

Abstract
Background
Genetic cardiac diseases are the main trigger of sudden cardiac death (SCD) in young adults. Hypertrophic cardiomyopathy (HCM) is the most prevalent cardiomyopathy and accounts for 0.5 to 1% of SCD cases per year.
Methods
Herein, we report a family with a marked history of SCD focusing on one SCD young adult case and one pediatric case with HCM.
Results
For the deceased young adult, postmortem whole‐exome sequencing (WES) revealed a missense variant in the ACTN2 gene: c.355G > A; p.(Ala119Thr) confirming the mixed hypertrophic/dilated cardiomyopathy phenotype detected in the autopsy. For the pediatric case, WES allowed us the identification of a novel frameshift variant in the LZTR1 gene: c.1745delT; p.(Val582Glyfs*10) which confirms a clinical suspicion of HCM related to Noonan syndrome.
Conclusion
The present study adds further evidence on the pathogenicity of ACTN2: p. Ala119Thr variant in SCD and expands the mutational spectrum of the LZTR1 gene related to Noonan syndrome.
Keywords: ACTN2 gene, HCM/DCM, LZTR1 gene, Noonan syndrome, postmortem whole‐exome sequencing, sudden cardiac death
Our study reveals a missense variant in ACTN2 gene confirming the mixed hypertrophic/dilated cardiomyopathy phenotype detected in the autopsy. For the pediatric case, we identify of a novel frameshift variant in LZTR1 gene which confirms a clinical suspicion of HCM related to Noonan syndrome.
1. INTRODUCTION
Genetic cardiac diseases are the main trigger of sudden cardiac death (SCD) in young adults (35 years and under) (Brion et al., 2015; Isbister & Semsarian, 2019). SCD is defined as an unexpected death of an apparently healthy person, not attributable to an extra‐cardiac cause, usually within 1 hr of symptom onset (Isbister & Semsarian, 2019).
Hypertrophic cardiomyopathy is the most leading cause of SCD in young adults. SCD in hypertrophic cardiomyopathy (HCM) is caused mainly by ventricular arrhythmias (O'Mahony et al., 2013). In pediatric cases, HCM is mostly associated with syndromes such as Noonan syndrome (Gelb et al., 2015). HCM is defined by increased left ventricular wall thickness in the absence of other loading conditions. The disease is mostly inherited in an autosomal dominant manner (Sabater‐Molina et al., 2018).
Postmortem genetic screening aims to identify the underlying cause of death. For SCD with structural heart pathologies such as HCM or dilated cardiomyopathy (DCM), the autopsy can guide the genetic test and its interpretation.
In the present study, we report a family with a clinical and genetic investigation of one SCD case and a pediatric case with HCM and clinical suspicion of Noonan syndrome.
2. MATERIALS AND METHODS
2.1. Whole‐exome sequencing
Peripheral blood samples were collected and genomic DNA was extracted by standard techniques.
Subsequently, whole‐exome sequencing was performed using the TWIST “Mechanical Fragmentation and Twist Universal Adapter System” (Twist Bioscience). All DNA preparations and library quality control protocols were performed according to the manufacturers' instructions. The DNA libraries were subjected to paired‐end sequencing using the Illumina NextSeq500 sequencing platform (Illumina, San Diego, CA, USA). Raw fastQ files were aligned to the hg19 reference human genome (University of California Santa Cruz, UCSC) using BWA software. Variant calling workflow was performed according to the GATK best practices. The output files were annotated using ANNOVAR software. Variant annotation process and exome analysis were performed with VarAFT software (http://varaft.eu/).
2.2. Variant prioritization
Variant prioritization was performed with the Variant Annotation and Filtering Tool (VarAFT), version 2.17–2 (https://varaft.eu/). To pinpoint putatively pathogenic and causal variants, we adopted the following filtering strategy: we first excluded variants with a minor allele frequency (MAF) >1% in the gnomAD database (http://gnomad.broadinstitute.org/). Then, we removed non‐coding and synonymous variants. Subsequently, the remaining variants were filtered based on their in silico pathogenicity prediction. Thus, variants predicted as polymorphisms according to UMD‐Predictor (http://umd‐predictor.eu/), SIFT (http://sift.jcvi.org/), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), or Mutation Taster (http://www.mutationtaster.org/) were excluded. The obtained variants were subsequently analyzed based on their clinical relevance.
