

Abstract
Background
Metabolic dysfunction has been suggested to be involved in the pathophysiology of amyotrophic lateral sclerosis (ALS). This study aimed to investigate the potential role of metabolic biomarkers in the progression of ALS and understand the possible metabolic mechanisms.
Methods
Fifty‐two patients with ALS and 24 normal controls were included, and blood samples were collected for analysis of metabolic biomarkers. Basal anthropometric measures, including body composition and clinical features, were measured in ALS patients. The disease progression rate was calculated using the revised ALS functional rating scale (ALSFRS‐R) during the 6‐month follow‐up.
Results
ALS patients had higher levels of adipokines (adiponectin, adipsin, resistin, and visfatin) and other metabolic biomarkers [C‐peptide, glucagon, glucagon‐like peptide 1 (GLP‐1), gastric inhibitory peptide, and plasminogen activator inhibitor type 1] than controls. Leptin levels in serum were positively correlated with body mass index, body fat, and visceral fat index (VFI). Adiponectin was positively correlated with the VFI and showed a positive correlation with the ALSFRS‐R and a negative correlation with baseline disease progression. Patients with lower body fat, VFI, and fat in limbs showed faster disease progression during follow‐ups. Lower leptin and adiponectin levels were correlated with faster disease progression. After adjusting for confounders, lower adiponectin levels and higher visfatin levels were independently correlated with faster disease progression.
Interpretation
The current study found altered levels of metabolic biomarkers in ALS patients, which may play a role in ALS pathogenesis. Adiponectin and visfatin represent potential biomarkers for prediction of disease progression in ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is a lethal neurodegenerative disease characterized by progressive loss of motor neurons. Most patients present with dysarthria, dysphagia, or muscle weakness at the beginning, which progresses to paralysis, severe disability, and eventual death within 2–5 years. There are many studies and hypotheses on the pathophysiological process of this disease, but the underlying mechanism remains unclear. Accumulating evidence suggests defective energy metabolism in ALS patients, including hypermetabolism, mitochondrial dysfunction, and insufficient energy supply, 1 , 2 , 3 which contributes to weight loss and a poor prognosis. 2 , 4
Animal experimental studies have demonstrated the crucial role of energy metabolism disturbance in the pathogenesis of ALS. 5 , 6 , 7 , 8 Transgenic ALS mice showed impaired glucose homeostasis due to altered glucose uptake in skeletal muscle and impaired glucose tolerance, 5 , 6 along with increased lipolysis and lipid peroxidation. 7 , 8 Based on these findings, dietary therapy, such as a high‐energy/high‐fat diet and intake of medium‐chain fatty acids or beta‐hydroxybutyrate, has shown therapeutic potential in animal models and ALS patients. 9 , 10
Lipid metabolism disorders have been widely reported in patients with ALS, presenting with hypercholesterolemia, hypertriglyceridemia, and other mixed dyslipidemias. 11 , 12 Genome‐wide association studies also suggested the contribution of low‐density lipoprotein cholesterol and total cholesterol risk alleles in the occurrence of ALS, 13 but controversy still exists concerning the prognostic role of dyslipidemia in ALS patients. 3 , 14 , 15
Adipokines are a group of factors released or secreted by adipose tissue and have many physiological functions, such as fat distribution, energy expenditure, appetite and satiety regulation, insulin secretion and sensitivity, and inflammation. 16 Given that some adipokines, such as leptin and adiponectin, can cross the blood–brain barrier and function in the brain, understanding the role of adipokines in the pathophysiology of ALS may illustrate the mechanism of metabolic dysfunction in ALS. 17 Adipokines may not only play a pivotal role in the energy metabolism of neurodegenerative disorders, 18 , 19 but also impact brain function and homeostasis through neuroinflammation or other signaling pathways. 17 , 20
As impaired energy homeostasis is one of the major features of ALS, understanding the underlying mechanism might provide potential therapeutic directions and strategies. Thus, we aimed to measure the levels of specific adipokines (adiponectin, adipsin, leptin, resistin, and visfatin) and other metabolic factors in ALS patients. We further explored the correlation between metabolic factors and clinical features and the potential impact on disease progression, hoping to understand the underlying metabolic mechanism in ALS.
Methods
Participants
Fifty‐two subjects were recruited between October 2020 and January 2022 among patients newly diagnosed with ALS in the Neurology Department of Peking Union Medical College Hospital. All the patients met the revised El Escorial diagnostic criteria of clinically definite, probable, or laboratory‐supported probable ALS 21 and underwent neuropsychological assessments to exclude cognitive or behavioral dysfunction. The study also included 24 healthy participants to compare adipokines and other metabolic biomarkers. All the participants denied a history of malignant disease, acute infection, acute or severe metabolic disease, or dieting. This study was approved by the ethics committees of Clinical Research of Peking Union Medical College Hospital (Beijing, China) and was conducted according to the ethical guidelines of the Declaration of Helsinki. All the participants gave their written informed consent.
Demographic data and a medical history of metabolic disorders, such as diabetes or dyslipidemia, were obtained from all participants during the first visit. Clinical features were also obtained from ALS patients at diagnosis, including the age of onset, disease duration, site of onset, and revised ALS functional rating scale (ALSFRS‐R) score. The reduction in ALSFRS‐R since symptom onset was also calculated by the formula ΔFRS = (48 − baseline ALSFRS‐R)/disease duration (months; time since disease onset). Appetite was measured using the Council on Nutrition Appetite Questionnaire (CNAQ), with a score ≤28 defined as loss of appetite. 22 Anthropometric characteristics were obtained at baseline, including body weight, height, body mass index (BMI), waist‐hip ratio (WHR), and body composition. Parameters of body composition were measured by a body composition analyzer (TongFang Health Technology, Beijing, China) 23 with the direct segmental multifrequency bioelectrical impedance analysis method, providing detailed information on fat‐free mass (FFM), fat mass (FM), visceral fat index (VFI), and fat in limbs (subcutaneous fat).
All patients with ALS were followed up for 6 months in outpatient clinics or by telephone. Adverse events, including tracheostomy, ventilator dependence, and death, were recorded. Neurological function at the 6‐month follow‐up was evaluated using ALSFRS‐R scoring, and the rates of disease progression (DPR) during follow‐up was calculated using the ALSFRS‐R at baseline: DPR = (ALSFRS‐R at follow‐up − ALSFRS‐R at baseline)/6 (months).
