

Abstract
Irregular nuclear shapes are a hallmark of human cancers. Recent studies suggest that alterations to chromatin regulators may cause irregular nuclear morphologies. Here we screened an epigenetic small molecule library consisting of 145 compounds against chromatin regulators for their ability to revert abnormal nuclear shapes that were induced by gene knockdown in noncancerous MCF10A human mammary breast epithelial cells. We leveraged a previously validated quantitative Fourier approach to quantify the elliptical Fourier coefficient (EFC ratio) as a measure of nuclear irregularities, which allowed us to perform rigorous statistical analyses of screening data. Top hit compounds fell into three major mode of action categories, targeting three separate epigenetic modulation routes: 1) histone deacetylase inhibitors, 2) bromodomain and extraterminal domain protein inhibitors, and 3) methyl-transferase inhibitors. Some of the top hit compounds were also efficacious in reverting nuclear irregularities in MDA-MB-231 triple negative breast cancer cells and in PANC-1 pancreatic cancer cells in a cell-type-dependent manner. Regularization of nuclear shapes was compound-specific, cell-type specific, and dependent on the specific molecular perturbation that induced nuclear irregularities. Our approach of targeting nuclear abnormalities may be potentially useful in screening new types of cancer therapies targeted toward chromatin structure.
INTRODUCTION
Unlike the smooth ellipsoidal nuclei of most normal epithelial cells, cancer cell nuclei tend to have blebs, folds, and invaginations which give them an irregular appearance. Such irregularities are useful for cancer diagnosis in clinical settings (Gil et al., 2002; Kashyap et al., 2018). Alterations to nuclear structure contribute to cancer progression and malignancy (Zink et al., 2004). As such, targeting nuclear structure is a promising approach for the development of new therapeutics.
Despite the extensive use of abnormal nuclear morphology by pathologists for cancer diagnosis, the mechanisms underlying cancer nuclear dysmorphia have remained poorly understood. At least two competing hypotheses have been proposed for explaining abnormal nuclear shapes in human cancers: a down-regulation of the nuclear lamins (Denais and Lammerding, 2014) and alterations to chromatin structure (Stephens et al., 2017b). Nuclear lamins underlie the nuclear envelope and impart mechanical stiffness to the nucleus at large extensions (Stephens et al., 2017a). As lamins are down-regulated in many cancers, the resulting softening of the nucleus may account for irregular cancer nuclear shapes (Lammerding et al., 2004; Lammerding et al., 2006; Tocco et al., 2018; Kent et al., 2019). Recent studies suggest, alternatively, that alterations to chromatin and epigenetic regulators may cause abnormal nuclear shapes (Imbalzano et al., 2013; Furusawa et al., 2015; Schreiner et al., 2015; Stephens et al., 2017a; Stephens et al., 2018; Senigagliesi et al., 2019; Tamashunas et al., 2020). Thus nuclear shape irregularities may, to an extent, be a readout of abnormal chromatin structure in cells.
An early study showed that knockdown of the chromatin remodeling enzyme BRG-1 (encoded by SMARCA4) increased the incidence of folds, bulges, and invaginations in MCF10A nuclei (Imbalzano et al., 2013). Consistent with a role for chromatin structures and not lamins in generating nuclear irregularities, increasing euchromatin or decreasing heterochromatin by treatment with small molecule compounds increased nuclear blebbing in HT1080 fibrosarcoma cells without altering lamins (Stephens et al., 2018). Given that small molecule compounds targeting chromatin can induce abnormalities in nuclear shape (Stephens et al., 2018), we reasoned that modulating chromatin regulators with small molecule compound libraries could be a promising strategy to revert nuclear shape abnormalities in cancer cells.
Here we leveraged a recently developed quantitative nuclear shape-based screen (Tamashunas et al., 2020) to perform a small molecule drug screen toward nuclear shape. We had previously conducted a high-throughput RNAi screen with the goal of expanding the list of epigenetic chromatin regulators that impact nuclear morphology. To quantify nuclear irregularities in an unbiased manner, we developed and used an automated elliptical Fourier analysis technique to quantify the elliptic Fourier coefficient (EFC) ratio (Tamashunas et al., 2020). The EFC ratio is a quantitative measure of the irregularity of the nuclear contour; lower values of EFC indicate high irregularity. Evaluation of nuclear irregularity with the EFC ratio allowed a statistical analysis across different gene-knockdown conditions. Top hits included TP53 which is associated with mostly loss-of-function mutations in more than 36% of human breast cancers and metastasis-associated protein encoded by the gene MTA2.
Guided by these previous results, here we reasoned that small molecule compounds that target chromatin regulators could be used to revert nuclear shape abnormalities induced by a single molecular perturbation such as a reduction in the expression of TP53 or MTA2. To this end, we used siRNA to deplete TP53 or MTA2 expression in noncancerous MCF10A human breast epithelial cells, which caused a reduction in the mean EFC ratio (indicative of irregular nuclear shapes). We next screened an epigenetic small molecule compound library for its effect on the induced irregular shapes in TP53- or MTA2-siRNA transfected MCF10A cells. Guided by the results of these screens, we next performed a targeted study of the effects of top hits on the mean nuclear EFC ratio in two human cancer cell lines with inherently irregular nuclear morphology that is not induced by any single molecular perturbation.
RESULTS
A Fourier approach for quantifying nuclear irregularities
To avoid observer bias associated with manual classification (Imbalzano et al., 2013) and also to enable quantitative small molecule compound screening applications targeted at nuclear morphology, we used elliptical Fourier analysis (Diaz et al., 1989; Lammerding et al., 2006) to quantify nuclear irregularities. Described in Tamashunas et al. (2020), our approach was to approximate the nuclear contour segmented from lamin A/C immunostained cells with a series of 15 harmonic ellipses and to calculate an EFC ratio. To exclude spurious fragments of nuclei from the analysis, we only included nuclei with EFC ratios above a value of 2 (Tamashunas et al., 2020). This approach yields a superior numerical separation compared with the solidity metric for quantifying nuclear irregularities (Tamashunas et al., 2020). We started out by confirming that depletion of TP53 or MTA2 expression using RNAi in cultured human mammary gland MCF10A epithelial cells induced abnormal nuclear shapes as measured by a statistically significant decrease in the EFC ratio compared with the vehicle controls. Fluorescent images of lamin A/C immunostained nuclei were collected. Each nucleus in the field of view was segmented, and an EFC ratio was calculated. Figure 1A shows a typical segmentation of nuclei in the field of view with corresponding EFC ratios overlaid on each nucleus. Depletion of TP53 or MTA2 expression caused a significant decrease in the EFC ratios compared with control (gene-knockdown validation results are shown in Supplemental Figure S1).
