

Abstract
Human gut microbial dynamics are highly individualized, making it challenging to link microbiota to health and to design universal microbiome therapies. This individuality is typically attributed to variation in host genetics, diets, environments, and medications, but it could also emerge from fundamental ecological forces that shape microbiota more generally. Here we leverage extensive gut microbial time series from wild baboons—hosts who experience little interindividual dietary and environmental heterogeneity—to test whether gut microbial dynamics are synchronized across hosts or largely idiosyncratic. Despite their shared lifestyles, baboon microbiota were only weakly synchronized. The strongest synchrony occurred among baboons living in the same social group, likely because group members range over the same habitat and simultaneously encounter the same sources of food and water. However, this synchrony was modest compared to each host’s personalized dynamics. In support, host-specific factors, especially host identity, explained, on average, more than 3 times the deviance in longitudinal dynamics compared to factors shared with social group members and 10 times the deviance of factors shared across the host population. These results contribute to mounting evidence that highly idiosyncratic gut microbiomes are not an artifact of modern human environments, and that synchronizing forces in the gut microbiome (e.g., shared environments, diets, and microbial dispersal) are not strong enough to overwhelm key drivers of microbiome personalization, such as host genetics, priority effects, horizontal gene transfer, and functional redundancy.
Introduction
Mammalian gut microbiotas are highly complex, dynamic ecosystems. From these dynamics emerge a set of life-sustaining services for hosts, which help them digest food, process toxins, and resist pathogens. Despite their importance, our understanding of how gut microbial communities change over time within hosts, especially the collective dynamics of microbiotas from hosts in the same population, is poor1, 2. This gap exists in part because we lack time series data that track gut microbiotas longitudinally across many hosts living together in the same population. As a result, it has been difficult to answer key questions. For example, when host populations encounter shifting environments and resources, does each host’s microbiota respond similarly—i.e., in synchrony—or idiosyncratically to these changes? Further, what factors predict synchronized versus idiosyncratic microbiota?
Answering these questions is important because synchronized gut microbial communities, if and when they occur1, could help explain shared microbiota-associated traits in host populations, such as patterns of disease susceptibility3, 4. A high degree of synchrony may also suggest that similar ecological principles govern changes in microbial composition across hosts5. Further, there is theoretical justification to expect coordinated microbial dynamics, as host populations and their microbiotas can be considered a ‘microbiome metacommunity’ (see e.g.,6, 7, 8, 9). Metacommunity theory predicts that synchrony will arise across microbiotas if their hosts experience similar environmental conditions and/or high rates of microbial dispersal between hosts10, 11. In support, fruit bats living in the same colony exhibit coordinated fur microbiota, due to shared environments and microbial dispersal1.
However, even in the presence of such synchronizing forces, there are many reasons to expect that hosts in a microbiome metacommunity will exhibit idiosyncratic (i.e., individualized) microbial compositions and dynamics. First, idiosyncratic dynamics are expected when the same microbes in different hosts respond in different ways to environmental fluctuations, chance events, or interactions with other microbes12, 13, 14, 15. These forces are likely important in microbiotas where priority effects, functional redundancy, and horizontal gene flow can cause the same microbe to play different ecological roles and exhibit different environmental responses in different hosts16, 17, 18, 19. Second, several cross-sectional studies, in both humans and animals, find that individual hosts exhibit distinctive gut microbiotas, and host identity explains a large fraction of population-wide microbiome taxonomic variation1, 20, 21, 22, 23, 24, 25. Further, some longitudinal studies in humans and animals find personalized gut microbial dynamics1, 24, 26, 27, 28. This personalization is usually attributed to interpersonal differences in diet, medications, and lifestyle27, 29, 30, 31. If correct, then idiosyncratic microbiome dynamics may be simply explained by a lack of shared environmental drivers rather than distinct microbiome responses to shared environments (but see 27). In contrast, if personalized dynamics persist even when hosts share the same environment, then (i) host-specific dynamics may not be solely attributable to interpersonal differences in lifestyles; (ii) predicting the dynamics of microbial taxa in individual hosts may prove difficult; and (iii) microbiome interventions that rely on manipulating taxa may face challenges beyond heterogeneity in lifestyles, and instead may be related to conserved ecological principles across microbiomes.
Results and Discussion
Baboon gut microbiota show seasonal and annual cycles
We tested for gut microbial synchrony using 17,265 16S rRNA gene sequencing-based microbiome profiles from 600 baboons living in 12 social groups over a 14-year span32 (Fig. 1A; Supplementary Fig. 1). The baboons were members of the well-studied Amboseli baboon population33, who experience shared diets, environments, and opportunities for between-host microbial dispersal. All groups used an overlapping ~60 km2 range (Fig. 1B; Supplementary video 1;32), and all baboons experienced the same seasonal changes in rainfall and temperature. Seasonal weather patterns drive a rotating set of baboon foods, including grass corms in the dry season and growing grass blades and grass seed heads in the wet season32, 34, 35 (Figs. 1C and D).
Fig. 1. Baboons in Amboseli experience shared environments at multiple scales.
