

Transforming growth factor beta (TGF-β) is a multifunctional cytokine that possesses immune stimulatory and inhibitory functions, influences T-lymphocyte differentiation, and promotes cell proliferation and wound healing.1 TGF-β signaling activates the transcription factor SMAD3, which increases IL-4 receptor expression associated with type 2–skewed immune responses. (Figure 1). Dysregulated TGF-β signaling has been implicated in human syndromes with co-existent connective tissue abnormalities and atopy (e.g., Ehlers-Danlos syndrome, Loeys-Dietz syndrome).2–4
Figure 1. TGF-β signaling and ERBIN regulation.
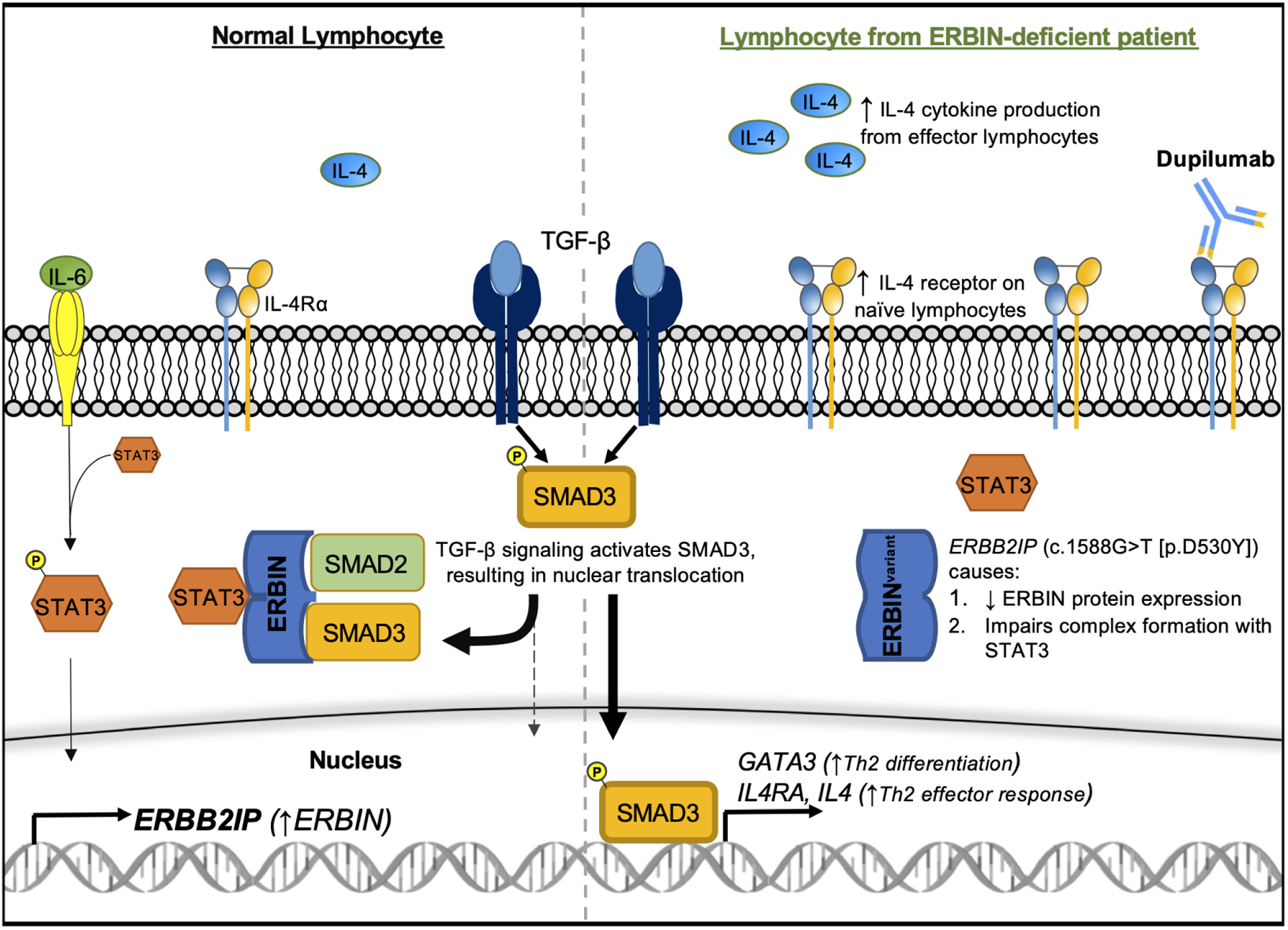
TGF-β activates SMAD3, which translocates into the nucleus to promote Th2 skewing of lymphocyte responses. The STAT3-ERBIN complex negatively regulates TGF-β signaling by sequestering cytoplasmic SMAD3. The ERBB2IP variant (c.1588G>T [p.D530Y]) impairs STAT3-ERBIN-SMAD2/3 complex formation, increasing TGF-β signaling through SMAD3. Dupilumab targets IL-4Rα and can block IL-4–mediated effects of dysregulated TGF-β signaling. P, phosphorylation
Protection against aberrant type 2 responses may be partially mediated by IL-6–driven STAT3 activation, which opposes Th2 skewing in T-lymphocytes. Patients with genetic defects leading to impaired IL-6 receptor or STAT3 signaling develop elevated IgE, atopy, and immunodeficiency.5 Lyons et al. demonstrated that IL-6–induced STAT3 activation inhibited TGF-β signaling by increasing the transcription of ERBB2-interacting protein (ERBIN), a protein scaffold that complexes with STAT3 to sequester cytoplasmic SMAD2/3 (Figure 1). Three family members with ERBIN deficiency (carrying a novel, disease-segregating ERBB2IP [c.1588G>T, p.D530Y] variant) had elevated IgE, atopy, and connective tissue abnormalities (e.g., hypermobility, aortic root dilation) associated with increased TGF-β signaling and activation of the type 2–skewing GATA-3/IL-4/IL-4Rα axis in naïve T and B lymphocytes.6
