

Abstract
Background
Cardiomyopathy is a known complication of organic acidemias but generally thought to be secondary to poor metabolic control.
Methods
Our patient was found through biochemical testing and Sanger sequencing to harbor an Icelandic founder mutation: NM_052845.4(MMAB):c.571C > T(p.Arg191Trp), leading to an early presentation (4 h after birth) of cblB‐type methylmalonic acidemia (MMA). Biochemical testing of this patient suggested B‐12‐responsiveness and thus the patient was treated with cyanocobalamin throughout life. Informed parental consent was obtained for this report.
Results
Our patient had three metabolic decompensations in her life (at birth, at 1 month, and at 5 months). The first decompensation was probably linked to stress of delivery, second to rhinovirus infection, and third by co‐infection of norovirus and enterovirus. At 3 months, the patient was noted to be tachypneic, although this was attributed to her underlying metabolic acidosis. At 5 months and 10 days, the patient was admitted with minor flu‐like symptoms but developed severe diarrhea in hospital and upon rehydration had cardiac decompensation and was found to have undiagnosed dilated cardiomyopathy. Although, patient was treated aggressively with dextrose, hemodialysis, levocarnitine, and vasoactive agents, there was limited response to medications to treat cardiac failure, and eventually the patient passed away before turning 6 months old.
Conclusions
Other than these three mild decompensations, patient had very good metabolic control, thus demonstrating that even without frequent metabolic decompensation, cardiomyopathy can be an observed phenotype in cblB‐type MMA even very early in life, suggesting that this phenotype may be independent of metabolic control.
Keywords: cblB, dilated cardiomyopathy, heart failure, MMAB
Early heart failure in a patient with methylmalonic acidemia (MMA). The patient succumbed to heartfailure due to dilated cardiomyopathy despite regular control as recommended for MMA patients.

1. INTRODUCTION
Methylmalonic acidemias (MMA) are a group of inborn errors of metabolism associated with an elevated methylmalonic acid concentration in the blood and urine. These are caused by an enzymatic defect disrupting normal amino acid metabolism. This leads to toxic build‐up of substances with potential serious decompensation events and metabolic crises. Isolated MMA results from the failure to convert methylmalonyl‐CoA into succinyl‐CoA from propionyl‐CoA in the mitochondrial matrix, without hyperhomocysteinemia, homocystinuria, hypomethioninemia, or variations in other metabolites, such as malonic acid (Manoli et al., 2016).
MMA is a genetically heterogeneous disorder inherited in an autosomal recessive manner. One type is the cblB‐type, caused by biallelic pathogenic variants in MMAB gene that encodes ATP:cob(I)alamin adenosyltransferase, which is involved in the synthesis of adenosylcobalamin (AdoCbl), a coenzyme for methylmalonyl‐CoA mutase (Online Mendelian Inheritance in Man, OMIM®, n.d.).
Diminished activity of cob(I)transferase results in the disorder that usually appears in newborns presenting with vomiting, metabolic acidosis, lethargy, failure to thrive, hyperammonia, methylmalonic acidemia, and methylmalonic aciduria (Manoli et al., 2016).
While cardiac complications are well‐known in propionic acidemia, a related organic acidemia, only few case reports exist of cardiomyopathies in some forms of MMA and disorders of cobalamin deficiency, particularly in Cobalamin C (CblC) deficiency (De Bie et al., 2009; Prada et al., 2011; Razzaghy‐Azar et al., 2007; Valayannopoulos et al., 2009).
However, cardiac complications are not listed in OMIM's clinical synopsis for MMA, cblB–type (Online Mendelian Inheritance in Man, OMIM®, n.d.). At least one patient with MMA, type cblB and hypertrophic cardiomyopathy (HCM) has been reported so far (Prada et al., 2011). Given the paucity of cases with MMA and cardiac complications, the mechanistic basis is unknown although most providers consider this a consequence of poor metabolic control. Here, we report a patient with MMA, type cblB, which despite early diagnosis and careful metabolic control succumbed to severe heart failure in the first year of life. Thus, this may indicate that in some individuals MMA may lead to a cardiac phenotype irrespective of metabolic control. This also, is a reminder of early cardiac surveillance among other regular recommended observation and monitoring for patients living with MMA.
