

Abstract
Neutrophils, the most abundant leukocyte population in humans, constantly patrol the body for foreign cells, including pathogens and cancer cells. Once neutrophils are activated, they engage distinct metabolic pathways to fulfill their specialized antipathogen functions. In this review, we examine current research on the metabolism of neutrophil differentiation and antipathogen responses. We also discuss how tumor-associated neutrophils (TANs) can be educated by cytokines and by the nutrient-restrictive milieu of the tumor microenvironment (TME) to suppress antitumor immunity, promote cancer progression, and contribute to biological heterogeneity among tumors. Last, we discuss the clinical implications of circulating neutrophils and infiltrating TANs and consider how targeting TAN metabolism may synergize with cancer immunotherapy.
Introduction
Neutrophils are an essential component of the innate immune system (see Glossary), providing vigilant protection against pathogens. Continuous production of neutrophils by granulopoiesis in the bone marrow is augmented by bacterial and inflammatory factors that promote the rapid differentiation and mobilization of mature neutrophils destined for sites of infection and chronic inflammation [1]. Activated neutrophils eliminate microbes through the release of proteolytic enzymes, engulfment of pathogens through phagocytosis, and a specialized metabolic response called the oxidative burst, a dramatic, transient production of reactive oxygen species (ROS) that participate in pathogen killing [2]. These physiological responses can be subverted in chronic inflammatory diseases, in which the inappropriate recruitment of neutrophils produces tissue damage. Abnormal neutrophil responses also contribute to cancer. Some tumors secrete inflammatory cytokines into the circulation, promoting granulopoiesis and recruitment of TANs; TAN metabolism is highly influenced by the milieu of the TME in ways that alter their biological properties [3]. Thus, while neutrophils have been traditionally considered as homogeneous, short-lived, predominantly glycolytic cells, recent studies have identified subpopulations of TANs with distinct metabolic states that contribute to tumor heterogeneity and, in some cases, cancer progression.
Advancements in immune checkpoint inhibitors and cellular immunotherapy in cancer have stimulated investigation into how the TME impacts immune cell functions, particularly of cytotoxic T cells. There is great interest in understanding how cancer cells and T cells compete for nutrients, and whether this competition prevents T cells from identifying and killing tumor cells. By comparison, we still know relatively little about the metabolic properties of TANs and other immune cells the presence of which in the TME counteracts immunotherapy. In this review, we discuss the metabolic pathways required for physiological neutrophil function, metabolic interplay between TANs and other cells in the TME, and the context-dependent roles of TANs in primary and metastatic tumors.
Metabolism of Neutrophil Differentiation
Neutrophils originate from the bone marrow, where upwards of 1011 neutrophils are produced daily to serve as a patrolling force seeking out pathogens. Neutrophils arise from hematopoietic stem cells and multipotent progenitors through a process termed ‘granulopoiesis’ [4]. Granulopoiesis is regulated by granulocyte colony-stimulating factor (G-CSF), the major cytokine driving differentiation of this lineage [5]. G-CSF signals predominantly through three transcription factors, PU.1, CEBPα, and GFI1, to drive neutrophil differentiation/maturation [6].
Distinct metabolic transitions occur during the differentiation of hematopoietic stem cells into myeloid precursors [7]. While hematopoietic stem cells are glycolytic, due in large part to their residence within hypoxic tissues [8], myeloid precursors engage oxidative phosphorylation more readily (Figure 1). The mechanism for this change involves decreased expression of pyruvate dehydrogenase kinase (PDK), a negative regulator of pyruvate dehydrogenase (PDH) [9]. Upon myeloid commitment, PDH catalyzes the conversion of pyruvate into acetyl-CoA and promotes entry of glucose-derived carbons into the tricarboxylic acid (TCA) cycle (Figure 1).
Figure 1. Metabolism of Granulopoiesis.
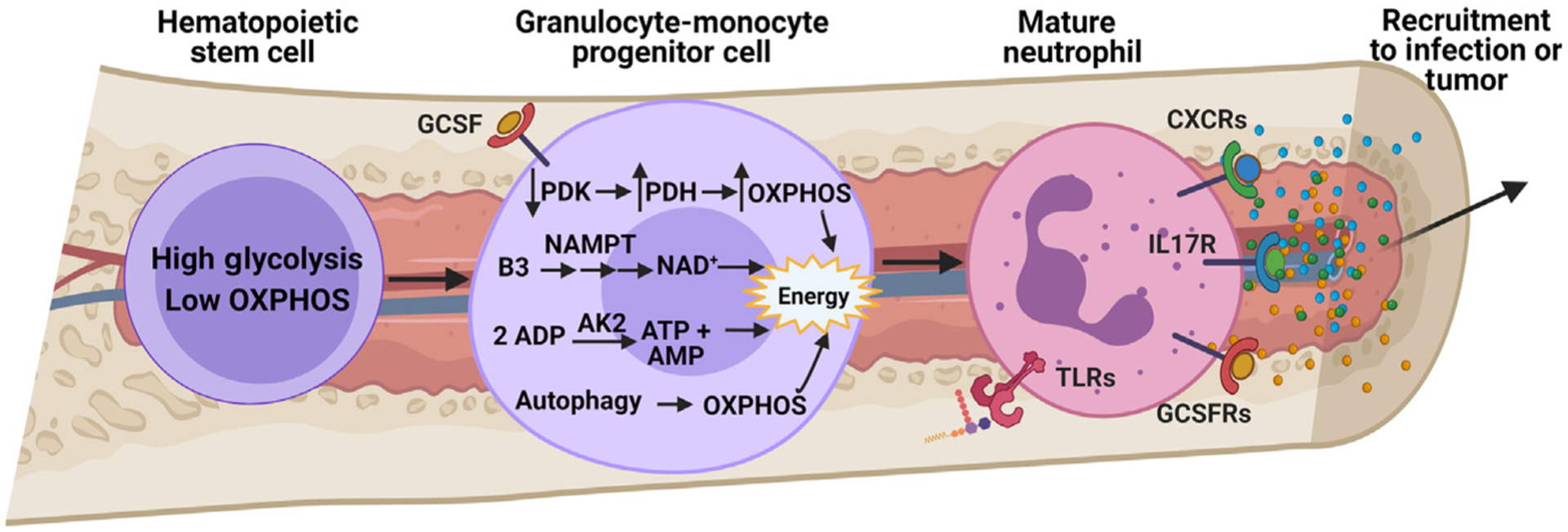
Granulopoiesis is a highly regulated differentiation process, leading to the generation of mature neutrophils from hematopoietic stem cells [4]. Granulocyte colony-stimulating factor (G-CSF) is the major cytokine promoting neutrophil differentiation in the bone marrow and also regulates salvage NAD+ synthesis through upregulation of nicotinamide phosphoribosyltransferase (NAMPT) [12]. This increase in NAD+ production coincides with engagement of oxidative phosphorylation (OXPHOS) as a result of increased oxygen availability. Improved OXPHOS function also occurs due to suppression of pyruvate dehydrogenase kinase (PDK), leading to the conversion of pyruvate to acetyl-CoA by pyruvate dehydrogenase (PDH) [9]. Regulation of adenylate nucleotides (AMP, ADP, and ATP) in the mitochondria by adenylate kinase-2 (AK2) is also necessary for proper granulopoiesis, wherein mutations in the gene encoding AK2 lead to reticular dysgenesis, a rare genetic disorder resulting in severe combined immunodeficiency [10]. Autophagy also supports mitochondrial respiration during granulopoiesis [14]. Emergency granulopoiesis by pathogen-derived byproducts also influences mature neutrophil metabolism through Toll-like receptors (TLRs) to engage the pentose phosphate pathway (PPP) and prime neutrophils for an oxidative burst once they reach the site of infection. In a similar fashion, tumor microenvironment (TME)-derived cytokines, such as G-CSF, chemokine (C-X-C motif) ligand 1/2 (CXCL1/2), or interleukin 17 (IL-17), can not only skew differentiation of hematopoietic cells toward granulocyte precursors and mature neutrophils [89], but also promote tissue-specific recruitment to support tumor progression and metastasis [90–92]. More research is needed to understand whether these TME-derived cytokines manipulate granulopoiesis-related metabolism or metabolically prime mature neutrophils toward an N2, protumor phenotype even before reaching a primary tumor or metastatic sites. Abbreviation: GCSFR, granulocyte colony-stimulating factor receptor.
