

Abstract
Background and Objective
Small cell lung cancer (SCLC) and Ewing’s sarcoma (ES) at the disseminated stage are not amenable to therapy and have a dismal prognosis with low survival rates. Despite representing different tumor entities, treatment for both malignancies relies on cytotoxic chemotherapy that has not considerably changed for the past decades. The genomic background has been extensively studied and found to comprise inactivation of p53 and RB1 in case of SCLC and EWSR1/FLI1 rearrangement in case of ES resulting in aggressive tumors in adults with heavy tobacco consumption and as bone tumor in juveniles, respectively. New therapeutic modalities are urgently needed to improve the outcomes of both tumor entities, especially in patients with metastatic disease or recurrences. This review summarizes the common cell biologic and clinical characteristics of difficult-to-treat SCLC and ES and discusses their refractoriness and options to improve the therapeutic efficacy.
Methods
PubMed and Euro PMC were searched from January 1st, 2012 to January 16th, 2022 using the following key words: “SCLC”, “Ewing´s sarcoma”, “Genomics” and “Chemoresistance” as well as own work.
Key Content and Findings
Therapy of SCLC and ES involves the use of undirected cytotoxic drugs in multimodal chemotherapy and administration of topotecan for 2nd line SCLC regimens. Despite highly aggressive chemotherapies, outcomes are dismal for patients with disseminated tumors. A host of unrelated drugs and targeted therapeutics have failed to result in progress for the patients and the underlying mechanisms of chemoresistance are still not clear. Identification of chemoresistance-reversing modulators in vitro and patient-derived xenografts of SCLC and ES has not translated into new therapies.
Conclusions
The global chemoresistance of SCLC and ES may be explained by physiological resistance at the tumor level and formation of larger spheroids that contain quiescent and hypoxic tumor cells in regions that occlude therapeutics. This type of chemoresistance is difficult to overcome and prevent the accumulation of effective drug concentration at the tumor cell level to a significant degree leaving therapeutic interventions of any kind ineffective.
Keywords: Small cell lung cancer (SCLC), Ewing’s sarcoma (ES), chemoresistance, spheroids
Introduction
Small cell lung cancer (SCLC) and Ewing’s sarcoma (ES) represent highly aggressive malignancies that show extensive metastatic potential, high chemoresistance in advanced stages and a dismal prognosis. Treatment comprises an arsenal of conventional cytotoxic drugs in multimodal combination regimens due to the failure of novel drugs and targeted therapy for the last decades. Clearly, SCLC and ES are different tumor entities deriving from neuroendocrine and neuroendocrine/mesenchymal precursors, respectively. Whereas ES affects a very small population of mostly juveniles, SCLC is responsible for the death of a considerable fraction of lung cancer patients. In order to develop novel approaches, a comparison of the biological characteristics of SCLC and ES may help to identify the key mechanisms of chemoresistance. The distinct genomic changes found in these tumors converge to a phenotype distinguished by high proliferation, early dissemination and profound refractoriness to present regimens of chemotherapy, either primary or in advanced stages or in relapses. Chemotherapy regimens are similar for SCLC and ES with very low survival rates in metastatic patients.
The rationale of the present review is to compare the current knowledge of the cell biological characteristics of SCLC and ES and to derive from both tumor entities, distinguished by a dismal prognosis, clues in regard to the mechanisms of chemoresistance and possible improvements of the therapeutic regimens. In detail, characterization of the genomic landscape of both SCLC and ES has not led to therapeutic advances and this review aims at the discussion of the cell physiological-mediated chemoresistance at the tumor level as key barrier of effective treatment. I present the following article in accordance with the Narrative Review reporting checklist (available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-58/rc).
Methods
PubMed and Euro PMC were searched from January 1st, 2012 to January 16th, 2022 using the following key words: “SCLC”, “Ewing’s sarcoma”, “Genomics” and “Chemoresistance” as well as own work (Table 1).
Table 1. Details of the search method for this narrative review.
| Items | Specification |
|---|---|
| Date of search | 16.01.2022 |
| Databases and other sources searched | PubMed, Euro PMC, Own Work |
| Search terms used | “SCLC”, “Ewing´s sarcoma”, with “Genomics” and “Chemoresistance” |
| Timeframe | 01.01.2012–16. 01. 2022 |
| Inclusion and exclusion criteria | Original publications, reviews and abstracts |
| Selection process | Selection by author |
| Any additional considerations, if applicable | None |
Characteristics of SCLC
Lung cancer is leading cancer mortality worldwide, with 1.8 million deaths in 2018 and the histological lung cancer subtype SCLC is responsible for at least 200,000 deaths globally each year (1). SCLC is linked to tobacco consumption, occurring with a lag time of about 30 years of smoking, and has a dismal prognosis with 5-year survival rate for all stages of 7% (2-4). The SCLC tumor mutational profiling reveals smoking signature as direct evidence for the initiation of SCLC by tobacco carcinogens (5). The US Surveillance, Epidemiology, and End Results (SEER) SCLC registry data verify that for the 1983–2012 period the overall survival (OS) shows no improvement (6). Only one-third of the SCLC patients present with localized disease, where cure may be achieved. Based on the expression of transcription factors, four major types of SCLC were defined as ASCL1 (SCLC-A), NEUROD1 (SCLC-N), POU2F3 (SCLC-P) or YAP1 (SCLC-Y) subtypes (7). Almost all cases of SCLC exhibit concurrent inactivation of the TP53 and RB1 suppressor genes (5). Rare cases of SCLC may develop through histological NSCLC-SCLC transformation of tyrosine kinase inhibitor (TKI)-treated lung adenocarcinomas in never-smokers (8).
The diagnosis of SCLC is usually established on biopsy or cytological samples leaving little tissue for research (9). In parallel to cytokeratins, pan-neuroendocrine markers are tested, with synaptophysin (SYP) and chromogranin A (CHGA) as first-choice markers, and CD56 and neuron-specific enolase (NSE) as additions with limited specificity. The mutations of RB1 and TP53 in SCLC result in RB1 loss and p53 overexpression in almost all samples (10). SCLC is thought to acquire distinct metastatic ability early in the course of tumor progression that affects predominantly the contralateral lung, the brain, liver, adrenal glands and bone (11).
Genomics of SCLC
Besides the loss of p53 and RB1 in SCLC, amplified genes of the MYC family members, MYC, MYCL and MYCN, are found in a subset of primary tumors and in patient-derived as well as circulating tumor cell (CTC)-derived xenografts (12,13). Furthermore, the loss of RB-related p107 or p130 proteins, alterations in the PTEN pathway and NOTCH receptors and of the chromatin regulator CREBBP as well as a high expression of Bcl-2 have all been held responsible for promoting growth and survival in SCLC. High expression of the stem cell transcription factor SOX2 has been suggested to cause plasticity in SCLC cells lineages (12). Chromatin modifiers are frequently mutated in SCLC, pointing to epigenetic regulation to cell-fate alterations (14). SCLC tumors are highly heterogenous and evasion of subpopulations of cancer cells may be an important mechanism that causes refractoriness to therapy (15,16). Targeting of plasticity of SCLC tumors may hinder the emergence of clinical resistance but the regulation of transcriptional changes in SCLC cells is poorly characterized (11).
SCLC CTCs
In accordance with the high metastatic capacity, the concentration of CTCs in SCLC exceeds that of solid tumor with exception of inflammatory breast cancer by a wide margin (17). This high number of CTCs in SCLC lends itself to the study of the biology of metastatic seeding, including genomic preconditions and tumor cell heterogeneity (18,19). The isolation of CTCs from the blood of SCLC patients can compensate for the lack of tumor material of advanced tumors. Furthermore, this property of SCLC has been taken advantage of to establish a number of permanent SCLC CTC cell lines for detailed characterization (20,21). Adhesion between CTCs in small clusters of several cells has been suggested as important aspect of cell survival during metastasis, most likely by increasing removal of CTCs from the circulation and homing in small capillaries (22). The most distinguishing feature of permanent SCLC CTCs lines in vitro is the spontaneous formation of large spheroids, termed tumorospheres, a process that in other cancer cell lines has to be enforced by nonadherent cell culture conditions (20). However, both clinical and basic research are in need of controlled clinical trials collecting tumor samples to characterize drivers of SCLC and corresponding inhibitors. An alternative source of SCLC cells are pleural effusions that contain considerable numbers of tumor cells, especially at the advanced stage (21). Mouse models of SCLC using patient-derived xenografts (PDX) are limited by non-orthotopic growth of the tumor cells in a murine microenvironment and poor representation of the human tissues (23).
