

Abstract
Traumatic brain injury (TBI) is a leading cause of neurologic disability; the most common deficits affect prefrontal cortex-dependent functions such as attention, working memory, social behavior, and mental flexibility. Despite this prevalence, little is known about the pathophysiology that develops in frontal cortical microcircuits after TBI. We investigated whether alterations in subtype-specific inhibitory circuits are associated with cognitive inflexibility in a mouse model of frontal lobe contusion in both male and female mice that recapitulates aberrant mental flexibility as measured by deficits in rule reversal learning. Using patch-clamp recordings and optogenetic stimulation, we identified selective vulnerability in the non-fast-spiking and somatostatin-expressing (SOM+) subtypes of inhibitory neurons in layer V of the orbitofrontal cortex 2 months after injury. These subtypes exhibited reduced intrinsic excitability and a decrease in their synaptic output onto pyramidal neurons, respectively. By contrast, the fast-spiking and parvalbumin-expressing interneurons did not show changes in intrinsic excitability or synaptic output, respectively. Impairments in non-fast-spiking/SOM+ inhibitory circuit function were also associated with network hyperexcitability. These findings provide evidence for selective disruptions within specific inhibitory microcircuits that may guide the development of novel therapeutics for TBI.
SIGNIFICANCE STATEMENT TBI frequently leads to chronic deficits in cognitive and behavioral functions that involve the prefrontal cortex, yet the maladaptive changes that occur in these cortical microcircuits are unknown. Our data indicate that alterations in subtype-specific inhibitory circuits, specifically vulnerability in the non-fast-spiking/somatostatin-expressing interneurons, occurs in the orbitofrontal cortex in the context of chronic deficits in reversal learning. These neurons exhibit reduced excitability and synaptic output, whereas the other prominent inhibitory population in layer V, the fast-spiking/parvalbumin-expressing interneurons as well as pyramidal neurons are not affected. Our work offers mechanistic insight into the subtype-specific function of neurons that may contribute to mental inflexibility after TBI.
Keywords: disinhibition, optogenetics, orbitofrontal cortex, selective vulnerability, somatostatin inhibitory neurons, traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of chronic neurologic disability (Engberg and Teasdale, 2004; Ponsford et al., 2008; Wilson et al., 2017). Damage often affects the frontal lobes, and thus the most frequent cognitive sequelae are deficits in prefrontal cortex (PFC)-dependent functions, such as attention, working memory, social behavior, mental flexibility, and task switching (Vilkki et al., 1994; Wallesch et al., 2001; Fujiwara et al., 2008; Struchen et al., 2008; Stuss, 2011; Spikman et al., 2012). Despite this prevalence, little is known about the chronic neuronal and network dysfunction associated with cognitive deficits after TBI, especially in frontal lobe circuits.
Cortical hyperexcitability has been identified in other regions of the neocortex adjacent to injury (Ding et al., 2011; Cantu et al., 2015; Carron et al., 2016). Loss of inhibitory function is postulated to contribute to these network changes because of a reduction in synaptic inhibition and decreases in expression of several immunohistochemical markers of interneurons, which have been reported particularly in severe TBI models (Buriticá et al., 2009; Cantu et al., 2015; Carron et al., 2016; Nichols et al., 2018). However, the direct intrinsic and synaptic function of specific interneuron subtypes has not been systemically evaluated and compared.
Two major classes of GABAergic interneurons mediating inhibition of cortical output, particularly in deeper cortical layers, are parvalbumin-expressing (PV+) fast-spiking (FS) cells (45–60%), and somatostatin-expressing (SOM+) low-threshold spiking or regular spiking cells (20–50%; Rudy et al., 2011; Naka and Adesnik, 2016). These neurons have distinct roles in the cortical microcircuit. PV+ neurons receive convergent thalamic input, synapse onto the perisomatic domains of pyramidal neurons, and have depressing synaptic inputs, supporting feedforward inhibition (Willems et al., 2018). This inhibition constrains the timing of temporal summation in pyramidal neurons (Pouille and Scanziani, 2001), affects stimulus tuning (Wang et al., 2004a; Zhu et al., 2015), increases the dynamic range of neuronal ensembles (Pouille et al., 2009), and mediates gamma oscillations during cognition (Cho et al., 2015). In contrast, SOM+ cells do not receive thalamic inputs, target dendrites instead of perisomatic regions, and have facilitating synapses (Naka and Adesnik, 2016). The combination of these properties promotes increasing SOM+ cell-mediated inhibition with increasing pyramidal neuron activity, producing feedback inhibition. Although PV+ cells also provide an immediate feedback inhibition, SOM+ cells have been found to support a more delayed feedback pathway (Silberberg and Markram, 2007), which contributes to expansion of the input response range in sensory cortices (Murayama et al., 2009) as well as to synchronization of lower frequency theta and beta oscillations between circuits (Chen et al., 2017; Abbas et al., 2018). Changes in orbitofrontal cortex (OFC) inhibitory neurons, particularly PV+ neurons, have been identified with reversal learning deficits in a sex-selective model of early life stress (Goodwill et al., 2018). How TBI alters these distinct inhibitory microcircuits in ways that may affect cortical circuit function and cognition has not been studied before.
We developed a mouse model of frontal lobe contusion that recapitulates cognitive inflexibility observed in humans after trauma (Chou et al., 2016). These mice exhibit deficits in reversal learning, a cognitive ability requiring OFC function (Bissonette et al., 2008). Interestingly, this type of contusion does not lesion the OFC, nor is the OFC in a directly pericontusional location. Thus, an immediately apparent mechanism for this deficit is unclear from the gross histology. The goal of this study was to understand if deficits in inhibition might be identified in the OFC at chronic time points after TBI when reversal learning impairments are observed. We used transgenic mouse lines and optogenetic techniques to systematically evaluate and compare the function of different subtypes of inhibitory interneurons in this model.
Materials and Methods
Experimental model
All experiments were conducted in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco (AN184326-02B).
Male and female mice were bred in house using the following strains: B6.129P2-Pvalbtm1(cre)Arbr/J (PV-Cre, catalog #017320, The Jackson Laboratory; RRID:IMSR_JAX:017320), Ssttm2.1(cre)Zjh/J (SOM-Cre, catalog #013044, The Jackson Laboratory; RRID:IMSR_JAX:013044), Tg(I12b-cre)1Jlr (DlxI12b-Cre, from John Rubenstein; Potter et al., 2009), B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J (Ai14, catalog #007914, The Jackson Laboratory; RRID:IMSR_JAX:007914), and B6.Cg-Gt(ROSA)26Sortm32(CAG-COP4*H134R/EYFP)Hze/J (Ai32, catalog #024109, The Jackson Laboratory; RRID:IMSR_JAX:024109). Mice were group housed in environmentally controlled conditions with a 12 h light/dark cycle at 21 ± 1°C, ∼50% humidity, and provided with food and water ad libitum. Mice were 12–13 weeks of age at the time of surgery. Both males and females were used, and results from both sexes were pooled. We did not observe any influence or association of sex on the experimental outcomes.
Surgical procedures
Mice were randomly assigned to a TBI or sham surgery group. They were anesthetized and maintained under anesthesia at 2–2.5% isoflurane. TBI, specifically frontal lobe contusion, was achieved using a controlled cortical impact (CCI) surgery, performed as previously described (Chou et al., 2016). Briefly, mice were secured in a stereotaxic frame. A midline incision exposed the skull followed by a ∼2.5 mm diameter craniectomy at +2.34 mm anteroposterior and +1.62 mm mediolateral with respect to bregma. After the craniectomy, contusion was induced using a 2 mm convex tip attached to an electromagnetic impactor (Leica). The contusion depth was set to 1.25 mm from dura with a velocity of 4.0 m/s sustained for 300 ms. Following impact, the scalp was sutured. These injury parameters were chosen to induce contusion over the right frontal lobe but not result in a large cavity. In terms of tissue loss, when compared with the classic severe parietal lobe CCI (Smith et al., 1995; Bondi et al., 2014), these injury parameters result in a more moderate injury. Sham animals were subjected to identical parameters excluding the craniectomy and impact.
Slice preparation
Sagittal brain slices (250 µm) including the orbitofrontal cortex (between approximately +0.7 mm to approximately +1.7 mm from bregma) were prepared from mice that underwent TBI or a sham procedure ∼2–3 months prior. Mice were anesthetized with Euthasol (0.1 ml/25 × g; NDC-051311-050-01, Virbac), and transcardially perfused with an ice-cold sucrose solution containing the following (in mm): 210 sucrose, 1.25 NaH2PO4, 25 NaHCO3, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 7 dextrose, 1.3 ascorbic acid, 3 sodium pyruvate (bubbled with 95% O2/5% CO2, pH ∼7.4). Mice were then decapitated, and the brain was isolated in the same sucrose solution and cut on a slicing vibratome (VT1200S, Leica Microsystems). Slices were incubated in a holding solution, composed of the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 2 MgCl2, 10 dextrose, 1.3 ascorbic acid, 3 sodium pyruvate (bubbled with 95% O2/5% CO2, pH ∼7.4 at 36°C for 30 min and then at room temperature for at least 30 min until recording).
Intracellular recording
Whole-cell recordings were obtained from these slices in a submersion chamber with a heated (32–34°C) artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 10 dextrose (bubbled with 95% O2/5% CO2, pH ∼7.4). Patch pipettes (3-6 MΩ) were manufactured from filamented borosilicate glass capillaries (catalog #BF100-58-10, Sutter Instruments) and filled with an intracellular solution containing the following (in mm): 135 K-Gluconate, 5 KCl, 10 HEPES, 4 NaCl, 4 MgATP, 0.3 Na3GTP, 7 2K-phosphcreatine, and 1–2% biocytin. For recording of quantal IPSCs (qIPSCs), a cesium-based intracellular solution was instead used containing the following (in mm): 135 CsMeS, 5 CsCl, 10 HEPES, 4 NaCl, 4 MgATP, 0.3 Na3GTP, 7 2K-phosphcreatine, 2 Qx314Br, and 1–2% biocytin. Neurons were visualized using infrared microscopy with a 40× water-immersion objective (Olympus). Pyramidal neurons and dTomato-expressing inhibitory neurons located in layer V were targeted for patching. Recordings were made using a MultiClamp 700B (Molecular Devices) amplifier, which was connected to the computer with a Digidata 1440A ADC (Molecular Devices) and recorded at a sampling rate of 20 kHz with pClamp software (Molecular Devices). We did not correct for the junction potential, but pipette capacitance was appropriately compensated before each recording. Cells were excluded if the access resistance was or rose to above 20 MΩ during recording.
