

Abstract
Objective
Inhibition of dihydroorotate dehydrogenase suppresses magnetic resonance imaging brain lesions and disease activity in multiple sclerosis but has limiting tolerability. We assessed the safety and efficacy of vidofludimus calcium, a novel, selective dihydroorotate dehydrogenase inhibitor, in patients with relapsing‐remitting multiple sclerosis.
Methods
This double‐blind, 24 weeks, placebo‐controlled, phase 2 trial (EMPhASIS) enrolled patients 18–55 years with relapsing‐remitting multiple sclerosis. Eligible patients were randomly assigned (1:1:1) to once‐daily vidofludimus calcium (30 mg or 45 mg) or placebo. The primary endpoint was the cumulative number of combined unique active lesions to week 24 between vidofludimus calcium 45 mg and placebo (clinicalTrials.gov number NCT03846219; EudraCT 2018–001896‐19).
Results
After 24 weeks, the mean cumulative number of combined unique active lesions was 6.4 (95% CI: 2.8–13.9) with placebo compared to 2.4 (95% CI: 1.1–4.9) with vidofludimus calcium 45 mg (rate ratio 0.38, 95% CI: 0.22–0.64; p = 0.0002); the rate ratio between vidofludimus calcium 30 mg and placebo was 0.30 (95% CI: 0.17–0.53; p < 0.0001). Treatment‐emergent adverse events occurred in 30 (44%) of patients assigned placebo and 60 (43%) of patients assigned vidofludimus calcium. Serious adverse events occurred in one (1%) assigned placebo and two (1%) assigned vidofludimus calcium. No increased incidence of infectious, hepatic, or renal treatment‐emergent adverse events or serious adverse events was observed.
Interpretation
Treatment with vidofludimus calcium led to a reduction in new magnetic resonance imaging lesions in patients with relapsing‐remitting multiple sclerosis and was well tolerated with a favorable safety profile. Assessment in longer, larger trials is justified.
Introduction
The immunopathogenesis of multiple sclerosis is characterized by pathogenic T and B cells that infiltrate the central nervous system and cause multifocal inflammation, demyelination, and neurodegeneration. Active and rapidly dividing lymphocytes meet their pyrimidine needs by highly expressing dihydroorotate dehydrogenase (DHODH), 1 , 2 a mitochondrial enzyme involved in the rate‐limiting step of de novo pyrimidine synthesis, whereas resting lymphocytes are able to meet their pyrimidine needs through an DHODH‐independent salvage pathway. 3 , 4 Hence, DHODH blockage is of significant clinical interest in pathologies mediated by activated pathogenic lymphocyte populations. The role of DHODH in the pathogenesis of multiple sclerosis was demonstrated in the TEMSO and TOWER trials of teriflunomide, the only approved DHODH inhibitor in Europe and the United States for relapsing forms of multiple sclerosis. 5 , 6 Use of teriflunomide can be limited by its side effect profile: diarrhea, alopecia, neutropenia, and elevated liver enzymes. 7
Vidofludimus calcium is a novel, orally available, highly selective inhibitor of DHODH in development for immune‐mediated inflammatory diseases including relapsing multiple sclerosis, ulcerative colitis, 8 primary sclerosing cholangitis, 9 and coronavirus disease 2019. 10 , 11 Vidofludimus calcium has a half‐life of approximately 30 h that allows for once‐daily oral dosing and rapid wash‐out at treatment discontinuation, 12 exhibited submicromolar potency against human DHODH, and inhibited T‐lymphocyte proliferation and secretion of interleukin (IL)‐17 and IFN‐γ in vitro. 13 Vidofludimus calcium demonstrated dose‐dependent activity in experimental autoimmune encephalomyelitis in rodents. 13 More than 800 healthy volunteers or patients with immune‐related conditions have been exposed to vidofludimus in completed or ongoing trials. In the COMPONENT trial, patients with active rheumatoid arthritis treated with 35 mg vidofludimus‐free acid once‐daily for 13 weeks had an overall similar safety profile as compared to placebo. 14 This included no notable increases in diarrhea, alopecia, neutropenia, or liver enzyme elevations.
In light of these findings, vidofludimus calcium may be both effective in treating relapsing multiple sclerosis and potentially have a strong safety and tolerability profile. Here we report the results of a phase 2, multi‐center, double‐blind, randomized‐controlled trial (EMPhASIS) that investigated the safety and efficacy of vidofludimus calcium as compared to placebo in patients with relapsing‐remitting multiple sclerosis.
Methods
Study design and participants
The EMPhASIS trial is a randomized, double‐blind, placebo‐controlled, phase 2 trial conducted at 38 medical centers in Bulgaria, Poland, Romania, and Ukraine (Table S1). The EMPhASIS trial consists of two cohorts: cohort one, which evaluated the efficacy and safety of 30 mg or 45 mg vidofludimus calcium compared to placebo; and cohort two, which evaluates the efficacy and safety of 10 mg vidofludimus calcium compared to placebo. The data presented in this report describe cohort one. Cohort two is currently ongoing, still blinded, and will be published separately.
The doses of 30 and 45 mg vidofludimus calcium were selected based on available in vitro, pharmacokinetic, and clinical data. The IC50 for in vitro inhibition of cytokine release in human lymphocytes was ~5–8 μM, which is approximately equivalent to serum trough concentrations following 20 mg vidofludimus calcium or higher observed in humans at steady‐state. 12 Clinical trial experience in other autoimmune diseases suggested the lowest active dose was between 20 mg and 35 mg. 14 , 15 Repeated doses up to 50 mg vidofludimus calcium were safe and well tolerated in Phase 1 trials. 12
Participants aged 18–55 years with a diagnosis of relapsing‐remitting multiple sclerosis meeting the 2017 McDonald criteria 16 were potentially eligible for the study. Additional inclusion criteria included an Expanded Disability Status Scale (EDSS) score of 0–4 and evidence of multiple sclerosis disease activity, defined as at least two relapses in the last 24 months (or at least one relapse in the last 12 months) before randomization and one or more multiple sclerosis‐related gadolinium‐enhancing brain lesions in the last 6 months before informed consent. Key exclusion criteria were as follows: any multiple sclerosis type other than relapsing‐remitting multiple sclerosis; a relapse within 30 days before screening or during the screening period; a history of nephrolithiasis or an underlying condition with a strong association of nephrolithiasis, or gout; or other clinically significant medical illnesses or laboratory abnormalities. The following treatments had to be discontinued for at least 12 months prior to screening: any intravenous immunoglobulin, mitoxantrone, cytotoxic, or immunosuppressive therapy. Patients were excluded if they had any previous or current use of natalizumab, alemtuzumab, daclizumab, ocrelizumab, anti‐CD4 therapy, rituximab, belimumab, or any DHODH inhibitor (e.g., teriflunomide and leflunomide). A list of the full inclusion and exclusion criteria is provided in Table S2.