Additional clues about which metabolic pathways are required for granulopoiesis come from human genetics and model systems. Reticular dysgenesis is a rare form of severe combined immunodeficiency caused by mutations in the gene encoding adenylate kinase-2 (AK2). This enzyme regulates levels of adenylate nucleotides (AMP, ADP, and ATP) in the mitochondria (Figure 1). Reticular dysgenesis presents at birth, often with fatal septicemia attributed to a compromised innate immunity [10]. In experimental systems, silencing AK2 impairs granulocyte differentiation but not the differentiation of other immune cells, including monocytes/macrophages, indicating a particular importance for AK2 in granulopoiesis [11].
De novo synthesis of NAD and NAD-dependent signaling also supports granulopoiesis. Expression of NAMPT, the rate-limiting enzyme in salvage NAD synthesis, is regulated by G-CSF (Figure 1). Granulocyte differentiation can be stimulated in culture by providing CD34+ bone marrow progenitor cells with exogenous NAMPT [12]. Dosing healthy humans with vitamin B3 (niacin, an analog of the NAMPT substrate nicotinamide) is sufficient to enhance the numbers of mature neutrophils in the circulation [12].
Autophagy is activated when essential nutrients are lacking, leading to a recycling of intracellular macromolecules and organelles for nutrition [13]. Deletion of the core autophagy gene Atg7 in neutrophils impairs granulopoiesis. Atg7-deficient cells display increased glycolysis and suppressed oxidation of mitochondrial fuels, including fatty acids. Stimulating mitochondrial respiration and ATP production by providing fatty acids and pyruvate promotes granulopoiesis in Atg7-deficient neutrophils [14]. These findings indicate a key role for mitochondrial respiration of alternative fuels in allowing cells to execute the granulopoiesis program under G-CSF stimulation (Figure 1).
Similarly to emergency granulopoiesis induced by pathogen-derived cytokines, cancer cells can also impact neutrophil biology in the bone marrow. Cancer cells produce a variety of inflammatory cytokines that not only skew bone marrow differentiation toward producing neutrophils over other innate immune cell types, but also promote the recruitment and infiltration of TANs out of circulation into the TME (Box 1 and Figure 1). It is still unknown whether tumor-derived cytokines, such as G-CSF, chemokine (C-X-C motif) ligand 1/2 (CXCL1/2), or interleukin 17 (IL-17), can induce metabolic changes in neutrophils and neutrophil progenitors residing within the bone marrow, or whether reprogrammed metabolic pathways are required for neutrophils to exit the bone marrow and travel into the TME.
Box 1. Recruitment of Neutrophils to Tumor.
Chemokine (C-X-C Motif) Ligands
Similar to pathogens, tumors secrete chemokines and inflammatory factors to mobilize neutrophils from the bone marrow and recruit them into the TME. Chemokine (C-X-C motif) ligands (CXCLs) attract neutrophils from the circulation to areas of inflammation, including tumors [93]. Neutrophils express the cognate receptors of CXCL ligands, including CXCR1/2 [94] (see Figure 2 in the main text). Tumor-derived CXCL2 stimulates the recruitment of immunosuppressive neutrophils that suppress antitumor immunity [95]. Furthermore, CXCR2 is required for neutrophil recruitment, and CXCR2 inhibition synergizes with anti-PD-1 checkpoint blockade in murine models of rhabdomyosarcoma and lung adenocarcinoma [91,92].
Colony-Stimulating Factors
CSF ligands are key molecules for the differentiation and mobilization of neutrophils. G-CSF stimulates neutrophils to leave the bone marrow and enter the circulation, and is frequently secreted by tumors [96] (see Figure 2 in the main text). In a murine breast cancer model, G-CSF secretion reprograms bone marrow niches, skewing them toward increased neutrophil differentiation. These tumor-educated neutrophils take on an immunosuppressive, tumor-promoting phenotype, characterized by increased G-CSF-dependent ROS production [89]. Furthermore, G-CSF induces these suppressive neutrophils to home to the lung during tumor development and promote tumor progression and metastasis [97,98]. Granulocyte-macrophage colony-stimulating factor (GM-CSF) induces neutrophil migration by activating multiple signaling cascades, including mTOR and p70 S6 kinase [99]. In gastric cancer, tumor-derived GM-CSF recruits neutrophils, prolongs their activation and survival, and, by activating JAK/STAT signaling, upregulates expression of PD-L1, thereby suppressing effector T cell function [100].
IL-17
Interleukin-17 (IL-17) also participates in recruiting neutrophils into the TME. Overexpression of IL-17 in a murine lung adenocarcinoma model enhanced neutrophil localization to the lung and dramatically altered the lung cytokine profile, leading to poor response to immune checkpoint blockade. Human non-small cell lung cancers with oncogenic KRAS express higher levels of IL-17 and G-CSF compared with tumors with other oncogenic drivers, such as mutant epidermal growth factor receptor (EGFR) [101]. Similar to cancer cells, γδ T cells in the TME also express IL-17. When stimulated by IL-1β, γδ T cells expand and activate protumor neutrophils through IL-17 secretion, promoting metastasis [102]. Expansion of γδ T cells can be activated in the lung by local microbiota, resulting in enhanced IL-17 secretion and infiltration of neutrophils that promote tumorigenesis [90].
Metabolism of Pathogen Responses
Phagocytosis and Oxidative Burst
The metabolic responses that allow neutrophils to respond to pathogens are relevant to cancer because they illustrate how metabolism is linked to neutrophil function. We review these pathways here. Microbial products, or pathogen-associated molecular patterns (PAMPs), are detected by neutrophil pattern recognition receptors, such as Toll-like receptors (TLRs). Circulating PAMPs recruit neutrophils to areas of infection and activate their defensive functions [15]. Neutrophils engulf bacteria into a specialized organelle called the phagolysosome, which destroys the microbes through the massive, localized production of superoxide, classically termed the ‘oxidative burst’ [16] (Figure 2). This reaction is catalyzed by NADPH oxidase, a multisubunit enzyme that consumes oxygen and NAPDH to produce superoxide and other oxygen radicals. This five-protein complex contains a transmembrane catalytic center, comprising NOX-2/gp91phox and gp22phox, which resides on the membrane of the phagosome in the resting state. The other three regulatory proteins that complete the NADPH oxidase complex are p40phox, p47phox, and p67phox, which reside in the cytoplasm under normal physiological conditions. When neutrophils are activated by pathogen-derived cytokines, these regulatory proteins translocate to the plasma membrane, where they combine with gp91phox and gp22phox to complete formation of an active NADPH oxidase complex [17]. The full complex catalyzes the transfer of electrons from NADPH to molecular oxygen, generating superoxide anions to kill pathogens.
Figure 2. Metabolic Requirements for Pathogen Response.
