

Abstract
Objective:
Review the studies that investigate the mechanisms underlying imatinib-resistant gastrointestinal stromal tumors (GIST).
Background:
GIST are the most common mesenchymal tumors of the gastrointestinal (GI) tract and the most common sarcoma in humans. GIST are thought to be arise from interstitial cells of Cajal (ICC), pacemaker and neuromodulator cells in the GI tract, as well as “fibroblast”-like cells, which are another type of interstitial cells of the gut wall and also known as telocyte or platelet-derived growth factor-alpha (PDGFRA)-positive cells. The majority of GIST harbor gain-of-function mutations in either KIT or PDGFRA, and these gain-of-function mutations are mutually exclusive and most often heterozygous. GIST are responsive to the KIT/PDGFRA tyrosine kinase inhibitor (TKI), imatinib, the standard first-line drug for advanced and metastatic GIST. However, imatinib alone does not eradicate GIST despite an initial clinical benefit, and more than 90% of GIST harbor imatinib-resistance. Although second and third-generation TKIs have been developed and are currently in clinical use, they are not curative for refractory and metastatic GIST due to the emergence of clones with drug-resistant mutations. Eradication of drug-resistant GIST will cure patients with refractory GIST. Several mechanisms may contribute to refractory GIST. These mechanisms are secondary mutations in KIT and/or PDGFRA, alternative activation of tyrosine kinases, stem cells for GIST and cellular quiescence, a reversible nonproliferating state in which cells retain the ability to reenter cell proliferation.
Methods:
We review our current optimal treatment approach for managing patients with advanced and refractory GIST.
Conclusions:
This review explores the novel and potential therapeutic approaches to combat drug-resistant GIST.
Keywords: Gastrointestinal stromal tumors (GIST), interstitial cells of Cajal (ICC), imatinib, stem cells, quiescence
Introduction
As rare cancer that develops in the gastrointestinal (GI) tract, gastrointestinal stromal tumors (GIST) are the most common GI sarcoma in human (1–3). GIST are thought to arise from a rare and specialized cell type in the muscle layers of the GI tract, the interstitial cells of Cajal (ICC), or the progenitor cells of ICC (1,4). ICC are widely distributed throughout the entire GI tract and serve as pacemaker cells to control GI motility (5). Although GIST can arise in any part of the GI tract, their most common site is the stomach, probably due to Helicobacter pylori (6), a common type of bacteria that can survive in the stomach. The second most common site of GIST in the small intestine and fewer than 10% of GIST are found in the colon or rectum (Figure 1). GIST can also occur in the esophagus, mesentery, or omentum, but the incidence of GIST at these sites is less than 1%. Metastatic GIST occur most often in the liver and peritoneum (2,3). GIST usually occur in older individuals, with equal distribution among men and women. Although GIST can occur in children (known as pediatric GIST), these cases are rare and account for just 1–2% of all GIST (3). Morphologically, GIST can be divided into 3 subtypes (2). The majority of GIST (approximately 70%) can be characterized by a uniform population of spindle cells with ovoid nuclei. About 20% are composed of epithelioid cells, and these are mainly observed in pediatric GIST. The remaining 10% of GIST display a mixture of these 2 morphologies. v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), also known as CD117, is expressed in the majority of GIST (1,2,7). CD34, a transmembrane glycophosphoprotein expressed by vascular endothelium and some fibroblasts, is also expressed in 50–80% of GIST (2). Another transmembrane glycoprotein, CD44, is also expressed in GIST and ICC or ICC-stem cells (ICC-SC), suggesting that CD44 is an ICC/GIST lineage marker (8,9). Although KIT is also expressed in mast cells in the GI tract, anoctamin 1 (ANO1), which was first discovered on GIST-1 (DOG-1), is predominantly expressed in GIST, including KIT-negative GIST (10,11), indicating that ANO1 is a better, more reliable diagnostic marker for GIST. Other markers that are used to diagnose GIST include protein kinase C theta (PKCθ), smooth muscle actin, G protein-coupled receptor 20 (GPR20), desmin, and S100 protein although their expressions are not specific to GIST Ets variant 1 (ETV1) is a transcription factor required for oncogenic transcriptional program of GIST (12). ETV1 is highly and selectively expressed in GIST and the subtypes of ICC that can give rise to GIST (7,12). Through in situ hybridization, the diagnostic utility of ETV1 messenger RNA was reported in patients with GIST even after imatinib treatment (13). The utility of ETV1 immunohistochemistry is unknown and remains to be explored. Pathologically, the diagnosis of GIST relies on a combination of morphology and immunohistochemistry. The size of GIST may vary, and small GIST may cause no serious symptoms due to their low growth rate. Larger GIST require attention, with possible GIST symptoms being GI motility-related disorders and dysfunctions, including abdominal pain, nausea, vomiting, loss of appetite, dysphagia, and weight loss (2,3). For patients with a localized GIST, surgery is the standard treatment and should be performed unless the tumors are too large to be resected or they have spread throughout the body. GIST are generally resistant to conventional chemotherapy and radiotherapy routinely used for other cancers (3). The gain-of-function mutations in KIT are a key oncogenic drive in more than 80% of GIST, and a subpopulation of GIST have gain-of-function mutations in platelet-derived growth factor-alpha (PDGFRA), which is closely related to type III receptor tyrosine kinase (1,14). These gain-of-function mutations are mutually exclusive and most often heterozygous. These discoveries have led to the development of imatinib (Gleevec) for the targeted therapy used to prevent GIST. Imatinib inhibits tyrosine kinase receptors, including those of KIT, PDGFRA, ABL, and BCR-ABL. Imatinib remains as the frontline therapy for patients with advanced, unresectable, refractory, or metastatic GIST, except those GIST with PDGFRA D842V mutation, which do not respond to imatinib treatment. Imatinib is occasionally used to shrink uresectable large GIST before surgery and prevent GIST recurrence after surgery. Imatinib treatment generally continues as long as imatinib is effective. Imatinib can cause mild side effects of mainly GI motor-related dysfunctions, and serious side effects from imatinib are uncommon and generally well tolerated (3). Most GIST are initially responsive to frontline imatinib treatment; however, complete eradication of GIST is rare (15,16). Eventually, almost all GIST gain resistance to imatinib, and imatinib-treated patients progress and die because of the emergence of clones with imatinib-resistant mutations. Although subsequent lines of TKIs have been developed to mitigate imatinib-resistant GIST, these newer TKIs also have limited efficacy in this context (3). Therefore, these refractory GIST might potentially have different resistant and/or escape mechanisms other than imatinib-resistant mutations. This review provides an overview of recent advancements in our understanding of GIST, particularly drug-resistant, refractory GIST.
Figure 1.
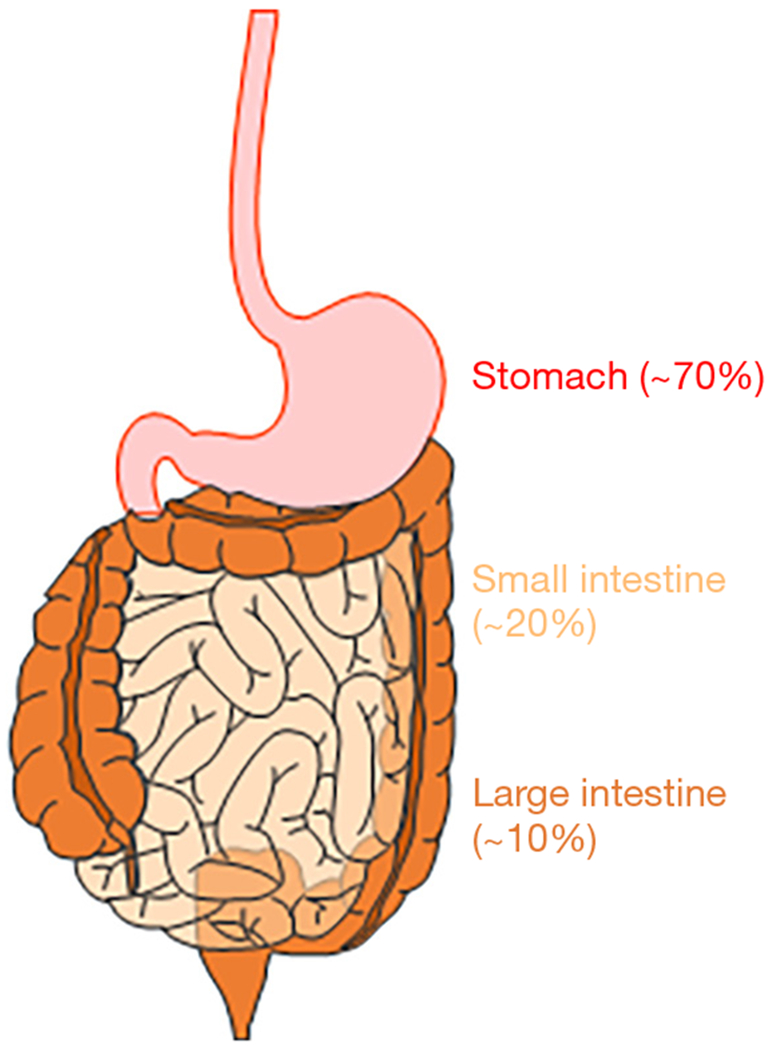
Primary GIST and metastatic sites with relative frequencies. The majority of GIST arise in the stomach and small intestine (jejunum and ileum). GIST also arise in the rectum and large intestine, but this is rare. GIST, gastrointestinal stromal tumors.