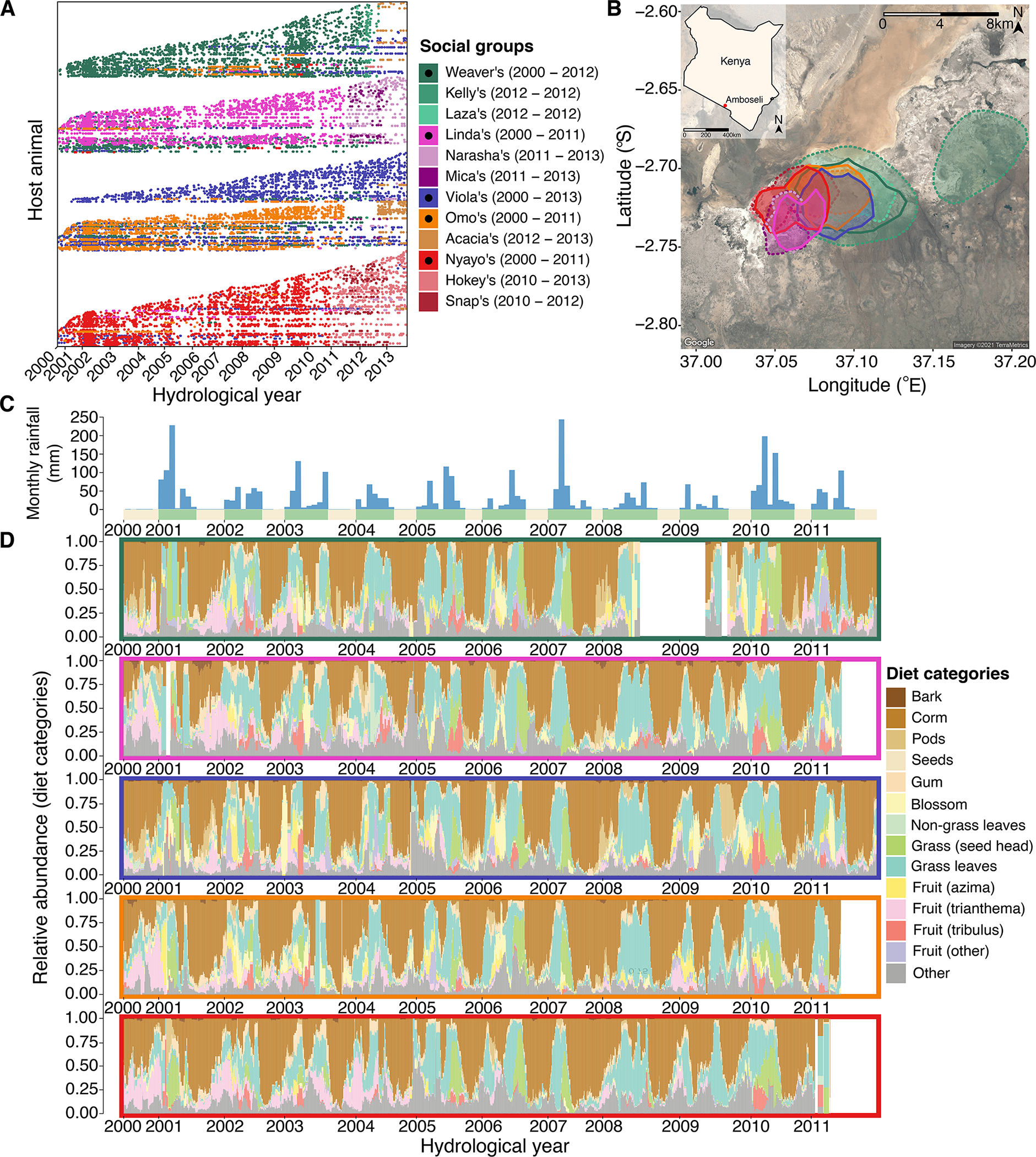
(A) Our microbiota time series consisted of 17,265 16S rRNA gene sequencing gut microbial profiles. Each point represents a microbiota sample, plotted by the date it was collected (x-axis). Each row (y-axis) corresponds to a unique individual host. Samples were collected from 600 wild baboons living in 5 original social groups (indicated by dark colors marked with black dots in the legend) and 7 groups that fissioned/fused from these original groups (no black dots). (B) All baboon groups ranged over a shared ~60 km2 area, and the social groups had largely overlapping home ranges. Ranges are shown as 90% kernel densities over the sampling period specific to each group; 5 original social groups are shown with solid borders, fission and fusion products with dashed borders. (C) Monthly rainfall amounts (blue bars, in mm) with yellow and green stripes along the x-axis representing dry and wet seasons, with the width of the green stripes reflecting the number of months within the focal year that had at least 1 mm rainfall. (D) Temporal shifts in diet from the years 2000 – 2013, shown as the relative abundance of diet components in the 5 original social groups over 30-day sliding windows prior to each sample collection date. Colors correspond to the 13 most common food types, while the grey bars correspond to other or unknown food types. Colored boxes around each panel reflect each of the 5 original, most extensively sampled social groups (colors as in plots A and B). The white bars indicate time periods where no diet data were collected.
We began by visualizing annual and inter-annual fluctuations across all 17,265 samples over the 14-year span of the data. Consistent with prior research on primates36, 37, 38, we found population-wide, cyclical shifts in microbiome community composition across seasons and years (Fig. 2). This wet-dry seasonal cyclicity was primarily observable in the first principal component (PC1) of a principal component analysis (PCA) of clr-transformed ASV read counts (Figs. 2A, 2B; Supplementary Figs. 2–4; PC1 explains 16.5% of the variance in microbiome composition). PC1 exhibited its lowest values during the dry season, and highest values during the wet season, mirroring monthly rainfall (Fig. 2B; Supplementary Fig. 4). PC1 was also linked to annual rainfall across years, exhibiting especially low values throughout 2008 and 2009, corresponding to the worst drought in the Amboseli ecosystem in 50 years (Figs. 2A, 2B). We also observed small, but statistically significant seasonal differences in PC2 and PC3 (8.4% and 3.7% of variation in community composition; Fig. 2C; Supplementary Figs. 2–4) and in measures of alpha diversity (Fig. 2C; Supplementary Figs. 4, 5), as has been reported in other ecosystems39. Together, these seasonal changes are probably caused by seasonal shifts in plant phenology and its effects on diet (Fig. 1D), as well as the effects of rainfall and other weather variables on bacterial exposures from the environment (e.g., soil communities and sources of drinking water).
Fig. 2. Baboons show population-wide, cyclical shifts in microbiome community composition across seasons and years.
