

ABSTRACT
Mitotic kinesin-like protein 2 (MKLP2; also known as KIF20A) is a motor protein with a well-established function in promoting cytokinesis. However, our results with siRNAs targeting MKLP2 and small-molecule inhibitors of MKLP2 (MKLP2i) suggest that it also has a function earlier in mitosis, prior to anaphase. In this study, we provide direct evidence that MKLP2 facilitates chromosome congression in prometaphase. We employed live imaging to observe HeLa cells with fluorescently tagged histones treated with MKLP2i and discovered a pronounced chromosome congression defect. We show that MKLP2 facilitates error correction, as inhibited cells have a significant increase in unstable, syntelic kinetochore–microtubule attachments. We find that the aberrant attachments are accompanied by elevated Aurora kinase (A and B) activity and phosphorylation of the downstream target HEC1 (also known as NDC80) at Ser55. Finally, we show that MKLP2 inhibition results in aneuploidy, confirming that MKLP2 safeguards cells against chromosomal instability.
This article has an associated First Person interview with the first author of the paper.
KEY WORDS: Aurora kinase, KIF20A, MKLP2, Chromosome congression, Motor kinesin
Summary: Inhibition of the motor protein MKLP2 results in a mitotic arrest characterized by the presence of pole-proximal chromosomes with unstable syntelic kinetochore–microtubule attachments.
INTRODUCTION
Organisms require accurate and reliable segregation of their genetic material to grow and replace damaged cells. This process, known as mitosis, is a well-regulated, multi-step event with checkpoints in place to ensure accurate chromosome distribution to daughter cells. However, errors can occur and can lead to daughter cells that have gains or losses of whole chromosomes, a cellular feature known as aneuploidy (Ben-David and Amon, 2020). Mitotic kinesin-like protein 2 (MKLP2; also known as KIF20A) is a motor protein that harnesses energy generated through ATP hydrolysis to travel along dynamic microtubules during mitosis. MKLP2 is overexpressed in multiple cancers and was one of the highest ranking genes to associate with functional aneuploidy in cancer cells across 12 different cancer data sets (Carter et al., 2006). However, this association cannot be explained by our current understanding of MKLP2. At present, there is a well-established body of evidence to indicate that MKLP2 is required at the onset of anaphase to promote cytokinesis, primarily via interaction with the chromosomal passenger complex (CPC, comprising AURKB, INCENP, survivin (BIRC5) and borealin) (Adriaans et al., 2020; Hümmer and Mayer, 2009; Kitagawa et al., 2014; Landino et al., 2017). Loss of MKLP2 allows formation of the ingression furrow between daughter cells but inhibits its completion, resulting in one binucleated cell instead of two daughter cells. Binucleation could explain the association of MKLP2 with polyploidy, multiple copies of the entire haploid genome, but does not explain the association with aneuploidy.
We observed that inhibition of MKLP2 with paprotrain (Tcherniuk et al., 2010), a first-generation small-molecule uncompetitive ATP inhibitor of MKLP2 (referred to here as MKLP2i1), caused a significant mitotic arrest that was not previously characterized. Therefore, the purpose of this study was to delineate the role of MKLP2 prior to anaphase and determine whether the observed arrest was caused by an inability of chromosomes to align at the poles (a chromosome congression defect) or cohesion fatigue, both of which could contribute to chromosomal instability. We report here that MKLP2 functions in early mitosis to promote chromosome congression via correction of syntelic attachments and regulation of Aurora kinase A (AURKA) and B (AURKB). Our experiments treating cells with second (MKLP2i2) and third (MKLP2i3) generation MKLP2 small-molecule inhibitors (Ferrero et al., 2019; Labrière et al., 2016) revealed lagging chromosomes, chromosome bridges and daughter cells with significantly distinct chromosome numbers, confirming that MKLP2 safeguards cells against aneuploidy.
RESULTS
MKLP2 deficiency leads to prolonged mitosis
In order to understand whether loss of MKLP2 has an effect prior to the onset of anaphase, we transfected HeLa cells with siRNAs targeting MKLP2 (siMKLP2) and quantified the mitotic index after release from double thymidine block (DTB) (Fig. 1A). HeLa cells transfected with siMKLP2 exhibited reductions in MKLP2 (Fig. 1B) and an elevated mitotic index that remained significantly higher 6 hours after cells peaked in mitosis, when the index of control siRNA (siCtrl)-transfected cells had declined, indicating MKLP2-deficient cells feature prolonged mitosis. To validate this finding, we tracked individual HeLa H2B–GFP cells as they entered and exited mitosis. Treatment with paprotrain, a first-generation small-molecule inhibitor of MKLP2 ATPase activity (MKLP2i1, data not shown), or the second-generation inhibitor MKLP2i2 revealed a dose-dependent increase in duration of mitosis such that 11 µM MKLP2i2 severely arrested cells (average 800 min) in metaphase (Fig. 1C). While tracking cells, we also observed a dose-dependent decrease in the percentage of cells undergoing normal division (Fig. 1D,E). The highest incidence of binucleation occurred at 3.7 µM MKLP2i2, whereas cells treated with 11 µM MKLP2i2 exhibited significantly more abnormal divisions (mitosis leading to three daughter cells or mitosis where cells exit mitosis with no apparent segregation of DNA). To confirm that the mitotic arrest was specific to MKLP2, we performed an experiment in HeLa cells synchronized through a DTB where MKLP2 siRNAs were transfected or small-molecule MKLP2 inhibitors were added in parallel prior to mitosis. We confirmed that all modes of perturbing MKLP2 function increase the mitotic index in a dose-dependent manner (Fig. 1F), and found that MKLP2i3 (Pouletty, 2019) is ∼300- to 1000-fold more potent than MKLP2i2 and MKLP2i1, respectively. Therefore, we used this drug in all subsequent experiments. To further examine the impact of the inhibitor, we asked whether ectopic MKLP2 would mitigate the effects of MKLP2i3. Indeed, expression of MKLP2 reduced the increase in mitotic index triggered by MKLP2 inhibition, an effect that was lost at increased drug concentration (Fig. 1G).
Fig. 1.

MKLP2 deficiency leads to prolonged mitosis. (A) Mitotic index (percentage of cells in mitosis) in HeLa cells transfected with the siRNAs indicated in B after release from DTB. *P<0.05, n>200 cells/condition from two independent experiments (two-way ANOVA with Tukey post-hoc test). (B) Western blot of representative MKLP2 knockdown from A. (C) Dose-dependent effect of MKLP2i2 on duration of mitosis in HeLa histone H2B–GFP cells ****P<0.0001, n>200 cells/condition from three independent experiments (unpaired two-tailed t-test). (D) Images portraying cell fates following treatment with MKLP2i2 for analysis quantified in C and E. Top rows, histone H2B–GFP channel; bottom rows, phase contrast and GFP merge. (E) Percentage of cells from C and D with indicated fates at the corresponding MKLP2i2 treatment dose. (F) Mitotic index in HeLa cells 14 h after release from DTB in the presence of various doses of MKLP2i1, MKLP2i2, MKLP2i3 and siMKLP2 (n=50 cells/experiment from three independent experiments). (G) Graph depicting mitotic index of HeLa cells transfected with either pCS2 flag (empty vector) or pCS2 MKLP2 in between DTB. Cell were treated with either 20 nM MKLP2i3 or 40 nM MKLP2i3 6 h before entering mitosis. The mitotic index was quantified 14 h after release from DTB. **P<0.01, n=50 cells/experiment from two independent experiments (unpaired two-tailed t-test). Results in A, F and G are mean±s.e.m. Mean is highlighted in C.