The study was approved as required by applicable regulatory authorities and an independent ethics committee. The study was conducted in accordance with the International Council for Harmonization Guideline for Good Clinical Practice (ICH GCP) and the Declaration of Helsinki as well as in keeping with applicable local laws and regulations. A steering committee was established by the funder who monitored the conduct of the trial and regularly reviewed safety data. The steering committee remained blinded throughout the study until database lock; however, unblinded safety analyses could be requested by the steering committee at any time, if necessary. A risk management system in line with the ICH GCP Guideline E6 (R2) was used to specify, track, and control risks associated with the study. This included the evaluation of adverse events and serious adverse events by medical monitors; risk assessment meetings were conducted every 3 months. Ethics oversight was provided by local ethics committees and all participants provided written informed consent before any study‐related procedures were conducted. A placebo was considered ethically justified because access to relapsing MS therapies is limited in the countries where this study was conducted. The study is registered with ClinicalTrials.gov (NCT03846219) and EudraCT (2018–001896‐19).
Randomization and masking
Participants were randomly assigned in a 1:1:1 ratio to receive vidofludimus calcium (30 mg or 45 mg) or matching placebo (with identical appearance, shape, color, labeling, and packaging) using a centralized interactive web response system based on randomization lists provided by an independent, unblinded team of biostatisticians (FGK Clinical Research GmbH). Randomization was stratified by the presence or absence of gadolinium‐positive lesions and magnetic resonance imaging (MRI) field strength.
The study sponsor, trial participants, study physicians, central MRI assessors, and all other personnel directly involved in the conduct of the trial were masked to the treatment assignments during the 24‐week treatment period. To achieve blinding for the evaluation of clinical endpoints, a treating physician oversaw the medical management of each patient and a separate, evaluating physician performed all standardized neurological examinations needed for EDSS. Patients and the treating physicians were instructed to not exchange any treatment‐related information or adverse events with the evaluating physician, who was restricted to speak to the patient for the purposes of the EDSS evaluation only. The evaluating physician did not have access to electronic case report forms, MRI scans, laboratory reports, or any other source documents.
Procedures
Participants took vidofludimus calcium (30 or 45 mg) or placebo once daily in the morning in a fasted state (before breakfast) for 24 weeks. To minimize the risk of changes to uric acid during the first 7 days of dosing, the assigned dose (containing vidofludimus calcium 15 mg, vidofludimus calcium 22.5 mg, or placebo) was taken on days 0–6 and then the full dose thereafter.
Participants underwent standardized brain MRI scans at baseline and every 6 weeks after the first day of dosing until week 24 (Table S3). MRI scans were centrally analyzed by a blinded and independent MRI center. Key clinical assessments included EDSS score (assessed at baseline, days 0 and 6, and weeks 6, 12, 18, and 24) and the Treatment Satisfaction Questionnaire for Medication 17 (assessed at baseline, week 6, and week 24), which were likewise done at any unscheduled visit due to a suspected relapse. In the case of suspected or protocol‐defined confirmed relapse, corticosteroid treatment was offered at the discretion of the investigator. Pharmacokinetic and pharmacodynamic measurements were also collected.
Adverse events were assessed at all scheduled (screening, baseline, day 7, and weeks 6, 12, 18, and 24) and unscheduled clinic visits. Routine clinical chemistries, hematology, liver function tests, and urinalysis were collected at all scheduled visits. Vital signs and electrocardiography were conducted at screening and regularly scheduled intervals. Adverse events of special interest included: red blood cells in urine, hematuria, and retroperitoneal colicky pain with suspected or confirmed nephrolithiasis.
Patients were required to discontinue treatment if at least one of the protocol‐defined criteria for liver events was met: alanine aminotransferase (ALT) or aspartate aminotransferase (AST) greater than eight times the upper limit of normal (ULN); ALT or AST greater than five times the ULN for more than 2 weeks; ALT or AST greater than three times the ULN and total bilirubin great than two times the ULN (or international normalized ratio of greater at 1·5 times the ULN); ALT or AST greater than three times the ULN with concomitant fatigue, nausea, vomiting, right upper quadrant pain or tenderness, fever, rash, or eosinophilia; or indirect bilirubin greater than three times the ULN.
Outcomes
The primary endpoint was the difference in the cumulative number of combined unique active lesions (gadolinium‐enhancing lesions and new/enlarging T2 lesions, without double‐counting the same lesion) up to week 24 between vidofludimus calcium 45 mg and placebo in the intention‐to‐treat population (i.e., weeks 6–24). The key secondary was the same outcome between vidofludimus calcium 30 mg and placebo. A sensitivity analysis of the primary endpoint and key secondary endpoint was done in the per‐protocol population and the modified intention‐to‐treat population. In addition, prespecified subgroup analyses of the primary endpoint and key secondary endpoint in the intention‐to‐treat population were done based on baseline demographics (gender, age, weight, country), MRI characteristics (gadolinium‐enhancing lesions strata), and clinical characteristics (EDSS score, duration of disease, and relapse history).