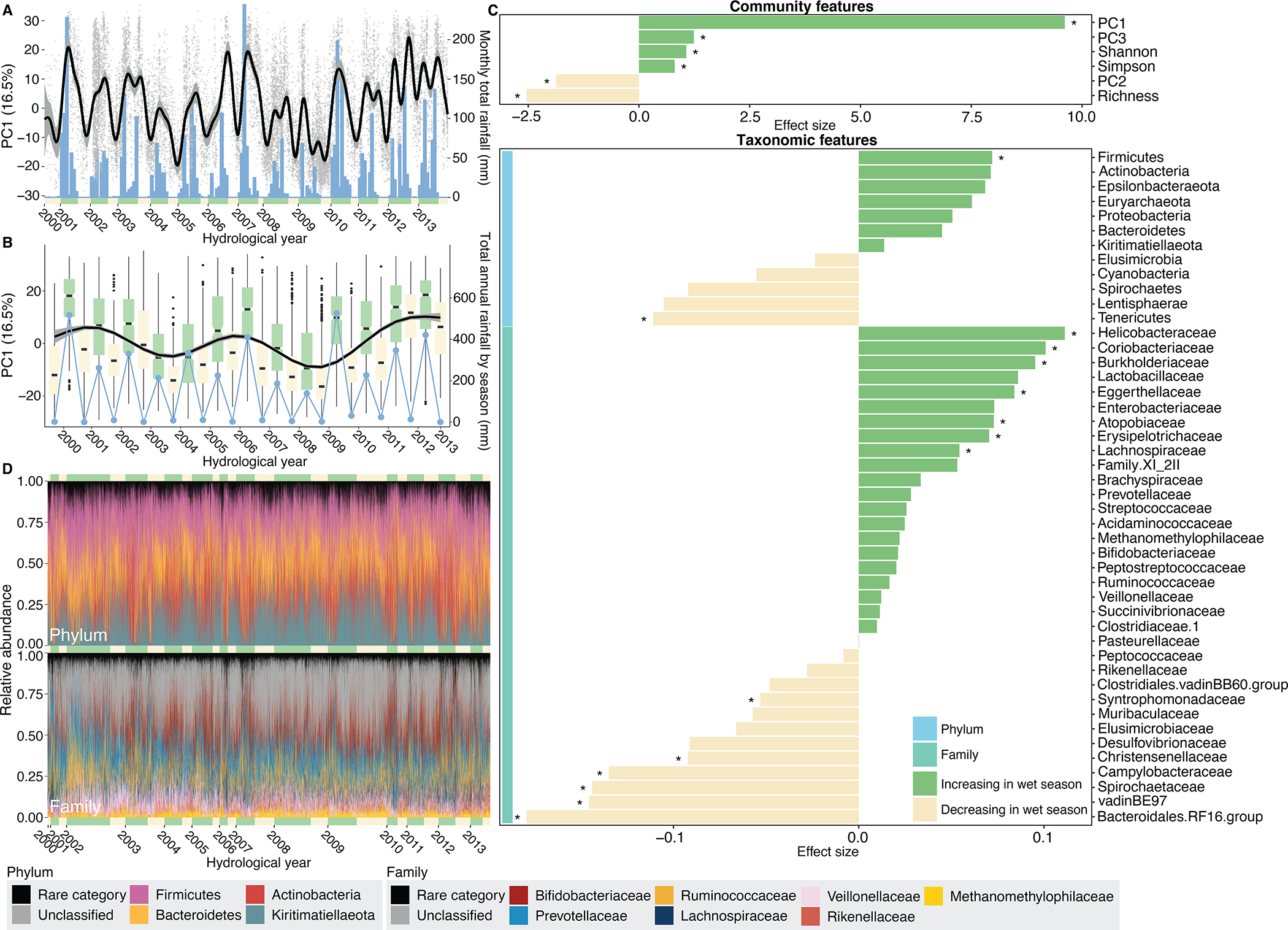
(A) Changes in microbiome PC1 mirror monthly rainfall across all 14 years. The grey points show values of PC1 for each of the 17,265 samples (y-axis) on the dates they were collected (x-axis). The thin black line and grey band are a plate regression spline and 95% simultaneous confidence interval for daily changes in microbiome PC1. Blue bars show monthly rainfall (right-hand y-axis), and the yellow and green bars along the x-axis represent dry and wet seasons, respectively, with the width reflecting the number of months within the focal year with at least 1 mm rainfall. (B) Changes in microbiome PC1 on an annual scale across all 14 years (N=17,265 samples). The box plots [box and whiskers indicate the median, 25th/75th percentile and 1.5 × interquartile range (IQR)] show the distribution of PC1 in wet (green) and dry (yellow) seasons. The thin black line and grey band are a plate regression spline and 95% simultaneous confidence interval for annual changes in microbiome PC1. Blue points show total annual rainfall (right-hand y-axis). (C) The effect of season varies across 52 features of the microbiome, including six community features (top panel) and 46 taxa (bottom panel; 12 phyla: light blue vertical bar; 34 families: turquoise vertical bar; for 341 ASVs, see Supplementary Fig. 13). Each horizontal bar shows the effect of season from linear mixed models for each feature. Asterisks indicate features that changed significantly between the wet and dry seasons (N=17,265 samples; FDR threshold=0.05). See Supplementary Fig. 6 and 7 for feature-specific smooths and Supplementary Fig. 8 and Supplementary Table 3 for results for ASVs. (D) Bar plots showing the relative abundance of ASVs colored by four most common microbial phyla (above) and the seven most common families (below) across all 17,265 samples. Green and yellow bars along the x-axes represent wet and dry seasons, with the width corresponding to the number of samples in the focal hydrological year and season. 22.9% of ASVs (78 of 341) could not be assigned to a known family (“unclassified”, shown in grey). The abundance of ASVs unclassified to family in the lower plot is ~35% because one unclassified ASV was the second most abundant ASV in the data set, with a mean abundance of 16.9% across all samples (ASV#2, phylum Kiritimatiellaeota, order WCHB1-41; Supplementary Table 2).
In terms of individual microbiome taxa, 17% of phyla (2 of 12) and 38% of families (13 of 34) exhibited significant changes in abundance between the wet and dry seasons (Fig. 2C; Supplementary Table 1; linear models with false discovery rate (FDR) threshold=0.05). These changes were significant for the phyla Firmicutes and Tenericutes (Fig. 2C, 2D; Supplementary Fig. 6), and were especially pronounced for the families Helicobacteraceae, Coriobacteriaceae, Burkholderaceae, Bacteroidales RF16 group, vadinBE97, Spirochaetaceae, and Campylobacteraceae (Fig. 2C; Supplementary Fig. 7). 28% of ASVs also exhibited significant changes in abundance across seasons (97 of 341 ASVs; linear models with FDR threshold=0.05 for n=393 models; Supplementary Fig. 8; Supplementary Tables 2 and 3). However, most ASVs, families and phyla did not change in abundance, suggesting that many taxa play consistent roles in the gut throughout the year, including Kiritimatiellaeota, Elusomicrobia, Ruminococcacaceae, Clostridiaceae 1, and Rikenellaceae (Fig. 2C; Supplementary Figs. 6 and 7; Supplementary Table 1).
Baboon gut microbial dynamics are individualized
While the microbiome metacommunity exhibited cyclical, seasonal shifts in composition, microbiome dynamics across different baboons were only weakly synchronized. Instead, consistent with prior observations of microbiome personalization1, 20, 21, 22, 23, 24, 25, patterns of temporal autocorrelation indicated that each baboon exhibited largely individualized gut microbiome compositions and dynamics (Fig. 3). In support, ASV-level Aitchison similarity was much higher for samples collected from the same baboon within a few days of each other than for samples from different baboons over the same time span, regardless of whether those animals lived in the same or a different social group (Fig 3A, 3B; Kruskal-Wallis: p<2.2×10−16 for all comparisons). Likewise, a permanova of Aitchison similarities between 4,277 samples from the 56 best-sampled baboons (Supplementary Fig. 9) revealed that host identity explained 8.6% (p<0.001) of the variation in community composition, much larger than the variation explained by sampling day or month (R2=2.5% and 1.4%), group membership (2.2%), or the first three principal components of diet (0.04% to 2.4%; Supplementary Table 4; Supplementary Fig. 10).
Fig. 3. Baboons exhibit largely idiosyncratic gut microbial compositions and dynamics.
