

Graphical abstract

Keywords: SARS-CoV-2, GS-441524, Deuterated
Abstract
The COVID-19 caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is continuing to spread around the world. GS-441524 is the parent nucleoside of remdesivir which is the first drug approved for the treatment of COVID-19, and demonstrates strong activity against SARS-Cov-2 in vitro and in vivo. Herein, we reported the synthesis of a series of deuterated GS-441524 analogs, which had deuterium atoms up to five at the ribose and the nucleobase moieties. Compared to GS-441524, all the deuterated compounds showed similar inhibitory activities against SARS-CoV-2 in vitro.
Introduction
Currently, the world is suffering from the COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 is a new type of coronavirus and the viral infection can lead to the acute respiratory distress syndrome [1]. Up to now, COVID-19 has caused more than 6 million deaths and major economic losses worldwide, making it one of the deadliest epidemics in human history. Since the outbreak of the pandemic, great efforts have been made to find effective therapy for this disease. In October 2020, remdesivir received the approval by the Food and Drug Administration (FDA) for treating hospitalized patients with COVID-19, and was recently expanded to be used for non-hospitalized patients [2].
Remdesivir is a phosphoramidate prodrug that is first rapidly hydrolyzed to GS-704277, and then metabolized to the corresponding nucleoside monophosphate. At last, the nucleoside monophosphate is phosphorylated to the active nucleoside triphosphate form (GS-443902) (Scheme 1 ) [3]. The nucleoside monophosphate can also be dephosphorylated to produce GS-441524, which is the predominant and persistent metabolite of the nucleoside monophosphate in human blood plasma. GS-441524 is the parent nucleoside of remdesivir, which exhibited similar to or more potent activity than remdesivir against SARS-CoV-2 in vivo [4], [5]. Compared with remdesivir, GS-441524 has some advantages including easier synthesis, more safety, lower dose-related toxicity, and better oral administration potential, so scientists believe that it may has better therapeutic effect for the treatment of COVID-19 [5], [6].
Scheme 1.

The metabolic pathway of remdesivir.
Deuterium, the stable isotope of hydrogen, is often used to replace hydrogen in compounds, which finds multiple uses in protein crystallography, NMR, MS, medical imaging, elucidation of organic and bio-organic mechanism, etc [7]. Deuterated compounds are widely used as internal standards for bioanalytical methods, making important contributions to the pharmacokinetic studies of novel drugs [8]. Moreover, the introduction of deuterium in drugs has been recognized as a potential path to me-better drugs. The mass of deuterium is twice that of hydrogen, resulting in a little stronger carbon-deuterium bond. In some cases, this small change in bond-strength could impart advantages in efficacy, metabolism or safety to the deuterated drugs [9], [10], [11]. To achieve a favorable outcome, it is important to determine the deuterated position, which requires sufficient theoretical analysis and experimental verification. GS-441524 and its oral prodrug are promising antiviral agents for COVID-19. Notably, a deuterated, oral GS-441524 tri-isobutyrate ester prodrug (VV116) has entered phase III clinical trials for evaluating its efficacy against SARS-CoV-2 infection [12], [13]. In this study, we synthesized different deuterated GS-441524 analogs at different position on the ribose and nucleobase moieties, and evaluated their efficacy against SARS-CoV-2 in vitro.
Results and discussion
Chemistry
The synthetic route of the di-deuterated GS-441524 analog (6) was described in Scheme 2 . With GS-441524 as the starting material, firstly, it was protected with 2,2-dimethoxypropane to give the isopropylidene 2 in 58% yield. Oxidation of 2 with TEMPO gave intermediate 3 which was then reached with (trimethylsilyl)diazomethane to give the methylester 4 in 59% yield. Intermediate 4 was reduced by sodium borodeuteride (NaBD4) in anhydrous tetrahydrofuran and deuterated methanol to afford the deuterated intermediate 5. Further treatment of 5 with hydrochloric acid produced compound 6.
Scheme 2.

Synthesis of the di-deuterated GS-441524 analog. Reagents and conditions: (i) 2,2-dimethoxypropane, p-toluenesulfonic acid monohydrate, acetone, 45 °C, 2 h; (ii) TEMPO, iodobenzene diacetate, sodium bicarbonate (NaHCO3), acetonitrile/water, rt, 5 h; (iii) trimethylsilyldiazomethane, THF/methanol, rt, 1 h; (iv) NaBD4, anhydrous THF/deuterated methanol, rt, 1 h; (v) HCl, THF, 40 °C, 12 h.
The tri-deuterated GS-441524 analog (15) was prepared from compound 7, a known intermediate for the synthesis of remdesivir, as shown in Scheme 3 . Iodination of the starting material 7 with iodine in DMF furnished 8 in 58% yield. The synthesis of compound 9 had been recently reported using NaBD4 by metal-catalyzed coupling reaction or deuterium oxide (D2O) by quenching the in situ formed organometallic intermediate from 8 [12]. In this study, we found that the synthesis of 9 could be achieved by reducing 8 with triethylamine, palladium and deuterium gas. In this method, the yield of 9 could reach 92% and the deuterated ratio could up to 99%. Intermediate 9 was subjected to deprotection with BCl3 to afford the mono-deuterated GS-441524 (10) in 83% yield. Then, tri-deuterated GS-441524 analog could be prepared from the 10 following the reactions described in Scheme 2.
Scheme 3.

Synthesis of the tri-deuterated GS-441524 analog. Reagents and conditions: (i) iodine, DMF, rt, 12 h; (ii) triethylamine, Pd/C, D2, anhydrous THF, 60 °C, 1 h;(iii) BCl3, DCM, −40 °C, 1 h; (iv) 2,2-Dimethoxypropane, p-toluenesulfonic acid monohydrate, acetone,45 °C, 2 h; (v) TEMPO, iodobenzene diacetate, NaHCO3, acetonitrile/water, rt, 5 h; (vi) trimethylsilyldiazomethane, THF/methanol, rt, 1 h; (vii) NaBD4, anhydrous THF/deuterated methanol, rt, 1 h; (viii) HCl, THF, 40 °C, 12 h.
