

Abstract
The endoplasmic reticulum membrane protein complex subunit 10 (EMC10) is a highly conserved protein responsible for the post‐translational insertion of tail‐anchored membrane proteins into the endoplasmic reticulum in a defined topology. Two biallelic variants in EMC10 have previously been associated with a neurodevelopmental disorder. Utilizing exome sequencing and international data sharing we have identified 10 affected individuals from six independent families with five new biallelic loss‐of‐function and one previously reported recurrent EMC10 variants. This report expands the molecular and clinical spectrum of EMC10 deficiency, provides a comprehensive dysmorphological assessment and highlights an overlap between the clinical features of EMC10‐and EMC1‐related disease.
Introduction
The endoplasmic reticulum membrane protein complex (EMC) is a highly conserved, multifunctional, and multi‐subunit protein complex responsible for the post‐translational insertion of tail‐anchored membrane proteins into the endoplasmic reticulum (ER) in a defined topology. 1 The insertion of proteins into membranes is an essential cellular process influencing vesicular trafficking, apoptosis, signal transduction, and lipid biosynthesis. 2 The mammalian EMC is composed of 10 subunits, EMC1–10, 3 encoded by the corresponding EMC1 − 10 genes, two of which have currently been linked to human disease. Thus, biallelic variants in EMC1 have been linked to the cerebellar atrophy, visual impairment, and psychomotor retardation syndrome (MIM: 616875), 4 , 5 , 6 , 7 and hitherto two homozygous loss‐of‐function (LOF) variants in EMC10 have been associated with the neurodevelopmental disorder (NDD) with dysmorphic facies and variable seizures (MIM: 619264) in eight families. 8 , 9 , 10 The two previously reported disease‐causing EMC10 variants include a homozygous splice acceptor site variant c.679‐1G>A found in two siblings from a Saudi Arabian family and a recurrent homozygous frameshift variant c.287delG, p.Gly96AlafsTer9 identified in 13 affected individuals from seven families of Bedouin, Saudi Arabia, and the United Arab Emirates origin. 8 , 9 , 10
Herein, we present 10 affected individuals from six unrelated families harboring five previously unreported and one recurrent homozygous LOF variants in EMC10. The report expands the clinical spectrum of EMC10‐related NDD towards the more severe end, highlights an overlap between the clinical features of EMC10‐and EMC1‐related disease, and discovers new disease‐associated EMC10 LOF variants.
Methods
Participants' recruitment, data collection, and clinical and genetic investigation
By exome sequencing (ES) of families affected by undiagnosed NDD, data mining of DNA sequences from families with rare disorders aggregated across several diagnostic and research laboratories, and using GeneMatcher data sharing platform 11 six independent families reported here were identified. A uniform clinical proforma was distributed to collect clinico‐demographic details. Facial photographs and brain magnetic resonance imaging (MRI) scans were obtained from the present cases and were retrieved from the previous reports 8 , 9 for a review conducted by an experienced dysmorphologist and pediatric neuroradiologist (M. S. and F. A.). Parents and legal guardians of all affected individuals gave their consent for the publication of clinical and genetic information according to the Declaration of Helsinki, and the study was approved by the respective local Ethics Committees. Follow‐up details on the parathyroid gland function and levels of global developmental delay/intellectual disability (GDD/ID) were obtained from the cases reported in Shao et al. 9 Trio ES or solo ES with subsequent Sanger segregation analysis was carried out in DNA extracted from blood‐derived leukocytes in six families in four different centers following slightly different protocols (Table 1 for methods). ES data analysis, variant filtering, and prioritization were performed using in‐house implemented pipelines of the local genetic centers (Table 1 for methods). In brief, the bioinformatics filtering strategy included screening for only exonic and donor/acceptor splicing variants. In accordance with the pedigree and phenotype, priority was given to rare variants (<0.01% in public databases) that were fitting a recessive (homozygous or compound heterozygous) or a de novo model and/or variants in genes previously linked to NDD/ID and other neurological disorders.
Table 1.