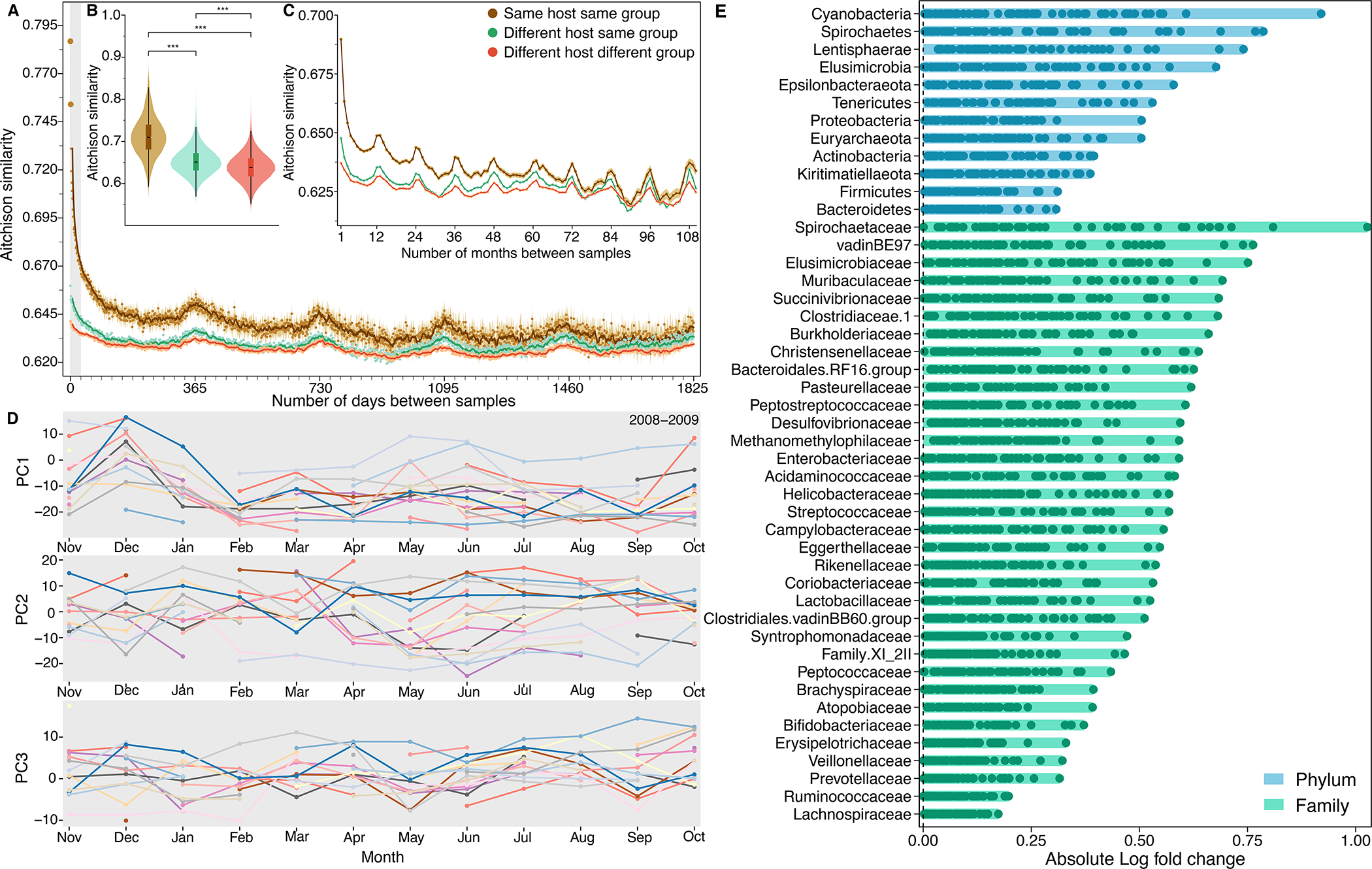
(A) Temporal autocorrelation in baboon gut microbiome communities for samples collected on the same day and up to 5-years (1,825 days) apart. Points show mean ASV-level Aitchison similarity (y-axis) between samples as a function of the number of days between sample collection (x-axis; small tick marks correspond to months). Lines depict moving averages (window size = 7 days) and their ribbons show 95% confidence intervals. The grey region on the left indicates samples collected within one month of each other. Brown points show average Aitchison similarity between samples collected from the same baboon (N=392,817 distinct sample pairs from 547 hosts with 2 or more samples); green points show similarity between samples from different baboons living in the same social group (N=16,391,761 distinct sample pairs); orange points show similarity between samples from different baboons living in different social groups (N=77,520,289 distinct sample pairs). (B) Average Aitchison similarity between pairs of samples collected within 10 days of each other. Samples from the same baboon are significantly more similar than samples collected from different baboons in the same or different social groups (Kruskal-Wallis; p = 2.22 × 10−16; N distinct sample pairs = 5,791 for within-host comparisons; 218,340 for different host same group; 779,054 for different host different group). Box and whiskers indicate the median, 25th/75th percentile and 1.5 × interquartile range (IQR). (C) Temporal autocorrelation in Aitchison similarity on monthly scales for samples collected up to 10 years apart (N distinct sample pairs=496,057 for within-host comparisons; 23,433,667 for different host same group; 114,170,919 for different host different group). (D) Microbiome dynamics for 174 samples from 17 baboons for which we had at least one sample from 10 or more months during the 2008–2009 hydrological year (Nov 2008 to Oct 2009). Panels show each individual’s mean values for microbiome PC1, PC2, and PC3; each colored line represents a distinct host. See Supplementary Fig. 14 for similar results during another densely sampled time period. Gaps indicate that the focal host did not have a sample in a given month. (E) Some taxa have more idiosyncratic abundances than others. Each horizontal bar shows a given taxon’s minimum and maximum absolute log fold change in abundance across the 56 best-sampled hosts (hosts are represented as points within the bars; see Supplementary Fig. 9 for information on the 4,277 samples from the 56 best-sampled hosts). Absolute fold changes were calculated, for a given taxon in a given host, as the taxon’s average clr-transformed abundance across all samples from that host, relative to the taxon’s grand mean in all hosts in the population. Hosts with large absolute fold changes for a given taxon therefore have abundances of that taxon that are either well above or below-average compared to its abundance in the host population at large (hosts with points close to zero exhibited taxonomic abundances typical of the population at large). For many taxa, hosts varied in their absolute log ratio values, indicating that they also deviated substantially from each other in the abundance of those taxa. Taxa (y-axis) are ordered (from top to bottom) by their highest absolute log ratio value across the 56 best-sampled hosts. Blue bars represent microbial phyla; green bars represent families. See Supplementary Fig. 15 for a longitudinal version of this analysis for the most and least idiosyncratic phyla and families.