The passive membrane and active action potential (AP) spiking characteristics were assessed by injection of a series of hyperpolarizing and depolarizing current steps with a duration of 250 ms from −250 pA to 700 pA (in increments of 50 pA). The resting membrane potential was the measured voltage of the cell after obtaining whole-cell configuration without current injection. A holding current was then applied to maintain the neuron at about −67 ± 3 mV before/after current injections. The input resistance was determined from the steady-state voltage reached during the −50 pA current injection. The membrane time constant was the time required to reach 63% of the maximum change in voltage for the −50 pA current injection.
AP properties including the half width, threshold, amplitude, rising slope, falling slope, and spike AHP were calculated based on the response to a current pulse that was 100 pA above the minimal level that elicited spiking, or if this injection had fewer than three action potentials, the first current injection with >3 action potentials. The AP threshold was defined as the voltage at which the third derivative of V (d3V/dt) was maximal just before the action potential peak. The AP amplitude was calculated by measuring the voltage difference between the peak voltage of the action potential and the spike threshold. The half width was determined as the duration of the action potential at half the amplitude. The spike AHP was the voltage difference between the AP threshold and the minimum voltage before the next AP. The rising and falling slopes of the AP were the maximum of the first derivative of V between the threshold to the peak amplitude and the peak amplitude to the minimum voltage before the next AP, respectively.
Action potential timing was detected by recording the time at which the positive slope of the membrane potential crossed −20 mV. From the action potential times, the instantaneous frequency for each action potential was determined (1/interspike interval). The adaptation index of each cell was the ratio of the first over the last instantaneous firing frequency in response to a current pulse that was 400 pA above the minimal level that elicited spiking. The AP rate as a function of current injection was examined by plotting the first instantaneous AP frequency versus current injection amplitude. The rheobase was then calculated as the intercept of the best linear fit of the first four positive values of this plot.
As noted above, we identified interneurons based on the expression of dTomato driven by a Cre-dependent fluorescent reporter mouse line (Ai14) crossed to a DlxI12b-Cre mouse (Potter et al., 2009). This strategy labels a majority of interneuron subtypes. Recorded interneurons were therefore subdivided into FS or non-FS groups based on the range of electrophysiological properties identified in a subset of these neurons that were successfully filled with biocytin and processed for immunohistochemistry with somatostatin and parvalbumin antibodies after recording. The criteria for the FS group was an adaptation index of <1.9 and a half width of <0.35 ms based on the eight histologically verified parvalbumin-expressing neurons. The criteria for the non-FS group was an adaptation index of >1.6 and a half width of 0.45–0.8 ms based on the 10 histologically verified somatostatin-expressing neurons.
Optogenetic stimulation
To measure optogenetically evoked SOM+- or PV+-mediated inhibitory postsynaptic currents (oIPSCs), SOM-Cre and PV-Cre mice were crossed with a ChR2 (channelrhodopsin-2)/EYFP (enhanced yellow florescent protein) reporter line (Ai32). Voltage-clamp recordings were attained from layer V pyramidal neurons in the OFC while the cell was held at 0 mV in voltage clamp. Ten-millisecond flashes of blue light, generated by a Lambda DG-4 high-speed optical switch with a 300 W Xenon lamp (Sutter Instruments) and an excitation filter set centered ∼470 nm, were delivered to the slice through a 40× objective (Olympus) to activate ChR2 at a range of light intensities from 40 uW/mm2 to 1 mW/mm2 (1–25% of the maximum intensity generated). At each light intensity, the light pulse was repeated five times with a 4 s interval in between each stimulus. The five oIPSCs were averaged, and the total charge/area under the curve (AUC), for 100 ms after the light pulse, was measured for each light intensity. Using a similar protocol, we also delivered trains of 5 ms light pulses at both 10 and 40 Hz stimulation frequencies. The AUC between light pulses was determined for each pulse in the train of pulses. The paired pulse ratio was calculated as the ratio of the response to the second pulse/first pulse.
To examine light-evoked qIPSCs specifically from SOM+ neurons, the calcium in the aCSF was replaced with an equal amount of strontium (2 mm SrCl2 instead of 2 mm CaCl2). Quantal IPSCs are similar to miniature IPSCs, whereby the frequency of qIPSCs is thought to reflect presynaptic properties, and the amplitude corresponds to postsynaptic changes of synapses, but they differ from mIPSCs in that they are evoked often by electrical or optogenetic stimulation of specific afferent connections or cell types. This allows dissection of the synaptic properties of specific inputs to a neuron, rather than measuring spontaneous activation of all synapses. This is possible with the replacement of calcium in the bath with strontium. Strontium is thought to induce asynchronous release through poorer binding affinity to calcium-binding proteins that regulate vesicular release causing slower vesicle docking and through a greater permeability through calcium channels leading to higher intracellular concentrations, and thus longer intervals to stimulate transmitter release (Xu-Friedman and Regehr, 1999). As described above, voltage-clamp recordings were obtained from layer V pyramidal neurons in SOM-CrexAi32 mice while activating ChR2 with a 10 ms light pulse delivered at the 5% light intensity (225 µW/mm2). The light pulse was repeated 30 times with a 10 s interval in between each stimulus. Analysis of quantal events was performed over an 800 ms window beginning 200 ms after the stimulus onset, a time window designed to eliminate synchronous synaptic responses. qIPSC frequency and amplitude were analyzed using a template-matching algorithm in ClampFit 10.7 (Molecular Devices; RRID:SCR_011323). The template was created using recordings from multiple pyramidal cells and included several hundred qIPSC events. The first 200 qIPSCs or all the events measured in the 30 sweeps from each cell were included for analysis. The mean frequency and amplitude for these events was calculated for each cell to compare between groups.
Network excitability
To evaluate network excitability in TBI and sham cortical networks, slices were incubated in aCSF without Mg2+ (0 mm MgCl2) and with increased K+ (5 mm KCl). In this solution, synchronized polysynaptic events occur with time, and the time to onset of the second clearly recurrent polysynaptic burst with an amplitude >2 mV was compared while recording in current clamp from layer V neurons while holding the membrane potential to about −65 to −70 mV with holding current. Activity was recorded for 45 min, and the occurrence of large paroxysmal depolarization events was also noted. These events were defined as input >20 mV and lasting >2 s. Some neurons were lost after the start of synchronized polysynaptic activity but before reaching a recording length of 45 min. These cells were included in the analysis of latency to onset of synchronized input but were excluded from the analysis for paroxysmal depolarizing shift (PDS) occurrence.
Immunohistochemistry
Brain slices obtained from electrophysiological recordings were drop-fixed in 4% paraformaldehyde in a phosphate-buffered solution (4–48 h), then rinsed with PBS with sodium azide (0.1 m PBS plus 0.05% NaN3) and stored at 4°C for 0–5 d until processing. Sections were then washed in PBS (3 × 10 min), incubated in a blocking solution (10% donkey serum diluted in 0.5% Triton X-100 in 0.1 m PBS) for 1 h, and then incubated with primary antibodies, somatostatin (1:1000; catalog #ab64053, Abcam; RRID:AB_1143012), and parvalbumin (1:8000; catalog #MAB1572, Millipore; RRID:AB_2174013), with 0.25% Triton X-100, and 5% donkey serum in 0.1 m PBS, at room temperature overnight, along with streptavidin-conjugated Pacific Blue (1:500; catalog #S11222, Thermo Fisher Scientific). The following day, sections were then rinsed with PBS (3 × 10 min) and incubated in secondary antibodies (1:500; Alexa 488 donkey anti-rabbit, catalog #A-21206, Invitrogen; RRID:AB_2535792 and 1:500; Alexa 647 donkey anti-mouse, catalog #ab181292, Abcam) at room temperature overnight. Finally, sections were rinsed and mounted in an aqueous medium. Images were obtained using a Zeiss Imager Z1 ApoTome microscope controlled by Zen software (Black Zen Software; RRID:SCR_018163).
For analysis of EYFP expression, slices were fixed as described above after recording optogenetically evoked IPSCs from both SOM-CrexAi32 and PV-CrexAi32 mice. These slices were incubated with a primary antibody to enhance expression of EYFP (anti-GFP, 1:500; catalog #11-476-C100, EXBIO Praha; RRID:AB_10735170), followed by secondary antibody (Alexa 488 donkey anti-rabbit, 1:500; catalog #A-21206, Invitrogen; RRID:AB_2535792) as described above. The OFC was imaged with a 20× objective, centered over layer V obtaining a z-stack of 15 µm with a 0.5 µm slice interval using the same exposure time across slices. Image stacks were flattened and analyzed using ImageJ (National Center for Microscopy and Imaging Research: ImageJ Mosaic Plug-ins; RRID:SCR_001935). Both the average intensity as well as the percentage area of staining after binary thresholding using the same threshold across images were calculated.
Experimental design and statistical analysis
All data were evaluated with GraphPad Prism 8 statistical software (RRID:SCR_002798). Mice were randomly assigned to experimental groups, and the different surgical procedures used within a given experiment were randomly distributed across these mice (and within a given surgical day). For the various analyses, we repeated the experiment in multiple cohorts of animals. Statistical significance between groups for most variables was determined using an unpaired t test with or without Welch's correction. For nonparametric data, a Mann–Whitney test was assessed to determine significance. The firing responses to increasing current injections and the oIPSC AUC with increasing light intensity or pulse number were analyzed as a repeated measures two-way ANOVA or mixed effects model. Post hoc multiple comparisons were assessed controlling for the false discovery rate using the method of Benjamini and Hochberg. A χ2 test evaluated the probability of PDS occurrence, and p values <0.05 were considered significant. All the statistical details of experiments can be found in the figure legends including the statistical tests used, exact value of n, and what n represents.
Results
Non-fast-spiking interneurons exhibit a selective reduction in intrinsic excitability at chronic time points after TBI
To investigate the neurophysiological changes that accompany the chronic reversal learning deficits that occur after frontal lobe contusion (Chou et al., 2016), we first assessed the intrinsic firing properties of layer V inhibitory neurons in the orbitofrontal cortex. Whole-cell patch-clamp recordings were obtained from mice 2–3 months after undergoing a frontal lobe controlled CCI or sham procedure. We visually identified interneurons using a transgenic mouse line that expresses a dTomato fluorescent reporter in inhibitory neurons (DlxI12b-Cre × Ai14). A subset of neurons was filled with biocytin and processed for immunohistochemistry with somatostatin and parvalbumin antibodies after recording (Fig. 1A,K; n = 10 somatostatin positive, n = 8 parvalbumin positive). The range of the action potential half-width and adaptation index in these immunohistochemically verified somatostatin- or parvalbumin-expressing neurons was then used to categorize recordings as FS (correlating with parvalbumin expression) or non-FS (correlating with somatostatin expression; Fig. 2).