Clinical features of the EMC10 cohort, genetic methods, and variant characteristics.
| Patient | Family 1 | Family 2 | Family 3 | Family 4 | Family 5 | Family 6 | Umair et al. (2020) | Shao et al. (2021) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 | S9 | S10 | 2 cases 1 family | 13 cases from 7 families | |||
| Variant annotation | Variant type | Frameshift | Frameshift | Stop gained | Stop gained | Frameshift | Splicing | Splicing | Splice acceptor | Frameshift | ||||
| Variant at the cDNA level (NM_206538.4) | c.543dup | c.66delC | c.66delC | c.259C>T | c.259C>T | c.259C>T | c.289C>T | c.287del | c.188‐2A>C | c.188‐2A>C | c.679‐1G>A | c.287delG | ||
| Variant at the protein level | p.Asn182GlnfsTer16 | p.Ser23ValfsTer82 | p.Ser23ValfsTer82 | p.Gln87Ter | p.Gln87Ter | p.Gln87Ter | p.Arg97Ter | p.Gly96AlafsTer9 | − | − | − | p.Gly96AlafsTer9 | ||
| Variant characterisitcs | Methods | Makrythanasis et al. 12 | Yaron et al. 13 | Ucuncu et al. 14 | Makrythanasis et al. 12 | Basel‐Salmon et al. 15 | Makrythanasis et al. 12 | |||||||
| Maximum allele frequency in variant databases 1 | <0.00001 | <0.000001 | <0.000001 | <0.00001 | <0.00001 | <0.00001 | <0.00001 | <0.00001 | <0.00001 | <0.00001 | <0.000001 | <0.0001 | ||
| ACMG classification | Pathogenic (PVS1, PM2) | Pathogenic (PVS1, PM2) | Pathogenic (PVS1, PM2) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3) | Pathogenic (PVS1, PM2, PP3, PP5) | Pathogenic (PVS1, PM2) | ||
| Epidemiological data | Ethnic group | Azerbaijani |
Bukharan Jewish |
Bukharan Jewish |
Egyptian | Egyptian | Egyptian | Persian | Bedouin | Egyptian | Egyptian | Saudi | Saudi, Arab, Bedouin | |
| Gender/current age | M/8 y 10 m | M/14.5 y | M/26 y | F/8 y | F/8 y | F/6 y | F/11.3 y | F/5 y | M/7 y 5 m | F/2 y 11 m | F, M/11, 14 y | F‐ 5, M‐8 | ||
| Age at last examination | 8 y 10 m | 14.5 y | 26 y | 7 y | 7 y | 9 y | 11 y | 4 y 7 m | 7 y 5 m | 2 y 11 m | 14 and 11 y | 3 m–27 y | ||
| Consanguinity/family history | +/− | +/+ | +/+ | +/+ | +/+ | +/+ | +/+ | +/− | +/+ | +/+ | +/+ | +/+ | ||
| Growth | At birth | Premature birth | − | + | + | + | + | − | − | − | − | − | − | NA |
| Congenital microcephaly | NA | NA | NA | −2 SD | −2 SD | −1 SD | − | NA | − | − | − | − | ||
| Low birth weight | − | + | + | − | − | − | − | − | − | − | − | − | ||
| At last exam‐n | Low weight | −2.68 SD | Yes | −1.34 SD | −1 SD | −1 SD | −1 SD | − | + | − | − | − | − | |
| Microcephaly (congenital or acquired) | NA | + | − | Congenital | Congenital | − | − | − | Acquired | Acquired | − | − | ||
| Development | Age at first symptoms | Infancy (3–4 m) | Infancy | Birth, infancy | 2.5 y | 2.5 | 3 y | 6 m | Infancy | 3–4 m | 3–4 m | 3 and 2 y | NA | |
| Type of progression (rapid, moderate, slow) | NP | NP | NP | Moderate | Moderate | Moderate | Slow | NP | NP | NP | NP | NP | ||