2. CLINICAL REPORT
Our patient was conceived naturally to non‐consanguineous parents with a previous healthy full sibling. Although pregnancy was uncomplicated, child was noted to have hypoglycemia (2.2 mmoL/L) shortly after delivery and found to have hyperammonemia (205 μmol/L) on the second day of life (Figure 1). Plasma carnitine profile was analyzed from the patient at 31 h after birth. Flow‐Injection Mass Spectrometry (FIA‐MSMS) analysis in plasma gave the following results that strongly indicated MMA: Free Carnitine (C0): 3,6 μmol/L (ref10‐50 μmol/L). Propionylcarnitine (C3): 13,7 μmol/L (ref < 1,0 μmol/L) and methylmalonic carnitine (C4DC) 0,9 μmol/L (ref < 0,15 μmol/L).
FIGURE 1.
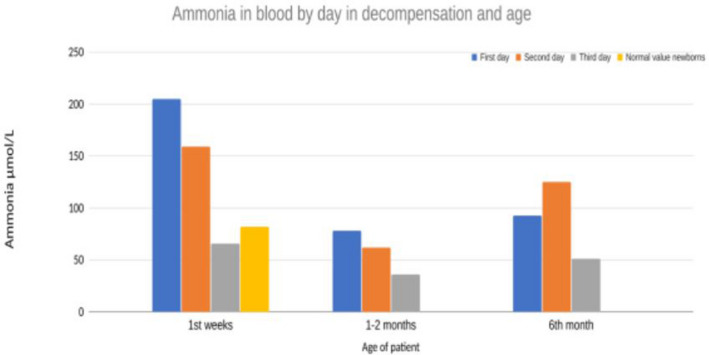
Plasma ammonia measurements in our patient for the first 3 days in all decompensations show that these decompensations were short‐lived (1–2 days) and relatively mild (highest value 200) (Madigan et al., 2020).
The patient was treated with intravenous (IV) 10% glucose 3 ml/kg/h with electrolytes, sodium bicarbonate 10 ml/h, levocarnitine (50 mg/kg/every 6 h), sodium benzoate, carbaglu, B‐12 vitamin daily, and l‐arginine following published organic acidemia guidelines (Baumgartner et al., 2014). Within 3 days ammonium levels normalized (P‐ammonium 66 μmol/L; ref < 85 (Madigan et al., 2020)) with maximum ammonium reaching 205 μmoL/L on the first day of life (Figure 1). An echocardiogram was performed on the third day of life (systolic pressure 70–80, heart rate 140, saturation 99% on room air). Echo revealed a structurally normal heart with a small PFO as could be expected at this time and good contractility of both ventricles. There was some dilatation of atria with leakage over atrioventricular valves, but this was resolved on a follow‐up examination.
After 3 weeks the patient was discharged on sodium bicarbonate, levocarnitine, D3 vitamin, Vibeden (cyanocobalamin) 1 mg/ml inj 1 mg intramuscular (IM) twice per week. Later switched to hydroxocobalamin (the preferred form for supplementation in MMA and genetic causes of Cbl deficiencies) 1 mg/0.7 ml which also had the benefit of being a smaller volume of injection IM.