Aitchison similarity among samples from the same baboon fell steeply for samples collected a few days to a few months apart, indicating that individualized dynamics are strongest for samples collected close in time (Figs. 3A–3C). At longer time scales (e.g., months and years), self-similarity was modest, but samples from the same baboon were significantly more similar to each other than they were to samples from different baboons, even for samples collected several years apart (Figs. 3A, 3C; Supplementary Figs. 11 and 12). Following the initial steep decline in self-similarity, community similarity rose again at 12-month intervals, both within and between hosts, reflecting synchronized, seasonal microbial dynamics across the host population. These small, 12-month peaks in similarity were visible even for samples collected more than 5 years apart, indicating that individual hosts and the population at large return to similar microbiome community states on 12-month cycles over several years (Fig. 3C). Hence, the patterns in Figs. 3A and 3C show both idiosyncratic and synchronized microbial dynamics: over short time scales, hosts are much more similar to themselves than they are to others, but on annual scales, all hosts are weakly synchronized across seasons.
The greater influence of individualized dynamics compared to synchronized dynamics can also be captured by comparing microbiome dynamics for deeply sampled hosts sharing the same habitat at the same time (Fig. 3D; Supplementary Fig. 13). For instance, during the 2008–2009 hydrological year, we collected nearly one sample per month from 17 baboons. When we aligned these time series, we observed little convergence to similar values within any given month and little evidence of shared changes in the top three principal components of ASV-level microbiome composition over time (Fig. 3D). Consequently, the microbiome of each baboon took a different path over the ordination space over the same 1-year span (Supplementary Fig. 13; see Supplementary Fig. 14 for similar results during 2007–2008).
Microbiome taxa varied in their contributions to individualized gut microbiome compositions (Fig. 3E; Supplementary Fig. 15). For the 56 best-sampled hosts (Supplementary Fig. 9), several phyla and families exhibited substantial variation in host mean clr-transformed abundance (across repeated samples for that host) compared to their mean clr-transformed abundance across all hosts. These taxa included the phyla Cyanobacteria, Spirochaetes, Lentisphaerae, and Elusimicrobia, and the families Spirochaetaceae, vadinBE97, Elusimicrobaceae, and Muribaculaceae (Fig. 3E; Supplementary Fig. 15). These highly variable taxa exhibited below-average abundances compared to less variable taxa (Supplementary Fig. 16). Hence, idiosyncratic dynamics may be more often linked to uncommon than common taxa, perhaps because uncommon taxa have greater functional variability across hosts.
To test whether individualized gut microbial dynamics could be explained by microbial dispersal limitation between hosts, we used the Sloan Neutral Community Model for Prokaryotes to estimate metacommunity-wide migration probabilities, m, for ASVs in each season and hydrological year40, 41. m provides a measure of dispersal limitation because it represents the probability that “vacancies” in a local community (a host’s microbiome) will be replaced by dispersal from the microbiome metacommunity (other hosts), as opposed to reproduction within a focal host’s community40, 41. We found little evidence that dispersal limitation contributed to idiosyncratic compositions and dynamics. The probability that a given ASV lost from a host’s microbiota would be replaced by an ASV from another host in the population was nearly 40% (average host population-wide m across seasons and years=0.373; range=0.332–0.416; Supplementary Fig. 17). These migration probabilities are generally lower than those Sieber et al.9 found for marine sponges sampled from the same location (m across sponge species: min=0.36; median=0.78; max=0.86), but much higher than for mice and nematodes, both in natural and laboratory populations (mice: mwild=0.11 and mlab=0.18; nematode: mwild=0.03 and mlab=0.01). Hence, dispersal limitation is low for baboon microbiota in Amboseli.
Interestingly, when we re-defined the microbiome metacommunity to be the host’s social group, instead of the whole host population, migration probabilities were similar (average m across groups=0.355; range=0.347 to 0.365; colored points on Supplementary Fig. 17). Hence, despite several studies that find microbiome compositional differences between hosts living in different social groups, including in the Amboseli baboons1, 42, 43, 44, 45, 46, social groups are not major barriers to microbial colonization between baboons, perhaps because of their overlapping home ranges, similar diets, and network connections via male dispersal (Fig. 1).
Shared environments lead to modest synchrony across hosts
To quantify the relative magnitude of idiosyncratic versus synchronized dynamics across the host population, social groups, and hosts, and to test whether synchrony varies for a set of common microbial taxa, we used generalized additive models (GAMs) to capture the nonlinear, longitudinal changes in 52 microbiome features (3 PCs of ASV-level community variation, 3 metrics of ASV-level alpha diversity, and clr-transformed relative abundances of 12 phyla and 34 families). For each feature, we ran three GAMs to measure the deviance explained in gut microbial dynamics by successive sets of parameters, reflecting the nested nature of our variables (Fig. 4A; x-axis of Fig. 4C; Supplementary Table 5). The population-level model (model P) captured factors experienced by the whole host population, including average rainfall and maximum daily temperature in the 30 days before sample collection and random effect splines to capture monthly and annual cyclicity in microbiome features (e.g., Figs. 2A and B; see Supplementary Fig. 18 for effects of time of day, which was not included). The group-level model (model P+G) included all the predictor variables in model P, and added a random effect spline for each social group, and variables to capture temporal changes in each group’s diet, home range use, and group size (Figs. 4A, 4C). The host-level model (model P+G+H) included all of the predictor variables in model P+G, and added a random effect spline for each host, and variables for host traits, including sex, age, and social dominance rank (Figs. 4A, 4C).
Fig. 4. Multilevel modeling identifies idiosyncratic microbial dynamics.