Figure 1.
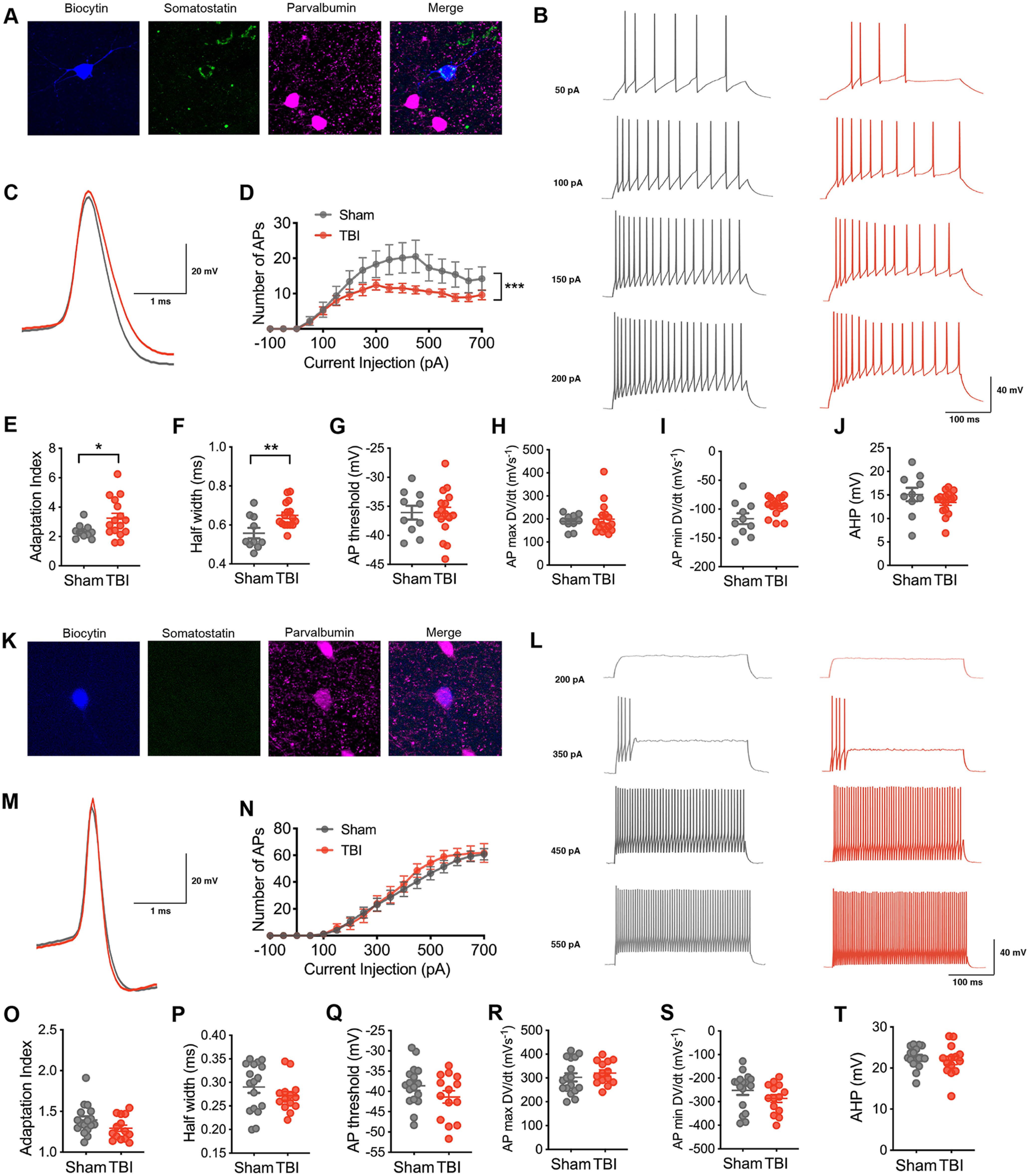
Selective vulnerability of intrinsic excitability in non-fast-spiking inhibitory neurons after chronic TBI. A, A non-fast-spiking interneuron in layer V of the orbitofrontal cortex that was filled with biocytin (blue) and later confirmed to express somatostatin (green) and not parvalbumin (pink). B, Representative current-clamp responses to depolarizing current steps in sham (gray) and TBI (red) mice. C, Average action potential shape. D, The number of action potentials plotted as a function of current injection (F(1,24) = 5.117, p = 0.0330 for TBI effect, and ***F(18,432) = 2.427, p = 0.0010 for interaction between current and TBI, repeated measures 2-way ANOVA). E, The adaptation index from current-clamp responses measured at 400 pA above spiking threshold (*p = 0.0226, unpaired t test with Welch's correction). F–J, The action potential (AP) half width, AP threshold, AP maximum rising slope, AP minimum falling slope, and AHP calculated from current-clamp responses 100 pA above spiking threshold (**p = 0.0053, unpaired t test; p = 0.9529, unpaired t test; p = 0.7366, Mann–Whitney test; p = 0.0530, unpaired t test with Welch's correction; p = 0.2323, unpaired t test, respectively). K, A fast-spiking interneuron in layer V of the orbitofrontal cortex that was filled with biocytin (blue) and later confirmed to express parvalbumin (pink) and not somatostatin (green). L, Representative current-clamp responses to depolarizing current steps in sham (gray) and TBI (red) mice. M, Average action potential shape. N, The number of action potentials plotted as a function of current injection (F(1,29) = 0.2705, p = 0.6069 for TBI effect, repeated measures 2-way ANOVA). O, The adaptation index from current-clamp responses measured at 400 pA above spiking threshold (p = 0.0994, unpaired t test). P–T, The AP half width, threshold, maximum rising slope, and minimum falling slope, and AHP calculated from current-clamp responses 100 pA above spiking threshold (p = 0.2574, unpaired t test; p = 0.1614, unpaired t test; p = 0.4253, unpaired t test; p = 0.2010, unpaired t test; p = 0.5087, unpaired t test, respectively). Each neuron is represented with a symbol; solid lines indicate the mean ± SEM [n = 10 sham and 16 TBI non-fast-spiking neurons from 7 (sham) and 8 (TBI) animals/group; n = 17 sham and 14 TBI fast-spiking neurons from 9 (sham) and 7 (TBI) animals/group].
Figure 2.

Parameters defining FS and non-FS neurons. The range of the action potential half-width and adaptation index for all dTomato+ neurons recorded in the Dlx-I12b-CrexAi14 is presented from both sham (gray) and TBI (red) mice. Immunohistochemically verified cells are indicated with a diamond shape. The PV-expressing neurons defined the parameters of the FS group (dashed box), whereas the SOM-expressing neurons defined the parameters of the non-FS group (dotted box).
Based on the response to a series of current injections, we evaluated intrinsic excitability in each inhibitory firing pattern subtype. Non-FS neurons from TBI animals exhibited a reduction in the number of action potentials seen with increasing current injection when compared with non-FS neurons from sham animals (Fig. 1B,D). This reduction in excitability was associated with a higher adaptation index (Fig. 1B,E) and a wider action potential half width (Fig. 1C,F). Other action potential metrics and passive membrane properties were not affected (Fig. 1G,H,J, Table 1).
Table 1.
Intrinsic properties of layer V neurons
| Non-FS Sham | Non-FS TBI | p value | FS sham | FS TBI | p Value | PYR sham | PYR TBI | p Value | |
|---|---|---|---|---|---|---|---|---|---|
| Action potential properties | |||||||||
| Half width (m sec) | 0.56 (0.03) | 0.65 (0.02) | 0.0053 (unpaired t test) | 0.29 (0.01) | 0.27 (0.01) | 0.2574 (unpaired t test) | 0.86 (0.07) | 0.78 (0.04) | 0.2938 (unpaired t test) |
| Amplitude (mV) | 52.07 (2.90) | 55.00 (2.68) | 0.4808 (unpaired t test) | 53.75 (1.80) | 54.75 (2.01) | 0.7432 (unpaired t test) | 74.12 (2.26) | 75.79 (1.32) | 0.5154 (unpaired t test) |
| Threshold (mV) | −36.09 (1.18) | −36.18 (1.02) | 0.9529 (unpaired t test) | −38.61 (1.22) | −41.37 (1.52) | 0.1614 (unpaired t test) | −39.56 (1.38) | −37.53 (1.08) | 0.2491 (unpaired t test) |
| Rising slope (mV/s) | *191.8 (165.4–212.7) | *177.3 (162.4–236.5) | 0.7366 (Mann–Whitney) | 302.5 (17.21) | 320.5 (12.87) | 0.4253 (unpaired t test) | 301.8 (19.99) | 353.8 (13.73) | 0.0368 (unpaired t test) |
| Falling slope (mV/s) | −116.6 (9.38) | −94.68 (4.12) | 0.0530 (Welch's t test) | −252.2 (19.13) | −286.2 (16.77) | 0.2010 (unpaired t test) | −99.33 (11.03) | −92.38 (6.77) | 0.5855 (unpaired t test) |
| Spike AHP (mV) | 15.11 (1.41) | 13.42 (0.64) | 0.2323 (unpaired t test) | 22.61 (0.62) | 21.85 (1.01) | 0.5087 (unpaired t test) | 11.59 (0.69) | 15.59 (0.89) | 0.0015 (unpaired t test) |
| Adaptation index | 2.35 (0.16) | 3.25 (0.33) | 0.0226 (Welch's t test) | 1.40 (0.04) | 1.292 (0.04) | 0.0994 (unpaired t test) | *2.59 (2.51 - 3.94) | *2.43 (2.12 - 3.48) | 0.2777 (Mann–Whitney) |
| Rheobase (mV) | 69.76 (20.73) | 58.75 (16.76) | 0.6849 (unpaired t test) | 194.20 (31.67) | 194 (28.01) | 0.9971 (unpaired t test) | 86.31 (14.48) | 86.1 (12.32) | 0.9915 (unpaired t test) |
| Passive properties | |||||||||
| Resting potential (mV) | −61.3 (2.92) | −59.19 (1.77) | 0.5166 (unpaired t test) | −65.12 (1.40) | −66.71 (1.85) | 0.4899 (unpaired t test) | −64.53 (1.85) | −66.06 (1.48) | 0.5200 (unpaired t test) |
| Membrane resistance (MΩ) | 271.3 (37.06) | 279.2 (26.77) | 0.8618 (unpaired t test) | 140.6 (16.22) | 124.8 (13.34) | 0.4702 (unpaired t test) | 133.3 (16.37) | 130.8 (10.60) | 0.8961 (unpaired t test) |
| Tau (m sec) | 15.67 (2.18) | 19.08 (2.70) | 0.3826 (unpaired t test) | *7.55 (6.10 - 9.53) | *6.9 (5.79 - 8.54) | 0.4041 (Mann–Whitney) | 17.7 (1.77) | 20.97 (1.63) | 0.1836 (unpaired t test) |
Values are mean (SEM) or
*median (95% CI). Statistical test is indicated with associated p value. PYR= pyramidal neuron. Italics further denote p values. Bold values indicate significant p values.
