

Abstract
Background
Recessive mutations in SLC12A6 have been linked to hereditary motor sensory neuropathy with agenesis of the corpus callosum. Patients with early‐onset peripheral neuropathy associated with SLC12A6 heterozygous variants were reported in 2016. Only five families and three variants have been reported to date, and the spectrum is unclear. Here, we aim to describe the clinical and mutation spectra of SLC12A6‐related Charcot–Marie–Tooth (CMT) disease in Japanese patients.
Methods
We extracted SLC12A6 variants from our DNA microarray and targeted resequencing data obtained from 2598 patients with clinically suspected CMT who were referred to our genetic laboratory by neurological or neuropediatric departments across Japan. And we summarized the clinical and genetic features of these patients.
Results
In seven unrelated families, we identified one previously reported and three novel likely pathogenic SLC12A6 heterozygous variants, as well as two variants of uncertain significance. The mean age of onset for these patients was 17.5 ± 16.1 years. Regarding electrophysiology, the median motor nerve conduction velocity was 39.6 ± 9.5 m/sec. For the first time, we observed intellectual disability in three patients. One patient developed epilepsy, and her brain MRI revealed frontal and temporal lobe atrophy without changes in white matter and corpus callosum.
Conclusions
Screening for the SLC12A6 gene should be considered in patients with CMT, particularly those with central nervous system lesions, such as cognitive impairment and epilepsy, regardless of the CMT subtype.
Introduction
Charcot–Marie–Tooth (CMT) disease is the most common type of inherited peripheral neuropathy, with a common phenotype of progressive sensory‐motor or motor neuropathy, foot deformity, and altered tendon reflexes. The prevalence of CMT ranges from 9.7/100,000 to 82.3/100,000. 1 CMT is categorized into the demyelinating type and axonal type in accordance with the motor nerve conduction velocity (MNCV). Generally, CMT with a slow MNCV (median MNCV <38 m/sec) is classified as CMT1 (autosomal dominant inheritance) and CMT4 (autosomal recessive inheritance), while CMT without a slow MNCV (median MNCV >38 m/sec) is classified as CMT2. There is an intermediate type of CMT, with the MNCV value between CMT1 and CMT2 (median MNCV; 25–45 m/sec). 2 To date, more than 100 genes have been associated with CMT and related disorders (https://neuromuscular.wustl.edu/). Despite presenting with varying clinical phenotypes, both dominant and recessive mutations of multiple genes, such as MFN2, GDAP1, and MME, have been specifically linked to peripheral neuropathy. 3 , 4 , 5 , 6 , 7 , 8
The SLC12A (solute carrier family 12) family encodes K+‐Cl− co‐transporters (KCCs), and SLC12A6 encodes KCC3. KCCs play crucial roles in cell volume homeostasis, epithelial transport, and neuronal excitability. 9 Bi‐allelic SLC12A6 mutations are known to cause severe peripheral neuropathy with agenesis of the corpus callosum. 10 In 2016, a heterozygous de novo p. Thr991Ala variant in SLC12A6 was identified in a child with severe and progressive peripheral neuropathy via whole‐exome sequencing. 11 The mechanism of neuropathy due to SLC12A6 heterozygous variants was predicted to be gain‐of‐function changes in KCC3, resulting in abnormal cell volume homeostasis. 11 Additional reports of sensorimotor neuropathy caused by SLC12A6 heterozygous variants have recently been reported from Germany and China. 12 , 13
In this study, we analyze the comprehensive genetic data collection from a large‐scale case series of Japanese patients with CMT and identified heterozygous SLC12A6 mutations in multiple families.
Materials and Methods
Patients
We analyzed DNA samples from 2598 patients with clinically diagnosed CMT across Japan between 2007 and 2021. Before enrollment in this study, all patients with demyelinating CMT were confirmed negative for PMP22 duplication/deletion using fluorescence in situ hybridization or multiplex ligation probe amplification. Mutation screening was performed in chronological order using DNA microarray (419 patients), Illumina MiSeq (438 patients), and Ion Proton (1741 patients). SLC12A6 was included as a target gene in all aforementioned genetic analyses. Figure 1 depicts the flowchart of our study.
Figure 1.