As shown in Scheme 4 , the penta-deuterated GS-441524 analog (36) could be synthesized from diacetone-d-glucose 16, a common and cheap sugar material. At the beginning, intermediate 26 was prepared by reference to the approach reported by Chattopadhyaya et al. [14]. The starting material 16 was oxidized by 2-iodoxybenzoic acid to give the ketone 17, which was converted to compound 18 by H-D exchanged with D2O. Reduction of 18 with NaBD4 gave the C3 and C4 di-deuterated compound 19 which was protected with benzyl bromide (BnBr) to produce 20. The ethylene glycol derivatives 21 was smoothly prepared from 20 in the presence of aqueous acetic acid, and then oxidized by sodium periodate to afford the aldehyde 22. Reaction of 22 with Br2 in the presence of NaHCO3 resulted in the formation of the methylester 23, which was subjected to reduction by NaBD4, followed by introduction of a benzyl protecting group into C2 hydroxyl group to give 25. Treatment of 25 with hydrochloric acid in methanol furnished 26, which was protected with benzyl to provide the ribose derivatives 27. The resulting 27 was subjected to demethylation and oxidation to yield the lactone 29. The glycosylation step of 29 was achieved by utilizing the strategy initially developed by our group’s previous work consisting of three sequential steps (Weinreb amidation, O-TMS protection, and Grignard addition) [15]. The hemiacetal 32 was converted to the 1′-cyano derivative 33 in the presence of trimethylsilyl trifluoromethanesulfonate, trifluoromethanesulfonic acid and trimethylsilyl cyanide and then the iodoation of 33 was achieved by NIS and treatment with trifluoroacetic acid. Reduction of the iodo-intermediate 34 with deuterium gas in the presence of Pd/C and triethylamine yielded 35. The three benzyl protecting groups were removed using boron trichloride to afford the penta-deuterated GS-441524 analog (36).
Scheme 4.

Synthesis of the penta-deuterated GS-441524 analog. Reagents and conditions: (i) IBX, acetonitrile, 85 °C, 6.5 h; (ii) D2O, pyridine, 95 °C for 20 min, then 1 day, rt, repeated 3 times (iii) NaBD4, deuterated methanol/anhydrous THF, rt, 2 h; (iv) NaH, BnBr, DMF, rt, 1 h; (v) 80% aqueous acetic acid, 40 °C, 2 h; (vi) NaIO4, ethanol/water, rt, 1 h; (vii) NaHCO3, Br2, methanol/water, rt, 3 h; (viii) NaBD4, anhydrous THF/deuterated methanol, rt, 2 h; (ix) NaH, BnBr, DMF, rt, 2 h; (x) HCl, methanol, 65 °C, 1 h; (xi) NaH, BnBr, DMF, rt, 2 h; (xii) 80% acetic acid aqueous solution, 60 °C, 4 h; (xiii) I2, K2CO3, tert-butanol, 80 °C, 4 h; (xiv) N,O-dimethylhydroxylamine hydrochloride, 2 M i-PrMgCl,anhydrous THF, 0 °C, 1.5 h; (xv) imidazole, TMSCl, DCM, rt, 0.5 h; (xvi) TMSCl, 3 M MeMgBr, 1.3 M i-PrMgCl·LiCl, anhydrous THF, −10 to 0 °C for 1 h, then 0 °C for 1 h; (xvii) TMSOTf, TFMS, TMSCN, DCM, −70 °C, 1 h; (xviii) NIS, TFA, DMF, 50 °C, 1 h; (xix) triethylamine, Pd/C, D2, anhydrous THF, 60 °C, 1 h;(xx) BCl3, DCM, −40 °C, 4 h.
The tetra-deuterated GS-441524 (37) analog was obtained by removing the benzyl groups of 33 with boron trichloride (Scheme 5 ).
Scheme 5.

Synthesis of the tetra-deuterated GS-441524 analog. Reagents and conditions:(i) BCl3, DCM, −40 °C, 4 h.
In vitro anti‐SARS-Cov-2 activity
With these deuterated GS-441524 analogs in hand, we tested their in vitro anti-SARS-CoV-2 activities. The inhibition of SARS-CoV-2 replication was evaluated in Vero E6 cells with GS-441524 as the positive control, the results were summarized in Table 1 . As expected, all deuterated GS-441524 analogs maintained strong antiviral activity of RDV-N against SARS-CoV-2.
Table 1.
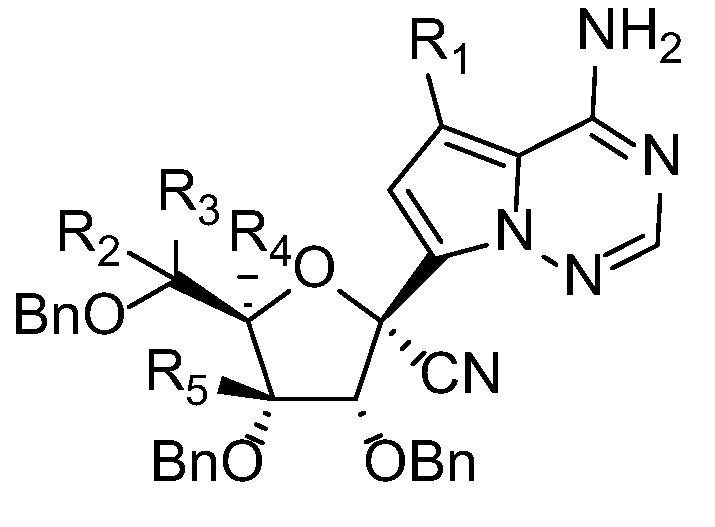
| Compound | R1 | R2 | R3 | R4 | R5 | EC50(μM) | CC50(μM) |
|---|---|---|---|---|---|---|---|
| 1 | H | H | H | H | H | 0.33 | >100 |
| 6 | H | D | D | H | H | 0.25 | >100 |
| 10 | D | H | H | H | H | 0.24 | >100 |
| 15 | D | D | D | H | H | 0.23 | >100 |
| 36 | D | D | D | D | D | 0.23 | >100 |
| 37 | H | D | D | D | D | 0.31 | >100 |
Conclusion
In conclusion, we disclosed the synthesis and antiviral evaluation of different deuterated GS-441524 analogs. All deuterated GS-441524 analogs displayed strong antiviral activity similar with GS-441524. These compounds are useful internal standards for VV116 and all prodrugs of GS-441524. Meanwhile, these synthetic routes may provide a method for the synthesis of other deuterated nucleosides.
