapted from ref.199, Springer Nature Limited.
The active sites of metalloproteins are generally fully asymmetric, as required for function. One approach to the de novo design of less symmetric proteins is to generate heterotetramers. A combinatorial approach can assess the effects of mutations on the oxidase activity of the diiron protein with a model substrate, such as 4-aminophenol.152 In this reaction, the diiron site alternates between the diferric and diferrous states, to oxidize the substrate and reduce O2 to peroxide, in a mechanism analogous to that for manganese catalases.153 To avoid formation of misassembled heterotetramers, a fully automated design algorithm was developed to consider favourable interhelical interactions, and interactions to destabilize other potential topologies termed “negative design.” Negative design is now a frequent consideration in the de novo design of proteins,43,154–157. Often “positive” design is engaged in the initial design, and the ability of the polypeptide to adopt any alternative folds is tested at the end of the design process using ab initio folding prediction calculations.158 These in silico experiments give a glimpse of a polypeptide chain’s folding preference derived in their apo-form. However, in cases where an explicit design criterion is to stabilize one tertiary structure over an alternative, these assessments can be built into the design process. A clear example is the design of the four-helix, antiparallel Zn2+-H+ transmembrane antiporter, Rocker.156 The high symmetry D2 version of this fold would generate a stable di-Zn2+ protein and prevent ion flux. In contrast, a design that preferred the lower-symmetry C2-state would allow for two symmetric energy wells, in which each well only had a single coordinated Zn2+, with the D2 state now a high-energy intermediate. In this way, Zn2+ could flow along a concentration gradient through the bundle and across the membrane. Thus, the two symmetry states are defined by different backbones and by different metal-binding constraints, whereby the favourability of the D2 vs the C2 state for a given polypeptide sequence could be assessed by molecular dynamics free-energy calculations.159
As discussed for three-helix bundles, symmetry can also be lowered by generating a single chain version of the protein (DFsc; DF single chain). By adding loop motifs, DFsc, such as the aforementioned heterotetrameric DF proteins, is suitable for ferroxidase reactivity, such as the oxidation of 4-aminophenol to the iminequinone.160 However, the loops instill considerably greater stability and, along with extending the helical chains, compensate for the destabilizing Gly mutations that are critical for substrate access. During design, it is important to consider the incorporation of an extended, well-packed hydrophobic core and appropriate loops to better stabilize the desired functional region of the metalloprotein.
Ultimately, a fully asymmetric binding site was designed by introducing a third His ligand, to mimic the active site of AurF.161 This substitution decreased ferroxidase reactivity and turned on aniline hydroxylation, akin to the native protein.162 These data provided evidence that these de novo designed enzymes are operating by the same rules that dictate the reactivity of native metalloenzymes supporting the contention that they can be used to discover fundamental structure–function relationships.
There has also been significant progress in the design of proteins that bind a variety of metal ion clusters120,163–165 that are structurally and functionally different from the diiron site featured in the DF proteins. In particular, several groups have adopted the well-known Cys-Xaa-Xbb-Cys (CXXC) binding motif, which is found in several unrelated proteins, to generate de novo designed ferrodoxin models166–169 that recapitulate the C2-symmetry of the natural active site first identified by Dayhoff in the 1970s.170 A related approach binds a Ni2+-(μ2-S•Cys)-[Fe4S4]2+ cluster in an attempt to model the A-cluster in carbon monoxide dehydrogenase.171 Moreover, a short heterochiral peptide with alternating L- and D-amino acids that uses the CXXC motif can form 4Fe-4S clusters that show robust, reversible electron transfer.172 This same CXXC motif has also been adapted to form other metalloclusters, including a multinuclear Cu+ binding site in four-helix bundles.173,174 In addition, by combining a CXXC motif with an HXXH motif on the two neighboring helices a binuclear, purple copper site can form that mimics the CuA site in cytochrome c oxidase.175 Designs also include four helix bundles that coordinate 4Fe-4S clusters without relying on the natural CXXC motif176–180 including examples that can assemble and transfer electrons in vivo. 179,180 It is important to note that these latter designs used physical force fields rather than statistical machine-learning based approaches.181,182
In all of the aforementioned metalloclusters, the metal incorporation and ligand composition were confirmed with spectroscopic data; however, high-resolution structures of the protein complexes were not obtained. Examples that have been structurally characterized are particularly notable and useful for elucidating design principles. For example, the careful incorporation of carboxylate and histidine ligands into a D2-symmetric four helix-bundle can stabilize a tetranuclear Zn cluster. This cluster was stabilized through a complex design of second and third-shell H-bonding interactions, demonstrating how the numerous ligands necessary to support clusters can be challenging to accommodate in such scaffolds.183,184
Proteins featuring multiple metals are also important in the context of protein–protein interface design. These interfaces can be stabilized and/or templated by the careful introduction of appropriate ligands on two or more different protein elements. Important work has been done in this area in the redesign of the surfaces of natural proteins both to bind xeno-biological metals185 and to generate reactive sites.186,187 Particularly interesting from the perspective of symmetry are the large protein assemblies that can be generated by the appropriate choice of metal and ligands on protein surfaces.32,186,188,189 The strategy of using metals to control protein–protein interfaces can also be employed in de novo designed proteins, such as to design helix-loop-helix motifs (that is, helical hairpins) that homodimerize in the presence of Zn2+ (Kd of 4 μM without Zn and 30 nm with Zn).164,190 This protein was later evolved to be a highly functional esterase191 and Diels alderase25 (see below).
In addition to direct ligation of metal ions by amino acids, many natural proteins feature biosynthetically generated cofactors to ligate metal ions. The canonical example, as discussed above, is heme B (Fe protoporphyrin IX). These cofactors are important in ligand sensing and transport, electron transfer, and substrate oxidation; this wide array of functionality demonstrates the influence of the protein microenvironment on tuning heme function. While there have been many successes in developing functional synthetic porphyrins,192,193 simplified de novo designed proteins provide excellent scaffolds to understand the complexities of natural hemoproteins. Moreover, it allows us to build on our understanding to develop new functionalities. However, before delving into designed proteins that bind porphyrins, we wish to also mention the success of hemes with covalently bound peptides, which recapitulate many aspects of larger proteins, including heme peroxidase activity and has been reviewed in the literature.16,194,195
As discussed, many natural heme-proteins contain bis-His binding sites within a four-helix bundle, therefore this strategy was applied to design de novo heme binding proteins, which presents the additional challenge of burying the bulky cofactor while maintaining a well-packed interior. The C2-symmetric bis-His binding motif can be used to prepare synthetic helix-turn-helix peptides that assemble into four-helix bundles.196,197 The Leu-rich α2 homodimeric peptide was used as the parent scaffold and a His residue was placed at a d-position — a bound heme cofactor would sit in the middle of the bundle with negatively charged propionate groups poised to interact favourably with Arg residues on the loop region. Additionally, two buried Leu residues were changed to smaller Val residues and another Leu was changed to an Ala to accommodate the bulky heme group. In the presence of stoichiometric heme, the peptides assembled into the desired 2:1 (peptide:heme) stoichiometry. Practical placement of ligating His residues along with carving out ample space was sufficient to convert a Leu-rich protein into a heme protein. Subsequently, design principles were used to engineer proteins that modulate the heme redox potential by over 100 mV, elucidating the motifs necessary to help control the thermodynamics and rates of electron transfer.198
Building upon this work, functional heme-binding maquettes were developed.198 Starting with the heptad repeat Leua-Glub-Gluc-Leud-Leue-Lysf-Lysg an a-position was replaced with His to bind two heme cofactors with bis-His ligation within a four-helix bundle.199 Heme in this D2-symmetric, coordinatively saturated ligand environment is not capable of binding external ligands, such as CO or O2; however, taking inspiration from the natural heme-protein neuroglobin, an entatic state was engineered into the scaffold. The binding of a single heme cofactor rotates the helices to achieve bis-His ligation, deliberately burying three destabilizing Glu residues, weakening one of the His ligands (Fig. 5c). Exposing this scaffold to O2 allows for the reversible formation of an oxy-ferrous heme species in which the displaced His ligand likely acts as a distal H-bond donor, as is often seen in natural hemoglobins. This H-bonding interaction is vital for stabilizing the oxy-ferrous heme (Fe2+-O2) without oxidizing Fe2+ to Fe3+ and releasing superoxide. Reducing the symmetry around the heme center stabilized this oxy-ferrous state for tens of seconds before oxidation occurs and showed that the complex globin fold is not necessary for reversible dioxygen binding. In addition, it showed the necessary design of purposeful instability around the metal site to elicit function.
Subsequently, the type C heme-binding maquettes could be assembled in vivo using the native biological machinery for cofactor insertion.200,201 Moreover, these maquettes served as malleable platforms for the development of catalytic systems that featured spectroscopic and mechanistic similarities to native enzymes.202 Given the lack of engineered substrate binding site, this protein is unsurprisingly active with many substrates, a feature that might have been characteristic of early metalloenzymes. Subsequently, this de novo platform could also perform abiological reactions, such as carbene transfer,203,204 which have been a recent focus for directed evolution work.205 This demonstration provides exciting support for the prospect that de novo proteins may provide excellent launching pads for optimization via evolutionary techniques (see below).
De novo designed proteins often use simplified symmetric scaffolds; however, the ability to move away from symmetry and design fully asymmetric de novo sequences provides a path to new-to-nature function. One initial strategy uses binary (polar/nonpolar) patterning of a helical bundle to develop combinatorial libraries of sequences.206–208 A general strategy for protein design follows the assumption that the ability of a sequence to form a secondary structure will suffice to drive a polypeptide to fold into a compact, native-like structure. Essentially, the formation of compactly folded structures does not require the explicit design of specific inter-residue contacts— only the sequence location, not the identity, of polar and nonpolar residues must be specified explicitly. Using this strategy, libraries of well-folded four-helix bundles were developed. While the scaffold had D2-symmetry (with respect to the helical backbone), these single chain proteins were fully asymmetric with respect to the sequence. They then applied this strategy to the design of heme proteins by placing His and Met at a buried position within the bundle.209 With this protein library, 15 of their 30 sequences were found to bind heme, the best of which bound heme with surprisingly strong affinity (KD = 0.7 μM). Moreover, some of these heme-binding sequences functioned as peroxidases, which suggests that binary patterning may have been a first step in the evolution of functional metalloenzymes.