| DD/ID (severity) | Mild‐to‐moderate | Mild‐to‐moderate | Mild‐to‐moderate | Moderate‐to‐severe | Moderate‐to‐severe | Moderate‐to‐severe | Mild‐to‐moderate | Mild‐to‐moderate | Mild‐to‐moderate | Mild‐to‐moderate | +/2 mild‐to‐moderate | +12/13 mild‐to‐moderate 2 | ||
| Failure to thrive | + | − | + | − | − | − | − | + | − | − | − | +4/13 | ||
| Regression in development | − | − | − | + | + | + | − | − | − | − | − | − | ||
| Non‐ambulatory | − | − | − | + | + | + | − | − | − | − | − | − | ||
| Neurological symptoms | Dysarthria | + | − | − | + | + | + | + | − | + | + | − | − | |
| Poor speech | + | + | − | + | + | + | + | + | − | − | − | − | ||
| Axial hypotonia | − | − | + | + | + | + | − | − | − | − | − | +3/5 2 | ||
| Peripheral hypotonia | + | − | + | − | − | − | − | + | + | + | − | − | ||
| Poor head control | − | − | − | + | + | + | + | − | − | − | − | − | ||
| Truncal ataxia | NA | − | − | + | + | + | − | − | − | − | − | − | ||
| Hyperkinetic/hypokinetic movement disorders | + | − | − | + | + | + | − | − | − | − | + | − | ||
| Ataxic/clumsy gait | − | − | − | + | + | + | + | − | − | − | − | − | ||
| Seizures, age of onset, type | − | − | − | +, 3 y.o., CP | +3, y.o., CP | +, 3 y.o., CP | +, 2 y.o., ST | − | − | − | +1, 3.5 y.o., FS | +6/13, infantile, MF, GTCS | ||
| Other systems and investigations | Facial dysmorphism | + | + | + | + | + | + | + | + | + | + | + | + | |
| Arachnodactyly | − | + | + | + | + | + | − | + | + | + | − | +3/13 | ||
| Elevated PTH | NA | + | + | NA | NA | NA | NA | NA | NA | NA | NA | +3/13 2 | ||
| Other features | UIH, CHD | NC | MHN | − | − | VSD | − | − | CD | CD | − |
+4/11 NC, +2/11 HN, +2/13 VSD, +5/13 UIH |
||
| Brain MRI findings | A Short and thin CC | Abnormal HP and IBA | Abnormal HP and IBA | Thin CC, VMA | Thin CC, VMA | Thin CC, VMA | Normal | FMC | Normal | NA | Normal |
Thin CC 3/13, Chiari I, 4/13, GMH 1/13 AM 3/13 |
||
S, subject; M, male; F, female; y, years; m, months; SD, standard deviation; NA, not available; NP, nonprogressive; CP, complex partial; ST, subclinical tonic; FS, febrile seizure; MF, multifocal seizures; GTCS, generalized tonic–clonic seizures; NC, nephrocalcinosis; MHN, mild hydronephrosis or hydroureter; CHD, congenital heart defect; PTH, parathyroid hormone; UIH, umbilical or inguinal hernias; CD, bilateral 5th digit clinodactyly; CC, corpus callosum; HP, hippocampi; IBA, initial brain atrophy; VMA, vermian atrophy; FMC, focal area of malformed cortex (thick and dysplastic) in the right frontal lobe; GMH, gray matter heterotopia; AM, abnormal myelination.
Database checked include GnomAD v3, gnomAD v2.1.1, TopMED Bravo, UKBiobank, Iranome, GME Variome, In‐house Database. The total number of alleles considered was ~1,314,000.
Follow‐up details.
Results
Clinical delineation
The main phenotypic features of the present cohort are summarized in (Fig. 1); Table 1 and Table S1. Detailed clinical history is provided in the Supplementary Case Reports.
Figure 1.