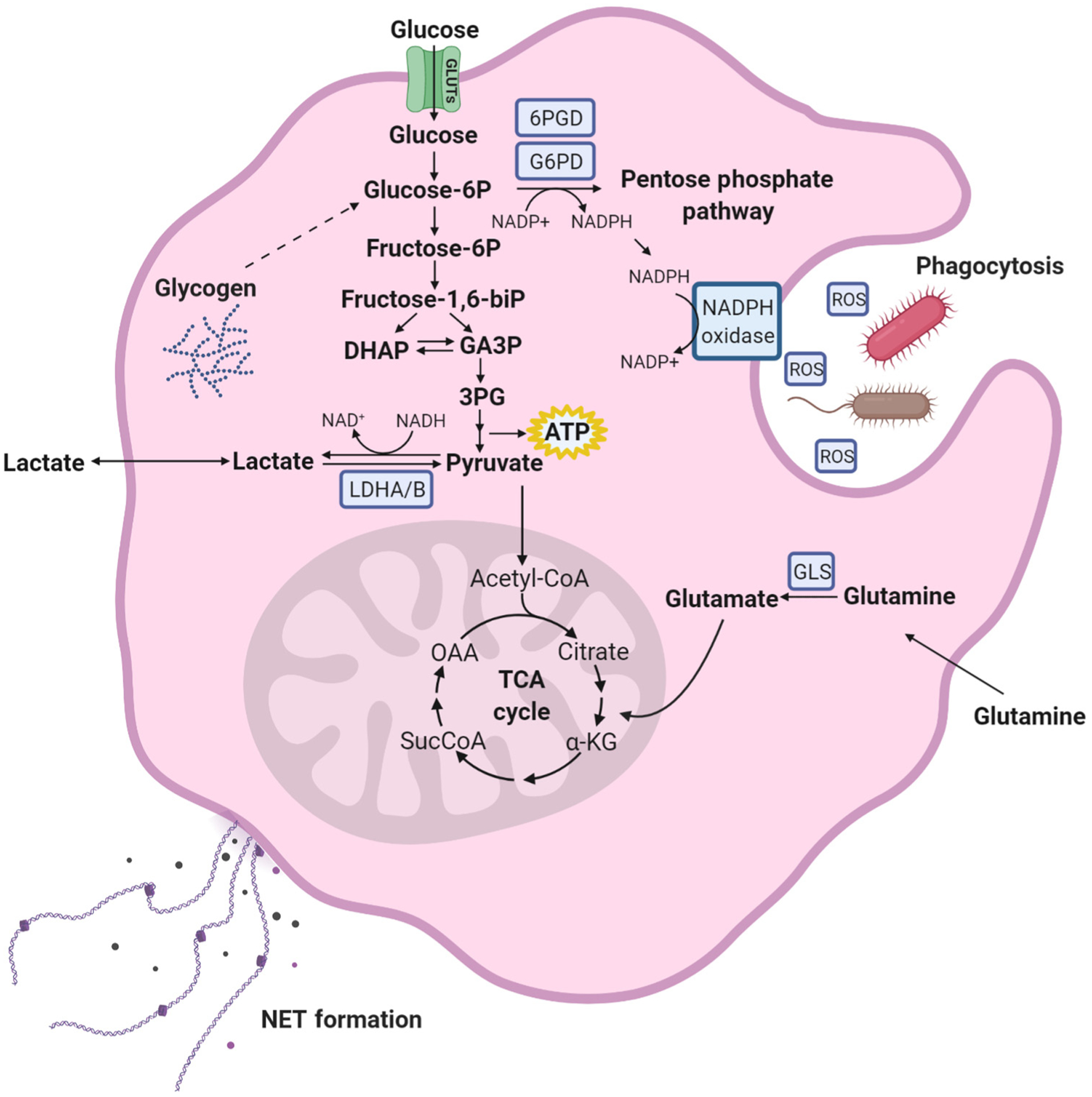
During infection, microbial byproducts are detected by neutrophils in the bone marrow, prompting granulopoiesis. Upon arriving to the site of infection, neutrophils engulf pathogens into the phagolysosome. Once engulfed, superoxide and other reactive oxygen species (ROS) are produced by the NADPH oxidase protein complex within the phagolysosome in a process termed the ‘oxidative burst’ [4]. NADPH, the substrate of NADPH oxidase, is produced by glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD) in the oxidative branch of the pentose phosphate pathway (PPP), and possibly by other enzymes, including NADPH-dependent isoforms of isocitrate dehydrogenase and malic enzyme. Shunting of carbons into the PPP is thought to be the predominant source of cytosolic NADPH in these cells. When glucose is limiting, neutrophils can maintain ATP production and carbon flux through the PPP by breaking down glycogen [18]. Glutamine uptake has also been shown to promote NAPDH oxidase activity in neutrophils, but whether this effect involves glutamine-dependent NAPDH production has not been fully examined. To suppress pathogen dissemination, neutrophils also secrete neutrophil extracellular traps (NETs), comprising granule proteins and DNA fragments. These NETs bind bacteria and degrade viral and bacterial proteins. Metabolically, similar to the ‘oxidative burst’, glycolysis and G6PD expression support NET formation [20]. Abbreviations: α-KG, α-ketoglutaric acid; GA3P, glyceraldehyde 3-phosphate; GLS, glutaminase; OAA, oxaloacetic acid; 3PG, 3-phosphoglyceric acid.
Data from early ex vivo studies indicate that glucose serves two important purposes during the oxidative burst. First, it feeds glycolysis to provide energy in the form of ATP. Neutrophils deprived of glucose maintain a glycolytic flux by degrading glycogen, thereby protecting their ATP supply at least temporarily [18]. Second, ROS generation through NAPDH oxidase requires production of NADPH. It is generally thought that most NADPH in neutrophils arises from the oxidative branch of the pentose phosphate pathway. This pathway initiates from glucose-6-phosphate (G6P) and produces NAPDH through two enzymatic reactions, glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (Figure 2). Other sugars, such as fructose, follow a different path of catabolism and do not compensate for glucose in experimental models of neutrophil responses [19,20]. Some mammalian cells contain other mechanisms to produce NADPH, including malic enzyme, isocitrate dehydrogenases-1 and −2, and the folate pathway. These pathways have not yet been studied thoroughly in neutrophils responding to pathogens [21].
Along with glucose, neutrophils consume high levels of glutamine, more so than monocytes and lymphocytes [22]. This abundant nutrient has been suggested to have a role in the oxidative burst [23]. The precise mechanism is unknown, but could involve production of NADPH for NADPH oxidases. It has been speculated that other fuels, particularly macromolecules, also support the oxidative burst in vivo, because glucose and other small-molecule nutrients are scarce in the purulent exudate that forms during inflammatory responses (Figure 2) [24].
Chronic granulomatous disease (CGD), a rare congenital immunodeficiency characterized by neutrophil dysfunction, is caused by mutations in the genes that encode the NADPH oxidase complex. In patients with CGD, mutations in any of the five proteins that comprise this complex lead to dramatic suppression of the oxidative burst, leaving patients with CGD susceptible to infections with bacteria and fungi that would normally be cleared by neutrophil phagocytosis [25]. In a recent clinical trial, lentiviral gene therapy was used to introduce a wild-type copy of the gene encoding the NOX-2/gp91phox subunit of NADPH oxidase into granulocyte progenitor cells from the bone marrow of patients with X-linked CGD [26]. Following transplantation of edited cells, most patients displayed persistence of neutrophils expressing NADPH oxidase, no new infections related to their underlying immunodeficiency, and reduced reliance on chronic antibiotic therapy [26].
Neutrophil Extracellular Trap Formation
At sites of infection, neutrophils release neutrophil extracellular traps (NETs), a mix of granule proteins and chromatin fibers that degrade virulence factors and bind bacteria [27]. NET formation is thought to be dependent on glycolysis, because, in cultured neutrophils, NET formation is suppressed by 2-deoxyglucose (2DG), a competitive inhibitor of glucose uptake and phosphorylation. Furthermore, in glucose-depleted conditions, only the addition of glucose and not pyruvate could rescue NET formation, suggesting that glycolysis per se rather than pyruvate oxidation regulates this process [20]. G6PD, which catalyzes an NADPH-generating step in the pentose phosphate pathway, is also necessary for NET formation (Figure 2) [19].
Functional and Metabolic Plasticity of TANs
Neutrophil Diversity in the TME
While the abundance of circulating neutrophils is significantly higher in humans than in mice (70% of white blood cells in human versus 30% or less in mouse) [28], both species have similarly heterogeneous populations of neutrophils, particularly in the pathological setting of cancer [15]. Initially, neutrophils were described to exhibit polarized states of N1 (antitumor) or N2 (protumor) phenotypes depending on the environmental context and exposure to specific tumor-derived cytokines [29], comparable with that of tumor-associated macrophages (TAMs) [30]. TANs have been further classified based on their buoyancy and the microscopic appearance of their nuclei. Using differential density centrifugation, low-density neutrophils could be subclassified morphologically into both mature (fully segmented nuclei) and immature (banded cell and ring nuclei) subsets, while high-density neutrophils exhibited mature morphology [31].