We present the following article in accordance with the narrative review checklist (available at https://dx.doi.org/10.21037/gist-21-10).
Pathology of GIST
A majority of GIST are believed to originate from the ICC lineage, while a subpopulation of GIST are thought to arise from telocytes or “fibroblast”-like cells or PDGFRA+ cells (1,14). GIST without KIT and PDGFRA mutations harbor mutations of succinate dehydrogenase (SDH) subunit (3). SDH is part of the tricarboxylic acid (TCA) cycle and the electron transfer chain. Inactivation of any of the SDH subunits results in the destabilization of these complexes and accumulation of succinate (17). Succinate accumulation activates pseudohypoxia signaling and insulin-like growth factor 1 (IGF1) via hypoxia-inducible factor 1 alpha (HIF1A) protein stabilization (17). The activation of HIF1A and IGF1 signaling with an energy metabolism defect is the key oncogenic mechanism in SDH-mutant GIST (3). However, the origin of these SDH-mutant GIST remains unknown. Recent reports found that v-Raf murine sarcoma viral oncogene homolog B1 (BRAF)-mutant GIST, which account for fewer than 1% of GIST, may be derived from GI smooth muscles (18). Different mutations are mutually exclusive and heterozygous in most cases. Ascertaining the mutational status of GIST is essential to determining current therapies’ choice and predicting the prognosis of patients with GIST (14,15).
Oncogenic mutations and tyrosine kinase switch in GIST
The vast majority of GIST harbor KIT or/and PDGFRA mutations, with both receptors being type III tyrosine kinase receptors (2). KIT and PDGFRA are structurally similar. Both receptors also have an extracellular domain, a transmembrane domain, a juxtamembrane domain, and 2 cytoplasmic kinase domains (Figure 2). Stem cell factor (SCF) is a KIT ligand, while PDGF is a ligand for PDGFRA. KIT and PDGFRA are activated upon dimerization with their ligand and lead to amplification of signaling cascades that include RAS/RAF/MEK/ETV1 and PI3K/AKT/mTOR pathways (3). In 1998, Hirota et al. were the first to discover KIT gain-of-function mutations like a key oncogenic driver in GIST (1). In 2003, the PDGFRA gain-of-function mutation was identified to be also another key oncogenic event in a subpopulation of GIST (14). These gain-of-function mutations constitutively activate the protein and downstream pathways without their ligands, leading to increased cell proliferation, adhesion, differentiation, survival, and resistance to apoptosis. Before 2000, surgery was the only successful treatment for primary localized GIST After these discoveries, the KIT/PDGFRA TKI, imatinib, was used in patients with advanced and metastatic GIST. Imatinib effectively suppresses GIST progression and improves the survival of patients with KIT- or PDGFRA-mutant GIST; however, those with PDGFRA exon 18 mutations, including D842V mutation, do not benefit, as GIST with this mutation are highly resistant to imatinib (15,16). Despite initial benefit, after several years of imatinib treatment, most patients ultimately develop disease progression due to heterogeneous acquired (or secondary) resistant mutation in KIT. These secondary KIT mutations involve the KIT-adenosine triphosphate (ATP)-binding pocket (encoded by exons 13 to 14) or the KIT activation loop (encoded by exons 17 to 18; Figure 2). Imatinib is ineffective against all of the common KIT secondary mutations (3). Imatinib also has secondary resistant mutations within PDGFRA encoded by exons 13 to 14 (Figure 2). These mutations limit the clinical benefit of imatinib and subsequent lines of treatment. For example, GIST with mutation in amino acid D816 (KIT activation loop encoded by exon 17) are resistant to all TKIs. To combat imatinib resistance, sunitinib (Sutent), a multi-TKI that blocks KIT, PDGFRA, and vascular endothelial growth factor (VEGF); and regorafenib (Stivarga), a TKI which blocks VEGF and epidermal growth factor receptor (EGFR), have been developed and used as the second- and third-line therapies, respectively (3). Recently, avapritinib (Ayvakit) was approved by the Food and Drug Administration (FDA) to treat GIST with PDGFRA exon 18 mutations (3). However, currently approved TKIs are still susceptible to the development of resistant mutations in KIT or/and PDGFRA, disease progression, and median disease-specific survival for patients with GIST are less than 5 years. In addition to TKI-resistant mutations with the KIT or PDGFRA genes, GIST may gain resistance to TKIs due to tyrosine kinase switch, which works as an “escape mechanism” (19). Furthermore, all TKIs have limited efficacy against wild-type (WT) GIST, a unique and minor subtype of GIST lacking gain-of-function mutations in KIT or PDGFRA (2). Therefore, it is critical to developing potential novel therapeutics for patients with refractory, TKIs-resistant GIST. These novel targets should not be related to KIT and PDGFRA, as most TKIs-resistant mutations occur within the KIT or PDGFRA genes themselves.