The intention‐to‐treat population was defined as all randomized patients who received at least 1 dose of placebo or vidofludimus calcium and were analyzed by the groups to which they were randomized. The safety population included all randomized patients who received at least 1 dose of placebo or vidofludimus calcium and was analyzed by the treatment actually assigned. The per‐protocol population was defined as all randomized patients receiving at least 1 dose of placebo or vidofludimus calcium and no violations of major protocol criteria during treatment. Major protocol violations are listed in the Supplementary Material (p. 19). The modified intention‐to‐treat population was defined as all randomized patients receiving at least 1 dose of placebo or vidofludimus calcium and having all MRI scans available for analysis.
Other secondary endpoints included: the difference in the cumulative number of combined unique active lesions up to week 24 between vidofludimus calcium 45 mg and vidofludimus calcium 30 mg; number or cumulative number of gadolinium‐enhancing, T1, or T2 lesions up to 24 weeks; annualized relapse rate from baseline until week 24; proportion of relapse‐free patients up to week 24; time‐to‐first relapse; change in EDSS score from baseline; proportion of patients with EDSS score progression; brain atrophy and other MRI endpoints; and safety and tolerability as assessed by the investigators. The annualized relapse rate was defined as the total number of confirmed relapses divided by the real exposure time in years assessed over the time during the double‐blind treatment period. EDSS progression was defined as an increase compared to baseline of at least 1.0 point for patients with a baseline EDSS score of 1.0 to 4.0 or of at least 1.5 points for patients with a baseline EDSS score of 0. Vidofludimus calcium trough concentrations (weeks 6 and 24) were used to assess the correlation between pharmacokinetics and MRI‐based assessments. The effects of vidofludimus calcium on serum neurofilament (baseline and week 24) were also analyzed.
Statistical analysis
The primary and secondary efficacy endpoints were analyzed using a generalized linear model with a negative binomial distribution and a logarithmic link function. Independent effects included in the model were as follows: treatment group, baseline volume of T2 lesions, MRI field strength, and baseline number of gadolinium‐enhancing lesions. The natural log transformation of the time from the first dose to the date of the last MRI assessment was included as an offset term to account for different lengths of treatment. Rate ratios (and corresponding 95% CIs) were derived from the model to compare the cumulative number of combined unique active lesions between groups. Analysis of the key secondary endpoint was conducted hierarchically to the primary endpoint. Both tests were performed at a significance level of 0·1 (one‐sided).
All other secondary efficacy analyses were reported using descriptive statistics and were considered exploratory. The mean number or cumulative number of gadolinium‐enhancing, T1, or T2 lesions were summarized descriptively and analyzed with a generalized linear model as described earlier but adjusted for MRI field strength and baseline number of gadolinium‐enhancing lesions only. The annualized relapse rate was compared between treatment groups and between the pooled vidofludimus calcium groups and placebo using a generalized linear model adjusted for baseline number of gadolinium‐enhancing lesions, which included the offset term for real exposure time. Time‐to‐first relapse up to 24 weeks was analyzed using a Cox proportional hazards model, with covariates for treatment group and baseline gadolinium‐enhancing lesions strata. The proportion of patients free of relapse was estimated using the Kaplan–Meier method. Statistical analyses were done using SAS version 9.4 (SAS Institute, Cary, NC, USA).
A sample size of 195 patients (65 patients per group) was designed to have 80% power on the primary endpoint assuming a mean rate of cumulative combined unique active lesions after 24 weeks of 4.5 and 8.0 for vidofludimus calcium 45 mg and placebo, respectively. The sample size calculation was based on a one‐side test for the ratio of two negative binomial rates assuming a negative binomial dispersion parameter of 1.7, a 25% withdrawal rate, and a 10% overall type I error rate. No interim analysis was planned or performed.
Role of the funding source
The funder contributed to study design, project oversight, medical oversight, interaction with the steering committee, data interpretation, and writing, review, and approval of the report. The funder contracted with FGK Clinical Research GmbH to gather, analyze, and write the report. Data interpretation was done by the funder and authors. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Of 284 patients screened between Jan 2, 2019 and Oct 4, 2019, 210 were randomized to receive placebo (n = 69), vidofludimus calcium 30 mg (n = 72), or vidofludimus calcium 45 mg (n = 69), one of whom withdrew consent and was not treated (vidofludimus calcium 30 mg group) (Fig. 1). Most of the patients who did not meet eligibility criteria were excluded because they received corticosteroids within 30 days of the screening MRI. Patients were from study centers in Bulgaria (n = 60 [29%]), Poland (n = 26 [12%]), Romania (n = 6 [3%]), and Ukraine (n = 118 [56%]). Two hundred and nine patients received at least 1 dose of placebo or vidofludimus calcium and comprised the intention‐to‐treat population and safety population. 198 (95%) of 209 patients completed the study and completion rates were similar between the placebo (n = 64 [93%]), vidofludimus calcium 30 mg (n = 69 [97%]), and vidofludimus calcium 45 mg (n = 65 [94%]) groups. In total, 11 (5%) of 209 patients discontinued the study treatment prematurely, which consisted of five (7%) patients assigned placebo, two (3%) assigned vidofludimus calcium 30 mg, and four (6%) assigned vidofludimus calcium 45 mg. Five discontinuations were attributable to treatment‐emergent adverse events in the placebo (n = 3 [4%]) and vidofludimus calcium (n = 2 [1%]) groups. The per‐protocol population consisted of 188 (90%) of 209 patients.
Figure 1.
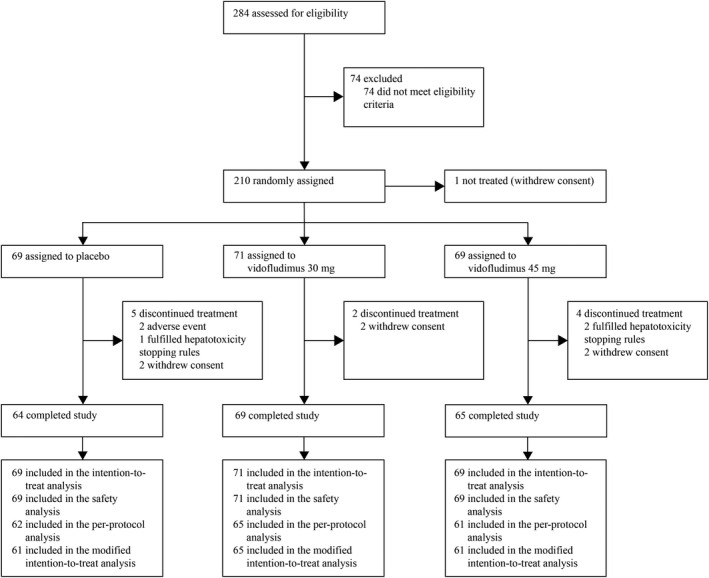
Trial profile.