Despite over two decades of work on de novo designed heme-proteins, high-resolution structures had not been solved prior to 2019. Atomic-level structural information allows us to assess the success of our designs. Moreover, natural metalloenzymes control their substrates very tightly to achieve both high rates and specificity, even small deviations from the desired geometry can result in sluggish catalysis.40 Thus, sub-Å accuracy is necessary, if we aspire to native-like catalysis. To address this issue of structural non-uniqueness, a strategy found in nature was applied to computationally design a structurally unique cofactor-binding protein (PS1). Starting from a D2-symmetric bundle backbone, an “Enfold” strategy was implemented to produce a well-packed protein (Fig. 6a–b).210 Instead of focusing on the symmetry of the structure, emphasis was placed on maintaining a well-packed apolar core distal from the binding site to ensure a well-structured protein. This folded core was treated as an extension of the primary and secondary-shell interactions with the cofactor and therefore the entire sequence was optimized in unison. To assure tight coupling between the fold of the core and the structure of the binding site, the amino acid sequence was designed using a sidechain repacking algorithm along with a flexible backbone design — both of which are implemented in the versatile computational design program Rosetta.9–11,211,212 A sequence was designed from scratch to bind an abiological Zn-porphyrin in an asymmetric binding site containing one His ligand (placed in a d-position). Remarkably, the first sequence designed not only folded and bound the cofactor, but it also yielded the first high-resolution NMR structure of a porphyrin-binding protein. The structure was in agreement with the design, with a helical backbone RMSD of 0.8 Å relative to the design. As designed, the structure of the apo-protein had a well-packed core that positioned a more flexible binding site for facile entry of the cofactor. This was consistent with ab initio folding calculations on the apo-protein sequence, that predicted the fold of the well-packed core with greater accuracy than the binding site. Indeed, the protein was so well-packed, that both the holo-and apo-proteins were hyper-stable, with Tm > 100 °C.
Fig. 6 |. Design strategy for well-structured porphyrin binding proteins.
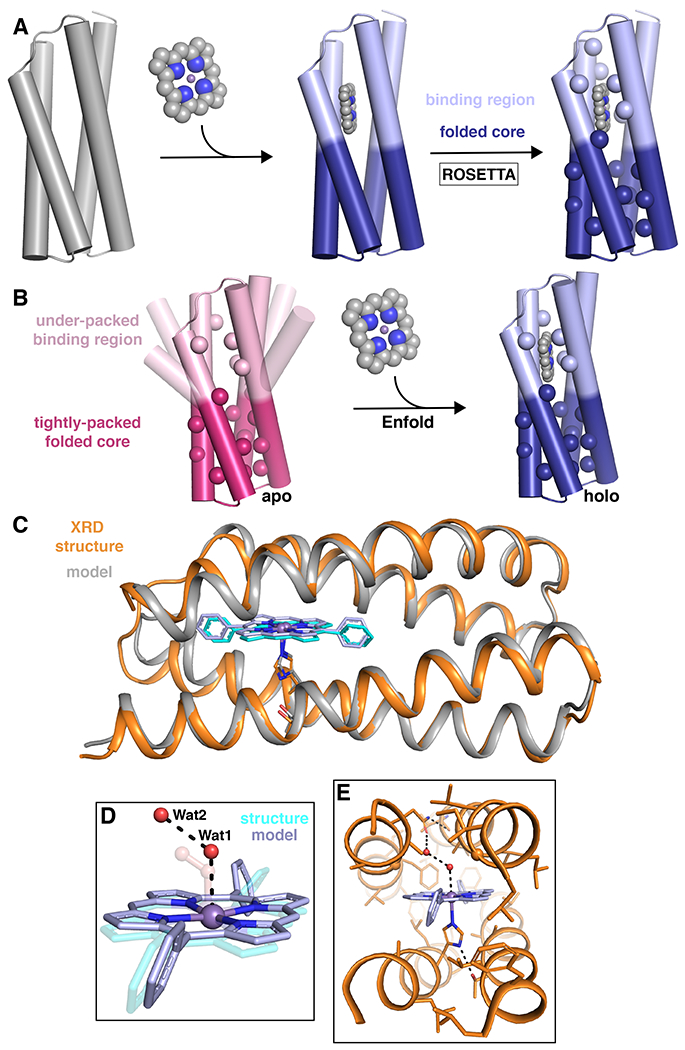
a |The general Enfold design strategy for PS1 in which the binding region and folded core regions are designed simultaneously to give an optimized sequence and backbone around the desired metallocofactor.210 b |An illustration of the Enfold strategy in which the under-packed binding site becomes well-structured on binding the metallocofactor to produce a well-folded, stable holo protein.210 c–e |Structural comparison of the designed model of MPP1 (gray) and the crystal structure (PDB: 7JRQ; orange).213 c |A cartoon representation showing an extremely good backbone match between the design and structure (0.6 Å all backbone RMSD). d |A comparison of the placement of two water molecules (Wat1 and Wat2; red spheres) relative to the dioxygen unit in the design (transparent) and e |extended H-bonding network from the binding site to the surface by the Wat1-Wat2 water network. XRD = X-ray diffraction. Parts a and b adapted from ref.210, Springer Nature Limited. Parts c–e reprinted with permission from ref.213, ACS.
Subsequently, a related backbone was used to build a multi-domain protein that included the porphyrin binding site from PS1 and the diiron binding site from the DF family of proteins (discussed above). This work used a bioinformatics approach based on Master searches (Box 3)4 to find the most designable links between two helical domains with significantly different architectures. The backbone changes that occur upon porphyrin binding were used to allosterically regulate the catalytic rate of the di-Fe sites.35
Box 3 |. Computational Protein Design Tools.
TOOLS FOR COILED COIL BACKBONE CONSTRUCTION
CCCP (Coiled coil Crick parameterization) – A tool to generate ideal coiled coil backbones from specified Crick parameters. It also allows for the extraction of parameters from helical bundles.51
CCBuilder 2.0 – A tool to model alpha-helical, coiled coil structures using Crick parameters for many oligomeric and topological states. It also allows for rapid visualization and minimization of helices with specific sequences.250
MASTER (Methods of accelerated search for tertiary ensemble representatives) – A fast, backbone RMSD-based structure search tool. The designer can use this tool to search backbone fragments to determine designability, to explore structure-sequence requirements of a motif, or to build loops to generate single-chain proteins.251
TOOLS FOR METAL BINDING SITE PREDICTION
SyPRIS – A computational design method used to locate clusters of backbone-specific positions capable of supporting symmetric coordination geometries.189
GaudiMM – A platform aimed at generating geometric candidates to perform hypothesis driven analysis of a metalloprotein’s conformational landscape.189
TOOLS FOR SIDECHAIN PACKING AND BINDING SITE DESIGN
COMBS (Convergent Motifs for Binding Sites) – A PDB-search algorithm using a defined structural unit called a van der Mer (vdM). This unit defines non-covalent interactions with key chemical groups in a ligand of interest to define its optimal position relative to the protein backbone.37,245
protCAD – Computational software that can design sequences for a given backbone. This tool uses physical force fields (that is, implicit solvent dielectric) to improve sequence and backbone design.181,182
FULLY AUTOMATED TOOLS FOR SEQUENCE AND STRUCTURE DESIGN
Rosetta – A widely used software suite that includes an expansive library of tools for computational modeling and analysis of protein structures. In metalloprotein design, it can design a sequence on a given backbone (or library of backbones) while constraining a metal binding site. It is well-maintained and has a thriving community of users and developers.9–11,211,212
TOOLS FOR PROTEIN FOLDING PREDICTION
Robetta and AlphaFold2 – Robetta has long been maintained as a server for ab initio protein folding predictions by the Rosetta community.252 In its most recent form (RoseTTAFold)253, it uses a machine-learned neural network similar to the simultaneously published AlphaFold2254 approach. Both of these tools have greatly improved predictions of protein folding from sequence and can generate predictive models and confidence metrics for de novo designed sequences.
Using the Enfold strategy, the protein, MPP1, could be designed to bind a synthetic Mn-diphenylporphyrin, and stabilize a high-valent Mn5+-oxo species to perform sulfoxidation chemistry.213 Moreover, this was the first crystallographically characterized porphyrin-binding protein, giving the exact position of the metal ion relative to the protein and location of aqua ligands and associated water molecules within the binding site (Fig 6c–e). As was the case for the DF proteins, it was important to introduce an access channel connecting the outside to the binding site. Therefore, to engineer function into the designed scaffold, a dioxygen unit was used in the open coordination site on the Mn center during design to ensure adequate space for two oxygen-atoms during catalysis. This further desymmetrized the bundle, maintaining access for oxidants and substrates. In fact, the crystal structure showed two structural water molecules sitting at the same Mn-O-O angle that was used in the design (Fig. 6d). This designed void likely had a destabilizing effect on the bundle, so highly designable loops were used to maximize backbone stability. Moreover, it shows the importance of substrate access as a design motif that can be implemented and tuned for a desired function. When considering catalytic function, both the metal and substrate binding sites should be explicitly designed to ensure the sequence properly accommodates the cofactors. In fact, MPP1 was highly specific, binding only the porphyrin of interest and preventing the strongly oxidizing Mn5+ species from deleterious reaction with the protein or the porphyrin ring. Taken together this work illustrates how the ability to design binding sites with sub-Å precision can be harnessed to program the function of O-atom transfer in a highly restrained environment.
Beta-sheet de novo metalloproteins
Most de novo designed proteins, including metalloproteins, are based on helical bundle motifs due to the deep understanding of the structure and folding of these proteins. However, other secondary structure elements, such as β-sheets, are also used extensively by nature in the construction of metalloproteins. The simplest β-strand containing motif is the β-hairpin, which features two β-strands connected by a reverse turn. This structure has been employed as a simple building block in the de novo design of metalloproteins.