Sanger sequencing and familial segregation were performed for the likely causative prioritized variants.
3. RESULTS AND DISCUSSION
3.1. Clinical findings
3.1.1. Subject III‐2
The index case (III‐2) died suddenly at the age of 28 years old. He collapsed at work and no clinical manifestations preceding the collapse were mentioned. The death occurred 1 hr later in the emergency room in the hospital. After being informed by the family of the sudden death, we were able, 4 days later, to collect a blood sample from the department of Legal Medicine for DNA extraction and genetic study.
Anamnesis revealed the occurrence of an episode of syncope one‐year before death not followed by cardiac investigations. The medico‐legal autopsy showed a 370 g heart with evidence of hypertrophic/dilated cardiomyopathy. The autopsy did not reveal any pattern of viral myocarditis or coronary artery disease. The lungs were extremely congested (1030 g combined weight). The pulmonary arteries were free of thromboembolism and hemorrhages.
Toxicological screening of standard toxics and drugs performed on blood, urine, and gastric samples was negative.
Genetic investigation of familial history revealed several cases of sudden death in the maternal branch: his mother died at the age of 28‐year‐old at 1 month postpartum (II‐2), his maternal uncle at 30‐year‐old (II‐4), maternal aunt at 42‐year‐old (II‐5), and his maternal grandmother at 43‐year‐old (I‐2). Sudden death events occurred during sleep (I‐2 and II‐5) and normal daily activities (II‐4 and III‐2). No exercise‐related sudden cardiac death was reported. However, several episodes of syncope were reported for Subjects II‐2 and II‐4.
The deceased index‐case (III‐2) has a daughter aged 2 years and 9 months (IV‐1) with congenital HCM and an apparently healthy son aged 18 months (IV‐2). The clinical and genetic examination of his children was not possible as they lived outside the country.
Family pedigree and genotypes are shown in Figure 1.
FIGURE 1.
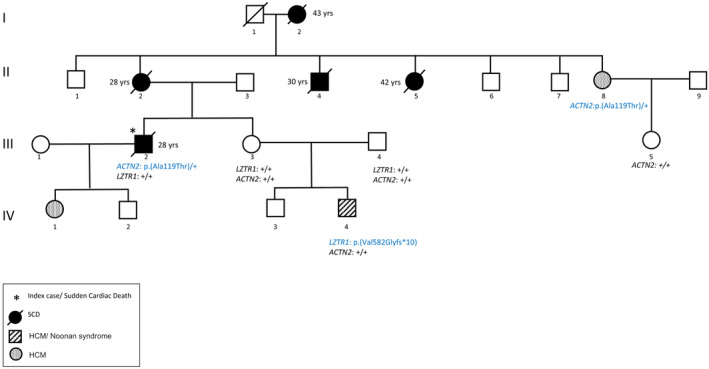
Pedigree of the family. Affected and/or suddenly deceased members are denoted by a filled circle, unaffected members are denoted by empty symbols. Ages at death are indicated beside each member. Genotypes are indicated below each member. (+) indicates the wild‐type allele. The GenBank reference sequence and version number used for the genes studied are NM_006767.3 for LZTR1 (OMIM number: 600574) and NM_001103.3 for ACTN2 (OMIM number: 102573)
3.1.2. Patient IV‐4
The patient, 3 years and 10 months of age, is the second child of apparently healthy and unrelated parents. He has a healthy 5‐year‐old brother. The mother and the father were, respectively, 22 and 29 years at conception. The patient was referred to genetic consultation at the age of 6 months because of HCM and familial history of SCD. Pregnancy was complicated by gestational diabetes, hydramnios, macrosomia, and HCM diagnosed prenatally on fetal morphology scan. Nuchal translucency was not evaluated. The child was delivered by the caesarian section at 35 weeks because of fetal distress. The Apgar scores were 7 at 1 min and 8 at 5 min. His birth measurements were above the 97th percentile (weight: 3800 g, height: 53 cm and head circumference: 36 cm). The patient was hospitalized at birth in the neonatal unit care for 14 days because of respiratory distress. Echocardiogram performed at 10 days confirmed the HCM diagnosis. Abdominal ultrasound was normal at 19 days. Testicular ultrasound performed at 19 days because of right testicular ectopia showed a right testicle of normal size and echostructure located at the lower part of the inguinal canal. The patient was then admitted to the department of pediatric cardiology at the age of 31 days for HCM exploration. Cardiac auscultation showed mill‐wheel murmur. An electrocardiogram and rhythmic Holter showed normal sinus rhythm and no conduction abnormalities. Chest X‐ray detected a cardiomegaly. Echocardiogram showed important interventricular septum hypertrophy (9–10 mm) and grade 2 mitral regurgitation. At 3 months, echocardiography showed concentric obstructive HCM with inter‐ventricular septum thickness at 12 mm and maximum gradient at 64 mmHg and grade 2 mitral regurgitation. Abdominal ultrasound at 2 months showed a mild hepatomegaly. Chromatographic analysis of amino acids and organic acids, performed at 3 months, were normal. The patient was treated with propranolol since the confirmation of his HCM at 10 days.