Through Sanger sequencing of the MMAB gene the patient was found to harbor an Icelandic founder pathogenic variant, MMAB: c.571C > T (p.Arg191Trp), in homozygous state. The parents both carried a single copy of the same variant. This was confirmed within first 2 months of life. Additionally, a comprehensive panel constituting of 54 genes related to organic acidemia/aciduria and cobalamin deficiency, and later whole‐genome sequencing, was run and did not detect any other pathogenic variants. Methylmalonic acid was decreased in plasma after 10 days of daily IM B‐12 vitamin injections and carnitine supplementation. Treatment was continued once the patient was in a stable phase as hydroxocobalamin injections IM twice a week. The MMA in blood went from 173000ug/l (day 2 of life) to 15900ug/l (day 11 of life) after treatment with B‐12 vitamin was started. The 90.8 percent decrease in MMA in blood pointed toward B‐12 responsiveness and thus the patient was kept on B‐12 (Manoli et al., 2016). Treatment was continued as high carbohydrate‐low‐protein diet, hydroxocobalamin, sodium bicarbonate, and levocarnitine (100 ml/kg/day). Regular monitoring of the patient through phone calls and appointments at the children's hospital with clinical examination, blood tests for monitoring p‐ammonia, kidney, and liver function and other recommended tests. These data suggested adequate metabolic control, measurements of PAA were representative and plasma ammonium was never found to be elevated other than the three occasions previously noted (Figure 1).
A certified dietician was involved from diagnosis and anabolism was maintained with sufficient calories from a combination of expressed breastmilk and a protein free formula (Energivit, Nutricia) from day 4 to 12 weeks old. Thereafter, our patient was fed expressed breastmilk alone until parents began supplementing with protein free weaning foods at 4 months of age. Apart from the three periods of decompensations, the patient had metabolic tolerance for protein at or above the FAO/WHO/UNU safe levels of protein intake for age by consuming protein from intact sources (breastmilk) throughout life.
The patient had a brief decompensation episode associated with viral illness/rhinovirus and was hospitalized for 1 week at 1 month of age (Figure 1).
On clinical examination at age 3 months and 3 days the patient showed intermittent tachypnea (RR ~ 88/min), hypotonic core muscles with visible neck lag, and delay in gross motor skills. The patient was afebrile and still feeding well with normal ammonia levels.
During periods of metabolic stability, the patient was contacted at least weekly by a registered dietician to monitor intake and discuss issues related to feeding. Weight gain was monitored when the patient came in for blood tests and at home in between those visits (maximum interval 18 days) and remained adequate until close to 4 months of life. From then on, feeding became increasingly difficult and weight fell from the 70th to the 33rd percentile in 6 weeks (from age 3 months 1 week to age 5 months 1 week). Ammonia levels were measured three times during that period with approximately 1 month interval and were 51, 63, and 49 μmol/L (see Table 1). At the latest measurement (at 4 months 26 days, 2 weeks before last admission) weight gain had only been 70 g over the past 4 weeks and clinical examination showed intermittent tachypnea (RR 80/min). In the weeks before and afterwards, advice on increasing energy density of feeds was provided along with other efforts such as an attempt to feed through a nasogastric tube (at week 22), which she tolerated poorly.
TABLE 1.