Experimental section
Chemistry
(3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile (2)
To a mixture of GS-441524 (0.5 g, 1.72 mmol) and 2,2-dimethoxypropane (0.89 g, 8.6 mmol) in acetone (10 mL), followed by the addition of p-toluenesulfonic acid monohydrate (0.59 g, 3.1 mmol) in one portion. The mixture was allowed to warm to 45 °C and stirred for about 2 h. The reaction mixture was then poured into saturated NaHCO3 solution (25 mL) and extracted with ethyl acetate (25 mL). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 2 (0.33 g, yield 58%) as a solid.
(3aS,4S,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxylic acid (3)
Compound 2 (0.25 g, 0.75 mmol) was dissolved in acetonitrile/water (V/V = 1/1, 15 mL), followed by the addition of TEMPO (0.047 g, 0.3 mmol), iodobenzene diacetate (1.06 g, 3.3 mmol) and NaHCO3 (0.25 g, 3 mmol). The mixture was stirred at room temperature for about 5 h. TLC showed that the reaction was completed. The reaction mixture was poured into 0.5 M NaHCO3 solution (20 mL) and extracted with ethyl acetate (20 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 3 (0.23 g, yield 89%) as a solid.
Methyl (3aS,4S,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxylate (4)
To a stirred solution of 3 (180 mg,0.52 mmol) in THF/methanol (V/V = 1/1, 8 mL) was added 2 M trimethylsilyldiazomethane in hexane solution (1.2 mL, 2.4 mmol). The mixture was stirred at room temperature for about 1 h. TLC showed that the reaction was completed. Acetic acid was then added dropwise to the reaction solution until no more bubbles. The solvent was evaporated under reduced pressure. The residue was added saturated NaHCO3 solution (15 mL) and extracted with ethyl acetate (20 mL). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 4 (0.11 g, yield 59%) as a solid.1H NMR (500 MHz, DMSO‑d6) δ 8.06–7.88 (m, 3H), 6.92 (d, J = 4.6 Hz, 1H), 6.89 (d, J = 4.6 Hz, 1H), 5.52 (d, J = 6.1 Hz, 1H), 5.37 (dd, J = 6.1, 2.2 Hz, 1H), 4.92 (d, J = 2.2 Hz, 1H), 3.40 (s, 3H), 1.62 (s,3H), 1.42 (s, 3H).
(3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-6-(hydroxymethyl-d2)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile (5)
To a solution of compound 4 (100 mg, 0.28 mmol) in anhydrous THF (4 mL) and deuterated methanol (1 mL), NaBD4 (47 mg, 1.12 mmol) was slowly added in the ice bath. The mixture was stirred at room temperature for about 1 h. TLC showed that the reaction was completed. The reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL × 2). The organic layer was evaporated and the residue was added methanol (10 mL). The solution was added dropwise 1 M HCl to a final pH of 2–3, stirred at room temperature for about 1–2 h and then added to pH of 7. The reaction mixture was poured into water (20 mL) and extracted with ethyl acetate (20 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 5 (60 mg, yield 64%) as a solid.
(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxymethyl-d2)tetrahydrofuran-2-carbonitrile (6)
To a stirred solution of 5 (60 mg, 0.18 mmol) in THF (2 mL), was added concentrated HCl (0.4 mL). The mixture was stirred at 40 °C for about 12 h. TLC showed that the reaction was completed. The reaction was added 1 M NaOH solution to pH of 7 and evaporated THF. The residue was filtered and the solid residue was slurried with distilled water (10 mL) and ethyl acetate (10 mL) to give 6 (34 mg, yield 64%) as a white solid. 1H NMR (500 MHz, DMSO‑d6) δ 8.03–7.82 (m, 3H), 6.92 (d, J = 4.5 Hz, 1H), 6.89 (d, J = 4.6 Hz, 1H), 6.10 (d, J = 6.3 Hz, 1H), 5.20 (d, J = 5.2 Hz, 1H), 4.89 (s, 1H), 4.66 (t, J = 5.7 Hz, 1H), 4.06 (d, J = 5.4 Hz, 1H), 4.00–3.93 (m, 1H). 13C NMR (126 MHz, DMSO‑d6) δ 156.10, 148.35, 124.35, 117.81, 117.01, 111.26, 101.27,85.80, 79.04, 74.70, 70.54. MS m/z = 294.0 [M + 1]+.