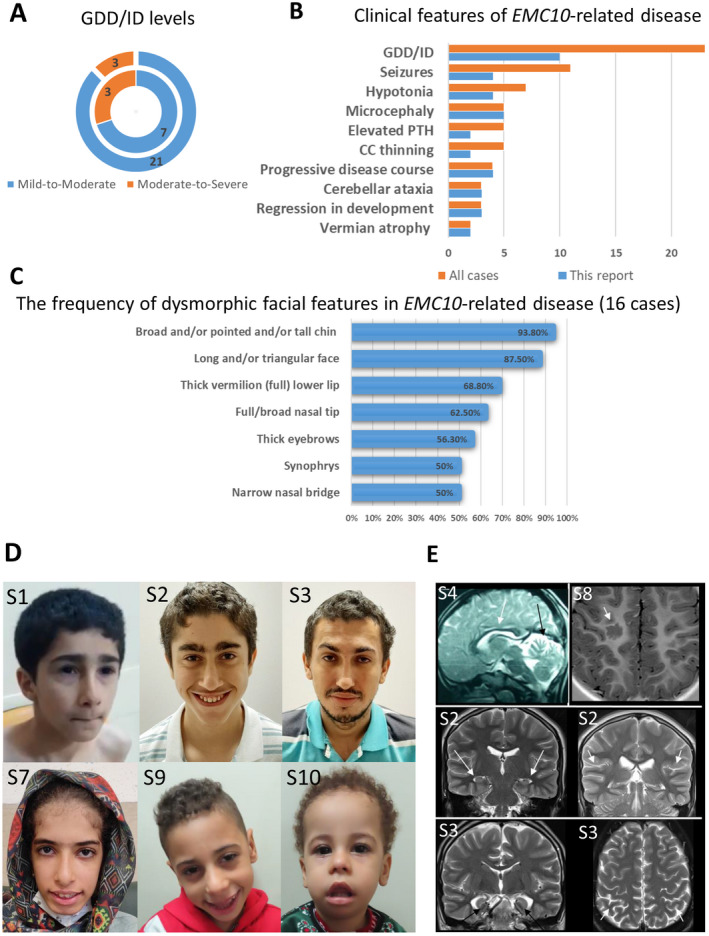
Clinical features of the cases with EMC10‐related NDD. (A) Levels of Global developmental delay/intellectual disability (GDD/ID): inner circle for cases reported here. Outer circles for all reported cases with biallelic EMC10 variants. (B) Clinical features of EMC10‐related NDD: for cases reported here and for all reported cases with biallelic EMC10 variants. PTH, parathyroid hormone; CC, corpus callosum. (C) The frequency of dysmorphic facial features in EMC10‐related disease (16 cases). This describes cases reported in the present study and all previously reported cases with available facial photos. (D) Facial photos of the affected individuals from this study: S1 – triangular face, thick eyebrows, narrow nasal bridge, pointed chin. S2 – long face, curly hair, low anterior hairline, right anterior cowlick, widow's peak, thick eyebrows (caterpillar‐like), synophrys, up‐slanting palpebral fissures, hypotelorism, lateral infraorbital creases, narrow nasal bridge, short philtrum, full nasal tip, tall and broad chin. S3 – Curly hair, prominent supraorbital ridges, thick and highly‐arched eyebrows, synophrys, up‐slanting right palpebral fissure, narrow nasal bridge, full nasal tip, short philtrum, full lips, and pointed chin. S7 – Long face, tall forehead, bifrontal narrowing, low columella, high anterior hairline, thick eyebrows, synophrys, up‐slanting palpebral fissures, narrow nasal bridge, full nasal tip, short philtrum, wide mouth, full lower lip, and broad chin. S9 – long face, curly hair, tall forehead, high anterior hairline, full cheeks, full nasal tip, low‐hanging columella, wide mouth, full lower lip, and broad chin. S10 ‐ long face, sparse and curly hair, high anterior hairline, tall forehead, bifrontal narrowing, full cheeks, large ear lobes, full nasal tip, tented upper lip, full and everted lower lip, midline depression over lower lip, broad chin, retrognathia. (E) Brain MRI scans. In the upper left row CC thinning (white arrows) and vermian atrophy (black arrow) in S4. On the right, dysplastic malformed cortex (arrowhead) is shown in S8. The middle and lower rows show mild brain atrophy and hippocampal dysmorphisms in two subjects from family 2 (S2 and S3). In the middle row white arrows point at small hippocampi while in the last row, black arrows show rounded and simplified hippocampal structure. In these affected individuals, mild cortical atrophy is demonstrated by the enlargement of cortical sulci (white arrowheads in the right middle and bottom rows). NDD, neurodevelopmental disorder.
The cohort is composed of 10 affected individuals including six females and four males, all of whom are currently alive with a mean age of 9.8 ± 6.5 years (range 5–26). All cohort members were born to consanguineous unions. Four cases were born prematurely with confirmed congenital microcephaly (2/10), low birth weight (2/10), and obstructive hydrocephalus (1/10). Failure to thrive was reported in 3/10 (30%) affected individuals. The disease was of infantile‐onset (≤12 months) in 7/10 and manifested at 2.5 and 3 years old in three siblings from family 3. GDD ranging from mild to severe was a common manifesting feature in the cohort. Concerning developmental domains, language acquisition skills were significantly delayed in all cohort members, three siblings from family 3 failed to acquire independent ambulation by the ages 6 and 7 years old, and S1 had difficulties with fine motor skills. The disease course was non‐progressive in 6/10 (60%) cases, and in others, the progression had either slow (family 4) or moderate (family 3) rates. Three siblings from family 3 had slow regression in motor skills.