Overall, baseline patient demographics, clinical, and MRI characteristics were comparable between groups although the vidofludimus calcium 30 mg group consisted of slightly fewer females (Table 1). 129 (62%) of 209 patients had no prior exposure to disease‐modifying therapy.
Table 1.
Baseline patient demographics, clinical and MRI characteristics (intention‐to‐treat population).
|
Placebo (n = 69) |
Vidofludimus calcium 30 mg (n = 71) |
Vidofludimus calcium 45 mg (n = 69) | |
|---|---|---|---|
| Age (years) | 37 (21–55) | 38 (18–55) | 36 (21–51) |
| Female | 46 (67%) | 40 (56%) | 50 (73%) |
| Race | |||
| White | 69 (100%) | 71 (100%) | 69 (100%) |
| Previous exposure to disease‐modifying drugs 1 | |||
| No exposure | 45 (65%) | 43 (61%) | 41 (59%) |
| Interferon or glatiramer acetate | 14 (20%) | 11 (15%) | 19 (28%) |
| Oral drugs | 10 (15%) | 17 (24%) | 8 (12%) |
| Biologics (antibodies) | 0 | 0 | 1 (1%) |
| Duration of disease (years) 2 | 3.61 (0.04–26.5) | 3.58 (0.02–24.6) | 3.49 (0.01–22.8) |
| EDSS score | 2.73 (0.90) | 2.65 (0.83) | 2.56 (0.96) |
| Number of relapses in the last 24 months | |||
| 1 | 35 (51%) | 32 (45%) | 35 (51%) |
| 2 | 25 (36%) | 28 (39%) | 28 (41%) |
| ≥3 | 9 (13%) | 11 (15%) | 6 (9%) |
| Gadolinium‐positive lesions | |||
| 0 | 33 (48%) | 39 (55%) | 33 (48%) |
| ≥1 | 36 (52%) | 32 (45%) | 36 (52%) |
| Number of gadolinium‐positive lesions | 1.2 (2.1) | 1.4 (2.4) | 0.9 (13) |
| Volume of T2 lesions per patient (cm3) | 11.980 (10.417) | 13.341 (15.079) | 13.918 (12.946) |
Data are n (%), mean (SD), or median (range). EDSS = Expanded Disability Status Scale.
Last treatment line.
Duration since time at which the diagnosis of multiple sclerosis was first documented.
In the primary analysis, the adjusted mean cumulative number of combined unique active lesions up to 24 weeks was 6.4 (95% CI 2.8–13.9) with placebo and 2.4 (95% CI 1.1–4.9) with vidofludimus calcium 45 mg (62% reduction; rate ratio 0.38, 95% CI 0.22–0.64; p = 0.0002; Table 2). The key secondary outcome was 13.2 (95% CI 6.6–26.4) with placebo and 4.0 (95% CI 2.2–7.2) with vidofludimus calcium 30 mg (70% reduction; rate ratio 0.30, 95% CI 0.17–0.53; p < 0.0001). Consistent results were observed in a pre‐planned sensitivity analysis of the per‐protocol population and the modified intention‐to‐treat population (Table S4). Prespecified univariate subgroup analyses also found fewer adjusted mean cumulative number of combined unique active lesions up to 24 weeks in either vidofludimus calcium groups as compared to placebo in most subgroups tested, although significance analysis was not conducted because they were exploratory outcomes (Fig. S1). No notable difference in the adjusted mean cumulative number of combined unique active lesions up to 24 weeks was observed between vidofludimus calcium 30 and 45 mg.
Table 2.
Cumulative number of combined unique active lesions up to 24 weeks (intention‐to‐treat population).
| Comparison | Cumulative CUA lesions | Rate ratio | 95% CI | p‐value |
|---|---|---|---|---|
| Primary endpoint (45 mg vs. placebo) | ||||
| Placebo (n = 69) | 6.3 (2.8–13.9) | – | – | – |
| Vidofludimus calcium 45 mg (n = 69) | 2.4 (1.1–4.9) | 0.38 | 0.22–0.64 | 0.0002 |
| Key secondary endpoint (30 mg vs. placebo) | ||||
| Placebo (n = 69) | 13.2 (6.6–26.4) | – | – | – |
| Vidofludimus calcium 30 mg (n = 71) | 4.0 (2.2–7.2) | 0.30 | 0.17–0.53 | <0.0001 |
| Secondary endpoint (45 mg vs. 30 mg) | ||||
| Vidofludimus calcium 45 mg (n = 69) | 4.4 (2.4–8.1) | ·· | ·· | ·· |
| Vidofludimus calcium 30 mg (n = 71) | 4.2 (25–7.1) | 0.96 | 0.56–1.63 | nc |
Data are adjusted mean (95% CI). CUA = combined unique active. nc = not calculated.
For comparison between treatment arms, the number of cumulative CUA lesions were adjusted for baseline T2 lesion volume, MRI field strength (1.5 or 3.0 Tesla), and baseline number of Gd+ lesions (0, ≥1) using a generalized linear model with a negative binomial distribution and a logarithmic link function. This ensured that these baseline and imaging factors did not influence inter‐group comparisons. Such adjustments lead to different number of lesions for treatment arms displayed in different analyses depending on the comparator used.