Figure 2.
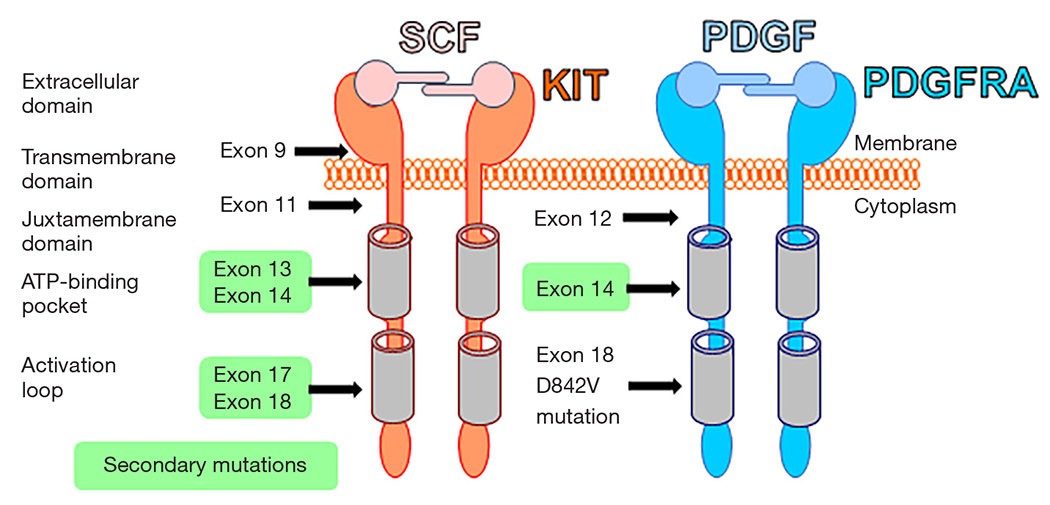
Oncogenic mutations in KIT and PDGFRA in GIST. KIT type mutations are the most common, followed by PDGFRA mutations. All mutations except KIT mutation in exon 17 and PDGFRA mutation exon 18 (D842V) are imatinib sensitive. Secondary mutations are shown in light green. KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; PDGFRA, platelet-derived factor-alpha; GIST, gastrointestinal stromal tumors.