Therapy of SCLC
Care of SCLC by surgery and adjuvant platinum-based chemotherapy is feasible for a few very early-stage disease patients but in most cases, patients present with metastatic disease that is treated with systemic chemotherapy with or without immunotherapy (24). In the beginning, disseminated SCLC is highly sensitive to platinum/etoposide combination therapy and response rates are well over 60% (25). However, for the majority of patients recurrent disease appears invariably leading to a median survival time of less than 2 years for patients with confined disease and of approximately 1 year for patients with extensive disease. Alterations in the local tumor stroma and in the immune microenvironment seem to contribute to SCLC tumorigenesis (2).
Most chemotherapeutics for the therapy of SCLC comprise DNA-targeting agents, such as cisplatin, DNA topoisomerase inhibitors, such as etoposide or topotecan, or γ-radiation that impairs either DNA synthesis, DNA replication or repair. Local treatment options include surgery and radiotherapy and in non-metastatic SCLC 5-year survival rates of 25–30% can be achieved. The standard 1st line chemotherapy regimen is cisplatin/etoposide (EP), which has not changed for the past three decades. Other chemotherapeutic drugs studied have activity in these patients but failed to show superiority. The topoisomerase I inhibitor topotecan is the single approved agent for 2nd line therapy of recurrent or metastatic SCLC, but combinations of doxorubicin/epirubicin with cyclophosphamide and vincristine (ACO/ECO) are likewise in use. The EP therapy for SCLC comprises five courses of etoposide 100 mg/m2 IV and cisplatin 75 mg/m2 IV on day 1, followed by oral etoposide 200 mg/m2 daily on days 2–4 (26,27). Topotecan is applied at a dosage of 1.5 mg/m2 IV daily for days 1–5 every 3 weeks. The CEV-regimen consists of five courses of epirubicin 50 mg/m2, cyclophosphamide 1,000 mg/m2, and vincristine 2 mg, all applied IV on day 1 every 21 days. Furthermore, a combination of cisplatin, etoposide, and irinotecan has been administered successfully to sensitive relapsed SCLC (28). Lurbinectedin, an alkylating agent that binds to DNA and affects transcription, was approved after exhibiting a 35% response rate in a phase II trial but failed to prolong the survival of SCLC patients (29).
Intracranial metastases occur in >50% of patients with SCLC and, therefore, prophylactic cranial irradiation (PCI) is applied for patients with extensive-stage disease and a good response to initial systemic therapy. Patients with advanced SCLC show frequently refractory intrathoracic disease after chemotherapy and disease progression. SCLC is distinguished by invariable relapses and up to 75% of patients with locally advanced disease and over 90% of patients with metastatic disease exhibit progressive disease within 2 years. For patients with metastatic disease with limited life expectancy treatment-associated toxicity has to be balanced against the symptomatic benefit of treatment. Due to smoking, SCLC cells have a high tumor mutational burden (TMB) that is expected to be linked to a favorable response to immunological T cell responses. However, the response to immune checkpoint blockade using monoclonal antibodies is limited to approximately 15% of patients with SCLC (30,31). The anti-PD1 monoclonal antibodies nivolumab and pembrolizumab received accelerated approval for third-line use in SCLC but were withdrawn later on. Recently, four SCLC subtypes were identified revealing a new 17% subtype showing unique expression of numerous immune checkpoints and human leukocyte antigens (HLAs) genes leading to the designation of Inflamed gene signature SCLC-I type. This SCLC-I tumors exhibited the greatest benefit from immunotherapy—chemotherapy combinations and should be enriched during recruitment for such therapy trials (32).
Chemoresistance of SCLC
Cellular resistance to drugs
The molecular mechanisms driving chemoresistance in SCLC remains uncharacterized (33). Treatment of tumor-bearing experimental animals with conventional chemotherapeutics reduced tumor volumes, but a complete regression of tumors was not achieved (34). CTC Copy Number Alteration (CAN) profiles of five patients with initially chemosensitive disease failed to show a chemorefractory CNA profile, proving that the initial chemoresistance differs from that of acquired chemoresistance (18). Pathway analysis on differentially upregulated proteins from an advanced SCLC cohort revealed overexpression of the HIF-1 signaling pathway (35). Analyses of transcriptomic data of SCLC cell lines confirmed upregulation of PI3K/AKT and HIF-1 pathways in chemoresistant SCLC cell lines. These data suggest that treatment-resistance in SCLC is characterized by coexisting subpopulations of cells with heterogeneous gene expression leading to multiple, concurrent resistance mechanisms (15).
Gains in the expression of the drug transporter ABCC1 and deletions in MYCL and mismatch repair proteins MSH2, and MSH6 have been identified in relapsed SCLC samples. Recurrences also exhibit frequent mutations and loss of heterozygosity in regulators of WNT signaling. In two SCLC cell lines, DNA copy number, mRNA and protein levels of the cell cycle regulator Wee1 were increased and high Wee1 expression predicted a better prognosis but Wee1 inhibitor AZD1775 failed in clinics (36). Cisplatin-resistant variants of SCLC NCI-H69 assayed in xenograft models in nude mice exhibited a higher expression of the MAPK regulator tribbles pseudokinase 2 (TRIB2) (37). Furthermore, protein ubiquitination and autophagy were more active in the SCLC H446/CDDP resistant cells (38). For example, minor or absent responses were obtained in clinical trials targeting antiapoptotic proteins like Bcl2 or Poly (ADP-ribose) polymerase (PARP) participating in DNA repair. These short lists of examples of markers of resistance of SCLC document variable and inconsistent findings in cell line models.
Thus, a host of inhibitors and modulators have been tried for the elimination of SCLC cells stemming from investigations using cell lines and patient-derived xenografts (39). So far, such findings could not translated successfully into clinical practice and SCLC has been designated as the “graveyard of drugs”. Some of these targets reported apply for minor population of patients only or did not play their suggested key role in vivo. An extensive review of the cellular targets and pathways for putative inhibition or killing of SCLC cells under discussion can be found in the recent review of Poirier et al. (39). Astonishingly, the mechanisms of chemoresistance for platinum compounds, topoisomerase I and II inhibitors as well as for camptothecines and other cytotoxic drugs that are known in great detail have not been fully reported in respect to their expression and function in SCLC cells (40,41).
Characteristics of ES
ES is a tumor triggered by a reciprocal translocation t(11;22)(q24;q12) involving EWSR1 and the ETS family transcription factor FLI1 (42,43). The N-terminal region of EWSR1 comprises a strong activation domain, causing aberrant transcription of a multitude of genes (44). Additionally, EWS-containing fusion proteins are known to regulate epigenetic determination, splicing, and metabolism of cells. ES originates from bony structures of the chest wall and far less often from soft tissue (45,46). Development of transgenic ES mouse models is impaired by the lethal action of EWSR1-FLI1 to most cells (47). Germline mutations in genes regulating DNA damage repair pathways were found to be associated with an increased risk of developing ES (48). Studies of the genomic landscape of ES revealed a scarcity of recurrent somatic mutations in this cancer (48,49). Accordingly, initial experience with immune checkpoint blockade for ES has yielded few if any responses, but still many trials check this approach. Immunotherapy for ES cells is hampered by a lack of HLA class I expression and an immunosuppressive tumor microenvironment characterized by myeloid-derived suppressor cells (MDSCs), fibrocytes, and M2-type macrophages (50). Mutation or downregulation of CDKN2A is reported in 10–30% of ES cases (51). Disease courses vary significantly between patients, attributed to a heterogeneity in the epigenetic profile of the individual ES tumors (52). Based on methylation profiles, ES can be separated from other cancer types and normal tissue. Novel therapeutic approaches for the therapy of ES involve inhibiting EWS-FLI1 itself, targeting DNA damage vulnerabilities or exploring immunotherapeutic strategies (53,54).