(2R,3R,4R,5R)-2-(4-amino-5-iodopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-carbonitrile (8)
To a solution of 7 (561 mg, 1.0 mmol) in DMF (5 mL), iodine (508 mg, 2 mmol) was slowly added. The mixture was stirred at room temperature for about 12 h. TLC showed that the reaction was completed. The reaction mixture was poured into mixed solution of sodium sulfite and sodium carbonate (30 mL) and extracted with ethyl acetate (30 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 8 (400 mg, yield 58%) as a solid. 1H NMR (500 MHz, DMSO‑d6) δ 7.98 (s, 1H), 7.40–7.22 (m, 15H), 6.86 (s, 1H), 4.91 (d, J = 11.7 Hz, 1H), 4.84–4.78 (m, 2H), 4.50 (q, J = 12.0 Hz, 4H), 4.41–4.35 (m, 1H), 4.12–4.08 (m, 1H), 3.72 (dd, J = 11.2, 2.8 Hz, 1H), 3.59 (dd, J = 11.2, 4.1 Hz, 1H).
(2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl)tetrahydrofuran-2-carbonitrile (9)
Under nitrogen protection, 8 (69 mg, 0.1 mmol) was added in anhydrous THF (5 mL). Triethylamine (20 mg, 0.2 mmol) and 10% Pd/C (7 mg) were added, the system was replaced by nitrogen gas for two times and filled with deuterium gas to normal pressure. The mixture was warmed to 60 °C and deuterium gas was pressurized to about 1 Mpa for about 30 min. Then, the reaction mixture was stirred for about 1 h. After the mixture was cooled to 20 °C, the system was replaced by nitrogen gas once and diluted with ethyl acetate. The reaction mixture was filtered, then washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 9 (52 mg, yield 92%) as a solid. 1H NMR (500 MHz, Methanol-d4) δ 7.65 (s, 1H), 7.34–7.16 (m, 15H), 6.84 (s, 1H), 4.95 (d, J = 5.1 Hz, 1H), 4.74 (q, J = 11.9 Hz, 2H), 4.57 (d, J = 11.9 Hz, 1H), 4.53–4.43 (m, 4H), 4.11 (t, J = 5.2 Hz, 1H), 3.73 (dd, J = 10.9, 4.1 Hz, 1H), 3.62 (dd, J = 10.9, 4.5 Hz, 1H).
(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-carbonitrile (10)
To a stirred solution of 9 (90 mg, 0.16 mmol) in DCM (10 mL), 1 M BCl3 (0.56 mL, 0.56 mmol) in DCM was added dropwise at −60 °C. The mixture was allowed to warm to −40 °C and stirred for about 1 h. TLC showed that the reaction was completed. The reaction solution concentrated and the residue was purified by column chromatography on silica gel using methanol and dichloromethane as the eluent to give 10 (38 mg, yield 83%) as a solid. 1H NMR (500 MHz, DMSO‑d6) δ 8.03–7.78 (m, 3H), 6.87 (s, 1H), 6.09 (d, J = 6.3 Hz, 1H), 5.19 (d, J = 5.2 Hz, 1H), 4.91(t, J = 5.7 Hz, 1H), 4.64 (t, J = 5.7 Hz, 1H), 4.09–4.02 (m, 1H), 3.99–3.92 (m, 1H), 3.68–3.59 (m, 1H), 3.55–3.47 (m, 1H). MS m/z = 293.0 [M + 1]+.
(3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-6-(hydroxymethyl)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile (11)
Following the above procedure of synthesizing 2, 11 was obtained from compound 10 (1.17 g, 4.0 mmol) with 2,2-dimethoxypropane (2.08 g, 20.0 mmol) and p-toluenesulfonic acid monohydrate (1.37 g, 7.2 mmol) in acetone (20 mL). Usual work-up to give crude product 11 (0.86 g, 65%) as a solid.
(3aS,4S,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxylic acid (12)
Following the above procedure of synthesizing 3, the reaction of compound 11 (225 mg, 0.68 mmol) with TEMPO (20 mg, 0.14 mmol), iodobenzene diacetate (660 mg, 2.04 mmol) and NaHCO3 (170 mg, 2.04 mmol) in acetonitrile/water (V/V = 1/1, 10 mL) afforded crude product 12, which could be used in the next step without further purification.
Methyl (3aS,4S,6R,6aR)-6-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-6-cyano-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carboxylate (13)
Following the above procedure of synthesizing 4, 13 was obtained from crude 12 with 2 M trimethylsilyldiazomethane hexane solution in THF/methanol (V/V = 1/1, 8 mL). Usual work-up and purification on silica gel using ethyl acetate and petroleum ether as the eluent to give 13 (60 mg, 24% over two steps) as a solid.
(3aR,4R,6R,6aR)-4-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-6-(hydroxymethyl-d2)-2,2-dimethyltetrahydrofuro[3,4-d][1,3]dioxole-4-carbonitrile (14)
Following the above procedure of synthesizing 5, the reaction of compound 13 (60 mg, 0.17 mmol) with NaBD4 (28 mg, 0.68 mmol) in anhydrous THF/deuterated methanol (V/V = 5/1, 6 mL) afforded crude product 14 as a solid.
(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-3,4-dihydroxy-5-(hydroxymethyl-d2)tetrahydrofuran-2-carbonitrile (15)
Following the above procedure of synthesizing 6 (0.05 g, 0.15 mmol), compound 15 was obtained from crude 14 with concentrated HCl (0.3 mL) in THF (1.5 mL). Usual work-up and the solid residue was slurried with distilled water (10 mL) and ethyl acetate (10 mL) to give 15 (30 mg, 68% over two steps) as a white solid.1H NMR (500 MHz, DMSO‑d6) δ 8.04–7.76 (m, 3H), 6.89 (s, 1H), 6.07 (d, J = 6.3 Hz, 1H), 5.18 (d, J = 5.2 Hz, 1H), 4.87 (s, 1H), 4.67 (t, J = 5.8 Hz, 1H), 4.07 (d, J = 5.3 Hz, 1H), 4.01–3.93 (m, 1H). m/z = 295.0 [M + 1]+.