In contrast, intrinsic excitability was not altered in the FS group with TBI. A similar number of action potentials was found at varying amplitudes of current injection in both experimental groups (Fig. 1L,N). Other action potential and passive membrane properties were not significantly changed.
For comparison, we also evaluated the excitability of layer V pyramidal neurons. Subtle differences in action potential shape were found with TBI in comparison to neurons from sham animals (Fig. 3C,H,J). However, these changes did not alter overall action potential production or intrinsic excitability (Fig. 3B,D).
Figure 3.
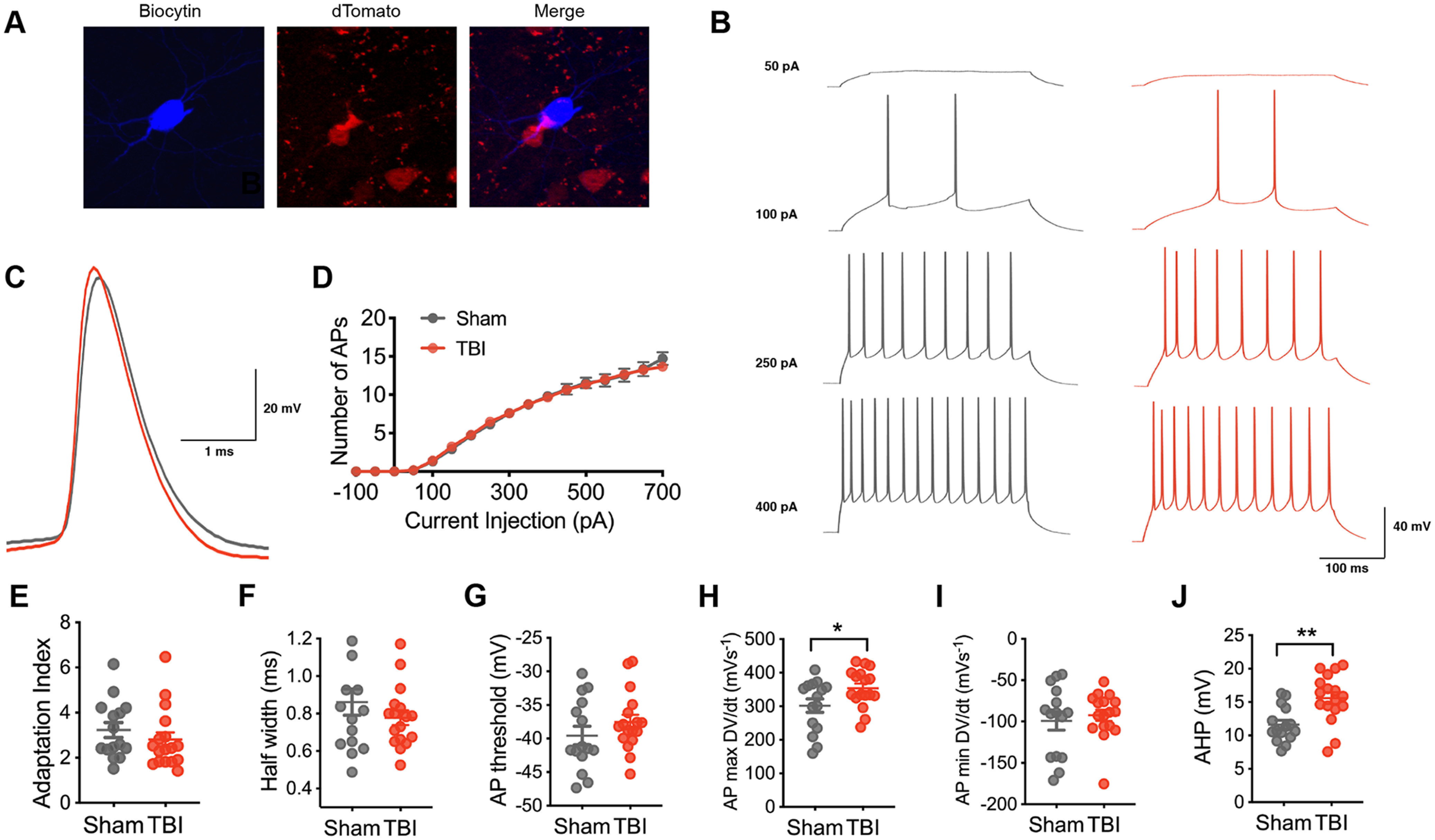
Intrinsic excitability in layer V pyramidal neurons after chronic TBI. A, A pyramidal neuron in layer V of the orbitofrontal cortex that was filled with biocytin (blue) and later confirmed to not express tdTomato (red). B, Representative current-clamp responses to depolarizing current steps in sham (gray) and TBI (red) mice. C, Average action potential shape. D, The number of action potentials plotted as a function of current injection (F(1,30) = 0.002, p = 0.9608 for TBI effect, repeated measures 2-way ANOVA). E, The adaptation index from current-clamp responses measured at 400 pA above spiking threshold (p = 0.2777, Mann–Whitney test). F–J, The AP half-width, AP threshold, AP maximum rising slope, AP minimum falling slope and AHP calculated from current-clamp responses 100 pA above spiking threshold (p = 0.2938, unpaired t test; p = 0.2491, unpaired t test; *p = 0.0368, unpaired t test; p = 0.5855, unpaired t test; **p = 0.0015, unpaired t test, respectively). Each neuron is represented with a symbol; solid lines indicate the mean ± SEM [n = 15 sham and 17 TBI layer V pyramidal neurons from 6 (sham) and 7 (TBI) animals/group].
These findings demonstrate selective deficits in inhibitory neuronal function, specifically in the non-fast-spiking interneuron subtype measured at chronic time points after frontal contusion injury.
Optogenetic stimulation reveals subtype-specific deficits in inhibitory synaptic input
Next, we examined whether this intrinsic deficit was associated with a selective deficit in synaptic inhibition onto layer V pyramidal neurons at chronic time points after contusion. Toward this end, we optogenetically stimulated SOM+ or PV+ inhibitory neurons with flashes of blue light (470 nm) across a high-power field and recorded oIPSCs in layer V pyramidal neurons (Fig. 4A,J). We expressed ChR2-EYFP in specific interneuron subtypes using transgenic mice (SOM-Cre × Ai32 and PV-Cre × Ai32 lines).
Figure 4.
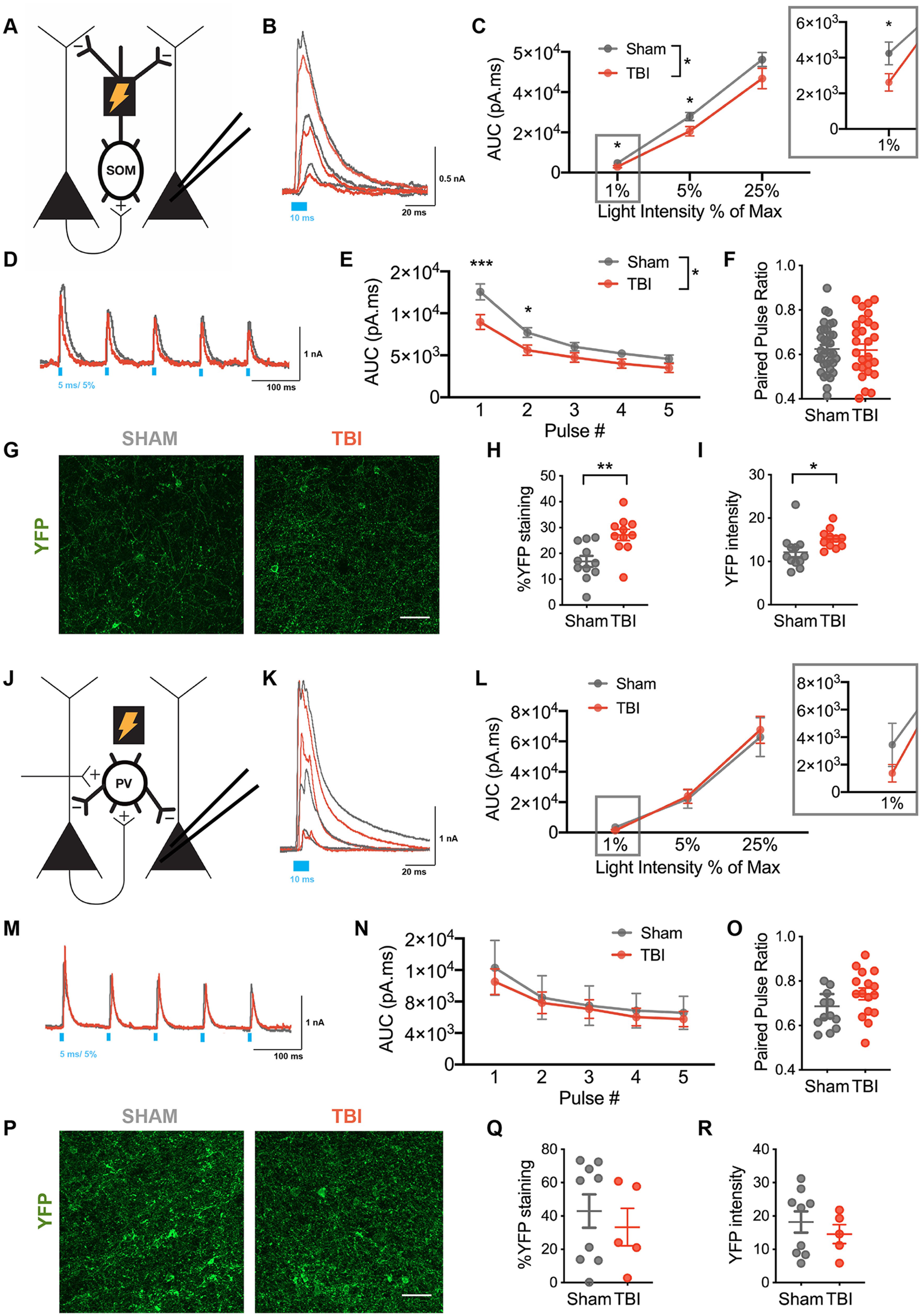
Selective reduced SOM+-mediated inhibitory synaptic output on layer V pyramidal neurons after TBI. A, Schematic of experimental design; voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing SOM+ interneurons. B, Example oIPSCs recorded from pyramidal neurons in sham (gray) and TBI (red) conditions in response to 10 ms light pulses of increasing intensity [40 μW (1%), 225 μW (5%), and 1 mW/mm2 (25%)]. C, Total charge (AUC) of oIPSC across light intensities (*F(1,62) = 4.665, p = 0.0347 for TBI effect, mixed effects model; *p < 0.05, post hoc tests controlling for the false discovery rate). D, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 10 Hz stimulation. E, Total charge (AUC) of oIPSCs elicited across pulse number (*F(1, 62) = 5.055, p = 0.0281 for TBI effect, repeated measures 2-way ANOVA; ***p < 0.0001, *p = 0.0189, post hoc tests controlling for the false discovery rate). F, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.8864, unpaired t test). G, Representative EYFP expression in layer V OFC in sham and TBI slices. H, Percentage area staining after binary thresholding (**p = 0.0032, unpaired t test). I, Average EYFP intensity (*p = 0.0386, unpaired t test). J, Schematic of experimental design. Voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing PV+ interneurons. K, Example oIPSCs recorded from pyramidal neurons in sham (gray) and TBI (red) conditions in response to 10 ms light pulses of