MKLP2 deficiency leads to chromosome misalignment in prometaphase
We next examined chromosome movement under high magnification in live HeLa mCherry–H2B cells synchronized in mitosis through a DTB. Treatment with MKLP2i3 resulted in a variety of abnormal mitotic phenotypes, including chromosome bridges, lagging chromosomes and a chromosome congression defect. This defect was characterized by the majority of cells properly congressing to the metaphase plate but with a proportion of chromosomes remained at the poles (Fig. 2A; Movies 1–3). We use the term ‘congression defect’ hereafter to refer to cells in this prometaphase arrest for greater than 100 min. We found that cells treated with MKLP2i3 exhibited a dose-dependent increase in the manifestation of the congression defect, where 11 nM MKLP2i3 caused a mitotic arrest that persisted for longer than the duration of the imaging experiment (over 480 min) so that we did not visualize the cells exiting mitosis (Fig. 2B). To further confirm that these phenotypes were associated with inhibition of MKLP2 and not an off-target effect of MKLP2i3, we generated MKLP2 mutants with ‘rigor’ point mutations within the switch II region (E413A) or P-loop region (G165E) of the motor domain, which have been previously shown to inhibit kinesin motor function (Browning et al., 2003). HeLa cells were co-transfected with siMKLP2 directed toward the MKLP2 3′ UTR and an H2B–GFP plasmid to track transfected cells, plus wild-type MKLP2 or either of the rigor mutants (Fig. 2C), and then synchronized with a DTB and imaged as in Fig. 2A. We found that MKLP2-depleted cells exhibited abnormal mitoses and the congression defect phenotype, with an arrest over 100 min, but were able to complete mitosis. As expected, expression of wild-type MKLP2 rescued the congression defects. We also observed the congression defect phenotype in cells transfected with siMKLP2 and the MKLP2 G165E mutant (20%) or MKLP2 E413A mutant (38%). However, unlike the siMKP2-depleted cells, the majority of the arrested cells transfected with the rigor mutants were not able to exit mitosis, indicating that MKLP2 activity is required to rescue the congression defect (Fig. 2C; Movies 4, 5). To further confirm the presence of a chromosome congression defect in MKLP2-deficient cells, we quantified misaligned chromosomes by determining the centromere distribution ratio, which reports on the ratio of centrosome-proximal centromeres to metaphase-plate-proximal centromeres (Stumpff et al., 2012). HeLa cells were synchronized in G2 with RO-3306, a CDK1 inhibitor, then released into DMSO or 33 nM MKLP2i3 and cells were fixed 30 min after release. We observed the congression defect phenotype in MKLP2i3-treated cells (Fig. 2D) and found a significant increase (P<0.001) in the centromere distribution ratio of MKLP2-deficient cells compared to vehicle-treated cells (Fig. 2E), confirming that MKLP2 inhibition impairs chromosome congression.
Fig. 2.

MKLP2 deficiency leads to chromosome misalignment in prometaphase. (A) Images depicting chromosome segregation events captured in HeLa H2B–mCherry cells following treatment with MKLP2i3 or DMSO. Arrowhead denotes lagging chromosome. (B) Quantification of live imaging experiments from A indicating percentage of cells with the following outcomes: normal mitosis, abnormal mitosis (mitosis leading to three daughter cells or mitosis where cells exit mitosis with no apparent segregation of DNA), congression defect with exit (prometaphase arrest >100 min from NEBD with eventual mitotic exit), or congression defect without exit (prometaphase arrest >100 min from NEBD without mitotic exit) (n>50 cells/condition). (C) Live imaging quantification of mitotic outcomes as in B in HeLa mCherry–H2B cells transfected with siMKLP2 plus one of the following: empty vector (EV), TAP–KIF20A (WT), TAP–KIF20A G165E (motor mutant), TAP–KIF20A E413A (rigor motor mutant) (n>50 cells/condition). (D) immunofluorescence of γ-tubulin and centromere proteins in HeLa cells synchronized at G2/M with RO-3306 then released into DMSO or 33 nM MKLP2i3 for 30 min. (E) Quantification of the centromere distribution ratio, calculated as the ratio of the sum of the centromere intensity of the outer quarters closest the poles to the sum of the intensity of the inner quarters, in HeLa cells treated with 33 nM MKLP2i3. ***P<0.001, n=100 from three independent experiments (unpaired two-tailed t-test). The mean is highlighted.
MKLP2 inhibition results in unstable, syntelic kinetochore–microtubule attachments
We hypothesized that the misaligned chromosomes seen in the congression defect phenotype were unable to reach the cell equator due to an inability to form stable kinetochore–microtubule (KT–MT) attachments. To test this idea, we employed the cold depolymerization assay to identify stable microtubules (DeLuca et al., 2016). Following G2 synchronization with RO-3306, we released cells into DMSO or 33 nM MKLP2i3 then incubated cells with cold medium. We then measured the intensity of the remaining cold-stable microtubules and found that mitotic cells in the presence of 33 nM MKLP2i3 exhibited significantly less (P<0.0001) tubulin compared to that seen in DMSO-treated cells (Fig. 3A,B). To determine whether unstable KT–MT attachments in MKLP2i3-treated cells are due to issues in correction of misaligned chromosomes, we employed the monastrol washout assay, where treatment with monastrol generates an abundance of syntelic attachments, which are then allowed to correct in a washout period. The cells do not exit mitosis due to the presence of the proteasome inhibitor MG132 (Lampson et al., 2004). We found that the majority of DMSO-treated cells were able to create bipolar spindles with aligned chromosomes after monastrol washout, whereas MKLP2i3-treated cells created bipolar spindles but with numerous misaligned chromosomes, resembling the congression defect phenotype (Fig. 3C,D). We next examined the KT–MT attachment status of non-congressed chromosomes in cells treated with MKLP2i3 alone. Consistent with the monastrol washout experiment data, these analyses revealed that the majority (61%) of non-congressed chromosomes were syntelically attached and the remainder exhibited either monotelic attachments (27%) or a lack of detectable attachment (Fig. 3E,F). Altogether, these experiments indicate that MKLP2 inhibition causes a defect in the formation of stable, amphitelic KT–MT attachments with an increase in the presence of syntelic attachments.
Fig. 3.

MKLP2 deficiency results in unstable syntelic MT–KT attachments. (A) Immunofluorescence of cold-stable microtubules in HeLa cells treated with DMSO or 33 nM MKLP2i3. (B) Quantification of relative tubulin intensity from cells in A. ****P<0.0001, n=100 from three independent experiments (unpaired two-tailed t-test). (C) Immunofluorescence depicting effect of DMSO or 33 nM MKLP2i3 on chromosome congression after monastraol washout. (D) Quantification of cells from C. ****P<0.0001, n=100 from three independent experiments (unpaired two-tailed t-test). (E) Representative immunofluorescence image of one HeLa cell treated with 33 nM MKLP2i3 depicting syntelic attachments evident in different planes of focus. MIP, maximum intensity projection. (F) Table depicting percentage of types of KT–MT attachments characterized from E (n=100 KT–MT attachments quantified over ten different cells). Results in D are mean±s.e.m. Mean is highlighted in B.
Aurora kinase A and B activities are upregulated in MKLP2-inhibited cells
The canonical error correction pathway is mediated by centromere-enriched AURKB-catalyzed phosphorylation of HEC1 (also known as NDC80), a component of the NDC80 complex that serves as the main link between kinetochores and microtubules (Krenn and Musacchio, 2015; Lampson and Cheeseman, 2011). AURKB phosphorylation of the HEC1 N-terminal ‘tail’ (including S55, denoted pHEC1 hereafter) domain causes destabilization of mal-oriented KT–MT attachments, thus providing a fresh opportunity for microtubules to make correct amphitelic attachments, where each sister kinetochore is attached to microtubules emanating from opposite poles (Wimbish and DeLuca, 2020). A distinct but complementary pathway, known as pole-based error correction, is an AURKA-mediated process that promotes proper alignment of chromosomes located near the pole, which are more likely to form syntelic attachments, via microtubule motor-based transportation of chromosomes to the cell equator (Chmátal et al., 2015; DeLuca, 2017; Ye et al., 2015).