Chemotherapy of ES
Tumor shrinkage in response to neoadjuvant chemotherapy and radiotherapy are employed to prevent amputation for ES patients, provided that a good response to chemotherapy with >90% necrosis is achieved (55). Prior to the 1970s, treatment of ES tumors with surgery and/or RT resulted in universal recurrences in nearly all patients (56,57). Subsequently trials established active agents against ES, comprising either of two combination therapies, consisting of vincristine and cyclophosphamide, or of vincristine, actinomycin-D, cyclophosphamide and doxorubicin (VACA) combinations (57). Various VACA-based regimens in form of adjuvant or neoadjuvant regimens achieved 5-year overall survivals between 49% and 79% (58-61). Short courses of VACA with higher dose intensity in three-week cycles resulted in equivalent or superior outcomes compared to lengthy regimens (62-65). Ifosfamide was established as an active agent against ES in the 1980s improving the response rates up to 25–94% for ifosfamide single drug or ifosfamide and etoposide (IE regimen), respectively (66). Subsequent trials incorporated IE into VACA-based chemotherapy therapies (62,63,67). The 5-year OS in these trials ameliorated to 60–70% for patients with non-metastatic disease. Now, the standard chemotherapy regimen for ES comprises vincristine (1.4 mg/m2, maximum 2 mg), doxorubicin (75 mg/m2), and cyclophosphamide (1.2 g/m2) (VDC) on day 1, alternating with ifosfamide (1.8 g/m2 days 1–5) and etoposide (100 mg/m2, days 1–5) (IE) every 3 weeks (68,69). Patients with localized disease currently have an overall event free survival of 60–70% (56,70,71). In the quarter of patients with primary metastatic disease the overall event free survival is as low as 20–30% (67,72). Addition of IE to VDC showed no benefit for patients with metastatic ES and reduction of IE cycles decreased toxicity (73,74). Nevertheless, approximately 23% of all ES patients present with intrinsic drug resistance (75). In North America, VDC/IE is the standard chemotherapy regimen for ES but recent European trials have combined VIDE induction with further VAI/VAC consolidation therapy (76). VDC/IE comprises vincristine, adriamycin, and cyclophosphamide alternating with ifosfamide and etoposide (VDC/IE) delivered every two weeks (70,77,78). For patients with metastatic disease, vincristine, actinomycin, cyclophosphamide, and adriamycin (VACA) alternating with IE did not improve outcomes for patients over VACA alone (72). Similarly, there was no improvement in survival with dose-intensified VDC/IE or with high-dose melphalan, etoposide, total body irradiation (TBI), and autologous stem-cell transplantation (ASCT) and local control (79,80).
Chemoresistance of ES
For the past 40 years, there has been no major progress in the successful treatment of metastatic and recurrent ES mainly because poor drug efficacy and high tumor resistance (81). So far, the mechanisms of drug resistance in ES are not clear (82). Most chemotherapeutics administered for ES damage tumor cell DNA. Cyclophosphamide and ifosfamide are alkylating agents producing DNA cross-links in tumor cells or alkylate guanines, causing DNA base mismatch (83). Doxorubicin and etoposide inhibit topoisomerase II indispensable for DNA replication and introduce DNA strand breaks. When tumor cells cannot repair DNA damage caused by chemotherapeutic agents, apoptosis is activated. However, cancer cells inhibit apoptosis by interfering with the proapoptotic pathway and by increasing the expression of antiapoptotic proteins (84). Increased drug efflux may play a role, but for ES the relationship between P-gp expression and prognosis is uncertain (75,85). Cytostatics may be inactivated by derivatization by cytochromes such as CYP3A4 that is otherwise expressed mainly in the liver and intestines (86). It metabolizes anticancer drugs administered to treat ES, including cyclophosphamide, doxorubicin, vincristine, ifosfamide, topotecan and etoposide. However, no correlation was found between CYP3A4 expression and prognosis. Similarly, glutathione S-transferases such as GSTM4 specifically expressed in ES may detoxify chemotherapeutics. It is controlled by EWS-FLI1 through a regulatory element in the GSTM4 promoter region and participates in ES tumorigenesis and drug resistance (87).
Analyses of the genomic landscape of ES demonstrate a scarcity of characteristic mutations (49). No recurrent aberrations have been found that include kinase mutations or gene amplifications. Minor consistent genetic abnormalities comprise deletions of STAG2 (17%) or mutations/deletions of TP53 (10%). Kinase inhibitors have been tested in ES trials, but none of these inhibitors has shown significant activity in phase 2 studies, including agents directed to aurora kinase A (88), c-kit (89), or insulin growth factor-1 receptor (IGF-1R) kinase. MET signaling has been demonstrated as requirement for ES tumorigenesis and VEGF signaling for growth and dissemination (90). Time to relapse is the key prognostic factor for ES, linking relapse within the first two years to an OS of less than 10% and the other patients to an OS of approximately 30%. (91). Newer camptothecin combinations are commonly administered for early relapses comprising, for example, for the rEECur trial topotecan and cyclophosphamide and irinotecan and temozolomide (92). Topotecan single agent was not active but yielded a response in 30% of patients with relapsed ES in combination with cyclophosphamide (93,94).
Histology of ES
ES tumors consist of small round cells, most likely derived from neural crest or primordial bone marrow-derived mesenchymal stem cells, and with highest incidence at a median age of 15 years (95). These uniform small blue round cells exhibit cell surface expression of CD99/MIC2 and reveal glycogen deposition in their cytoplasm (96,97). However, CD99 is a very sensitive but poorly specific diagnostic marker for ES in approximately 95% of ES cases (98-101). Detection of EWSR1 rearrangements by FISH or of ES-specific gene fusions by RT-PCR are used for diagnosis (102). Although circulating tumor cells (CTCs) have been demonstrated in ES no correlation to survival has been established (103,104).
Dissemination of ES
The frequency of somatic mutations in ES is extremely low with on average of only 6–11 somatic mutations, most likely indicating a tumor initiation at an early age (49,105). Genetic differences of metastases from the primary tumor seem to preceed the first clinical presentation by 1–2 years (43). Obviously, cells carrying subclonal mutations at diagnosis become dominant at relapse, and recurrences accumulate new mutations (105). Chemotherapy-associated mutational signatures in advanced ES indicate that new mutations drive metastasis in ES (43). Accordingly, the mutation rates increased 3fold from primary tumors to recurrences (105). Intratumoral DNA-methylation showed increased heterogeneity in metastatic disease compared to localized tumors (52). In the rare cases of ES rebiopsies, similar numbers of divergent genes were reported, but the observed changes were no consistent from patient to patient. Frequent alterations of CXCR4 expression point to upregulation of this chemokine due to growth factor deprivation, hypoxia, and intratumoral pressure in the TME (106).
Depletion of the EWS-Fli1 fusion product led to decreased proliferation but increased migration, invasion and dissemination of ES possibly due to epithelial-mesenchymal transition (EMT), including upregulation of the expression of hypoxia-related genes (105,106). EWS-FLI1low ES cells exhibited higher chemoresistance, upregulated immune checkpoint proteins and started angiogenesis controlled by Wnts (107). These metastatic EWSR1-FLI1low tumor cells seem to exist in primary ES in low frequencies of 1–2% as part of the heterogenous SCLC tumors (108,109). Due to a shortness of untreated primary tumor material from diagnosis, most of these studies employed primary patient-derived lines and organoid models (110,111).
Discussion: physiological chemoresistance of SCLC and ES
SCLC has been unresponsive to a host of diverse and unrelated anticancer drugs and in ES the majority of patients responded to highly aggressive multimodal chemotherapy, except for resistant patients and patients experiencing recurrent disease. During the last decades for both malignancies no new chemotherapeutics have been introduced into clinics that would have led to superior outcomes. The reasons for this high chemoresistance have not been traced back to molecular mechanisms at the cellular level that would have been reversible by appropriate resistance modifiers. Successful cytotoxic anticancer therapy requires the delivery of sufficient concentrations of the active agent into the tumor tissue. Tumors restrict the release of compounds by irregular vascularization, cell-cell adhesion, increased intralesional pressure and cancer niches exhibiting hypoxic, acidic and nutrient-depleted regions. This type of chemoresistance is labeled as physiological resistance at the tumor tissue level (112,113).
Cellular aggregation, cell-cell adhesion, IL-6, IGF-1, oxygen and nutrient supply, as well as interstitial acidification might be related to anticancer drug resistance (114,115). Angiogenesis plays vital roles in tumor growth and metastasis. Hypoxia and slow tumor cell growth are associated with poor response to chemotherapy and rapid recurrence of tumors (115,116). Tumor interstitial fluid pressure is higher than that of normal tissue and this characteristic impedes drug delivery to the target tumor tissue (117). As malignant tumor cells are remote from blood vessels, various anticancer drugs are unevenly distributed in them (115). ES exhibits resistance both to traditional chemotherapeutic agents and targeted drugs. The mechanisms of drug resistance are highly complex and influenced by numerous and diverse intracellular and extracellular factors (118,119).