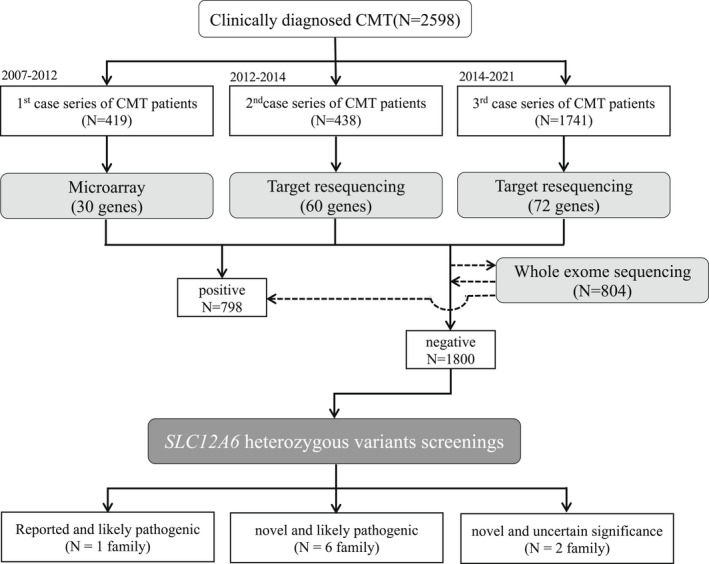
Flow chart of our study. Using DNA microarray and target resequencing, we analyzed 2598 patients with clinically diagnosed CMT, and extracted all SLC12A6 (NM_133647) variants from the 1800 test‐negative patients. CMT, Charcot–Marie–Tooth.
Genomic DNA extraction
We extracted genomic DNA from peripheral blood or saliva using a Gentra Puregene Blood Kit (QIAGEN, Valencia, CA) or Oragene DNA self‐collection kit (DNA Genotek, Ottawa, Ontario, Canada) according to the manufacturer's instructions.
Microarray chip sequencing
For the initial 419 patients enrolled between 2007 and 2012, we performed mutation screening of 30 genes using a customized MyGeneChip® CustomSeq® Resequencing Array (Affymetrix, Inc., Santa Clara, CA). The detailed procedure has previously been described. 14
Targeted resequencing
Since 2012, mutation screening has been conducted to target 60 and 72 known/candidate CMT‐related genes using Illumina Miseq (Illumina Inc., San Diego, CA) and Ion Proton (ThermoFisher Scientific, Inc., Waltham, MA), respectively. The detailed sequencing and analysis methods have previously been described. 15
Data analysis and variant interpretation
We extracted all SLC12A6 (NM_133647) variants from all aforementioned studies and compared them to a global control database (gnomAD; https://gnomad.broadinstitute.org), a Japanese control database (jMorp; https://jmorp.megabank.tohoku.ac.jp/202109/), and our in‐house control database. Furthermore, we performed six lines of computational analysis, namely, SIFT/PROVEAN(http://provean.jcvi.org/index.php), PolyPhen‐2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Assessor (http://mutationassessor.org/r3/), FATHMM (http://fathmm.biocompute.org.uk), and Condel (https://bbglab.irbbarcelona.org/fannsdb/), to predict the pathogenicity of these variants. Protein stability analysis was also performed using DynaMut (http://biosig.unimelb.edu.au/dynamut/) and iMutant (https://folding.biofold.org/i‐mutant/i‐mutant2.0.html). All variants were validated using Sanger sequencing and interpreted according to ACMG‐AMP (American College of Medical Genetics and Genomics and the Association for Molecular Pathology) standards and guidelines. 16
Ethics statement
This study was approved by the Institutional Review Board of Kagoshima University. All patients and family members provided informed consent for participation in the study and genetic analysis.
Results
Clinical findings
Table 1 summarizes the clinical and electrophysiological findings of seven cases with likely pathogenic SLC12A6 heterozygous variants. The families are depicted in Figure 2A. Their CMT neuropathy scores (CMTNS) are shown in Table S1, and the clinical and electrophysiological features of two cases with a variant of uncertain significance are shown in Data S1 and Figure 1.
Table 1.
Clinical and electrophysiological features of current and reported CMT patient with SLC12A6 heterozygous variants.