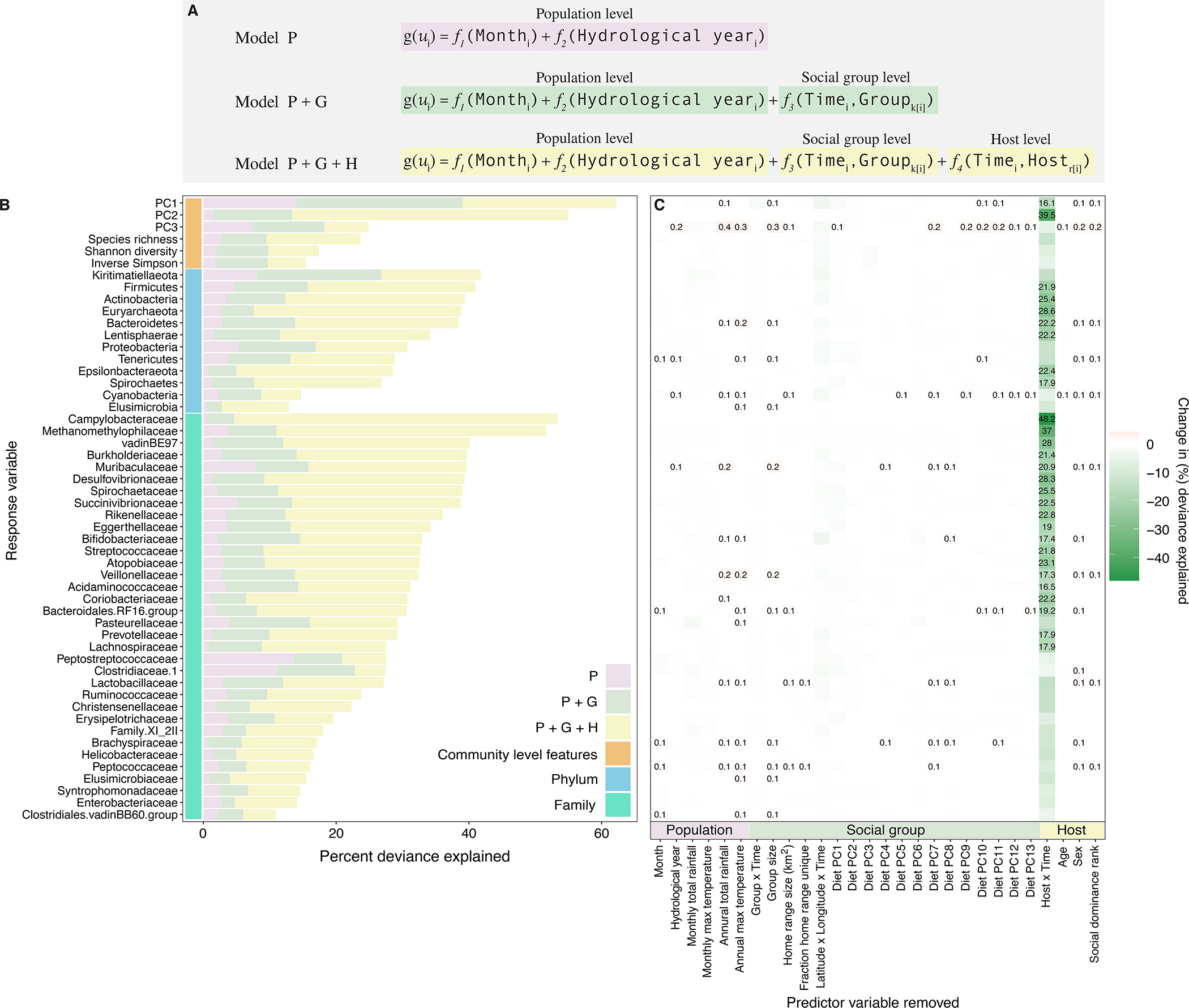
(A) We fit three hierarchical GAMs to 52 microbiome features measured in 4,277 samples from the 56 best-sampled baboons, all of whom lived in the 5 social groups sampled the longest (between 2002 and 2010; median=72.5 samples per host; minimum=48 samples; maximum=164 samples; Supplementary Fig. 9). Each model contained successive sets of predictor variables reflecting population-level factors (pink), group-level factors (green) and host-level factors (yellow). The factors at each level are listed at the bottom of panel C and defined in Supplementary Table 5). Panel (B) shows, for each microbiome feature (i.e., response variable), the deviance explained by model P and the successive sets of predictor variables added in model P+G and model P+G+H, respectively (Supplementary Table 6; percent deviance is a measure of goodness-of-fit for nonlinear models and is analogous to the unadjusted R2 for linear models). Panel (C) shows the loss in deviance explained for model P+G+H as we successively removed each predictor variable in turn from model P+G+H, keeping the model otherwise intact (Supplementary Table 7). Losses in deviance are shown in green, and we only provide numeric values for losses in deviance > 15%. Gains in deviance are shown in red; we only show numeric values for gains > 0.1%.
Consistent with our autocorrelation analyses (Fig. 3), comparing the deviance explained for each microbiome feature across the three models revealed stronger idiosyncratic than synchronized dynamics for most microbiome features (Figs. 4B, 4C). Host-specific factors, especially host identity, explained, on average, 10 times the deviance in the longitudinal dynamics of microbiome features, compared to factors shared across all hosts and more than three times the deviance by factors shared with group members. Specifically, model P only explained on average 3.3% (range=0.46%–14.0%) of the deviance across all 52 microbiome features (pink bars in Fig 4B; Supplementary Table 6), compared to 8.1% on average after adding group-level factors to the population-level model (increase from model P to model P+G; range=2%–25%; green bars in Fig. 4B; Supplementary Table 6), and 30.1% of the deviance after including host-level dynamics (model P+G+H; range=11.0%–62.2%) for the same set of features (yellow bars in Fig. 4B; Supplementary Table 6). Importantly, the added deviance for model P+G+H compared to model P or model P+G was not caused by including more parameters. Randomizing host identity and host-level traits across samples, while keeping each sample’s annual, seasonal, and group identity intact, led to a substantial drop in deviance explained compared to the real data (Supplementary Fig. 19).
44 of the 52 microbiome features exhibited greater gains in deviance by adding host-level factors to model P+G, compared to adding group-level factors to model P. Of these 44, 22 features gained more than 20% deviance explained between model P+G and model P+G+H (Fig. 4B; Supplementary Table 6). Three of the most common phyla, Actinobacteria, Bacteroidetes, and Firmicutes all gained >20% deviance explained between model P+G and model P+G+H (Actinobacteria=27.1%; Bacteroidetes=24.6%, and Firmicutes=25.2%; Fig. 4B; Supplementary Table 6). The most idiosyncratic features (i.e., those that gained >30% deviance explained by adding host-level factors), were microbiome PC2, the phylum Euryarchaeota, and the families Campylobacteraceae, Methanomethylophilaceae and Desulfovibrionaceae (Fig. 4B; Supplementary Table 6). Even the most synchronous feature, microbiome PC1 (14% deviance explained by the P model), gained 23.2% deviance explained when adding host-level factors to the P+G model.
Removing covariates from model P+G+H one at a time, while keeping all other covariates intact, revealed that host identity explained nearly all of the deviance in our models (Fig. 4C; Supplementary Table 6; average loss in deviance explained by removing host identity=17.3% versus 0.2% deviance for all other factors). Beyond host identity, the next most important factor was the geographic area where the group traveled in the 30 days prior to sample collection, which explained 1% of the deviance, on average, across all 52 features (Supplementary Fig. 20; Supplementary Table 6). All other individual predictor variables had only minor effects on deviance explained (Supplementary Fig. 20; Supplementary Table 6).
To investigate whether some of the idiosyncrasy we observed was due to host genetic effects, we tested for a relationship between the deviance explained by each GAM and the narrow-sense heritability (h2) of microbiome taxon abundance as estimated by Grieneisen et al.32. We found that higher levels of deviance explained by model P+G+H were predicted by higher taxon heritability (Pearson correlation: R=0.37, p=0.016; Fig. 5A). In contrast, we found no such effect at the population or group level, as expected since genotype is a property of individual hosts, not groups or populations (model P+G: R=0.047, p=0.76; model P: R=0.0085, p=0.96; Fig. 5B). We explained substantially more deviance by adding the host level to model P+G for microbiome taxa with h2 > 0.05 than we did for taxa with very low h2 values (model P+G+H: min=16.0, median=32.6, max=53.4 vs model P+G: min=4.6, median=11.1, max=26.8; Fig. 5B). Hence, some idiosyncrasy in gut microbiome dynamics is likely a consequence of differences in host genotype. However, because h2 estimates cannot be mapped directly onto estimates of deviance explained in GAMs, direct estimates of genetic versus environmental effects on host dynamics remain an important topic for future work.