Lower adjusted mean cumulative number of combined unique active lesions up to 24 weeks in either vidofludimus calcium groups compared to placebo was evident as early as week 6 and continued through week 24 (Fig. 2A). This observation was similar when assessing the adjusted mean cumulative number of gadolinium‐enhancing lesions up to 24 weeks (Fig. 2B). The cumulative number of new gadolinium‐enhancing lesions at week 24 was 13·0 (95% CI: 6.8–24.5) with placebo, 4.5 (95% CI: 2.7–7.7) with vidofludimus calcium 30 mg, and 4.0 (95% CI: 2.1–7.8) with vidofludimus calcium 45 mg. The adjusted mean annualized relapse rate was 0.53 (95% CI: 0.32–0.89) with placebo, 0.39 (95% CI: 0.22–0.69) with vidofludimus calcium 30 mg, and 0.48 (95% CI: 0.28–0.82) with vidofludimus calcium 45 mg. The rate ratio between vidofludimus calcium 30 mg versus placebo and vidofludimus calcium 45 mg versus placebo was 0.72 (95% CI: 0.34–1.55) and 0.90 (95% CI: 0.44–1.86), respectively. The hazard ratio for relapse up to 24 weeks between vidofludimus calcium 30 mg versus placebo and vidofludimus calcium 45 mg versus placebo was 0.58 (95% CI: 0.26–1.29) and 0.78 (95% CI: 0.37–1.59), respectively. Median change in neurofilament light chain in serum from baseline to week 24 was 6.5%, −17.0%, and −20.5% for placebo, vidofludimus calcium 30 mg, and vidofludimus calcium 45 mg, respectively. All other secondary efficacy outcomes are provided in the Supplementary Material.
Figure 2.
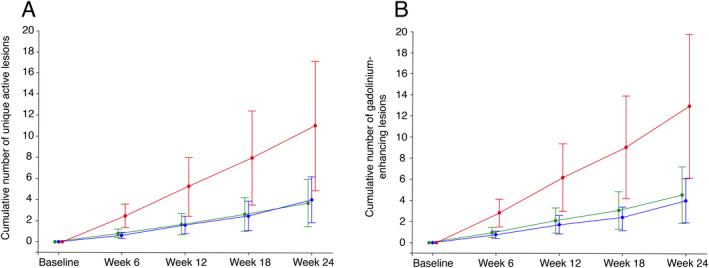
Cumulative number of combined unique active lesions (A) and gadolinium‐enhancing lesions (B) from baseline to Week 24 (intention‐to‐treat population). Red: placebo (n = 69); green: vidofludimus calcium 30 mg (n = 71); blue: vidofludimus calcium (n = 69). Data are presented as adjusted mean with the upper and lower limits of the 95% CIs. MRI = magnetic resonance imaging. Estimates were adjusted for baseline volume of T2 lesions, MRI field strength (1.5 or 3.0 Tesla), and baseline number of gadolinium‐enhancing lesions (0 or ≥1) using a generalized linear model with a negative binomial distribution and a logarithmic link function. [Colour figure can be viewed at wileyonlinelibrary.com]
Both doses of vidofludimus calcium were well tolerated. A similar proportion of patients experienced at least one treatment‐emergent adverse event in the placebo (44%), vidofludimus calcium 30 mg (45%), and vidofludimus calcium 45 mg (41%) groups, which were predominantly mild or moderate in severity (Table 3). Alopecia, fatigue, rash, and cystitis were treatment‐emergent adverse events that occurred in greater than 1% of vidofludimus calcium‐treated patients that were not recorded in placebo‐treated patients (1%–4%). Serious adverse events were low (0%–3%). Three patients experiencing four serious adverse events occurred in the vidofludimus calcium 30 mg (n = 2) and the placebo group (n = 1) and were considered unrelated to treatment: hydronephrosis and ureterolithiasis (vidofludimus calcium 30 mg), open fracture (vidofludimus calcium 30 mg), and squamous cell carcinoma of the cervix (placebo). The two most common treatment‐emergent adverse events, headache, and nasopharyngitis, occurred with similar frequency between the placebo and either vidofludimus calcium groups. No remarkable cardiovascular, hematological, infectious, or malignancy‐related adverse events were noted. Adverse events of special interest occurred in one (1%) patient treated with placebo (hematuria) and one (1%) patient treated with any dose of vidofludimus calcium (ureterolithiasis). Renal treatment‐emergent events occurred in one (1%) patient treated with placebo and three (2%) patients treated with any dose of vidofludimus calcium. No changes in serum uric acid were observed in any treatment group over 24 weeks. No deaths occurred during the trial.
Table 3.
Summary of treatment‐emergent adverse events (safety population).
|
Placebo (n = 69) |
Vidofludimus calcium 30 mg (n = 71) | Vidofludimus calcium 45 mg (n = 69) | |
|---|---|---|---|
| Any treatment‐emergent adverse event 1 | 30 (44%) | 32 (45%) | 28 (41%) |
| Number of treatment‐emergent adverse events | 62 | 69 | 59 |
| Treatment‐emergent adverse events occurring in >5% of total patients by preferred term 2 | |||
| Headache | 4 (6%) | 3 (4%) | 4 (6%) |
| Nasopharyngitis | 3 (4%) | 3 (4%) | 5 (7%) |
| Treatment‐emergent adverse events occurring in 2%–5% of total patients by preferred term 2 | |||
| Upper respiratory tract infection | 3 (4%) | 2 (3%) | 0 |
| Viral respiratory tract infection | 3 (4%) | 0 | 2 (3%) |
| Treatment‐emergent adverse events occurring in >1 to <2% of total patients by preferred term 2 | |||
| Back pain | 2 (3%) | 1 (1%) | 0 |
| ALT increased | 2 (3%) | 1 (1%) | 0 |
| Influenza | 2 (3%) | 0 | 1 (1%) |
| Liver enzymes increased | 1 (1%) | 1 (1%) | 2 (3%) |
| Nausea | 1 (1%) | 1 (1%) | 2 (3%) |
| Bronchitis | 1 (1%) | 0 | 2 (3%) |
| Alopecia | 0 | 3 (4%) | 1 (1%) |
| Fatigue | 0 | 2 (3%) | 2 (3%) |
| Rash | 0 | 2 (3%) | 2 (3%) |
| Cystitis | 0 | 1 (1%) | 3 (4%) |
| Treatment‐emergent adverse events by severity | |||
| Mild | 23 (33%) | 29 (41%) | 21 (30%) |
| Moderate | 8 (12%) | 11 (16%) | 15 (23%) |
| Severe | 1 (1%) | 0 | 0 |
| Serious adverse events | 1 (1%) | 2 (3%) | 0 |
| Treatment discontinuation for any reason | 5 (7%) | 2 (3%) | 4 (6%) |
| Treatment‐emergent adverse events leading to treatment discontinuation | 3 (4%) | 0 | 2 (3%) |
ALT = alanine aminotransferase.