| Kahle et al. (2016) | Park et al. (2019) | Shi et al. (2021) | This report | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 1 | Patient 2 | Patient 3 | Patient 1 | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | |
| Mutation | c.2971A>G | c.620G>A | c.620G>A | c.2036A>G | c.620G>A | c.620G>A | c.865G>A | c.865G>A | c.1731_1733del | c.1731_1733del | c.1731_1733del | c.2036A>C |
| p. T991A | p.R207H | p.R207H | p.Y679C | p.R207H | p.R207H | p.E289L | p.E289L | p.F578del | p.F578del | p.F578del | p.Y679S | |
| Ethnicity | American | German | German | German | Chinesse | Japanese | Japanese | Japanese | Japanese | Japanese | Japanese | Japanese |
| Onset age | 9 month | 1–2 y.o | 1–2 y.o | 1–2 y.o | 27 y.o | 1 y.o | 11 y.o | 10 y.o | 19 y.o | 40 y.o | 39 y.o | 3 y.o |
| Exam age | 10 y.o | 11 y.o | 11 y.o | 15 y.o | 31 y.o | 8 y.o | 13 y.o | 40 y.o | 19 y.o | 52 y.o | 47 y.o | 26 y.o |
| Genotype | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | Sporadic | AD | AD | AD | AD | AD | Sporadic |
| Gender | Male | Male | Male | Female | Male | Female | Male | Female | Female | Female | Female | Female |
| Distal weakness | + | + | + | − | + | + | + | + | + | + | + | + |
| Distal atrophy | + | + | + | + | + | + | − | − | + | + | + | + |
| Distal leg MMT | 1–2 | 4− | 4+ | NA | 0 | 4 | 4 | NA | 2 | NA | 3 | 1 |
| Sensory disturbance | − | − | + | + | + | + | + | NA | + | + | − | + |
| DTR | Absent | Decrease | Absent | Brisk | Absent | Absent | Decrease | Absent | Absent | n.a | Absent | Absent |
| Cognitive impaiment | − | − | − | NA | − | − | Poor school record | − | IQ71 | − | − | IQ46 |
| Other symptom | − | Hemolytic anemia | No seizure EEG; burst of genernlised spike and polyspike and wave | Spasticity celiac disease short stature migrane bladder and bowel incontinence | CK427 | Low vision | CK533 | − | CK1162 | − | − | Epilepsy hemolytic anemia |
| Median CMAP (mV) | 0.2 | 1.2 | 3.7–7.3 | Normal | 1.6/5.4 | n.a | 6.2 | 2.5 | 5.65 | 5.5 | 5.4 | 0.6/0.2 |
| Median MNCV (m/sec) | 31 | 33 | 32–35 | Normal | 41/41.8 | 31/42 | 40.3 | 38 | 39.3 | 53 | 54.5 | 33.6/25.1 |
| Median SNAP (μV) | 9 | 3.5 | NE | Normal | 1.0/1.37 | n.a | NE | NE | 1.3 | 4 | NE | 2.0/1.0 |
| Median SCV (m/sec) | 44 | 21 | NE | Normal | 32.4/30.7 | n.a | NE | NE | 38.6 | 39 | NE | 40.9/40.0 |
| Tibial CMAP (mV) | NA | 3.5 | NE | 33–40 | 0.07/0.31 | n.a | 2.3 | 0.3 | 0.13 | 2.1 | 0.13 | 0.14/0.05 |
| Tibial MNCV (m/sec) | NA | 23 | NE | 1–1.9 | NA | 21/24 | 39.8 | 29 | 41.6 | 46 | 69.2 | 13.8/21.1 |
| Sural SNAP (μV) | 8 | NE | NE | NE | NE | n.a | NE | NE | 0.3 | NE | NE | 1.0/2.0 |
| Sural SCV (m/sec) | 27 | NE | NE | NE | NE | n.a | NE | NE | 25.2 | NE | NE | 34.1/32.3 |
| Brain MRI | Normal | Normal | Normal | Normal | Normal | NA | NA | NA | NA | NA | NA | brain atrophy |
MMT, manual muscle test; DTR, deep tendon reflex; NA, not available; AD, autosomal dominant; CK, Creatine kinase (U/L); IQ, intelligence quotient; MNCV, Motor nerve conduction velocity; CMAP, Compound motor action potential; SCV, sensory nerve conduction velocity, SNAP, sensory nerve action potential; NE, not evoked; Normal range, median CMAP >3.1 mV; median MNCV >49.6 m/sec; median SNAP >7.0 μV; median SCV >47.2 m/sec; tibial CMAP >4.4 mV; tibial MNCV >41.7 m/sec; sural SNAP >5.0 μV; sural SCV >40.8 m/sec.
Figure 2.