In order to understand how MKLP2 inhibition regulates KT–MT attachment and error correction, we sought to determine the relative levels of active AURKB, AURKA and the downstream target pHEC1. Immunofluorescence analysis of phosphorylated AURKB and AURKA (pAURKB and pAURKA) revealed significant increases (P<0.0001) in the active states of both kinases in MKLP2i3-treated cells (Fig. 4A–D). Because both kinases regulate the downstream target HEC1 through phosphorylation (DeLuca et al., 2011; Ye et al., 2015), we examined the impact of MKLP2 inhibition to pHEC1 using a phospho-specific antibody against pS55 in HeLa cells. Compared to control cells, MKLP2i3 treatment caused a significant increase in pHEC1 staining (Fig. 4E,F; P<0.0001). In an attempt to tease out contributions of the Aurora kinases to impaired error correction in MKLP2i3-treated cells, we synchronized HeLa cells with a DTB and treated cells with MKLP2i3 in combination with barasertib (an AURKB inhibitor) or alisertib (an AURKA inhibitor) to determine whether decreasing specific AURK activity could rescue the mitotic arrest. Although we did not observe a rescue of the MKLP2i-induced mitotic arrest with barasertib (Fig. S1A), a low dose (12.5 nM) of alisertib reduced the duration of the mitotic arrest, as evidenced by a lower mitotic index 18 h after release from the DTB (Fig. 4G). Importantly, no impact of alisertib on the increased mitotic index induced by nocodazole or paclitaxel was observed (Fig. S1B). These results suggest that elevated AURKA activity is at least partially responsible for the MKLP2i3-induced arrest although the contribution might be indirect. Notably, the lack of a rescue with barasertib does not rule out the contributions of AURKB, since AURKB inhibition on its own induces a moderate mitotic arrest and inhibition of AURKB is known to prevent correction of erroneous attachments (Gurden et al., 2016; Kapoor et al., 2006).
Fig. 4.

Aurora kinase A and B activities are upregulated in MKLP2 inhibited cells. (A) Immunofluorescence staining for pAURKB and with anti-centromere antibody (ACA) in mitotically synchronized HeLa cells with and without 33 nM MKLP2i3. (B) Relative pAURKB levels after 33 nM MKLPi3 treatment pAURKB intensities were normalized to ACA. ****P<0.0001, n=100 from three independent experiments (unpaired two-tailed t-test). (C) Images of immunofluorescence staining for pAURKA and total AURKA in mitotically synchronized HeLa cells with and without 33 nM MKLP2i3. (D) Graph depicting increased pAURKA in cells treated with MKLP2i3. pAURKA intensities were normalized to AURKA. ****P<0.0001, n=100 from three independent experiments (unpaired two-tailed t-test). (E) Images of immunofluorescence staining for pHEC1 and total HEC1 in mitotically synchronized HeLa cells with and without 33 nM MKLP2i3. (F) pHEC1 levels in cells treated with 33 nM MKLP2i3. pHEC1 intensities were normalized to HEC1. ****P<0.0001, n=100 from three independent experiments (unpaired two-tailed t-test). (G) Mitotic index of HeLa cells synchronized with DTB and treated with DMSO or 33 nM MKLP2i3 with or without 12.5 nM alisertib (an AURKA inhibitor). Mitotic index quantified 14 h after release from DTB. ***P<0.001, n>100 cells from three independent experiments (unpaired two-tailed t-test). Results in G are mean±s.e.m. Mean is highlighted in B,D,F.
MKLP2 loss causes aneuploidy and exacerbates chromosomal instability
To determine whether congression defects caused by MKLP2 deficiency contribute to aneuploidy, we employed satellite enumeration probes to quantify individual chromosomes via fluorescence in situ hybridization (FISH). We treated HeLa cells with 33 nM MKLP2i3 for 48 h, then probed for chromosomes 2, 7, 10 or 15. We quantified the mode of each chromosome (e.g. most HeLa cells contained three copies of chromosome 2), then determined the proportion of cells that exhibited a chromosome number distinct from the mode (modal deviation) for chromosomes 2, 7, 10 and 15. Although increased modal deviation was found for all chromosomes, we found significant changes in modal deviation for chromosomes 2, 7 and 15 (Fig. 5A,B). This data suggests that the association of MKLP2 with functional aneuploidy observed by Carter et al. (2006) is due to MKLP2 expression limiting chromosomal instability (CIN), the gain or loss of chromosomes at a higher incidence than normal. We hypothesized that cells with CIN would be more dependent on MKLP2 for proper chromosome congression. As excessive CIN is detrimental, we reasoned that cells with CIN would thus be more sensitive to MKLP2 loss than cells without CIN. To gain insight into this possibility we took advantage of the Cancer Dependency Map, which encompasses data from RNAi and CRISPR-mediated loss of function screens as well as genomic information across hundreds of cell lines (https://depmap.org/). We compared the gene effect score for MKLP2 in colorectal cancer (CRC) cell lines with CIN (microsatellite stable, MSS) or without CIN (microsatellite unstable, MSI) from both RNAi and CRISPR screens. A gene effect score of −1 is the median of all essential genes, whereas 0 is the median score of non-essential genes. Thus, a low score indicates a higher likelihood that the gene of interest is essential for a given cell line (Barretina et al., 2012; Dempster et al., 2021; Meyers et al., 2017; Tsherniak et al., 2017). These analyses revealed that cell lines exhibiting CIN (MSS) were significantly more sensitive to loss of MKLP2 compared to CRC cell lines without CIN (Fig. 5C,D), suggesting that cells with ongoing CIN require MKLP2 to facilitate chromosome congression and limit excessive CIN.
Fig. 5.
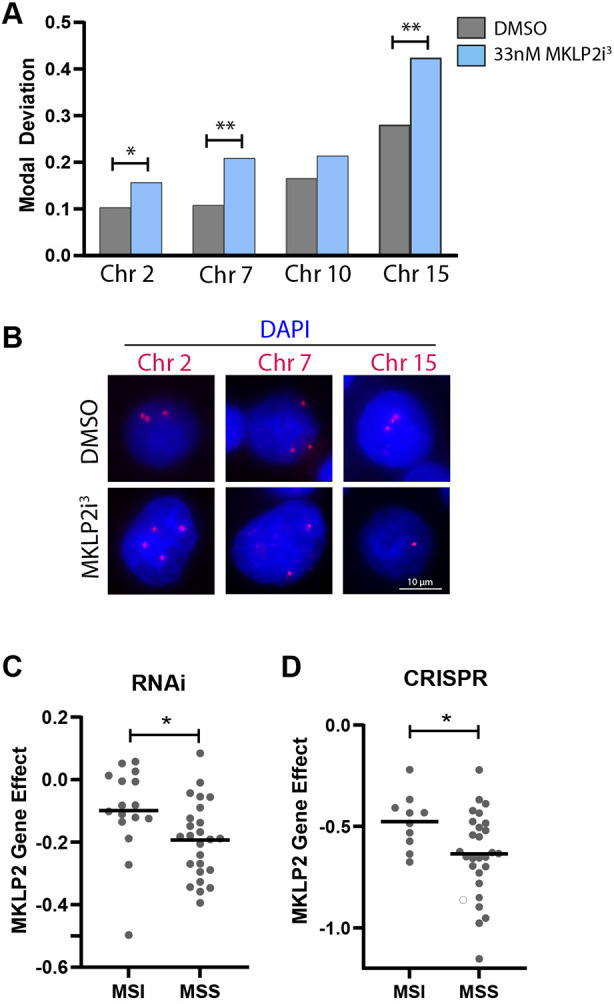
MKLP2 loss causes aneuploidy and exacerbates CIN. (A) Graph depicting fraction of cells with chromosome number deviating from the mode (three chromosomes/cell) for four chromosomes (2, 7, 10 and 15). *P<0.05, **P<0.01, n>200 cells/condition from two independent experiments (Chi squared test). (B) Images of chromosome enumeration probes (red) from A. (C) Gene effect scores of CRC cell lines with CIN (MSS, n=25) and without CIN (MSI, n=16) when MKLP2 was depleted via RNAi. *P<0.05 (unpaired two-tailed t-test). (D) Gene effect scores of CRC cell lines with CIN (MSS, n=27) and without CIN (MSI, n=10) when MKLP2 was depleted via CRISPR. *P<0.05 (unpaired two-tailed t-test). Mean is highlighted in C,D.
DISCUSSION
In this manuscript, we report that MKLP2 functions in early mitosis to facilitate congression of syntelically attached pole-based chromosomes. It is somewhat curious that a role for MKLP2 in early mitosis has not yet been described given our 20 year knowledge of MKLP2 function in cytokinesis and the occurrence of binucleation with its depletion (Neef et al., 2003). However, our findings complement previously published data demonstrating an increase in the percentage of mitotic cells and misaligned chromosomes with siMKLP2 in the breast cancer cell line MCF7 (Khongkow et al., 2016). Likewise, a previous report indicated a mild mitotic delay in siMKLP2-transfected HeLa cells (Neef et al., 2003). MKLP2 homologs in other organisms have also been implicated in early mitosis. For example, loss of the Drosophila homolog Subito, which is also involved in spindle function in meiosis and mitosis, results in an elevated mitotic index (Cesario et al., 2006) and fission yeast Klp9 promotes error correction at metaphase (Choi and McCollum, 2012). We propose that the role of MKLP2 in early mitosis requires a smaller amount of functional enzyme than is required for its role in cytokinesis. Indeed, the early mitotic phenotype becomes more pronounced with increasing concentration of MKLP2 inhibition, whereas binucleation is apparent at lower doses (Figs 1C and 2B). Therefore, RNAi might not deplete sufficient amounts of the protein to reveal the phenotype consistently.