FIGURE 1:

Representative nuclear shapes and EFC ratio differences in control vs. TP53- or MTA2-siRNA transfected MCF10A cells. (A) Shapes of lamin A/C stained nuclei with corresponding EFC ratios overlaid on each nucleus in a representative image of control (nontarget siRNA transfected, left), TP53-siRNA transfected (middle) and MTA2-siRNA transfected (right) MCF10A epithelial cells at 120 h post-siRNA treatment without drug treatment (vehicle-control 0.5% DMSO only). Scale bars represent 50 µm. (B) Mean EFC ratios differences (+ SEM) for the three siRNA treatment groups without compound treatment (with vehicle control 0.5% DMSO only). Between-group analysis was done using two-tailed Mann–Whitney U test in GraphPad. P values were <0.0001 (depicted as **** in the graph); the statistical comparison included 32601 nuclei for nontarget siRNA, 25362 nuclei for TP53 siRNA, and 6191 nuclei for MTA2 siRNA.
Small molecule screen toward nuclear abnormalities induced by siRNA transfection targeting TP53 or MTA2
We next performed a high-throughput screen on cells transfected with TP53 siRNA or MTA2 siRNA against the Cayman Epigenetic Screening Library (ESL) (Cayman Chemicals) at a 1 µM final test concentration with the goal of discovering compounds that create a statistically significant increase in the EFC ratio in knockdown cells (Supplemental Table S1 contains a list of screened compounds). The data set consisted of EFC ratios quantified from nuclei imaged in at least three replicate wells containing cells per drug treatment for each gene-knockdown condition and control. Because the distribution of the measured EFC ratios was not Gaussian, we analyzed log-transformed data that showed normality (see Supplemental Figures S2–S5). This allowed us to then model the log-transformed data using a two-way ANOVA model of interactions between drug and gene knockdown (see Materials and Methods). To aid comparison across EFC values for different groups, we focused on treatment (interaction) contrasts obtained from the ANOVA model. The resulting test statistics involved differences in means of the log-transformed data, which were then converted into a ratio of geometric means of the nontransformed data. We accounted for multiplicity in the p values for the treatment contrasts computed from the ANOVA model using the Benjamini-Hochberg false discovery adjustment procedure (Benjamini and Hochberg, 1995).
Treatment of cells containing irregular nuclear shapes induced by gene knockdown with small molecule compound libraries (each compound was treated at 1 µM concentration, which was presumed to be sufficient to engage its corresponding epigenetic target without significant toxicity based on cellular viability data; data not shown) had the following three outcomes (Figure 2; data are organized according to compound functions): 1) there was no effect of the treatment on nuclear irregularity (i.e., treatment mean EFC ratio in gene knockdown/DMSO mean EFC ratio in gene knockdown was indistinguishable from 1, in gray), 2) worsening of nuclear irregularity (i.e., treatment mean EFC ratio in gene knockdown/DMSO mean EFC ratio in gene knockdown was significantly less than 1, in shades of orange), and 3) improvement in the gene-knockdown-induced nuclear irregularity (i.e., treatment mean EFC ratio under gene knockdown/DMSO mean EFC ratio under gene knockdown was significantly greater than 1, in shades of blue). Small molecules from the epigenetic libraries tended to increase EFC ratios (i.e., make nuclear shapes regular) in TP53-knockdown cells to a larger extent than in MTA2-knockdown cells. Compounds that improved the nuclear EFC ratios/shapes in both gene-depletion conditions (compounds highlighted in blue) were selected as hits for dose-response validation. Five of the 14 selected hits belonged to broad-range Zn2+-dependent (class I/II/IV) HDAC inhibitors. Only two of the tested compounds made nuclear shapes more irregular (lowered EFC ratios) in MTA2-knockdown (orange) cells. Collectively, these results demonstrate the first successful targeting of nuclear shape irregularities with a small molecule compound library against epigenetic pathways.
FIGURE 2:

Analysis of the extent to which small molecule compound libraries alter nuclear morphological abnormalities in MCF10A cells transfected with siRNA toward TP53 or MTA2. Heat maps show within-group comparisons of small molecule compound library effects on nuclear EFC ratios in TP53- or MTA2–knockdown cells. Comparisons are shown of treatment vs. DMSO effects within gene-knockdown datasets. The treatments are grouped by the small molecule functions; cells were imaged for each treatment condition from four different wells (four technical replicates). For each small molecule treatment (1 µM dose), the ratio of geometric means of nuclear EFC ratios for that treatment group to the DMSO group for a given knockdown condition (TP53 or MTA2) is displayed as a heat map. Ratios of geometric means statistically significantly greater than 1 are displayed in shades of blue (improvement of nuclear shapes), those statistically significantly less than 1 are displayed in shades of orange (worsening of nuclear shapes), and those not statistically significantly different from 1 are displayed as gray. Compound names highlighted in blue indicate that the ratio of geometric means was statistically significantly different from 1 in both gene-knockdown conditions. BET, bromodomain and extraterminal domain; DMT, DNA methyltransferase; HDM, histone demethylase; HMT, histone methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; PARP, poly adenosine diphosphate-ribose polymerase. Statistical comparisons were performed with tests of contrasts on an omnibus two-way ANOVA model (see Materials and Methods). The number of nuclei per treatment, adjusted p values, and estimates of contrasts are provided in Supplemental Table S2.
Analysis of reversion of abnormal nuclear morphologies by small molecule treatment
We then investigated the extent to which drug treatment was able to revert the irregular nuclear shape induced by gene knockdown to nuclear shapes in control MCF10A epithelial cells. To test this, we compared the nuclear EFC ratio in control cells transfected with nontarget siRNA and treated with DMSO with cells transfected with TP53 or MTA2 siRNA and treated with small molecule compounds. Our analysis revealed the following outcomes: 1) in the case of a few compounds, there was complete reversion of the gene knockdown-induced nuclear irregularity (gray bar in Figure 3), and 2) in most cases, there continued to be some irregularity in the nuclear shape in cells transfected with TP53 siRNA or MTA2 siRNA despite drug treatment (orange bars in Figure 3). Notably, complete reversion of irregularity was observed in TP53-knockdown cells by four different Zn2+-dependent HDAC inhibitors—LAQ824, oxamflatin, SB939, and trichostatin A.