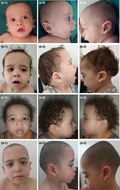
Clinical evaluation by geneticists at 6 months showed weight at 6 kg500 (3rd–10th percentiles), height at 61 cm (< 3rd percentiles) and head circumference at 44 cm (50th–75th percentiles). His facial dysmorphism included large forehead, frontal bossing, bitemporal narrowing, shallow orbital ridge, hypertelorism, epicanthus, down‐slanting palpebral fissures, depressed root of nose, anteverted nares, posteriorly rotated ears with thickened and pointed helix, detached ear lobes, smooth long philtrum, thin upper lip, thick lower lip, microretrognathia, and a short neck. Cutaneous abnormalities were present including sparse eyebrows and eyelashes, loose skin, deep palmoplantar creases, and occipital hemangioma (Figures 2 and 3). Thoracic examination showed a mild pectus excavatum. Heart auscultation indicated systolic murmur. Neurologic examination showed normal axial and peripheral tonus. Examination of the external genitalia showed right cryptorchidism. The diagnosis of Noonan syndrome (NS) was suspected as key features of this syndrome were present, namely the characteristic facies, the HCM, the pectus excavatum, the cryptorchidism, and the cutaneous abnormalities.
FIGURE 2.
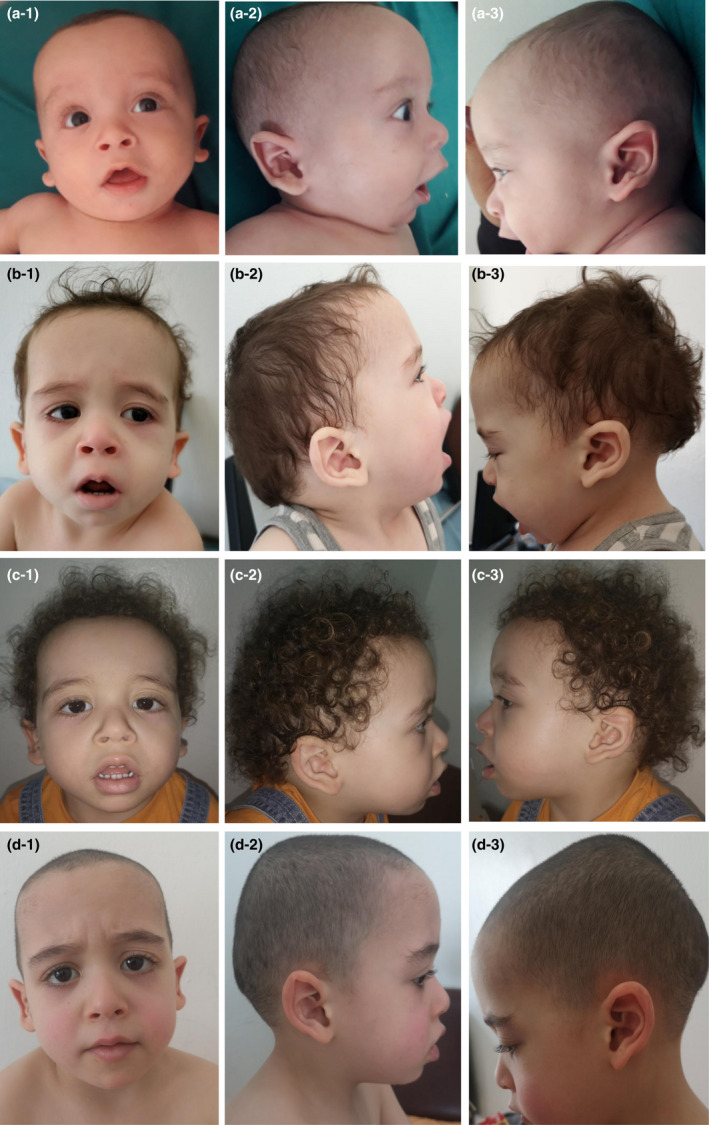
Photos of the patient at age of (a) 6 months (b) 16 months (c) 2 years and 3 months (d) and 3 years and 10 months. Note the large forehead, frontal bossing, bitemporal narrowing, sparse eyebrows and eyelashes, hypertelorism, epicanthus, down‐slanting palpebral fissures, depressed root of nose, anteverted nares, posteriorly rotated ears with thickened and pointed helix, detached ear lobes, smooth long philtrum, thick lower lip, microretrognathia, short neck (a‐d), and sparse and curly hair (c)
FIGURE 3.