The first de novo metalloproteins to use this motif were rubredoxin mimics. The β-hairpin motif is pseudo-C2 symmetric and provides a highly organized primary and secondary coordination sphere in the natural protein. With de novo design it has been shown that these functional qualities can be reproduced despite new-to-nature sequences, if this tertiary structure is maintained.166–169,172
β-hairpin motifs can also be adapted to the de novo design of metalloproteins not typically encountered in this fold. In particular, initial de novo designed, membrane-bound beta-hairpin motifs for heme binding yielded mini-peptides (that is, 8 residues) featuring a single His ligand.214 Later designs were extended to feature a pair of β-hairpins each donating a ligating His.215,216 By combining more β-hairpins (up to twelve β-sheets), ensembles of multiple hemes could be assembled in a controlled fashion.217 Structural characterization of these β-hairpin proteins by NMR provides insight into how flexibility and coordination number can affect functional properties such as electron transfer or peroxidase activity.218
The edges of β-strands are often considered to be “sticky” and, thus, can lead to the aggregation of β-sheets into an amyloid structure. By controlling the nature of aggregation, short peptides can generate large scaffolds. Ligating residues (that is, His) can be introduced into these peptides to create catalytically active amyloids, including Zn-dependent hydrolases219 and Cu-dependent oxidases.220 High resolution structures of these amyloids were determined by solid-state NMR and showed that in addition to the translational symmetry inherent to the amyloid, the stacked sheets packed back-to-back generating an C2 symmetry axis (Fig. 7a–b).37 The introduction of a hydrophobic Phe into such peptides also allowed them to bind hemin and mediate catalytic cycloproponation.221 These results demonstrate the versatility of simple scaffolds for generating metalloenzymes.
Fig. 7 |. Beta-sheet containing designed metalloproteins.
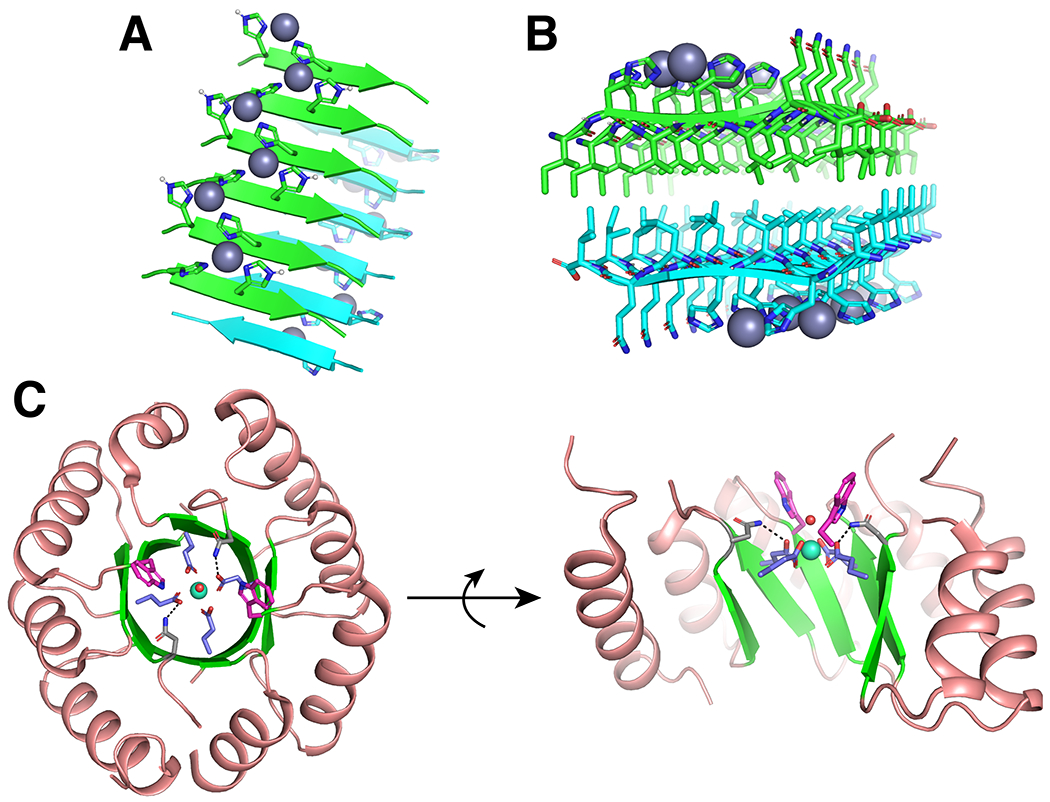
a, b |Two views of the solid-state NMR structure (PDB: 5UGK) of Zn2+ bound catalytic amyloids.37 a |Each Zn2+ ion is coordinated by three His nitrogens between two neighboring strands. b |Two beta-sheets stack anti-parallel with hydrophobic residues facing each other forming a C2-symmetry axis along the fibril axis (perpendicular to the beta-strands). c |Two views of the crystal structure (PDB: 6ZV9) of Tb3+ bound designed TIM barrel with a C2 symmetry axis.226 Tb3+ is shown as a cyan sphere with a coordinated water (red sphere), alpha helices are shown in salmon and beta-sheets are shown in green. Coordinating Glu residues are shown in blue, second-shell H-bonding Asn are shown in grey with H-bonds as black dashed lines, and Trp “antenna” are shown in pink.
Alternative, beta-sheet based architectures may also prove useful in the context of de novo protein design. Ancestrally reconstructed beta-propellor proteins can be useful in the stabilization of unusual Cd chloride nanocrystals.222–224 The high symmetry of beta-propellor proteins and the open pore generated by their funnel-like shape provide an opportunity for further metalloprotein design. Moreover, a number of common natural protein folds combine beta-sheets with alpha-helices. One such example is the triose phosphate isomerase (TIM) barrel, which features 8 external alpha helices and 8 internal beta-sheets creating a pseudo-C8 axis running down the center of the barrel. Recently, the de novo design of a TIM barrel with C4 symmetry225 was extended to the design of a metalloprotein (Fig. 7c).226 In this case, the Tb3+ metalloprotein features a C4 coordination geometry consisting of four Glu ligands with a symmetry that lowers to C2 on consideration of the secondary coordination sphere (Fig. 7c).
Outlook
This Review describes the extent to which we have progressed in achieving an active, working knowledge of metalloproteins, by formulating and executing a set of chemical and engineering principles. As we have come to understand metalloproteins, we have become increasingly successful in designing them from scratch — rather than by modifying natural proteins. Designed proteins test our hypothetical understanding of metalloprotein function and can ultimately serve as starting points to design useful catalysts. In the above sections, we focused on simple parametric protein backbones to illustrate the principles of metalloprotein design. Nonetheless, we have already seen that by beginning with a hypothesis concerning the metal, the geometry of first/second shell-ligands, and solvent/substrate accessibility a metalloprotein can be imbued with the desired functionality. Indeed, a simple diiron site can be systematically modified to catalyse two- or four-electron chemistry, resulting in drastically different products starting from similar substrates.17,162 In addition, the midpoint potentials, binding and reactivity of hemes can be modulated over a wide range to create multiple functions.198 However, we are just scratching the surface of what is possible, given the remarkable versatility of proteins and their ability to create myriad ligand geometries, dynamics and auxiliary binding sites. Surely, there is much more to be accomplished by a new generation of protein designers and inorganic chemists.
Even within the geometric space of helical bundles, metalloprotein designers have not strayed far from the idealized left-handed bundles and coiled coils. Other geometries such as right-handed coiled coils or straight bundles offer opportunities for the design of novel binding sites.49,51,76,77 De novo design of metalloproteins outside of the context of coiled coils opens up even more possibilities. This is emphasized by the design of rubredoxin mimics that use the pseudo-C2 symmetric beta-hairpin motif of the natural protein.166–169,172 The highly organized primary and secondary coordination sphere provided by the beta-hairpin motif improves the reversibility and O2-stability of these rubredoxin mimics compared to those designed in helical bundles, which demonstrates the importance of tertiary structure on function. These successes and the recent de novo design of natural topologies such as TIM barrels (including a metalloprotein)225,226 and beta barrels,39,227 in addition to non-native folds228,229, should encourage metalloprotein designers to explore more diverse folds and, consequently, active sites.
As we have discussed, deviations from ideality are often used by natural proteins to shape the active site or to correctly position ligands.71,73,74 Nonetheless, design strategies for incorporating deviations, such as pi-bulges or substrate-access channels, are thus far limited, but hold potential for dramatically increasing the design space. One possible approach is to start from an ideal backbone but introduce constraints (that is, metal-ligand bond distances or angles) that will strain the local backbone, thereby introducing the deviation. Alternatively, a bioinformatics approach could be used to position a deviation, for example a pi-bulge in an ideal backbone, such as an alpha-helix, before the design begins. Using the Enfold strategy to offset the instability of these deviations, a well-packed core can be added distal from the active site. Statistical scoring methods to designing proteins will inherently score such designs poorly because of their relative infrequent occurrence in natural proteins; however, as we have stressed, such deviations are often key to function. Thus, alternative approaches to assess the stability of non-ideal secondary structures, such as the use of molecular dynamics or methods that use physics-based scoring functions, may be particularly valuable to assess if such geometries are reasonable.
In addition to new topologies, new active sites can be explored. Already there has been success in the de novo design of active sites featuring xeno-biological metals including lanthanides44 and titanium,230 as well as a demonstration that helical bundle formation can be used to guide the reactivity of di-rhodium metallopeptides.231,232 However, by adapting rules derived from synthetic inorganic chemistry, an abundance of xeno-biological active sites should be readily accessible. Moreover, the broad success in designing proteins for non-natural heme derivatives35,196,197,199,210,213 suggests that de novo protein design could be used to tailor the reactivity of a broad range of organometallic species. Lastly, improvements in both peptide synthesis233 and protein expression234–236 increasingly enables the use of non-canonical amino acids as building blocks in active sites.237–239
We anticipate that future metalloprotein design efforts will be targeted at the final tier of our design hierarchy, the use of metal ions to functionalize complex organic substrates in a regio- and stereoselective fashion. While some progress towards this aim has been made, such as building shape-selective channels capable of binding apolar substrates proximal to metal sites,151,213,240 much work remains to be done. Unlike natural proteins, most de novo metalloenzymes remain highly promiscuous, because the flexibility that allows them to attain high activity also results in ill-defined active site pockets. One approach to achieve better specificity is to evolve these flexible scaffolds to discover new and unexpected tertiary structures, ligand environments, and binding sites. In early work, a closed-shell Zn2+-binding structural site was added between two helix-loop-helix motifs.164 Serendipitously, the resulting proteins had rather malleable tertiary structures, with metal-binding sites not fully anticipated by design that nevertheless presented open ligation sites for hydrolytic catalysis.190 Using directed evolution, accessible sequence landscapes were searched to find proteins that catalysed Zn2+-dependent esterase reactions.191 This resulted in the change of one of the ligands in the designed protein164 to a new position in the tertiary structure and a significant remodeling of the substrate-binding pockets. These findings illustrate not only the power of directed evolution, but also the relative facility with which new activities can be discovered beginning with only a rough draft of a protein.33,241,242 Moreover, the features discovered through evolutionary screening can be added to the design toolbox and applied to future designs.