Cancer stem cells in GIST
Cancer growth is constantly fueled by a small number of stem cells (SCs) in dedicated niches of cancers, and therapy that targets cancer SCs initially provided hope of a cure for patients with advanced and refractory cancers (20,21). A growing body of evidence supports the existence of cancer stem cells in many cancers, although the means to their identification and eradication are still not clear, and better therapeutic methods remain to be established (22). Previously, KITlowCD34+CD44+ cells were identified as ICC-SC in murine gastric musculature (9). ICC-SC represent small portion of epithelioid-shaped cells that are clonogenic and self-renewing (Figure 3). Unlike mature KIT+ICC, ICC-SC survival does not depend on KIT signaling due to these cells’ low KIT expression (4). Thus ICC-SC are highly resistant to the KIT blockade, including that induced by imatinib (Figure 4). When transplanted into athymic NCr-nu/nu mice, transplanted ICC-SC were found to give rise to malignant, KIT+ solid tumors, indicating that ICC-SC are truly GIST precursors or SCs (4). Although imatinib can reduce the proportion of KIT+ cells, imatinib treatment has not affected KITlow GIST SCs in KitK641E/+ mice, a model of human familial GIST (4), indicating that imatinib may not eliminate KITlow GIST stem cells due to their KITlow status. These findings support the previously described “cancer stem cell model” of imatinib-resistant GIST (15). In patients with GIST, tumors occasionally recur, probably due to the KITlow stem cell pool of GIST following the cessation of imatinib treatment. This is due to a failure of imatinib to eradicate KITlow GIST SCs. However, our understanding of GIST SCs in humans is still limited and identification of GIST SCs in humans will require further studies. Imatinib resistance has also been correlated with the recurrence of imatinib-resistant leukemia-initiating cells (LICs) in chronic myeloid leukemia (23). Although this imatinib-resistant SC model in chronic myeloid leukemia is similar to the GIST SC model, as originally proposed in mouse GIST (4,15), it remains unclear whether this model can be applied to human GIST. Importantly, LICs upregulate wingless-type MMTV integration site (Wnt) signaling during imatinib treatment, and inhibition of Wnt signaling with β-catenin inhibitor has been shown to sensitize LICs to imatinib’s cytotoxic effect (24). Very recently, Wnt/β-catenin signaling has been found to be highly expressed in ICC-SC and to be capable of knocking down β-catenin to prevent proliferation of ICC-SC (25), indicating that Wnt/β-catenin signaling is a key survival factor for ICC-SC and a possible origin of GIST precursors. Taken together, these findings suggest that the utility of inhibitors of Wnt/β-catenin signaling is effective for imatinib-resistant GIST via the targeting of their SCs.
Figure 3.
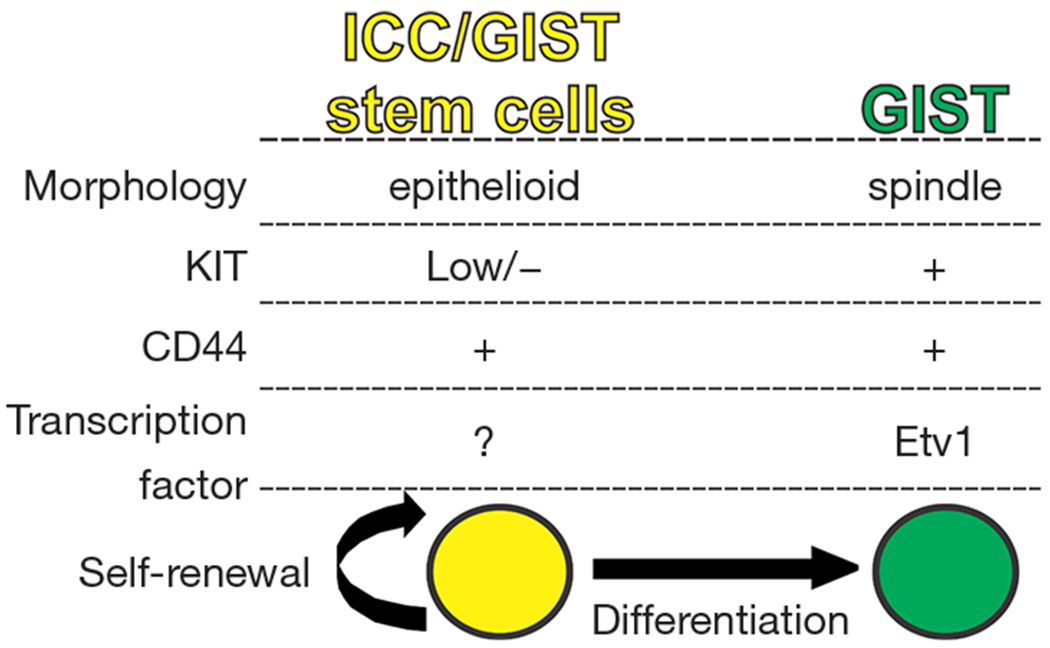
Proposed model of ICC/GIST stem cells and GIST. + : present; −: absent; low: low expression. GIST, gastrointestinal stromal tumors; KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog; ICC, interstitial cells of Cajal; Etv1, Ets variant 1.
Figure 4.