Histopathological features of ES include the occurrence of small round tumor cells with a low cytoplasmic fraction and granular nuclear chromatin with normal nucleoli (120). With a median of 80 mitoses per mm2 ES shows an accelerated proliferation correlating with numerous apoptotic figures and eventually extensive necrosis. Densely packed tumor cells typically appear sheet-like with occasionally rosettes or groups of cells detached by stroma, or as parallel structures at the periphery of nests. In order to test novel therapeutics, classical 2D culture is widely used but may not truly reflect the cellular characteristics observed under in vivo conditions (119,121). ES cells growing on 3D scaffolds under shear stress showed an increased production of IGF1 and an altered response to IGF1R inhibitors (122). Cocultured ES and mesenchymal stromal cells (MSCs) showed mutually stimulation of proliferation and altered responses to various inhibitors (123). Thus, the high resistance of ES to chemotherapy agents seems in part due to their particular MSCs origin but also to its complex TME (124). The coculture of these ES spheroids in the bone matrix tissue simulated typical tumor characteristics, including the induction of a hypoxic and glycolytic phenotype and the triggering of angiogenesis. At aggregate sizes exceeding 400 µm, A673 ES spheroids displayed a gradient of necrotic cells from the interior of the spheroids to viable proliferating cells located at their peripheral rim. Another example is presented in Figure 1. showing spheroids of different sizes spontaneously formed during regular cell culture of the KAL ES cell line that has been initiated from a soft tissue metastasis of a relapsing patient (121). The distinct layers of larger spheroids and the gradients of oxygen and nutrients as well as the localization of hypoxic and necrotic cell regions are indicated in fig.1. Cancer cells in the interior of the spheroids are protected from chemotherapeutics and as whole this specific cell assembly displays high resistance.
Figure 1.
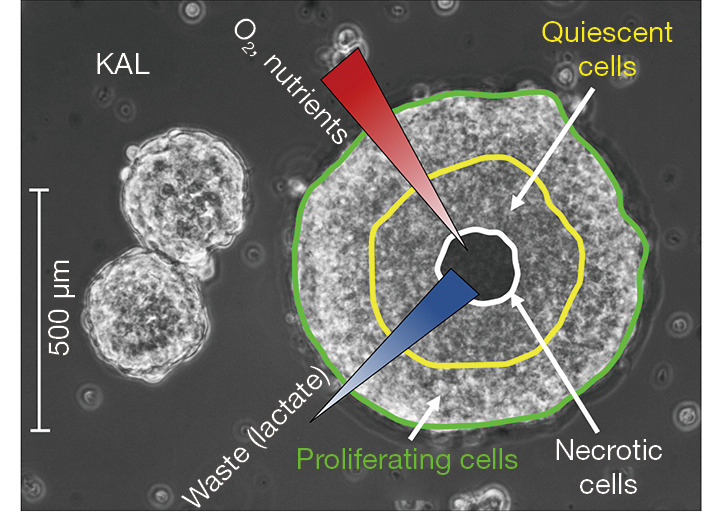
ES cell line KAL spheroids. KAL ES cells derived from a cutaneous metastasis form dense spheroids in tissue culture. Three KAL spheroids of different sizes as observed in light microscopy are shown in Figure 1. This growth occurs spontaneously and needs not to be enforced by nonadherent culture conditions. Spheroids exceeding a diameter of 400 µm are beyond the diffusion limit of oxygen and contain hypoxic and necrotic cells in their core with associated gradients of nutrients and accumulating waste products. Such spheroids inhibit the penetration of drugs, quiescent cells in the deeper layer of the aggregate are less sensitive to chemotherapy and cells in the more hypoxic regions are refractory to irradiation. ES, Ewing’s sarcoma.
A similar investigation studied ES tumor cell lines under anchorage-independent conditions on agar-coated flasks resulting in marked up-regulation of E-cadherin and rapid formation of multicellular spheroids in suspension (125,126). Spheroid formation was accompanied by inhibition of cell proliferation and reduced expression of cyclin D1, the major D-type cyclin in ES (82). These cultures exhibited marked chemoresistance to diverse cytotoxic agents, including carboplatin, compared with 2D cultures, which could be reversed by suppression of E-cadherin. Identical results were observed with etoposide, melphalan, doxorubicin, or topotecan. Downregulation of ErbB4 expression by siRNA blocked Akt-mediated activation and cell survival and restored chemosensitivity of the spheroids. In conclusion, ES tumors exhibit signs of special cell arrangements in tumor tissues in combination with markers of hypoxia and the putatively corresponding ES spheroids demonstrate increased drug resistance.
Similar to ES, SCLC proved to be resistant to a host of chemotherapeutic drugs by mechanisms possibly related to physiological resistance at the tumor tissue level. SCLC tumors are covered by an extended stroma of extracellular matrix (ECM) and high levels of ECM correlated with a dismal prognosis (127). ECM via β1 integrin-mediated PI3-kinase activation makes SCLC cells resistant to proliferation arrest and apoptosis induced by DNA damage. The excessive numbers of circulating tumor cells (CTCs) in SCLC allowed for the in vitro expansion of 5 CTC cell lines (BHGc7, 10, 16, 26 and UHGc5) from blood samples of patients with recurrent tumors (20). These cell lines exhibit the typical SCLC markers and all established CTCs lines develop spontaneously very large multicellular aggregates, termed tumorospheres. Ki67 as proliferation marker and carbonic anhydrase 9 (CAIX) as hypoxic marker staining of tumorosphere sections revealed quiescent and hypoxic cells located at the inner layers and in the core, respectively. Accordingly, chemosensitivity tests of CTC either as single cell suspensions or as tumorospheres proved increased resistance of the clusters against chemotherapeutics commonly used for treatment of SCLC (20). A report by Lee et al. showed the formation of aggregates by short-term cultivated SCLC CTCs consisting of large-sized, round-shaped spheroids, small-sized cohesive irregular or round spheroids and “grape-like” spheroids (128). Chemoresistance against cisplatin and etoposide has been observed among the expanded CTCs. The expression of NOTCH pathway components, such es DLL3 (delta-like protein 3) has been described as prominent feature of SCLCs and targeted clinically using a specific antibody carrying a highly toxic payload, named Rovalpituzumab tesirine (21). However, despite promising preclinical and phase I trial activity, this antibody drug conjugate (ADC) failed in several advanced phase II-III clinical trials exhibiting low activity and high toxicity (129). DLL3 remains still an important target for SCLC but is has to be investigated whether the 3D-arrangement of SCLCs hamper the efficacy of ADCs. Similar approaches testing a range of diverse ADCs are ongoing for sarcomas (130). Therefore, despite all differences SCLC and ES display similar tumor tissue arrangements and cancer cells that spontaneously form large and highly chemoresistent spheroids.
Summary
SCLC and ES share a dismal prognosis, especially in advanced disseminated stage due to low response to systemic chemotherapy linked to largely unknown mechanisms of chemoresistance. Both tumors exhibit high proliferation and metastatic potential triggered by tumor suppressor inactivation/mutations and a genetic rearrangement that result in complex alteration of the transcription of a host of genes. Whereas aggressive combination chemotherapy can save the majority of ES patients, almost all SCLC patients succumb to the disease within several years. Although actionable genetic defects were identified for SCLC and ES, the proposed inhibitors or modulators failed to improve outcomes in patients so far. A short comparison of the characteristics of SCLC and ES is shown in Table 2. Findings from genetic and histological studies point to a special arrangement of cells in tumor tissue leading to low-perfused hypoxic conditions. Primary cell cultures are distinguished by spontaneous formation of spheroids/tumorospheres with high chemoresistance against a host of chemically unrelated drugs, triggered by special cellular mechanisms. Thus, efficient chemotherapy of SCLC and ES will require new approaches to increase the delivery of drugs to reach sufficient concentrations of drugs for the totality of tumor cells. Part of the failure to advance therapy of SCLC and ES has to be ascribed to the impairment of the delivery of the host of compounds studied in clinical trials. In relation to genetic studies and investigations using permanent 2D cell line models, research on complex tumor tissues and on improved drug delivery is lagging behind. Future research may concentrate on cell biology, cell-cell adhesion, organization and methods to disassemble 3D spheroids to aid drug delivery in clinics. So far, the heterogenous ES evades the most intensive conventional therapy and metastatic disease remains a major challenge for this tumor and SCLC (131).