While this review focuses on the functional and metabolic state of N2, protumor neutrophils, other work indicates how N1 neutrophils exert their ant-tumor capacity. Some tumor-derived cytokines upregulate expression of the proto-oncogene MET in TANs, leading to increased production of reactive nitrogen species, which kill tumor cells [32]. These TANs collaborate with natural killer cells and cytotoxic T cells to promote tumor cell killing through inflammasome release [33], and can suppress seeding of organs by metastatic cancer cells through H2O2-mediated tumor cell cytotoxicity [34].
Nutrients and Enzymes Contributing to TAN Function in the TME
We currently know less about metabolic reprogramming in TANs compared with neutrophil pathogen responses, but this is now an active area of investigation. Rapidly growing tumors tend to outgrow their blood supply, leading to nutrient depletion and localized cytokine accumulation, both of which may influence TAN metabolism. While glucose is critical to allow neutrophils to respond to pathogens, glucose limitations in the TME may require that TANs turn to other substrates. Indeed, several lines of evidence support this view. c-Kit+ TANs engage in mitochondrial respiration, oxidizing glutamine and fatty acids to generate reducing equivalents for ROS production [35]. This is relevant because the TAN-mediated oxidative burst is thought to promote T cell apoptosis and suppress antitumor T cell proliferation and inflammatory signaling [36,37]. As with ROS production, NET formation by TANs in the TME is also supported by glutamine and fatty acids. NETs physically shield tumor cells from cytotoxic CD8+ T cells and natural killer cells, making the metabolic pathways supporting NET formation vital to the immunosuppressive environment of the TME. Immature, low-density TANs engage in mitochondrial respiration fueled by glutamine catabolism and fatty acid oxidation, and blocking these pathways suppresses NET formation [31] (Figure 3, Key Figure).
Altogether, these observations position glutamine and fatty acid oxidation by TANs as reprogrammed and possibly targetable metabolic activities that promote tumor progression by interfering with cytotoxic T cell responses. The involvement of glutamine in supporting ROS and NET production is interesting in part because of the availability of clinical and experimental drugs that inhibit glutamine metabolism. In the context of TAN metabolism and immunotherapy, JHU-083, a drug that broadly inhibits many pathways of glutamine catabolism in the TME, induces neutrophil apoptosis in both the tumor and circulation, and improves antitumor immunity in mice [38]. Given the versatility of glutamine as a carbon and nitrogen source in many different cells within the TME, it will be interesting to determine precisely which metabolic effects are responsible for these changes in TAN function.
While fatty acid oxidation in TANs supports their immunosuppressive capacity against T cells [39], there are also nonoxidative functions of fatty acids that influence TAN behavior. Fatty acid uptake through fatty acid transport protein 2 (FATP2) is upregulated in TANs compared with naïve neutrophils through a granulocyte-macrophage colony-stimulating factor (GM-CSF)/STAT5 (signal transducer and activator of transcription 5) signaling cascade. FATP2 facilitates the uptake of arachidonic acid and supports the synthesis of the inflammatory mediator prostaglandin E2. FATP2 deletion or inhibition reduces tumor growth and synergizes with immunotherapy [40]. A second study found that surface expression of lectin-type oxidized low-density lipoprotein (LDL) receptor-1 (LOX-1), which binds to oxidized lipoproteins, is expressed by TANs but not by naïve neutrophils, and that the abundance of circulating LOX-1+ neutrophils correlates with poor outcomes in patients [41]. In vitro, LOX-1+ neutrophils are more potent in suppressing T cell function compared with LOX-1− neutrophils isolated from the same patients with cancer [41]. Given that LOX-1 signaling in endothelial cells promotes NAPDH oxidase activity [42], it is possible that a similar signaling mechanism is present in LOX-1+ TANs, leading to immunosuppressive, tumor-promoting ROS production (Figure 3).
TANs also express metabolic enzymes with immunosuppressive effects. Inducible nitric oxide synthase (iNOS), frequently expressed by inflammatory TANs, converts arginine and NAPDH into citrulline and nitric oxide. Nitric oxide reacts with superoxide to produce peroxynitrite, which induces T cell toxicity in the TME [37]. TANs also secrete arginase (ARG1), which converts arginine to ornithine. Arginine is required for T cell function and its depletion suppresses antitumor immunity [43] (Figure 3). Transforming growth factor beta (TGF-β) signaling drives iNOS and ARG1 expression in TANs and loss of TGF-β receptor signaling in myeloid cells (macrophage and neutrophils) suppresses tumor metastasis [44].
Hypoxia
Hypoxia alters tumor cell metabolism and contributes to T cell dysfunction [45]. However, contrary to other kinds of immune cell, hypoxic conditions may be advantageous to neutrophils, perhaps because of their high capacity for glycolysis. Hypoxia promotes neutrophil degranulation, leading to lung epithelial cell injury [46]. In most cell types, hypoxia leads to the stabilization of oxygen-labile α-subunits of the hypoxia-inducible transcription factors (HIFs), activating the expression of genes that allow cells to adapt to hypoxia [45]. This response is required to support several aspects of neutrophil function. While deleting HIF-1α blunts ATP production in peritoneal neutrophils [47], HIF/NF-κB signaling in human and mouse neutrophils prolongs survival under hypoxia [48]. Neutrophils from patients with heterozygous germline mutations in the gene encoding Von Hippel–Lindau (VHL) display an extended lifespan, delayed apoptosis, and enhanced phagocytic capability relative to cells with wild-type VHL [49]. HIF-2α also contributes to neutrophil inflammatory function. Gain-of-function mutations in HIF-2α within neutrophils prolong cell survival and exacerbate acute lung injury in mice. In humans, HIF-2α is chronically upregulated in circulating neutrophils and in neutrophils from lung biopsies of patients with inflammatory arthritis and chronic obstructive pulmonary disease (COPD) [50]. Metabolically, under conditions of hypoxia and limited glucose, airway neutrophils scavenge glutamine from the extracellular space and use it to fuel central carbon metabolism [51].
Hypoxia signaling is at the root of interactions between cancer cells and neutrophils and may explain some of the tumor-promoting effects of neutrophils in the TME. In response to hypoxia, tumor cells secrete neutrophil-recruiting cytokines in a HIF-2α-dependent manner [52]. HIF-1α promotes expression of the immune checkpoint protein programmed death ligand 1 (PD-L1) on cancer cells and myeloid-derived suppressor cells, leading to suppressed cytotoxic T cell activation [53]. ARG2, an alternative isoform to ARG1, is a transcriptional target of HIF-2α [54]. TANs can also promote HIF signaling in cancer cells by establishing a hypoxic niche through disruption of local vasculature. This in turn promotes lung cancer cell invasion through expression of Snail [55]. Interestingly, when the uterine epithelial TME was reoxygenated in mice housed in hyperoxia chambers, neutrophil recruitment was suppressed and the neutrophils that did arrive were repolarized and exerted an antitumor effect. This effect was caused by increased neutrophil-derived ROS and matrix metalloproteinase 9 (MMP9) secretion, leading to tumor cell sloughing and apoptosis [56].
Acidosis
Glycolytic tumor cells secrete lactate through monocarboxylate transport proteins MCT1 and MCT4. Co-transport of lactate with protons results in acidification of the TME. This process is enhanced in regions distant from the vasculature [57]. Highly proliferative tumors with heightened glycolysis inhibit antitumor effector T cell functions in part by establishing an acidic microenvironment, which suppresses cytokine production and T cell survival [58,59]. Conversely, neutrophils appear to be amenable to acidic environments. At sites of infection, the microbial-neutrophil milieu is acidic until the infection is resolved [60]. Neutrophil apoptosis is suppressed at pH 6 relative to pH 7.4, indicating that they contain specialized mechanisms to survive acidity. While their antimicrobial capacity is suppressed at low pH, possibly because of less effective NADPH oxidase function, acidic conditions enhance neutrophil chemotaxis and endocytosis [61]. Lactate alone has not been shown to polarize TANs, but it stimulates the polarization of TAMs toward an M2, protumor phenotype; in these cells, lactate is sufficient to promote the expression of protumor factors VEGF and ARG1 [62]. In addition to the effects of lactate on macrophages, low pH also induces NF-κB-dependent expression of the immunosuppressive protein iNOS [63]. Further study is needed to determine whether the polarizing effect of lactate and an acidic TME is common to all myeloid cell types. Additionally, lactate serves as a direct energy source for tumor cells and can be shuttled between cancer cells and fibroblasts within the TME [64,65]. Whether TANs can also oxidize and/or shuttle lactate in the TME still needs to be tested.