We present the clinical course of the 16-year-old male proband with ERBIN deficiency who presented with refractory eosinophilic gastrointestinal disease (EGID), uncontrolled eosinophilic respiratory disease, recurrent infections and hypogammaglobulinemia, peripheral eosinophilia, and elevated IgE. After failure of traditional therapies, we postulated that targeted treatment of the excessive IL-4R expression with IL-4R blockade would be an effective therapeutic strategy.
Dupilumab-induced remission of refractory eosinophilic gastrointestinal disease (EGID)
The patient’s past medical history revealed a progressive course of atopy and ineffectiveness and/or complications of standard therapies. The patient was diagnosed with eosinophilic esophagitis (an EGID, 6 yo), followed years later by eosinophilic gastritis (an EGID) and peripheral eosinophilia (9 yo). Multiple swallowed steroids (budesonide, fluticasone) initially induced histologic remission of his EGID. However, this regimen was complicated by esophageal candidiasis and iatrogenic adrenal insufficiency; necessary discontinuation of topical steroids led to EGID recurrence.
Given the persistent hypereosinophilia (blood eosinophils 1600–4450 cells/mcL for >6 months) and refractory gastrointestinal disease, mepolizumab (hypereosinophilic syndrome dosage: 300 mg every 4 weeks) was trialed. Prior to mepolizumab, tissue histology showed severe gastric eosinophilic inflammation with eosinophil degranulation and active eosinophilic esophagitis (Figure 2). After 5 months of mepolizumab, the blood absolute eosinophil count (AEC) and gastrointestinal tissue eosinophilia were markedly decreased (72% and 75% reduction, respectively); however, the gastrointestinal disease remained histologically active, and clinical symptoms failed to improve (Figure 2). Given the incomplete histologic response and poor clinical improvement, mepolizumab was discontinued, which caused an acute exacerbation of gastrointestinal symptoms, loss of asthma control, and worsening rhinitis symptoms. Severe eosinophilia (AEC 6850 cells/mcL) with elevated IL-5 (55 pg/mL; normal <1 pg/mL) and IL-4 (19 pg/mL; normal <8 pg/mL) levels in the peripheral blood developed.