Measurement of adipokines and other metabolic biomarkers
Blood samples were collected from all 76 participants using a standardized protocol. Then, the blood was centrifuged for 10 min at 3000g and 4°C to obtain serum. The samples were transferred immediately into containers and stored at −80°C until further use. The specific adipokines (adiponectin, adipsin, leptin, resistin, and visfatin) and other metabolic biomarkers [C‐peptide, insulin, ghrelin, gastric inhibitory peptide (GIP), glucagon‐like peptide 1 (GLP‐1), glucagon, and plasminogen activator inhibitor type 1 (PAI‐1)] were analyzed using magnetic bead‐based multiplex assays (2‐plex Bio‐Plex Pro Human Adiponectin and Adipsin assay and 10‐plex Bio‐Plex Pro Human Diabetes assay from Bio–Rad, Ref. 171A7001 M and 171A7002 M, CA). For adiponectin and adipsin assays, samples were prepared with a dilution factor of 1:2500 due to the high physiological concentrations previously found in humans. For assays other than adiponectin and adipsin (10‐plex Bio‐Plex Pro Human Diabetes assay, Ref. 171A7001 M), serum samples were diluted fourfold (1:4) for analysis. Measurements were performed on a BioPlex 200 system (Bio–Rad Laboratories, CA), and all the analyses were performed in duplicate. All procedures and measurements strictly followed the instructions and directions. The interassay coefficients of variation (CVs) and intraassay %CVs were both <10%. Magnetic bead‐based multiplex assays offer best‐in‐class performance in a single experiment with good sensitivity and reproducibility 24 and have been widely used in a variety of studies. 25
Statistical analysis
All statistical analyses were carried out with IBM Statistical Package for the Social Sciences (SPSS) version 22.0 (Armonk, NY). Categorical variables are expressed as numbers (percentage) and were compared via chi‐squared analysis or Fisher's test. Continuous variables that were normally distributed are expressed as the mean ± SD, while nonparametric variables are expressed as the median (interquartile range). Student's t‐test or a Mann–Whitney test was used for comparisons between two groups. Spearman correlation tests were performed to assess the association of metabolic biomarkers with body composition and clinical features. The association between various adipokines and the rate of disease progression during follow‐ups was further identified by multivariate linear regression analysis to correct for potential confounding factors, including the age of onset and disease duration. A p value lower than 0.05 was considered statistically significant. We used the step‐down Bonferroni correction (Holm) and Benjamini Hochberg (BH) method to adjust for multiple comparisons.
Results
Comparison of metabolic biomarkers between patients and controls
Baseline demographic data and the medical history of metabolic disorders are shown in Table 1. The mean age at enrolment was 50.40 ± 10.90 years old in patients with ALS, of which 29 (55.8%) were men and 23 (44.2%) were women. Age and sex were similar between patients and controls. Few participants had a history of diabetes or hyperlipidemia.
Table 1.
Comparison of baseline characteristics and metabolic biomarkers in patients and controls.
| Patients (n = 52) | Controls (n = 24) | p value | p Holm | p BH | |
|---|---|---|---|---|---|
| Age (years) | 50.40 ± 10.90 | 47.17 ± 9.50 | 0.215 | ||
| Sex (n, %) | 0.139 | ||||
| Male | 29 (55.8) | 9 (37.5) | |||
| Female | 23 (44.2) | 15 (62.5) | |||
| Diabetes (n, %) | 5 (9.6) | 1 (4.2) | 0.388 | ||
| Hyperlipidemia (n, %) | 2 (3.8) | 3 (12.5) | 0.175 | ||
| Metabolic biomarkers | |||||
| Adiponectin (pg/mL) | 4945.8 (4579.8, 5194.9) | 4417.5 (3488.5, 5064.1) | 0.007** | 0.049* | 0.014* |
| Adipsin (pg/mL) | 305.5 (157.4, 1853.8) | 115.0 (87.0, 218.0) | <0.001*** | 0.0005*** | 0.0003*** |
| C‐peptide (pg/mL) | 2167.8 (1493.6, 3454.8) | 1395.0 (818.8, 2443.9) | 0.012* | 0.072 | 0.021* |
| Ghrelin (pg/mL) | 784.5 (463.5, 1025.9) | 819.5 (555.4, 1055.4) | 0.531 | 1.062 | 0.58 |
| GIP (pg/mL) | 42.0 (37.3, 49.0) | 31.3 (26.1, 36.5) | <0.001*** | <0.001*** | <0.001*** |
| GLP‐1 (pg/mL) | 140.5 (130.5, 161.0) | 119.8 (91.4, 136.8) | <0.001*** | 0.0005*** | 0.0002*** |
| Glucagon (pg/mL) | 51.5 (48.0, 57.8) | 46.0 (36.0, 57.0) | 0.031* | 0.155 | 0.047* |
| Insulin (pg/mL) | 1259.8 (802.5, 2749.3) | 881.3 (384.8, 2241.3) | 0.146 | 0.438 | 0.175 |
| Leptin (pg/mL) | 3647.5 (2073.4, 7069.3) | 4030.3 (2487.6, 6615.8) | 0.884 | 0.884 | 0.884 |
| PAI‐1 (pg/mL) | 7791.8 (6731.3, 8286.1) | 7129.0 (4630.1, 8050.4) | 0.038* | 0.152 | 0.051 |
| Resistin (pg/mL) | 3377.8 (2475.9, 4509.8) | 2441.0 (2031.5, 3528.0) | 0.005** | 0.04* | 0.012* |
| Visfatin (pg/mL) | 397.3 (336.5, 481.8) | 292.5 (193.5, 377.8) | <0.001*** | 0.0006*** | 0.0002*** |
Values of metabolic biomarkers were shown as medians and interquartile ranges. GIP, gastric inhibitory peptide; GLP‐1, glucagon‐like peptide 1; PAI‐1, Plasminogen activator inhibitor‐1. p Holm = Corrected p values by step‐down Bonferroni method. p BH = Corrected p values by Benjamini Hochberg method.
p < 0.05.
p < 0.01.
p < 0.001.
When comparing adipokines in patients and controls (Table 1), we found significant differences in the levels of adiponectin, adipsin, resistin, and visfatin between the two groups (p < 0.01). The median level of leptin was similar between ALS patients and normal controls (3647.5 pg/mL vs. 4030.3 pg/mL, p = 0.884). The median level of insulin was also similar in the two groups, while C‐peptide was higher in ALS patients than in controls (p < 0.05). ALS patients had higher levels of glucagon, GLP‐1, GIP, and PAI‐1 than controls (p < 0.05), while the level of ghrelin was similar between groups (784.5 pg/mL vs. 819.5 pg/mL, p = 0.531).
The differences in metabolic biomarkers between patients and controls were still significant after correcting for multiple comparisons. The levels of adiponectin, adipsin, resistin, visfatin, GIP, and GLP‐1 were significantly higher in patients than in controls after Bonferroni step‐down correction (p Holm < 0.05). The levels of adiponectin, adipsin, resistin, visfatin, glucagon, GIP, and GLP‐1 were significantly higher in patients than in controls after Benjamini Hochberg correction (p BH < 0.05).
Correlations between different biomarkers and anthropometric characteristics
Correlations between different metabolic biomarkers were analyzed and are shown in Figure 1. Significant correlations were found between leptin and GIP, glucagon, and PAI‐1 (p < 0.05). Resistin was positively correlated with both GLP‐1 and visfatin (p < 0.05). Positive correlations were also found between visfatin and ghrelin and glucagon (p < 0.05).
Figure 1.