Overview of laboratory results in last admission and the previous months
| Reference | 3 months 3 days | 3 months 28 | 4 months 26 days | 5 months 9 days/ first day of last admission | Day 2 of last admission | Day 3 of last admission/ before CRRT | Day 6 of last admission | Day 10 of last admission/2 days after CRRT was stopped | One day before passing | |
|---|---|---|---|---|---|---|---|---|---|---|
| aB‐pH | 7.35–7.45 | 7.04 | 7.42 | 7.41 | ||||||
| aB‐pCO2 (mm Hg) | 34–45 | 62 | 35 | 42 | ||||||
| aB‐Bicarbonate (mmol/L) | 22–27 | 16 | 22 | 26 | ||||||
| aB‐Lacate (mmol/L) | 0.38–1.34 | 9,4 | 1 | 1,2 | ||||||
| vB‐pH | 7.32–7.43 | 7,41 | 7,34 | |||||||
| vB‐pCO2 (mm Hg) | 40–50 | 42 | 40 | |||||||
| vB‐Bicarbonate (mmol/L) | 22–27 | 26 | 20 | |||||||
| vB‐Lactate (mmol/L) | 0.5–1.7 | 1.8 | 3,5 | |||||||
| P‐Ammonia (μmol/L) | 11–49 | 51 | 63 | 49 | 68 | 125 | 39 | 71 | ||
| Free carnitine (C0) (μumol/L) | 10–50 | 310 | ||||||||
| Acylcarnitine (C2) (μmol/L) | 5–25 | 26,3 | ||||||||
| Propionylcarnitine (C3) (μmol/L) | 0,2‐1,0 | 95,5 | ||||||||
| Glycine (μmol/L) | 120–350 | 185 | ||||||||
| Alanine (μmol/L) | 100–350 | 173 | ||||||||
| Troponin‐T (ng/L) | <14 | 283 | 821 | 98 |
A few days later the patient presented with sweating despite being afebrile, 1 day of vomiting and increased work of breathing, difficulty feeding and was admitted to the hospital and NICU. A stool sample was positive for enterovirus (echovirus 18) and weak‐positive for norovirus. A cardiac ultrasound was obtained that showed dilated cardiomyopathy (DCM) and severe heart failure. (Figure 2). Highly elevated pro‐BNP (~193,000 ng/L) on day 2 of admission supported the diagnosis of severe heart failure. It decreased to ~3400 ng/L over 2 days but remained high throughout life. Troponin‐T peaked at 821 ng/L on day 3 of admission.
FIGURE 2.
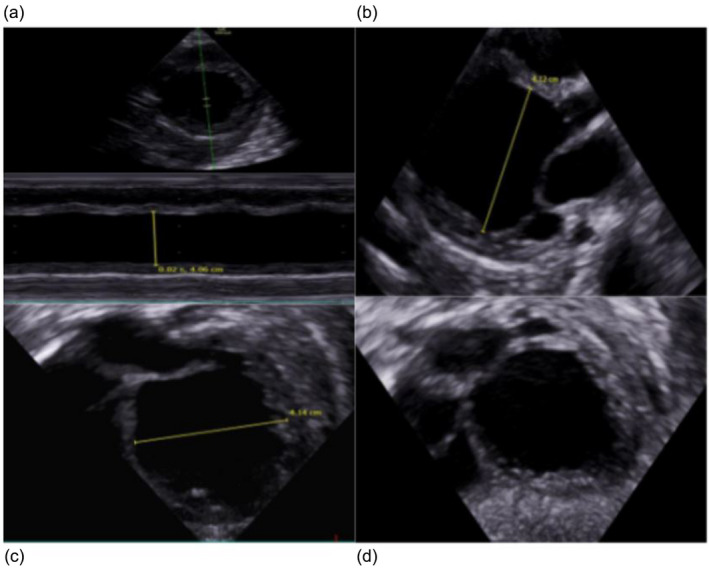
Echocardiography shows dilated cardiomyopathy in our patient at 5 months. (a) Left ventricle (LV) m mode showing severe reduced contractility and LV dilatation. (b) PLAX 1: End diastole. (c) PLAX 2: End diastole LV diameter. (d) Subcostal 4ch: End diastole showing very dilated LV.
In the setting of severe heart failure, concern for hemodynamic instability, acute rise of ammonia and lethargy a decision was made to intubate the patient to decrease metabolic demands and start continuous renal replacement therapy (CRRT) for ammonia clearance. CRRT treatment was initiated without complications and decreased blood ammonia levels to normal range as well as normalizing metabolic acidosis (Figure 1 and Table 1). Electrolytes remained stable and no arrhythmia was seen on the cardiac monitor. However, cardiac function only marginally improved despite maximum support including vasopressors and heart failure treatment (including milrinone noradrenaline, dopamine, dobutamine, levosimendan, and captopril), medical management of metabolic crisis (dextrose, L‐carnitine, N‐acetylcysteine, vitamin B1 and B12, multivitamin mixture and TPN with lipids, and breast milk through NG‐tube) and CRRT. Two days after CRRT acylcarnitine and free carnitine was measured and was elevated, likely due to abundant supplementation of carnitine. (Table 1). Plasma‐amino‐acids were within normal limits. A decision was made to withdraw support and several days later the patient passed away before turning 6 months old. At the time of death, there was no metabolic dysregulation. (Figure 1).