FIGURE 3:

Analysis of the extent to which small molecule compound libraries revert nuclear morphology to control in MCF10A cells transfected with siRNA toward TP53 or MTA2. Heat maps show between-group comparisons of drug library effects on nuclear EFC ratios in TP53- or MTA2-knockdown cells. Comparisons are shown of treatment in gene knockdown vs. DMSO treatment in cells transfected with nontargeting scrambled siRNA control. The treatments are grouped by small molecule functions; cells were imaged for each treatment condition from four different wells (four technical replicates). For each treatment, the ratio of geometric means of lamin A/C staining-based nuclear EFC ratios for that treatment group for a given gene-knockdown condition to the DMSO group in cells transfected with scrambled siRNA is displayed as a heat map. Ratios of geometric means statistically significantly less than 1 are displayed in shades of orange; those not statistically significantly different from 1 are displayed in gray. BET, bromodomain and extraterminal domain; DMT, DNA methyltransferase; HDM, histone demethylase; HMT, histone methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; PARP, poly adenosine diphosphate-ribose polymerase. Statistical comparisons were performed with tests of contrasts on an omnibus two-way ANOVA model (see Materials and Methods). The number of nuclei per treatment, adjusted p values and estimates of contrasts are provided in Supplemental Table S3.
Concordance of EFC ratio with projected nuclear area
In cultured cells, the process of cell-spreading drives the unwrinkling of the nuclear lamina and nuclear flattening (Cosgrove et al., 2021; Dickinson et al., 2022). As such, the regularization of the nuclear shape abnormalities could be a consequence of changes to cell spreading which in turn will impact the projected two-dimensional nuclear cross-sectional area. In a previous paper, we reported that nuclear EFC ratio does not correlate on average with the degree of cell spreading nor with nuclear cross-sectional area (Tamashunas et al., 2020). Here we additionally assessed the utility of incorporating projected nuclear area (in pixels) as a secondary measure of nuclear geometry for specific treatment conditions (rather than average values) in our screening study. First, we performed a correlation analysis assessing concordance between nuclear EFC ratios and nuclear areas for different treatment and gene-knockdown combinations; the associated Spearman correlation coefficients are displayed in Figure 4. Most of the correlation coefficients were small and positive across treatment conditions for MTA2-knockdown cells (range of 0.13 to 0.38). For p53-knockdown cells, in contrast, area and EFC ratio were anticorrelated for some conditions, positively correlated for others, and not correlated for still others. In all cases, any correlations were again small (range of –0.07 to 0.2). Second, we considered an analogous, parallel omnibus two-way ANOVA analysis with log nuclear areas (instead of log EFC ratios) as the response. The resulting findings from the model are displayed in Supplemental Figures S8 and S9, which are analogs of Figures 2 and 3, respectively. Consistent with the correlation analysis, there are only modest similarities in the effects of treatment conditions on EFC ratios and on projected nuclear areas in MTA2-knockdown cells, and little to no correspondence between the two in p53-knockdown cells. Overall, changes to nuclear projected area were found to be not explanatory of the observed trends in EFC ratios.
FIGURE 4:

Correlation analysis of nuclear projected area and nuclear EFC ratio. Heat maps show Spearman correlation coefficients between nuclear projected area and EFC ratios for each treatment in TP53- or MTA2-knockdown cells. The treatments are grouped by small molecule functions; cells were imaged for each treatment condition from four different wells (four technical replicates). Correlation coefficients less than 0 are displayed in shades of orange, and those above 0 are displayed in shades of blue.
Validation of top hits by dose response analysis
We next chose the top 14 hits that significantly reduced nuclear irregularity in TP53- and MTA2-knockdown cells (names are highlighted in blue in Figure 2). Since two class I-selective HDAC inhibitors were among the 14 hits (apicidin and pyroxamide), for further validation we also included FK228, an FDA-approved class I HDAC inhibitor, and our marine-derived preclinical class I HDAC inhibitor largazole (Hong and Luesch, 2012; Al-Awadhi et al., 2020) to probe for potential HDAC class selectivity. In total, we measured 32 different drug-dose curves. We performed an analysis of the dependence of the nuclear EFC ratios on drug dose for a given knockdown condition (TP53 or MTA2) while controlling for numbers of nuclei. Most of the top hits were found to reduce nuclear irregularity significantly over a range of drug doses (Figure 5). This was true both for TP53- and MTA2-knockdown cells. Of the 28 different drug-dose curves for the 14 original hits, only 3 curves (bromosporine, CPI−203, and 5-Azacytidine treatment for MTA2-knockdown cells) showed no improvement in nuclear irregularity at any dose. Thus our larger drug screen was deemed to have an 89% success rate. All but one drug (bromosporine) increased the EFC ratio in TP53-depleted cells. Furthermore, while apicidin showed normalizing effects on nuclear shape under both conditions, the other two class I HDAC inhibitors (largazole and FK228) only reduced TP53-knockdown-induced irregularity but not in MTA2-depleted cells, suggesting both common and differential effects for different inhibitors of the same class. Cell viability assays carried out in parallel confirmed over 50% survival of cells at 1 µM concentration (Supplemental Figure S6).
FIGURE 5:

Dependence of nuclear shape and the EFC ratio on the compound dose in a targeted screen of drugs in TP53- and MTA2-knockdown MCF10A cells. (A) For each compound, the corresponding panel plots the log of the ratio of geometric means of nuclear EFC ratios (y axis) at different doses (x axis) for that drug group to the DMSO group for a given knockdown condition (TP53 in the gray line or MTA2 in the green line). Cells were imaged for each treatment condition from four different wells (four technical replicates). Ratios of geometric means statistically significantly greater than 1 are displayed with blue dots; those statistically significantly less than 1 are displayed with brown dots; those not statistically significantly different from 1 are displayed with gray dots. (B) Representative nuclear shapes in TP53-knockdown MCF10A cells for select hits that showed improvement at doses lower than the dose tested in the screen (1 µM) compared with the vehicle control (DMSO). Overlaid image of nuclei stained with DAPI (blue) and immunostained for lamin A/C (green) are shown. Scale bars represent 25 µm. Statistical comparisons were performed with tests of contrasts on an omnibus three-way ANOVA model (see Materials and Methods). The number of nuclei per treatment, adjusted p values, and estimates of contrasts are provided in Supplemental Table S4.