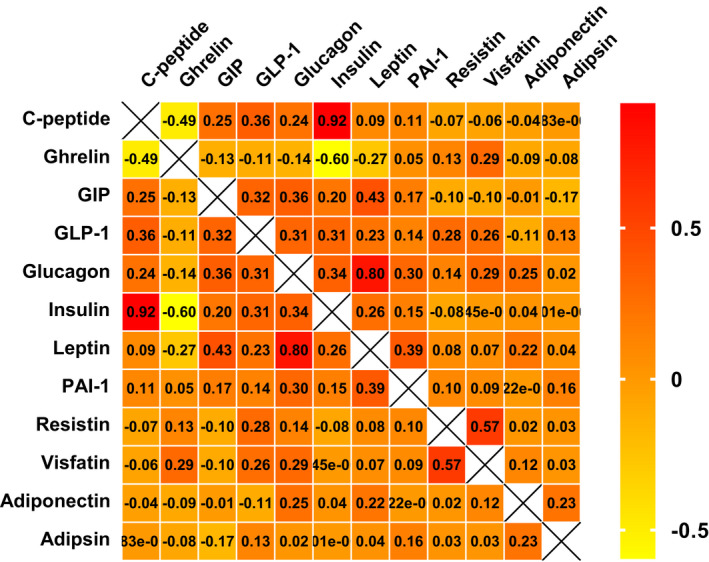
Correlation between different metabolic biomarkers. Yellow indicated a negative correlation, red indicated a positive correlation, and orange indicated the correlation was not significant. Correlation coefficients were shown for each cell. Rows and columns were labeled with each metabolic biomarker.
The detailed anthropometric characteristics are shown in Table 2. Compared with men, women had lower body weight and FFM (p < 0.001), with a higher level of leptin (6269.0 pg/mL vs. 3303.5 pg/mL, p < 0.001), and glucagon (58.0 pg/mL vs. 50.5 pg/mL, p = 0.001). The differences in leptin and glucagon were still significant between male and female patients after correcting for multiple comparisons (p corrected < 0.05). Other metabolic biomarkers were similar in males and females. Furthermore, we analyzed the association between metabolic biomarkers and anthropometric characteristics in patients with ALS. Leptin was positively associated with BMI, WHR (p = 0.034, r = 0.295), FM, VFI, and fat in limbs (p < 0.001, r = 0.532) (Fig. 2A–C). Glucagon and PAI‐1 were also positively associated with BMI, FM, and fat in limbs (p = 0.004, r = 0.389; p = 0.033, r = 0.295; p = 0.031, r = 0.299 in PAI‐1) (Fig. 2D and E). No significant correlation was found between visfatin and VFI (p = 0.063, r = 0.260) or WHR (p = 0.127, r = 0.214). A positive association was found between VFI and adiponectin (p = 0.036, r = 0.291), glucagon (p < 0.001, r = 0.586), and GIP (p = 0.06, r = 0.263) (Fig. 2F and G). However, no significant correlation was found between metabolic biomarkers and FFM.
Table 2.
Anthropometric and clinical characteristics in ALS patients.
| Patients (n = 52) | Male patients (n = 29) | Female patients (n = 23) | p value | p Holm | p BH | |
|---|---|---|---|---|---|---|
| Anthropometric characteristics | ||||||
| Height (cm) | 167.23 ± 6.96 | 171.83 ± 5.06 | 161.43 ± 4.08 | <0.001*** | <0.001*** | <0.001*** |
| Weight (kg) | 66.78 ± 11.81 | 72.33 ± 11.96 | 59.78 ± 7.00 | <0.001*** | <0.001*** | <0.001*** |
| BMI (kg/m2) | 23.83 ± 3.12 | 24.42 ± 3.35 | 23.10 ± 2.69 | 0.13 | 0.39 | 0.182 |
| WHR | 0.91 ± 0.06 | 0.91 ± 0.04 | 0.92 ± 0.08 | 0.759 | 0.759 | 0.759 |
| FFM (kg) | 48.97 ± 9.54 | 55.38 ± 7.53 | 40.88 ± 4.02 | <0.001*** | <0.001*** | <0.001*** |
| FM (kg) | 17.81 ± 4.92 | 16.95 ± 5.22 | 18.9 ± 4.37 | 0.158 | 0.316 | 0.184 |
| VFI | 9.87 ± 2.02 | 9.48 ± 2.03 | 10.36 ± 1.92 | 0.12 | 0.48 | 0.21 |
| Clinical characteristics | ||||||
| Onset age (years) | 48.71 ± 11.44 | 49.14 ± 11.90 | 48.17 ± 11.07 | 0.766 | ||
| Disease duration | 12 (8, 19) | 11 (8, 19) | 13 (9, 20) | 0.417 | ||
| Site of onset (n, %) | 0.927 | |||||
| Bulbar | 11 (21.2) | 6 (20.7) | 5 (21.7) | |||
| Spinal | 41 (78.8) | 23 (79.3) | 18 (78.3) | |||
| ALSFRS‐R | 37.04 ± 6.32 | 38.24 ± 5.21 | 35.52 ± 7.32 | 0.124 | ||
| CNAQ score (n = 19/14) | 28.61 ± 3.21 | 28.95 ± 3.06 | 28.14 ± 3.46 | 0.486 | ||
| Metabolic biomarkers | ||||||
| Glucagon (pg/mL) | 51.5 (48.0, 57.8) | 50.5 (45.8, 53.0) | 58.0 (49.0, 74.0) | 0.001** | 0.011* | 0.006** |
| Insulin (pg/mL) | 1259.8 (802.5, 2749.3) | 1213.5 (806.5, 2562.3) | 1267.5 (694.5, 3919.0) | 0.905 | 0.91 | 0.905 |
| Leptin (pg/mL) | 3647.5 (2073.4, 7069.3) | 3303.5 (1634.8, 3951.0) | 6269.0 (3588.0, 10444.0) | <0.001*** | 0.004** | 0.004** |
Values of anthropometric characteristics, onset age, ALSFRS‐R, and CNAQ score were shown as mean ± standard deviation. Values of disease duration were shown as medians and interquartile ranges. BMI, body mass index; WHR, waist‐hip rate; FFM, fat‐free mass; FM, fat mass; VFI, visceral fat index; ALSFRS‐R, revised ALS Functional Rating Scale; CNAQ, the Council on Nutrition Appetite Questionnaire. p Holm = Corrected p values by step‐down Bonferroni method. p BH = Corrected p values by Benjamini Hochberg method.
p < 0.05.
p < 0.01.
p < 0.001.
Figure 2.
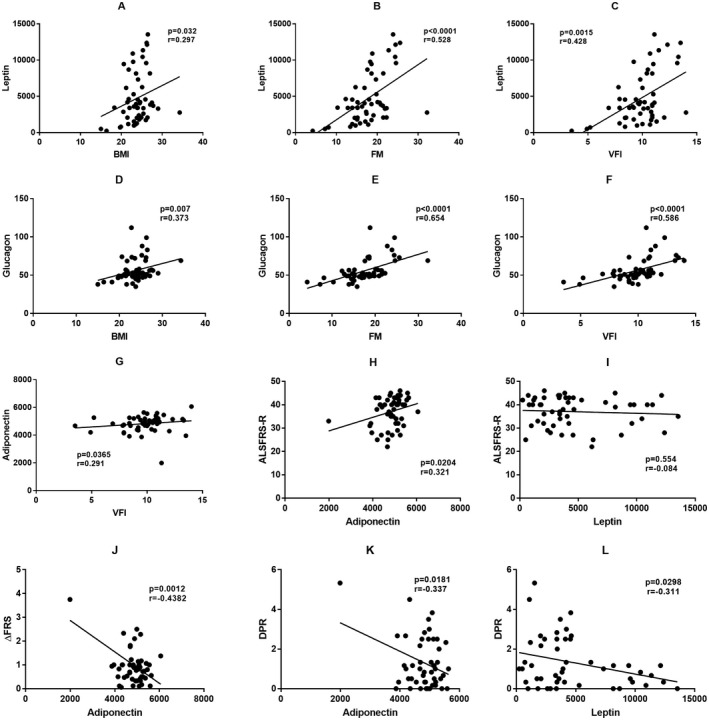
Correlation of metabolic biomarkers with anthropometric and clinical features. Leptin was positively correlated with BMI (A), FM (B), and VFI (C). Glucagon was positively correlated with BMI (D), FM (E), and VFI (F). Adiponectin was positively correlated with VFI (G). Adiponectin was positively correlated with ALSFRS‐R (H), while no significant correlation was found between leptin and ALSFRS‐R (I). Adiponectin was negatively correlated with ΔFRS (J) and DPR (K), and leptin was also negatively correlated with DPR (L). BMI, body mass index; FM, fat mass; VFI, visceral fat index; ALSFRS‐R, revised ALS Functional Rating Scale; ΔFRS, slope of reduction in ALSFRS‐R at baseline since symptom onset; DPR, rate of disease progression during follow‐ups.