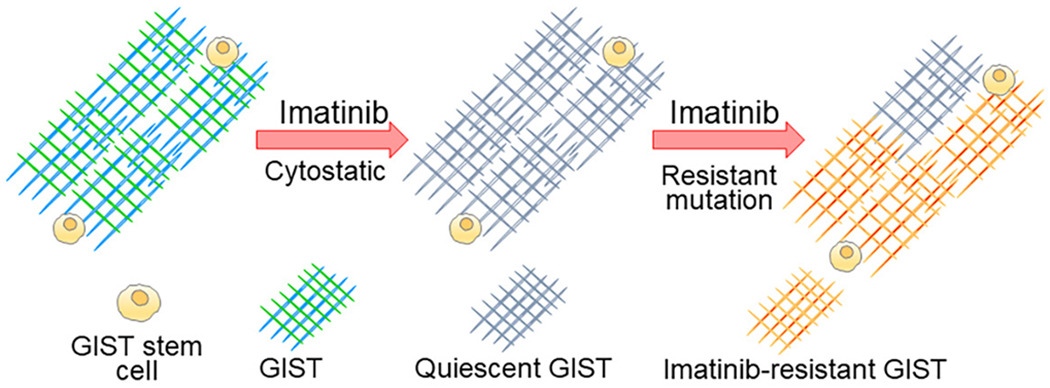
Hypothetical stem cell and quiescent model of imatinib resistance in GIST. Imatinib does not affect GIST stem cells due to their KITlow/− expression. Although GIST are initially sensitive to imatinib, imatinib’s effect is cytostatic, as GIST are in a quiescent state during imatinib treatment. During quiescence, imatinib-treated GIST can harbor imatinib-resistant mutations in KIT or/and PDGFRA, leading to the emergence of imatinib-resistant GIST. GIST, gastrointestinal stromal tumors; KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog.
Epithelial-mesenchymal transition and mesenchymal-epithelial transition in GIST
Most epithelial cancer cells can acquire a mesenchymal transcriptional program that facilitates cancer initiation, invasion, metastasis, and resistance to therapy (22). This process is known as an epithelial-mesenchymal transition (EMT), and EMT is essential to sustain epithelial cancer stemness (22,26). However, this EMT concept may not apply to GIST, since GIST are originally mesenchymal cancers. Of note, ICC-SC and GIST SCs are thought to be epithelioid phenotypes (4,7,27), suggesting that the transcriptional program maintaining GIST SCs is different from that of epithelial cancer SCs. For example, CD133, a pentaspan membrane glycoprotein used as a cancer SC biomarker in epithelial cancers, was reported to be highly expressed in GIST, but not ICC-SC and GIST stem cells (8). Of note, epithelioid GIST with reduced KIT expression have been reported in relapse GIST patients who had undergone imatinib treatment (28), suggesting that mesenchymal GIST can adopt the KITlow epithelioid phenotype to gain imatinib resistance. This mesenchymal-epithelial transition (MET)-associated morphological change was also observed in GIST culture in vitro after long-term exposure of imatinib (19). Taken together, these and other studies suggest that MET may be essential for the maintenance of stem cells in GIST and imatinib resistance. Whether the MET could be druggable targets in refractory GIST remains unclear and will require further clinical studies.