increasing intensity [40 μW (1%), 225 μW (5%), and 1 mW/mm2 (25%)]. L, Total charge (AUC) of oIPSC across light intensities (F(1,31) = 0.036, p = 0.8501 for TBI effect, repeated measures 2-way ANOVA). M, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 10 Hz stimulation. N, Total charge (AUC) of oIPSC elicited across pulse number (F(1,31) = 1.289, p = 0.2649 for TBI effect, repeated measures 2-way ANOVA). O, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.0771; unpaired t test). P, Representative EYFP expression in layer V OFC in sham and TBI slices. Q, Percentage area staining after binary thresholding (p = 0.5545 unpaired t test). R, Average EYFP intensity (p = 0.4649, unpaired t test). Circles represent mean; solid lines indicate SEM in C, E, L, N. Each neuron/slice is represented with a symbol, and solid lines indicate the mean ± SEM in F, H, I, O, Q, R [n = 34 sham and 30 TBI neurons from 7 (sham) and 6 (TBI) animals/group for oIPSC data from SOM+ stimulation; n = 12 sham and 11 TBI slices for immunohistochemistry for EYFP expression in SOM+ neurons from 4 (sham) and 4 (TBI) animals/group; n = 16 sham and 17 TBI neurons from 4 (sham) and 4 (TBI) animals/group for oIPSC data from PV+ stimulation; n = 9 sham and 5 TBI slices for immunohistochemistry for EYFP expression in PV+ neurons from 3 (sham) and 3 (TBI) animals/group].
During SOM-specific stimulation, oIPSCs were significantly reduced in neurons from TBI compared with sham animals as measured by the total charge elicited with light stimulation (Fig. 4B,C). Interestingly, post hoc evaluation suggested that this reduction was most notable at lower light intensities, specifically 1% and 5% of the maximum light intensity. In addition to assessing the response to a unitary light pulse, we also determined the response to repetitive stimulation at various frequencies (10 and 40 Hz). Similar to the unitary pulse, repetitive light stimulation at the 5% light intensity demonstrated a lower response in neurons from TBI animals, most prominent with the first few pulses of light (Figs. 4D,E, 5B,C). Also similar to the unitary pulse data, a TBI-associated reduction in response was less apparent at a higher light intensity (25%; Fig. 6A–C). To assess whether these synaptic deficits were driven by changes in presynaptic release probability, we measured the frequency-dependent attenuation of oIPSCs with the paired-pulse ratio (PPR) at both 10 and 40 Hz stimulation. No change in the PPR was identified at either frequency to support TBI-induced chronic changes in the presynaptic release probability (Figs. 4F, 5D, 6D).
Figure 5.
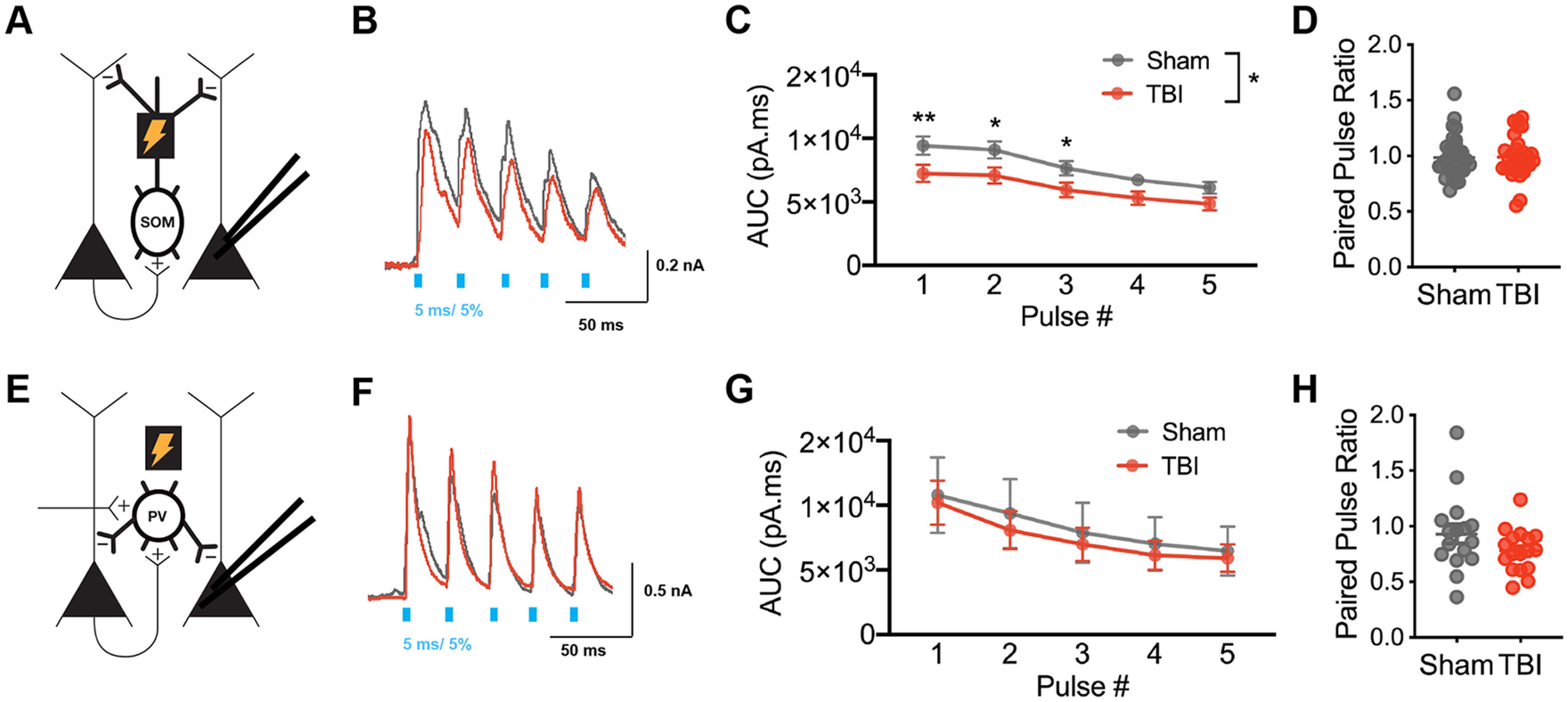
Selective reduced SOM+-mediated inhibitory synaptic input in layer V pyramidal neurons after TBI with 40 Hz repetitive stimulation. A, Schematic of experimental design; voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing SOM+ interneurons. B, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 40 Hz stimulation. C, Total charge (AUC) of oIPSCs elicited across pulse number (*F(1,62) = 4.524, p = 0.0374 for TBI effect, repeated measures 2-way ANOVA; **p = 0.0093, * p < 0.05 post hoc tests controlling for the false discovery rate). D, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.9064, unpaired t test). E, Schematic of experimental design; voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing PV+ interneurons. F, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 40 Hz stimulation. G, Total charge (AUC) of oIPSC elicited across pulse number (F(1,31) = 1.000, p = 0.7540 for TBI effect, repeated measures 2-way ANOVA). H, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.1364; unpaired t test with Welch's correction). Circles represent mean, solid lines indicate SEM in C, G. Each neuron/slice is represented with a symbol, and solid lines indicate mean ± SEM in D, H [n = 34 sham and 30 TBI neurons from 7 (sham) and 6 (TBI) animals/group for oIPSC data from SOM+ stimulation; n = 16 sham and 17 TBI neurons from 4 (sham) and 4 (TBI) animals/group for oIPSC data from PV+ stimulation].
Figure 6.