Correlation of metabolic biomarkers with clinical features and disease progression
Of 52 patients with ALS, 11 (21.2%) had bulbar onset, and 41 (78.8%) had spinal onset (Table 2). Compared with patients with spinal onset, those with bulbar onset had an older age of onset (57.36 ± 9.72 years vs. 48.54 ± 10.54 years, p = 0.014) and a higher rate of medical history of hyperlipidemia (18.2% vs. 0%, p = 0.041). Serum levels of glucagon and PAI‐1 were slightly higher in patients with bulbar onset (p < 0.05, p corrected >0.05), while anthropometric features, including BMI and FM, were similar between the two groups. Six patients had a positive family history of ALS, five patients carried SOD1 mutations, and one patient carried an FUS mutation (Table S1). No difference in metabolic factors was found between patients with and without a family history. We also studied the association between metabolic factors and the severity of the disease. A positive association between ALSFRS‐R and adiponectin was found (p < 0.05, Fig. 2H), but no association was found with other biomarkers (Fig. 2I). However, the association was not significant after correcting for multiple comparisons (p corrected >0.05). The median change in the ALSFRS‐R from symptom onset to baseline (ΔFRS) was 0.83 (IQR: 0.48–1.05). Moreover, we studied the association between metabolic biomarkers and changes in the ALSFRS‐R from symptom onset to baseline (ΔFRS). A significant association was observed between ΔFRS and adiponectin (r = −0.428, p = 0.0012, p corrected = 0.014 < 0.05; Fig. 2J), but significant associations between ΔFRS and other metabolic factors were not observed.
Thirty‐three patients with ALS completed the CNAQ questionnaire, and 12 (36.4%) suffered from loss of appetite. Patients with good appetite had higher body weight and FFM, but no difference in BMI or body fat was found between patients with and without loss of appetite. Correlation analysis indicated a positive correlation between PAI‐1 and CNAQ scores (r = 0.366, p = 0.036), while no correlation was found in other metabolic biomarkers. Linear regression also did not reveal a significant correlation between metabolic factors and CNAQ scores, while a higher ALSFRS‐R score was correlated with a better appetite (β = 0.464, p = 0.025, 95% CI: 0.035–0.487).
During the 6‐month follow‐up, one patient died from respiratory failure, one patient underwent tracheostomy, and two patients withdrew from the study. All living patients were evaluated for ALSFRS‐R during follow‐ups, and DPR was calculated for prognostic analysis. The median DPR was 1.00 during follow‐ups (IQR: 0.33–2.42, ranging from 0 to 5.33), by which patients were divided into two groups to study the factors contributing to disease progression (Table S2). Patients with rapid disease progression had lower baseline FM (15.73 ± 4.76 kg) and VFI (9.00 ± 2.13 kg) values than those with slow progression (19.39 ± 3.49 kg, 10.63 ± 1.40 kg, p corrected < 0.05). No significant difference in CNAQ scores was found between patients with fast or slow disease progression. DPR was negatively associated with leptin and adiponectin (p < 0.05, Fig. 2K and L), while no association was observed between other metabolic factors and DPR. However, no significant correlation was observed between metabolic factors and DPR after Bonferroni step‐down correction or Benjamini Hochberg correction (p corrected > 0.05). Multiple regression analysis showed that visfatin and adiponectin were independently associated with DPR after adjusting for confounding factors, including onset age, disease duration, FM, and VFI (p < 0.05, Table 3). Considering the effect of sex on metabolism, we performed multiple regression analysis in male and female patient subgroups. However, no significant association between metabolic factors and disease progression was found in either subgroup.
Table 3.
Multiple regression analyses of disease progression rate.
| Variables | B | Beta | p value | 95% CI |
|---|---|---|---|---|
| Onset age | 0.032 | 0.292 | 0.073 | −0.003, 0.068 |
| Disease duration | −0.01 | −0.228 | 0.133 | −0.022, 0.003 |
| Leptin | −0.000086 | −0.241 | 0.177 | −0.00021, 0.000041 |
| Resistin | −0.000147 | −0.142 | 0.346 | −0.00046, 0.000165 |
| Visfatin | 0.003 | 0.395 | 0.015* | 0.001, 0.006 |
| Adiponectin | −0.001 | −0.334 | 0.017* | −0.001, −0.000135 |
| Adipsin | −0.000148 | −0.1 | 0.463 | −0.001, 0.000256 |
| FM | 0.05 | 0.176 | 0.617 | −0.15, 0.25 |
| VFI | −0.147 | −0.226 | 0.506 | −0.59, 0.296 |
FM, fat mass; VFI, visceral fat index.
p < 0.05.
Discussion
Recently, growing evidence has indicated that metabolic dysfunction is correlated with the pathological process of neurodegenerative diseases, such as Alzheimer's disease and ALS, in which adipokines and other metabolic biomarkers may play a potential role in the onset and progression of neurological symptoms. 19 , 26 This study found significant differences in the serum levels of adipokines and other metabolic biomarkers between ALS patients and normal controls. The levels of adiponectin, adipsin, resistin, and visfatin and some feeding‐related peptides, including GLP‐1 and GIP, were significantly higher in patients with ALS than in controls, indicating the involvement of metabolic dysfunction in ALS.