Upon the most recent follow‐up at a mean age of 9.5 ± 6.2 years, 5/10 (50%) cases had microcephaly and 3/10 (20%) cases had small weights. Congenital heart disease was found in 2/10 (20%) cases. GDD/ID ranging from mild to severe was present in all cohort members. Most of the cases (9/10, 90%) had limited language abilities. Mild axial (4/10, 40%) and peripheral (5/10, 50%) hypotonia, truncal ataxia (3/10, 30%), dysarthria (7/10, 70%) with ataxic or clumsy gait (4/10, 40%), and hyperkinetic movement disorders (4/10, 40%) were frequent neurological findings. Complex partial seizures controlled by sodium valproate were reported in siblings from family 3. Their electroencephalography showed bilateral frontotemporal epileptogenic dysfunction. The proband from family 4 had subclinical tonic seizures from the age of 2 years responsive to carbamazepine.
Facial photographs were reviewed for six affected individuals from four families. This included five children and one adult (Fig. 1D). Published photographs of 10 previously reported patients were also reviewed. 8 , 9 These included eight children, one adolescent and one adult from five families. The dysmorphic features were described based on terminology recommended by Elements of Morphology. Where this classification had no descriptive term available for a dysmorphic feature seen in a patient, HPO terminology was used instead. Detailed facial dysmorphic features for all affected individuals whose photographs were reviewed are shown in Table S2 and dysmorphic features are tabulated using HPO codes in Table S3 to generate the frequency of each feature. Figure 1C shows the most frequently seen facial dysmorphic features of EMC10‐related NDD. Additionally, arachnodactily and bilateral fifth digit clinodactyly were present in 8/10 (80%) and 2/10 (20%) subjects, respectively.
Siblings from Family 2 had repeatedly (S2) or transiently (S3) elevated parathyroid hormone levels with nephrocalcinosis (S2), osteoporosis (S2), hyperuricemia (S3), and hydronephrosis (S3). Three cases from Shao et al. report 9 were confirmed to have elevated parathyroid hormone levels. Plasma and urine metabolic tests available from the presently reported seven cases were unremarkable.
Full brain MRI exams were available in six cases, while in two subjects from family 3 only midline sagittal slices could be reviewed. The abnormal findings include a thin corpus callosum (CC) (2/8), abnormal hippocampi (small and/or rounded) and initial brain atrophy (2/6), vermian atrophy (2/6), and a focal cortex malformation (1/6) (Fig. 1C). Two subjects from family 4 and family 6 showed normal findings.
Genetic findings
We identified five new and one previously reported recurrent homozygous LOF EMC10 (NM_206538.4) variants in six consanguineous families as follows: c.543dup; p.Asn182GlnfsTer16 in family 1, c.66delC; p.Ser23ValfsTer82 in family 2, c.259C>T; p.Gln87Ter in family 3, c.289C>T; p.Arg97Ter in family 4; c.287del; p.Gly96AlafsTer9 in family 5 and c.188‐2A>C in family 6 (Table 1). These variants reside within sizeable regions of homozygosity and are segregated with the disease in the families (Fig. 2A). The location of the variants did not indicate any cluster sites in the gene nor in the protein (Fig. 2B–C). Inspection of multiple public and private variant databases including gnomAD (v2.1.1 and v3.1.2), TopMED, Iranome, GME Variome, UK Biobank, and our in‐house database of ~25,000 exomes (total number of alleles ~1,314,000) revealed that the variants are absent or with extremely rare allele frequency reported ranging from <0.0001 to <0.000001. Moreover, according to ACMG classification, all reported variants were considered pathogenic (Table 1). No unaffected individuals reported homozygous for any of these variants. Upon further re‐analysis of the exomes, no additional clinically relevant variants were identified in any of the families.
Figure 2.