Key Figure
Neutrophil Metabolism in the Tumor Microenvironment (TME)
Figure 3.
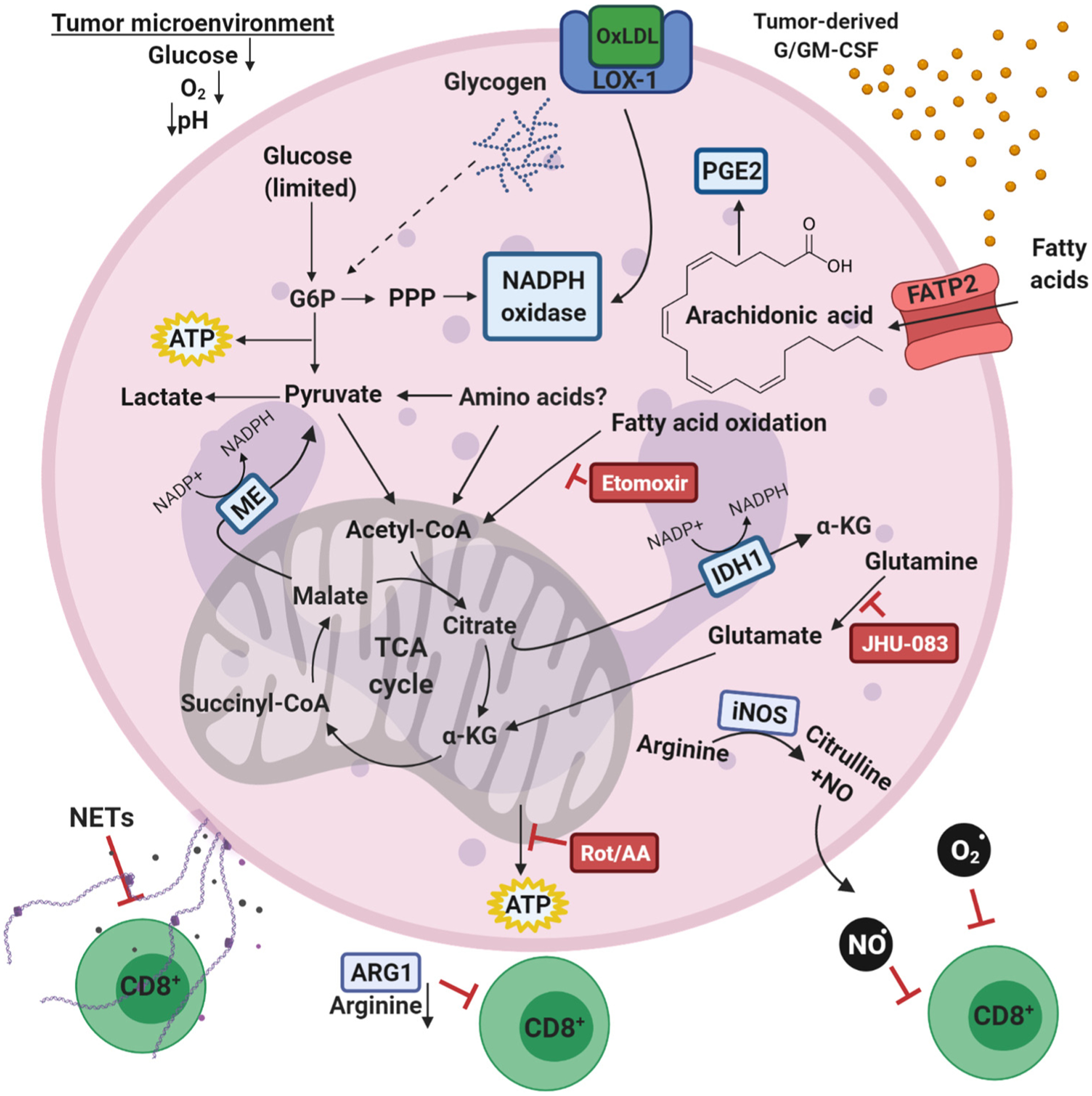
Within the TME, rapidly dividing tumors cells may outcompete immune and stromal cells for essential nutrients, forcing metabolic adaptations for cell function and survival. In the TME, if glucose availability declines, alternative nutrients must supply the critical pathways that allow tumor-associated neutrophils (TANs) to support tumor progression. TANs utilize fatty acid oxidation to power immunosuppression of antitumor T cells [39]. Tumor-derived granulocyte-macrophage colony-stimulating factor (GM-CSF) also promotes expression of fatty acid transport protein 2 (FATP2), through which TANs take up fatty acids, such as arachidonic acid, for prostaglandin-mediated inflammatory signaling [40]. TANs uniquely express cell surface receptor lectin-type oxidized low-density lipoprotein (OxLDL) receptor-1 (LOX-1), which may stimulate NADPH oxidase [41]. In glucose-depleted conditions, c-KIT+ and low-density TANs exhibit increased mitochondrial respiration and are sensitive to either electron transport chain or fatty acid oxidation inhibitors, such as rotenone/antimycin A and etomoxir, respectively [31,35]. Furthermore, in some glucose-limited settings, NADPH oxidase activity can be supported by NADPH production outside of the pentose phosphate pathway (PPP), likely through either malic enzyme (ME) or isocitrate dehydrogenase (IDH) [35]. Pharmacological inhibition of glutamine catabolism by JHU-083 in the TME decreases TAN abundance, positioning glutamine as a regulator of TAN survival [38]. TANs also secrete metabolites and metabolic enzymes to suppress antitumor immunity. They directly suppress T cell inflammatory signaling by releasing proteases, NADPH oxidase-derived reactive oxygen species (ROS) and reactive nitrogen species produced by inducible nitric oxide synthase (iNOS) [37,103]. They also deplete the local environment of arginine, an essential nutrient for T cell function, by secreting arginase-1 (ARG1) [37]. TANs can also suppress cytotoxic T cell function through the release of neutrophil extracellular traps (NETs), which can form a physical barrier between tumor cells and antitumor T cells, further disrupting antitumor immunity [104]. Abbreviations: α-KG, α-ketoglutaric acid; PGE2, prostaglandin E2, NO, nitric oxide; Rot/AA, rotenone and Antimycin A; TCA, tricarboxylic acid, electron transport chain inhibitors.
Neutrophil Involvement in Cancer Progression and Metastasis
Evidence from multiple sources indicates that TANs promote the acquisition of properties characteristic of advanced malignancy. TANs secrete MMPs, which degrade the extracellular matrix and promote malignant cell invasion within primary tumors. MMP2 and MMP-9 expressed by TANs and other myeloid cells promote squamous cell carcinoma in mice [66]. Recruitment of CXCR2-positive TANs drives the development of carcinogen-induced colon carcinomas by suppressing cytotoxic CD8+ T cells [67]. TANs also promote progression of established tumors [68].
In some models, depletion of TANs markedly reduced the growth of otherwise highly aggressive tumors, even in immunodeficient mice lacking a functional T cell compartment [69]. In these studies, the influence of metabolic reprogramming was not specifically studied, but it appears reasonable to speculate that metabolism either in TANs or cancer cells contributes to cancer progression. For example, in a genetic model of lung cancer driven by oncogenic Kras, neutrophil elastase (NE) promotes PI3K activation by degrading its negative regulator insulin receptor substrate-1 (IRS-1) [70]. Given that PI3K signaling is a major regulator of cellular nutrient uptake and metabolism, the proliferation of lung cancer cells exposed to NE in these models likely involves activation of multiple PI3K-stimulated metabolic pathways.