In this study, serum metabolic biomarkers were found to be related to detailed body composition in ALS patients, which is partly in line with previous findings from healthy individuals and patients with different diseases. 27 , 28 In this study, we found that women had a higher level of leptin than men, and serum leptin was positively correlated with body fat, VFI, and BMI. It should be noted that the serum levels of leptin and BMI showed a weak positive correlation, while leptin was highly correlated with total fat. The relationship between leptin and fat in limbs was more significant than the relationship between leptin and VFI, indicating the prominent contribution of subcutaneous fat to the leptin serum concentration. 29 Previous epidemiological studies found sex‐dependent differences in the development, clinical features, and prognosis of ALS, which is generally explained by the protective role of sex hormones. 30 It has been reported that female patients are more susceptible to developing bulbar involvement and cognitive impairment, especially older women. 31 Since leptin functions in diet regulation and can be affected by sex hormones, we assumed that the sex‐related differences in leptin and fat mass may be due to the effect of sex hormones in ALS, 32 warranting further investigation of the correlation between diet, body composition, sex hormones, and metabolism in ALS to understand the mechanism. Adiponectin was also positively correlated with VFI, which might indicate a correlation between adiponectin and visceral adipose tissue. In addition to adipokines, other metabolic biomarkers, such as glucagon, were found to be correlated with body composition, which may indicate extensive involvement of metabolic dysfunction in ALS. Since impaired glucose metabolism and homeostasis in ALS are correlated with abnormal insulin reactions, 6 , 33 abnormalities in glucagon levels, and altered glucagon sensitivity 34 may also contribute to the impaired energy metabolism in ALS. The increased circulating level of glucagon and its relationship with body composition (BMI, FM, and VFI) that we observed may be explained by the role of glucagon in regulating lipid metabolism 35 , 36 and increased energy production from lipid nutrients in ALS. 37 , 38 However, the significance of this observation and the precise underlying mechanism is still unknown. Although adipokines are secreted mostly from adipocytes, the interaction between adipocytes and other metabolic peptides and its role in nutritional status in ALS, including altered body composition, have not yet been fully elucidated and require further research.
Although patients with ALS had a lower level of leptin than controls, we did not find a significant difference between patients and controls, which is inconsistent with previous research. Ahmed et al. 39 showed an increased level of leptin in ALS patients compared with controls, while Nagel et al. 40 found that ALS patients had significantly lower leptin levels than controls. A previous animal experiment found an upregulation of leptin mRNA levels in adipose tissue of ALS‐related genetic mutant mice compared with levels in wild‐type mice, which continued to increase as the disease progressed. 41 Contrary to the results at the transcriptional level, the plasma level of leptin was lower in mutant mice than in control mice and further decreased at the end stage of the disease. 41 Due to the significant association between leptin and adipose tissue, the conflicting results of these studies could be attributed to differences in the study populations, such as race 42 and body composition. Considering the muscle and fat wasting that occur during the course of ALS, the difference in disease duration may also be responsible for the different findings of serum leptin levels in ALS patients in previous studies and our results. However, whether the altered leptin levels in ALS patients are secondary to changes in body composition or serve as the primary trigger factor in the pathogenesis of ALS remains unclear. A population‐based study in Germany found that increased leptin was negatively correlated with the risk of ALS in the general population, while the correlation was not significant after adjusting for BMI, 40 indicating that the leptin correlation resulted from the influence of nutritional status. Moreover, an increasing number of studies have shown that leptin plays a neuroprotective role in neurodegenerative diseases, including Alzheimer's disease and Parkinson's disease. 43 Thus, we hypothesized that the changes in serum leptin in ALS patients may be secondary to the nutritional status or pathological state in ALS, but the altered serum leptin may be further involved in multiple signaling pathways and play a pivotal role in physiological and pathological processes in ALS. Further longitudinal studies with larger sample sizes and longer follow‐up periods are needed to investigate the longitudinal changes in leptin and body composition in ALS patients and confirm a causal relationship between leptin and nutritional status in ALS.
In addition, we found a weak correlation between serum leptin and DPR during follow‐ups, and the relevance tended to be lost after correcting for multiple comparisons or adjusting for covariates. Nagel et al. 40 also suggested that leptin concentrations were positively correlated with the survival rate in ALS patients, indicating protective effects of leptin in patients with ALS. It has been widely demonstrated that the serum leptin concentration is strongly correlated with body weight or BMI, which was also confirmed in this study. As higher body weight and BMI have been found to be associated with a lower risk of ALS and better prognosis in ALS patients, 44 , 45 The positive association between leptin and prognosis in ALS has been attributed to the protective effect of nutritional status. However, some previous studies found that the association of leptin with the DPR and survival in ALS patients remained significant after adjusting for BMI. 40 Thus, there are likely other mechanisms underlying the potential role of leptin in the pathological process of ALS. The variable results further suggest that the function of leptin is complex.
Increasing evidence has revealed that leptin has a wide range of functions in energy homeostasis, fertility, inflammation, and autoimmunity. 46 , 47 Leptin can also be transported across the blood–brain barrier and bind to leptin receptors in the hypothalamus, causing reduced appetite and food intake. 48 In addition, a leptin gene knockout mouse model with mutant SOD1 showed increased weight and fat mass, improved neurological function, and enhanced survival, 49 indicating the contribution of leptin to energy expenditure. Loss of appetite is widely present in ALS patients; however, no correlation between serum leptin and appetite was found in this study. One possible explanation is that leptin functions in different physiological processes by binding to leptin receptors in different tissues, and leptin in cerebrospinal fluid, rather than serum leptin, may be correlated with appetite and have a negative effect on the prognosis of ALS. The loss of leptin receptors in the hypothalamus due to pathological hypothalamic atrophy and loss of orexin neurons in ALS patients 50 could be another explanation for the conflicting results. In addition, numerous studies have found that circulating leptin has a role beyond energy regulation and can modulate innate immunity and inflammation, including inhibiting the function and proliferation of regulatory T lymphocytes (Tregs). 51 Notably, reduced and dysfunctional Tregs have been reported in ALS patients, and restored Treg function is associated with delayed disease progression and prolonged survival. 52 Thus, the immunomodulatory effects of serum leptin may be another interpretation of its protective role in ALS. There have been relatively few explorations on the role of leptin in ALS, and the functional complexity of leptin in peripheral tissue and the brain may hinder understanding. Future research will be required to confirm our hypothesis and further determine the mechanism underlying the association between leptin and the pathogenesis of ALS.
Similarly, several studies have reported that serum adiponectin concentrations are significantly elevated in patients with ALS. 40 , 53 Our study demonstrated a weakly positive correlation between adiponectin concentrations and ALSFRS‐R scores, suggesting that adiponectin may serve as a marker to reflect the severity of the disease. Moreover, the baseline serum levels of adiponectin were negatively correlated with both the prebaseline DPR and the DPR during follow‐ups. Although the correlation was weak between adiponectin and DPR during follow‐ups, the result was still significant after adjusting for covariates. Our findings contrast with those of previous studies demonstrating that adiponectin does not affect the mortality of ALS patients. 40 This discrepancy may be explained by the heterogeneity of clinical features and racial differences. Although the correlation between adiponectin and disease progression and prognosis in ALS patients has been sparsely researched, previous studies on the biological functions of adiponectin provide some evidence suggesting that adiponectin is beneficial in ALS. As one of the most abundant adipokines secreted by adipocytes, adiponectin functions in multiple physiological processes, including insulin sensitization, glucose regulation, lipid metabolism, and anti‐inflammatory and antiapoptotic activities. 54 , 55 , 56 Animal studies initially found that adiponectin can increase glucose uptake and enhance fatty acid oxidation in skeletal muscle cells through the AMPK signaling pathway, which was further confirmed in human tissue. 57 , 58 Since energy metabolism dysfunction, such as impaired glucose and increased lipid utilization, has been demonstrated in the skeletal muscle of ALS patients, 4 , 38 adiponectin may be a potential therapeutic target in the future.