Effect of small molecules on cancer nuclear abnormalities
Of the validated 16 compounds (including largazole and FK228) that increased nuclear EFC ratios, we performed a smaller screen for concentration dependency of effects on EFC ratios that spontaneously exist in two different cancer cell types: the highly invasive, metastatic MDA-MB-231 breast cancer cells and the PANC-1 pancreatic carcinoma cancer cells. The subset of compounds for testing against cancer cells was selected based on their effectiveness in reducing nuclear irregularity at doses lower than 1 µM in the MCF10A siRNA-knockdown assay. Several compounds increased nuclear irregularity (reduced EFC ratio) in MDA-MB-231 cells, while others decreased it (Figure 6). Dose-response analysis for cell viability carried out in parallel confirmed over 50% survival of cells at 1 µM concentration (Supplemental Figure S7).
FIGURE 6:

Dependence of nuclear shape and the EFC ratio on the drug dose in a targeted screen of drugs in MDA-MB-231 breast cancer cells and PANC-1 pancreatic cancer cells. (A) For each compound, the corresponding panel plots the log of the ratio of geometric means of nuclear EFC ratios (y axis) at different doses (x axis) for that drug group to the DMSO group for a given cancer cell line (MDA-MB-231 in the green line, PANC-1 in the gray line). Cells were imaged for each treatment condition from four different wells (four technical replicates). Ratios of geometric means statistically significantly greater than 1 are displayed with blue dots; those statistically significantly less than 1 are displayed with brown dots; those not statistically significantly different from 1 are displayed with gray dots. (B) Representative nuclear shapes in MDA-MB-231 or PANC-1 cancer cells for best hits that showed improvement in both cell lines compared with vehicle control (DMSO). Overlaid image of nuclei stained with DAPI (blue) and immunostained for lamin A/C (green) are shown for the lowest tested doses that showed visual improvement of nuclei (MDA-MB-231 cells; largazole at 32 nM; FK228 at 320 pM and UNC642 at 1 uM; PANC-1 cells, largazole at 100 nM; FK228 at 3.2 nM and UNC642 at 3.2 uM). Scale bars represent 25 µm. Statistical comparisons were performed with tests of contrasts on an omnibus three-way ANOVA model (see Materials and Methods). The number of nuclei per treatment, adjusted p values and estimates of contrasts are provided in Supplemental Table S5.
The majority of the compounds reduced nuclear irregularity in PANC-1 cells. These results demonstrate the first successful targeting of cancer nuclear irregularities by epigenetic compounds and demonstrate the compound-dependent effects on nuclear phenotype.
DISCUSSION
Nuclear shape irregularities are a common feature of human cancers (Zink et al., 2004), but whether they can be targeted by small molecule compound libraries has not been tested. In this paper, we tested the hypothesis that nuclear irregularity could be modulated with small molecule compounds that target chromatin regulators. We performed a small molecule compound screen on normal and cancer cells, collected images on a high-throughput microscope, and quantified nuclear irregularities with a recently validated computational method (Tamashunas et al., 2020) that calculates an EFC ratio as a measure of nuclear irregularity. Statistical analysis revealed several compounds across different conditions which consistently decreased nuclear irregularities both when irregularities were induced by gene knockdown in normal cells and in cancer cells that naturally contain nuclear irregularities.
While several compounds had similar effects independent of gene-knockdown condition (TP53 or MTA2) or even cell type (MDA-MB-231 cells or PANC-1 cells), many compounds did not have similar effects across different conditions. This shows that impact of compounds on nuclear shape is cell-type specific. Also, more compounds tended to reduce nuclear irregularity on depletion of TP53 in mammary epithelial cells than on depletion of MTA2. Therefore effects of compounds on nuclear shape also depend on the specific molecular perturbation that causes the nuclear shape abnormalities. These results suggest collectively that shape regularization is likely caused by specific molecular effects of the compounds rather than nonspecific effects on cellular pathways. The compounds may impact nuclear shape through distinct mechanisms (Lele et al., 2018). Changes to chromatin state may result in an increase in the nuclear size at constant area which is predicted to smoothen the nuclear contour. The relative lack of correlation between nuclear projected area and EFC ratio does not support this mechanism. Alternatively, changes in chromatin state may alter nuclear mechanical properties; stiffening the nucleus may promote shape regularization and softening it may result in altered shapes. Finally, the compounds may have indirect effects on the levels of nuclear lamins which may in turn impact nuclear shape.
Previous studies have shown that treating HT1080 human fibrosarcoma cells or mouse embryonic fibroblasts with HDAC inhibitors caused nuclear blebbing due to decompaction of chromatin and a softening of the nucleus (Stephens et al., 2018). Consistent with these findings, the two compounds that inhibit HDAC—salermide (SIRT = class III inhibitor) and panobinostat (pan-class I/II inhibitor)—increased nuclear irregularity (Figure 2). On the other hand, several HDAC inhibitors also consistently tended to increase the EFC ratio across a range of conditions. The irregularities we measured in nuclei in this study were from lamin A/C-stained images. Thus we focused specifically on irregularities of the nuclear lamina, while in Stephens et al. (2018), the focus was on nuclear blebs that form independently of lamins. Therefore, the type of nuclear irregularity studied likely differs between our work and that reported in Stephens et al. (2018). Also, effects of compounds tend to be cell-type specific as we have shown here, which may also partially account for these differences. Furthermore, we used both cancer cells and “normal” (noncancer) cells where HDAC inhibition may produce variable phenotypic outcomes, such as preferential activity for cancer cell growth inhibition compared with nontransformed cells. However, we provided insights into the class I selectivity, which was consistently observed in TP53-depleted MCF10A cells. Differential effects of compounds with a similar pharmacological profile at the target level may provide opportunities for complementary cell-type-specific applications.