Alternatively, achieving stereo- and enantio-selective metalloenzymes may be possible by design processes that explicitly consider the target substrate from the initial design. Few metalloprotein designs have thus far incorporated substrates. The aforementioned Mn-porphyrin protein, MPP1, which was structurally characterized to have water molecules occupying the positions intended for the O2 ligand, is an initial step in this direction.213 Moreover, it speaks to the importance of the development of general strategies to handle water in computational de novo design, which are currently in their infancy.243,244 A notable success in this final design tier was to use the transition state of the Zn-mediated Diels–Alder transformation in a recent redesign of the MID1sc protein. Impressively, the first generation designs show moderate catalytic activity, which were dramatically enhanced via directed evolution.25 We anticipate that advances in the de novo design of small molecule binding proteins245 will soon be incorporated into metalloenzyme design to improve initial design models. Moreover, these design strategies could be coupled to molecular dynamics simulations to improve in silico assessment of the feasibility of accessing a desired transition state. These designs could then be further improved by experimental validation aided by modern methods of gene synthesis coupled to an appropriate activity screen allowing, in principle, thousands of individual designs to be evaluate.242
Advances in computation, gene synthesis, structural biology, and evolution have dramatically improved our abilities to de novo design proteins. The varied and important functions of metalloproteins remain an important measuring stick for our understanding of the underlying structure-function principles of proteins. We anticipate that further advances will make the de novo design of functional metalloproteins increasingly routine, opening up attractive possibilities in a variety of areas including green catalysis, sensing, and therapeutics.
Key points.
The metalloprotein designer must first consider the desired function, select an appropriate active site to achieve it, and the tertiary structure to support it.
Design approaches can be usefully drawn from either the inorganic chemistry literature or from a bioinformatics approach.
There must be a unifying element to the local symmetry of the protein tertiary structure and the metal active site.
The design must balance the energetics of protein folding with metal-ligand binding in order to achieve the desired coordination geometry.
The introduction of asymmetry is a key strategy for introducing function into metalloproteins and must be compensated for by the introduction of stabilizing elements elsewhere in the design.
The design space beyond coiled coils remains sparsely studied and offers opportunities for more diverse active sites and, hence, functions.
Acknowledgements
The authors acknowledge support from the National Institutes of Health grants GMR35 122603, F32GM130029, and F32GM139379. We also thank our many coworkers who have contributed to the field, as well as reviewers for helpful comments and suggestions.
Glossary:
- Allostery
a biological phenomenon, in which regulation occurs at a distal site often triggered by a ligand-binding event, such as a metal ion.
- Rotamers
preferred orientations of an amino acid side chain relative to the main chain.
- Maquette
simple peptide models that can be progressively altered to test the characteristics of their construction that have been commonly studied in the de novo design of proteins.
Footnotes
Competing interests
The authors declare no competing interests.
References:
- 1.Winkler JR & Gray HB Electron flow through metalloproteins. Chem. Rev 114, 3369–3380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Theil EC & Raymond KN Transition-metal storage, transport, and biomineralization in Bioinorganic chemistry (eds. Beritini I, Gray HB, Lippard SJ, & Valentine JS) (University Science Books, Mill Valley, CA, 1994). [Google Scholar]
- 3.Shimizu T et al. Gaseous O2, NO, and CO in signal transduction: Structure and function relationships of heme-based gas sensors and heme-redox sensors. Chem. Rev 115, 6491–6533 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Liu J et al. Metalloproteins containing cytochrome, iron–sulfur, or copper redox centers. Chem. Rev 114, 4366–4469 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm RH, Kennepohl P & Solomon EI Structural and functional aspects of metal sites in biology. Chem. Rev 96, 2239–2314 (1996). [DOI] [PubMed] [Google Scholar]
- 6.Regan L Protein design: Novel metal-binding sites. Trends Biochem. Sci 20, 280–285 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Hellinga HW The construction of metal centers in proteins by rational design. Fold Des. 3, R1–R8 (1998). [DOI] [PubMed] [Google Scholar]
- 8.Yu F et al. Protein design: Toward functional metalloenzymes. Chem. Rev 114, 3495–3578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alford RF et al. The Rosetta all-atom energy function for macromolecular modeling and design. J. Chem. Theory Compu 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leman JK et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 17, 665–680 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leaver-Fay A et al. Rosetta3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vallee BL & Williams RJ Metalloenzymes: The entatic nature of their active sites. Proc. Natl Acad. Sci. U. S. A 59, 498–505 (1968). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gomes CM & Wittung-Stashede P Metal ions, protein folding, and conformational states: An introduction in Protein folding and metal ions: Mechanisms, biology and disease (CRC Press, Boca Raton, Fl, 2011). [Google Scholar]
- 14.Handel TM, Williams SA & DeGrado WF Metal ion-dependent modulation of the dynamics of a designed protein. Science 261, 879–885 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Mocny CS & Pecoraro VL De novo protein design as a methodology for synthetic bioinorganic chemistry. Acc. Chem. Res 48, 2388–2396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nastri F et al. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev 45, 5020–5054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lombardi A, Pirro F, Maglio O, Chino M & DeGrado WF De novo design of four-helix bundle metalloproteins: One scaffold, diverse reactivities. Acc. Chem. Res 52, 1148–1159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dudev T & Lim C Principles governing Mg, Ca, and Zn binding and selectivity in proteins. Chem. Rev 103, 773–788 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Pinter TBJ, Koebke KJ & Pecoraro VL Catalysis and electron transfer in de novo designed helical scaffolds. Angew. Chem. Int. Ed 59, 7678–7699 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosseinzadeh P & Lu Y Design and fine-tuning redox potentials of metalloproteins involved in electron transfer in bioenergetics. Biochim. Biophys. Acta. Bioenerg 1857, 557–581 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muñoz Robles V et al. Toward the computational design of artificial metalloenzymes: From protein–ligand docking to multiscale approaches. ACS Catal. 5, 2469–2480 (2015). [Google Scholar]
- 22.Berman HM et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burley SK et al. RCSB protein data bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 49, D437–D451 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groom CR, Bruno IJ, Lightfoot MP & Ward SC The Cambridge structural database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater 72, 171–179 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basler S et al. Efficient lewis acid catalysis of an abiological reaction in a de novo protein scaffold. Nat. Chem 13, 231–235 (2021). [DOI] [PubMed] [Google Scholar]; This article demonstrates a successful design around a computational transition state, theozyme, in order to achieve a novel reaction.
- 26.Liang AD, Serrano-Plana J, Peterson RL & Ward TR Artificial metalloenzymes based on the biotin-streptavidin technology: Enzymatic cascades and directed evolution. Acc. Chem. Res 52, 585–595 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lewis JC Beyond the second coordination sphere: Engineering dirhodium artificial metalloenzymes to enable protein control of transition metal catalysis. Acc. Chem. Res 52, 576–584 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Roelfes G LmrR: A privileged scaffold for artificial metalloenzymes. Acc. Chem. Res 52, 545–556 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oohora K, Onoda A & Hayashi T Hemoproteins reconstituted with artificial metal complexes as biohybrid catalysts. Acc. Chem. Res 52, 945–954 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Natoli SN & Hartwig JF Noble-metal substitution in hemoproteins: An emerging strategy for abiological catalysis. Acc. Chem. Res 52, 326–335 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirts EN, Bhagi-Damodaran A & Lu Y Understanding and modulating metalloenzymes with unnatural amino acids, non-native metal ions, and non-native metallocofactors. Acc. Chem. Res 52, 935–944 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Churchfield LA & Tezcan FA Design and construction of functional supramolecular metalloprotein assemblies. Acc. Chem. Res 52, 345–355 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Reetz MT Directed evolution of artificial metalloenzymes: A universal means to tune the selectivity of transition metal catalysts? Acc. Chem. Res 52, 336–344 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Polizzi NF et al. Photoinduced electron transfer elicits a change in the static dielectric constant of a de novo designed protein. J. Am. Chem. Soc 138, 2130–2133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pirro F et al. Allosteric cooperation in a de novo-designed two-domain protein. Proc. Natl. Acad. Sci. U. S. A 117, 33246–33253 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regan L & Clarke ND A tetrahedral zinc(II)-binding site introduced into a designed protein. Biochemistry 29, 10878–10883 (1990). [DOI] [PubMed] [Google Scholar]; This is the first example of a de novo designed metalloprotein.
- 37.Lee M et al. Zinc-binding structure of a catalytic amyloid from solid-state NMR. Proc. Natl Acad. Sci. U.S.A 114, 6191–6196 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zanghellini A et al. New algorithms and an in silico benchmark for computational enzyme design. Protein Sci. 15, 2785–2794 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dou J et al. De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiss G, Çelebi-Ölçüm N, Moretti R, Baker D & Houk KN Computational enzyme design. Angew. Chem. Int. Ed 52, 5700–5725 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Ho SP & DeGrado WF Design of a 4-helix bundle protein: Synthesis of peptides which self-associate into a helical protein. J. Am. Chem. Soc 109, 6751–6758 (1987). [Google Scholar]
- 42.Richardson JS & Richardson DC The de novo design of protein structures. Trends Biochem. Sci 14, 304–309 (1989). [DOI] [PubMed] [Google Scholar]
- 43.Grigoryan G, Reinke AW & Keating AE Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature 458, 859–864 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slope LN & Peacock AFA De novo design of xeno-metallo coiled coils. Chem. – Asian J 11, 660–666 (2016). [DOI] [PubMed] [Google Scholar]
- 45.Crick F The fourier transform of a coiled-coil. Acta Crystallogr. 6, 685–689 (1953). [Google Scholar]
- 46.Crick F The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 6, 689–697 (1953). [Google Scholar]
- 47.North B, Summa CM, Ghirlanda G & DeGrado WF Dn-symmetrical tertiary templates for the design of tubular proteins. J. Mol. Biol 311, 1081–1090 (2001). [DOI] [PubMed] [Google Scholar]
- 48.DeGrado WF, Summa CM, Pavone V, Nastri F & Lombardi A De novo design and structural characterization of proteins and metalloproteins. Annu. Rev. Biochem 68, 779–819 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Lupas AN, Bassler J & Dunin-Horkawicz S The structure and topology of α-helical coiled coils in Fibrous proteins: Structures and mechanisms (eds. Parry DAD & Squire JM) 95–129 (Springer International Publishing, Cham, 2017). [Google Scholar]
- 50.Lupas AN & Bassler J Coiled coils – a model system for the 21st century. Trends Biochem. Sci 42, 130–140 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Grigoryan G & DeGrado WF Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol 405, 1079–1100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article describes the mathematical parameters that define an ideal coiled coil and their relationship to stability and designability.