We also observed a positive correlation between visfatin and the DPR after adjusting for covariates, indicating a potential prognostic role of visfatin in ALS. Visfatin, also named nicotinamide phosphoribosyltransferase, was originally known for its secretion from visceral adipose tissue, 59 which was also confirmed in our study by the finding that visfatin had a weakly positive correlation with VFI. Increased visceral fat has been reported in ALS patients, 60 which may explain the elevated level of visfatin observed in this study, but the exact function of visfatin in ALS progression remains unclear. We found that patients with a lower baseline VFI had a rapidly progressive course at follow‐up, suggesting that energy stores in visceral fat may delay ALS progression. Although visfatin was positively correlated with VFI, higher visfatin was associated with a faster rate of progression, suggesting that the positive correlation between visfatin and DPR may be independent of visceral fat. Moreover, studies have shown inconsistent results in the correlation between visfatin and visceral fat. 61 , 62 Sitticharoon et al. 63 suggested that serum visfatin levels were negatively correlated with weight gain, while no correlation was found between visfatin and BMI. These results indicate that visfatin may be correlated with changes in body weight rather than actual fat distribution, which may indirectly explain the positive correlation between serum visfatin level and the DPR in ALS patients. In addition to metabolic regulation, previous studies have demonstrated that visfatin is involved in inflammation, autoimmunity, and apoptosis, 64 showing potential prognostic and therapeutic value in several inflammatory diseases and cancer types. 65 Thus, further studies are needed to explore the role of visfatin and its underlying mechanism in the pathological process of ALS.
Our study shed some light on the understanding of metabolic dysfunction in ALS by investigating serum metabolic biomarkers and further exploring their prognostic value in patients. However, there was no detailed information on body composition in controls, and the difference in body composition may affect the comparison of serum metabolic biomarkers between groups. Although we analyze the correlation between body composition and serum metabolic biomarkers in ALS patients, it is still unknown whether this result was similar to the correlation in healthy controls. Confounding factors, including body composition, were adjusted for when exploring the effect of metabolic factors on the rate of progression in ALS patients. In addition, the absence of data on diet limits the investigation of the underlying mechanism of metabolic biomarkers in ALS. Although it is widely known that leptin is involved in the regulation of appetite and food intake, the interaction of leptin and other biomarkers in energy metabolism has not been fully clarified. Moreover, the rather small sample size and short‐term follow‐up limit the exploration of prognostic value, and we will continue to follow these patients and expand the sample size to further analyze the effect of metabolic biomarkers on the survival of patients with ALS. Furthermore, the type I error rates may be relatively high in this study due to the multiple comparisons, for which we performed some statistical analysis corrections. Overall, this study produced some interesting findings, which we hope will arouse interest in further study of metabolic biomarkers in the future.
Conclusions
In conclusion, this study showed altered adipokines in patients with ALS, which were associated with disease severity and prognosis. Our findings suggest that adipokines may serve as useful biomarkers in ALS, providing new insight for additional studies of metabolic dysfunction in ALS and possible therapeutic strategies for ALS in the future.
Author Contributions
J. L. and L. C. developed the concept and designed the study. J. L., X. S., and D. S. were responsible for data acquisition and analysis. X. Y., Q. L., and L. C. were responsible for access and verification of the data. J. L. prepared the figures and wrote the draft. M. L. and L. C. were responsible for the interpretation of the data and critical revision of the manuscript.
Conflict of Interest
The authors declare that they have no competing interests.
Supporting information
Table S1. Metabolic factors and anthropometric characteristics in familial ALS patients.
Table S2. Clinical features and baseline anthropometric characteristics in patients categorized by disease progression during follow‐ups.
Acknowledgments
The authors would like to thank all the patients and their families for their support and understanding. Many thanks to Miss Chen Wang, who generously provided her assistance in the measures of body composition.
Funding Information
This work was supported by CAMS Innovation Fund for Medical Sciences (Grant number: 2021‐I2M‐1‐003), Strategic Priority Research Program of the Chinese Academy of Sciences “Biological basis of aging and therapeutic strategies” (Grant number: XDB39040100), Chinese Academy of Medical Science Neuroscience Center Fund “Molecular diagnosis and pathogenesis of ALS” (Grant number: 2014xh0601_A322102), and “Molecular diagnosis and neural network of ALS” (Grant number: 20141001_A322104).
Funding Statement
This work was funded by CAMS Innovation Fund for Medical Sciences grant 2021‐I2M‐1‐003; Chinese Academy of Medical Science Neuroscience Center Fund grants 20141001_A322104 and 2014xh0601_A322102; Strategic Priority Research Program of the Chinese Academy of Sciences grant XDB39040100.
Reference
- 1. Jesus P, Fayemendy P, Nicol M, et al. Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. Eur J Neurol. 2018;25:97‐104. doi: 10.1111/ene.13468 [DOI] [PubMed] [Google Scholar]
- 2. Ioannides ZA, Ngo ST, Henderson RD, McCombe PA, Steyn FJ. Altered metabolic homeostasis in amyotrophic lateral sclerosis: mechanisms of energy imbalance and contribution to disease progression. Neurodegener Dis. 2016;16:382‐397. doi: 10.1159/000446502 [DOI] [PubMed] [Google Scholar]
- 3. Dupuis L, Corcia P, Fergani A, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70:1004‐1009. doi: 10.1212/01.wnl.0000285080.70324.27 [DOI] [PubMed] [Google Scholar]
- 4. Tefera TW, Borges K. Metabolic dysfunctions in amyotrophic lateral sclerosis pathogenesis and potential metabolic treatments. Front Neurosci. 2016;10:611. doi: 10.3389/fnins.2016.00611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stallings NR, Puttaparthi K, Dowling KJ, et al. TDP‐43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS One. 2013;8:e71793. doi: 10.1371/journal.pone.0071793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pradat PF, Bruneteau G, Gordon PH, et al. Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:166‐171. doi: 10.3109/17482960902822960 [DOI] [PubMed] [Google Scholar]