(3aR,5R,6aS)-5-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyldihydrofuro[2,3-d][1,3]dioxol-6(5H)-one (17)
To a solution of 16 (10.4 g, 40 mmol) in acetonitrile (200 mL), IBX (16.8 g, 60 mmol) was added. The mixture was warmed to 85 °C and stirred for about 6.5 h. TLC showed that the reaction was completed. After cooling to room temperature, the mixture was stirred in the ice bath for about 0.5 h. The reaction mixture was filtered, and the organic layer was evaporated to give crude product 17 and directly to the next step.
(3aR,5R,6aS)-5-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyldihydrofuro[2,3-d][1,3]dioxol-6(5H)-one-5-d (18)
Compound 17 (10.3 g, 40 mmol) was dissolved in pyridine (200 mL), followed by the addition of D2O (40 mL) in one portion. The mixture was warmed to 95 °C and stirred for about 20 min. After cooling to room temperature, the reaction mixture was stirred for 24 h at room temperature. The solvent was evaporated, and repeated the above operation twice again. At last, the solution was evaporated to give crude product 18 and directly to the next step.
(3aR,5S,6R,6aR)-5-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5,6-d2-6-ol (19)
To a solution of compound 18 (10.3 g, 39.73 mmol) in anhydrous THF (20 mL) and deuterated methanol (20 mL), NaBD4 (2.494 g, 59.59 mmol) was slowly added in the ice bath. The mixture was stirred at room temperature for about 2 h. TLC showed that the reaction was completed. The reaction mixture was poured into saturated NH4Cl solution (50 mL), concentrated the solution and extracted with ethyl acetate (60 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 19 (7.17 g, 68% over three steps). 1H NMR (500 MHz, DMSO‑d6) δ 5.65 (d, J = 3.6 Hz, 1H), 5.06 (s, 1H), 4.45 (d, J = 3.6 Hz, 1H), 4.23 (t, J = 7.2 Hz, 1H), 3.93 (t, J = 7.4 Hz, 1H), 3.81 (t, J = 7.8 Hz, 1H), 1.44 (s, 3H), 1.32 (s, 3H), 1.27 (s, 3H), 1.26 (s, 3H).
(3aR,5R,6R,6aR)-6-(benzyloxy)-5-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5,6-d2 (20)
Compound 19 (7.17 g, 27.34 mmol) was dissolved in DMF (30 mL), followed NaH (1.64 g, 41.02 mmol) was slowly added in the ice bath. The mixture was stirred at 0 °C for about 15 min. Then, BnBr (5.61 g, 32.81 mmol) was added dropwise, and about 0.5 h later, the mixture was warmed to room temperature and stirred for about 1 h. TLC showed that the reaction was completed. The reaction mixture was poured into ice water (40 mL), and then warmed to room temperature, extracted with ethyl acetate (40 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 20 and directly to the next step.
(S)-1-((3aR,5R,6R,6aR)-6-(benzyloxy)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl-5,6-d2)ethane-1,2-diol (21)
Crude compound 20 (10.36 g) was dissolved in 80% aqueous acetic acid (80 mL) and stirred at 40 °C for about 2 h. TLC showed that the reaction was completed. The reaction mixture was cooled to room temperature and extracted with petroleum ether (80 mL × 2). The organic layer was washed with saturated NaHCO3 solution, brine, dried over Na2SO4, and evaporated to give crude product 21 and directly to the next step.
(3aR,5S,6R,6aR)-6-(benzyloxy)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5,6-d2-5-carbaldehyde (22)
To a stirred solution of 21 (7.92 g, 25.37 mmol) in ethanol/water (V/V = 1, 40 mL), a solution of sodium periodate (5.43 g, 25.37 mmol) in water (15 mL) was added slowly. The mixture was stirred at room temperature for about 1 h. TLC showed that the reaction was completed. The residue was filtered, and the solution was concentrated. The resulting solution was diluted with ethyl acetate (150 mL), then washed with brine, dried over Na2SO4, and evaporated to give crude product 22.
Methyl (3aR,5S,6S,6aR)-6-(benzyloxy)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5-carboxylate-5,6-d2 (23)
To a solution of 22 (6.30 g, 22.48 mmol) in methanol/water (V/V = 9/1, 45 mL), NaHCO3 (37.8 g, 450 mmol) was slowly added, followed by the slow addition of Br2 (14.4 mg, 90 mmol) in methanol/water (V/V = 3/1, 45 mL). The mixture was stirred at room temperature for about 3 h. TLC showed that the reaction was completed. The reaction mixture was poured into saturated NaS2O3/NaHCO3 (1/1) solution (40 mL) and extracted with tert-butyl methyl ether (80 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 23 and directly to the next step.
((3aR,5R,6R,6aR)-6-(benzyloxy)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxol-5-yl-5,6-d2)methan-d2-ol (24)
To a solution of compound 23 (5.96 g, 19.19 mmol) in anhydrous THF/deuterated methanol (V/V = 3/1, 20 mL), NaBD4 (1.21 g, 28.79 mmol) was slowly added in the ice bath. The mixture was stirred at room temperature for about 2 h. TLC showed that the reaction was completed. The reaction mixture was poured into saturated NH4Cl solution (40 mL), concentrated the solution and extracted with ethyl acetate (60 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 24 (4.06 g, 52% over 5 steps).1H NMR (500 MHz, DMSO‑d6) δ 7.35 (d, J = 4.3 Hz, 4H), 7.32–7.26 (m, 1H), 5.72 (d, J = 3.7 Hz, 1H), 4.72–4.67 (m, 2H), 4.64 (d, J = 11.8 Hz, 1H), 4.50 (d, J = 11.8 Hz, 1H), 1.45 (s, 3H), 1.29 (s, 3H).