There are several lines of evidence to support our premise that only a small amount of MKLP2 is needed to promote congression. First, there are two documented mechanisms to prevent MKLP2 binding to the central spindle in early mitosis – CDK1 phosphorylation (Kitagawa et al., 2014) and Mad2 binding (Lee et al., 2010). This double-layered regulation suggests that the small fraction of MKLP2 protein that circumvents these regulations in prometaphase is sufficient to facilitate chromosome congression. Notably, similar mechanisms negatively regulate the association of the CPC with microtubules prior to anaphase, yet microtubule-dependent roles of the CPC in early mitosis have recently been identified (Trivedi et al., 2019; Wheelock et al., 2017). Indeed, MKLP2 localization to spindle poles in early mitosis has been demonstrated (Krupina et al., 2016; Lee et al., 2010), and we previously found that MKLP2 and components of the CPC associated with microtubules in early mitotic extracts via mass spectrometry (Torres et al., 2011), verifying that microtubule-associated pools of MKLP2 exist prior to the onset of anaphase. Second, compound heterozygosity consisting of a premature stop codon and a rigor mutant causes defective development and death, whereas heterozygosity for either mutant has no phenotype, suggesting that a small amount of MKLP2 is sufficient for viability whereas complete loss of function is not (Louw et al., 2018). Third, examination of the Cancer Dependency Map data via the DepMap Portal revealed that partial loss of MKLP2 function via RNAi is generally tolerated, whereas total loss of function achieved via CRISPR indicates that MKLP2 has an essential function (compare gene effect scores for RNAi and CRISPR-mediated loss of MKLP2 in Fig. 5C). Fourth, comparison of cells treated with a moderate dose of MKLP2i1 with cells expressing an MKLP2 mutant showed different amounts of CPC relocalization due to differing levels of inhibition, confirming that the degree of MKLP2 inhibition results in distinct phenotypes (Adriaans et al., 2020).
The prevalence of monotelic and syntelic KT–MT attachments in chromosomes located at the poles due to MKLP2i3 treatment (Fig. 3F) suggests a defect in syntelic error correction or an impediment in the ability to form proper amphitelic attachments. Because AURKA activity was elevated in MKLP2i3-treated cells (Fig. 4B,C) and AURKA regulates chromosome congression, contributes to segregation fidelity by promoting chromosome oscillation and is directly involved in syntelic error correction (Chmátal et al., 2015; DeLuca et al., 2018; Iemura et al., 2021; Poser et al., 2019; Ye et al., 2015), it follows that altered AURKA supports the congression defect. One possibility is that the antagonistic regulation of CENP-E by the Ser/Thr phosphatase protein phosphatase 1 (PP1) and AURKA (Kim et al., 2010) could explain how excessive AURKA activity caused by inhibition of MKLP2 might prevent PP1 binding to CENP-E and the dephosphorylation cascade necessary to create stable KT–MT attachments.
How exactly MKLP2 impacts AURK activities is still an unanswered question. The fact that the G165E and E413A MKLP2 rigor mutants recapitulate the congression defect (Fig. 2C) not only confirms the specificity of our finding to MKLP2, but also suggests that the motor activity of MKLP2 is important to chromosome congression and regulating AURK activities. MKLP2 has processive plus-end-directed motor activity (Adriaans et al., 2020), suggesting that, under normal conditions, the MKLP2 motor moves substrates towards the cell equator to negatively regulate AURK activity. In support of this idea, MKLP2 rigor mutants concentrate at the spindle poles and MKLP2i1 has a similar impact (Adriaans et al., 2020; Krupina et al., 2016). Interestingly, AURKA interacts with and is activated by the CPC (Katayama et al., 2008), similar to AURKB. The AURKA–CPC complex is thought to mediate phosphorylation of AURKA substrates at the kinetochore and/or centromere throughout mitosis (DeLuca et al., 2018). Little is known about the regulation of this complex, but it appears that although it does not require microtubules to form, proper localization does require microtubules (DeLuca et al., 2018). Thus, an attractive possibility is that MKLP2 regulates AURKA in a fashion analogous to AURKB through transportation of the AURKA–CPC complex, with loss of MKLP2 function leading to an accumulation of active AURKA at the poles (Adriaans et al., 2020; Serena et al., 2020). Consistent with our hypothesis that only a small fraction of MKLP2 is required for this function, it has been difficult to visualize this pool of AURKA–CPC at endogenous levels (DeLuca et al., 2018). Alternatively, evidence suggests that MKLP2 interacts with BRCA1, which negatively regulates AURKA activity (Ertych et al., 2016; Hill et al., 2014). Further work is required to determine how these mechanisms might contribute to regulation of AURKA activity by MKLP2.
AURKB misregulation might also contribute to the MKLP2i-induced congression defect (Figs 3A–D, 4E,F). MKLP2 promotes relocalization of AURKB-CPC from centromeres to the central spindle in anaphase and it is tempting to speculate that it could regulate the accumulation of AURKB activity at kinetochores and/or centromeres similarly. Indeed, MKLP2 has been proposed to contribute to UBASH3B-mediated relocalization of AURKB from chromosome arms to centromeres in early mitosis, and microtubules are important for proper AURKB function (Krupina et al., 2016; Trivedi et al., 2019; Wheelock et al., 2017). Thus, MKLP2 might function by fine-tuning specific pools of AURKB to promote the fidelity of mitotic progression. In support of this idea, Subito, the Drosophila homolog, interacts with inter-centromere microtubules, which have been implicated in regulation of AURKB to control error correction. Although we did find a significant increase in pAURKB activity in MKLP2i3-treated cells (Fig. 4C,D), we cannot conclude whether increased pAURKB destabilizing KT–MT attachments contributes to congression failure of pole-proximal chromosomes or is simply a consequence of the persistence of aberrant attachments due to impaired pole-based error correction. A rescue experiment similar to the alisertib experiment was performed with the AURKB inhibitor barasertib (Fig. S1A), and did not show rescue of the mitotic delay caused by MKLP2i. However, AURKB inhibition in itself induces a moderate mitotic arrest and prevents error correction (Gurden et al., 2016; Kapoor et al., 2006), confounding the interpretation. Further studies will be needed to more completely characterize the contributions of AURKB in the MKLP2i-mediated chromosome congression defect. Further examination of the relationship between MKLP2 and AURKB in early mitosis is necessary.
In addition to potential impacts on AURK activities, MKLP2 might also play a more direct role in facilitating the formation of amphitelic attachments. Kinesin-6 family members are known to interact with and bundle anti-parallel microtubules, for example MKLP1 and MKLP2 at the central spindle in late mitosis. Drosophila Subito utilizes this ability to promote acentrosomal spindle assembly in meiotic oocytes (Jang et al., 2007). Interestingly, microtubules nucleate near centromeres during the early mitosis of mammalian cells (Sikirzhytski et al., 2018). Many of these chromosome-proximal microtubules are captured by kinetochores and promote biorientation by interacting indirectly with centrosomal microtubules. Recent work has demonstrated that bundles of short, anti-parallel microtubules mediate this connection to the spindle and promote amphitelic attachment (Renda et al., 2022). PRC1, an MKLP2-interacting protein, is a key component in the formation of these bundles and biophysical characterization of MKLP2 supports that it might organize or sense tension within microtubule bundles (Atherton et al., 2017). It is thus possible that MKLP2 could contribute to the formation of amphitelic attachments by enhancing bundling of non-centrosomal microtubules. As Aurora kinase activities also promote the formation of microtubules by chromosomes and contribute to spindle formation (Andrews et al., 2004; Godek et al., 2015; Lan et al., 2004; Sampath et al., 2004; van Heesbeen et al., 2017), it is worth noting that this potential regulation of microtubules and the impacts on Aurora kinases discussed above are not mutually exclusive.