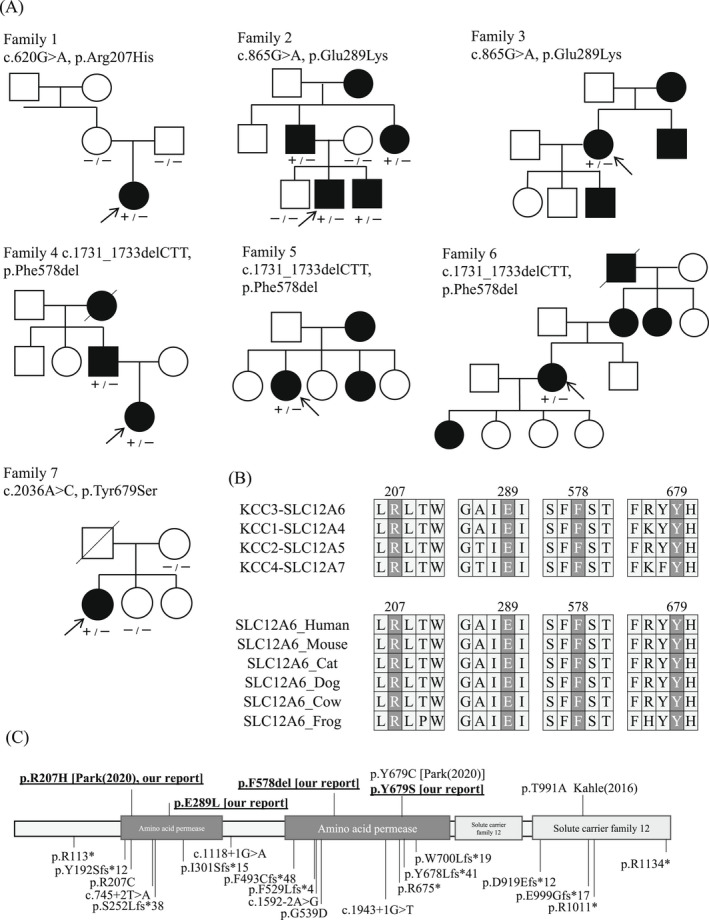
Genetic findings of seven families and variants review from literatures. (A) Seven families with SLC12A6 heterozygous variants in our study. Arrows indicate proband. −: wild type, +: variant positive (B) The variant residues detected in our study are highly conserved in human KCC family members and across species. (C) Location of SLC12A6 heterozygous and homozygous variants. Heterozygous variants are labeled above the protein, and variants detected in our study are underlined. Reported homozygous variants are labeled below the protein diagram.
Family 1 (p.Arg207His)
The proband was an eight‐years‐old girl with no family history of neuromuscular disease. Frequent falling was noted since she was a year old. Her first hospital visit was at 8 years old due to a foot deformity. She was diagnosed with CMT, which caused distal muscle weakness, muscle atrophy, decreased vibration sensation, and loss of tendon reflexes. She also had progressive vision loss. Based on her nerve conduction studies (NCS), she was classified as CMT‐intermediate type (right median MNCV 30.9 m/sec and left median MNCV 41.9 m/sec).
Family 2 (p.Glu289Lys)
The proband was a 13‐year‐old boy with a positive family history of autosomal dominant inheritance. He developed a steppage gait at the age of 11 and experienced abnormal sensations a year later. At 13 years of age, his physical examination revealed weakness and wasting of distal muscles, decreased vibration sensation, and loss of tendon reflexes. He was not mentally retarded but had a poor academic record. His creatine kinase (CK) level was high (533 IU/mL [male >59, <248 IU/mL]), and his NCS revealed sensory dominant axonal neuropathy (median MNCV 40.3 m/sec, CMAP (compound motor action potential) 6.2 mV, SNAP (sensory nerve action potential) not evoked).
Family 3 (p.Glu289Lys)
The proband was a 40‐year‐old woman, and similar symptoms were observed in her mother, younger sister, and son, indicating autosomal dominant inheritance. Starting at 10 years of age, she had been falling frequently and had difficulty in running. At the age of 40 years, she developed severe muscle atrophy and weakness in her extremities and became wheelchair‐bound. Her NCS results indicated CMT‐intermediate type (median MNCV 38.0 m/sec, CMAP 2.5 mV, SNAP not evoked).
Family 4 (p.Phe583del)
The proband was a 19‐year‐old girl with an autosomal dominant family history. She was referred to the hospital for hand atrophy, and physical examination revealed weakness and wasting of distal muscles, distal dominant sensory disturbance, and loss of tendon reflexes. Her intelligence quotient (IQ) was 71, which was at the borderline of mental function. High CK levels (1191 IU/mL) were observed, and her NCS indicated an axonal type (median MNCV 39.3 m/sec, CMAP 5.65 mV, SNAP 1.3 μV).
Family 5 (p.Phe583del)
The proband was a 52‐year‐old woman with an autosomal dominant family history. She developed claudication after the age of 40, and her lower limb muscle weakness and gait disturbance worsened when she was 49. At 52 years of age, physical examination revealed distal dominant weakness and atrophy, as well as decreased vibration sensation. Her NCS indicated sensory dominant axonal neuropathy (median MNCV 53 m/sec, CMAP 5.5 mV, SNAP 4 μV).