(3aR,5R,6R,6aR)-6-(benzyloxy)-5-((benzyloxy)methyl-d2)-2,2-dimethyltetrahydrofuro[2,3-d][1,3]dioxole-5,6-d2 (25)
Compound 24 (4.06 g, 14.27 mmol) was dissolved in DMF (15 mL), followed NaH (856.4 mg, 21.41 mmol) was slowly added in the ice bath. The mixture was stirred at 0 °C for about 15 min. Then, BnBr (2.93 g, 17.13 mmol) was added dropwise, and about 0.5 h later, the mixture was warmed to room temperature and stirred for about 2 h. TLC showed that the reaction was completed. The reaction mixture was poured into ice water (20 mL), and after warm to room temperature, extracted with ethyl acetate (20 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 25 and directly to the next step.
(2S,3R,4S,5R)-4-(benzyloxy)-5-((benzyloxy)methyl-d2)-2-methoxytetrahydrofuran-4,5-d2-3-ol (26)
To a stirred solution of 25 (4.64 g, 12.38 mmol) in methanol (20 mL), was added concentrated HCl (645 mg, 2.48 mmol). The mixture was stirred at 65 °C for about 1 h. TLC showed that the reaction was completed. The reaction was slowly added NaHCO3 to pH of 7 and extracted with ethyl acetate (40 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 26 (3.45 g, 69% over two steps).
(2R,3R,4R,5S)-3,4-bis(benzyloxy)-2-((benzyloxy)methyl-d2)-5-methoxytetrahydrofuran-2,3-d2 (27)
Compound 26 (4.29 g, 12.30 mmol) was dissolved in DMF (15 mL), followed NaH (738 mg, 18.45 mmol) was slowly added in the ice bath. The mixture was stirred at 0 °C for about 15 min. Then, BnBr (2.52 g, 14.76 mmol) was added dropwise, and about 0.5 h later, the mixture was warmed to room temperature and stirred for about 2 h. TLC showed that the reaction was completed. The reaction mixture was poured into ice water (40 mL), and after warm to room temperature, extracted with ethyl acetate (40 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 27 (3.90 g, 91%).1H NMR (500 MHz, DMSO‑d6) δ 7.40–7.24 (m, 15H), 4.92 (d, J = 4.3 Hz, 1H), 4.63–4.42 (m, 6H), 3.89 (d, J = 4.3 Hz, 1H), 3.29 (s, 3H).
(2S,3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)tetrahydrofuran-4,5-d2-2-ol (28)
Compound 27 (3.90 g, 8.89 mmol) was dissolved in 80% acetic acid aqueous solution (50 mL). The mixture was stirred at 60 °C for about 4 h. TLC showed that the reaction was completed. The reaction was extracted with ethyl acetate (40 mL × 3). The organic layer was washed with saturated NaHCO3 solution, brine, dried over Na2SO4, and evaporated to give crude product 28.
(3R,4R,5R)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)dihydrofuran-2(3H)-one-4,5-d2 (29)
To a solution of 28 (3.62 g, 8.53 mmol) in tert-butanol (30 mL), iodine (6.5 g, 25.6 mmol) and K2CO3 (3.53 g, 25.6 mmol) were slowly added. The mixture was stirred at 80 °C for about 4 h. TLC showed that the reaction was completed. The reaction mixture was poured into saturated NaS2O3 solution (25 mL) and extracted with ethyl acetate (35 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 29 (3.1 g, 83% over two steps).1H NMR (500 MHz, DMSO‑d6) δ 7.40–7.18 (m, 15H), 4.73–4.59 (m, 4H), 4.57 (s, 1H), 4.48 (s, 2H).
(2R,3R)-2,3,5-tris(benzyloxy)-4-hydroxy-N-methoxy-N-methylpentanamide-3,4,5,5-d4 (30)
To a mixture of 29 (3.87 g, 9.16 mmol) and N,O-dimethylhydroxylamine hydrochloride (1.61 g, 16.5 mmol) in anhydrous THF (20 mL), followed by the slowly addition of 2 M i-PrMgCl THF solution (16.5 mL, 33 mmol) in an ice bath. The mixture was stirred at 0 °C for about 1.5 h, and TLC showed that the reaction was completed. The mixture was poured into saturated NH4Cl aqueous solution (50 mL) and extracted with ethyl acetate (75 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and evaporated to give crude product 30 and directly to the next step.
(2R,3S)-2,3,5-tris(benzyloxy)-N-methoxy-N-methyl-4-((trimethylsilyl)oxy)pentanamide-3,4,5,5-d4 (31)
Compound 30 (9.16 mmol) was dissolved in DCM (50 mL) and cooled to −20 °C. Imidazole (1.12 g, 16.49 mmol) was added, followed by the slow addition of TMSCl (1.19 g, 10.99 mmol). The mixture was allowed to warm to room temperature and stirred for about 0.5 h. TLC showed that the reaction was completed. The reaction mixture was diluted with DCM (30 mL) and washed with water. The organic layer was separated and concentrated. The obtained oil product was dissolved in tert-butyl methyl ether (60 mL), which was subsequently washed with water and brine. The organic layer was separated, dried over Na2SO4, and evaporated to give crude product 31, which could be used in the next step without further purification.
(2S,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)tetrahydrofuran-4,5-d2-2-ol (32)
Compound 4-amino-7-iodopyrrolo[2,1-f][1,2,4]triazine (2.38 g, 9.16 mmol) was dissolved in anhydrous THF (14 mL) and cooled to −10 °C. TMSCl (1.99 g, 18.32 mmol) was added, and about 10 min later, 3 M MeMgBr (6.11 mL, 18.32 mmol) in ether was added, and after about 20 min, 1.3 M i-PrMgCl·LiCl (9.87 mL, 12.82 mmol) in THF was added. Then, the reaction mixture was stirred at −10 to 0 °C for about 1 h and TLC showed that the material was completely converted into the grignard reagent. Then, compound 31 was dissolved in anhydrous THF (7 mL) was slowly added. The mixture was stirred at 0 °C for 1 h. The solution was added dropwise 1 M HCl to a final pH of 2–3, stirred at room temperature for about 1 h, diluted with water (30 mL) and extracted with ethyl acetate (60 mL × 2). The organic layer was washed with saturated NaHCO3 solution, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 32 (1.96 g, 39% over three steps).