Our study provides evidence that MKLP2 ensures chromosome congression by promoting the formation of stable KT–MT attachments. This previously undescribed function provides a mechanism for the association of MKLP2 with aneuploidy and also a rationale for the use of MKLP2 inhibitors as anti-mitotic treatments in cancer. Important follow-up work would focus on identifying the cargoes and pathways of MKLP2 in prometaphase in an attempt to delineate the mechanism by which MKLP2 impacts Aurora kinase activities and chromosome alignment, which will be important for further understanding why some cells are more sensitive to loss of MKLP2 than others.
MATERIALS AND METHODS
Experimental model and subject details
HeLa (female; ATCC) and HeLa mCherry–H2B GFP–tubulin cells (female; gift from Dr Katharine Ullman, Huntsman Cancer Institute, Salt Lake City, UT, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM), 10% fetal bovine serum (FBS; Avantor), and 1% penicillin and streptomycin except when conducting live imaging experiments when they were maintained in Leibovitz without Phenol Red, 10% FBS (Avantor), 1% penicillin and streptomycin. Cell culture reagents were sourced from Corning unless indicated otherwise. Cell lines were verified with microsatellite genotyping by The Ohio State University Comprehensive Cancer Center Genomics Shared Resource and Mycoplasma tested negative with SIGMA Lookout Mycoplasma PCR Detection Kit.
Cell synchronization
For experiments shown in Figs 2D,E; 3A,B,E; and 4A–F, cells were synchronized in G2 with a single thymidine treatment (2 mM) for 20 h followed by release into medium containing 5 µM CDK1 inhibitor, RO-3306, for 7 h, then released into DMSO or drug. Experiments began when control cells were just entering metaphase – 30 min after release from RO-3306. For experiments shown in Figs 2A–C and 4G, cells were synchronized with a double thymidine block (DTB) consisting of 2 mM thymidine for 18 h, followed by release in normal medium for 6 h, followed by 2 mM thymidine for 18 h, release in normal medium. Cells entered mitosis ∼10–12 h after release. Cells were rinsed three times with phosphate-buffered serum (PBS), which was warmed to improve synchronizations.
Cloning
The pCS2 TAP MKLP2 plasmid was generated by obtaining MKLP2 cDNA from the Harvard PlasmID repository and ligating into the pCS2 TAP vector [pCS2-TAP (tandem affinity purification) was generated by subcloning the Flag-TEV-S peptide and gateway cassette from pGLAP2 (Torres et al., 2009) into the BamHI and StuI sites of pCS2+ (Turner and Weintraub, 1994)] via gateway technology. To generate the G165E p-loop and E413A switch II MKLP2 motor mutants, primers noted in Table S1 were used with NEB Q5 Hot Start Master Mix to amplify pCS2 TAP MKLP2 plasmid. Mutations were confirmed with sequencing.
Immunofluorescence
For immunofluorescence cells were seeded onto round coverslips in one well of a 24-well plate. When washing or adding reagents, instead of directly adding to the well, we found that moving coverslips into wells that had been prefilled with wash/primary/block etc. greatly minimized loss of mitotic cells. All immunofluorescence experiments were imaged with Invitrogen EVOS M7000 at 100×. Cells from Fig. 3 (all panels) were imaged in a z-stack.
For immunofluorescence detection of centromeres and γ-tubulin, cells were fixed in 4% paraformaldehyde (PFA) for 20 min, then ice-cold methanol stored at −20°C for 10 min, blocked in 20% goat serum at room temperature (RT) for 1 h, then incubated with a human anti-centromere antibody {cat. no 15-234, Antibodies Inc; 1:500 in AbDil [1% BSA, 0.1% Triton X-100, 0.1% sodium azide in Tris-buffed saline (TBS), pH 7.4]} overnight at 4°C. After a wash, cells were incubated with anti-tubulin antibody (cat. no T5326; Sigma; 1:1000 in AbDil) for 1 h at RT. Corresponding secondary antibodies were also diluted 1:500 in AbDil and incubated with cells for 1 h at RT. Details of antibodies and their catalog numbers are provided in Table S1. Cells were counterstained with Hoechst 33342 (Sigma) at 1:10,000 during a 5 min wash and then mounted with Fluoromount (Southern Biotech). All washes were undertaken three times for 5 min in TBS.
For cold-induced microtubule stabilizing immunofluorescence for α-tubulin, 30 min after HeLa cells synchronized in mitosis were released into medium containing DMSO or 50 nM compound 38 (MKLP2 inhibitor synthesized by WuXi, China), medium was replaced with ice-cold DMEM (containing either drug or DMSO) and placed on ice for 10 min. Cells were washed once with PHEM buffer (60 mM PIPES, 25 mM HEPES, 20 mM EGTA, 8 mM MgSO4, pH 7.0) for 5 min, then permeabilized in 0.5% Triton X-100 in PHEM buffer for 4 min at RT. Cells were fixed in 4% PFA for 20 min at RT, then blocked in 10% goat serum diluted in PHEM at RT for 1 h. Rat anti-α-tubulin antibody (cat. no MA1-80017, clone YL1/2, Thermo Fisher Scientific) was diluted 1:200 in 5% goat serum diluted in PHEM and incubated at RT for 1 h. Corresponding secondary antibody was diluted 1:300 and incubated at RT for 1 h. Cells were counterstained with Hoechst 33342 (Sigma) 1:10,000 during a 5 min wash. All washes were undertaken three times for 5 min with PHEM. Relative spindle intensity was measured by quantifying α-tubulin staining with FIJI software and normalized to DAPI intensity.
For determining the level of pHEC1 Ser55 normalized to total HEC1, cells were pre-extracted with pre-warmed BRB80 (80 mM PIPES pH 6.8, 5 mM EGTA, 1 mM MgCl2, 0.5% Triton X-100) for 2 min at 37°C. Cells were then fixed in 4% PFA for 15 min at room temperature, washed twice for 5 min in PBS with 0.1% Triton X-100 (PBST), and blocked in blocking buffer (PBST with 3% BSA) for 30 min at room temperature. Primary antibodies for GTX pHec1 Ser55 (cat. no GTX70017, GeneTex) and GTX Hec1 9G3.23 (cat. no GTX70268, GeneTex) were diluted 1:3000 in blocking buffer and incubated overnight at 4°C. The next morning cells were washed twice and incubated with secondary antibodies (1:500 diluted in blocking buffer) for 1 h at room temperature. Cells were washed three times in PBST, counterstained with Hoechst and mounted using Fluoromount (Southern Biotech). Mitotic cells were imaged with a 100× objective using an EVOS Cell Imaging System (Thermo Fisher Scientific). Intensities were measured in FIJI by splitting channels, background subtracting, then encircling foci contained within the chromosomal area to specify area to be measured. The intensity of pHEC1 was normalized to total HEC1 intensity in three biological replicates (25 cells per condition).
For immunofluorescence to detect the nature of KT–MT attachments, cells were synchronized in RO-3306 for 20 h, then released into medium containing DMSO or MKLP2i3 for 26 min. Cells were then pre-extracted in microtubule-stabilizing buffer (80 mM PIPES pH 6.8, 1 mM MgCl2, 1 mM EDTA, 5 mM EGTA and 0.5% Triton X-100) for 30 s at room temperature. To fix cells, they were transferred to wells containing ice-cold methanol for 3 min, then rehydrated by incubating with PBS twice for 3 min. Cells were blocked in AbDil (0.1% Triton X-100, 2% BSA and 0.1% NaAzide) for 10 min. Kinetochores were stained first with (Hec1 9G3.23 1:250 in AbDil) for 1 h at room temperature, followed by two 5 min washes in TBS with 0.1% Triton X-100 (TBST) and secondary antibody (1:500 in AbDil) for 30 min at room temperature. Next, tubulin was stained by incubating with primary antibody (tubulin Yl1/2, 1:500 in AbDil) for 30 min at room temperature, followed by two 5 min washes in TBST and secondary antibody (1:500 in AbDil). Cells were washed twice with TBST for 5 min, then mounted onto slides with ProLong Gold Anti-Fade with NucBlue mountant and allowed to cure in the dark ON.