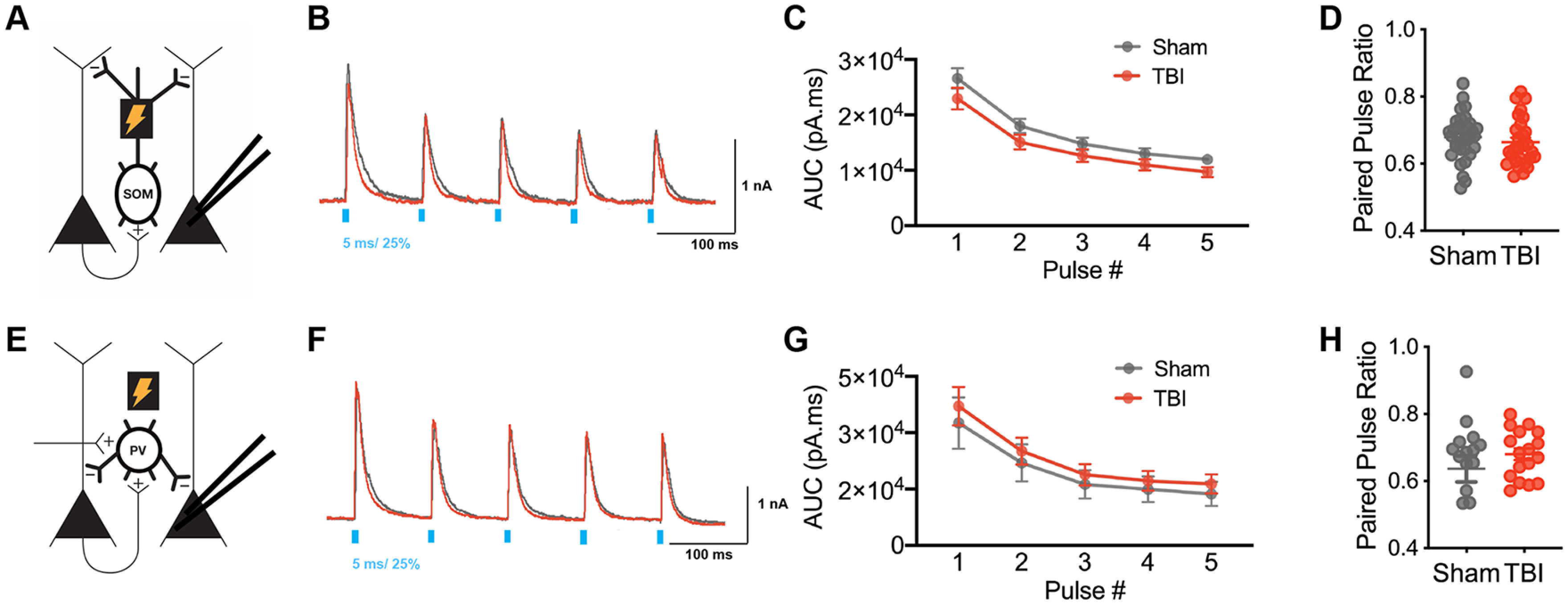
SOM+- and PV+-mediated inhibitory synaptic input in layer V pyramidal neurons with 10 Hz repetitive stimulation at 25% light intensity. A, Schematic of experimental design; voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing SOM+ interneurons. B, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 10 Hz stimulation at 25% light intensity. C, Total charge (AUC) of oIPSCs elicited across pulse number (F(1,58) = 2.225, p = 0.1412 for TBI effect, repeated measures 2-way ANOVA). D, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.4160, unpaired t test). E, Schematic of experimental design. Voltage-clamp recordings were obtained from layer V pyramidal neurons while activating ChR2-expressing PV+ interneurons. F, Example oIPSCs in sham (gray) and TBI (red) conditions elicited with 10 Hz stimulation at 25% light intensity. G, Total charge (AUC) of oIPSC elicited across pulse number (F(1,31) = 0.3038, p = 0.5855 for TBI effect, repeated measures 2-way ANOVA). H, The paired-pulse ratio determined as the ratio of pulse 2/pulse 1 (p = 0.3199; unpaired t test with Welch's correction). Circles represent mean; solid lines indicate SEM in C, G. Each neuron/slice is represented with a symbol, and solid lines indicate mean ± SEM in D, H [n = 34 sham and 30 TBI neurons from 7 (sham) and 6 (TBI) animals/group for oIPSC data from SOM+ stimulation; n = 16 sham and 17 TBI neurons from 4 (sham) and 4 (TBI) animals/group for oIPSC data from PV+ stimulation].
In contrast, during PV-specific stimulation, TBI did not alter the total charge of oIPSCs, whether with unitary (Fig. 4K,L) or repetitive light stimulation (Figs. 4M,N, 5E–G, 6E–G) at all tested light intensities.
To rule out potential confounding reductions in ChR2-EYFP expression, we confirmed that EYFP fluorescence was not lower after TBI. Indeed, EYFP expression was not reduced in somatostatin neurons; surprisingly, it was increased in the TBI compared with sham group, both the percentage area of staining after binary thresholding, as well as the average intensity (Fig. 4G–I). Thus, the functional impairment measured in somatostatin-specific synaptic inhibition may be even greater than our data suggest given the TBI-associated compensation of increased EYFP expression. EYFP expression in the PV+ population was not different for TBI versus sham groups (Fig. 4P–R).
These data support selective deficits in SOM+ neuronal function at chronic time points after TBI, specifically SOM-specific reduction in synaptic inhibition in the orbitofrontal cortex.
TBI-induced reduction of SOM-mediated inhibition is also partially mediated by changes in quantal size and frequency
To further probe how changes in synaptic function might contribute to reduced synaptic output from SOM+ interneurons after TBI, we analyzed qIPSCs during optogenetically evoked asynchronous release (Fig. 7A). qIPSCs differ from miniature IPSCs in that they are evoked by activation of specific afferent connections or cell-types, in this case SOM+ neurons, allowing dissection of the synaptic properties of SOM-specific inputs to a neuron, rather than measuring spontaneous activation of all synapses. We replaced calcium with strontium in the extracellular solution to desynchronize the evoked release of neurotransmitter, allowing analysis of quantal events from stimulated synapses. Similar to miniature IPSCs, the frequency of qIPSCs is thought to reflect presynaptic properties, whereas the amplitude corresponds to postsynaptic changes.
Figure 7.

TBI reduces both quantal size and frequency of SOM-mediated inhibition. A, Example optogenetically evoked qIPSCs elicited with strontium replacing calcium in the aCSF, recorded from pyramidal neurons in sham (gray) and TBI (red) conditions after 10 ms light stimulation of SOM+ neurons. The yellow line denotes window of analysis after stimulation. B, qIPSC frequency (*p = 0.0159; unpaired t test). C, qIPSC amplitude (**p = 0.0086; Welch's t test). Each neuron is represented with a symbol, and solid lines indicate mean ± SEM [n = 11 sham and 12 TBI neurons from 3 (sham) and 3 (TBI) animals/group].
SOM-specific optogenetic stimulation revealed that TBI altered both qIPSC frequency and amplitude at SOM+ inhibitory synapses when compared with sham recordings. Specifically, both the amplitude and frequency were reduced in TBI compared with sham cohorts (Fig. 7B,C). This suggests that changes in both presynaptic and postsynaptic factors may contribute to the reduced inhibitory output from SOM+ interneurons onto pyramidal neurons after TBI.
Cortical network excitability is associated with a reduction of SOM-mediated inhibition after TBI
To assess whether non-FS/SOM-specific deficits in inhibition in the OFC correlate with a hyperexcitable cortical network, we measured the onset time to synchronized network input and the likelihood of large paroxysmal depolarization events in TBI and sham slices incubated in aCSF with 0 mm Mg2+ and 5 mm K+. In this solution, we observed the gradual development of polysynaptic synchronized events (>2 mV in amplitude) within current-clamp recordings over time. In some cases, this progressed further to large, prolonged events (>20 mV in amplitude, >2 s in duration) with overlaid high-frequency spiking activity, termed paroxysmal depolarizations (PDSs; Fig. 8A).
Figure 8.

Frontal lobe contusion induces network hyperexcitability in the orbitofrontal cortex. A, Representative current-clamp recordings from layer V pyramidal neurons of the OFC in a sham (gray) and TBI (red) animal showing synchronized input induced with a 0 mm Mg2+/5 mm K+ ACSF. Transition to large paroxysmal depolarizations with higher frequency components only occurred in the TBI animal in this example. B, The latency from 0 mm Mg2+/5 mm K+ ACSF application until the start of synchronized inputs (*p = 0.0257, unpaired t test). Each neuron is represented with a symbol, and solid lines indicate mean ± SEM [n = 10 sham and 6 TBI neurons from 3 (sham) and 3 (TBI) animals/group]. C, The number of slices that progressed to large paroxysmal depolarizations in TBI compared with sham slices [p = 0.0261, χ2 test; n = 7 sham and 6 TBI neurons from 3 (sham) and 3 (TBI) animals/group].
Slices from animals that underwent TBI 2–3 months prior had a significantly shortened latency to the onset of synchronized synaptic inputs compared with slices from sham animals (Fig. 8B). In addition, all slices from animals that received TBI exhibited PDS events within 45 min of incubation in this hyperexcitable solution, whereas only three of the seven sham slices showed this activity. These data support the hypothesis that TBI-induced non-FS/SOM-specific disinhibition may alter the network balance of excitation and inhibition required for OFC function.
Discussion
Here, we dissected inhibitory circuit function in the orbitofrontal cortex using a mouse model of frontal lobe contusion that exhibits deficits in reversal learning. Our findings demonstrate selective vulnerability in the non-fast-spiking and somatostatin-expressing subtypes of inhibitory neurons at chronic time points after TBI. Specifically, these subtypes exhibited a reduction in excitability and a decrease in their associated synaptic output onto pyramidal neurons, respectively. In contrast, fast-spiking and parvalbumin-expressing neurons did not show changes in intrinsic excitability or synaptic output onto pyramidal neurons, respectively. This reduction in SOM-inhibitory output was not solely because of changes in intrinsic excitability as both the amplitude and frequency of quantal IPSCs originating from SOM interneurons were diminished after TBI. Finally, this selective vulnerability of non-fast-spiking and SOM+ neuronal function identified in the OFC with chronic TBI was associated with a hyperexcitable network. These findings support selective vulnerability of inhibitory populations in TBI and a possible target for therapeutic development.
Careful dissection of cortical inhibitory neuron subtypes and their circuit function has not been previously performed in the context of TBI, especially at chronic time points when persistent neurologic deficits occur. Assessment of intrinsic firing patterns in particular has been limited. PV+/FS neuronal intrinsic function has been evaluated several weeks after a severe frontoparietal pediatric TBI induced with a controlled cortical impact as well as at 24 h after a central fluid percussion injury. Similar to our data, neither study found a change in intrinsic excitability (Nichols et al., 2018; Vascak et al., 2018), but the SOM+/non-FS population was not evaluated. PV+ neurons in the dentate gyrus also did not show changes in intrinsic membrane properties ∼1 week after lateral fluid percussion injury (Folweiler et al., 2020).
A general reduction in synaptic inhibition has been observed in the cortex after TBI. Spontaneous IPSCs are reduced in frequency in layer V pyramidal neurons 2–4 weeks after a severe CCI over the sensorimotor cortex (Cantu et al., 2015). Interestingly, this reduction in sIPSC frequency has also been noted in FS interneurons in a severe pediatric CCI (Nichols et al., 2018) and acutely in mild TBI (Vascak et al., 2018). Although the authors of these studies attribute this reduction to a loss of PV+ neurons or axonal processes, FS neurons also receive inhibition from somatostatin neurons (Cottam et al., 2013; Xu et al., 2013). These findings may thus be compatible with our finding showing selective reduction in subtype-specific inhibition onto layer V pyramidal neurons.