Metastasis involves a multistep process that includes invasion into surrounding tissues, intravasation into the blood stream, survival while circulating, extravasation to a secondary site, and eventual outgrowth [71]. Recent work indicates that several specific metabolic hurdles confront potentially metastatic cancer cells, keeping the overall efficiency of metastasis low. Chief among these metabolic challenges is the excessive oxidative stress encountered by cancer cells after escaping from the primary tumor, particularly during their exposure to the circulation [72,73]. Activating systems to produce NADPH allows cancer cells to counteract ROS, and this promotes metastatic efficiency in mouse models of melanoma [73,74]. With this metabolic challenge in mind, the association of circulating tumor cells with neutrophils is interesting. On the one hand, the propensity for neutrophils to produce ROS would appear to promote culling of cancer cells from the circulation, reducing metastatic efficiency. On the other hand, cancer cell–TAN associations stimulate tumor cell proliferation and enhance metastasis in some models [75]. These findings suggest a supportive role for TANs in the context of circulating tumor cells. It is possible that the close contact between these two cell types culminates in mechanisms to mitigate ROS in the cancer cells, perhaps by transferring substrates with antioxidant functions.
Little is known about the metabolic processes that promote the reactivation and growth of dormant cancer cells at metastatic sites, but interesting recent work positions TANs and NETs as key players in this process. Specifically, in mouse models of breast and prostate cancer, dormant cancer cells seeded in the lung re-establish their proliferative capacity through extracellular matrix remodeling mediated by the NET-associated proteins NE and MMP-9 [76]. The findings imply that the metabolic processes enabling TANs to produce NETs contribute to the transition between dormancy and formation of macrometastatic lesions, at least in the lung.
Concluding Remarks
Neutrophils rely on highly regulated metabolic activities to support granulopoiesis and specialized cellular functions during acute, self-limited immunological responses. Under states of chronic inflammation and cancer, aberrant neutrophil metabolism can promote tissue dysfunction and damage. Understanding context-dependent metabolism of neutrophils may provide therapeutic targets to improve clinical outcomes in several diseases. The preponderance of evidence suggests that TANs promote cancer progression through multiple mechanisms, adding to the complex biology of tumor heterogeneity. We need to further understand how context-specific cues determine interactions among cancer cells, TANs, and other immune and stromal cells in the TME (see Outstanding Questions). Clearly, the N1/N2 polarization of TANs is context dependent and understanding the metabolic differences between these two states may further distinguish distinct TAN populations and provide therapeutic avenues to repolarize TANs. Continued research on surface markers and gene expression signatures associated with neutrophil polarization states will complement TAN metabolic studies to help explain the mechanisms that govern the heterogeneous biological properties of neutrophils in cancer [29].
Outstanding Questions.
What metabolic features distinguish between multipotent progenitors, neutrophil progenitors, and mature neutrophils during emergency granulopoiesis induced by infection or tumor progression?
How do metabolic signatures differ among circulating neutrophils, neutrophils at sites of infection, and neutrophils in the TME?
Would it be more therapeutically beneficial to prevent neutrophil recruitment to the tumor or to repolarize neutrophils already within the tumor? Can targeting specific metabolic pathways repolarize TANs into a phenotype that promotes antitumor immunity?
Does the recruitment of neutrophils to tumors by tumor-derived cytokines require metabolic changes before entry into the TME? If so, can recruitment be suppressed by blocking these pathways?
How similar are the metabolic challenges encountered by neutrophils in the TME and in purulent exudates? Can defining the nutrient milieu in these different settings help us broadly define metabolic strategies in neutrophils?
Do alternative carbon sources (glycogen, fatty acids, glutamine, and amino acids) provide energy and reducing power in neutrophils when glucose is limiting?
Do TANs release metabolites that benefit cancer cells? Is there other metabolic crosstalk between TANs and cancer cells that is mutually beneficial?
The major foreseeable limitation in studying neutrophil metabolism in vivo is the variable life span of these cells. The half-life of a neutrophil can change dramatically depending on its tissue of residence [77] and no study to date has accurately measured the half-life of a neutrophil within a tumor. Metabolic profiling of TANs and other tumor-infiltrating immune cells will clearly benefit from better methods to assess immune cell metabolism in vivo. Such methods will likely include continued improvements in single-cell gene expression techniques, as well as high-fidelity approaches to rapidly examine metabolomics and metabolic flux phenotypes in neutrophils purified from native microenvironments, including the TME [78,79].
The circulating neutrophil-to-lymphocyte (NLR) ratio has been assessed as a clinical biomarker of cancer severity and therapy response [80]. High ratios correlate with poor clinical outcome and recurrence in several different cancer types [81–83], but this predictive value is not shared across all cancer types [84,85]. Examining tumor-infiltrating neutrophils either within the tumor or in the surrounding stroma may provide better prognostic value than the NLR ratio in the blood. In a cohort of patients with non-small cell lung cancer treated with immunotherapy, neutrophil content within the tumor predicted poor T cell infiltration and immune checkpoint blockade failure [86].
Clinical trials have begun targeting neutrophil recruitment to tumors (NCT03177187 and NCT03161431). Both these trials are testing the efficacy of small-molecule inhibitors of CXCR2 in combination with other cancer therapies, including immunotherapy, in advanced cancers. Clinical trials specifically targeting the metabolism of TANs are currently limited to testing arginase inhibitors as part of a combination therapy in multiple myeloma (NCT03837509). Targeting critical metabolic nodes specifically in TANs may prove to be difficult. Therapeutic targeting of G6PD, a critical enzyme for NAPDH oxidase function and NET formation, is limited by a lack of highly specific small-molecule inhibitors for clinical studies. Recently, a novel G6PD inhibitor was shown to deplete NAPDH in neutrophils, neutralizing the oxidative burst [87]. While this result in neutrophils is promising, additional experiments showed that other immune cell types, such as T cells, also depend on G6PD to maintain their intracellular NADPH pool. Depletion of NADPH in T cells suppressed their cytokine production, which could have negative consequences when attempting to mount a robust antitumor immune response in vivo [87]. To specifically target drug delivery to neutrophils and other protumor myeloid cells, particulate drug carriers, such as liposomes, can be used to naturally target cells of the mononuclear phagocytic system [88]. Modifying these liposomes to express ligands specifically recognized by neutrophils to activate phagocytosis, such as N-formyl-Met-Leu-Phe (fMLP), could provide a targeted way to deliver metabolic inhibitors to suppress or repolarize TANs.
The concept emerging from these metabolic studies in the TANs is that neutrophils display substantial metabolic flexibility, enabling a surprisingly large number of nutrients to support neutrophil function. Although most studies to this point have focused on abundant extracellular nutrients, such as glucose, glutamine, and fatty acids, it will be fascinating to determine how neutrophils utilize a panoply of other physiological nutrients, and how the importance of these fuel sources changes in the TME. Connecting these pathways to specific mechanisms by which TANs influence tumor cell growth and antitumor immunity may provide opportunities to target TAN metabolism for therapeutic benefit.
Highlights.
Circulating neutrophils act as the first line of defense against foreign pathogens
Inflammatory cytokines secreted from areas of chronic inflammation and primary tumors can promote granulopoiesis in the bone marrow and recruit neutrophils out of circulation into the tumor microenvironment (TME).
Glycolysis and the pentose phosphate pathway are metabolically necessary for pathogen responses, while fatty acid oxidation and oxidative phosphorylation are heightened in neutrophils in the TME.
Targeting the metabolism of immunosuppressive tumor-associated neutrophils (TANs) could synergize with cancer immunotherapy to improve therapeutic response.
Acknowledgments
We thank Jessica Moreland and Jessica Hook for helpful discussions on neutrophil biology. We also thank our funding sources: National Cancer Institute (NCI) Pre to Postdoctoral Fellow Transition Award (K00CA212230) to T.J.R. and NCI R35 Outstanding Investigator Award (R35CA22044901) to R.J.D. R.J.D. is also supported by the Howard Hughes Medical Institute.