For immunofluorescence of pAURKB, centromeres (Fig. 4A), pAURKA, and total AURKA (Fig. 4C), cells were fixed in 4% PFA for 20 min at room temperature, then permeabilized in 0.5% Triton X-100 for 2 min. After a brief rinse in PBS, cells were blocked in 5% BSA (in PBS) for 30 min, then incubated with primary antibody overnight at 4°C (all primary antibodies were prepared in 5% BSA at 1:1000 dilution). The next day coverslips were washed three times with PBS (5 min each) then secondary antibodies (1:500 diluted in 5% BSA) were incubated in the dark for 1 h at room temperature then washed three times (5 min each) with PBS. Coverslips were mounted onto slides with Invitrogen ProLong Gold Anti-Fade with NucBlue mountant and allowed to cure in the dark ON.
Live imaging
For the live-cell imaging shown in Fig. 2A,B, HeLa H2B–mCherry GFP–tubulin cells were synchronized with a double thymidine block and released into normal DMEM. For the experiment shown in Fig. 2B with various doses of MKLP2i3, the medium was replaced with Leibovitz medium containing 33 nM MKLP2i3 6 h after the second release. For live imaging experiments with transfection of the MKLP2 rigor mutants (Fig. 2C), MKLP2 plasmids (G165E or E413A) or siRNAs directed toward MKLP2 3′ UTR were transfected 1 h after release from first thymidine block with LT1 according to MIrus Bio protocol. The transfection mixture containing siRNAs directed toward the MKLP2 3′ UTR and RNAiMax (prepared according to Thermo Fisher Scientific protocol) was overlaid onto cells after addition of medium containing thymidine 6 h after release from the first thymidine block. Cells were imaged using-wide field imaging on Nikon TiE microscope and captured on an Andor Ultra EMCCD camera. Cells were imaged every 5 min for 10 h using a 60× or 20× objective and 1 µm step size (total steps=14). 2D projections of z-stacks were generated using smooth manifold extraction (Shihavuddin et al., 2017) and assembled into movies using a script that is available upon request.
For the live imaging data shown in Fig. 1C–E, cells were released from double thymidine block into various doses of MKLP2i2, then imaged in a single plane of focus with the IncuCyte ZOOM Live-Cell Analysis System with a 20× objective every 5 min for 48 h.
FISH with Cytocell satellite enumeration probes
To determine aneuploidy following treatment with MKLP2i3, we performed fluorescence in situ hybridization with Cytocell satellite enumeration probes (Oxford Gene Technology). To harvest, trypsinized cells were centrifuged (200 g) for 3 min, then the supernatant was aspirated leaving ∼100–150 μl in the bottom of the tube, which was used to resuspend cells. Pre-warmed (37°C) hypotonic 0.075 M KCl (6 ml) was added slowly to each sample, then samples were incubated in a 37°C water bath for 15 min. Ten drops of freshly prepared Carnoys solution (1:3 glacial acetic acid:methanol) was added to each sample, then centrifuged at 100 g for 10 min. Supernatant was carefully aspirated leaving ∼100–150 μl for resuspension, then 8 mls Carnoys was added and cells were fixed for 30 min at 4°C. Samples were centrifuged at 100 g for 3 min, and supernatant aspirated leaving ∼100–150 μl for cell resuspension. Cells (10 µl Carnoy's resuspension) were dropped onto clean slides warmed to 65°C on a hot plate. Cells dried to slide by leaving them on the hot plate for 15 s, then a pap marker was used to outline the dried suspension circumference on the underside of the slide. Cells were denatured by incubating in 2× SSC for 2 min followed by a brief protein digestion (1:20 proteinase K: 2× SSC) for 12 min at 37°C. Cells were incubated once more in 2× SSC for 2 min, followed by dehydration in an ethanol series (70%, 85% and 100%) for 2 min each. Slides were air dried by leaning up and resting on a kimwipe. Slides and probes mixture (3 µl probe plus 7 µl Cytocell hybridization solution) were warmed in a 37°C incubator, then 10 µl probe in hybridization solution was overlaid onto each cell sample. A coverslip was applied and the edges were sealed with rubber cement. Slides were placed on a 75°C hot plate for 2 min, then incubated overnight at 37°C in the dark. The next day rubber cement was carefully and completely removed, then slides were incubated in 0.4× SSC for 2 min at 72°C. Next slides were immersed in 2× SSC, 0.05% Tween-20 for 30 s, followed by the ethanol series again (70%, 85%, 100%) for 2 min each. Slides were allowed to air dry on a kimwipe. Prior to applying the coverslip, 5 µl diluted Hoechst 33342 (1 µl Hoechst 33342 in 8 ml PBS) and 5 µl Fluoromount (Southern Biotech) were applied to the cell area. Slides were allowed to dry at room temperature overnight in the dark. Images were captured with an Invitrogen EVOS M7000 imager.
Quantification and statistical analysis
Image analysis was performed in ImageJ/FIJI software. For calculation of the centromere distribution ratios in Fig. 2E,F, cells were imaged in a plane of focus that captured both centrosomes using a Leica DM5500B fluorescence microscope as described previously (Stumpff et al., 2012). Centromere intensity per cell quarter was measured using FIJI software. The centromere distribution ratio (r=γ1+γ2/ε) was then calculated for each cell by dividing the sum of the intensity of the outer quarters closest to the poles by the sum of the intensity of the inner quarters. Mitotic index refers to the percentage of cells in mitosis [cells in mitosis (prophase through telophase), divided by the total number of cells×100]. To quantify cells arrested for at least 100 min in prometaphase for the live imaging experiments, the individual smooth manifold extraction (SME) projections were inspected for signs of mitosis. Cells in mitosis for over 20 projections (images taken 5 min apart) were considered to have the congression defect when they were arrested in prometaphase for 100 min with or without obvious chromosome misalignment. Z-stack images of cells from immunofluorescence experiments (Fig. 4A–F) were obtained on an EVOS M7000 Imaging System. The stacks were minimally processed in ImageJ/FIJI using the background subtraction feature. Intensities were measured for Fig. 4A–F in FIJI using the freehand tool to specify the area of interest (Fig. 4A,B,E,F, chromosomes; Fig. 4C,D, centrosomes). For analysis of syntelically attached chromosomes, immunofluorescence slides were imaged on an EVOS M7000 Imaging System and deconvolved using Celleste 3D Deconvolution. KT–MT attachments were visually tracked and verified by examining each Z-slice. Throughout the manuscript, statistical analysis was performed using Graph Pad9 software using Chi squared, two-way ANOVA with Tukey's post-hoc test and two-tailed unpaired t-tests as indicated in the figure legends. Error bars represent the s.e.m.
Supplementary Material
Acknowledgements
The authors thank the Ohio State University Comprehensive Cancer Center Genomics Shared Resource for technical support. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Advancing Translational Sciences or the NIH. We thank Dr Katharine Ullman for gifting us the HeLa mCherry H2B GFP tubulin cells. M.S.S. thanks Paula Monsma for training and advice for live-cell imaging experiments, and Jennifer Morse for expertise with the Cytocell satellite enumeration probe experiments.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: M.S.S., M.K.S.; Methodology: M.S.S.; Software: D.T.; Formal analysis: M.S.S., M.K.S.; Investigation: L.S., B.R.S., C.S., A.E.T., L.K.; Resources: M.K.S.; Data curation: C.S., M.K.S.; Writing - original draft: M.S.S., M.K.S.; Writing - review & editing: M.S.S., M.K.S.; Supervision: A.C., M.K.S.; Project administration: M.K.S.; Funding acquisition: A.C., M.K.S.
Funding
This work was supported by an American Brain Tumor Association Basic Research Fellowship sponsored by an anonymous corporate partner (M.S.S.); National Institutes of Health grants R01GM108743 (M.K.S.), R01GM112895 (M.K.S.), R01CA227874-03 (A.C.), R01CA1145128-06 (A.C.), R01NS104332 (A.C.), R01CA227874 (A.C.), R01CA188228-06 (A.C.), P30NS104177 (OSU Neuroscience Imaging Core), P30CA016058, NCI-CCSC P30CA16059 (OSU Medicinal Chemistry Shared Resource) and UL1TR002733 (National Center for Advancing Translational Sciences); and Ohio State University, Comprehensive Cancer Center, Department of Radiation Oncology start-up funds (M.K.S.). Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/article-lookup/doi/10.1242/jcs.259560.