It is possible that some hits may have been missed through the initial screen since we performed the primary screen at a single concentration. The screening dose of 1 µM was selected based on a previous toxicity screen in cancer cell lines (data not shown) so that over 80% of the tested compounds will display less than 50% cytotoxicity. This, however, may be a limitation of the hit discovery for some of the compounds, as it may fall below the EC50 dose. This may explain the relatively low hit rate (14 hits out of 145 compounds) for such a focused library. Also, compounds that were efficacious in noncancerous cells need not be effective in cancer cells given the complex genetic background of cancer cells.
Our results suggest that treatment with small molecule compounds may be an effective approach to normalize cancer nuclear morphology caused by loss of tumor suppressors like p53. It is known that the absence of p53 increases the frequency of spontaneous rupture of the nuclear envelope (Yang et al., 2017). Nuclear envelope rupture in turn causes DNA damage (Denais et al., 2016) and promotes invasive migration (Nader et al., 2021). Reverting abnormal shapes caused by loss of p53 with the pharmacological approach developed here may help prevent cancer nuclear ruptures and prevent invasive cancer cell behaviors. Abnormal nuclear shapes tend to be correlated with a mechanical softening of nuclei (Denais and Lammerding, 2014), which in turn correlates with the ability of cancer cells to invade metastatically (Vortmeyer-Krause et al., 2020). Thus normalizing nuclear shape may reduce the ability of cancer cells to invade surrounding interstitial tissue and eventually enter the circulation. Also, nuclear shape abnormalities may be a useful phenotypic cellular marker in screening efficacy of small molecules targeted toward chromatin structure in cellular screens. Parsing the extent to which nuclear shape regularization impacts nuclear rupture, nuclear stiffness, DNA damage, and cancer cell invasion will be important to future efforts that therapeutically target nuclear morphological abnormalities in cancer.
MATERIALS AND METHODS
Cell culture
Human mammary gland fibrocystic disease epithelial cells, MCF10A (ATCC CRL-10317), were cultured in DMEM/F12 (Corning, Cat#: 10-092-CV) media supplemented as described previously (Tamashunas et al., 2020). Cell cultures were maintained in T-75 flasks prior to experiments. For the high-throughput screen, cells were dissociated from the flasks using 0.05% Trypsin-EDTA (Life Technologies, Cat#: 25-300-054), counted, and seeded in 50 μl/well fresh media in 384-well Cell Carrier Ultra black-wall, optically clear bottom plates (PerkinElmer, Cat#: 6057308). Cells were left to attach and acclimate overnight before transfection. Human breast adenocarcinoma MDA-MB-231 cells (ATCC, HTB-26) and human pancreatic adenocarcinoma cells PANC1 (ATCC, CRL-1469) were cultured in L–glutamine-enriched DMEM (Life Technologies cat#: 11965118) supplemented with 10% fetal bovine serum (Sigma) and 1% antibiotic/antimycotic (Life Technologies, cat#: 15240-062).
Screening optimization
The assay was previously developed and optimized in MCF-10A cells for screening of epigenetic targets through an siRNA screen (Tamashunas et al., 2020). Here we optimized the screen in MCF10A cells for MTA2- and TP53-gene knockdown using Dharmafect 1 transfection reagent (Dharmacon, cat#: T-2001-03) and siRNAs for TP53 and MTA2 followed by drug treatment for 72 h in 384-well format. siGENOME SMARTpool siRNA for TP53 (cat#: M-003329-03-0005) and MTA2 (cat#: M-008482-00-0005), as well as a nontargeting siRNA control (Pool#1; cat#: D-001206-13-05), were purchased from Dharmacon.
Seeding cell density was optimized to allow for optimal imaging at 120 h after initiation of the experiment (48 h of siRNA transfection followed by 72 h of drug treatment) with no overlap in nuclei, while at least 80% confluence was observed in control-treated wells. Cells were fixed, stained with DAPI nuclear stain, and evaluated for density at 144 h postseeding (corresponding to 24 h attachment, + 48 h transfection + 72 h drug treatment) in 384-well CellCarrier Ultra plates (PerkinElmer) at 100, 250, 400, or 500 cells/well. Best image density at 144 h was observed at seeding density of 400 cell/well.
Final concentration of Dharmafect 1 and siRNA concentrations were determined where 2, 1, 0.5, 0.2, and 0.1% of Dharmafect 1 and 12.5, 25, and 50 nM final concentrations of TP53 and MTA2 siRNAs were tested for nuclear morphology effects and cytotoxicity at 120 h posttransfection. Based on the optimization experiments 1% Dharmafect 1 and 25 nM siRNA concentrations were selected for use in the final screen for both targets, as well as for the nontargeting siRNA control, as this combination gave best transfection-to-cytotoxicity ratio. Gene knockdown at 72 h posttransfection for both siRNA targets was confirmed by RT-PCR as described previously (Tamashunas et al., 2020) at 25 and 50 nM final concentrations but not at 12.5 nM (Supplemental Figure S1).
Cayman ESL screen, transfection, fluorescence staining, high-throughput imaging, and nuclear morphometric image analysis
Based on the optimization experiments described above, siRNA transfections were carried out at 1% final concentration of Dharmafect 1 and 25 nM final concentration of siRNAs in OptiMEM serum-free media (Thermo Fisher Scientific) per the manufacturer’s protocols. Posttransfection, cells were incubated for 48 h to allow siRNA-mediated interference and turnover of endogenous proteins.