Treatment‐emergent adverse events were defined as any event not present prior to the first dose of placebo or vidofludimus calcium or any event already present that worsened in either intensity or frequency following treatment.
Patients were counted only once by preferred term.
Hepatic treatment‐emergent adverse events were recorded in three (4%) patients treated with placebo and in six (4%) patients treated with any dose of vidofludimus calcium. Rises in ALT or AST greater than five times the ULN occurred in two (3%) patients with placebo, one (1%) patient with vidofludimus calcium 30 mg, and three (4%) patients with vidofludimus calcium 45 mg. Of the five patients who discontinued treatment due to treatment‐emergent adverse events, one in the placebo group and two in the vidofludimus calcium 45 mg group met hepatotoxicity stopping rules. One additional patient in the placebo group experienced an ALT increase of greater than eight times the ULN and was discontinued at the investigator's discretion before hepatotoxicity stopping rules were applied. All occurrences were mild or moderate, did not occur with clinically significant increases in bilirubin, and resolved following treatment discontinuation.
Discussion
A once‐daily oral dose of vidofludimus calcium 30 or 45 mg resulted in fewer combined unique active MRI lesions over 24 weeks when compared with placebo. Suppression of MRI lesions was evident by week 6 and continued through week 24. Pre‐specified subgroup analyses found a similar treatment effect for both doses of vidofludimus calcium irrespective of the subgroups assessed, although these findings should be interpreted with caution due to the limited sample size. Other MRI outcomes, including the cumulative number of T1, T2, and gadolinium‐enhancing lesions up to 24 weeks favored vidofludimus calcium and were consistent with study's primary findings.
Both doses of vidofludimus calcium were well‐tolerated; zero (0%) and two (3%) patients treated with vidofludimus calcium 30 and 45 mg, respectively, discontinued treatment due to a treatment‐emergent adverse event as compared to three (4%) patients in the placebo group. The incidence of the two most common treatment‐emergent adverse events—nasopharyngitis and headache—was low and similar between vidofludimus calcium‐ and placebo‐treated patients. Moreover, no increased incidence in liver, renal, or infection‐related treatment‐emergent adverse events was observed compared to placebo. No changes in hematologic laboratory values, as measured by neutrophil, lymphocyte, and leukocyte count, were observed with vidofludimus calcium. This observation is consistent with the mechanism of action of vidofludimus calcium and its biological selectivity toward activated T and B cells with high metabolic requirements for DHODH‐mediated pyrimidine synthesis.
Trends favoring vidofludimus calcium were noted in clinical outcomes (i.e., annualized relapse rate) and clinically relevant biomarkers (i.e., neurofilament light chain), although the study was not powered for these outcomes. Coupled with the short study duration and relatively low sample size, drawing definitive conclusions on non‐MRI outcomes in this context is limited. Longer studies in a larger patient population are needed to demonstrate the effect of vidofludimus calcium on clinical outcomes. No notable differences between vidofludimus calcium 30 and 45 mg on MRI outcomes were evident and both doses of vidofludimus calcium had a comparable safety profile. The reason for the lack of measurable difference between doses remains unclear and suggests that evaluation of a lower dose may be helpful. A second cohort in the EMPhASIS trial is currently ongoing and will evaluate vidofludimus calcium 10 mg versus placebo.
Vidofludimus calcium is being developed with the goal to demonstrate robust efficacy comparable to other first‐line disease‐modifying therapies while also being safe and well‐tolerated. Indeed, the 62%–70% reduction in combined unique active lesions compared to placebo observed in the EMPhASIS trial is comparable to that seen in many other phase 2 trials of disease‐modifying therapies approved for multiple sclerosis. 18 , 19 , 20 , 21 , 22 , 23 Safety liabilities associated with available therapies, including lymphopenia (teriflunomide, 5 , 6 , 20 dimethyl fumarate, 24 , 25 and sphingosine‐1‐phosphate [S1P] inhibitors 18 , 19 , 26 ), increased liver enzymes (teriflunomide 5 , 6 , 20 and S1P inhibitors 19 , 26 ), and risk of infections (S1P inhibitors and CD20 inhibitors 26 , 27 ), are well documented in clinical trials and typically require ongoing monitoring during treatment. 26 In the TOWER and TEMSO trials investigating teriflunomide, ALT increases occurred in 11–14% and diarrhea in 11–18% of teriflunomide‐treated patients, which was higher than the placebo arm. 5 , 6 This contrasts with the present study which found that ALT increases in 1% and diarrhea in 0% of vidofludimus‐treated patients, indicating a potential safety advantage. This difference may be because vidofludimus calcium is a selective DHODH inhibitor without off‐target effects on kinases and thus spares generalized effects such as immunosuppression. 12 , 14 The absence of other safety differences between placebo‐ and vidofludimus calcium‐treated patients as measured by hepatic adverse events (4% vs. 4%, respectively), renal adverse events (1% and 2%, respectively), infections, and white blood cell counts are consistent with a favorable safety profile, although a further study in larger, longer phase 3 trials is needed.
In conclusion, vidofludimus calcium suppressed the development of new MRI lesions and demonstrated a favorable safety and tolerability profile. Based on these positive findings, confirmatory phase 3 trials are underway.
Authors’ Contributions
RJF, AM, CW, and NdS contributed to the study concept and study design. RJF was the coordinating investigator. VG, KR, PSB, NT, IS, and other study investigators (Table S1) acquired the data. AM contributed to the statistical analysis. JS contributed to safety analysis, medical monitoring, and medical oversight. AM and CW contributed to data analysis and data interpretation. AM completed and signed the clinical study report. All authors directed the content and editing of the report (the initial draft was supported by a medical writer) and approved the final version of the report. All authors had full access to the data and affirm the integrity of the data and analyses.