Fig. 5. Microbiome taxon heritability is associated with idiosyncratic dynamics.
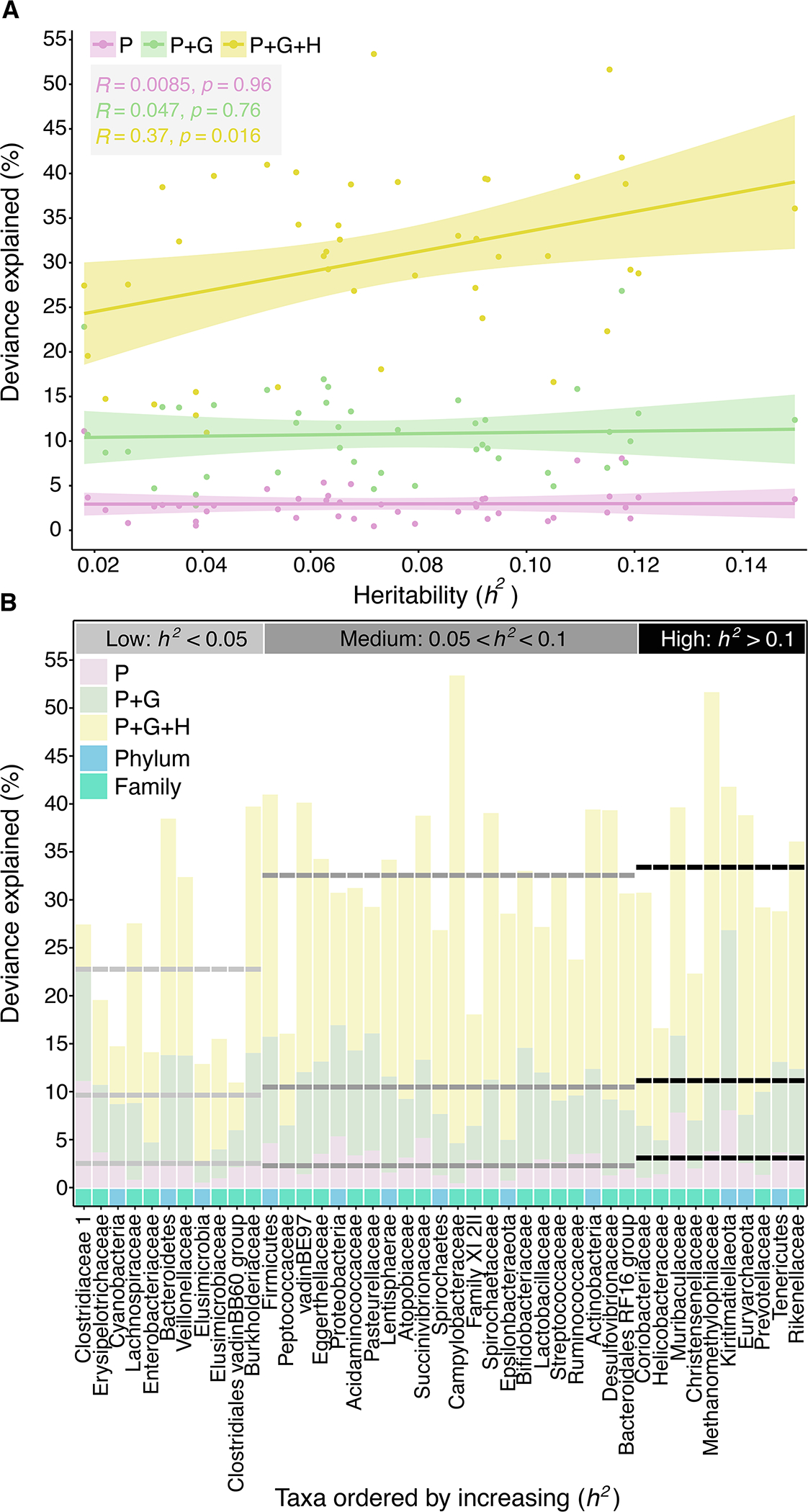
(A) Deviance explained (y-axis) by the phylum and family level GAMs (from Fig. 4) plotted against the focal taxon’s heritability estimate (h2; x-axis). Pink, green and yellow denote model P, model P+G and model P+G+H, respectively. Each regression line is plotted with its 95% confidence interval. (B) Deviance explained (y-axis) across the model hierarchy (pink: model P; green: model P+G; yellow: model P+G+H) for each taxonomic feature (i.e., at the phylum and family level; x-axis). The x-axis is ordered by increasing heritability with light blue and turquoise squares representing phyla and families, respectively. Horizontal dashed lines show the average deviance explained per model for taxa with low heritability estimates (h2 <0.05; light gray); medium heritability estimates (0.05< h2< 0.1; dark gray); and high heritability estimates (h2 ≥0.1; black).
The strongest synchrony is among social group members
Previous research finds that hosts in the same social group have more similar gut microbiota than hosts in different groups1, 42, 43, 44, 47. Likewise, in our current data set, several taxa exhibited abundances that were, on average, higher or lower within a given group compared to their average abundance in the host population at large (Supplementary Figs. 21 ans 22). Hence, we tested whether shared social group membership is linked to greater microbiome synchrony than hosts in different groups. In support, the patterns of temporal autocorrelation in Fig. 3A showed that hosts in the same group have more similar microbiomes than those in different groups, especially for samples collected within 10 days of each other (Fig. 3B; Kruskal-Wallis: p <2.2×10−16). Likewise, samples from the same group occupy similar ordination space over time (Supplementary video 2). While small, these group-level similarities were detectable, even for samples collected more than 2 years apart (Fig. 3C; Supplementary Fig. 11A). The addition of group-level splines to our GAMs led to gains in deviance that explained more than 10% for 15 of 52 microbiome features, including all three microbiome PCs, five phyla, and seven families (Figs. 4B, 4C; Supplementary Table 6).
Gut microbial congruence among group members could also be linked to shared behaviors and environments: baboons in the same group eat the same foods at the same time, travel as a unit across the landscape, and may be grooming partners that are frequently in physical contact32, 48, 49, 50, 51, 52 (Figs. 1B, 1D). Indeed, after host identity, the next most important variable in model P+G+H was the group’s home range in the 30 days before sample collection (Supplementary Fig. 20; Supplementary Table 7). Despite previous evidence that grooming partners have similar microbiota42, we did not find evidence for this pattern in our data (Supplementary Fig. 23). Samples collected from individuals with strong grooming bonds were not more similar than samples from animals with weak or no grooming relationships (Supplementary Fig. 24). However, the lack of a grooming effect in this data set should be interpreted with caution. Our prior research on this topic42 characterized microbial communities using shotgun metagenomic sequencing from >90% of social network members, all within 30 days of each other. Such data provide higher taxonomic resolution and more accurate estimates of abundance than 16S data, and may more accurately capture transmission between hosts.