MCF10A cells, seeded at 400/well in 50 µl of media per well in 384-well plates, were left to attach overnight. Two 384-well plates for each siRNA condition were transfected with TP53-, MTA2-, or nontargeting siRNA as described above. The final volume/well after transfection was 55.56 µl. The transfected cells were incubated at 37°C, 5% CO2 for 48 h and then treated with the Cayman ESL library of small epigenetic modulators (Cayman, cat#:11076) in quadruplicates using the JANUS liquid handling system (Perkin Elmer) (200 nL pin-tool, 96-to-384 protocol) at a final concentration of 1 µM. The library consists of two compound plates. The screening of each compound plate was performed in a separate experiment and contained its own vehicle controls and was tested on all three siRNA transfection conditions simultaneously in the same experiment. After treatment cells were incubated for an additional 72 h at 37°C, 5% CO2 and then fixed for 30 min at room temperature with paraformaldehyde (PFA) at a final concentration of 4% (by adding 50 µl/well 8% PFA dilution in calcium/magnesium-free Dulbecco’s modified phosphate-buffered solution [DPBS]). After fixation the plates were washed with DPBS at 100 µl/well and permeabilized with 50 µl/well permeabilization buffer (DPBS supplemented with 1 mg/ml bovine serum albumin and 0.2% Triton X-100) for 30 min at room temperature. After permeabilization cells were washed again with 100 µl/well DPBS and treated with 25 µl/well anti-Lamin A mouse monoclonal antibody (Abcam; #ab8980) at 1:1000 dilution in permeabilization buffer overnight at 4°C. The next day, plates were washed 2× for 5 min with 100 µl/well of DPBS –/– and stained with 25 µl/well of a mixture of goat anti-mouse AlexaFluor 488 (Invitrogen, A-11001) secondary antibody at 1:500, and DAPI (Invitrogen; #D1306) at 1:200 dilution in permeabilization buffer, for 2 h at room temperature. After an additional wash with DPBS, cells were left in 100 µl/well DPBS for imaging. Plates were imaged at 40× magnification (35 fields/well) using Operetta high-throughput imaging system (Perkin Elmer) and images were exported for further analysis.
Nuclear images were evaluated for EFC ratios using MATLAB (version 2019b; 9.7.0) and a custom-developed script described previously (Tamashunas et al., 2020).
Follow-up studies
The 14 hits identified in the ESL library screen were tested in a dose-response experiment on TP53- and MTA2-siRNA transfected MCF10A cells or nontarget siRNA transfected MCF10A cells. Seeding, transfection, and treatment of MCF10A cells were performed as described for the screening. For each drug, cells were seeded and transfected in parallel in two separate 384-well black-wall clear bottom plates. PerkinElmer CellCarrier Ultra plates were used for imaging and Greiner black, clear bottom plates for performing viability analysis. At 48 h posttransfection, cells in both plate types were treated simultaneously with threefold dilutions of the tested drugs and incubated for an additional 72 h. Cells destined for imaging were stained and imaged as described for the screen experiment, while the cells in the Grainer plates were used for viability assay using PrestoBlue Cell Viability Reagent, (ThermoFisher Scientific, cat#:A13261), according to the manufacturer’s recommendation.
PANC-1 and MDA-MB-231 cells were seeded at 1200/well or 1500/well, respectively, in 55 µl of media, and left to attach overnight. Next day cells were treated with selected inhibitors at threefold dilutions in a dose-response manner. Cells were left to incubate for 72 h and then were fixed and stained as described for the screen experiment. Viability was also checked in parallel at the same doses using the ATP-lite OneStep toxicity assay (PerkinElmer, cat#: 6016739) according to the manufacturer’s recommendation.
Statistical analyses
Statistical analysis of drug effects on nuclear morphological abnormalities induced by gene knockdown ( Figures 2 and 3).
The data consisted of nuclear EFC ratios measured from cells receiving either DMSO or a treatment (a member of the compound libraries) each under one of three possible gene conditions viz. scrambled cells (no knockdown), TP53 knockdown, and MTA2 knockdown (in quadruplicate technical replicates). Our interest was in comparing treatment versus DMSO effects and we focused on these comparisons under two settings: (A) within group comparison: comparing the effects when both the treatment and DMSO are applied to cells under similar gene-knockdown conditions; (B) between group comparison: comparing the effects when the treatment is administered to a siRNA-based gene-knockdown condition while DMSO is applied to a scrambled siRNA transfection condition.
Nuclear EFC ratio is a positive quantity, and a study of the empirical distributions of the logarithms of the observed EFC ratios under different treatment-knockdown combinations clearly indicated normality (based on histograms and qq-plots). At the outset, we thus log-transformed the data on nuclear EFC ratios. Subsequently, to aid formal quantification of the type-A and type-B comparisons described above, we considered an omnibus two-way ANOVA model with interactions (MODEL-1) of the form:
Here (a) yijk denotes the (natural) logarithm of the nuclear EFC ratio of the cell in the k-th replicate which receives the i-th treatment in the j-th gene-knockdown condition (with i = 0 denoting DMSO and j = 0 denoting no knockdown [scramble]); (b) the model parameters μ, αiβj, and yij denote the mean/overall effect, the main effect of the i-th treatment, the main effect of the j-th gene-knockdown condition, and the interaction effect of the i-th treatment when administered under the j-th gene-knockdown condition, respectively; and (c) ∈ijk are independently and identically distributed random normal noise.
Under MODEL-1 we considered general interaction contrasts of the form τij –τ0j’ with τij = μ + αi +βj + γij = E(yijk), quantifying the expected difference in the log EFC ratios between cells receiving treatment i under j-th gene-knockdown condition and those receiving DMSO under j’-th gene-knockdown condition; j,j’ = 0,1,2 and i ≥ 1. Note that, letting j = j’ aids within group comparisons (type-A) and letting j| = 1,2 and j’ = 0 aids between group comparisons (type B).
The above MODEL-1 was first fit and then the subsequent treatment contrasts were estimated using the method of maximum likelihood. To aid statistical inference on these effects, we conducted tests of individual hypotheses concerning statistical significance of each contrast under consideration and then performed Benjamini-Hochberg false discovery rate (FDR) adjustments (Benjamini and Hochberg, 1995) to the resulting p values obtained from individual tests to account for multiplicity. Significances of all hypotheses were finally determined at level 0.05 of the adjusted p values. The estimated treatment contrasts were reverted into the original scale via exponentiation, thus producing a ratio of geometric means of nuclear EFC ratios.
We visualized the results through the heat maps plotted in Figures 2 and 3. The cell colors represent the effect sizes (in original nuclear EFC ratio scale) of the corresponding contrast; the contrasts that were not statistically significantly different from zero (FDR adjusted p value 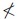 0.05) were plotted in gray. The compounds are grouped by their functions.
0.05) were plotted in gray. The compounds are grouped by their functions.
Statistical analysis of dose effects of targeted drugs on nuclear morphological abnormalities induced by gene knockdown ( Figure 5).
Here the data consisted of nuclear EFC ratios measured from cells receiving either DMSO or a treatment each under one of two possible gene-knockdown conditions, viz., TP53 and MTA2 (in quadruplicate technical replicates). Several doses of the treatments were considered, and our interest was in comparing the effects of these treatment-specific doses, relative to DMSO, in stabilizing the nuclei. The number of cells for each knockdown + treatment + dose combination was measured separately in each image and was used as a control variable in our analysis.