Table 2. Comparison of the most important characteristics of SCLC and ES.
| Characteristic | SCLC | ES |
|---|---|---|
| Genomics | p53 and RB1 inactivation | EWS1-Fli translocation |
| Number of cases/year | 250,000 worldwide | 200–250 USA |
| Risk factor | Smoking | ? |
| Origin | Neuroendocrine carcinoma | Neuroendocrine/epithelial lung |
| 5-year survival | 7% all stages | 70%/23% (primary/metastatic ES) |
| Mutation rate | High | Low |
| Markers | CKs, NSE, SYN, CHGA, CD56, p53 | CD99, EWS1-Fli proteins |
| Metastasis | All patients | Approximately 23% |
| CTCs | High numbers | Scarce |
| Therapy | 1st line Platinum/etoposide; 2nd line Topotecan/ECO | Vincristine-doxorubicin-cyclo-phosphamide (VDC) and Ifosfamide-etoposide (IE) |
| Response rates | 60% initial, 2nd very low | 77% initial |
| Immunotherapy | 1st, 2nd Combination chemotherapy, 3rd low response | Low responses |
| Cellular resistance | ABCx? GST? BCl2? PARP? | P-gp? CYP3A4? GSTM4? |
| Physiological resistance | Spheroid/tumorospheres | 3D spheroids |
| Hypoxia | HIF1α positive | HIF1α positive |
| Targeted therapy | Ineffective | Ineffective |
?, not clear. SCLC, small cell lung cancer; ES, Ewing’s sarcoma; RB1, Retinoblastoma1; EWS1, Ewing’s sarcoma gene 1; CK, cytokeratin; NSE, neuron-specific enolase; CHGA, chromogranin A; ECO, epirubicin-cyclophosphamide-vincristine; ABC, ATP binding cassette; GST, glutathione S-transferases; BCL2, B-cell lymphoma 2; PARP, poly (ADP-ribose)-polymerase; P-gp, P-glycoprotein; CYP3A4, cytochrome P450 3A4; GSTMA4, glutathione S-transferase µA4.
Supplementary
The article’s supplementary files as
Acknowledgments
The help of MSc. Barbara Rath and MSc. Sandra Stickler in the preparation of this manuscript is gratefully acknowledged. Furthermore, I wish to thank Dr. T. Hohenheim for enduring endorsement.
Funding: None.
Ethical Statement: The author is accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Footnotes
Reporting Checklist: The author has completed the Narrative Review reporting checklist. Available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-58/rc
Conflicts of Interest: The author has completed the ICMJE uniform disclosure form (available at https://tlcr.amegroups.com/article/view/10.21037/tlcr-22-58/coif). The author has no conflicts of interest to declare.
References
- 1.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 2.Huang R, Wei Y, Hung RJ, et al. Associated Links Among Smoking, Chronic Obstructive Pulmonary Disease, and Small Cell Lung Cancer: A Pooled Analysis in the International Lung Cancer Consortium. EBioMedicine 2015;2:1677-85. 10.1016/j.ebiom.2015.09.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tariq S, Kim SY, Monteiro de Oliveira Novaes J, et al. Update 2021: Management of Small Cell Lung Cancer. Lung 2021;199:579-87. 10.1007/s00408-021-00486-y [DOI] [PubMed] [Google Scholar]
- 4.Rudin CM, Brambilla E, Faivre-Finn C, et al. Small-cell lung cancer. Nat Rev Dis Primers 2021;7:3. 10.1038/s41572-020-00235-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George J, Lim JS, Jang SJ, et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015;524:47-53. 10.1038/nature14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang S, Tang J, Sun T, et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci Rep 2017;7:1339. 10.1038/s41598-017-01571-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudin CM, Poirier JT, Byers L, et al. Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat Rev Cancer 2019;19:289-97. 10.1038/s41568-019-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rath B, Plangger A, Hamilton G. Non-small cell lung cancer-small cell lung cancer transformation as mechanism of resistance to tyrosine kinase inhibitors in lung cancer. Cancer Drug Resist 2020;3:171-8. 10.20517/cdr.2019.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Righi L, Volante M, Papotti M. Small-Cell Carcinoma of the Lung: What We Learned about It? Acta Cytol 2021. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 10.Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer 2017;17:725-37. 10.1038/nrc.2017.87 [DOI] [PubMed] [Google Scholar]
- 11.Yang D, Denny SK, Greenside PG, et al. Intertumoral Heterogeneity in SCLC Is Influenced by the Cell Type of Origin. Cancer Discov 2018;8:1316-31. 10.1158/2159-8290.CD-17-0987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Little CD, Nau MM, Carney DN, et al. Amplification and expression of the c-myc oncogene in human lung cancer cell lines. Nature 1983;306:194-6. 10.1038/306194a0 [DOI] [PubMed] [Google Scholar]
- 13.Udagawa H, Umemura S, Murakami I, et al. Genetic profiling-based prognostic prediction of patients with advanced small-cell lung cancer in large scale analysis. Lung Cancer 2018;126:182-8. 10.1016/j.lungcan.2018.11.014 [DOI] [PubMed] [Google Scholar]
- 14.Gardner EE, Lok BH, Schneeberger VE, et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 2017;31:286-99. 10.1016/j.ccell.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stewart CA, Gay CM, Xi Y, et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat Cancer 2020;1:423-36. 10.1038/s43018-019-0020-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khoogar R, Li F, Chen Y, et al. Single-cell RNA profiling identifies diverse cellular responses to EWSR1/FLI1 downregulation in Ewing sarcoma cells. Cell Oncol (Dordr) 2022;45:19-40. 10.1007/s13402-021-00640-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu HH, Liu YT, Feng Y, et al. Circulating tumor cells (CTCs)/circulating tumor endothelial cells (CTECs) and their subtypes in small cell lung cancer: Predictors for response and prognosis. Thorac Cancer 2021;12:2749-57. 10.1111/1759-7714.14120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter L, Rothwell DG, Mesquita B, et al. Molecular analysis of circulating tumor cells identifies distinct copy-number profiles in patients with chemosensitive and chemorefractory small-cell lung cancer. Nat Med 2017;23:114-9. 10.1038/nm.4239 [DOI] [PubMed] [Google Scholar]
- 19.Hamilton G, Hochmair M, Rath B, et al. Small cell lung cancer: Circulating tumor cells of extended stage patients express a mesenchymal-epithelial transition phenotype. Cell Adh Migr 2016;10:360-7. 10.1080/19336918.2016.1155019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klameth L, Rath B, Hochmaier M, et al. Small cell lung cancer: model of circulating tumor cell tumorospheres in chemoresistance. Sci Rep 2017;7:5337. 10.1038/s41598-017-05562-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rath B, Plangger A, Krenbek D, et al. Rovalpituzumab tesirine resistance: analysis of a corresponding small cell lung cancer and circulating tumor cell line pair. Anticancer Drugs 2022;33:300-7. 10.1097/CAD.0000000000001267 [DOI] [PubMed] [Google Scholar]
- 22.Hou JM, Krebs MG, Lancashire L, et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol 2012;30:525-32. 10.1200/JCO.2010.33.3716 [DOI] [PubMed] [Google Scholar]
- 23.Ferone G, Lee MC, Sage J, et al. Cells of origin of lung cancers: lessons from mouse studies. Genes Dev 2020;34:1017-32. 10.1101/gad.338228.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamilton G, Rath B, Ulsperger E. A review of the role of surgery for small cell lung cancer and the potential prognostic value of enumeration of circulating tumor cells. Eur J Surg Oncol 2016;42:1296-302. 10.1016/j.ejso.2016.04.063 [DOI] [PubMed] [Google Scholar]
- 25.Chu X, Han C, Su C. Treatment of small cell lung cancer: recent advances. Curr Opin Oncol 2022;34:83-8. 10.1097/CCO.0000000000000804 [DOI] [PubMed] [Google Scholar]