- 7. Manzo E, O'Conner AG, Barrows JM, Shreiner DD, Birchak GJ, Zarnescu DC. Medium‐chain fatty acids, Beta‐hydroxybutyric acid and genetic modulation of the carnitine shuttle are protective in a drosophila model of ALS based on TDP‐43. Front Mol Neurosci. 2018;11:182. doi: 10.3389/fnmol.2018.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Angelova PR, Esteras N, Abramov AY. Mitochondria and lipid peroxidation in the mechanism of neurodegeneration: finding ways for prevention. Med Res Rev. 2021;41:770‐784. doi: 10.1002/med.21712 [DOI] [PubMed] [Google Scholar]
- 9. Olivan S, Martinez‐Beamonte R, Calvo AC, et al. Extra virgin olive oil intake delays the development of amyotrophic lateral sclerosis associated with reduced reticulum stress and autophagy in muscle of SOD1G93A mice. J Nutr Biochem. 2014;25:885‐892. doi: 10.1016/j.jnutbio.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 10. Dupuis L, Oudart H, Rene F, et al. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: benefit of a high‐energy diet in a transgenic mouse model. Proc Natl Acad Sci USA. 2004;101:11159‐11164. doi: 10.1073/pnas.0402026101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chelstowska B, Baranczyk‐Kuzma A, Kuzma‐Kozakiewicz M. Dyslipidemia in patients with amyotrophic lateral sclerosis ‐ a case control retrospective study. Amyotroph Lateral Scler Frontotemporal Degener. 2021;22:195‐205. doi: 10.1080/21678421.2020.1832119 [DOI] [PubMed] [Google Scholar]
- 12. Huang R, Guo X, Chen X, et al. The serum lipid profiles of amyotrophic lateral sclerosis patients: a study from south‐West China and a meta‐analysis. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:359‐365. doi: 10.3109/21678421.2015.1047454 [DOI] [PubMed] [Google Scholar]
- 13. Chen X, Yazdani S, Piehl F, Magnusson PKE, Fang F. Polygenic link between blood lipids and amyotrophic lateral sclerosis. Neurobiol Aging. 2018;67:202.e1‐202.e6. doi: 10.1016/j.neurobiolaging.2018.03.022 [DOI] [PubMed] [Google Scholar]
- 14. Dedic SI, Stevic Z, Dedic V, et al. Is hyperlipidemia correlated with longer survival in patients with amyotrophic lateral sclerosis? Neurol Res. 2012;34:576‐580. doi: 10.1179/1743132812Y.0000000049 [DOI] [PubMed] [Google Scholar]
- 15. Rafiq MK, Lee E, Bradburn M, McDermott CJ, Shaw PJ. Effect of lipid profile on prognosis in the patients with amyotrophic lateral sclerosis: insights from the olesoxime clinical trial. Amyotroph Lateral Scler Frontotemporal Degener. 2015;16:478‐484. doi: 10.3109/21678421.2015.1062517 [DOI] [PubMed] [Google Scholar]
- 16. Fasshauer M, Bluher M. Adipokines in health and disease. Trends Pharmacol Sci. 2015;36:461‐470. doi: 10.1016/j.tips.2015.04.014 [DOI] [PubMed] [Google Scholar]
- 17. Parimisetty A, Dorsemans AC, Awada R, Ravanan P, Diotel N, Lefebvre d’Hellencourt C. Secret talk between adipose tissue and central nervous system via secreted factors‐an emerging frontier in the neurodegenerative research. J Neuroinflammation. 2016;13:67. doi: 10.1186/s12974-016-0530-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Procaccini C, Santopaolo M, Faicchia D, et al. Role of metabolism in neurodegenerative disorders. Metabolism. 2016;65:1376‐1390. doi: 10.1016/j.metabol.2016.05.018 [DOI] [PubMed] [Google Scholar]
- 19. Letra L, Santana I. The influence of adipose tissue on brain development, cognition, and risk of neurodegenerative disorders. Adv Neurobiol. 2017;19:151‐161. doi: 10.1007/978-3-319-63260-5_6 [DOI] [PubMed] [Google Scholar]
- 20. Forny‐Germano L, De Felice FG, Vieira M. The role of leptin and adiponectin in obesity‐associated cognitive decline and Alzheimer's disease. Front Neurosci. 2018;12:1027. doi: 10.3389/fnins.2018.01027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron D . El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 22. Wilson MM, Thomas DR, Rubenstein LZ, et al. Appetite assessment: simple appetite questionnaire predicts weight loss in community‐dwelling adults and nursing home residents. Am J Clin Nutr. 2005;82:1074‐1081. doi: 10.1093/ajcn/82.5.1074 [DOI] [PubMed] [Google Scholar]
- 23. Chen W, Jiang H, Yang JX, et al. Body composition analysis by using bioelectrical impedance in a young healthy Chinese population: methodological considerations. Food Nutr Bull. 2017;38:172‐181. doi: 10.1177/0379572117697534 [DOI] [PubMed] [Google Scholar]
- 24. Hansenova Manaskova S, van Belkum A, Endtz HP, et al. Comparison of non‐magnetic and magnetic beads in bead‐based assays. J Immunol Methods. 2016;436:29‐33. doi: 10.1016/j.jim.2016.06.003 [DOI] [PubMed] [Google Scholar]
- 25. Graham H, Chandler DJ, Dunbar SA. The genesis and evolution of bead‐based multiplexing. Methods. 2019;158:2‐11. doi: 10.1016/j.ymeth.2019.01.007 [DOI] [PubMed] [Google Scholar]
- 26. Cai H, Cong WN, Ji S, Rothman S, Maudsley S, Martin B. Metabolic dysfunction in Alzheimer's disease and related neurodegenerative disorders. Curr Alzheimer Res. 2012;9:5‐17. doi: 10.2174/156720512799015064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Geisler C, Schweitzer L, Muller MJ. Functional correlates of detailed body composition in healthy elderly subjects. J Appl Physiol (1985). 2018;124:182‐189. doi: 10.1152/japplphysiol.00162.2017 [DOI] [PubMed] [Google Scholar]
- 28. Park AJ, Battaglino RA, Nguyen NMH, Morse LR. Associations between lean mass and leptin in men with chronic spinal cord injury: results from the FRASCI‐muscle study. PLoS One. 2018;13:e0198969. doi: 10.1371/journal.pone.0198969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tinggaard J, Hagen CP, Christensen AN, et al. Anthropometry, DXA, and leptin reflect subcutaneous but not visceral abdominal adipose tissue on MRI in 197 healthy adolescents. Pediatr Res. 2017;82:620‐628. doi: 10.1038/pr.2017.138 [DOI] [PubMed] [Google Scholar]
- 30. Pape JA, Grose JH. The effects of diet and sex in amyotrophic lateral sclerosis. Rev Neurol (Paris). 2020;176:301‐315. doi: 10.1016/j.neurol.2019.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trojsi F, D'Alvano G, Bonavita S, et al. Genetics and sex in the pathogenesis of amyotrophic lateral sclerosis (ALS): is there a link? Int J Mol Sci. 2020;21:10.3390/ijms21103647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giagulli VA, Castellana M, Pelusi C, et al. Androgens, body composition, and their metabolism based on sex. Front Horm Res. 2019;53:18‐32. doi: 10.1159/000494900 [DOI] [PubMed] [Google Scholar]
- 33. Perurena OH, Festoff BW. Reduction in insulin receptors in amyotrophic lateral sclerosis correlates with reduced insulin sensitivity. Neurology. 1987;37:1375‐1379. doi: 10.1212/wnl.37.8.1375 [DOI] [PubMed] [Google Scholar]
- 34. McDonald TS, Kumar V, Fung JN, Woodruff TM, Lee JD. Glucose clearance and uptake is increased in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis through an insulin‐independent mechanism. FASEB J. 2021;35:e21707. doi: 10.1096/fj.202002450R [DOI] [PubMed] [Google Scholar]