Figure 2. Patient characteristics by treatment stage.
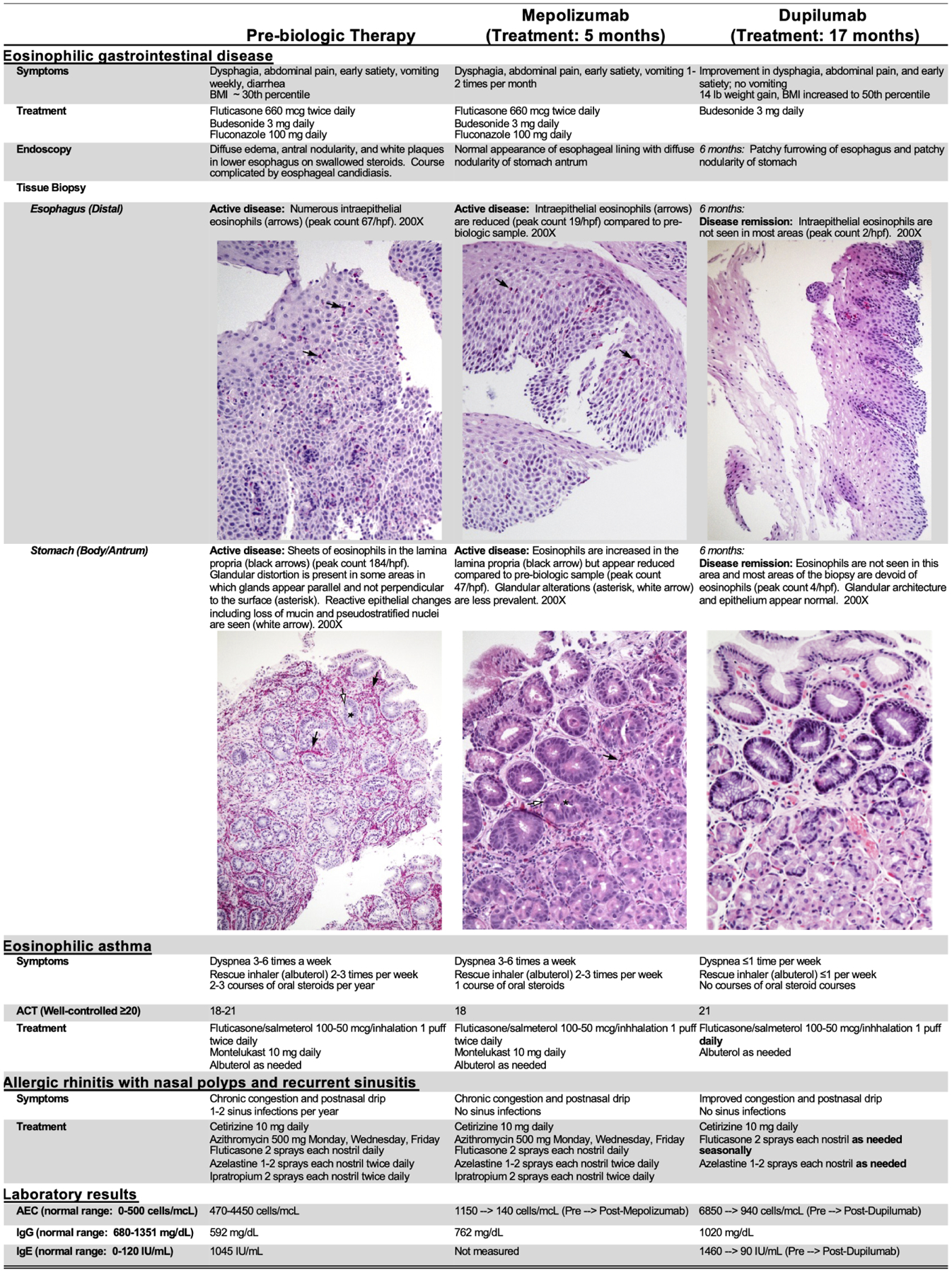
ACT = Asthma Control Test is a subjective scoring system filled out by the patient, 20–25 suggests controlled asthma, 16–19 suggests partially controlled asthma, and less than 16 suggests poorly controlled asthma; AEC = Absolute Eosinophil Count; IgG = Immunoglobulin G; IgE = Immunoglobulin E; BMI = Body Mass Index; hpf = High powered field
Because IL-5–directed therapy was ineffective, we hypothesized that IL-4/13 blockade might be therapeutic and initiated dupilumab (300 mg, every 2 weeks). After 4 weeks of dupilumab, the patient reported improvement of his gastrointestinal symptoms, and his blood eosinophilia began downtrending. After 6 months of dupilumab, he reported nearly complete resolution of gastrointestinal symptoms and significant weight gain (6.4 kg/ 14 lbs; body mass index 50th percentile); AEC decreased to 970 cells/mcL, and peripheral blood IL-5 levels normalized (<1 pg/mL). Esophageal and gastric biopsies demonstrated histologic remission of eosinophilic inflammation (Figure 2). Swallowed fluticasone was subsequently discontinued. The 12-month follow-up endoscopy for the course (dupilumab, crushed enteric budesonide) showed minimal esophageal eosinophilic inflammation (2 eosinophils/ high-power field) and no recurrence of gastric disease.
Dupilumab-induced remission of eosinophilic respiratory disease
Similar to the EGIDs (eosinophilic esophagitis, eosinophilic gastritis), asthma and chronic rhinitis progressively worsened during the patient’s teenage years, with poor asthma symptom control, development of nasal polyps, and recurrent sinusitis. Treatment of his respiratory disease was complicated by iatrogenic adrenal insufficiency. The patient experienced weekly asthma symptoms