- 26.Hagmann R, Hess V, Zippelius A, et al. Second-Line Therapy of Small-Cell Lung Cancer: Topotecan Compared to a Combination Treatment with Adriamycin, Cyclophosphamide And Vincristine (ACO) - a Single Center Experience. J Cancer 2015;6:1148-54. 10.7150/jca.13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundstrøm S, Bremnes RM, Kaasa S, et al. Second-line chemotherapy in recurrent small cell lung cancer. Results from a crossover schedule after primary treatment with cisplatin and etoposide (EP-regimen) or cyclophosphamide, epirubicin, and vincristin (CEV-regimen). Lung Cancer 2005;48:251-61. [DOI] [PubMed] [Google Scholar]
- 28.Goto K, Ohe Y, Shibata T, et al. Combined chemotherapy with cisplatin, etoposide, and irinotecan versus topotecan alone as second-line treatment for patients with sensitive relapsed small-cell lung cancer (JCOG0605): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol 2016;17:1147-57. 10.1016/S1470-2045(16)30104-8 [DOI] [PubMed] [Google Scholar]
- 29.Trigo J, Subbiah V, Besse B, et al. Lurbinectedin as second-line treatment for patients with small-cell lung cancer: a single-arm, open-label, phase 2 basket trial. Lancet Oncol 2020;21:645-54. 10.1016/S1470-2045(20)30068-1 [DOI] [PubMed] [Google Scholar]
- 30.Ready NE, Ott PA, Hellmann MD, et al. Nivolumab Monotherapy and Nivolumab Plus Ipilimumab in Recurrent Small Cell Lung Cancer: Results From the CheckMate 032 Randomized Cohort. J Thorac Oncol 2020;15:426-35. 10.1016/j.jtho.2019.10.004 [DOI] [PubMed] [Google Scholar]
- 31.Hamilton G, Rath B. Immunotherapy for small cell lung cancer: mechanisms of resistance. Expert Opin Biol Ther 2019;19:423-32. 10.1080/14712598.2019.1592155 [DOI] [PubMed] [Google Scholar]
- 32.Gay CM, Stewart CA, Park EM, et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021;39:346-360.e7. 10.1016/j.ccell.2020.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wagner AH, Devarakonda S, Skidmore ZL, et al. Recurrent WNT pathway alterations are frequent in relapsed small cell lung cancer. Nat Commun 2018;9:3787. 10.1038/s41467-018-06162-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taromi S, Kayser G, von Elverfeldt D, et al. An orthotopic mouse model of small cell lung cancer reflects the clinical course in patients. Clin Exp Metastasis 2016;33:651-60. 10.1007/s10585-016-9808-8 [DOI] [PubMed] [Google Scholar]
- 35.Jin Y, Chen Y, Tang H, et al. Activation of PI3K/AKT Pathway Is a Potential Mechanism of Treatment Resistance in Small Cell Lung Cancer. Clin Cancer Res 2022;28:526-39. 10.1158/1078-0432.CCR-21-1943 [DOI] [PubMed] [Google Scholar]
- 36.Zhao X, Kim IK, Kallakury B, et al. Acquired small cell lung cancer resistance to Chk1 inhibitors involves Wee1 up-regulation. Mol Oncol 2021;15:1130-45. 10.1002/1878-0261.12882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang Y, Yu D, Perez-Soler R, et al. TRIB2 contributes to cisplatin resistance in small cell lung cancer. Oncotarget 2017;8:109596-608. 10.18632/oncotarget.22741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma K, Li S, Huo X, et al. Exploring the mechanism of cisplatin resistance by transcriptome sequencing and reversing the chemoresistance by autophagy inhibition in small cell lung cancer. Biochem Biophys Res Commun 2020;533:474-80. 10.1016/j.bbrc.2020.09.023 [DOI] [PubMed] [Google Scholar]
- 39.Poirier JT, George J, Owonikoko TK, et al. New Approaches to SCLC Therapy: From the Laboratory to the Clinic. J Thorac Oncol 2020;15:520-40. 10.1016/j.jtho.2020.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olszewski U, Hamilton G. A better platinum-based anticancer drug yet to come? Anticancer Agents Med Chem 2010;10:293-301. 10.2174/187152010791162306 [DOI] [PubMed] [Google Scholar]
- 41.Hamilton G, Klameth L, Rath B, et al. Synergism of cyclin-dependent kinase inhibitors with camptothecin derivatives in small cell lung cancer cell lines. Molecules 2014;19:2077-88. 10.3390/molecules19022077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lawlor ER, Sorensen PH. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit Rev Oncog 2015;20:155-71. 10.1615/CritRevOncog.2015013553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anderson ND, de Borja R, Young MD, et al. Rearrangement bursts generate canonical gene fusions in bone and soft tissue tumors. Science 2018;361:eaam8419. 10.1126/science.aam8419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bailly RA, Bosselut R, Zucman J, et al. DNA-binding and transcriptional activation properties of the EWS-FLI-1 fusion protein resulting from the t(11;22) translocation in Ewing sarcoma. Mol Cell Biol 1994;14:3230-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shamberger RC, Grier HE. Ewing’s sarcoma/primitive neuroectodermal tumor of the chestwall. Semin Pediatr Surg 2001;10:153-60. 10.1053/spsu.2001.24699 [DOI] [PubMed] [Google Scholar]
- 46.Schuck A, Ahrens S, Paulussen M, et al. Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2003;55:168-77. 10.1016/S0360-3016(02)03797-5 [DOI] [PubMed] [Google Scholar]
- 47.Minas TZ, Surdez D, Javaheri T, et al. Combined experience of six independent laboratories attempting to create an Ewing sarcoma mouse model. Oncotarget 2017;8:34141-63. 10.18632/oncotarget.9388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brohl AS, Patidar R, Turner CE, et al. Frequent inactivating germline mutations in DNA repair genes in patients with Ewing sarcoma. Genet Med 2017;19:955-8. 10.1038/gim.2016.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tirode F, Surdez D, Ma X, et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov 2014;4:1342-53. 10.1158/2159-8290.CD-14-0622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales E, Olson M, Iglesias F, et al. Role of immunotherapy in Ewing sarcoma. J Immunother Cancer 2020;8:e000653. 10.1136/jitc-2020-000653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maitra A, Roberts H, Weinberg AG, et al. Aberrant expression of tumor suppressor proteins in the Ewing family of tumors. Arch Pathol Lab Med 2001;125:1207-12. 10.5858/2001-125-1207-AEOTSP [DOI] [PubMed] [Google Scholar]
- 52.Sheffield NC, Pierron G, Klughammer J, et al. DNA methylation heterogeneity defines a disease spectrum in Ewing sarcoma. Nat Med 2017;23:386-95. 10.1038/nm.4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stegmaier K, Wong JS, Ross KN, et al. Signature-based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med 2007;4:e122. 10.1371/journal.pmed.0040122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grünewald TGP, Cidre-Aranaz F, Surdez D, et al. Ewing sarcoma. Nat Rev Dis Primers 2018;4:5. 10.1038/s41572-018-0003-x [DOI] [PubMed] [Google Scholar]
- 55.Gerrand C, Bate J, Seddon B, et al. Seeking international consensus on approaches to primary tumour treatment in Ewing sarcoma. Clin Sarcoma Res 2020;10:21. 10.1186/s13569-020-00144-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bacci G, Picci P, Gherlinzoni F, et al. Localized Ewing’s sarcoma of bone: Ten years’ experience at the Istituto Ortopedico Rizzoli in 124 cases treated with multimodal therapy. Eur J Cancer Clin Oncol 1985;21:163-73. 10.1016/0277-5379(85)90168-3 [DOI] [PubMed] [Google Scholar]
- 57.Chan RC, Sutow WW, Lindberg RD, et al. Management and results of localized Ewing’s sarcoma. Cancer 1979;43:1001-6. [DOI] [PubMed] [Google Scholar]
- 58.Donaldson SS, Torrey M, Link MP, et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma: End results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125-35. 10.1016/S0360-3016(98)00191-6 [DOI] [PubMed] [Google Scholar]
- 59.Hayes FA, Thompson EI, Meyer WH, et al. Therapy for localized Ewing’s sarcoma of bone. J Clin Oncol 1989;7:208-13. 10.1200/JCO.1989.7.2.208 [DOI] [PubMed] [Google Scholar]
- 60.Rosen G, Caparros B, Nirenberg A, et al. Ewing’s sarcoma: Ten-year experience with adjuvant chemotherapy. Cancer 1981;47:2204-13. [DOI] [PubMed] [Google Scholar]
- 61.Zucker JM, Henry-Amar M, Sarrazin D, et al. Intensive systemic chemotherapy in localized Ewing’s sarcoma in childhood. A historical trial. Cancer 1983;52:415-23. [DOI] [PubMed] [Google Scholar]