- 35. Anoop S, Misra A, Bhatt SP, Gulati S, Mahajan H, Prabakaran G. High plasma glucagon levels correlate with waist‐to‐hip ratio, suprailiac skinfold thickness, and deep subcutaneous abdominal and intraperitoneal adipose tissue depots in nonobese Asian Indian males with type 2 diabetes in North India. J Diabetes Res. 2017;2017:2376016. doi: 10.1155/2017/2376016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abbas E, Ahmed Siddiqui I, Khan MS, Perveen K, Butt A, Fawwad A. Fasting glucagon level in type 2 diabetes and impaired glucose tolerance and its association with diabetes‐associated clinical parameters: a study from Karachi, Pakistan. Cureus. 2021;13:e13430. doi: 10.7759/cureus.13430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dodge JC, Treleaven CM, Fidler JA, et al. Metabolic signatures of amyotrophic lateral sclerosis reveal insights into disease pathogenesis. Proc Natl Acad Sci USA. 2013;110:10812‐10817. doi: 10.1073/pnas.1308421110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Steyn FJ, Li R, Kirk SE, et al. Altered skeletal muscle glucose‐fatty acid flux in amyotrophic lateral sclerosis. Brain Commun. 2020;2:fcaa154. doi: 10.1093/braincomms/fcaa154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahmed RM, Phan K, Highton‐Williamson E, et al. Eating peptides: biomarkers of neurodegeneration in amyotrophic lateral sclerosis and frontotemporal dementia. Ann Clin Transl Neurol. 2019;6:486‐495. doi: 10.1002/acn3.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagel G, Peter RS, Rosenbohm A, et al. Adipokines, C‐reactive protein and amyotrophic lateral sclerosis ‐ results from a population‐ based ALS registry in Germany. Sci Rep. 2017;7:4374. doi: 10.1038/s41598-017-04706-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferrer‐Donato A, Contreras A, Frago LM, et al. Alterations in leptin signaling in amyotrophic lateral sclerosis (ALS). Int J Mol Sci. 2021;22:10.3390/ijms221910305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hunma S, Ramuth H, Miles‐Chan JL, et al. Do gender and ethnic differences in fasting leptin in Indians and Creoles of Mauritius persist beyond differences in adiposity? Int J Obes (Lond). 2018;42:280‐283. doi: 10.1038/ijo.2017.213 [DOI] [PubMed] [Google Scholar]
- 43. Zou X, Zhong L, Zhu C, et al. Role of leptin in mood disorder and neurodegenerative disease. Front Neurosci. 2019;13:378. doi: 10.3389/fnins.2019.00378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moglia C, Calvo A, Grassano M, et al. Early weight loss in amyotrophic lateral sclerosis: outcome relevance and clinical correlates in a population‐based cohort. J Neurol Neurosurg Psychiatry. 2019;90:666‐673. doi: 10.1136/jnnp-2018-319611 [DOI] [PubMed] [Google Scholar]
- 45. Jawaid A, Murthy SB, Wilson AM, et al. A decrease in body mass index is associated with faster progression of motor symptoms and shorter survival in ALS. Amyotroph Lateral Scler. 2010;11:542‐548. doi: 10.3109/17482968.2010.482592 [DOI] [PubMed] [Google Scholar]
- 46. Bluher M, Mantzoros CS. From leptin to other adipokines in health and disease: facts and expectations at the beginning of the 21st century. Metabolism. 2015;64:131‐145. doi: 10.1016/j.metabol.2014.10.016 [DOI] [PubMed] [Google Scholar]
- 47. La Cava A. Leptin in inflammation and autoimmunity. Cytokine. 2017;98:51‐58. doi: 10.1016/j.cyto.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guzman A, Hernandez‐Coronado CG, Rosales‐Torres AM, et al. Leptin regulates neuropeptides associated with food intake and GnRH secretion. Ann Endocrinol (Paris). 2019;80:38‐46. doi: 10.1016/j.ando.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 49. Lim MA, Bence KK, Sandesara I, et al. Genetically altering organismal metabolism by leptin‐deficiency benefits a mouse model of amyotrophic lateral sclerosis. Hum Mol Genet. 2014;23:4995‐5008. doi: 10.1093/hmg/ddu214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gabery S, Ahmed RM, Caga J, Kiernan MC, Halliday GM, Petersén Å. Loss of the metabolism and sleep regulating neuronal populations expressing orexin and oxytocin in the hypothalamus in amyotrophic lateral sclerosis. Neuropathol Appl Neurobiol. 2021;47:979‐989. doi: 10.1111/nan.12709 [DOI] [PubMed] [Google Scholar]
- 51. Alti D, Sambamurthy C, Kalangi SK. Emergence of leptin in infection and immunity: scope and challenges in vaccines formulation. Front Cell Infect Microbiol. 2018;8:147. doi: 10.3389/fcimb.2018.00147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thonhoff JR, Simpson EP, Appel SH. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol. 2018;31:635‐639. doi: 10.1097/WCO.0000000000000599 [DOI] [PubMed] [Google Scholar]
- 53. Ngo ST, Steyn FJ, Huang L, et al. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2015;357:22‐27. doi: 10.1016/j.jns.2015.06.053 [DOI] [PubMed] [Google Scholar]
- 54. Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol. 2018;8:1031‐1063. doi: 10.1002/cphy.c170046 [DOI] [PubMed] [Google Scholar]
- 55. Yadav A, Kataria MA, Saini V, Yadav A. Role of leptin and adiponectin in insulin resistance. Clin Chim Acta. 2013;417:80‐84. doi: 10.1016/j.cca.2012.12.007 [DOI] [PubMed] [Google Scholar]
- 56. Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784‐1792. doi: 10.1172/JCI29126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu Y, Sweeney G. Adiponectin action in skeletal muscle. Best Pract Res Clin Endocrinol Metab. 2014;28:33‐41. doi: 10.1016/j.beem.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 58. Kumari R, Kumar S, Kant R. An update on metabolic syndrome: metabolic risk markers and adipokines in the development of metabolic syndrome. Diabetes Metab Syndr. 2019;13:2409‐2417. doi: 10.1016/j.dsx.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 59. Fukuhara A, Matsuda M, Nishizawa M, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426‐430. doi: 10.1126/science.1097243 [DOI] [PubMed] [Google Scholar]
- 60. Lindauer E, Dupuis L, Muller HP, et al. Adipose tissue distribution predicts survival in amyotrophic lateral sclerosis. PLoS One. 2013;8:e67783. doi: 10.1371/journal.pone.0067783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Berndt J, Kloting N, Kralisch S, et al. Plasma visfatin concentrations and fat depot‐specific mRNA expression in humans. Diabetes. 2005;54:2911‐2916. doi: 10.2337/diabetes.54.10.2911 [DOI] [PubMed] [Google Scholar]
- 62. Willers SM, Brunekreef B, Abrahamse‐Berkeveld M, et al. Serum visfatin and leptin in relation to childhood adiposity and body fat distribution: the PIAMA birth cohort study. Ann Nutr Metab. 2015;66:63‐71. doi: 10.1159/000369979 [DOI] [PubMed] [Google Scholar]
- 63. Sitticharoon C, Nway NC, Chatree S, Churintaraphan M, Boonpuan P, Maikaew P. Interactions between adiponectin, visfatin, and omentin in subcutaneous and visceral adipose tissues and serum, and correlations with clinical and peripheral metabolic factors. Peptides. 2014;62:164‐175. doi: 10.1016/j.peptides.2014.10.006 [DOI] [PubMed] [Google Scholar]
- 64. Dahl TB, Holm S, Aukrust P, Halvorsen B. Visfatin/NAMPT: a multifaceted molecule with diverse roles in physiology and pathophysiology. Annu Rev Nutr. 2012;32:229‐243. doi: 10.1146/annurev-nutr-071811-150746 [DOI] [PubMed] [Google Scholar]
- 65. Stofkova A. Resistin and visfatin: regulators of insulin sensitivity, inflammation and immunity. Endocr Regul. 2010;44:25‐36. doi: 10.4149/endo_2010_01_25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Metabolic factors and anthropometric characteristics in familial ALS patients.
Table S2. Clinical features and baseline anthropometric characteristics in patients categorized by disease progression during follow‐ups.