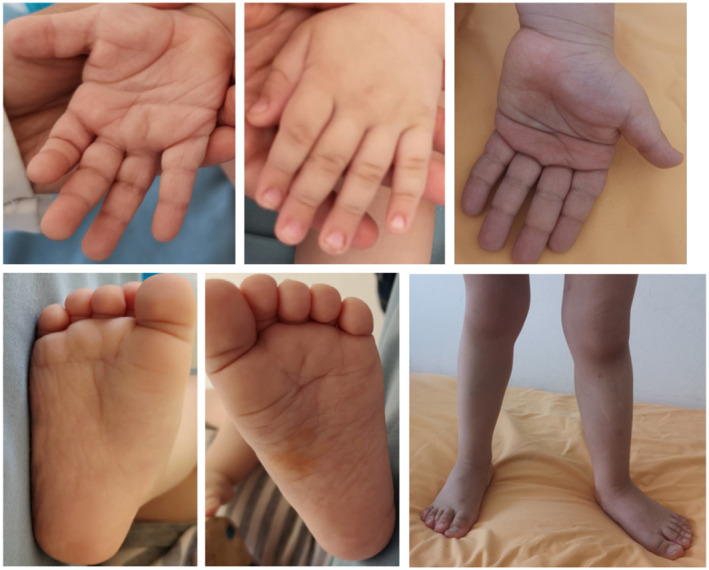
Photos of extremities showing deep palmoplantar creases and genu valgum
Parental examination showed no facial dysmorphism suggestive of NS and no short stature: the father was 1 m 68 cm tall and the mother was 1 m 61 cm tall. Echocardiography of the mother (III‐3) and the older brother (IV‐3) was normal.
R‐banded chromosome analysis on cultured peripheral blood lymphocytes from the patient was normal 46, XY. Molecular analysis of RASopathies was indicated.
At the age of 19 months, the patient underwent mitral valvuloplasty. His echocardiogram before the cardiac surgery showed severely dysplastic mitral valve with possible cleft at the anterior leaflet, severe concentric LV hypertrophy with significant reduction of the endocavitary volumes and obliteration of the systolic cavity (End‐Diastolic Diameter (EDD) = 29 mm; intraventricular septum thickness in diastole (IVSd) = 8.4 mm; left ventricular posterior wall in end‐diastole (LPWd) = 1.63 mm). Postoperatively, the patient was placed on propranolol, furosemide, captopril, and spironolactone. His personal history was complicated by several lower respiratory infections that required hospitalizations.
At the age of 2 years and 3 months, genetic evaluation revealed motor developmental delay as he walked at the age of 2 years and bisyllabic speech. The weight was 15 kg (90th percentiles), the height 86,6 cm (10th–25th percentiles), and the head circumference 51 cm (50th–75th percentiles). The facial dysmorphism was more suggestive of NS with accentuated down‐slanting palpebral fissures, moderate bulbous tip with anteverted nares, lip thickening, full cheeks, webbed and short neck, and sparse and curly hair (Figure 1c). Echocardiogram showed HCM and severe residual mitral regurgitation (mean mitral valve pressure gradient = 4 mmHg). Abdominal echography was normal.