Quiescence in GIST
Cellular quiescence is a state of reversible arrest in the G0 stage of the cell cycle, with quiescent cells remaining alive and metabolically active (21). Cancer cell quiescence is essential for several types of cancers to acquire additional mutations and to become resistant to chemotherapeutic agents, as most chemotherapeutic agents specifically target proliferating cells (21). These “dormant” cancer cells are thought to cause refractory cancers and cancer progression (20). Therefore, targeting quiescent cell populations in cancer could be a valuable approach to treating cancer. Although imatinib effectively suppresses GIST proliferation and stimulates GIST death, a considerable proportion of the GIST survives (29,30), suggesting that imatinib’s effect is more cytostatic than cytotoxic. After cessation of imatinib treatment, GIST can re-enter the cell division cycle, leading to progression (Figure 4). Imatinib was found to be able to accumulate the cyclin-dependent kinase (CDK) inhibitor p27Kip1, a major regulator of quiescence [also known as cyclin-dependent kinase inhibitor 1B (CDKN1B)], by downregulating the F-box protein SKP2 in GIST in vitro and in vivo (29). Furthermore, DREAM complex (DP, p130/RBL2, E2F4, and MuvB) was identified in imatinib-induced GIST cell quiescence (31). Molecular and pharmacological inhibition of dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1A), a critical player in DREAM complex formation, can mitigate imatinib-induced GIST quiescence and increase the susceptibility of GIST to imatinib (31). This fact provides insight into the mechanism underlying the relationship between cellular quiescence and imatinib resistance although this mechanism in imatinib-treated human GIST with different mutations is needed to verified. However, due to their side effects, drugs targeting cellular quiescence have yet to be implemented in the clinic as standard care for imatinib-resistant GIST. Additional druggable targets and pathways remain to be identified and explored. Cellular senescence, a permanent state of cell cycle arrest induced by various external stimuli, including cellular stress and DNA damage, is a key driver of aging and aging-associated diseases (32). Cellular senescence is an essential cancer suppressive mechanism that prevents cell division with oncogenic activation and promotes immune clearance of these cells. Previous reports indicate that imatinib does not induce senescence in imatinib-sensitive GIST cell lines, as assessed by senescence-associated beta-galactosidase (SA-β-gal) activity (29), the most widely used senescence marker, and senescence-associated marker p16INK4A [also known as cyclin-dependent kinase 2A (CDKN2A)], suggesting no involvement of cellular senescence when GIST become imatinib resistant (29). Surprisingly, a recent paper also demonstrated no convincing evidence for the up-regulation of canonical markers or mediators of cellular senescence in ICC and ICC-SC in the stomachs of aged mice and progenic klotho mice (25), which function as model of accelerated aging (33), indicating no involvement of senescence in age-related ICC/ICC-SC loss. Although we cannot explain these findings, one possibility is that the susceptibility of ICC/ICC-SC and GIST to cellular senescence may be different from that of other tissues and cancers.
Conclusions and future directions
Despite the remarkable advancements made in the clinical management of advanced and relapsed GIST over the past two decades following the introduction of imatinib, critical issues remain to be resolved. One key issue is still imatinib resistance and disease persistence in GIST. To counter imatinib resistance, subsequent lines of TKIs targeting KIT and PDGFRA have been developed, but these TKIs have demonstrated limited clinical benefit. As described in this review, imatinib resistance involves multiple escape mechanisms, including tyrosine kinase switch, the independence of GIST SCs from KIT, cellular plasticity involving MET, and cellular quiescence to gain mutation. Hence, future strategies should focus on targeting escape pathways in GIST to eradicate therapy-resistant GIST and their precursors susscessfully.
Funding:
This work was supported in part by the National Institutes of Health grants R01 DK121766 (YH), P30 DK084567 (Mayo Clinic Center for Cell Signaling in Gastroenterology), Mayo Clinic Center for Biomedical Discovery Pilot Award (YH), the American Gastroenterology Association-Allergan Foundation Pilot Research Award in Gastroparesis (YH). The funding agencies had no role in the study analysis or writing of the manuscript. Its contents are solely the responsibility of the authors.
Footnotes
Reporting Checklist: The authors have completed the narrative review checklist. Available at https://dx.doi.org/10.21037/gist-21-10
Conflicts of Interest: Both authors have completed the ICMJE uniform disclosure form (available at https://dx.doi.org/10.21037/gist-21-10). The authors have no conflicts of interest to declare.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
References
- 1.Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998;279:577–80. [DOI] [PubMed] [Google Scholar]
- 2.Foo WC, Liegl-Atzwanger B, Lazar AJ. Pathology of gastrointestinal stromal tumors. Clin Med Insights Pathol 2012;5:23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blay JY, Kang YK, Nishida T, et al. Gastrointestinal stromal tumours. Nat Rev Dis Primers 2021;7:22. [DOI] [PubMed] [Google Scholar]