(2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)tetrahydrofuran-2-carbonitrile-4,5-d2 (33)
To a solution of 32 (1.96 g, 3.52 mmol) was dissolved in DCM (20 mL), trimethylsltrifluoromethanesulphonate (1.57 g, 7.04 mmol) was slowly added dropwise in −70 °C, and about 30 min later, trifluoromethanesulfonic acid (1.06 g, 7.04 mmol) was slowly added dropwise. After about 30 min, trimethylsilyl cyanide (1.40 g, 14.08 mmol) was slowly added dropwise and the mixture was stirred for about 1 h. TLC showed that the reaction was completed. Saturated NaHCO3 solution was poured into the reaction and controlled the reaction temperature under −10 °C. Then the mixture was stirred for about 0.5 h. The organic phase was separated and dried over Na2SO4. To the organic phase, silica gel (200–300 mesh, 1 g) was added and the solution was stirred for about 1 h. The mixture was filtered and concentrated under vacuum. The residue was dissolved in toluene and stirred at 80 °C for about 10 min. Then the solution was cooled to room temperature and the mixture was filtered to give 33 (1.54 g, 77%).
(2R,3R,4R,5R)-2-(4-amino-5-iodopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)tetrahydrofuran-2-carbonitrile-4,5-d2 (34)
To a stirred solution of 33 (738 mg, 1.30 mmol) in DMF (10 mL) was slowly added NIS (323 mg, 1.44 mmol) and trifluoroacetic acid (30 mg, 0.26 mmol) in the ice bath. The mixture was stirred at 50 °C for about 1 h. TLC showed that the reaction was completed. The reaction mixture was poured into mixed solution of sodium sulfite and sodium carbonate (30 mL) and extracted with ethyl acetate (30 mL × 2). The organic layer was washed with water, brine, dried over Na2SO4, and concentrated. The residue was slurried with isopropyl ether to give 34 (467 mg, 52%).
(2R,3R,4R,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-3,4-bis(benzyloxy)-5-((benzyloxy)methyl-d2)tetrahydrofuran-2-carbonitrile-4,5-d2 (35)
To a solution of 34 (890 mg, 1.29 mmol) in anhydrous THF (10 mL), triethylamine (261 mg, 2.58 mmol) and 10% Pd/C (89 mg) was added. The system was replaced by nitrogen gas for two times and filled with deuterium gas to normal pressure. The reaction was warmed to 60 °C and deuterium gas is pressurized to about 1 Mpa for about 30 min. Then, the reaction mixture was stirred for about 1 h. After the mixture was cooled to 20 °C, the system was replaced by nitrogen gas once and diluted with ethyl acetate. The reaction mixture was filtered, then washed with water, brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography on silica gel using ethyl acetate and petroleum ether as the eluent to give 35 (653 mg, yield 89%) as a solid. 1H NMR (500 MHz, DMSO‑d6) δ 8.00–7.78 (m, 3H), 7.37–7.22 (m, 15H), 6.75 (s, 1H), 4.90 (s, 1H), 4.84 (d, J = 11.8 Hz, 1H), 4.77 (d, J = 11.7 Hz, 1H), 4.58–4.46 (m, 4H).
(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl-5-d)-3,4-dihydroxy-5-(hydroxymethyl-d2)tetrahydrofuran-2-carbonitrile-4,5-d2 (36)
Following the above procedure of synthesizing 10, the reaction of compound 35 (653 mg, 1.15 mmol) with 1 M BCl3 (3.8 mL, 3.80 mmol) in DCM (13 mL) afforded 36 (281 mg, 82%).1H NMR (500 MHz, DMSO‑d6) δ 8.02–7.79 (m, 3H), 6.88 (s, 1H), 6.08 (d, J = 6.3 Hz, 1H), 5.16 (s, 1H), 4.87 (s, 1H), 4.64 (d, J = 6.3 Hz, 1H). MS m/z = 297.0 [M + 1]+.
(2R,3R,4S,5R)-2-(4-aminopyrrolo[2,1-f][1,2,4]triazin-7-yl)-3,4-dihydroxy-5-(hydroxymethyl-d2)tetrahydrofuran-2-carbonitrile-4,5-d2 (37)
Following the above procedure of synthesizing 10, the reaction of compound 33 (52 mg, 0.09 mmol) with 1 M BCl3 (0.36 mL, 0.36 mmol) in DCM (2 mL) afforded 37 (16 mg, 62%).1H NMR (400 MHz, DMSO‑d6) δ 8.07–7.69 (m, 3H), 6.91 (d, J = 4.5 Hz, 1H), 6.88 (d, J = 4.5 Hz, 1H), 6.08 (d, J = 6.2 Hz, 1H), 5.16 (s, 1H), 4.87 (s, 1H), 4.64 (d, J = 4.6 Hz, 1H). MS m/z = 296.0 [M + 1]+.
Biological studies
Antiviral activity assays in Vero E6 cells
Vero E6 cells were firstly added to 48-well plates (50,000 cells/well). Cells were allowed to adhere for 16–18 h and treated with medium containing graded concentrations of 100 μL/well of nucleoside analogs for 1 h. SARS-CoV-2 was then directly added into cells at an MOI of 0.01. After an hour, the supernatant was discarded, and the infected cells were washed with prewarmed phosphate-buffered saline (PBS) and treated with fresh medium containing graded concentrations of 200 μL/well of the nucleoside analogs. Antiviral activities were assessed by quantitative real-time polymerase chain reaction (qRT-PCR) quantification of viral copy numbers in the cell supernatants 24 h post-infection. The IC50 values of nucleoside analogs were calculated according to the number of virus copies with Graphpad Prism software 8.0. At least three independent experiments were performed.