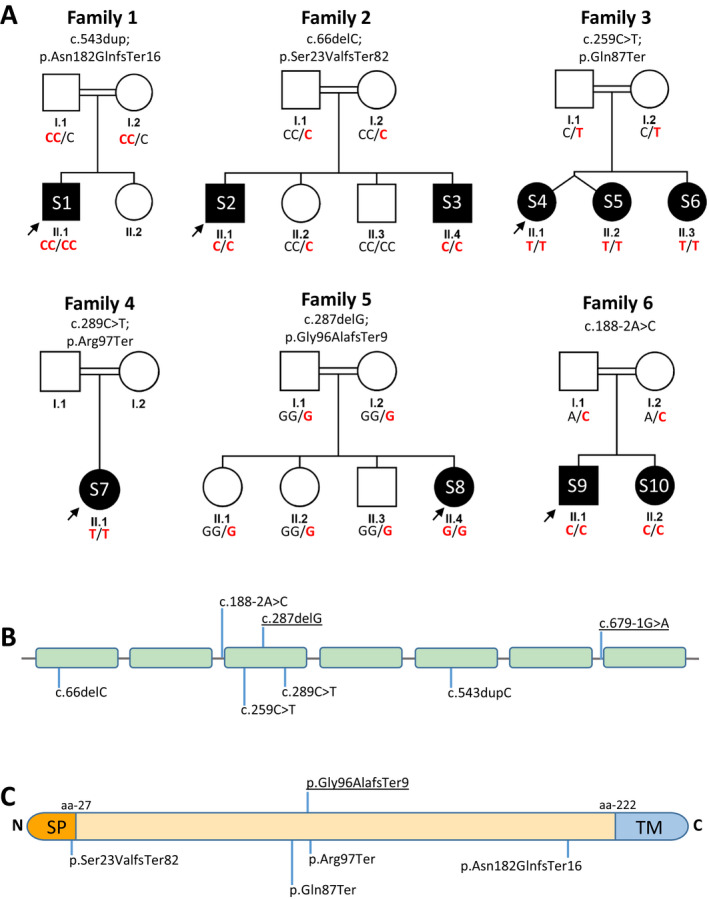
Pedigrees and genetic data. (A) Pedigrees of the six families described. The variant is indicated by the bold red letter. Square = male; circle = female; black filled symbol = affected individual; white symbols = unaffected individuals. Double lines indicate consanguinity. (B) Schematic representation of the seven exons in the EMC10 gene. Previously described variants are underlined. (C) Schematic diagram of the EMC10 protein. The orange section represents N terminal signal peptide (SP), the yellow section represents the topological domain, and the blue section shows the C terminal transmembrane domain (TM). 22
Discussion
The EMC has been thought to be implicated in several cellular processes though its primary function remains debatable. 16 The EMC family was first identified in yeast as a multi‐protein transmembrane complex, where it was thought to be an ER‐mitochondria tether that interacts with the outer membrane protein Tom5 of the Translocase of the mitochondrial outer membrane complex (TOM). 16 , 17 Defective EMC has been suggested to decrease, but not abolish, the insertion and proper function of multiple transmembrane proteins. 9 EMC has also been implicated in the folding of multipass membrane proteins. 18 Several recent studies have shown that impaired ER‐mitochondria crosstalk could be involved in neurodegenerative diseases. 19
EMC10 was first identified as a disease‐associated gene in two independent studies utilizing ES in individuals with GDD. 8 , 9 The recurrent EMC10 c.287delG, p.Gly96AlafsTer9 variant identified in seven families significantly reduced EMC10 RNA expression and resulted in an unstable truncated EMC10 protein. Staining for EMC10 in postmortem human infant brain showed colocalization with neuronal markers MAP2 and NeuN, markers specifically expressed in neuronal perikarya and dendrites and the nucleus of post‐mitotic neurons, respectively. 9 , 20
Here, we report 10 affected individuals from six unrelated families with five previously unreported and one reported recurrent biallelic LOF EMC10 variants. Replicating the features described in the previous EMC10 cohort, the current cases exhibited uniform GDD/ID (with prominent delay in verbal milestones) and facial dysmorphism, and variably presented with failure to thrive, axial hypotonia, seizures, microcephaly, CC thinning, and cardiac and renal pathology. The dysmorphic facial feature most commonly shared between the previous and present EMC10 cohorts was a long and/or triangular face. In contrast, expanding the clinical spectrum of the EMC10‐related NDD, the cases reported here presented with dysarthria, persistent peripheral hypotonia, movement disorders, gait ataxia, and vermian atrophy together with noticeable growth impairment, and hyperparathyroidism. Furthermore, the progressive disease course and regression in motor skills seen in the current cohort appear to expand the phenotype severity of the EMC10‐related NDD towards the more severe end. Being based on the cumulative phenotypic analysis of all available cases with defective EMC10, Figure 1B depicts the most common clinical features of EMC10‐related NDD.