- 52.Kamtekar S & Hecht MH The four-helix bundle: What determines a fold? FASEB J. 9, 1013–1022 (1995). [DOI] [PubMed] [Google Scholar]
- 53.Schneider JP, Lombardi A & DeGrado WF Analysis and design of three-stranded coiled coils and three-helix bundles. Fold Des. 3, R29–R40 (1998). [DOI] [PubMed] [Google Scholar]
- 54.Moutevelis E & Woolfson DN A periodic table of coiled-coil protein structures. J. Mol. Biol 385, 726–732 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Chothia C, Levitt M & Richardson D Helix to helix packing in proteins. J. Mol. Biol 145, 215–250 (1981). [DOI] [PubMed] [Google Scholar]
- 56.Kellis JT, Nyberg K, Săil D. a. & Fersht AR Contribution of hydrophobic interactions to protein stability. Nature 333, 784–786 (1988). [DOI] [PubMed] [Google Scholar]
- 57.Matthews BW Studies on protein stability with T4 lysozyme in Adv. Protein chem (eds. Anfinsen CB, Richards FM, Edsall JT, & Eisenberg DS) 249–278 (Academic Press, 1995). [DOI] [PubMed] [Google Scholar]
- 58.Harbury PB, Zhang T, Kim PS & Alber T A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262, 1401–1407 (1993). [DOI] [PubMed] [Google Scholar]
- 59.Bryson JW et al. Protein design: A hierarchic approach. Science 270, 935–941 (1995). [DOI] [PubMed] [Google Scholar]
- 60.Betz SF & DeGrado WF Controlling topology and native-like behavior of de novo-designed peptides: Design and characterization of antiparallel four-stranded coiled coils. Biochemistry 35, 6955–6962 (1996). [DOI] [PubMed] [Google Scholar]
- 61.Betz SF, Liebman PA & DeGrado WF De novo design of native proteins: Characterization of proteins intended to fold into antiparallel, ROP-like, four-helix bundles. Biochemistry 36, 2450–2458 (1997). [DOI] [PubMed] [Google Scholar]
- 62.Barlow DJ & Thornton JM Ion-pairs in proteins. J. Mol. Biol 168, 867–885 (1983). [DOI] [PubMed] [Google Scholar]
- 63.Schneider JP, Lear JD & DeGrado WF A designed buried salt bridge in a heterodimeric coiled coil. J. Am. Chem. Soc 119, 5742–5743 (1997). [Google Scholar]
- 64.Marqusee S & Sauer RT Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in λ repressor. Protein Sci. 3, 2217–2225 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O’Shea EK, Lumb KJ & Kim PS Peptide ‘velcro’: Design of a heterodimeric coiled coil. Curr. Biol 3, 658–667 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Lavigne P et al. Interhelical salt bridges, coiled-coil stability, and specificity of dimerization. Science 271, 1136–1138 (1996). [DOI] [PubMed] [Google Scholar]
- 67.Sindelar CV, Hendsch ZS & Tidor B Effects of salt bridges on protein structure and design. Protein Sci. 7, 1898–1914 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grigoryan G & Keating AE Structural specificity in coiled-coil interactions. Curr. Opin. Struct. Biol 18, 477–483 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Predki PF, Agrawal V, Brünger AT & Regan L Amino-acid substitutions in a surface turn modulate protein stability. Nat. Struct. Biol 3, 54–58 (1996). [DOI] [PubMed] [Google Scholar]
- 70.Efimov AV Patterns of loop regions in proteins. Curr. Opin. Struct. Biol 3, 379–384 (1993). [Google Scholar]
- 71.Brown JH, Cohen C & Parry DAD Heptad breaks in α-helical coiled coils: Stutters and stammers. Proteins: Struct. Funct., Bioinf 26, 134–145 (1996). [DOI] [PubMed] [Google Scholar]
- 72.Hartmann MD et al. A coiled-coil motif that sequesters ions to the hydrophobic core. Proc. Natl. Acad. Sci. U. S. A 106, 16950–16955 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schmidt NW, Grigoryan G & DeGrado WF The accommodation index measures the perturbation associated with insertions and deletions in coiled-coils: Application to understand signaling in histidine kinases. Protein Sci. 26, 414–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pinter TBJ et al. Making or breaking metal-dependent catalytic activity: The role of stammers in designed three-stranded coiled coils. Angew. Chem. Int. Ed 59, 20445–20449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Woolfson DN, Bartlett GJ, Bruning M & Thomson AR New currency for old rope: From coiled-coil assemblies to α-helical barrels. Curr. Opin. Struct. Biol 22, 432–441 (2012). [DOI] [PubMed] [Google Scholar]
- 76.Lupas A Coiled coils: New structures and new functions. Trends Biochem. Sci 21, 375–382 (1996). [PubMed] [Google Scholar]
- 77.Huang P-S et al. High thermodynamic stability of parametrically designed helical bundles. Science 346, 481–485 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Agnew C et al. Correlation of in situ mechanosensitive responses of the Moraxella catarrhalis adhesin UspA1 with fibronectin and receptor CEACAM1 binding. Proc. Natl. Acad. Sci. U.S.A 108, 15174–15178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xia D et al. Crystal structure of the cytochrome bc1 complex from bovine heart mitochondria. Science 277, 60–66 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hunte C, Palsdottir H & Trumpower BL Protonmotive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 545, 39–46 (2003). [DOI] [PubMed] [Google Scholar]
- 81.Berry EA & Walker FA Bis-histidine-coordinated hemes in four-helix bundles: How the geometry of the bundle controls the axial imidazole plane orientations in transmembrane cytochromes of mitochondrial complexes II and III and related proteins. J. Biol. Inorg. Chem 13, 481–498 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yun CH, Wang Z, Crofts AR & Gennis RB Examination of the functional roles of 5 highly conserved residues in the cytochrome b subunit of the bc1 complex of Rhodobacter sphaeroides. J. Biol. Chem 267, 5901–5909 (1992). [PubMed] [Google Scholar]
- 83.Saribaş AS, Ding H, Dutton PL & Daldal F Substitutions at position 146 of cytochrome b affect drastically the properties of heme bL and the QO site of Rhodobacter capsulatus cytochrome bc1 complex. Biochim. Biophys. Acta, Bioenerg 1319, 99–108 (1997). [DOI] [PubMed] [Google Scholar]
- 84.Schneider S, Marles-Wright J, Sharp KH & Paoli M Diversity and conservation of interactions for binding heme in b-type heme proteins. Nat. Prod. Rep 24, 621–630 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Reedy CJ & Gibney BR Heme protein assemblies. Chem. Rev 104, 617–649 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Nordlund P & Eklund H Di-iron—carboxylate proteins. Curr. Opin. Struct. Biol 5, 758–766 (1995). [DOI] [PubMed] [Google Scholar]
- 87.Jasniewski AJ & Que L Dioxygen activation by nonheme diiron enzymes: Diverse dioxygen adducts, high-valent intermediates, and related model complexes. Chem. Rev 118, 2554–2592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wahlgren WY et al. Structural characterization of bacterioferritin from Blastochloris viridis. PLoS One 7, e46992 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Macedo S et al. The nature of the di-iron site in the bacterioferritin from Desulfovibrio desulfuricans. Nat. Struct. Biol 10, 285–290 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Acheson JF, Bailey LJ, Brunold TC & Fox BG In-crystal reaction cycle of a toluene-bound diiron hydroxylase. Nature 544, 191–195 (2017). [DOI] [PubMed] [Google Scholar]
- 91.Banerjee R, Jones JC & Lipscomb JD Soluble methane monooxygenase. Annu. Rev. Biochem 88, 409–431 (2019). [DOI] [PubMed] [Google Scholar]
- 92.Jones JC, Banerjee R, Shi K, Aihara H & Lipscomb JD Structural studies of the Methylosinus trichosporium OB3b soluble methane monooxygenase hydroxylase and regulatory component complex reveal a transient substrate tunnel. Biochemistry 59, 2946–2961 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rardin RL et al. Synthesis and characterization of the linear trinuclear complexes [MII3(O2CCH3)6(biphme)2], M = Mn, Fe. Angew. Chem., Int. Ed. Engl 29, 812–814 (1990). [Google Scholar]
- 94.Whittington DA & Lippard SJ Crystal structures of the soluble methane monooxygenase hydroxylase from methylococcus capsulatus (bath) demonstrating geometrical variability at the dinuclear iron active site. J. Am. Chem. Soc 123, 827–838 (2001). [DOI] [PubMed] [Google Scholar]
- 95.Wright JG, Natan MJ, MacDonnel FM, Ralston DM & O’Halloran TV Mercury(II)—thiolate chemistry and the mechanism of the heavy metal biosensor MerR. Prog. Inorg. Chem 38, 323–412 (1990). [Google Scholar]
- 96.Lovejoy B et al. Crystal structure of a synthetic triple-stranded alpha-helical bundle. Science 259, 1288–1293 (1993). [DOI] [PubMed] [Google Scholar]
- 97.Neil KT & DeGrado WF A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250, 646–651 (1990). [DOI] [PubMed] [Google Scholar]
- 98.Dieckmann GR et al. De novo design of mercury-binding two- and three-helical bundles. J. Am. Chem. Soc 119, 6195–6196 (1997). [Google Scholar]
- 99.Chakraborty S, Touw DS, Peacock AFA, Stuckey J & Pecoraro VL Structural comparisons of apo- and metalated three-stranded coiled coils clarify metal binding determinants in thiolate containing designed peptides. J. Am. Chem. Soc 132, 13240–13250 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article highlights the interplay between protein structure and metal binding using crystallographically determined structures.