Cytotoxicity assays in Vero E6 cells
Vero E6 cells were pre-seeded to a 96-well plate (20,000 cells/well) and treated with medium containing gradient concentrations of 100 μL/well of nucleoside analogs next day. After 24 h, the CC50 values of nucleoside analogs were determined with the CCK8 detection kit and calculated with Graphpad Prism software 8.0.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by Shanghai Municipal Science and Technology Major Project and the Lingang Laboratory (No. LG202103-04-04).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.tetlet.2022.154012.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
No data was used for the research described in the article.
References
- 1.Sharma A., Tiwari S., Deb M.K., Marty J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): a global pandemic and treatment strategies. Int. J. Antimicrob. Agents. 2020;56(2) doi: 10.1016/j.ijantimicag.2020.106054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rubin D., Chan-Tack K., Farley J., Sherwat A. FDA approval of remdesivir — a step in the right direction. N. Engl. J. Med. 2020;383:2598–2600. doi: 10.1056/NEJMp2032369. [DOI] [PubMed] [Google Scholar]
- 3.Li Y., Cao L., Li G., Cong F., Li Y., Sun J., Luo Y., Chen G., Li G., Wang P., Xing F., Ji Y., Zhao J., Zhang Y., Guo D., Zhang X. Remdesivir metabolite GS-441524 effectively inhibits SARS-CoV-2 infection in mouse models. J. Med. Chem. 2022;65:2785–2793. doi: 10.1021/acs.jmedchem.0c01929. [DOI] [PubMed] [Google Scholar]
- 4.Pruijssers A.J., George A.S., Schäfer A., Leist S.R., Gralinksi L.E., Dinnon K.H., Yount B.L., Agostini M.L., Stevens L.J., Chappell J.D., Lu X., Hughes T.M., Gully K., Martinez D.R., Brown A.J., Graham R.L., Perry J.K., Du Pont V., Pitts J., Ma B., Babusis D., Murakami E., Feng J.Y., Bilello J.P., Porter D.P., Cihlar T., Baric R.S., Denison M.R., Sheahan T.P. Remdesivir Inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. Cell Rep. 2020;32:107940. doi: 10.1016/j.celrep.2020.107940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yan V.C., Muller F.L. Advantages of the parent nucleoside GS-441524 over remdesivir for covid-19 treatment. ACS Med. Chem. Lett. 2020;11:1361–1366. doi: 10.1021/acsmedchemlett.0c00316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie J., Wang Z. Can remdesivir and its parent nucleoside GS-441524 be potential oral drugs? an in vitro and in vivo DMPK assessment. Acta Pharm. Sinica B. 2021;11:1607–1616. doi: 10.1016/j.apsb.2021.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gant T.G. Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem. 2014;57:3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 8.Higashi T., Ogawa S. Isotope-coded ESI-enhancing derivatization reagents for differential analysis, quantification and profiling of metabolites in biological samples by LC/MS: A review. J. Pharm. Biomed. Anal. 2016;130:181–193. doi: 10.1016/j.jpba.2016.04.033. [DOI] [PubMed] [Google Scholar]
- 9.Cargnin S., Serafini M., Pirali T. A primer of deuterium in drug design. Future Med. Chem. 2019;11:2039–2042. doi: 10.4155/fmc-2019-0183. [DOI] [PubMed] [Google Scholar]
- 10.Pirali T., Serafini M., Cargnin S., Genazzani A.A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 2019;62:5276–5297. doi: 10.1021/acs.jmedchem.8b01808. [DOI] [PubMed] [Google Scholar]
- 11.Timmins G.S. Deuterated drugs: where are we now? Expert Opin. Ther. Pat. 2014;24:1067–1075. doi: 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xie Y., Yin W., Zhang Y., Shang W., Wang Z., Luan X., Tian G., Aisa H.A., Xu Y., Xiao G., Li J., Jiang H., Zhang S., Zhang L., Xu H.E., Shen J. Design and development of an oral remdesivir derivative VV116 against SARS-CoV-2. Cell Res. 2021;31:1212–1214. doi: 10.1038/s41422-021-00570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qian H.-J., Wang Y., Zhang M.-Q., Xie Y.-C., Wu Q.-Q., Liang L.-Y., Cao Y., Duan H.-Q., Tian G.-H., Ma J., Zhang Z.-B., Li N., Jia J.-Y., Zhang J., Aisa H.A., Shen J.-S., Yu C., Jiang H.-L., Zhang W.-H., Wang Z., Liu G.-Y. Safety, tolerability, and pharmacokinetics of VV116, an oral nucleoside analog against SARS-CoV-2, in Chinese healthy subjects. Acta Pharmacol. Sin. 2022 doi: 10.1038/s41401-022-00895-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Földesi A., Chattopadhyaya J. Studies towards the large scale chemical synthesis of the precursors of ribonucleosides-3′,4′,5′,5″- 2H 4 and -2′,3′,4′,5′,5″- 2H 5. Nucleosides Nucleotides Nucleic Acids. 2003;22:2093–2104. doi: 10.1081/ncn-120026632. [DOI] [PubMed] [Google Scholar]
- 15.Xie Y., Hu T., Zhang Y., Wei D., Zheng W., Zhu F., Tian G., Aisa H.A., Shen J. Weinreb amide approach to the practical synthesis of a key remdesivir intermediate. J. Organic Chem. 2021;86:5065–5072. doi: 10.1021/acs.joc.0c02986. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data was used for the research described in the article.