Family 6 (p.Phe583del)
This family also has autosomal dominant history, and the proband was a 47‐year‐old woman. She has experienced lower limb muscle weakness since the age of 39. Physical examination revealed distal muscle weakness, muscle atrophy, and loss of tendon reflexes, but no sensory disturbance. Her NCS was an axonal type (median MNCV 54.5 m/sec, CMAP 5.4 mV), and no response was recorded on SNAP (SNAP).
Family 7 (p.Tyr679Ser)
The proband was a 26‐year‐old woman with no family history of neuromuscular disease. She was diagnosed with clubfoot at the age of three. Her motor function was low, and her academic record was poor. She developed epilepsy at the age of 12. Physical examination revealed weakness and wasting of distal muscles, decreased pinprick and vibration sensation, and loss of tendon reflexes. Her IQ was 46, indicating mild mental retardation. Based on her NCS, she was classified as a CMT‐demyelinating type (right median MNCV 33.6 m/sec, CMAP 0.6 mV, SNAP 2.0 μV and left median MNCV 25.1 m/sec, CMAP 0.2 mV, SNAP 1.0 μV). Her brain MRI revealed frontal and temporal lobe atrophy, but no white matter or corpus callosum abnormalities were observed (Fig. 3). Sural nerve biopsy showed marked loss of myelinated nerve density and thinning myelin sheath, suggesting mixed axonal and demyelinating changes (Fig. S2).
Figure 3.
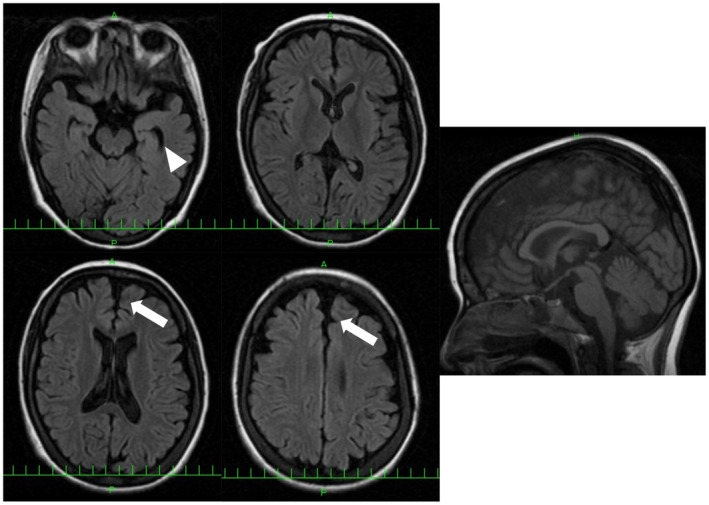
Brain MRI of patient 7 with p.Tyr679Ser variant. FLAIR images show frontal (arrow) and temporal lobe atrophy (arrow head). White matter change and corpus callosum change are not observed. [Colour figure can be viewed at wileyonlinelibrary.com]
Genetic findings
From seven unrelated families with CMT, we identified heterozygous variants in SLC12A6, including a previously reported variant (p.Arg207His) and three likely pathogenic variants (p.Glu289Lys, p.Phe578del, and p.Thr679Ser). Additionally, two other novel variants, p.Thr580Lys and p.Ser647Pro, were detected and classified as having uncertain significance due to a lack of agreement of their family members in segregation analysis. All these novel variants were absent in the public control databases and our in‐house control database. We further evaluated the novel variants using six computational analyses (SIFT, POLYPHEN2, PROVEAN, Mutation Assessor, FATHMM, and Condel) and protein stability analyses (Dynamut and i‐Mutant). All five novel missense variants were predicted to have a damaging effect in multiple analyses. However, the p.Phe578del variant could only be evaluated using PROVEAN, indicating a deleterious effect. All variants were also highly conserved across species and in other KCC family members, such as SLC12A4, SLC12A5, and SLC12A7 (Fig. 2B). We reviewed and summarized the described SLC12A6 heterozygous and homozygous variants in Figure 2C. Table 2 shows all the genetic findings and ACMG criteria.
Table 2.