and 2–3 exacerbations/year, requiring oral steroids while taking ICS plus long-acting β2-agonist (ICS-LABA) combination therapy and montelukast. Similarly, his allergic rhinitis remained poorly controlled, leading to recurrent sinusitis (5–6 episodes/year) and 4 surgical procedures to treat refractory sinusitis and nasal polyposis despite ongoing management (intranasal fluticasone, azelastine, and ipratropium; oral cetirizine; prophylactic azithromycin). With mepolizumab, there had been no significant improvements in upper or lower respiratory symptoms. Conversely, his asthma control significantly improved within 6–8 weeks of dupilumab therapy. After >17 months of dupilumab, he reported minimal asthma symptoms and had no oral steroid courses, the ICS-LABA dose was decreased, and montelukast discontinued (Figure 2). His FEV1 was normal prior to biologic therapy and did not change with dupilumab. He did report improved upper respiratory symptoms, with subsequent discontinuation of azithromycin and ipratropium, and no recurrence of nasal polyps nor sinus infections.
Humoral immunity normalized with IL-4 blockade
Due to recurrent infections, the patient had serial immunologic evaluations. The patient developed intermittent hypogammaglobulinemia (IgG 385–791 mg/dL) of unclear etiology during adolescence in the context of robust vaccine responses and without gastrointestinal protein loss. He also demonstrated persistently elevated IgE (peak level 1460 IU/mL). Remarkably, his IgG titers increased during dupilumab therapy (IgG 941–1020 mg/dL), and IgE dramatically decreased to 90 IU/mL. We postulate that the hypogammaglobulinemia may have been, at least partially, due to the excessive type 2 response; however, a reduction in corticosteroid use could have also contributed to the observed IgG increase.