At last evaluation at the age of 3 years and 10 months, anamnesis revealed an improvement in language since the integration of the nursery school at the age of 3 years. Examination showed a normal language, a weight of 16 kg (50th percentiles), a height of 99 cm (25th–50th percentiles), a head circumference of 51.5 cm (50th–75th percentiles), the same facial dysmorphism and cutaneous abnormalities, right cryptorchidism and genu valgum (Figure 3). The patient was on propranolol and furosemide.
3.1.3. Genetic findings
Whole‐exome sequencing was performed for the deceased patient (III‐2) and the patient (IV‐4) with HCM and NS suspicion.
For the latter case (IV‐4), a novel frameshift variant in the LZTR1 gene was identified. The LZTR1:c.1745delT; p.(Val582Glyfs*10) variant (NM_006767.3; OMIM number: 600574) is absent in all public databases as well as in our in‐house database. We have checked the European Network on Noonan Syndrome and related disorders for the LZTR1 gene and the identified variant is not reported (https://nseuronet.com/). The LZTR1:c.1745del T; p.(Val582Glyfs*10) occurred de novo at a heterozygous state. Both parents (III‐3 and III‐4) do not harbor the variant. According to the American College of Medical Genetics and Genomics (ACMG) guidelines for interpretation of sequence variants (Richards et al., 2015), the LZTR1:c.1745del T; p.(Val582Glyfs*10) variant was classified as pathogenic.
LZTR1 (leucine zipper‐like transcription regulator 1), located at 22q11.21, encodes a protein member of the BTB‐Kelch superfamily, located in the Golgi apparatus and involved in apoptosis (Nacak et al., 2006) and ubiquitination (Lu & Pfeffer, 2014; Stogios & Privé, 2004). LZTR1 was first identified as a causative gene of NS by Yamamoto et al. in 2015. Its association with the RAF1/SHOC2/PPP1CB complex involved in the RAS/MAPK signaling pathway was subsequently demonstrated by Umeki et al. (2019). Thus, LZTR1 is associated with Noonan syndrome 10 (OMIM 616564) with an autosomal dominant pattern of inheritance and with Noonan syndrome 2 (OMIM 605275) with an autosomal recessive model of inheritance. Regardless the genotype of LZTR1 carriers, 79,4% to 100% of patients exhibited cardiac defects (Umeki et al., 2019). In particular, HCM was found in 48, 3% to 71.4% of cases (Johnston et al., 2018; Umeki et al., 2019). In patient IV‐4, according to the clinical examination and WES results, congenital HCM was imputed to Noonan syndrome and not to the family history of SCD.
For the index patient (III‐2), we identified a variant in the ACTN2 gene previously reported as causative of diverse cardiac phenotypes such as HCM, DCM, left ventricular non‐compaction, and sudden cardiac death (Bagnall et al., 2014; Chiu et al., 2010; Haywood et al., 2016). The (ACTN2): c.355G > A; p.(Ala119Thr) variant (NM_001103.3; OMIM number: 102573) was found in the deceased patient at a heterozygous state.
The index patient's daughter (IV‐1) had congenital HCM. However, genetic testing was not possible for her and her brother, as she lived outside the country.
Familial segregation in subjects II‐8, III‐3, and III‐5 revealed the presence of p.(Ala119Thr) variant in the maternal aunt (II‐8) with HCM and supra‐ventricular excitability disorder and its absence in subjects III‐3 and III‐5 with normal electrocardiogram and echocardiography.
The ACTN2 p.(Ala119Thr) variant is located in the highly conserved actin‐binding CH1 domain. This variant has been reported in 2 large families with marked clinical heterogeneity including DCM, HCM, idiopathic ventricular fibrillation, LVNC, and sudden unexplained death (Bagnall et al., 2014; Chiu et al., 2010; Good et al., 2020; Haywood et al., 2016). Of note, in the previously reported cases, there have been two sudden cardiac deaths in young people aged under 35 years, associated with hypertrophic cardiomyopathy in one case (Bagnall et al., 2014).