- 62.Craft AW, Cotterill SJ, Bullimore JA, et al. Long-term results from the first UKCCSG Ewing’s Tumour Study (ET-1). United Kingdom Children’s Cancer Study Group (UKCCSG) and the Medical Research Council Bone Sarcoma Working Party. Eur J Cancer 1997;33:1061-9. 10.1016/S0959-8049(97)00043-9 [DOI] [PubMed] [Google Scholar]
- 63.Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing’s sarcoma: The Second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study. J Clin Oncol 1998;16:3628-33. 10.1200/JCO.1998.16.11.3628 [DOI] [PubMed] [Google Scholar]
- 64.Burgert EO, Jr, Nesbit ME, Garnsey LA, et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: Intergroup study IESS-II. J Clin Oncol 1990;8:1514-24. 10.1200/JCO.1990.8.9.1514 [DOI] [PubMed] [Google Scholar]
- 65.Evans RG, Nesbit ME, Gehan EA, et al. Multimodal therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bones: A report from the second intergroup study. J Clin Oncol 1991;9:1173-80. 10.1200/JCO.1991.9.7.1173 [DOI] [PubMed] [Google Scholar]
- 66.Kung FH, Pratt CB, Vega RA, et al. Ifosfamide/etoposide combination in the treatment of recurrent malignant solid tumors of childhood. A Pediatric Oncology Group Phase II study. Cancer 1993;71:1898-903. [DOI] [PubMed] [Google Scholar]
- 67.Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: Final results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin Oncol 2001;19:1818-29. 10.1200/JCO.2001.19.6.1818 [DOI] [PubMed] [Google Scholar]
- 68.Bailey K, Cost C, Davis I, et al. Emerging novel agents for patients with advanced Ewing sarcoma: a report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Res 2019;8:F1000 Faculty Rev-493. [DOI] [PMC free article] [PubMed]
- 69.Juergens C, Weston C, Lewis I, et al. Safety assessment of intensive induction with vincristine, ifosfamide, doxorubicin, and etoposide (VIDE) in the treatment of Ewing tumors in the EURO-E.W.I.N.G. 99 clinical trial. Pediatr Blood Cancer 2006;47:22-9. 10.1002/pbc.20820 [DOI] [PubMed] [Google Scholar]
- 70.Womer RB, West DC, Krailo MD, et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: a report from the Children's Oncology Group. J Clin Oncol 2012;30:4148-54. 10.1200/JCO.2011.41.5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jain S, Kapoor G. Chemotherapy in Ewing’s sarcoma. Indian J Orthop 2010;44:369-77. 10.4103/0019-5413.69305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Miser JS, Goldsby RE, Chen Z, et al. Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy–a report from the Children’s Oncology Group. Pediatr Blood Cancer 2007;49:894-900. 10.1002/pbc.21233 [DOI] [PubMed] [Google Scholar]
- 73.Flores G, Grohar PJ. One oncogene, several vulnerabilities: EWS/FLI targeted therapies for Ewing sarcoma. J Bone Oncol 2021;31:100404. 10.1016/j.jbo.2021.100404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang L, Yu Y. Drug resistance in recurrent and metastatic Ewing sarcoma. Authorea Preprints; 2021. 10.22541/au.163476822.21319119/v1 [DOI]
- 75.Roundhill E, Burchill S. Membrane expression of MRP-1, but not MRP-1 splicing or Pgp expression, predicts survival in patients with ESFT. Br J Cancer 2013;109:195-206. 10.1038/bjc.2013.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le Deley MC, Paulussen M, Lewis I, et al. Cyclophosphamide compared with ifosfamide in consolidation treatment of standard-risk Ewing sarcoma: results of the randomized noninferiority Euro-EWING99-R1 trial. J Clin Oncol 2014;32:2440-8. 10.1200/JCO.2013.54.4833 [DOI] [PubMed] [Google Scholar]
- 77.Nesbit ME, Jr, Gehan EA. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term followup of the First Intergroup study. J Clin Oncol 1990;8:1664-74. 10.1200/JCO.1990.8.10.1664 [DOI] [PubMed] [Google Scholar]
- 78.Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694-701. 10.1056/NEJMoa020890 [DOI] [PubMed] [Google Scholar]
- 79.Miser JS, Krailo MD, Tarbell NJ, et al. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide–a Children’s Cancer Group and Pediatric Oncology Group study. J Clin Oncol 2004;22:2873-76. 10.1200/JCO.2004.01.041 [DOI] [PubMed] [Google Scholar]
- 80.Meyers PA, Krailo MD, Ladanyi M, et al. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing’s sarcoma does not improve prognosis. J Clin Oncol 2001;19:2812-20. 10.1200/JCO.2001.19.11.2812 [DOI] [PubMed] [Google Scholar]
- 81.Biswas B, Bakhshi S. Management of Ewing sarcoma family of tumors: Current scenario and unmet need. World J Orthop 2016;7:527-38. 10.5312/wjo.v7.i9.527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang J, Hu S, Schofield DE, et al. Selective usage of D-Type cyclins by Ewing's tumors and rhabdomyosarcomas. Cancer Res 2004;64:6026-34. 10.1158/0008-5472.CAN-03-2594 [DOI] [PubMed] [Google Scholar]
- 83.Bukowski K, Kciuk M, Kontek R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int J Mol Sci 2020;21:3233. 10.3390/ijms21093233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ahmed AA, Zia H, Wagner L. Therapy resistance mechanisms in Ewing’s sarcoma family tumors. Cancer Chemother Pharmacol 2014;73:657-63. 10.1007/s00280-014-2392-1 [DOI] [PubMed] [Google Scholar]
- 85.Perri T, Fogel M, Mor S, et al. Effect of P-glycoprotein expression on outcome in the Ewing family of tumors. Pediatr Hematol Oncol 2001;18:325-34. 10.1080/088800101300312591 [DOI] [PubMed] [Google Scholar]
- 86.Zia H, Murray GI, Vyhlidal CA, et al. CYP3A isoforms in Ewing’s sarcoma tumours: an immunohistochemical study with clinical correlation. Int J Exp Pathol 2015;96:81-6. 10.1111/iep.12115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhuo R, Kosak KM, Sankar S, et al. Targeting Glutathione S-transferase M4 in Ewing sarcoma. Front Pediatr 2014;2:83. 10.3389/fped.2014.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mossé YP, Fox E, Teachey DT, et al. A Phase II Study of Alisertib in Children with Recurrent/Refractory Solid Tumors or Leukemia: Children’s Oncology Group Phase I and Pilot Consortium (ADVL0921). Clin Cancer Res 2019;25:3229-38. 10.1158/1078-0432.CCR-18-2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bond M, Bernstein ML, Pappo A, et al. A phase II study of imatinib mesylate in children with refractory or relapsed solid tumors: a Children's Oncology Group study. Pediatr Blood Cancer 2008;50:254-8. 10.1002/pbc.21132 [DOI] [PubMed] [Google Scholar]
- 90.Fleuren ED, Roeffen MH, Leenders WP, et al. Expression and clinical relevance of MET and ALK in Ewing sarcomas. Int J Cancer 2013;133:427-36. 10.1002/ijc.28047 [DOI] [PubMed] [Google Scholar]
- 91.Leavey PJ, Mascarenhas L, Marina N, et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer 2008;51:334-8. 10.1002/pbc.21618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McCabe MG, Kirton L, Khan M, et al. Results of the second interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J Clin Oncol 2020;38:11502. 10.1200/JCO.2020.38.15_suppl.11502 [DOI] [Google Scholar]
- 93.Hunold A, Weddeling N, Paulussen M, et al. Topotecan and cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer 2006;47:795-800. 10.1002/pbc.20719 [DOI] [PubMed] [Google Scholar]
- 94.Hawkins DS, Bradfield S, Whitlock JA, et al. Topotecan by 21-day continuous infusion in children with relapsed or refractory solid tumors: a Children's Oncology Group study. Pediatr Blood Cancer 2006;47:790-4. 10.1002/pbc.20739 [DOI] [PubMed] [Google Scholar]
- 95.Eaton BR, Claude L, Indelicato DJ, et al. Ewing sarcoma. Pediatr Blood Cancer 2021;68 Suppl 2:e28355. 10.1002/pbc.28355 [DOI] [PubMed] [Google Scholar]
- 96.Ent W, Sand LGL, Hogendoorn PCW. Molecular genetics of Ewing sarcoma, model systems and finding novel (immuno-) therapeutic targets. J Transl Genet Genom 2018;2:10. [Google Scholar]
- 97.Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients - The INFORM pilot study. Eur J Cancer 2016;65:91-101. 10.1016/j.ejca.2016.06.009 [DOI] [PubMed] [Google Scholar]