3. DISCUSSION
Cardiomyopathy has been reported in other patients with MMA. Previously there have been few reports of cardiac complications in patients with MMA (De Bie et al., 2009; Prada et al., 2011; Razzaghy‐Azar et al., 2007; Valayannopoulos et al., 2009) and regular echocardiographic and electrocardiographic surveillance has been recommended, both for patients with propionic and methylmalonic aciduria (Fraser & Venditti, 2016).
While a case of HCM with MMA type cblB, has been reported, we found no reports on associated DCM. The founder mutation reported is known in Iceland and in the last 5 years, two patients have been diagnosed with this genotype. The carrier frequency is around one in every 270 Icelanders (personal communication, deCODE genetics). In our institution, we plan on incorporating regular screening with echocardiograms from birth, even without obvious symptoms or metabolic derangements. Reviewing our case, given the severity of symptoms of DCM at diagnosis, it is likely that the onset of heart disease was at an early age in the setting of metabolic stability. That indicates that metabolic control is not a dominating factor in these patients, and this could be another or a newly discovered rare but specific phenotype. Thus, our data suggest that MMA may cause both but makes it less likely that the cardiac phenotype is secondary to the metabolic dysfunction (at least dysfunction we can measure).
Enterovirus is known to cause cardiomyopathy and in this case, the virus could be a potential cause of cardiomyopathy, although the clinical progress in our case with relatively little improvement after initial event speaks against this. Alternatively, perhaps the occurrence of cardiomyopathy in MMA and related conditions requires a second hit in another gene to occur. A screening of pathogenic variants in known genes causing cardiomyopathy had been undertaken in our patient as a part of whole‐genome sequencing. The pathophysiology of cardiomyopathy and cardiac arrhythmias in the setting of MMA and other organic acidemias is unclear but accumulation of toxic metabolites, oxidative stress, and mitochondrial dysfunction may play a role (Park et al., 2020). However, our findings further support prior recommendations of regular screening with echocardiography (Fraser & Venditti, 2016). Finally, we treated our patient throughout her life with B12, but while writing this report a new publication has been released that suggests that this variant is not responsive to B12 treatment (Forny et al., 2021).
4. CONCLUSIONS
Patients with MMA type cblB should be screened like other causes of MMA with yearly evaluation with ECG or echocardiography, or more frequently if there are additional cardiac concerns. This case suggests that poor metabolic control is not the culprit of the cardiac phenotypes although obviously further research is needed to solidify this idea.
AUTHOR CONTRIBUTIONS
DA, VKS, and EP drafted. DA and IR contributed to figures. HTB, VKS, LF, ARE, GS, and HV critically revised the article. All authors approved of the final version submitted.
CONFLICT OF INTEREST
Author(s) declare no conflict of interest although HTB is a consultant for Mahzi therapeutics.
ETHICAL COMPLIANCE
We went through a clinical informed consent process and parents have given their written approval for this particular case report. A separate IRB license for this study was not sought as single cases are exempt.
ACKNOWLEDGMENTS
The authors would like to thank the family of the patient for allowing us to write up the clinical course from this patient. The authors also would like to thank Herdís Gísladóttir, genetics nurse at the Department of Genetics and Molecular Medicine, Landspítali hospital.