Genetic findings and interpretation of SLC12A6 heterozygous variants.
| Nucleotide change | c.620G>A | c.865G>A | c.1731_1733del | c.2036A>C | ||||
| Amino acid change | p.Arg207His | p.Glu289Lys | p.Phe578del | p.Tyr679Ser | ||||
| Zygosity | Heterozygous | Heterozygous | Heterozygous | Heterozygous | ||||
| Alelle frequency‐gnomAD | 0 | 0 | 0 | 0 | ||||
| Alelle frequency‐jMorp | 0 | 0 | 0 | 0 | ||||
| Our control | – | – | – | – | ||||
| SIFT/prediction | 0 | Damaging | 0 | Damaging | – | – | 0 | Damaging |
| PROVEAN/prediction | −4.57 | Deleterious | −3.9 | Deleterious | −12.78 | Deleterious | −8.72 | Deleterious |
| Polyphen2/prediction | 0.998 | Damaging | 1 | Damaging | – | – | 0.995 | Damaging |
| MutationAssesor/prediction | 4.025 | Damaging | 4.27 | Damaging | – | – | 3.265 | Damaging |
| FATHMM/prediction | −5.25 | Damaging | −5.12 | Damaging | – | – | −5.1 | Damaging |
| Condel/prediction | 0.73 | Damaging | 0.746 | Damaging | – | – | 0.673 | Damaging |
| Dynamut | ||||||||
| ΔΔG/prediction | −1.285 kcal/mol | Destabilizing | 0.841 kcal/mol | Stabilizing | – | – | −2.425 kcal/mol | Destabilizing |
| ΔΔG (ENCOM)/prediction | −0.127 kcal/mol | Destabilizing | −0.108 kcal/mol | Destabilizing | – | – | −0.747 kcal/mol | Destabilizing |
| iMutant | ||||||||
| RI/stability | 8 | Decrease | 0 | Decrease | – | – | 8 | Decrease |
| ACMG | ||||||||
| Population data | PS4_Supporting PM2 | PS4_Supporting PM2 | PS4_Moderate PM2 | PS4_Supporting PM2 | ||||
| In sillico data | PP3 | PP3 | PP3 | |||||
| De novo/segregation data | PM6 | PP1_Moderate | PP1 | |||||
| Other data | PS3 | PM4 | PM5 | |||||
| Criteria | Pathogenic | Likely pathogenic | Likely pathogenic | Likely pathogenic | ||||
In silico analysis cut off: SIFT <0.05; PP2 >0.9; PROVEAN < −2.5; MA >1.9; FATHMM < −1.5; Condel >0.47.
Discussion
Using a series of high‐throughput sequencing systems, we identified one previously reported (p.Arg207His) and three novel SLC12A6 heterozygous variants (p.Glu289Lys, p.Phe578del, and p.Thr679Ser) in seven patients from our large case series of Japanese patients with CMT. Their CMT phenotypes were diverse, including demyelinated, axonal, and intermediate types. Furthermore, various central nervous system (CNS) involvements were observed.
KCC3, which is encoded by SLC12A6, is expressed in both the CNS and peripheral nervous system. 17 Loss of KCC3 function affects both the CNS and peripheral nervous system, with serious consequences manifesting as corpus callosum agenesis and severe sensorimotor neuropathy. 10 In a previous report, patients with the de novo SLC12A6 p.Thr991Ala heterozygous variant had early‐onset and progressive motor‐predominant axonal neuropathy without sensory neuropathic symptoms. 11 Since then, CMT patients with other mutations in SLC12A6 (p.Arg207His heterozygous or p.Tyr679Cys heterozygous) have been reported. 12 , 13 We summarized the clinical and electrophysiological findings from previous reports and our report (Table 1). In our report, the mean age of onset was 17.5 ± 16.1 years (range; 1–40), and the inheritance pattern was sporadic for two patients and autosomal dominant for the others. Patients with the p.Phe578del variant in SLC12A6 developed CMT symptoms at an age later than that of other patients, indicating a less damaging effect.
Regarding electrophysiology, the median MNCV was 39.6 ± 9.5 m/sec (range 25.0–54.5) in our cases, not much different than previous reports of 34.7 ± 4.6 m/sec (range 31.0–41.8). 11 , 12 , 13 CMT caused by SLC12A6 heterozygous variants may be classified as demyelinating, axonal, or intermediate type. This is consistent with the mixed axonal and demyelinating changes in sural nerve pathology of patient 7 in this study. Therefore, genetic analysis of the SLC12A6 gene should be considered in all types of CMT. Interestingly, we observed intellectual disability in three patients (42.9%). Patients 4 (IQ 71) and 6 (IQ 46) had their IQs confirmed. Patient 2 was also suspected of having mental retardation due to poor academic performance, but IQ was not confirmed. Additionally, the proband of family 7 was diagnosed with epilepsy (generalized tonic seizure). Park et al. reported a case with EEG abnormality but no convulsions. 12 More importantly, to the best of our knowledge, this is the first report of a CMT patient with a heterozygous SLC12A6 variant developing brain atrophy. Actually, various CNS involvements have been described only in patients with biallelic SLC12A6 variants until now. Our findings indicate that CMT with SLC12A6 heterozygous variants may also affect the CNS, broadening the current phenotypic spectrum. Additionally, hyperCKemia has been observed in multiple patients in our study and previous reports, 13 which might also be a feature of SLC12A6‐related CMT.