- 98.Hamilton G, Fellinger EJ, Schratter I, et al. Characterization of a human endocrine tissue and tumor-associated Ewing's sarcoma antigen. Cancer Res 1988;48:6127-31. [PubMed] [Google Scholar]
- 99.Fellinger EJ, Garin-Chesa P, Su SL, et al. Biochemical and genetic characterization of the HBA71 Ewing's sarcoma cell surface antigen. Cancer Res 1991;51:336-40. [PubMed] [Google Scholar]
- 100.Ambros IM, Ambros PF, Strehl S, et al. MIC2 is a specific marker for Ewing's sarcoma and peripheral primitive neuroectodermal tumors. Evidence for a common histogenesis of Ewing's sarcoma and peripheral primitive neuroectodermal tumors from MIC2 expression and specific chromosome aberration. Cancer 1991;67:1886-93. [DOI] [PubMed] [Google Scholar]
- 101.Baldauf MC, Orth MF, Dallmayer M, et al. Robust diagnosis of Ewing sarcoma by immunohistochemical detection of super-enhancer-driven EWSR1-ETS targets. Oncotarget 2017;9:1587-601. 10.18632/oncotarget.20098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sorensen PH, Liu XF, Delattre O, et al. Reverse transcriptase PCR amplification of EWS/FLI-1 fusion transcripts as a diagnostic test for peripheral primitive neuroectodermal tumors of childhood. Diagn Mol Pathol 1993;2:147-57. 10.1097/00019606-199309000-00002 [DOI] [PubMed] [Google Scholar]
- 103.Grohs JG, Zoubek A, Jugovic D, et al. Intraoperative dissemination of tumour cells in patients with Ewing tumours detected by RT-PCR. Int Orthop 2004;28:222-5. 10.1007/s00264-004-0551-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zoubek A, Kovar H, Kronberger M, et al. Mobilization of tumour cells during biopsy in an infant with Ewing sarcoma. Eur J Pediatr 1996;155:373-6. 10.1007/BF01955264 [DOI] [PubMed] [Google Scholar]
- 105.Crompton BD, Stewart C, Taylor-Weiner A, et al. The genomic landscape of pediatric Ewing sarcoma. Cancer Discov 2014;4:1326-41. 10.1158/2159-8290.CD-13-1037 [DOI] [PubMed] [Google Scholar]
- 106.Krook MA, Nicholls LA, Scannell CA, et al. Stress-induced CXCR4 promotes migration and invasion of ewing sarcoma. Mol Cancer Res 2014;12:953-64. 10.1158/1541-7786.MCR-13-0668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chaturvedi A, Hoffman LM, Jensen CC, et al. Molecular dissection of the mechanism by which EWS/FLI expression compromises actin cytoskeletal integrity and cell adhesion in Ewing sarcoma. Mol Biol Cell 2014;25:2695-709. 10.1091/mbc.e14-01-0007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aynaud MM, Mirabeau O, Gruel N, et al. Transcriptional Programs Define Intratumoral Heterogeneity of Ewing Sarcoma at Single-Cell Resolution. Cell Rep 2020;30:1767-1779.e6. 10.1016/j.celrep.2020.01.049 [DOI] [PubMed] [Google Scholar]
- 109.Franzetti GA, Laud-Duval K, van der Ent W, et al. Cell-to-cell heterogeneity of EWSR1-FLI1 activity determines proliferation/migration choices in Ewing sarcoma cells. Oncogene 2017;36:3505-14. 10.1038/onc.2016.498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brodin BA, Wennerberg K, Lidbrink E, et al. Drug sensitivity testing on patient-derived sarcoma cells predicts patient response to treatment and identifies c-Sarc inhibitors as active drugs for translocation sarcomas. Br J Cancer 2019;120:435-43. 10.1038/s41416-018-0359-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Fong EL, Lamhamedi-Cherradi SE, Burdett E, et al. Modeling Ewing sarcoma tumors in vitro with 3D scaffolds. Proc Natl Acad Sci U S A 2013;110:6500-5. 10.1073/pnas.1221403110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weiswald LB, Bellet D, Dangles-Marie V. Spherical cancer models in tumor biology. Neoplasia 2015;17:1-15. 10.1016/j.neo.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hamilton G, Rath B. Applicability of tumor spheroids for in vitro chemosensitivity assays. Expert Opin Drug Metab Toxicol 2019;15:15-23. 10.1080/17425255.2019.1554055 [DOI] [PubMed] [Google Scholar]
- 114.Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett 2017;387:61-8. 10.1016/j.canlet.2016.01.043 [DOI] [PubMed] [Google Scholar]
- 115.Nikolaou M, Pavlopoulou A, Georgakilas AG, et al. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis 2018;35:309-18. 10.1007/s10585-018-9903-0 [DOI] [PubMed] [Google Scholar]
- 116.Mohammad IS, He W, Yin L. Insight on Multidrug Resistance and Nanomedicine Approaches to Overcome MDR. Crit Rev Ther Drug Carrier Syst 2020;37:473-509. 10.1615/CritRevTherDrugCarrierSyst.2020025052 [DOI] [PubMed] [Google Scholar]
- 117.He B, Sui X, Yu B, et al. Recent advances in drug delivery systems for enhancing drug penetration into tumors. Drug Deliv 2020;27:1474-90. 10.1080/10717544.2020.1831106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kilic M, Kasperczyk H, Fulda S, et al. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007;26:2027-38. 10.1038/sj.onc.1210008 [DOI] [PubMed] [Google Scholar]
- 119.Gaebler M, Silvestri A, Haybaeck J, et al. Three-Dimensional Patient-Derived In Vitro Sarcoma Models: Promising Tools for Improving Clinical Tumor Management. Front Oncol 2017;7:203. 10.3389/fonc.2017.00203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grünewald TGP, Cidre-Aranaz F, Surdez D, et al. Ewing sarcoma. Nat Rev Dis Primers 2018;4:5. 10.1038/s41572-018-0003-x [DOI] [PubMed] [Google Scholar]
- 121.Hofbauer S, Hamilton G, Theyer G, et al. Insulin-like growth factor-I-dependent growth and in vitro chemosensitivity of Ewing's sarcoma and peripheral primitive neuroectodermal tumour cell lines. Eur J Cancer 1993;29A:241-5. 10.1016/0959-8049(93)90183-G [DOI] [PubMed] [Google Scholar]
- 122.Santoro M, Lamhamedi-Cherradi SE, Menegaz BA, et al. Flow perfusion effects on three-dimensional culture and drug sensitivity of Ewing sarcoma. Proc Natl Acad Sci U S A 2015;112:10304-9. 10.1073/pnas.1506684112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Santoro M, Menegaz BA, Lamhamedi-Cherradi SE, et al. Modeling Stroma-Induced Drug Resistance in a Tissue-Engineered Tumor Model of Ewing Sarcoma. Tissue Eng Part A 2017;23:80-9. 10.1089/ten.tea.2016.0369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Munoz-Garcia J, Jubelin C, Loussouarn A, et al. In vitro three-dimensional cell cultures for bone sarcomas. J Bone Oncol 2021;30:100379. 10.1016/j.jbo.2021.100379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kang HG, Jenabi JM, Zhang J, et al. E-cadherin cell-cell adhesion in ewing tumor cells mediates suppression of anoikis through activation of the ErbB4 tyrosine kinase. Cancer Res 2007;67:3094-105. 10.1158/0008-5472.CAN-06-3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lawlor ER, Scheel C, Irving J, et al. Anchorage-independent multi-cellular spheroids as an in vitro model of growth signaling in Ewing tumors. Oncogene 2002;21:307-18. 10.1038/sj.onc.1205053 [DOI] [PubMed] [Google Scholar]
- 127.Hodkinson PS, Mackinnon AC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. Int J Radiat Biol 2007;83:733-41. 10.1080/09553000701570204 [DOI] [PubMed] [Google Scholar]
- 128.Lee HL, Chiou JF, Wang PY, et al. Ex Vivo Expansion and Drug Sensitivity Profiling of Circulating Tumor Cells from Patients with Small Cell Lung Cancer. Cancers (Basel) 2020;12:3394. 10.3390/cancers12113394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uprety D, Remon J, Adjei AA. All That Glitters Is Not Gold: The Story of Rovalpituzumab Tesirine in SCLC. J Thorac Oncol 2021;16:1429-33. 10.1016/j.jtho.2021.07.012 [DOI] [PubMed] [Google Scholar]
- 130.Polito L, Calafato G, Bortolotti M, et al. Antibody Conjugates for Sarcoma Therapy: How Far along Are We? Biomedicines 2021;9:978. 10.3390/biomedicines9080978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Riggi N, Suvà ML, Stamenkovic I. Ewing's Sarcoma. N Engl J Med 2021;384:154-64. 10.1056/NEJMra2028910 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The article’s supplementary files as