Agnarsdóttir, D. , Sigurjónsdóttir, V. K. , Emilsdóttir, A. R. , Petersen, E. , Sigfússon, G. , Rögnvaldsson, I. , Franzson, L. , Vernon, H. , & Bjornsson, H. T. (2022). Early cardiomyopathy without severe metabolic dysregulation in a patient with cblB‐type methylmalonic acidemia. Molecular Genetics & Genomic Medicine, 10, e1971. 10.1002/mgg3.1971
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Baumgartner, M. R. , Hörster, F. , Dionisi‐Vici, C. , Haliloglu, G. , Karall, D. , Chapman, K. A. , Huemer, M. , Hochuli, M. , Assoun, M. , Ballhausen, D. , Burlina, A. , Fowler, B. , Grünert, S. C. , Grünewald, S. , Honzik, T. , Merinero, B. , Pérez‐Cerdá, C. , Scholl‐Bürgi, S. , Skovby, F. , … Chakrapani, A. (2014). Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet Journal of Rare Diseases, 9(1), 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bie, I. , Nizard, S. D. P. , & Mitchell, G. A. (2009). Fetal dilated cardiomyopathy: An unsuspected presentation of methylmalonic aciduria and hyperhomocystinuria, cblC type. Prenatal Diagnosis, 29(3), 266–270. 10.1002/pd.2218 [DOI] [PubMed] [Google Scholar]
- Forny, P. , Plessl, T. , Frei, C. , Bürer, C. , Froese, D. S. , & Baumgartner, M. R. (2021). Spectrum and characterization of bi‐allelic variants in MMAB causing cblB‐type methylmalonic aciduria. Human Genetics. 10.1007/s00439-021-02398-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, J. L. , & Venditti, C. P. (2016). Methylmalonic and propionic acidemias: Clinical management update. Current Opinion in Pediatrics, 28(6), 682–693. 10.1097/MOP.0000000000000422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madigan, T. , Block, D. R. , Carey, W. A. , Kaemingk, B. D. , & Patel, R. (2020). Proposed plasma ammonia reference intervals in a reference Group of Hospitalized Term and Preterm Neonates. The Journal of Applied Laboratory Medicine, 5(2), 363–369. 10.1093/jalm/jfz001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoli, I. , Sloan, J. L. , & Venditti, C. P. (2016). Isolated methylmalonic acidemia (Vol. 42) GeneReviews. [Google Scholar]
- Online Mendelian Inheritance in Man, OMIM® . Johns Hopkins University, Baltimore, MD. MIM number: {251110}: {03/09/2021}: World Wide Web URL: https://omim.org.
- Park, K. C. , Krywawych, S. , Richard, E. , Desviat, L. R. , & Swietach, P. (2020). Cardiac complications of propionic and other inherited organic acidemias. Frontiers in Cardiovascular Medicine, 7, 617451. 10.3389/fcvm.2020.617451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prada, C. E. , Al Jasmi, F. , Kirk, E. P. , Hopp, M. , Jones, O. , Leslie, N. D. , & Burrow, T. A. (2011). Cardiac disease in methylmalonic acidemia. The Journal of Pediatrics, 159(5), 862–864. 10.1016/j.jpeds.2011.06.005 [DOI] [PubMed] [Google Scholar]
- Razzaghy‐Azar, M. , Shakiba, M. , & Tafreshi, R. (2007). Heart failure in a patient with methylmalonic acidemia. Molecular Genetics and Metabolism, 92, 188. 10.1016/j.ymgme.2007.05.008 [DOI] [PubMed] [Google Scholar]
- Valayannopoulos, V. , Hubert, L. , Benoist, J. F. , Romano, S. , Arnoux, J. B. , Chrétien, D. , Kaplan, J. , Fakhouri, F. , Rabier, D. , Rötig, A. , Lebre, A. S. , Munnich, A. , de Keyzer, Y. , & de Lonlay, P. (2009). Multiple OXPHOS deficiency in the liver of a patient with CblA methylmalonic aciduria sensitive to vitamin B(12). Journal of Inherited Metabolic Disease, 32(2), 159–162. 10.1007/s10545-009-1023-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.