Our research supports that both changes in intrinsic and synaptic function underlie the reduction of SOM-mediated inhibition to pyramidal neurons. The non-FS neurons, presumably enriched for SOM+ neurons, demonstrate decreased excitability with underlying increases in AP half-width and adaptation index, supporting changes in AP repolarization and afterhyperpolarization. AP repolorization in SOM+ neurons is known to depend on both Kv1 channels, as well as BK Ca2+-activated K+ channels, the latter of which is not involved in FS interneuron repolarization (Casale et al., 2015). Certainly decreases in calcium-signaling genes have been noted in neurons after chronic TBI (Witcher et al., 2021), which might suggest different calcium dynamics occur that could affect BK-channel function. These channels are also widely modulated by numerous signaling molecules, including heme, which increases after trauma (Sancho and Kyle, 2021). Other types of potassium channels such as the M-type potassium channel are known to be involved with spike-frequency adaptation and AP repolarization and can be highly affected by neuromodulation (Cooper and Jan, 2003). Noncanonical mechanisms should also be considered. Tonic inhibition can paradoxically increase the gain specifically in non-FS/SOM+ neurons, putatively through enhanced AP depolarization and interaction with potassium currents (Bryson et al., 2020). Reductions in tonic inhibition have been identified in other regions after TBI and should be considered (Parga Becerra et al., 2021).
In addition to intrinsic action potential production, both the frequency and amplitude of SOM-mediated qIPSCs were reduced after TBI. Although we cannot completely exclude that a reduction in amplitude could affect the ability to identify events and artifactually reduce the detection frequency, these findings are potentially consistent with changes in both presynaptic and postsynaptic aspects of SOM-mediated inhibition. A reduction in inhibitory synapses has been found after TBI (Vascak et al., 2017), and a differential effect on GABA(A) receptor subunit types has been reported (Raible et al., 2012; Guerriero et al., 2015), which might correspond to changes in synapse number and/or quantal size. Why these changes appear limited to SOM+ synapses in our data are unclear. Axonal branches of SOM+ Martinotti neurons extend throughout the layers with extensive horizontal arborization, especially in layer I (Wang et al., 2004b). Perhaps this anatomy is more sensitive to compression after TBI, selectively reducing more distal dendritic input and the number of SOM+ synapses to pyramidal neurons. Interestingly, calretinin-expressing interneurons, some of which are SOM+ Martinotti neurons, (Cauli et al., 2014; Nigro et al., 2018), are reported to selectively remodel neurite structure after injury (Brizuela et al., 2017), suggesting a capacity for maladaptive plasticity that selectively occurs in a subset of SOM+ neurons. Further dissection of specific presynaptic and post synaptic mechanisms that mediate SOM inhibition is needed to refine a more intricate understanding of inhibitory networks after TBI.
Previous research has often focused on a loss of inhibitory neurons after TBI as measured with immunohistochemistry (Buriticá et al., 2009; Cantu et al., 2015; Nichols et al., 2018). Comparison of immunohistochemistry with transgenic mouse lines suggests that some of this reduction is secondary to changes in the expression of markers rather than an actual loss of neurons (Nichols et al., 2018). In our model, at chronic time points, we did not document a loss of PV+ or SOM+ neurons or processes as measured by expression of YFP in Cre-dependent transgenic mouse lines. Interestingly, although PV+ YFP expression was not affected, there was a paradoxical increase in SOM+ neuronal YFP expression. This finding might suggest a compensatory mechanism that is, however, ineffective in fully correcting the chronic functional deficits observed here.
In general, impairment in SOM+ function is associated with pathologic network activity and behavioral deficits. Mice lacking DLX1, a transcription factor important for the development of interneuron subsets, show a reduction of SOM+ but not PV+ cells, develop delayed-onset epilepsy, and have deficits in behavioral inhibition and associative fear learning (Cobos et al., 2005; Mao et al., 2009). In epileptic circuits, loss of inhibitory input specifically to the dendritic compartment rather than perisomatic has been noted (Cossart et al., 2001), suggesting a selective dysfunction of SOM+ inhibition. Specific manipulation of SOM+ neurons through selective mutation of MeCP2 leads to repetitive stereotyped behaviors as well as seizures, indicating a specific role for SOM interneurons in Rett syndrome (Ito-Ishida et al., 2015).
It should be emphasized that non-fast-spiking interneurons and somatostatin neurons are not a homogeneous population. Our definition of non-fast-spiking interneurons is based on the half-width and adaptation index of known somatostatin-expressing neurons, thus we believe this subtype to be enriched for in this group. However, other interneurons, such as 5HT3aR- and VIP-expressing interneurons, can exhibit action potential properties that would also include them in this group (Lee et al., 2010). Although these subtypes occur more frequently in supragranular locations, some of these neurons may be included in our analysis, and we cannot exclude the possibility that these subtypes might also be affected by TBI. In addition, exclusively SOM+ neurons represent a diverse group with heterogenous morphology and function. The classic SOM+ neuron, thought to compose the majority of deeper layer SOM+ cells, is the Martinotti cell (MC). This subtype is characterized by translaminar ascending axon collaterals, especially in layer I, and low-threshold or regular spiking firing behavior (Riedemann, 2019). Other SOM+ neurons are often referred to as non-MCs and include morphologically defined basket cells, double-bouquet cells, and long-range inhibitory projection cells among others (Riedemann, 2019). Basket cells may behave more similarly to PV+ neurons, exhibiting innervation around pyramidal cell bodies and quasi-fast spiking physiology (Naka et al., 2019; Riedemann, 2019). Non-MCs also have higher spike thresholds compared with MC neurons (Nigro et al., 2018; Naka et al., 2019). Our data suggest more deficits in SOM-specific input to pyramidal neurons at lower light intensities, which should correspond to neurons with lower thresholds for action potential production. This might be indicative of specific dysfunction within MC cell populations after TBI.
In summary, we have established that non-FS and SOM+ neurons are particularly vulnerable to TBI. Why these non-FS/SOM+ interneurons, and exactly which subpopulation, are particularly vulnerable is a question of great interest. The intrinsic ability for plasticity (Brizuela et al., 2017), unique axonal structure (Wang et al., 2004b), and/or interaction with neuroinflammatory mediators (Um, 2017) needs to be fully investigated to inform potential subtype-targeted therapies for cognitive dysfunction and other sequelae of TBI.
Footnotes
A.L.N. was supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke (Grant K08NS114170) and the University of California, San Francisco Clinical and National Center for Advanced Translational Sciences at National Institutes of Health (Grant TL1 TR001871). S.R. was supported by National Institutes of Health–National Institute on Aging (Grant R01AG056770).
The authors declare no competing financial interests.
References
- Abbas AI, Sundiang MJM, Henoch B, Morton MP, Bolkan SS, Park AJ, Harris AZ, Kellendonk C, Gordon JA (2018) Somatostatin interneurons facilitate hippocampal-prefrontal synchrony and prefrontal spatial encoding. Neuron 100:926–939.e3. 10.1016/j.neuron.2018.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissonette GB, Martins GJ, Franz TM, Harper ES, Schoenbaum G, Powell EM (2008) Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J Neurosci 28:11124–11130. 10.1523/JNEUROSCI.2820-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi CO, Cheng JP, Tennant HM, Monaco CM, Kline AE (2014) Old dog, new tricks: the attentional set-shifting test as a novel cognitive behavioral task after controlled cortical impact injury. J Neurotrauma 31:926–937. 10.1089/neu.2013.3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizuela M, Blizzard CA, Chuckowree JA, Pitman KA, Young KM, Dickson T (2017) Mild traumatic brain injury leads to decreased inhibition and a differential response of calretinin positive interneurons in the injured cortex. J Neurotrauma 34:2504–2517. 10.1089/neu.2017.4977 [DOI] [PubMed] [Google Scholar]
- Bryson A, Hatch RJ, Zandt BJ, Rossert C, Berkovic SF, Reid CA, Grayden DB, Hill SL, Petrou S (2020) GABA-mediated tonic inhibition differentially modulates gain in functional subtypes of cortical interneurons. Proc Natl Acad Sci U S A 117:3192–3202. 10.1073/pnas.1906369117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buriticá E, Villamil L, Guzmán F, Escobar MI, García-Cairasco N, Pimienta HJ (2009) Changes in calcium-binding protein expression in human cortical contusion tissue. J Neurotrauma 26:2145–2155. 10.1089/neu.2009.0894 [DOI] [PubMed] [Google Scholar]
- Cantu D, Walker K, Andresen L, Taylor-Weiner A, Hampton D, Tesco G, Dulla CG (2015) Traumatic brain injury increases cortical glutamate network activity by compromising GABAergic control. Cereb Cortex 25:2306–2320. 10.1093/cercor/bhu041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carron SF, Alwis DS, Rajan R (2016) Traumatic brain injury and neuronal functionality changes in sensory cortex. Front Syst Neurosci 10:47. 10.3389/fnsys.2016.00047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casale AE, Foust AJ, Bal T, McCormick DA (2015) Cortical interneuron subtypes vary in their axonal action potential properties. J Neurosci 35:15555–15567. 10.1523/JNEUROSCI.1467-13.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauli B, Zhou X, Tricoire L, Toussay X, Staiger JF (2014) Revisiting enigmatic cortical calretinin-expressing interneurons. Front Neuroanat 8:52. 10.3389/fnana.2014.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Zhang Y, Li X, Zhao X, Ye Q, Lin Y, Tao HW, Rasch MJ, Zhang X (2017) Distinct inhibitory circuits orchestrate cortical beta and gamma band oscillations. Neuron 96:1403–1418.e6. 10.1016/j.neuron.2017.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS (2015) Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/-) mice. Neuron 85:1332–1343. 10.1016/j.neuron.2015.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou A, Morganti JM, Rosi S (2016) Frontal lobe contusion in mice chronically impairs prefrontal-dependent behavior. PLoS One 11:e0151418. 10.1371/journal.pone.0151418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JL (2005) Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci 8:1059–1068. 10.1038/nn1499 [DOI] [PubMed] [Google Scholar]