Separately for each reagent, we considered the following two omnibus-nested three-way ANOVA regression models:
and MODEL – 2:
Here i indexes the two gene-knockdown conditions (TP53 and MTA2), j indexes the treatments (with j = 0 being DMSO), k indexes doses (nested within treatments; DMSO has only one dose), h indexes image numbers, and l indexes individual observations obtained from an individual image. Moreover, yi,j,k,h.l denotes the natural logarithm of the nuclear EFC ratio of the (i, j, k, h ,l ) combination; αi is the effect of the i-th gene knockdown; βj denotes the main effect of the j treatment; yj,k is the effect of the k-th dose of j-th treatment; δi,j is the interaction effect of knockdown-i and treatment j; ki,j,k is that of dose k in treatment-j and knockdown i; vi,j,k,h is the logarithm of the number of cells observed in the h-th image under the (i,j,k) combination and τ is the corresponding regression coefficient; and ∈ is independent and identically distributed zero mean random normal noise.
We were interested in understanding how the dose level affects the efficacy of a given treatment, and whether this effect was significantly different from DMSO. To this end, we tested the statistical significance of contrasts of the form 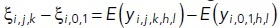 = overall effect of (j,k) treatment-dose combination under i-th gene-knockdown condition minus the overall effect of DMSO under the i-th gene-knockdown condition. Through their estimates obtained from fitted omnibus Model 2, we constructed t tests for all such individual contrasts, and subsequently adjusted the p values using Benjamini-Hochberg FDR adjustment procedure (Benjamini and Hochberg, 1995) to determine significance of each estimated contrasts. These estimates along with 95% confidence intervals are then displayed as point and error bars (color coded by 5% FDR-adjusted significance status) for various doses in Figure 5.
= overall effect of (j,k) treatment-dose combination under i-th gene-knockdown condition minus the overall effect of DMSO under the i-th gene-knockdown condition. Through their estimates obtained from fitted omnibus Model 2, we constructed t tests for all such individual contrasts, and subsequently adjusted the p values using Benjamini-Hochberg FDR adjustment procedure (Benjamini and Hochberg, 1995) to determine significance of each estimated contrasts. These estimates along with 95% confidence intervals are then displayed as point and error bars (color coded by 5% FDR-adjusted significance status) for various doses in Figure 5.
Statistical analysis of dose effects of targeted drugs on nuclear morphological abnormalities in cancer cells ( Figure 6).
Here the data consisted of nuclear EFC ratios measured from cells receiving either DMSO or a treatment each, for two cancer cell lines, viz., MDA-MB-231 and PANC1 (in quadruplicate technical replicates). Several doses of the treatments were considered and our interest lay in comparing the effects of these treatment-specific doses, relative to DMSO, in stabilizing the nuclei. The number of cells for each cell type + treatment + dose combination was measured separately in each image, and they were used as a control variable, similarly to the analysis performed for dose specific effects of targeted drugs under gene knockdown.
We considered the following two omnibus nested three-way ANOVA regression models:
and MODEL – 3:
Here i indexes the 2 cancer cell types (MDA and PANC1), j indexes the treatments (with j = 0 being DMSO), k indexes doses (nested within treatments; DMSO has only one dose), h indexes image numbers, and l indexes individual observations obtained from an individual image. Moreover, yi,j,k,h,l denotes the natural logarithm of the nuclear EFC ratio of the (i, j, k, h, l) combination; αi is the effect of the i-th cell type; βj denotes the main effect of the j treatment; yj,k is the effect of the k-th dose of j-th treatment; δi,j is the interaction effect of cell type-i and treatment j; ki,j,k is that of dose k in treatment-j, and cell type i; vi,j,k,h is the logarithm of the number of cells observed in the h-th image under the (i, j, k) combination and τ is the corresponding regression coefficient; and ∈ are independent and identically distributed zero mean random normal noise.
We were interested in understanding how the dose level affects the efficacy of a given treatment, and whether this effect was significantly different from DMSO. To this end, we tested the statistical significance of contrasts of the form 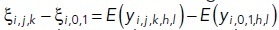 = overall effect of (j, k) treatment-dose combination under i-th cancer cell type minus the overall effect of DMSO under the i-th cancer cell type. Through their estimates obtained from the fitted omnibus Model 3, we constructed t tests for all such individual contrasts (separately for each model), and subsequently adjusted the p values using Benjamini-Hochberg FDR adjustment procedure (Benjamini and Hochberg, 1995) to determine significance of each estimated contrasts. These estimates along with 95% confidence intervals are then displayed as point and error bars (color coded by 5% FDR-adjusted significance status) for various doses and are plotted in Figure 6.
= overall effect of (j, k) treatment-dose combination under i-th cancer cell type minus the overall effect of DMSO under the i-th cancer cell type. Through their estimates obtained from the fitted omnibus Model 3, we constructed t tests for all such individual contrasts (separately for each model), and subsequently adjusted the p values using Benjamini-Hochberg FDR adjustment procedure (Benjamini and Hochberg, 1995) to determine significance of each estimated contrasts. These estimates along with 95% confidence intervals are then displayed as point and error bars (color coded by 5% FDR-adjusted significance status) for various doses and are plotted in Figure 6.
Statistical computing environment
All statistical analyses were performed in R v4.0.0(Team, 2021). The tidyverse set (Wickham et al., 2019) of packages were used for data preprocessing and for data/results visualization, and the emmeans (Lenth, 2021) package was used for computations involving treatment contrasts.
Supplementary Material
Acknowledgments
We acknowledge support from National Institutes of Health U01 CA225566 (T.P.L.), CPRIT established investigator award Grant # RR200043 (T.P.L.), UF Health Cancer Center (H.L.), Debbie and Sylvia DeSantis Chair Professorship (H.L.), R01CA172310 (H.L.), R50CA211487 (R.R.), and UFHCC and ASHANDS funding for salary support (K.R.A.) and equipment (high content imaging). We thank Andrew Tamashunas for his help with validation of gene-knockdown experiments.