- 100.Suzuki K, Hiroaki H, Kohda D & Tanaka T An isoleucine zipper peptide forms a native-like triple stranded coiled coil in solution. Protein Eng. Des., Sel 11, 1051–1055 (1998). [DOI] [PubMed] [Google Scholar]
- 101.Dieckmann GR et al. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. J. Mol. Biol 280, 897–912 (1998). [DOI] [PubMed] [Google Scholar]
- 102.Peacock AFA, Stuckey JA & Pecoraro VL Switching the chirality of the metal environment alters the coordination mode in designed peptides. Angew. Chem. Int. Ed 48, 7371–7374 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ghosh D & Pecoraro VL Understanding metalloprotein folding using a de novo design strategy. Inorg. Chem 43, 7902–7915 (2004). [DOI] [PubMed] [Google Scholar]
- 104.Ghosh D, Lee K-H, Demeler B & Pecoraro VL Linear free-energy analysis of mercury(II) and cadmium(II) binding to three-stranded coiled coils. Biochemistry 44, 10732–10740 (2005). [DOI] [PubMed] [Google Scholar]
- 105.Boyle AL et al. Selective coordination of three transition metal ions within a coiled-coil peptide scaffold. Chem. Sci 10, 7456–7465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lee SC, Lo W & Holm RH Developments in the biomimetic chemistry of cubane-type and higher nuclearity iron–sulfur clusters. Chem. Rev 114, 3579–3600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tsui EY, Kanady JS & Agapie T Synthetic cluster models of biological and heterogeneous manganese catalysts for O2 evolution. Inorg. Chem 52, 13833–13848 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Buchwalter P, Rosé J & Braunstein P Multimetallic catalysis based on heterometallic complexes and clusters. Chem. Rev 115, 28–126 (2015). [DOI] [PubMed] [Google Scholar]
- 109.Wei KY et al. Computational design of closely related proteins that adopt two well-defined but structurally divergent folds. Proc. Natl. Acad. Sci. U. S. A 117, 7208–7215 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Falke JJ, Drake SK, Hazard AL & Peersen OB Molecular tuning of ion binding to calcium signaling proteins. Q. Rev. Biophys 27, 219–290 (1994). [DOI] [PubMed] [Google Scholar]
- 111.Lee K-H, Matzapetakis M, Mitra S, Marsh ENG & Pecoraro VL Control of metal coordination number in de novo designed peptides through subtle sequence modifications. J. Am. Chem. Soc 126, 9178–9179 (2004). [DOI] [PubMed] [Google Scholar]
- 112.Lee K-H, Cabello C, Hemmingsen L, Marsh ENG & Pecoraro VL Using nonnatural amino acids to control metal-coordination number in three-stranded coiled coils. Angew. Chem. Int. Ed 45, 2864–2868 (2006). [DOI] [PubMed] [Google Scholar]
- 113.Ruckthong L, Deb A, Hemmingsen L, Penner-Hahn JE & Pecoraro VL Incorporation of second coordination sphere D-amino acids alters Cd(II) geometries in designed thiolate-rich proteins. J. Biol. Inorg. Chem 23, 123–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ruckthong L, Stuckey JA & Pecoraro VL How outer coordination sphere modifications can impact metal structures in proteins: A crystallographic evaluation. Chem. – Eur. J 25, 6773–6787 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iranzo O, Chakraborty S, Hemmingsen L & Pecoraro VL Controlling and fine tuning the physical properties of two identical metal coordination sites in de novo designed three stranded coiled coil peptides. J. Am. Chem. Soc 133, 239–251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kiyokawa T et al. Binding of Cu(II) or Zn(II) in a de novo designed triple-stranded α-helical coiled-coil toward a prototype for a metalloenzyme. J. Pept. Res 63, 347–353 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Tanaka T et al. Two-metal ion, Ni(II) and Cu(II), binding α-helical coiled coil peptide. J. Am. Chem. Soc 126, 14023–14028 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Supuran Claudiu T. Structure and function of carbonic anhydrases. Biochem. J 473, 2023–2032 (2016). [DOI] [PubMed] [Google Scholar]
- 119.Horrell S, Kekilli D, Strange RW & Hough MA Recent structural insights into the function of copper nitrite reductases. Metallomics 9, 1470–1482 (2017). [DOI] [PubMed] [Google Scholar]
- 120.Zastrow ML, Peacock AFA, Stuckey JA & Pecoraro VL Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem 4, 118–123 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work demonstrates that de novo metalloenzymes can recapitulate native-like activity in non-natural folds.
- 121.Zastrow ML & Pecoraro VL Influence of active site location on catalytic activity in de novo-designed zinc metalloenzymes. J. Am. Chem. Soc 135, 5895–5903 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Koebke KJ & Pecoraro VL Development of de novo copper nitrite reductases: Where we are and where we need to go. ACS Catal. 8, 8046–8057 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tegoni M, Yu F, Bersellini M, Penner-Hahn JE & Pecoraro VL Designing a functional type 2 copper center that has nitrite reductase activity within α-helical coiled coils. Proc. Natl. Acad. Sci. U. S. A 109, 21234–21239 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yu F, Penner-Hahn JE & Pecoraro VL De novo-designed metallopeptides with type 2 copper centers: Modulation of reduction potentials and nitrite reductase activities. J. Am. Chem. Soc 135, 18096–18107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koebke KJ et al. Modifying the steric properties in the second coordination sphere of designed peptides leads to enhancement of nitrite reductase activity. Angew. Chem. Int. Ed 57, 3954–3957 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Koebke KJ et al. Methylated histidines alter tautomeric preferences that influence the rates of Cu nitrite reductase catalysis in designed peptides. J. Am. Chem. Soc 141, 7765–7775 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Suzuki K, Hiroaki H, Kohda D, Nakamura H & Tanaka T Metal ion induced self-assembly of a designed peptide into a triple-stranded α-helical bundle: A novel metal binding site in the hydrophobic core. J. Am. Chem. Soc 120, 13008–13015 (1998). [Google Scholar]
- 128.Berwick MR et al. De novo design of Ln(III) coiled coils for imaging applications. J. Am. Chem. Soc 136, 1166–1169 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors achieve a functional, xeno-biological active site via the application of rules from protein structure and inorganic chemistry.
- 129.Berwick MR et al. Location dependent coordination chemistry and MRI relaxivity, in de novo designed lanthanide coiled coils. Chem. Sci 7, 2207–2216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Slope LN et al. Tuning coordination chemistry through the second sphere in designed metallocoiled coils. Chem. Commun 56, 3729–3732 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Teare P et al. pH dependent binding in de novo hetero bimetallic coiled coils. Dalton Trans. 47, 10784–10790 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Ogihara NL et al. Design of three-dimensional domain-swapped dimers and fibrous oligomers. Proc. Natl. Acad. Sci. U. S. A 98, 1404–1409 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Roy A, Sarrou I, Vaughn MD, Astashkin AV & Ghirlanda G De novo design of an artificial bis[4Fe-4S] binding protein. Biochemistry 52, 7586–7594 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Roy A et al. A de novo designed 2[4Fe-4S] ferredoxin mimic mediates electron transfer. J. Am. Chem. Soc 136, 17343–17349 (2014). [DOI] [PubMed] [Google Scholar]
- 135.Sommer DJ, Roy A, Astashkin A & Ghirlanda G Modulation of cluster incorporation specificity in a de novo iron-sulfur cluster binding peptide. Pept. Sci 104, 412–418 (2015). [DOI] [PubMed] [Google Scholar]
- 136.Beinert H, Kennedy MC & Stout CD Aconitase as iron–sulfur protein, enzyme, and iron-regulatory protein. Chem. Rev 96, 2335–2374 (1996). [DOI] [PubMed] [Google Scholar]
- 137.Nautiyal S, Woolfson DN, King DS & Alber T A designed heterotrimeric coiled coil. Biochemistry 34, 11645–11651 (1995). [DOI] [PubMed] [Google Scholar]
- 138.Lombardi A, Bryson JW & DeGrado WF De novo design of heterotrimeric coiled coils. Pept. Sci 40, 495–504 (1996). [DOI] [PubMed] [Google Scholar]
- 139.Kashiwada A, Hiroaki H, Kohda D, Nango M & Tanaka T Design of a heterotrimeric α-helical bundle by hydrophobic core engineering. J. Am. Chem. Soc 122, 212–215 (2000). [Google Scholar]
- 140.Kashiwada A, Ishida K & Matsuda K Lanthanide ion-induced folding of de novo designed coiled-coil polypeptides. Bull. Chem. Soc. Jpn 80, 2203–2207 (2007). [Google Scholar]
- 141.Tolbert AE et al. Heteromeric three-stranded coiled coils designed using a Pb(II)(Cys)3 template mediated strategy. Nat. Chem 12, 405–411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Walsh STR, Cheng H, Bryson JW, Roder H & DeGrado WF Solution structure and dynamics of a de novo designed three-helix bundle protein. Proc. Natl. Acad. Sci. U. S. A 96, 5486–5491 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chakraborty S et al. Design of a three-helix bundle capable of binding heavy metals in a triscysteine environment. Angew. Chem. Int. Ed 50, 2049–2053 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Plegaria JS, Dzul SP, Zuiderweg ERP, Stemmler TL & Pecoraro VL Apoprotein structure and metal binding characterization of a de novo designed peptide, α3div, that sequesters toxic heavy metals. Biochemistry 54, 2858–2873 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tebo AG et al. Development of a rubredoxin-type center embedded in a de dovo-designed three-helix bundle. Biochemistry 57, 2308–2316 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Koebke KJ et al. Clarifying the copper coordination environment in a de novo designed red copper protein. Inorg. Chem 57, 12291–12302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Plegaria JS, Herrero C, Quaranta A & Pecoraro VL Electron transfer activity of a de novo designed copper center in a three-helix bundle fold. Biochim. Biophys. Acta, Bioenerg 1857, 522–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Plegaria JS et al. De novo design and characterization of copper metallopeptides inspired by native cupredoxins. Inorg. Chem 54, 9470–9482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Summa CM, Lombardi A, Lewis M & DeGrado WF Tertiary templates for the design of diiron proteins. Curr. Opin. Struct. Biol 9, 500–508 (1999). [DOI] [PubMed] [Google Scholar]
- 150.Lombardi A et al. Retrostructural analysis of metalloproteins: Application to the design of a minimal model for diiron proteins. Proc. Natl. Acad. Sci. U. S. A 97, 6298–6305 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper is an early example of how complicated natural metalloproteins can be distilled to a few principles and reconstructed in a simpler, de novo scaffold.