Standardized and accurate variant evaluation is essential for an effective genetic diagnosis. Therefore, disease‐specific ACMG‐AMP guidelines for consistent and accurate variant classification have been developed for several diseases. However, there are no specific ones for CMT. Therefore, we used the expert panel of RASopathy, another clinically and genetically heterogeneous disease, which is comparable to CMT, as a reference for the classification. 18 The classification method is summarized in Table S2. All previously reported (p.Arg207His) and novel variants (p.Glu289Lys, p.Phe578del, and p.Thr679Ser) detected in our study were absent in the global, Japanese, and in‐house databases (PM2; ACMG criteria). The p.Arg207His variant of family 1 was not found in her unaffected parents and thus was considered as de novo (PM6). Additionally, a functional study (PS3) revealed that this variant has a complete loss of function. 12 We classified this variant as pathogenic. Among the novel variants, the p.Glu289Lys variant was identified from two families (PS4‐supporting) and was found co‐segregated from multiple affected/unaffected family members of family 2 (PP1‐moderate), and a deleterious effect was indicated by several in silico analyses (PP3). Whereas, p.Phe578del, detected in three probands (PS4‐moderate), would produce a shorter protein (PM4). Otherwise, p.Tyr679Ser locates at the residue where another pathogenic variant (p.Tyr679Cys) has been reported (PM5). 12 Taken together, we interpreted these three novel variants as likely pathogenic.
Meanwhile, p.Thr580Lys and p.Ser647Pro were discovered in two families. Computational analysis revealed that they had deleterious effects and were highly conserved across species and in other KCC family members, such as SLC12A4, SLC12A5, and SLC12A7 (Fig. S1). However, the segregation study was insufficient, and we were unable to perform functional studies due to technical and material limitations. As a result, we classified these two variants as having uncertain significance, and further studies are required to clarify their pathogenicity.
Conclusively, we described one previously reported and three novel SLC12A6 heterozygous variants in Japanese CMT patients with/without CNS involvement. Mutation screening of SLC12A6 should be considered in all patients with CMT, regardless of the clinical subtype. Our findings broaden the clinical spectrum of SLC12A6 heterozygous variants, manifesting with motor and sensory polyneuropathies and CNS dysfunctions. In the clinic, it is recommended to pay more attention to CNS lesions, particularly in patients showing cognitive impairment and epilepsy.
Author Contributions
MA, YH, JY, and HT contributed to the concept and design of the study. MA, YH, JY, and AY contributed to the analysis and interpretation of data. MA and HT produced the first draft of the manuscript. All authors provided input into subsequent drafts and reviewed and approved the final version for submission.
Conflict of Interest
All authors declare that there is no conflict of interest.
Supporting information
Figure S1. Clinical and genetic findings of variants of uncertain significance.
Figure S2. Sural nerve pathology from patient 7 (SLC12A6 p.Tyr679Ser).
Table S1. CMT neuropathy score (CMTNS) of seven CMT probands with SLC12A6 heterozygous variants.
Table S2. ACMG criterion of our report.
Data S1. Clinical summary of probands with variants of uncertain significance in SLC12A6.
Acknowledgments
The authors appreciate Tomoko Ohnishi at Kagoshima University, for her great technical assistance. The authors are supported by Enago (www.enago.jp) for reviewing the English in this report. We appreciate the Division of Gene Research, Research Support Centre, Kagoshima University, for the use of their facilities. This work was supported by Grants‐in‐Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (201610002B). This research is also supported by the Research program for conquering intractable disease from Japan agency for Medical Research and development (AMED) (201442014A, 201442071A, 17929553) and JSPS KAKENHI Grant Numbers JP18H02742, JP20K16604, JP21K15702, JP21H02842.
Funding Information
This work was supported by Grants‐in‐Aid from the Research Committee of Ataxia, Health Labour Sciences Research Grant, the Ministry of Health, Labour and Welfare, Japan (201610002B). This research is also supported by the Research program for conquering intractable disease from Japan Agency for Medical Research and Development (AMED) (201442014A, 201442071A, 17929553) and JSPS KAKENHI Grant Numbers JP18H02742, JP20K16604, JP21K15702, JP21H02842.