To our knowledge, in addition to the p.(Ala119Thr) mutation, one other variant (p.Leu320Arg) within the ACTN2 gene has been associated with sudden cardiac death. This variant was found in a Chinese family with DCM, ventricular tachycardia, and SCD (Fan et al., 2019).
As a further example of the clinical heterogeneity seen in ACTN2 mutations carriers, the p.(Met228Thr) mutation reported by Girolami et al. was found in a large Italian family with members affected by HCM, early onset of supraventricular arrhythmias and LVNC (Girolami et al., 2014). Moreover, the p.(Glu628Gly) variant reported by Chiu et al., was found in a woman with moderate HCM and his son with asymmetric septal hypertrophy. However, her second son who also carried the variant was clinically unaffected (Chiu et al., 2010).
Thus, a clear‐cut genotype–phenotype correlation of ACTN2 mutations is lucky and the penetrance of ACTN2 variants remains poorly assessed.
The phenotypic pleiotropy of ACTN2 variants is suggestive of a complex disease mechanism altering the cardiac function in the structural and arrhythmogenic levels. Indeed, the ACTN2 gene encodes the α‐ actinin‐2 cross‐linking protein which is highly expressed in the cardiac sarcomeric Z‐disc that anchors and crosslinks the myofibrillar actin filaments. It plays several structural and functional roles in the sarcomere and the contractile apparatus (Lornage et al., 2019; Prondzynski et al., 2019). Moreover, ACTN2 is involved in ion‐channel organization in the cardiomyocytes Z‐disc (Bagnall et al., 2014; Prondzynski et al., 2019). Thus, disruption of α‐actinin‐2 protein may impact the electrical activity in the heart as well as sarcomere organization which may explain the different clinical phenotypes of the ACTN2: c.355G > A; p.(Ala119Thr) variant. Haywood et al. showed that ACTN2 p.(Ala119Thr) mutant has reduced F‐actin binding activity and altered Z‐disk localization (Haywood et al., 2016). Furthermore, depletion of Actn2 in zebrafish embryos leads to lateral alignment defects in the Z‐disc and morphological changes in the ventricle through a mechanical force‐dependent mechanism (Yang & Xu, 2012).
Indeed, clinical heterogeneity is a feature of HCM. Patients with ACTN2 pathogenic variants showed diverse hypertrophy distribution including septal, apical, concentric, and biventricular hypertrophy (Bagnall et al., 2014; Chiu et al., 2010). Strikingly, patients with mild hypertrophy had serious clinical outcomes ranging from resuscitated cardiac arrest, severe heart failure, and SCD (Bagnall et al., 2014; Chiu et al., 2010). Moreover, some patients had progressing cardiomyopathy from hypertrophic to dilated phenotype (Chiu et al., 2010). In our index patient, the autopsy showed evidence of a mixed hypertrophic/dilated cardiomyopathy. This combination of HCM/DCM may be explained by the multifunctional role of ACTN2 in both sarcomere and cytoskeleton. Our study adds an additional case with the ACTN2: p.(Ala119Thr) variant which may aid a deeper understanding of ACTN2‐related cardiac phenotypes and SCD risk assessment.
For families with SCD members, the clinical and genetic diagnosis is of greatest importance as it allows the identification of at‐risk family members and helps to reduce anxiety. In the current family, the mother (III‐3) presented heart‐focused anxiety that increased after the announcement of the HCM diagnosis of her child (IV‐4) and the sudden death of her brother (III‐2). Sanger sequencing in the mother did not show the ACTN2: p.(Ala119Thr) variant, which helped to reassure her. Genetic counseling and testing for the ACTN2 variant were proposed for the III‐3 maternal family.
The current study highlights the utility of postmortem WES in the identification of causative gene variant underlying SCD and points out the importance of clinical examination which allowed us to suspect a syndromic HCM occurring in the same family, namely the Noonan syndrome confirmed then by the identification of a novel frameshift variant in LZTR1gene.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
ETHICAL COMPLIANCE
This study was conducted according to the principles of the Declaration of Helsinki and to the ethical standards of the first author's institutional review board. All patients described in this study (or their parents/legal representative) gave informed consent to participate in this study.
ACKNOWLEDGMENTS
We would like to thank the family for their collaboration. This work was supported by the Tunisian Ministry of Public Health, and the “Association Française contre les Myopathies‐AFM Telethon” [MoThARD‐Project], and the “Institut National de la Santé et de la Recherche Médicale” to S.Z. H.J. received Postdoctoral fellowships from the “Association Française contre les Myopathies‐AFM Telethon”.