Glossary
- Hypoxia-inducible factors (HIFs)
transcription factors that activate the expression of genes required to adapt to low-oxygen conditions. HIFs comprise two subunits, including an oxygen-labile alpha subunit targeted for degradation when oxygen is abundant.
- Immunotherapy
treatments that activate immune cells, particularly cytotoxic T cells, to kill tumor cells. A common subset of immunotherapy includes immune checkpoint inhibitors (also known as checkpoint blockade therapy) that block PD-1 or CTLA4.
- Innate immune system
components of the immune system that provide initial, immediate, and often nonspecific responses to infection. Through secretion of cytokines and antigen presentation, the innate immune system recruits and activates the adaptive immune system for a more specialized response to the pathogen. Neutrophils, through phagocytosis and NET production, function as components of innate immunity.
- Neutrophil extracellular trap (NET)
mixture of proteins and nucleic acids secreted by neutrophils to bind microbial pathogens, limiting their dissemination and contributing to their containment.
- Oxidative burst
metabolic response to pathogens involving the rapid generation of superoxide, a reactive oxygen molecule by NADPH oxidase.
- Phagocytosis
cellular process by which cells physically engulf nutrients or pathogens for internal breakdown.
- Tumor-associated neutrophils (TANs)
neutrophils recruited from the circulation into the tumor microenvironment.
- Tumor microenvironment (TME)
internal milieu of solid tumors, including malignant cells (cancer cells) as well as immune cells, stromal cells (fibroblasts, pericytes, and endothelial cells) and the extracellular matrix. The pH, nutrient availability, and cell–cell interactions in the TME may differ substantially from non-malignant organs and may contribute to metabolic heterogeneity.
Footnotes
Declaration of Interests
R.J.D. is a scientific advisor for Agios Pharmaceuticals and Vida Ventures. T.JR has no competing interests to declare.
References
- 1.Borregaard N (2010) Neutrophils, from marrow to microbes. Immunity 33, 657–670 [DOI] [PubMed] [Google Scholar]
- 2.Fine N et al. (2020) The neutrophil: constant defender and first responder. Front. Immunol 11, 571085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar S and Dikshit M (2019) Metabolic insight of neutrophils in health and disease. Front. Immunol 10, 2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hidalgo A et al. (2019) The neutrophil life cycle. Trends Immunol 40, 584–597 [DOI] [PubMed] [Google Scholar]
- 5.Yvan-Charvet L and Ng LG (2019) Granulopoiesis and neutrophil homeostasis: a metabolic, daily balancing act. Trends Immunol. 40, 598–612 [DOI] [PubMed] [Google Scholar]
- 6.Zhang DE et al. (1997) Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc. Natl. Acad. Sci. U. S. A 94, 569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shyh-Chang N et al. (2013) Stem cell metabolism in tissue development and aging. Development 140, 2535–2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suda T et al. (2011) Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310 [DOI] [PubMed] [Google Scholar]
- 9.Takubo K et al. (2013) Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoenig M et al. (2018) Recent advances in understanding the pathogenesis and management of reticular dysgenesis. Br. J. Haematol 180, 644–653 [DOI] [PubMed] [Google Scholar]
- 11.Six E et al. (2015) AK2 deficiency compromises the mitochondrial energy metabolism required for differentiation of human neutrophil and lymphoid lineages. Cell Death Dis. 6, e1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skokowa J et al. (2009) NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat. Med 15, 151–158 [DOI] [PubMed] [Google Scholar]
- 13.Levy JMM et al. (2017) Targeting autophagy in cancer. Nat. Rev. Cancer 17, 528–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riffelmacher T et al. (2017) Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 47, 466–480 e465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silvestre-Roig C et al. (2019) Neutrophil diversity in health and disease. Trends Immunol. 40, 565–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cowland JB and Borregaard N (2016) Granulopoiesis and granules of human neutrophils. Immunol. Rev 273, 11–28 [DOI] [PubMed] [Google Scholar]
- 17.Nguyen GT et al. (2017) Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front. Cell. Infect. Microbiol 7, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borregaard N and Herlin T (1982) Energy metabolism of human neutrophils during phagocytosis. J. Clin. Invest 70, 550–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Azevedo EP et al. (2015) A metabolic shift toward pentose phosphate pathway is necessary for amyloid fibril- and phorbol 12-myristate 13-acetate-induced neutrophil extracellular trap (NET) formation. J. Biol. Chem 290, 22174–22183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodriguez-Espinosa O et al. (2015) Metabolic requirements for neutrophil extracellular traps formation. Immunology 145, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanukoglu I and Rapoport R (1995) Routes and regulation of NADPH production in steroidogenic mitochondria. Endocr. Res 21, 231–241 [DOI] [PubMed] [Google Scholar]
- 22.Pithon-Curi TC et al. (2004) Glucose and glutamine utilization by rat lymphocytes, monocytes and neutrophils in culture: a comparative study. Cell Biochem. Funct 22, 321–326 [DOI] [PubMed] [Google Scholar]
- 23.Pithon-Curi TC et al. (2002) Glutamine plays a role in superoxide production and the expression of p47phox, p22phox and gp91phox in rat neutrophils. Clin. Sci. (Lond.) 103, 403–408 [DOI] [PubMed] [Google Scholar]
- 24.Weisdorf DJ et al. (1982) Granulocytes utilize different energy sources for movement and phagocytosis. Inflammation 6, 245–256 [DOI] [PubMed] [Google Scholar]
- 25.Arnold DE and Heimall JR (2017) A review of chronic granulomatous disease. Adv. Ther 34, 2543–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kohn DB et al. (2020) Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med 26, 200–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brinkmann V et al. (2004) Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 [DOI] [PubMed] [Google Scholar]
- 28.Mestas J and Hughes CC (2004) Of mice and not men: differences between mouse and human immunology. J. Immunol 172, 2731–2738 [DOI] [PubMed] [Google Scholar]
- 29.Fridlender ZG et al. (2009) Polarization of tumor-associated neutrophil phenotype by TGF-beta: ‘N1’ versus ‘N2’ TAN. Cancer Cell 16, 183–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Najafi M et al. (2019) Macrophage polarity in cancer: a review. J. Cell. Biochem 120, 2756–2765 [DOI] [PubMed] [Google Scholar]
- 31.Hsu BE et al. (2019) Immature low-density neutrophils exhibit metabolic flexibility that facilitates breast cancer liver metastasis. Cell Rep. 27, 3902–3915 [DOI] [PubMed] [Google Scholar]
- 32.Finisguerra V et al. (2015) MET is required for the recruitment of anti-tumoural neutrophils. Nature 522, 349–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Riise RE et al. (2015) TLR-stimulated neutrophils instruct NK cells to trigger dendritic cell maturation and promote adaptive T cell responses. J. Immunol 195, 1121–1128 [DOI] [PubMed] [Google Scholar]
- 34.Granot Z et al. (2011) Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20, 300–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rice CM et al. (2018) Tumour-elicited neutrophils engage mitochondrial metabolism to circumvent nutrient limitations and maintain immune suppression. Nat. Commun 9, 5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gelderman KA et al. (2006) T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc. Natl. Acad. Sci. U. S. A 103, 12831–12836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michaeli J et al. (2017) Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFalpha and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 6, e1356965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh MH et al. (2020) Targeting glutamine metabolism enhances tumor specific immunity by modulating suppressive myeloid cells. J. Clin. Invest 130, 3865–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hossain F et al. (2015) Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res 3, 1236–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veglia F et al. (2019) Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 569, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Condamine T et al. (2016) Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol 1, aaf8943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sugimoto K et al. (2009) LOX-1-MT1-MMP axis is crucial for RhoA and Rac1 activation induced by oxidized low-density lipoprotein in endothelial cells. Cardiovasc. Res 84, 127–136 [DOI] [PubMed] [Google Scholar]
- 43.Gallina G et al. (2006) Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Invest 116, 2777–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pang Y et al. (2013) TGF-beta signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 3, 936–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schito L and Semenza GL (2016) Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2, 758–770 [DOI] [PubMed] [Google Scholar]