Funding Statement
This work was funded by Japan Agency for Medical Research and Development grants 17929553, 201442014A, and 201442071A; JSPS grants JP18H02742, JP20K16604, JP21H02842, and JP21K15702; Ministry of Health, Labour and Welfare, Japan grant 201610002B; Health Labour Sciences Research Grant; Research Committee of Ataxia.
References
- 1. Braathen GJ, Sand JC, Lobato A, Høyer H, Russell MB. Genetic epidemiology of Charcot‐Marie‐Tooth in the general population. Eur J Neurol. 2011;18:39‐48. [DOI] [PubMed] [Google Scholar]
- 2. Stojkovic T. Hereditary neuropathies: an update. Rev Neurol (Paris). 2016;172(12):775‐778. [DOI] [PubMed] [Google Scholar]
- 3. Zuchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36:449‐451. [DOI] [PubMed] [Google Scholar]
- 4. Polke JM, Laura M, Pareyson D, et al. Recessive axonal Charcot‐Marie‐Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology. 2011;77:168‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baxter RV, Ben Othmane K, Rochelle JM, et al. Ganglioside‐induced differentiation‐associated protein‐1 is mutant in Charcot‐Marie‐Tooth disease type 4A/8q21. Nat Genet. 2002;30:21‐22. [DOI] [PubMed] [Google Scholar]
- 6. Auranen M, Ylikallio E, Toppila J, Somer M, Kiuru‐Enari S, Tyynismaa H. Dominant GDAP1 founder mutation is a common cause of axonal Charcot‐Marie‐Tooth disease in Finland. Neurogenetics. 2013;14:123‐132. [DOI] [PubMed] [Google Scholar]
- 7. Higuchi Y, Hashiguchi A, Yuan J, et al. Mutations in MME cause an autosomal‐recessive Charcot‐Marie‐Tooth disease type 2. Ann Neurol. 2016;79:659‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Auer‐Grumbach M, Toegel S, Schabhuttl M, et al. Rare variants in MME, encoding metalloprotease neprilysin, are linked to late‐onset autosomal‐dominant axonal polyneuropathies. Am J Hum Genet. 2016;99:607‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gagnon KB, Delpire E. Molecular physiology of SPAK and OSR1: two Ste20‐related protein kinases regulating ion transport. Physiol Rev. 2012;92:1577‐1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howard HC, Mount DB, Rochefort D, et al. The K‐Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosum. Nat Genet. 2002;32:384‐392. [DOI] [PubMed] [Google Scholar]
- 11. Kahle KT, Flores B, Bharucha‐Goebel D, et al. Peripheral motor neuropathy is associated with defective kinase regulation of the KCC3 cotransporter. Sci Signal. 2016;9:ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Park J, Flores BR, Scherer K, et al. De novo variants in SLC12A6 cause sporadic early‐onset progressive sensorimotor neuropathy. J Med Genet. 2020;57(4):283‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shi J, Zhao F, Pang X, et al. Whole‐exome sequencing identifies a heterozygous mutation in SLC12A6 associated with hereditary sensory and motor neuropathy. Neuromuscul Disord. 2021;31(2):149‐157. [DOI] [PubMed] [Google Scholar]
- 14. Hashiguchi A, Higuchi Y, Nomura M, et al. Neurofilament light mutation causes hereditary motor and sensory neuropathy with pyramidal signs. J Peripher Nerv Syst. 2014;19:311‐316. [DOI] [PubMed] [Google Scholar]
- 15. Yoshimura A, Yuan JH, Hashiguchi A, et al. Genetic profile and onset features of 1005 patients with Charcot‐Marie‐Tooth disease in Japan. J Neurol Neurosurg Psychiatry. 2019;90:195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pearson MM, Lu J, Mount DB, Delpire E. Localization of the K(+)‐Cl(−) cotransporter, KCC3, in the central and peripheral nervous systems: expression in the choroid plexus, large neurons and white matter tracts. Neuroscience. 2001;103:481‐491. [DOI] [PubMed] [Google Scholar]
- 18. Gelb BD, Cavé H, Dillon MW, et al. ClinGen's RASopathy Expert Panel consensus methods for variant interpretation. Genet Med. 2018;20:1334‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clinical and genetic findings of variants of uncertain significance.
Figure S2. Sural nerve pathology from patient 7 (SLC12A6 p.Tyr679Ser).
Table S1. CMT neuropathy score (CMTNS) of seven CMT probands with SLC12A6 heterozygous variants.
Table S2. ACMG criterion of our report.
Data S1. Clinical summary of probands with variants of uncertain significance in SLC12A6.