Kraoua, L. , Jaouadi, H. , Allouche, M. , Achour, A. , Kaouther, H. , Ahmed, H. B. , Chaker, L. , Maazoul, F. , Ouarda, F. , Zaffran, S. , M’rad, R. (2022). Molecular autopsy and clinical family screening in a case of sudden cardiac death reveals ACTN2 mutation related to hypertrophic/dilated cardiomyopathy and a novel LZTR1 variant associated with Noonan syndrome. Molecular Genetics & Genomic Medicine, 10, e1954. 10.1002/mgg3.1954
Funding informationThis research received no specific grant from any funding agency, commercial, or not‐for‐profit sector
Stéphane Zaffran and Ridha M'rad contributed equally to this work.
REFERENCES
- Bagnall, R. D. , Molloy, L. K. , Kalman, J. M. , & Semsarian, C. (2014). Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Medical Genetics, 15, 99. 10.1186/s12881-014-0099-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion, M. , Sobrino, B. , Martinez, M. , Blanco‐Verea, A. , & Carracedo, A. (2015). Massive parallel sequencing applied to the molecular autopsy in sudden cardiac death in the young. Forensic Science International Genetics, 18, 160–170. 10.1016/j.fsigen.2015.07.010 [DOI] [PubMed] [Google Scholar]
- Chiu, C. , Bagnall, R. D. , Ingles, J. , Yeates, L. , Kennerson, M. , Donald, J. A. , Jormakka, M. , Lind, J. M. , & Semsarian, C. (2010). Mutations in alpha‐actinin‐2 cause hypertrophic cardiomyopathy: A genome‐wide analysis. Journal of the American College of Cardiology, 55, 1127–1135. 10.1016/j.jacc.2009.11.016 [DOI] [PubMed] [Google Scholar]
- Fan, L.‐L. , Huang, H. , Jin, J.‐Y. , Li, J.‐J. , Chen, Y.‐Q. , & Xiang, R. (2019). Whole‐exome sequencing identifies a novel mutation (p.L320R) of Alpha‐Actinin 2 in a Chinese family with dilated cardiomyopathy and ventricular tachycardia. Cytogenetic and Genome Research, 157, 148–152. 10.1159/000496077 [DOI] [PubMed] [Google Scholar]
- Gelb, B. D. , Roberts, A. E. , & Tartaglia, M. (2015). Cardiomyopathies in Noonan syndrome and the other RASopathies. Progress in Pediatric Cardiology, 39, 13–19. 10.1016/j.ppedcard.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girolami, F. , Iascone, M. , Tomberli, B. , Bardi, S. , Benelli, M. , Marseglia, G. , Pescucci, C. , Pezzoli, L. , Sana, M. E. , Basso, C. , Marziliano, N. , Merlini, P. A. , Fornaro, A. , Cecchi, F. , Torricelli, F. , & Olivotto, I. (2014). Novel α‐actinin 2 variant associated with familial hypertrophic cardiomyopathy and juvenile atrial arrhythmias: A massively parallel sequencing study. Circulation Cardiovascular Genetics, 7, 741–750. 10.1161/CIRCGENETICS.113.000486 [DOI] [PubMed] [Google Scholar]
- Good, J.‐M. , Fellmann, F. , Bhuiyan, Z. A. , Rotman, S. , Pruvot, E. , & Schläpfer, J. (2020). ACTN2 variant associated with a cardiac phenotype suggestive of left‐dominant arrhythmogenic cardiomyopathy. HeartRhythm Case Reports, 6, 15–19. 10.1016/j.hrcr.2019.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haywood, N. J. , Wolny, M. , Rogers, B. , Trinh, C. H. , Shuping, Y. , Edwards, T. A. , & Peckham, M. (2016). Hypertrophic cardiomyopathy mutations in the calponin‐homology domain of ACTN2 affect Actin binding and cardiomyocyte Z‐disc incorporation. The Biochemical Journal, 473, 2485–2493. 10.1042/BCJ20160421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbister, J. , & Semsarian, C. (2019). Sudden cardiac death: An update. Internal Medicine Journal, 49, 826–833. 10.1111/imj.14359 [DOI] [PubMed] [Google Scholar]