Akin to defective EMC1 presentation, we report signs of cerebellar ataxia with detectable loss of cerebellar volume, persistent hypotonia, movement disorders, CC thinning, and anomalies in the hippocampi in our cohort. 4 , 5 , 6 , 7 The presence of truncal ataxia in the EMC10 cohort might replicate the abnormal gait observed in the homozygous emc10‐knockout mouse models characterized by the International Mouse Genotyping Consortium (https://www.mousephenotype.org/data/genes/MGI:1916933). 21 The EMC1 cohort has shown progressive brain atrophy, most pronounced in the cerebellum, suggesting a neurodegenerative process. 4 The presence of motor regression, progressive disease course, and supratentorial brain atrophy in the current cohort might also imply underlying neurodegeneration. The described expansion of the defective EMC10 phenotype suggests that clinical features might overlap between EMC1 and EMC10 defects.
Given that EMC10 is a ubiquitously expressed gene and knockout mouse (emc10−/−) models showed decreased bone mineral content (https://www.mousephenotype.org/data/genes/MGI:1916933), 21 the hyperparathyroidism together with osteoporosis and nephrocalcinosis observed in F2:S1, F2:S2, and the three previously reported cases might be linked to dysfunctional EMC10 presentation.
Regarding the genotype–phenotype correlation, cases with stop gained variants had more severe symptoms including regression combined with persisting axial hypotonia, seizures, and ataxic gait.
In conclusion, this report expands the genetic and clinical spectrum of EMC10 deficiency, provides a comprehensive dysmorphological assessment, and highlights an overlap between the clinical features of EMC10‐ and EMC1‐related NDD.
Conflict of Interest
The authors have no conflict of interest to report.
Web Resources
gnomAD, https://gnomad.broadinstitute.org/.
Ensemble, https://www.ensembl.org/index.html.
Uniprot, https://www.uniprot.org/.
GeneMatcher, https://genematcher.org/.
OMIM, https://www.omim.org/.
Iranome, http://www.iranome.ir/.
Varnomen, http://varnomen.hgvs.org/.
Varsome, https://varsome.com/.
Database of Genome Variants, http://dgv.tcag.ca/.
UKBB, https://www.ukbiobank.ac.uk/.
GME Variome, http://igm.ucsd.edu/gme/data‐browser.php.
TOPMed, https://bravo.sph.umich.edu/freeze8/hg38/gene/snv/EMC10.
Supporting information
Data S1. Supplementary case reports.
Table S1. Extended clinical table.
Table S2. Detailed facial dysmorphic features for all subjects whose photographs were reviewed.
Table S3. Frequency of each dysmorphic feature in EMC10 cohort with available facial photos.
Acknowledgments
We thank all patients and relatives for consent to be part of the study. Families 1 and 4 were collected as part of the SYNaPS Study Group collaboration funded by The Wellcome Trust and strategic award (Synaptopathies) funding (WT093205 MA and WT104033AIA) and research was conducted as part of the Queen Square Genomics group at University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. We are also grateful to Queen Square genomics at the Institute of Neurology University College London, supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre, for the bioinformatics support. For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission. Open access funding enabled and organized by ProjektDEAL.
Funding Information
This research was funded in part, by the Wellcome Trust [WT093205MA, WT104033AIA, and the Synaptopathies Strategic Award, 165908]. This study was funded by the Medical Research Council (MRC) (MR/S01165X/1, MR/S005021/1, G0601943). D. D. S. was supported by the National Institute of Neurological Disorders and Stroke (NINDS) Neurology Resident Research Education Program (R25 NS070682). For the purpose of Open Access, the author has applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Funding Statement
This work was funded by Medical Research Council grants G0601943, MR/S005021/1, and MR/S01165X/1; National Institute of Neurological Disorders and Stroke (NINDS) Neurology Resident Research Education Program grant R25 NS070682; Wellcome Trust grants 165908, WT093205MA, and WT104033AIA.