- 4.Bardsley MR, Horváth VJ, Asuzu DT, et al. Kitlow stem cells cause resistance to Kit/platelet-derived growth factor alpha inhibitors in murine gastrointestinal stromal tumors. Gastroenterology 2010;139:942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanders KM, Kito Y, Hwang SJ, et al. Regulation of Gastrointestinal Smooth Muscle Function by Interstitial Cells. Physiology (Bethesda) 2016;31:316–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kagihara J, Matsuda B, Young KL, et al. Novel association between Helicobacter pylori infection and gastrointestinal stromal tumors (GIST) in a multi-ethnic population. Gastrointest Stromal Tumor 2020;3:1. [Google Scholar]
- 7.Hayashi Y, Bardsley MR, Toyomasu Y, et al. Platelet-Derived Growth Factor Receptor-α Regulates Proliferation of Gastrointestinal Stromal Tumor Cells With Mutations in KIT by Stabilizing ETV1. Gastroenterology 2015;149:420–32.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Guo T, Zhang L, et al. CD133 and CD44 are universally overexpressed in GIST and do not represent cancer stem cell markers. Genes Chromosomes Cancer 2012;51:186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorincz A, Redelman D, Horváth VJ, et al. Progenitors of interstitial cells of cajal in the postnatal murine stomach. Gastroenterology 2008;134:1083–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, et al. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol 2009;296:G1370–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dailey DD, Ehrhart EJ, Duval DL, et al. DOG1 is a sensitive and specific immunohistochemical marker for diagnosis of canine gastrointestinal stromal tumors. J Vet Diagn Invest 2015;27:268–77. [DOI] [PubMed] [Google Scholar]
- 12.Chi P, Chen Y, Zhang L, et al. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 2010;467:849–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang BG, Lee HE, Kim WH. ETV1 mRNA is specifically expressed in gastrointestinal stromal tumors. Virchows Arch 2015;467:393–403. [DOI] [PubMed] [Google Scholar]
- 14.Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol 2008;26:5352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ordog T, Zörnig M, Hayashi Y. Targeting Disease Persistence in Gastrointestinal Stromal Tumors. Stem Cells Transl Med 2015;4:702–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McDonnell MJ, Punnoose S, Viswanath YKS, et al. Gastrointestinal stromal tumours (GISTs): an insight into clinical practice with review of literature. Frontline Gastroenterol 2017;8:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gottlieb E, Tomlinson IP. Mitochondrial tumour suppressors: a genetic and biochemical update. Nat Rev Cancer 2005;5:857–66. [DOI] [PubMed] [Google Scholar]
- 18.Kondo J, Huh WJ, Franklin JL, et al. A smooth muscle-derived, Braf-driven mouse model of gastrointestinal stromal tumor (GIST): evidence for an alternative GIST cell-of-origin. J Pathol 2020;252:441–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahadevan D, Cooke L, Riley C, et al. A novel tyrosine kinase switch is a mechanism of imatinib resistance in gastrointestinal stromal tumors. Oncogene 2007;26:3909–19. [DOI] [PubMed] [Google Scholar]
- 20.Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23:1124–34. [DOI] [PubMed] [Google Scholar]
- 21.Cho IJ, Lui PP, Obajdin J, et al. Mechanisms, Hallmarks, and Implications of Stem Cell Quiescence. Stem Cell Reports 2019;12:1190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature 2013;501:328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corbin AS, Agarwal A, Loriaux M, et al. Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR-ABL activity. J Clin Invest 2011;121:396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry JM, Tao F, Roy A, et al. Overcoming Wnt-β-catenin dependent anticancer therapy resistance in leukaemia stem cells. Nat Cell Biol 2020;22:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayashi Y, Asuzu DT, Bardsley MR, et al. Wnt-induced, TRP53-mediated Cell Cycle Arrest of Precursors Underlies Interstitial Cell of Cajal Depletion During Aging. Cell Mol Gastroenterol Hepatol 2021;11:117–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer 2002;2:442–54. [DOI] [PubMed] [Google Scholar]
- 27.Bauer S, Yu LK, Demetri GD, et al. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res 2006;66:9153–61. [DOI] [PubMed] [Google Scholar]
- 28.Dudeja V, Armstrong LH, Gupta P, et al. Emergence of imatinib resistance associated with downregulation of c-kit expression in recurrent gastrointestinal stromal tumor (GIST): optimal timing of resection. J Gastrointest Surg 2010;14:557–61. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Perdreau SA, Chatterjee P, et al. Imatinib mesylate induces quiescence in gastrointestinal stromal tumor cells through the CDH1-SKP2-p27Kip1 signaling axis. Cancer Res 2008;68:9015–23. [DOI] [PubMed] [Google Scholar]
- 30.Bauer S, Duensing A, Demetri GD, et al. KIT oncogenic signaling mechanisms in imatinib-resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007;26:7560–8. [DOI] [PubMed] [Google Scholar]
- 31.Boichuk S, Parry JA, Makielski KR, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res 2013;73:5120–9. [DOI] [PubMed] [Google Scholar]
- 32.van Deursen JM. The role of senescent cells in ageing. Nature 2014;509:439–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997;390:45–51. [DOI] [PubMed] [Google Scholar]