- 151.Maglio O, Nastri F, Pavone V, Lombardi A & DeGrado WF Preorganization of molecular binding sites in designed diiron proteins. Proc. Natl. Acad. Sci. U. S. A 100, 3772–3777 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kaplan J & DeGrado WF De novo design of catalytic proteins. Proc. Natl. Acad. Sci. U. S. A 101, 11566–11570 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Law NA, Tyler Caudle M & Pecoraro VL Manganese redox enzymes and model systems: Properties, structures, and reactivity. Adv. Inorg. Chem 46, 305–440 (1998). [Google Scholar]
- 154.Davey James A., Damry Adam M., Euler Christian K., Goto Natalie K. & Chica Roberto A. Prediction of stable globular proteins using negative design with non-native backbone ensembles. Structure 23, 2011–2021 (2015). [DOI] [PubMed] [Google Scholar]
- 155.Leaver-Fay A, Jacak R, Stranges PB & Kuhlman B A generic program for multistate protein design. PLoS One 6, e20937 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Joh NH et al. De novo design of a transmembrane Zn2+-transporting four-helix bundle. Science 346, 1520–1524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is a clear case of applying the principle of negative design and a rare example of a de novo, membrane metalloprotein.
- 157.Löffler P, Schmitz S, Hupfeld E, Sterner R & Merkl R Rosetta:MSF: A modular framework for multi-state computational protein design. PLoS Comp. Biol 13, e1005600 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bradley P, Misura KM & Baker D Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–1871 (2005). [DOI] [PubMed] [Google Scholar]
- 159.Joh NH, Grigoryan G, Wu Y & DeGrado WF Design of self-assembling transmembrane helical bundles to elucidate principles required for membrane protein folding and ion transport. Philosophical Transactions of the Royal Society B: Biological Sciences 372, 20160214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Calhoun JR et al. Computational design and characterization of a monomeric helical dinuclear metalloprotein. J. Mol. Biol 334, 1101–1115 (2003). [DOI] [PubMed] [Google Scholar]
- 161.Choi YS, Zhang H, Brunzelle JS, Nair SK & Zhao H In vitro reconstitution and crystal structure of p-aminobenzoate N-oxygenase (AurF) involved in aureothin biosynthesis. Proc. Natl. Acad. Sci. U. S. A 105, 6858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Reig AJ et al. Alteration of the oxygen-dependent reactivity of de novo Due Ferri proteins. Nat. Chem 4, 900–906 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study highlights that natural and de novo proteins function via the same fundamental rules by showing that an active site alteration of the de novo scaffold recapitulates reactivity differences observed in natural proteins.
- 163.Chakraborty S, Iranzo O, Zuiderweg ERP & Pecoraro VL Experimental and theoretical evaluation of multisite cadmium(II) exchange in designed three-stranded coiled-coil peptides. J. Am. Chem. Soc 134, 6191–6203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Der BS et al. Metal-mediated affinity and orientation specificity in a computationally designed protein homodimer. J. Am. Chem. Soc 134, 375–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ghadiri MR & Case MA De novo design of a novel heterodinuclear three-helix bundle metalloprotein. Angew. Chem., Int. Ed. Engl 32, 1594–1597 (1993). [Google Scholar]
- 166.Lombardi A et al. Miniaturized metalloproteins: Application to iron–sulfur proteins. Proc. Natl. Acad. Sci. U. S. A 97, 11922–11927 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gibney BR, Mulholland SE, Rabanal F & Dutton PL Ferredoxin and ferredoxin–heme maquettes. Proc. Natl. Acad. Sci. U. S. A 93, 15041–15046 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kennedy ML & Gibney BR Proton coupling to [4Fe-4S]2+/+ and [4Fe-4Se]2+/+ oxidation and reduction in a designed protein. J. Am. Chem. Soc 124, 6826–6827 (2002). [DOI] [PubMed] [Google Scholar]
- 169.Nanda V et al. De novo design of a redox-active minimal rubredoxin mimic. J. Am. Chem. Soc 127, 5804–5805 (2005). [DOI] [PubMed] [Google Scholar]
- 170.Eck RV & Dayhoff MO Evolution of the structure of ferredoxin based on living relics of primitive amino acid sequences. Science 152, 363 (1966). [DOI] [PubMed] [Google Scholar]
- 171.Laplaza CE & Holm RH Helix–loop–helix peptides as scaffolds for the construction of bridged metal assemblies in proteins: The spectroscopic a-cluster structure in carbon monoxide dehydrogenase. J. Am. Chem. Soc 123, 10255–10264 (2001). [DOI] [PubMed] [Google Scholar]
- 172.Kim JD et al. Minimal heterochiral de novo designed 4Fe–4S binding peptide capable of robust electron transfer. J. Am. Chem. Soc 140, 11210–11213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xie F, Sutherland DEK, Stillman MJ & Ogawa MY Cu(I) binding properties of a designed metalloprotein. J. Inorg. Biochem 104, 261–267 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Kharenko OA, Kennedy DC, Demeler B, Maroney MJ & Ogawa MY Cu(I) luminescence from the tetranuclear Cu4S4 cofactor of a synthetic 4-helix bundle. J. Am. Chem. Soc 127, 7678–7679 (2005). [DOI] [PubMed] [Google Scholar]
- 175.Shiga D et al. Creation of a binuclear purple copper site within a de novo coiled-coil protein. Biochemistry 51, 7901–7907 (2012). [DOI] [PubMed] [Google Scholar]
- 176.Grzyb J et al. De novo design of a non-natural fold for an iron–sulfur protein: Alpha-helical coiled-coil with a four-iron four-sulfur cluster binding site in its central core. Biochim. Biophys. Acta, Bioenerg 1797, 406–413 (2010). [DOI] [PubMed] [Google Scholar]
- 177.Grzyb J et al. Empirical and computational design of iron-sulfur cluster proteins. Biochim. Biophys. Acta, Bioenerg 1817, 1256–1262 (2012). [DOI] [PubMed] [Google Scholar]
- 178.Nanda V et al. Structural principles for computational and de novo design of 4Fe–4S metalloproteins. Biochim. Biophys. Acta, Bioenerg 1857, 531–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jagilinki BP et al. In vivo biogenesis of a de novo designed iron–sulfur protein. ACS Synthetic Biology 9, 3400–3407 (2020). [DOI] [PubMed] [Google Scholar]
- 180.Mutter AC et al. De novo design of symmetric ferredoxins that shuttle electrons in vivo. Proc. Natl. Acad. Sci. U. S. A 116, 14557 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This article demonstrates that de novo proteins can perform essential functions in vivo.
- 181.Summa CM Computational methods and their applications for de novo functional protein design and membrane protein solubilization. (University of Pennsylvania, 2002). [Google Scholar]
- 182.Pike DH & Nanda V Empirical estimation of local dielectric constants: Toward atomistic design of collagen mimetic peptides. Biopolymers 104, 360–370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Chino M et al. Spectroscopic and metal binding properties of a de novo metalloprotein binding a tetrazinc cluster. Biopolymers 109, e23339 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zhang S-Q et al. De novo design of tetranuclear transition metal clusters stabilized by hydrogen-bonded networks in helical bundles. J. Am. Chem. Soc 140, 1294–1304 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhou L et al. A protein engineered to bind uranyl selectively and with femtomolar affinity. Nat. Chem 6, 236–241 (2014). [DOI] [PubMed] [Google Scholar]
- 186.Song WJ & Tezcan FA A designed supramolecular protein assembly with in vivo enzymatic activity. Science 346, 1525–1528 (2014). [DOI] [PubMed] [Google Scholar]
- 187.Song WJ, Yu J & Tezcan FA Importance of scaffold flexibility/rigidity in the design and directed evolution of artificial metallo-β-lactamases. J. Am. Chem. Soc 139, 16772–16779 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Golub E et al. Constructing protein polyhedra via orthogonal chemical interactions. Nature 578, 172–176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hansen WA & Khare SD Benchmarking a computational design method for the incorporation of metal ion-binding sites at symmetric protein interfaces. Protein Sci. 26, 1584–1594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Der BS, Edwards DR & Kuhlman B Catalysis by a de novo zinc-mediated protein interface: Implications for natural enzyme evolution and rational enzyme engineering. Biochemistry 51, 3933–3940 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Studer S et al. Evolution of a highly active and enantiospecific metalloenzyme from short peptides. Science 362, 1285–1288 (2018). [DOI] [PubMed] [Google Scholar]; This study highlights how de novo design of a malleable scaffold affords opportunities for using evolution to achieve remarkably active metalloenzymes from scratch.
- 192.Huang X & Groves JT Oxygen activation and radical transformations in heme proteins and metalloporphyrins. Chem. Rev 118, 2491–2553 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lu H & Zhang XP Catalytic C-H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev 40, 1899–1909 (2011). [DOI] [PubMed] [Google Scholar]
- 194.Leone L et al. Mimochrome, a metalloporphyrin-based catalytic swiss knife. Biotechnol. Appl. Biochem 67, 495–515 (2020). [DOI] [PubMed] [Google Scholar]
- 195.Rosenblatt MM, Wang J & Suslick KS De novo designed cyclic-peptide heme complexes. Proc. Natl. Acad. Sci. U.S.A 100, 13140–13145 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Choma CT et al. Design of a heme-binding four-helix bundle. J. Am. Chem. Soc 116, 856–865 (1994). [Google Scholar]
- 197.Robertson DE et al. Design and synthesis of multi-haem proteins. Nature 368, 425–432 (1994). [DOI] [PubMed] [Google Scholar]
- 198.Farid TA et al. Elementary tetrahelical protein design for diverse oxidoreductase functions. Nat. Chem. Biol 9, 826–833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Koder RL et al. Design and engineering of an O2 transport protein. Nature 458, 305–309 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work highlights the advantage of intentionally introducing strain into a de novo protein to allow switching between states.