References
- 1. Chitwood PJ, Juszkiewicz S, Guna A, et al. EMC is required to initiate accurate membrane protein topogenesis. Cell. 2018;175(6):1507‐1519.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guna A, Volkmar N, Christianson JC, Hegde RS. The ER membrane protein complex is a transmembrane domain insertase. Science. 2018;359(6374):470‐473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Christianson JC, Olzmann JA, Shaler TA, et al. Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol. 2011;14(1):93‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harel T, Yesil G, Bayram Y, et al. Monoallelic and biallelic variants in EMC1 identified in individuals with global developmental delay, hypotonia, scoliosis, and cerebellar atrophy. Am J Hum Genet. 2016;98(3):562‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eldomery MK, Coban‐Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cabet S, Lesca G, Labalme A, et al. Novel truncating and missense variants extending the spectrum of EMC1‐related phenotypes, causing autism spectrum disorder, severe global development delay and visual impairment. Eur J Med Genet. 2020;63(6):103897. [DOI] [PubMed] [Google Scholar]
- 7. Geetha TS, Lingappa L, Jain AR, et al. A novel splice variant in EMC1 is associated with cerebellar atrophy, visual impairment, psychomotor retardation with epilepsy. Mol Genet Genomic Med. 2018;6(2):282‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Umair M, Ballow M, Asiri A, et al. EMC10 homozygous variant identified in a family with global developmental delay, mild intellectual disability, and speech delay. Clin Genet. 2020;98(6):555‐561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shao DD, Straussberg R, Ahmed H, et al. A recurrent, homozygous EMC10 frameshift variant is associated with a syndrome of developmental delay with variable seizures and dysmorphic features. Genet Med. 2021;23(6):1158‐1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haddad‐Eid E, Gur N, Eid S, et al. The phenotype of homozygous EMC10 variant: a new syndrome with intellectual disability and language impairment. Eur J Paediatr Neurol. 2022;37:56‐61. [DOI] [PubMed] [Google Scholar]
- 11. Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36(10):928‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Makrythanasis P, Maroofian R, Stray‐Pedersen A, et al. Biallelic variants in KIF14 cause intellectual disability with microcephaly. Eur J Hum Genet. 2018;26:330‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yaron Y, Ofen Glassner V, Mory A, et al. Exome sequencing as a first‐tier test for fetuses with severe central nervous system structural anomalies. Ultrasound Obstet Gynecol. 2022. doi: 10.1002/uog.24885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ucuncu E, Rajamani K, Wilson SCW, et al. MINPP1 prevents intracellular accumulation of the chelator inositol hexakisphosphate and is mutated in pontocerebellar hypoplasia. Nat Commun. 2020;11(1):6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Basel‐Salmon L, Orenstein N, Markus‐Bustani K, et al. Improved diagnostics by exome sequencing following raw data reevaluation by clinical geneticists involved in the medical care of the individuals tested. Genet Med. 2019;21(6):1443‐1451. [DOI] [PubMed] [Google Scholar]
- 16. Wideman JG. The ubiquitous and ancient ER membrane protein complex (EMC): tether or not? F1000Res. 2015;4:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lahiri S, Chao JT, Tavassoli S, et al. A conserved endoplasmic reticulum membrane protein complex (EMC) facilitates phospholipid transfer from the ER to mitochondria. PLoS Biol. 2014;12(10):e1001969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Satoh T, Ohba A, Liu Z, et al. dPob/EMC is essential for biosynthesis of rhodopsin and other multi‐pass membrane proteins in drosophila photoreceptors. Elife. 2015;4:e06306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paillusson S, Stoica R, Gomez‐Suaga P, et al. There's something wrong with my MAM; the ER‐mitochondria Axis and neurodegenerative diseases. Trends Neurosci. 2016;39(3):146‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korzhevskii D, Karpenko M, Kirik O. Microtubule‐associated proteins as indicators of differentiation and the functional state of nerve cells. Neurosci Behav Physiol. 2012;42(3):215‐222. [Google Scholar]
- 21. Dickinson M, Flenniken A, Ji X, et al. High‐throughput discovery of novel developmental phenotypes. Nature. 2016;537:508‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reboll MR, Korf‐Klingebiel M, Klede S, et al. EMC10 (endoplasmic reticulum membrane protein complex subunit 10) is a bone marrow‐derived angiogenic growth factor promoting tissue repair after myocardial infarction. Circulation. 2017;136(19):1809‐1823. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary case reports.
Table S1. Extended clinical table.
Table S2. Detailed facial dysmorphic features for all subjects whose photographs were reviewed.
Table S3. Frequency of each dysmorphic feature in EMC10 cohort with available facial photos.