Abbreviations used:
- BET
bromodomain and extraterminal domain
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s modified Eagle medium
- DMSO
dimethyl sulfoxide
- DMT
DNA methyltransferase
- DPBS
Dulbecco’s modified phosphate-buffered solution
- EDTA
ethylenediaminetetraacetic acid
- EFC
Elliptical Fourier coefficient
- ESL
epigenetic screening library
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- HDM
histone demethylase
- HMT
histone methyltransferase
- PARP
poly adenosine diphosphate-ribose polymerase
- PFA
paraformaldehyde
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E21-10-0528) on March 24, 2022.
REFERENCES
- Al-Awadhi FH, Salvador-Reyes LA, Elsadek LA, Ratnayake R, Chen QY, Luesch H (2020). Largazole is a brain-penetrant class I HDAC inhibitor with extended applicability to glioblastoma and CNS diseases. ACS Chem Neurosci 11, 1937–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. R. Stat. Soc., B: Stat. Methodol. 57.1, 289–300. [Google Scholar]
- Cosgrove BD, Loebel C, Driscoll TP, Tsinman TK, Dai EN, Heo SJ, Dyment NA, Burdick JA, Mauck RL (2021). Nuclear envelope wrinkling predicts mesenchymal progenitor cell mechano-response in 2D and 3D microenvironments. Biomaterials 270, 120662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais C, Lammerding J (2014). Nuclear mechanics in cancer. Adv Exp Med Biol 773, 435–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K, Lammerding J (2016). Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz G, Zuccarelli A, Pelligra I, Ghiani A (1989). Elliptic fourier analysis of cell and nuclear shapes. Comput Biomed Res 22, 405–414. [DOI] [PubMed] [Google Scholar]
- Dickinson RB, Katiyar A, Dubell CR, Lele TP (2022). Viscous shaping of the compliant cell nucleus. APL Bioeng 6, 010901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa T, Rochman M, Taher L, Dimitriadis EK, Nagashima K, Anderson S, Bustin M (2015). Chromatin decompaction by the nucleosomal binding protein HMGN5 impairs nuclear sturdiness. Nat Commun 6, 6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil J, Wu H, Wang BY (2002). Image analysis and morphometry in the diagnosis of breast cancer. Microsc Res Tech 59, 109–118. [DOI] [PubMed] [Google Scholar]
- Hong J, Luesch H (2012). Largazole: from discovery to broad-spectrum therapy. Nat Prod Rep 29, 449–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbalzano KM, Cohet N, Wu Q, Underwood JM, Imbalzano AN, Nickerson JA (2013). Nuclear shape changes are induced by knockdown of the SWI/SNF ATPase BRG1 and are independent of cytoskeletal connections. PLoS One 8, e55628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashyap A, Jain M, Shukla S, Andley M (2018). Role of Nuclear Morphometry in Breast Cancer and its Correlation with Cytomorphological Grading of Breast Cancer: A Study of 64 Cases. J Cytol 35, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent IA, Zhang Q, Katiyar A, Li Y, Pathak S, Dickinson RB, Lele TP (2019). Apical cell protrusions cause vertical deformation of the soft cancer nucleus. J Cell Physiol 234, 20675–20684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT (2006). Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 281, 25768–25780. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lele TP, Dickinson RB, Gundersen GG (2018). Mechanical principles of nuclear shaping and positioning. J Cell Biol 217, 3330–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth RV (2021). Emmeans: Estimated Marginal Means, Aka Least-Squares Means. [Google Scholar]
- Nader GPF, Aguera-Gonzalez S, Routet F, Gratia M, Maurin M, Cancila V, Cadart C, Palamidessi A, Ramos RN, San Roman M, et al. (2021). Compromised nuclear envelope integrity drives TREX1-dependent DNA damage and tumor cell invasion. Cell 184, 5230–5246.e5222. [DOI] [PubMed] [Google Scholar]
- Schreiner SM, Koo PK, Zhao Y, Mochrie SG, King MC (2015). The tethering of chromatin to the nuclear envelope supports nuclear mechanics. Nat Commun 6, 7159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senigagliesi B, Penzo C, Severino LU, Maraspini R, Petrosino S, Morales-Navarrete H, Pobega E, Ambrosetti E, Parisse P, Pegoraro S, et al. (2019). The High Mobility Group A1 (HMGA1) Chromatin Architectural Factor Modulates Nuclear Stiffness in Breast Cancer Cells. Int J Mol Sci 20, 2733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Banigan EJ, Adam SA, Goldman RD, Marko JF (2017a). Chromatin and lamin A determine two different mechanical response regimes of the cell nucleus. Mol Biol Cell 28, 1984–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Banigan EJ, Marko JF (2017b). Separate roles for chromatin and lamins in nuclear mechanics. Nucleus 9, 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens AD, Liu PZ, Banigan EJ, Almassalha LM, Backman V, Adam SA, Goldman RD, Marko JF (2018). Chromatin histone modifications and rigidity affect nuclear morphology independent of lamins. Mol Biol Cell 29, 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamashunas AC, Tocco VJ, Matthews J, Zhang Q, Atanasova KR, Paschall L, Pathak S, Ratnayake R, Stephens AD, Luesch H, et al. (2020). High-throughput gene screen reveals modulators of nuclear shape. Mol Biol Cell 31, 1392–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team RC (2021). R: A Language and Environment for Statistical Computing, Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- Tocco VJ, Li Y, Christopher KG, Matthews JH, Aggarwal V, Paschall L, Luesch H, Licht JD, Dickinson RB, Lele TP (2018). The nucleus is irreversibly shaped by motion of cell boundaries in cancer and non-cancer cells. J Cell Physiol 233, 1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortmeyer-Krause M, Lindert Mt, Riet Jt, Boekhorst Vt, Marke R, Perera R, Isermann P, van Oorschot T, Zwerger M, Yang F, et al. (2020). Lamin B2 follows lamin A/C- mediated nuclear mechanics and cancer cell invasion efficacy. bioRxiv, 028969. [Google Scholar]
- Wickham H, Averick M, Bryan J, Chang W, McGowan LDA, François R, Grolemund G, Hayes A, Henry L, Hester J, et al. (2019). Welcome to the tidyverse. J Open Source Softw 4, 1686. [Google Scholar]
- Yang Z, Maciejowski J, de Lange T (2017). Nuclear Envelope Rupture Is Enhanced by Loss of p53 or Rb. Mol Cancer Res 15, 1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink D, Fischer AH, Nickerson JA (2004). Nuclear structure in cancer cells. Nat Rev Cancer 4, 677–687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.