- 46.Hoenderdos K et al. (2016) Hypoxia upregulates neutrophil degranulation and potential for tissue injury. Thorax 71, 1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cramer T et al. (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walmsley SR et al. (2005) Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J. Exp. Med 201, 105–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walmsley SR et al. (2006) Neutrophils from patients with heterozygous germline mutations in the von Hippel Lindau protein (pVHL) display delayed apoptosis and enhanced bacterial phagocytosis. Blood 108, 3176–3178 [DOI] [PubMed] [Google Scholar]
- 50.Thompson AA et al. (2014) Hypoxia-inducible factor 2alpha regulates key neutrophil functions in humans, mice, and zebrafish. Blood 123, 366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Watts ER et al. (2021) Hypoxia drives murine neutrophil protein scavenging to maintain central carbon metabolism. J. Clin. Invest Published online April 6, 2021. 10.1172/JCI134073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Triner D et al. (2017) Epithelial hypoxia-inducible factor 2alpha facilitates the progression of colon tumors through recruiting neutrophils. Mol. Cell. Biol 37, e00481–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Noman MZ et al. (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med 211, 781–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krotova K et al. (2010) Hypoxic upregulation of arginase II in human lung endothelial cells. Am. J. Physiol. Cell Physiol 299, C1541–C1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Faget J et al. (2017) Neutrophils and Snail orchestrate the establishment of a pro-tumor microenvironment in lung cancer. Cell Rep. 21, 3190–3204 [DOI] [PubMed] [Google Scholar]
- 56.Mahiddine K et al. (2020) Relief of tumor hypoxia unleashes the tumoricidal potential of neutrophils. J. Clin. Invest 130, 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Helmlinger G et al. (1997) Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat. Med 3, 177–182 [DOI] [PubMed] [Google Scholar]
- 58.Calcinotto A et al. (2012) Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 72, 2746–2756 [DOI] [PubMed] [Google Scholar]
- 59.Fischer K et al. (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 [DOI] [PubMed] [Google Scholar]
- 60.Edlow DW and Sheldon WH (1971) The pH of inflammatory exudates. Proc. Soc. Exp. Biol. Med 137, 1328–1332 [DOI] [PubMed] [Google Scholar]
- 61.Cao S et al. (2015) Extracellular acidification acts as a key modulator of neutrophil apoptosis and functions. PLoS One 10, e0137221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Colegio OR et al. (2014) Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bellocq A et al. (1998) Low environmental pH is responsible for the induction of nitric-oxide synthase in macrophages. Evidence for involvement of nuclear factor-kappaB activation. J. Biol. Chem 273, 5086–5092 [DOI] [PubMed] [Google Scholar]
- 64.Faubert B et al. (2017) Lactate metabolism in human lung tumors. Cell 171, 358–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fiaschi T et al. (2012) Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 72, 5130–5140 [DOI] [PubMed] [Google Scholar]
- 66.Coussens LM et al. (2000) MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 103, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Katoh H et al. (2013) CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 24, 631–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coffelt SB et al. (2016) Neutrophils in cancer: neutral no more. Nat. Rev. Cancer 16, 431–446 [DOI] [PubMed] [Google Scholar]
- 69.Pekarek LA et al. (1995) Inhibition of tumor growth by elimination of granulocytes. J. Exp. Med 181, 435–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Houghton AM et al. (2010) Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med 16, 219–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chaffer CL et al. (2016) EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 35, 645–654 [DOI] [PubMed] [Google Scholar]
- 72.Schafer ZT et al. (2009) Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Piskounova E et al. (2015) Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tasdogan A et al. (2020) Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Szczerba BM et al. (2019) Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 566, 553–557 [DOI] [PubMed] [Google Scholar]
- 76.Albrengues J et al. (2018) Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ballesteros I et al. (2020) Co-option of neutrophil fates by tissue environments. Cell 183, 1282–1297 [DOI] [PubMed] [Google Scholar]
- 78.Artyomov MN and Van den Bossche J (2020) Immunometabolism in the single-cell era. Cell Metab. 32, 710–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma EH et al. (2019) Metabolic profiling using stable isotope tracing reveals distinct patterns of glucose utilization by physiologically activated cd8(+) T cells. Immunity 51, 856–870 [DOI] [PubMed] [Google Scholar]
- 80.Shaul ME and Fridlender ZG (2019) Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol 16, 601–620 [DOI] [PubMed] [Google Scholar]
- 81.Li YW et al. (2011) Intratumoral neutrophils: a poor prognostic factor for hepatocellular carcinoma following resection. J. Hepatol 54, 497–505 [DOI] [PubMed] [Google Scholar]
- 82.Jensen HK et al. (2009) Presence of intratumoral neutrophils is an independent prognostic factor in localized renal cell carcinoma. J. Clin. Oncol 27, 4709–4717 [DOI] [PubMed] [Google Scholar]
- 83.Trellakis S et al. (2011) Polymorphonuclear granulocytes in human head and neck cancer: enhanced inflammatory activity, modulation by cancer cells and expansion in advanced disease. Int. J. Cancer 129, 2183–2193 [DOI] [PubMed] [Google Scholar]
- 84.Ino Y et al. (2013) Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 108, 914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ilie M et al. (2012) Predictive clinical outcome of the intratumoral CD66b-positive neutrophil-to-CD8-positive T-cell ratio in patients with resectable nonsmall cell lung cancer. Cancer 118, 1726–1737 [DOI] [PubMed] [Google Scholar]
- 86.Kargl J et al. (2019) Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight 4, e130850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ghergurovich JM et al. (2020) A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat. Chem. Biol 16, 731–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kelly C et al. (2011) Targeted liposomal drug delivery to monocytes and macrophages. J. Drug Deliv 2011, 727241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Casbon AJ et al. (2015) Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. U. S. A 112, E566–E575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jin C et al. (2019) Commensal microbiota promote lung cancer development via gammadelta T cells. Cell 176, 998–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Highfill SL et al. (2014) Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Sci. Transl. Med 6, 237ra267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun L et al. (2019) Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 4, e126853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cheng Y et al. (2019) Potential roles and targeted therapy of the CXCLs/CXCR2 axis in cancer and inflammatory diseases. Biochim. Biophys. Acta Rev. Cancer 1871, 289–312 [DOI] [PubMed] [Google Scholar]
- 94.Belperio JA et al. (2002) Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J. Clin. Invest 110, 1703–1716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chun E et al. (2015) CCL2 promotes colorectal carcinogenesis by enhancing polymorphonuclear myeloid-derived suppressor cell population and function. Cell Rep. 12, 244–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Semerad CL et al. (2002) G-CSF is an essential regulator of neutrophil trafficking from the bone marrow to the blood. Immunity 17, 413–423 [DOI] [PubMed] [Google Scholar]
- 97.Wculek SK and Malanchi I (2015) Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 528, 413–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kowanetz M et al. (2010) Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc. Natl. Acad. Sci. U. S. A 107, 21248–21255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khajah M et al. (2011) Granulocyte-macrophage colony-stimulating factor (GM-CSF): a chemoattractive agent for murine leukocytes in vivo. J. Leukoc. Biol 89, 945–953 [DOI] [PubMed] [Google Scholar]
- 100.Wang TT et al. (2017) Tumour-activated neutrophils in gastric cancer foster immune suppression and disease progression through GM-CSF-PD-L1 pathway. Gut 66, 1900–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Akbay EA et al. (2017) Interleukin-17A promotes lung tumor progression through neutrophil attraction to tumor sites and mediating resistance to PD-1 blockade. J. Thorac. Oncol 12, 1268–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Coffelt SB et al. (2015) IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bank U et al. (1999) Selective proteolytic cleavage of IL-2 receptor and IL-6 receptor ligand binding chains by neutrophil-derived serine proteases at foci of inflammation. J. Interf. Cytokine Res 19, 1277–1287 [DOI] [PubMed] [Google Scholar]
- 104.Teijeira A et al. (2020) CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity 52, 856–871 [DOI] [PubMed] [Google Scholar]