Damaging variants in ERBIN-encoding ERBB2IP manifest with connective tissue abnormalities and atopy associated with dysregulated TGF-β signaling and increased IL-4 receptor expression and type 2 immune responses. As ERBIN complexes with STAT3, there are reports of gastrointestinal disease improving with dupilumab therapy in patients with autosomal dominant hyper-IgE syndrome (AD-HIES) being treated with dupilumab for severe atopic dermatitis.7,8 This case highlights that specifically blocking type 2 signaling via anti–IL-4Rα therapy (dupilumab) in a patient with ERBIN deficiency was an effective strategy to treat co-existent allergic inflammation and illustrates a potential strategy for treating other patients with dysregulated TGF-β signaling and comorbid atopy. The impact of dupilumab on the patient’s connective tissue disorder remains unclear; given that aberrant TGF-β signaling is thought to directly contribute to the underlying connective tissue abnormality independent of its effect on the IL-4/IL-4Rα axis, improvement is not anticipated. This case illustrates how clarity regarding a patient’s underlying disease mechanism can effectively guide a targeted biologic therapy approach.9
Clinical Implications.
ERBIN deficiency causes dysregulated TGF-β signaling driving aberrant IL-4–associated type 2 immunity. Anti–IL-4/IL-13 receptor immunotherapy effectively treated an ERBIN–deficient patient with refractory severe atopy including eosinophilic gastrointestinal disease and may benefit other patients with dysregulated TGF-β signaling–associated atopy.
Acknowledgments:
The authors thank Shawna Hottinger for editorial assistance.
Funding:
This research was supported in part by the Division of Intramural Research of the National Institute of Allergy and Infectious Diseases, NIH. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Conflicts of Interest:
H.R.D., J.P.A., J.D.M., J.J.L., A.F.F., and K.A.R. have no relevant conflicts of interest to disclose. M.H.C. is a consultant for Allakos, Arena Pharmaceuticals, AstraZeneca, Bristol Myers Squibb (BMS), Calypso Biotech, EsoCap, GlaxoSmith Kline (GSK), Regeneron Pharmaceuticals, Inc., and Shire and has received research funding from Receptos/BMS, Regeneron Pharmaceuticals, Inc. and Shire. V.A.M. is a consultant for Shire/Takeda and Allakos. M.E.R. is a consultant for Pulm One, Spoon Guru, ClostraBio, Serpin Pharm, Allakos, Celgene, AstraZeneca, Adare/Ellodi Pharma, GlaxoSmith Kline, Regeneron/Sanofi, Revolo Biotherapeutics, and Guidepoint and has an equity interest in the first five listed and royalties from reslizumab (Teva Pharmaceuticals), PEESSv2 (Mapi Research Trust) and UpToDate. M.E.R. is an inventor of patents owned by Cincinnati Children’s Hospital. J.T.S. has received research funding from Knopp Biosciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol 2016;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacFarlane EG, Haupt J, Dietz HC, Shore EM. TGF-β Family Signaling in Connective Tissue and Skeletal Diseases. Cold Spring Harb Perspect Biol 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abonia JP, Wen T, Stucke EM, Grotjan T, Griffith MS, Kemme KA, et al. High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. J Allergy Clin Immunol 2013;132:378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006;355:788–98. [DOI] [PubMed] [Google Scholar]
- 5.Freeman AF, Milner JD. The Child with Elevated IgE and Infection Susceptibility. Curr Allergy Asthma Rep 2020;20:65. [DOI] [PubMed] [Google Scholar]
- 6.Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med 2017;214:669–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixit C, Thatayatikom A, Pappa H, Knutsen AP. Treatment of severe atopic dermatitis and eosinophilic esophagitis with dupilumab in a 14-year-old boy with autosomal dominant hyper-IgE syndrome. J Allergy Clin Immunol Pract. 2021. Jul 16. [DOI] [PubMed] [Google Scholar]
- 8.Lu C-W, Lee W-I, Chung W-H. Dupilumab for STAT3-Hyper-IgE Syndrome With Refractory Intestinal Complication. Pediatrics. 2021. Sep;148(3). [DOI] [PubMed] [Google Scholar]
- 9.Muraro A, Lemanske RF, Castells M, Torres MJ, Khan D, Simon HU, et al. Precision medicine in allergic disease-food allergy, drug allergy, and anaphylaxis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma and Immunology. Allergy 2017;72:1006–21. [DOI] [PubMed] [Google Scholar]