Conflict of Interest
RJF reports personal fees from AB Science, Biogen, Celgene, EMD Serono, Genentech, Genzyme, Immunic AG, Janssen Novartis, Sanofi, and TG Therapeutics; clinical trial contracts from Biogen, Novartis, and Sanofi.
HW reports grants and personal fees from AbbVie, Biogen, Merck Serono, and Sanofi Genzyme; personal fees from Actelion, Alexion, Evgen, F. Hoffmann‐La Roche, Gemeinnützige Hertie‐Stiftung, Immunic, Lundbeck, MedDay Pharmaceuticals, Novartis, Roche Pharma AG, Teva, and WebMD Global; and grants from Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, European Union, Fresenius Foundation, German Ministry for Education and Research (BMBF), GlaxoSmithKline, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, NRW Ministry of Education and Research, PML Consortium, RE Children's Foundation, and Swiss MS Society, outside the submitted work.
CW is a partner at Lycalis sprl and reports compensation for his organization for consulting from BMS, Celgene, Desitin, Immunic, Merck KGaA, Novartis, Roche, Synthon, Teva, and Viatris; and for speaking from Synthon and Viatris.
NdS has received honoraria from Biogen‐ Idec, Genzyme, Immunic, Merck, Novartis, Roche, Celgene and Teva for consulting services, speaking, and travel support. He serves on advisory boards for Merck, Novartis, Biogen‐Idec, Immunic, Roche, and Genzyme, and he has received research grant support from the Italian MS Society.
JS received honoraria for participation in advisory boards, consultancy, and lecturing from Biogen, BMS, Immunic, Merck, Novartis, Roche, and Sanofi.
VG: reports grants/personal fees from Medpace, PPD, PRA Health Sciences, PSI, Roche, Sanofi, and Verum.
KR is the president elect of the Polish Neurological Society.
PSB reports no declaration of interests.
NT reports no declaration of interests.
IS reports no declaration of interests.
AM is a shareholder and employee of trial sponsor, and a holder of patents for the drug under investigation.
Supporting information
Table S1 EMPhASIS study investigators.
Table S2. Study inclusions and exclusion criteria.
Table S3. Schedule of assessments.
Table S4. Sensitivity analysis on the cumulative number of combined unique active lesions up to 24 weeks.
Table S5. Cumulative number of combined unique active lesions at weeks 6–24 (1.5T only).
Table S6. Median percentage change from baseline in T2 and T1 lesion load at weeks 6, 12, 18, and 24 (intention‐to‐treat population).
Table S7. Adjusted mean cumulative number of gadolinium‐enhancing, T2, and T1 lesions up to weeks 6, 12, 18, and 24 (intention‐to‐treat population).
Table S8. Proportion of patients with or without new lesions at 24 weeks (intention‐to‐treat population).
Table S9. Proportion of relapse‐free patients and hazard ratio up to 24 weeks (intention‐to‐treat population).
Table S10. Absolute change in EDSS from baseline (intention‐to‐treat population).
Table S11. Proportion of patients with unconfirmed EDSS progression (intention‐to‐treat population).
Figure S1. Cumulative number of combined unique active MRI lesions based on prespecified demographic, clinical, and MRI subgroup analyses of 45 mg vidofludimus calcium versus placebo (A) and 30 mg vidofludimus calcium versus placebo (B).
Acknowledgments
We thank all investigators, study personnel, and patients as well as their families who participated in the EMPhASIS trial. The study was funded by Immunic AG (Gräfelfing, Germany). We also thank Jason Slizgi, Ph.D. (Los Angeles, CA, USA) for preparing the initial draft and revisions of the manuscript during peer review, under the authors' direction, and who was funded by Immunic AG.
Funding Information
The study was funded by Immunic AG (Gräfelfing, Germany).
Funding Statement
This work was funded by Immunic AG.
Data Availability Statement
Data will be shared with qualified researchers who submit a research proposal following approval by an independent review board and a signed data sharing agreement. Requests for data can be made 6 months after the indication studied has been approved in the United States and Europe with no expiration date on requests. De‐identified data, including the study protocol, statistical analysis plan, clinical study report, and case report forms will be provided in a secure sharing environment.
References
- 1. Rückemann K, Fairbanks LD, Carrey EA, et al. Leflunomide inhibits pyrimidine de novo synthesis in mitogen‐stimulated T‐lymphocytes from healthy humans. J Biol Chem. 1998;273(34):21682‐21691. [DOI] [PubMed] [Google Scholar]
- 2. Löffler M, Klein A, Hayek‐Ouassini M, Knecht W, Konrad L. Dihydroorotate dehydrogenase mRNA and protein expression analysis in Normal and drug‐resistant cells. Nucleosides Nucleotides Nucleic Acids. 2004;23:1281‐1285. [DOI] [PubMed] [Google Scholar]
- 3. Jameson SC. Maintaining the norm: T‐cell homeostasis. Nat Rev Immunol. 2002;2(8):547‐556. [DOI] [PubMed] [Google Scholar]
- 4. Fairbanks LD, Bofill M, Ruckemann K, Simmonds HA. Importance of ribonucleotide availability to proliferating T‐lymphocytes from healthy humans: disproportionate expansion of pyrimidine pools and contrasting effects of de novo synthesis inhibitors. J Biol Chem. 1995;270(50):29682‐29689. [PubMed] [Google Scholar]
- 5. O'Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of Oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293‐1303. [DOI] [PubMed] [Google Scholar]
- 6. Confavreux C, O'Connor P, Comi G, et al. Oral teriflunomide for patients with relapsing multiple sclerosis (TOWER): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Neurol. 2014;13(3):247‐256. [DOI] [PubMed] [Google Scholar]
- 7. Teriflunomide [package insert]. Genzyme Corporation; 2021. [Google Scholar]
- 8. Phase 2 Dose‐finding IMU‐838 for Ulcerative Colitis (CALDOSE‐1). ClinicalTrials.gov Identifier: NCT03341962. February 24, 2021. Accessed March 21, 2019. https://clinicaltrials.gov/ct2/show/NCT03341962
- 9. Vidofludimus Calcium for Primary Sclerosing Cholangitis (PSC). ClinicalTrials.gov Identifier: NCT03722576. November 25, 2020. Accessed March 21, 2019. https://clinicaltrials.gov/ct2/show/NCT03722576
- 10. A study to evaluate the efficacy, safety and tolerability of IMU‐838 as addition to investigator's choice of standard of care therapy, in patients with coronavirus disease 19 (COVID‐19). ClinicalTrials.gov Identifier: NCT04379271. July 1, 2020.