- Johnston, J. J. , van der Smagt, J. J. , Rosenfeld, J. A. , Pagnamenta, A. T. , Alswaid, A. , Baker, E. H. , Blair, E. , Borck, G. , Brinkmann, J. , Craigen, W. , Dung, V. C. , Emrick, L. , Everman, D. B. , van Gassen, K. L. , Gulsuner, S. , Harr, M. H. , Jain, M. , Kuechler, A. , Leppig, K. A. , … Biesecker, L. G. (2018). Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genetics in Medicine, 20, 1175–1185. 10.1038/gim.2017.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lornage, X. , Romero, N. B. , Grosgogeat, C. A. , Malfatti, E. , Donkervoort, S. , Marchetti, M. M. , Neuhaus, S. B. , Foley, A. R. , Labasse, C. , Schneider, R. , Carlier, R. Y. , Chao, K. R. , Medne, L. , Deleuze, J. F. , Orlikowski, D. , Bönnemann, C. G. , Gupta, V. A. , Fardeau, M. , Böhm, J. , & Laporte, J. (2019). ACTN2 mutations cause “multiple structured Core disease” (MsCD). Acta Neuropathologica, 137, 501–519. 10.1007/s00401-019-01963-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, A. , & Pfeffer, S. R. (2014). A cullinary ride across the secretory pathway: More than just secretion. Trends in Cell Biology, 24, 389–399. 10.1016/j.tcb.2014.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacak, T. G. , Leptien, K. , Fellner, D. , Augustin, H. G. , & Kroll, J. (2006). The BTB‐kelch protein LZTR‐1 is a novel Golgi protein that is degraded upon induction of apoptosis. The Journal of Biological Chemistry, 281, 5065–5071. 10.1074/jbc.M509073200 [DOI] [PubMed] [Google Scholar]
- O'Mahony, C. , Elliott, P. , & McKenna, W. (2013). Sudden cardiac death in hypertrophic cardiomyopathy. Circulation: Arrhythmia and Electrophysiology, 6, 443–451. 10.1161/CIRCEP.111.962043 [DOI] [PubMed] [Google Scholar]
- Prondzynski, M. , Lemoine, M. D. , Zech, A. T. , Horváth, A. , Di Mauro, V. , Koivumäki, J. T. , Kresin, N. , Busch, J. , Krause, T. , Krämer, E. , Schlossarek, S. , Spohn, M. , Friedrich, F. W. , Münch, J. , Laufer, S. D. , Redwood, C. , Volk, A. E. , Hansen, A. , Mearini, G. , … Carrier, L. (2019). Disease modeling of a mutation in α‐actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Molecular Medicine, 11, e11115. 10.15252/emmm.201911115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabater‐Molina, M. , Pérez‐Sánchez, I. , del Rincón, J. P. H. , & Gimeno, J. R. (2018). Genetics of hypertrophic cardiomyopathy: A review of current state. Clinical Genetics, 93, 3–14. 10.1111/cge.13027 [DOI] [PubMed] [Google Scholar]
- Stogios, P. J. , & Privé, G. G. (2004). The BACK domain in BTB‐kelch proteins. Trends in Biochemical Sciences, 29, 634–637. 10.1016/j.tibs.2004.10.003 [DOI] [PubMed] [Google Scholar]
- Umeki, I. , Niihori, T. , Abe, T. , Kanno, S.‐I. , Okamoto, N. , Mizuno, S. , Kurosawa, K. , Nagasaki, K. , Yoshida, M. , Ohashi, H. , Inoue, S. I. , Matsubara, Y. , Fujiwara, I. , Kure, S. , & Aoki, Y. (2019). Delineation of LZTR1 mutation‐positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1‐PPP1CB complexes. Human Genetics, 138, 21–35. 10.1007/s00439-018-1951-7 [DOI] [PubMed] [Google Scholar]
- Yang, J. , & Xu, X. (2012). α‐Actinin2 is required for the lateral alignment of Z discs and ventricular chamber enlargement during zebrafish cardiogenesis. The FASEB Journal, 26, 4230–4242. 10.1096/fj.12-207969 [DOI] [PMC free article] [PubMed] [Google Scholar]