- 200.Anderson JLR et al. Constructing a man-made c-type cytochrome maquette in vivo: Electron transfer, oxygen transport and conversion to a photoactive light harvesting maquette. Chem. Sci 5, 507–514 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Watkins DW et al. A suite of de novo c-type cytochromes for functional oxidoreductase engineering. Biochim. Biophys. Acta, Bioenerg 1857, 493–502 (2016). [DOI] [PubMed] [Google Scholar]
- 202.Watkins DW et al. Construction and in vivo assembly of a catalytically proficient and hyperthermostable de novo enzyme. Nat. Commun 8, 358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate the in vivo maturation of a de novo hemoprotein and its ability to perform catalysis.
- 203.Stenner R, Steventon JW, Seddon A & Anderson JLR A de novo peroxidase is also a promiscuous yet stereoselective carbene transferase. Proc. Natl. Acad. Sci. U. S. A 117, 1419 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Stenner R & Anderson JLR Chemoselective N–h insertion catalyzed by a de novo carbene transferase. Biotechnol. Appl. Biochem 67, 527–535 (2020). [DOI] [PubMed] [Google Scholar]
- 205.Liu Z & Arnold FH New-to-nature chemistry from old protein machinery: Carbene and nitrene transferases. Curr. Opin. Biotechnol 69, 43–51 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kamtekar S, Schiffer JM, Xiong H, Babik JM & Hecht MH Protein design by binary patterning of polar and nonpolar amino acids. Science 262, 1680–1685 (1993). [DOI] [PubMed] [Google Scholar]; This work presents an early strategy for developing libraries of proteins that can be readily interrogated for function.
- 207.Hecht MH, Das A, Go A, Bradley LH & Wei Y De novo proteins from designed combinatorial libraries. Protein Sci. 13, 1711–1723 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hecht MH, Zarzhitsky S, Karas C & Chari S Are natural proteins special? Can we do that? Curr. Opin. Struct. Biol 48, 124–132 (2018). [DOI] [PubMed] [Google Scholar]
- 209.Rojas NR et al. De novo heme proteins from designed combinatorial libraries. Protein Sci. 6, 2512–2524 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Polizzi NF et al. De novo design of a hyperstable non-natural protein–ligand complex with sub-Å accuracy. Nat. Chem 9, 1157–1164 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work highlights the importance of stabilizing the active site through design of a complementary well-folded region of a de novo protein.
- 211.Kaufmann KW, Lemmon GH, DeLuca SL, Sheehan JH & Meiler J Practically useful: What the Rosetta protein modeling suite can do for you. Biochemistry 49, 2987–2998 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Bender BJ et al. Protocols for molecular modeling with Rosetta3 and RosettaScripts. Biochemistry 55, 4748–4763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mann SI, Nayak A, Gassner GT, Therien MJ & DeGrado WF De novo design, solution characterization, and crystallographic structure of an abiological Mn-porphyrin-binding protein capable of stabilizing a Mn(V) species. J. Am. Chem. Soc 143, 252–259 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Mahajan M & Bhattacharjya S B-hairpin peptides: Heme binding, catalysis, and structure in detergent micelles. Angew. Chem. Int. Ed 52, 6430–6434 (2013). [DOI] [PubMed] [Google Scholar]
- 215.D’Souza A, Mahajan M & Bhattacharjya S Designed multi-stranded heme binding β-sheet peptides in membrane. Chem. Sci 7, 2563–2571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.D’Souza A, Wu X, Yeow EKL & Bhattacharjya S Designed heme-cage β-sheet miniproteins. Angew. Chem. Int. Ed 56, 5904–5908 (2017). [DOI] [PubMed] [Google Scholar]
- 217.D’Souza A, Torres J & Bhattacharjya S Expanding heme-protein folding space using designed multi-heme β-sheet mini-proteins. Commun. Chem 1, 78 (2018). [Google Scholar]
- 218.D’Souza A & Bhattacharjya S De novo-designed β-sheet heme proteins. Biochemistry 60, 431–439 (2021). [DOI] [PubMed] [Google Scholar]
- 219.Rufo CM et al. Short peptides self-assemble to produce catalytic amyloids. Nat. Chem 6, 303–309 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate the first de novo-designed, catalytic metalloamyloids.
- 220.Lengyel Z, Rufo CM, Moroz YS, Makhlynets OV & Korendovych IV Copper-containing catalytic amyloids promote phosphoester hydrolysis and tandem reactions. ACS Catal. 8, 59–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zozulia O & Korendovych IV Semi-rationally designed short peptides self-assemble and bind hemin to promote cyclopropanation. Angew. Chem. Int. Ed 59, 8108–8112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Voet ARD et al. Computational design of a self-assembling symmetrical β-propeller protein. Proc. Natl. Acad. Sci. U. S. A 111, 15102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Voet ARD, Noguchi H, Addy C, Zhang KYJ & Tame JRH Biomineralization of a cadmium chloride nanocrystal by a designed symmetrical protein. Angew. Chem. Int. Ed 54, 9857–9860 (2015). [DOI] [PubMed] [Google Scholar]
- 224.Vrancken JPM, Noguchi H, Zhang KYJ, Tame JRH & Voet ARD The symmetric designer protein pizza as a scaffold for metal coordination. Proteins: Struct. Funct., Bioinf 89, 945–951 (2021). [DOI] [PubMed] [Google Scholar]
- 225.Huang P-S et al. De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat. Chem. Biol 12, 29–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Caldwell SJ et al. Tight and specific lanthanide binding in a de novo TIM barrel with a large internal cavity designed by symmetric domain fusion. Proc. Natl. Acad. Sci. U. S. A 117, 30362–30369 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work is an excellent example of de novo design of a high-symmetry active site that does not use a coiled coil.
- 227.Vorobieva AA et al. De novo design of transmembrane β barrels. Science 371, eabc8182 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Pan X et al. Expanding the space of protein geometries by computational design of de novo fold families. Science 369, 1132–1136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Yang C et al. Bottom-up de novo design of functional proteins with complex structural features. Nat. Chem. Biol 17, 492–500 (2021). [DOI] [PubMed] [Google Scholar]
- 230.Paredes A, Loh BM, Peduzzi OM, Reig AJ & Buettner KM DNA cleavage by a de novo designed protein–titanium complex. Inorg. Chem 59, 11248–11252 (2020). [DOI] [PubMed] [Google Scholar]
- 231.Ball ZT Molecular recognition in protein modification with rhodium metallopeptides. Curr. Opin. Chem. Biol 25, 98–102 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ohata J & Ball ZT Rhodium at the chemistry–biology interface. Dalton Trans. 47, 14855–14860 (2018). [DOI] [PubMed] [Google Scholar]
- 233.Behrendt R, White P & Offer J Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci 22, 4–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Chin JW Expanding and reprogramming the genetic code of cells and animals. Annu. Rev. Biochem 83, 379–408 (2014). [DOI] [PubMed] [Google Scholar]
- 235.Mukai T, Lajoie MJ, Englert M & Söll D Rewriting the genetic code. Annu. Rev. Microbiol 71, 557–577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wang L Engineering the genetic code in cells and animals: Biological considerations and impacts. Acc. Chem. Res 50, 2767–2775 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yu Y, Hu C, Xia L & Wang J Artificial metalloenzyme design with unnatural amino acids and non-native cofactors. ACS Catal. 8, 1851–1863 (2018). [Google Scholar]
- 238.Lu Y Design and engineering of metalloproteins containing unnatural amino acids or non-native metal-containing cofactors. Curr. Opin. Chem. Biol 9, 118–126 (2005). [DOI] [PubMed] [Google Scholar]
- 239.Agostini F et al. Biocatalysis with unnatural amino acids: Enzymology meets xenobiology. Angew. Chem. Int. Ed 56, 9680–9703 (2017). [DOI] [PubMed] [Google Scholar]
- 240.Yoon JH et al. Uno Ferro, a de novo designed protein, binds transition metals with high affinity and stabilizes semiquinone radical anion. Chem. – Eur. J 25, 15252–15256 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Leveson-Gower RB, Mayer C & Roelfes G The importance of catalytic promiscuity for enzyme design and evolution. Nat. Rev. Chem 3, 687–705 (2019). [Google Scholar]
- 242.Chen K & Arnold FH Engineering new catalytic activities in enzymes. Nat. Catal 3, 203–213 (2020). [Google Scholar]
- 243.Pavlovicz RE, Park H & DiMaio F Efficient consideration of coordinated water molecules improves computational protein-protein and protein-ligand docking discrimination. PLoS Comp. Biol 16, e1008103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lai JK, Ambia J, Wang Y & Barth P Enhancing structure prediction and design of soluble and membrane proteins with explicit solvent-protein interactions. Structure 25, 1758–1770.e1758 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Polizzi NF & DeGrado WF A defined structural unit enables de novo design of small-molecule-binding proteins. Science 369, 1227–1233 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Huang LS, Cobessi D, Tung EY & Berry EA Binding of the respiratory chain inhibitor antimycin to the mitochondrial bc1 complex: A new crystal structure reveals an altered intramolecular hydrogen-bonding pattern. J. Mol. Biol 351, 573–597 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Håkansson K, Carlsson M, Svensson LA & Liljas A Structure of native and apo carbonic anhydrase II and structure of some of its anion-ligand complexes. J. Mol. Biol 227, 1192–1204 (1992). [DOI] [PubMed] [Google Scholar]
- 248.Zaytsev DV et al. Metal-binding properties and structural characterization of a self-assembled coiled coil: Formation of a polynuclear Cd–thiolate cluster. J. Inorg. Biochem 119, 1–9 (2013). [DOI] [PubMed] [Google Scholar]
- 249.Lahr SJ et al. Analysis and design of turns in α-helical hairpins. J. Mol. Biol 346, 1441–1454 (2005). [DOI] [PubMed] [Google Scholar]
- 250.Wood CW & Woolfson DN CCBuilder 2.0: Powerful and accessible coiled-coil modeling. Protein Sci. 27, 103–111 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Zhou J & Grigoryan G Rapid search for tertiary fragments reveals protein sequence-structure relationships. Protein Sci. 24, 508–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Raman S et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins: Struct. Funct., Bioinf 77, 89–99 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Baek M et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]