- 11. Hahn F, Wangen C, Häge S, et al. IMU‐838, a developmental DHODH inhibitor in phase II for autoimmune disease, shows anti‐SARS‐CoV‐2 and broad‐Spectrum antiviral efficacy in vitro. Viruses. 2020;12(12):1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Muehler A, Kohlhof H, Groeppel M, Vitt D. Safety, tolerability and pharmacokinetics of Vidofludimus calcium (IMU‐838) after single and multiple ascending Oral doses in healthy male subjects. Eur J Drug Metab Pharmacokinet. 2020;45(5):557‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Muehler A, Peelen E, Kohlhof H, Gröppel M, Vitt D. Vidofludimus calcium, a next generation DHODH inhibitor for the treatment of relapsing‐remitting multiple sclerosis. Mult Scler Relat Disord. 2020;43:102129. [DOI] [PubMed] [Google Scholar]
- 14. Muehler A, Kohlhof H, Groeppel M, Vitt D. The selective Oral immunomodulator Vidofludimus in patients with active rheumatoid arthritis: safety results from the COMPONENT study. Drugs R D. 2019;19(4):351‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herrlinger KR, Diculescu M, Fellermann K, et al. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: the ENTRANCE study. J Crohns Colitis. 2013;7(8):636‐643. [DOI] [PubMed] [Google Scholar]
- 16. Thompson AJ, Banwell BL, Barkhof F, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018;17(2):162‐173. [DOI] [PubMed] [Google Scholar]
- 17. Atkinson MJ, Sinha A, Hass SL, et al. Validation of a general measure of treatment satisfaction, the treatment satisfaction questionnaire for medication (TSQM), using a national panel study of chronic disease. Health Qual Life Outcomes. 2004;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1‐phosphate receptor modulator ozanimod in relapsing multiple sclerosis (RADIANCE): a randomised, placebo‐controlled, phase 2 trial. Lancet Neurol. 2016;15(4):373‐381. [DOI] [PubMed] [Google Scholar]
- 19. Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124‐1140. [DOI] [PubMed] [Google Scholar]
- 20. O'Connor PW, Li D, Freedman MS, et al. A phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66(6):894‐900. [DOI] [PubMed] [Google Scholar]
- 21. Kappos L, Gold R, Miller DH, et al. Efficacy and safety of oral fumarate in patients with relapsing‐remitting multiple sclerosis: a multicentre, randomised, double‐blind, placebo‐controlled phase IIb study. Lancet. 2008;372(9648):1463‐1472. [DOI] [PubMed] [Google Scholar]
- 22. Selmaj K, Li DKB, Hartung HP, et al. Siponimod for patients with relapsing‐remitting multiple sclerosis (BOLD): an adaptive, dose‐ranging, randomised, phase 2 study. Lancet Neurol. 2013;12(8):756‐767. [DOI] [PubMed] [Google Scholar]
- 23. Comi G, Filippi M, Wolinksy J; European/Canadian Gla . European/Canadian multicenter, double‐blind, randomized, placebo‐controlled study of the effects of glatiramer acetate on magnetic resonance imaging‐‐measured disease activity and burden in patients with relapsing multiple sclerosis. Ann Neurol. 2001;49(3):290‐297. [PubMed] [Google Scholar]
- 24. Gold R, Kappos L, Arnold DL, et al. Placebo‐controlled phase 3 study of Oral BG‐12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098‐1107. [DOI] [PubMed] [Google Scholar]
- 25. Fox RJ, Miller DH, Phillips JT, et al. Placebo‐controlled phase 3 study of Oral BG‐12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087‐1097. [DOI] [PubMed] [Google Scholar]
- 26. Jalkh G, Abi Nahed R, Macaron G, Rensel M. Safety of newer disease modifying therapies in multiple sclerosis. Vaccine. 2021;9(1):1‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luna G, Alping P, Burman J, et al. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 2020;77(2):184‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 EMPhASIS study investigators.
Table S2. Study inclusions and exclusion criteria.
Table S3. Schedule of assessments.
Table S4. Sensitivity analysis on the cumulative number of combined unique active lesions up to 24 weeks.
Table S5. Cumulative number of combined unique active lesions at weeks 6–24 (1.5T only).
Table S6. Median percentage change from baseline in T2 and T1 lesion load at weeks 6, 12, 18, and 24 (intention‐to‐treat population).
Table S7. Adjusted mean cumulative number of gadolinium‐enhancing, T2, and T1 lesions up to weeks 6, 12, 18, and 24 (intention‐to‐treat population).
Table S8. Proportion of patients with or without new lesions at 24 weeks (intention‐to‐treat population).
Table S9. Proportion of relapse‐free patients and hazard ratio up to 24 weeks (intention‐to‐treat population).
Table S10. Absolute change in EDSS from baseline (intention‐to‐treat population).
Table S11. Proportion of patients with unconfirmed EDSS progression (intention‐to‐treat population).
Figure S1. Cumulative number of combined unique active MRI lesions based on prespecified demographic, clinical, and MRI subgroup analyses of 45 mg vidofludimus calcium versus placebo (A) and 30 mg vidofludimus calcium versus placebo (B).
Data Availability Statement
Data will be shared with qualified researchers who submit a research proposal following approval by an independent review board and a signed data sharing agreement. Requests for data can be made 6 months after the indication studied has been approved in the United States and Europe with no expiration date on requests. De‐identified data, including the study protocol, statistical analysis plan, clinical study report, and case report forms will be provided in a secure sharing environment.