References
- Adriaans, I. E., Hooikaas, P. J., Aher, A., Vromans, M. J. M., van Es, R. M., Grigoriev, I., Akhmanova, A. and Lens, S. M. A. (2020). MKLP2 is a motile kinesin that transports the chromosomal passenger complex during anaphase. Curr. Biol. 30, 2628-2637. 10.1016/j.cub.2020.04.081 [DOI] [PubMed] [Google Scholar]
- Andrews, P. D., Ovechkina, Y., Morrice, N., Wagenbach, M., Duncan, K., Wordeman, L. and Swedlow, J. R. (2004). Aurora B regulates MCAK at the mitotic centromere. Dev. Cell 6, 253-268. 10.1016/S1534-5807(04)00025-5 [DOI] [PubMed] [Google Scholar]
- Atherton, J., Yu, I. M., Cook, A., Muretta, J. M., Joseph, A., Major, J., Sourigues, Y., Clause, J., Topf, M., Rosenfeld, S. S.et al. (2017). The divergent mitotic kinesin MKLP2 exhibits atypical structure and mechanochemistry. Elife 6, e27793. 10.7554/eLife.27793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina, J., Caponigro, G., Stransky, N., Venkatesan, K., Margolin, A. A., Kim, S., Wilson, C. J., Lehár, J., Kryukov, G. V., Sonkin, D.et al. (2012). The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603-607. 10.1038/nature11003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-David, U. and Amon, A. (2020). Context is everything: aneuploidy in cancer. Nat. Rev. Genet. 21, 44-62. 10.1038/s41576-019-0171-x [DOI] [PubMed] [Google Scholar]
- Browning, H., Hackney, D. D. and Nurse, P. (2003). Targeted movement of cell end factors in fission yeast. Nat. Cell Biol. 5, 812-818. 10.1038/ncb1034 [DOI] [PubMed] [Google Scholar]
- Carter, S. L., Eklund, A. C., Kohane, I. S., Harris, L. N. and Szallasi, Z. (2006). A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043-1048. 10.1038/ng1861 [DOI] [PubMed] [Google Scholar]
- Cesario, J. M., Jang, J. K., Redding, B., Shah, N., Rahman, T. and McKim, K. S. (2006). Kinesin 6 family member Subito participates in mitotic spindle assembly and interacts with mitotic regulators. J. Cell Sci. 119, 4770-4780. 10.1242/jcs.03235 [DOI] [PubMed] [Google Scholar]
- Chmátal, L., Yang, K., Schultz, R. M. and Lampson, M. A. (2015). Spatial regulation of kinetochore microtubule attachments by destabilization at spindle poles in meiosis I. Curr. Biol. 25, 1835-1841. 10.1016/j.cub.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, S. H. and McCollum, D. (2012). A role for metaphase spindle elongation forces in correction of merotelic kinetochore attachments. Curr. Biol. 22, 225-230. 10.1016/j.cub.2011.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca, J. G. (2017). Aurora a kinase function at kinetochores. Cold Spring Harb. Symp. Quant. Biol. 82, 91-99. 10.1101/sqb.2017.82.034991 [DOI] [PubMed] [Google Scholar]
- DeLuca, K. F., Herman, J. A. and DeLuca, J. G. (2016). Measuring kinetochore-microtubule attachment stability in cultured cells. Methods Mol. Biol. 1413, 147-168. 10.1007/978-1-4939-3542-0_10 [DOI] [PubMed] [Google Scholar]
- DeLuca, K. F., Lens, S. M. and DeLuca, J. G. (2011). Temporal changes in Hec1 phosphorylation control kinetochore-microtubule attachment stability during mitosis. J. Cell Sci. 124, 622-634. 10.1242/jcs.072629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca, K. F., Meppelink, A., Broad, A. J., Mick, J. E., Peersen, O. B., Pektas, S., Lens, S. M. A. and DeLuca, J. G. (2018). Aurora A kinase phosphorylates Hec1 to regulate metaphase kinetochore-microtubule dynamics. J. Cell Biol. 217, 163-177. 10.1083/jcb.201707160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster, J. M., Boyle, I., Vazquez, F., Root, D. E., Boehm, J. S., Hahn, W. C., Tsherniak, A. and McFarland, J. M. (2021). Chronos: a cell population dynamics model of CRISPR experiments that improves inference of gene fitness effects. Genome Biol. 22, 343. 10.1186/s13059-021-02540-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertych, N., Stolz, A., Valerius, O., Braus, G. H. and Bastians, H. (2016). CHK2-BRCA1 tumor-suppressor axis restrains oncogenic Aurora-A kinase to ensure proper mitotic microtubule assembly. Proc. Natl Acad. Sci. USA 113, 1817. 10.1073/pnas.1525129113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero, H., Corachán, A., Quiñonero, A., Bougeret, C., Pouletty, P., Pellicer, A. and Domínguez, F. (2019). Inhibition of KIF20A by BKS0349 reduces endometriotic lesions in a xenograft mouse model. Mol. Hum. Reprod. 25, 562-571. 10.1093/molehr/gaz044 [DOI] [PubMed] [Google Scholar]
- Godek, K. M., Kabeche, L. and Compton, D. A. (2015). Regulation of kinetochore-microtubule attachments through homeostatic control during mitosis. Nat. Rev. Mol. Cell Biol. 16, 57-64. 10.1038/nrm3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurden, M. D., Anderhub, S. J., Faisal, A. and Linardopoulos, S. (2016). Aurora B prevents premature removal of spindle assembly checkpoint proteins from the kinetochore: a key role for Aurora B in mitosis. Oncotarget 9, 19525-19542. 10.18632/oncotarget.10657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, S. J., Rolland, T., Adelmant, G., Xia, X., Owen, M. S., Dricot, A., Zack, T. I., Sahni, N., Jacob, Y., Hao, T.et al. (2014). Systematic screening reveals a role for BRCA1 in the response to transcription-associated DNA damage. Genes Dev. 28, 1957-1975. 10.1101/gad.241620.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hümmer, S. and Mayer, T. U. (2009). Cdk1 negatively regulates midzone localization of the mitotic kinesin Mklp2 and the chromosomal passenger complex. Curr. Biol. 19, 607-612. 10.1016/j.cub.2009.02.046 [DOI] [PubMed] [Google Scholar]
- Iemura, K., Natsume, T., Maehara, K., Kanemaki, M. T. and Tanaka, K. (2021). Chromosome oscillation promotes Aurora A-dependent Hec1 phosphorylation and mitotic fidelity. J. Cell Biol. 220, e202006116. 10.1083/jcb.202006116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang, J. K., Rahman, T., Kober, V. S., Cesario, J. and McKim, K. S. (2007). Misregulation of the kinesin-like protein Subito induces meiotic spindle formation in the absence of chromosomes and centrosomes. Genetics 177, 267-280. 10.1534/genetics.107.076091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor, T. M., Lampson, M. A., Hergert, P., Cameron, L., Cimini, D., Salmon, E. D., McEwen, B. F. and Khodjakov, A. (2006). Chromosomes can congress to the metaphase plate before biorientation. Science 311, 388-391. 10.1126/science.1122142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama, H., Sasai, K., Kloc, M., Brinkley, B. R. and Sen, S. (2008). Aurora kinase-A regulates kinetochore/chromatin associated microtubule assembly in human cells. Cell Cycle 7, 2691-2704. 10.4161/cc.7.17.6460 [DOI] [PubMed] [Google Scholar]
- Khongkow, P., Gomes, A. R., Gong, C., Man, E. P. S., Tsang, J. W. H., Zhao, F., Monteiro, L. J., Coombes, R. C., Medema, R. H., Khoo, U. S.et al. (2016). Paclitaxel targets FOXM1 to regulate KIF20A in mitotic catastrophe and breast cancer paclitaxel resistance. Oncogene 35, 990-1002. 10.1038/onc.2015.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., Holland, A. J., Lan, W. and Cleveland, D. W. (2010). Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell 142, 444-455. 10.1016/j.cell.2010.06.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa, M., Fung, S. Y., Hameed, U. F., Goto, H., Inagaki, M. and Lee, S. H. (2014). Cdk1 coordinates timely activation of MKlp2 kinesin with relocation of the chromosome passenger complex for cytokinesis. Cell Rep. 7, 166-179. 10.1016/j.celrep.2014.02.034 [DOI] [PubMed] [Google Scholar]