- Cooper EC, Jan LY (2003) M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol 60:496–500. 10.1001/archneur.60.4.496 [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4:52–62. 10.1038/82900 [DOI] [PubMed] [Google Scholar]
- Cottam JC, Smith SL, Häusser M (2013) Target-specific effects of somatostatin-expressing interneurons on neocortical visual processing. J Neurosci 33:19567–19578. 10.1523/JNEUROSCI.2624-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding MC, Wang Q, Lo EH, Stanley GB (2011) Cortical excitation and inhibition following focal traumatic brain injury. J Neurosci 31:14085–14094. 10.1523/JNEUROSCI.3572-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engberg AW, Teasdale TW (2004) Psychosocial outcome following traumatic brain injury in adults: a long-term population-based follow-up. Brain Inj 18:533–545. 10.1080/02699050310001645829 [DOI] [PubMed] [Google Scholar]
- Folweiler KA, Xiong G, Best KM, Metheny HE, Nah G, Cohen AS (2020) Traumatic brain injury diminishes feedforward activation of parvalbumin-expressing interneurons in the dentate gyrus. eNeuro 7:ENEURO.0195-19.2020. 10.1523/ENEURO.0195-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara E, Schwartz ML, Gao F, Black SE, Levine B (2008) Ventral frontal cortex functions and quantified MRI in traumatic brain injury. Neuropsychologia 46:461–474. 10.1016/j.neuropsychologia.2007.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwill HL, Manzano-Nieves G, LaChance P, Teramoto S, Lin S, Lopez C, Stevenson RJ, Theyel BB, Moore CI, Connors BW, Bath KG (2018) Early life stress drives sex-selective impairment in reversal learning by affecting parvalbumin interneurons in orbitofrontal cortex of mice. Cell Rep 25:2299–2307.e4. 10.1016/j.celrep.2018.11.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero RM, Giza CC, Rotenberg A (2015) Glutamate and GABA imbalance following traumatic brain injury. Curr Neurol Neurosci Rep 15:27. 10.1007/s11910-015-0545-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito-Ishida A, Ure K, Chen H, Swann JW, Zoghbi HY (2015) Loss of MeCP2 in parvalbumin-and somatostatin-expressing neurons in mice leads to distinct Rett syndrome-like phenotypes. Neuron 88:651–658. 10.1016/j.neuron.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hjerling-Leffler J, Zagha E, Fishell G, Rudy B (2010) The largest group of superficial neocortical GABAergic interneurons expresses ionotropic serotonin receptors. J Neurosci 30:16796–16808. 10.1523/JNEUROSCI.1869-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao R, Page DT, Merzlyak I, Kim C, Tecott LH, Janak PH, Rubenstein JL, Sur M (2009) Reduced conditioned fear response in mice that lack Dlx1 and show subtype-specific loss of interneurons. J Neurodev Disord 1:224–236. 10.1007/s11689-009-9025-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama M, Pérez-Garci E, Nevian T, Bock T, Senn W, Larkum ME (2009) Dendritic encoding of sensory stimuli controlled by deep cortical interneurons. Nature 457:1137–1141. 10.1038/nature07663 [DOI] [PubMed] [Google Scholar]
- Naka A, Adesnik H (2016) Inhibitory circuits in cortical layer 5. Front Neural Circuits 10:35. 10.3389/fncir.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naka A, Veit J, Shababo B, Chance RK, Risso D, Stafford D, Snyder B, Egladyous A, Chu D, Sridharan S, Mossing DP, Paninski L, Ngai J, Adesnik H (2019) Complementary networks of cortical somatostatin interneurons enforce layer specific control. Elife 8:e43696. 10.7554/eLife.43696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, Bjorklund GR, Newbern J, Anderson T (2018) Parvalbumin fast-spiking interneurons are selectively altered by paediatric traumatic brain injury. J Physiol 596:1277–1293. 10.1113/JP275393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro MJ, Hashikawa-Yamasaki Y, Rudy B (2018) Diversity and connectivity of layer 5 somatostatin-expressing interneurons in the mouse barrel cortex. J Neurosci 38:1622–1633. 10.1523/JNEUROSCI.2415-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parga Becerra A, Logsdon AF, Banks WA, Ransom CB (2021) Traumatic brain injury broadly affects GABAergic signaling in dentate gyrus granule cells. eNeuro 8:ENEURO.0055-20.2021. 10.1523/ENEURO.0055-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsford J, Draper K, Schönberger M (2008) Functional outcome 10 years after traumatic brain injury: its relationship with demographic, injury severity, and cognitive and emotional status. J Int Neuropsychol Soc 14:233–242. 10.1017/S1355617708080272 [DOI] [PubMed] [Google Scholar]
- Potter GB, Petryniak MA, Shevchenko E, McKinsey GL, Ekker M, Rubenstein JL (2009) Generation of Cre-transgenic mice using Dlx1/Dlx2 enhancers and their characterization in GABAergic interneurons. Mol Cell Neurosci 40:167–186. 10.1016/j.mcn.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouille F, Scanziani M (2001) Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science 293:1159–1163. 10.1126/science.1060342 [DOI] [PubMed] [Google Scholar]
- Pouille F, Marin-Burgin A, Adesnik H, Atallah BV, Scanziani M (2009) Input normalization by global feedforward inhibition expands cortical dynamic range. Nat Neurosci 12:1577–1585. 10.1038/nn.2441 [DOI] [PubMed] [Google Scholar]
- Raible DJ, Frey LC, Cruz Del Angel Y, Russek SJ, Brooks-Kayal AR (2012) GABA(A) receptor regulation after experimental traumatic brain injury. J Neurotrauma 29:2548–2554. 10.1089/neu.2012.2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedemann T (2019) Diversity and function of somatostatin-expressing interneurons in the cerebral cortex. Int J Mol Sci 20:2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, Fishell G, Lee S, Hjerling-Leffler J (2011) Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 71:45–61. 10.1002/dneu.20853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho M, Kyle BD (2021) The large-conductance, calcium-activated potassium channel: a big key regulator of cell physiology. Front Physiol 12:750615. 10.3389/fphys.2021.750615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberberg G, Markram H (2007) Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 53:735–746. 10.1016/j.neuron.2007.02.012 [DOI] [PubMed] [Google Scholar]
- Smith DH, Soares HD, Pierce JS, Perlman KG, Saatman KE, Meaney DF, Dixon CE, McIntosh TK (1995) A model of parasagittal controlled cortical impact in the mouse: cognitive and histopathologic effects. J Neurotrauma 12:169–178. 10.1089/neu.1995.12.169 [DOI] [PubMed] [Google Scholar]
- Spikman JM, Timmerman ME, Milders MV, Veenstra WS, van der Naalt J (2012) Social cognition impairments in relation to general cognitive deficits, injury severity, and prefrontal lesions in traumatic brain injury patients. J Neurotrauma 29:101–111. 10.1089/neu.2011.2084 [DOI] [PubMed] [Google Scholar]
- Struchen MA, Clark AN, Sander AM, Mills MR, Evans G, Kurtz D (2008) Relation of executive functioning and social communication measures to functional outcomes following traumatic brain injury. NeuroRehabilitation 23:185–198. [PubMed] [Google Scholar]
- Stuss DT (2011) Traumatic brain injury: relation to executive dysfunction and the frontal lobes. Curr Opin Neurol 24:584–589. 10.1097/WCO.0b013e32834c7eb9 [DOI] [PubMed] [Google Scholar]
- Um JW (2017) Roles of glial cells in sculpting inhibitory synapses and neural circuits. Front Mol Neurosci 10:381. 10.3389/fnmol.2017.00381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascak M, Sun J, Baer M, Jacobs KM, Povlishock JT (2017) Mild traumatic brain injury evokes pyramidal neuron axon initial segment plasticity and diffuse presynaptic inhibitory terminal loss. Front Cell Neurosci 11:157. 10.3389/fncel.2017.00157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascak M, Jin X, Jacobs KM, Povlishock JT (2018) Mild traumatic brain injury induces structural and functional disconnection of local neocortical inhibitory networks via parvalbumin interneuron diffuse axonal injury. Cereb Cortex 28:1625–1644. 10.1093/cercor/bhx058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilkki J, Ahola K, Holst P, Ohman J, Servo A, Heiskanen O (1994) Prediction of psychosocial recovery after head injury with cognitive tests and neurobehavioral ratings. J Clin Exp Neuropsychol 16:325–338. 10.1080/01688639408402643 [DOI] [PubMed] [Google Scholar]
- Wallesch CW, Curio N, Galazky I, Jost S, Synowitz H (2001) The neuropsychology of blunt head injury in the early postacute stage: effects of focal lesions and diffuse axonal injury. J Neurotrauma 18:11–20. 10.1089/089771501750055730 [DOI] [PubMed] [Google Scholar]
- Wang XJ, Tegner J, Constantinidis C, Goldman-Rakic PS (2004a) Division of labor among distinct subtypes of inhibitory neurons in a cortical microcircuit of working memory. Proc Natl Acad Sci U S A 101:1368–1373. 10.1073/pnas.0305337101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Toledo-Rodriguez M, Gupta A, Wu C, Silberberg G, Luo J, Markram H (2004b) Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J Physiol 561:65–90. 10.1113/jphysiol.2004.073353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems JGP, Wadman WJ, Cappaert NLM (2018) Parvalbumin interneuron mediated feedforward inhibition controls signal output in the deep layers of the perirhinal-entorhinal cortex. Hippocampus 28:281–296. 10.1002/hipo.22830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson L, Stewart W, Dams-O'Connor K, Diaz-Arrastia R, Horton L, Menon DK, Polinder S (2017) The chronic and evolving neurological consequences of traumatic brain injury. Lancet Neurol 16:813–825. 10.1016/S1474-4422(17)30279-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witcher KG, Bray CE, Chunchai T, Zhao F, O'Neil SM, Gordillo AJ, Campbell WA, McKim DB, Liu X, Dziabis JE, Quan N, Eiferman DS, Fischer AJ, Kokiko-Cochran ON, Askwith C, Godbout JP (2021) Traumatic brain injury causes chronic cortical inflammation and neuronal dysfunction mediated by microglia. J Neurosci 41:1597–1616. 10.1523/JNEUROSCI.2469-20.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jeong HY, Tremblay R, Rudy B (2013) Neocortical somatostatin-expressing GABAergic interneurons disinhibit the thalamorecipient layer 4. Neuron 77:155–167. 10.1016/j.neuron.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (1999) Presynaptic strontium dynamics and synaptic transmission. Biophys J 76:2029–2042. 10.1016/S0006-3495(99)77360-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Qiao W, Liu K, Zhong H, Yao H (2015) Control of response reliability by parvalbumin-expressing interneurons in visual cortex. Nat Commun 6:6802. 10.1038/ncomms7802 [DOI] [PubMed] [Google Scholar]