- Krenn, V. and Musacchio, A. (2015). The aurora B kinase in chromosome Bi-orientation and spindle checkpoint signaling. Front. Oncol. 5, 225. 10.3389/fonc.2015.00225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupina, K., Kleiss, C., Metzger, T., Fournane, S., Schmucker, S., Hofmann, K., Fischer, B., Paul, N., Porter, I. M., Raffelsberger, W.et al. (2016). Ubiquitin receptor protein UBASH3B drives aurora B recruitment to mitotic microtubules. Dev. Cell 36, 63-78. 10.1016/j.devcel.2015.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrière, C., Talapatra, S. K., Thoret, S., Bougeret, C., Kozielski, F. and Guillou, C. (2016). New MKLP-2 inhibitors in the paprotrain series: Design, synthesis and biological evaluations. Bioorg. Med. Chem. 24, 721-734. 10.1016/j.bmc.2015.12.042 [DOI] [PubMed] [Google Scholar]
- Lampson, M. A. and Cheeseman, I. M. (2011). Sensing centromere tension: Aurora B and the regulation of kinetochore function. Trends Cell Biol. 21, 133-140. 10.1016/j.tcb.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson, M. A., Renduchitala, K., Khodjakov, A. and Kapoor, T. M. (2004). Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 6, 232-237. 10.1038/ncb1102 [DOI] [PubMed] [Google Scholar]
- Lan, W., Zhang, X., Kline-Smith, S. L., Rosasco, S. E., Barrett-Wilt, G. A., Shabanowitz, J., Hunt, D. F., Walczak, C. E. and Stukenberg, P. T. (2004). Aurora B phosphorylates centromeric MCAK and regulates its localization and microtubule depolymerization activity. Curr. Biol. 14, 273-286. 10.1016/j.cub.2004.01.055 [DOI] [PubMed] [Google Scholar]
- Landino, J., Norris, S. R., Li, M., Ballister, E. R., Lampson, M. A. and Ohi, R. (2017). Two mechanisms coordinate the recruitment of the chromosomal passenger complex to the plane of cell division. Mol. Biol. Cell 28, 3634-3646. 10.1091/mbc.e17-06-0399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. H., McCormick, F. and Saya, H. (2010). Mad2 inhibits the mitotic kinesin MKlp2. J. Cell Biol. 191, 1069-1077. 10.1083/jcb.201003095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louw, J., Nunes Bastos, R., Chen, X., Verdood, C., Corveleyn, A., Jia, Y., Breckpot, J., Gewillig, M., Peeters, H., Santoro, M.et al. (2018). Compound heterozygous loss-of-function mutations in KIF20A are associated with a novel lethal congenital cardiomyopathy in two siblings. PLoS Genet. 14, e1007138. 10.1371/journal.pgen.1007138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers, R. M., Bryan, J. G., McFarland, J. M., Weir, B. A., Sizemore, A. E., Xu, H., Dharia, N. V., Montgomery, P. G., Cowley, G. S., Pantel, S.et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 49, 1779-1784. 10.1038/ng.3984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef, R. D., Preisinger, C., Sutcliffe, J., Kopajtich, R., Nigg, E. A., Mayer, T. U. and Barr, F. A. (2003). Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J. Cell Biol. 162, 863-876. 10.1083/jcb.200306009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser, E., Caous, R., Gruneberg, U. and Barr, F. A. (2019). Aurora A promotes chromosome congression by activating the condensin-dependent pool of KIF4A. J. Cell Biol. 219, e201905194. 10.1083/jcb.201905194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouletty, P. (2019). New derivatives of indole for the treatment of endometriosis. Biokinesis. US Patent US-20190275000-A1.
- Renda, F., Miles, C., Tikhonenko, I., Fisher, R., Carlini, L., Kapoor, T. M., Mogilner, A. and Khodjakov, A. (2022). Non-centrosomal microtubules at kinetochores promote rapid chromosome biorientation during mitosis in human cells. Curr. Biol. 32, 1049-1063. 10.1016/j.cub.2022.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath, S. C., Ohi, R., Leismann, O., Salic, A., Pozniakovski, A. and Funabiki, H. (2004). The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell 118, 187-202. 10.1016/j.cell.2004.06.026 [DOI] [PubMed] [Google Scholar]
- Serena, M., Bastos, R. N., Elliott, P. R. and Barr, F. A. (2020). Molecular basis of MKLP2-dependent Aurora B transport from chromatin to the anaphase central spindle. J. Cell Biol. 219, e201910059. 10.1083/jcb.201910059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihavuddin, A., Basu, S., Rexhepaj, E., Delestro, F., Menezes, N., Sigoillot, S. M., Del Nery, E., Selimi, F., Spassky, N. and Genovesio, A. (2017). Smooth 2D manifold extraction from 3D image stack. Nat. Commun. 8, 15554. 10.1038/ncomms15554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikirzhytski, V., Renda, F., Tikhonenko, I., Magidson, V., McEwen, B. F. and Khodjakov, A. (2018). Microtubules assemble near most kinetochores during early prometaphase in human cells. J. Cell Biol. 217, 2647-2659. 10.1083/jcb.201710094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpff, J., Wagenbach, M., Franck, A., Asbury, C. L. and Wordeman, L. (2012). Kif18A and chromokinesins confine centromere movements via microtubule growth suppression and spatial control of kinetochore tension. Dev. Cell 22, 1017-1029. 10.1016/j.devcel.2012.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherniuk, S., Skoufias, D. A., Labriere, C., Rath, O., Gueritte, F., Guillou, C. and Kozielski, F. (2010). Relocation of AuroraB and survivin from centromeres to the central spindle impaired by a kinesin-specific MKLP-2 inhibitor. Angew. Chemie Int. Ed. 49, 8228-8231. 10.1002/anie.201003254 [DOI] [PubMed] [Google Scholar]
- Torres, J. Z., Miller, J. J. and Jackson, P. K. (2009). High-throughput generation of tagged stable cell lines for proteomic analysis. Proteomics, 9, 2888-2891. 10.1002/pmic.200800873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres, J. Z., Summers, M. K., Peterson, D., Brauer, M. J., Lee, J., Senese, S., Gholkar, A. A., Lo, Y.-C., Lei, X., Jung, K.et al. (2011). The STARD9/Kif16a kinesin associates with mitotic microtubules and regulates spindle pole assembly. Cell 147, 1309-1323. 10.1016/j.cell.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi, P., Zaytsev, A. V., Godzi, M., Ataullakhanov, F. I., Grishchuk, E. L. and Stukenberg, P. T. (2019). The binding of Borealin to microtubules underlies a tension independent kinetochore-microtubule error correction pathway. Nat. Commun. 10, 682. 10.1038/s41467-019-08418-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak, A., Vazquez, F., Montgomery, P. G., Weir, B. A., Kryukov, G., Cowley, G. S., Gill, S., Harrington, W. F., Pantel, S., Krill-Burger, J. M.et al. (2017). Defining a cancer dependency map. Cell 170, 564-576. 10.1016/j.cell.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, D. L. and Weintraub, H. (1994). Expression of achaete-scute homolog 3 in Xenopus embryos converts ectodermal cells to a neural fate. Genes Dev. 8, 1434-1447. 10.1101/gad.8.12.1434 [DOI] [PubMed] [Google Scholar]
- van Heesbeen, R., Raaijmakers, J. A., Tanenbaum, M. E., Halim, V. A., Lelieveld, D., Lieftink, C., Heck, A. J. R., Egan, D. A. and Medema, R. H. (2017). Aurora A, MCAK, and Kif18b promote Eg5-independent spindle formation. Chromosoma 126, 473-486. 10.1007/s00412-016-0607-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock, M. S., Wynne, D. J., Tseng, B. S. and Funabiki, H. (2017). Dual recognition of chromatin and microtubules by INCENP is important for mitotic progression. J. Cell Biol. 216, 925-941. 10.1083/jcb.201609061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimbish, R. T. and DeLuca, J. G. (2020). Hec1/Ndc80 tail domain function at the kinetochore-microtubule interface. Front. Cell Dev. Biol. 8, 43. 10.3389/fcell.2020.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye, A. A., Deretic, J., Hoel, C. M., Hinman, A. W., Cimini, D., Welburn, J. P. and Maresca, T. J. (2015). Aurora a kinase contributes to a pole-based error correction pathway. Curr. Biol. 25, 1842-1851. 10.1016/j.cub.2015.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.