Conclusions
We find that gut microbial dynamics are both weakly synchronized across hosts and strongly idiosyncratic to individual hosts. Like members of a poorly coordinated microbial orchestra, microbial communities in different baboons are only weakly “in concert” across the host population. Instead, gut microbial dynamics are idiosyncratic at the level of individual hosts, and each baboon “player” approaches the gut microbial “song” differently. Our results contribute to mounting evidence that forces proposed to synchronize gut microbial metacommunities—shared environments, diets, and between-host microbial dispersal—can create modest synchrony among hosts, especially for hosts living in the same social unit. However, these forces are typically not strong enough to overwhelm powerful and well-known drivers of microbiome personalization, including host genetic effects, individual-level priority effects, horizontal gene transfer, and functional redundancy16, 17, 18, 19. Interestingly, these idiosyncratic dynamics were strong even for microbial phyla and families, whose dynamics reflect multiple microbial functions and interactions that potentially buffer them against large fluctuations in abundance. We expect that the personalized dynamics we observed will be even stronger for finer taxonomic levels, especially bacterial species or strains that exhibit a high degree of functional variability across hosts.
Understanding if hosts in the same social group or population exhibit shared microbiome dynamics may be useful to researchers interested in predicting individual microbiome changes, linking microbiome dynamics to health outcomes, and designing broadly effective microbiome interventions. These objectives have already been difficult to achieve, in part because of gut microbial personalization in humans and animals. For instance, predictive models of gut microbiome dynamics from one person fail when they are applied to other people27. Our results support the idea that microbiome predictions and interventions focused on specific taxa will require personalized approaches. Even then, “universal” microbiome therapies that work the same way for all hosts may be unattainable. Instead, interventions will likely work best when they are designed for host groups or populations that have shared compositions and dynamics. Functional redundancy and horizontal gene flow may also mean that functions will be more predictable than taxa, and prediction and intervention efforts that focus on microbiome functional traits (e.g., metabolite levels; the presence of specific pathways) will likely be less affected by gut microbiome personalization. Together, our results provide novel insights about the extent and ecological causes of microbiome personalization, and they indicate that personalized compositions and dynamics are not an artifact of modern human lifestyles.
Methods
All data collection procedures adhere to the regulations of the Institutional Animal Care and Use Committees of Duke and Notre Dame universities, and to the laws of Kenya. A complete description of our methods is in the Supplementary Materials and Methods, Sections 1A–1C.
Study subjects.
Our subjects were individual wild baboons studied by the Amboseli Baboon Research Project (ABRP) in Kenya33. Baboons are terrestrial primates that live in stable social groups, typically with 20 to 130 members. The 600 baboons in our data set lived in 12 social groups between April 2000 and September 2013 (5 original groups and 7 groups that were fission/fusion products from these original groups; Fig. 1A). ABRP collects detailed longitudinal data on rainfall and temperature; social group membership, ranging patterns and diet; and host traits such as age, sex, social relationships, and dominance rank (see Supplementary Methods Section A). The Amboseli ecosystem is a semi-arid savanna with a 5-month long dry season spanning June to October, during which very little rain falls. The remaining 7 months (November to May) constitute the wet season, which has highly variable rainfall (mean annual rainfall between 2000 and 2013 was 319 mm; range=140–559 mm).
Sample collection.
A large majority of the microbiota data we use here were published in Grieneisen et al.32, but we include data from 1,031 additional samples that were generated at the same time using the same methods (they were not included in the heritability analysis of Grieneisen et al.32 because we lack pedigree information for these hosts). The addition of these 1,031 samples led to a total of 17,265 samples in our study. These samples were collected from baboons who ranged in age from 7.4 months to 27.7 years, spanning these animals’ natural lifespans (Supplementary Fig. 1A). Each baboon was sampled a median of 19 times, and 124 baboons were sampled at least 50 times (Supplementary Fig. 1B). On average, these samples spanned 4.3 years of a baboon’s life (range=4 days to 13.2 years; Supplementary Fig. 1C), with a median of 35 days between consecutive samples (Supplementary Fig. 1D).
DNA extraction and sequencing.
DNA was extracted from each sample using MoBio and QIAGEN PowerSoil kits and subjected to 16S rRNA sequencing on the Illumina HiSeq 2500 platform (896,911,162 total sequencing reads; mean=51,913.6 reads per sample; range=1021–477,241; Supplementary Fig. 1E). We used DADA253 for sequence quality processing following the default protocol for large data sets. To allow us to compare the dynamics of individual taxa in different hosts, we filtered to taxa found in at least 20% of samples, resulting in 341 ASVs (mean=162 ASVs per sample; range=19–311 ASVs; Supplementary Fig. 1F; Supplementary Table 2). This filtering captured 92% of the reads and many of the same compositional properties of the data set when filtered to 5% prevalence (Supplementary Fig. 25). DNA concentration and ASV diversity were not predicted by time since sample collection (Supplementary Figs. 1G, 1H). As is typical for wild microbiota, 22.9% of the 341 ASVs could not be assigned to a known family (78 of 341), and 5.5% of ASVs could not be assigned to a known phylum (19 of 341; Supplementary Table 2). To address the compositional nature of our data, read counts were centered log-ratio (clr) transformed independently in each sample (including independent transforms for samples from the same individual), prior to all analyses54, 55.
Statistical analyses.
To test whether shared environmental conditions and host traits lead to similar gut microbial compositions and synchronized dynamics across the microbiome metacommunity, we first characterized patterns of temporal autocorrelation in ASV-level Aitchison similarity within and between hosts over time. Our expectation was that, if hosts or social groups exhibit idiosyncratic composition and dynamics, then samples collected close in time from the same baboon, or from baboons in the same group, should be more similar than they are to samples collected from different baboons living in different groups. Alternatively, if gut microbial dynamics are strongly synchronized, then samples collected close in time across the metacommunity should be compositionally similar, and samples collected from the same host should not be substantially more similar than samples from different baboons. These analyses were run in R (v 4.0.256) using custom-written functions (code and analyzed data are available on GitHub/OSF; see Data Statement).
To test whether dispersal limitation could explain microbiome idiosyncrasy, we estimated metacommunity-wide microbial migration probabilities in each season and year using the Sloan Neutral Community Model for Prokaryotes40, 41. This model assumes that each local community, defined as the ASV-level microbial composition of a single host in a given season-year, is the outcome of stochastic population dynamics and microbial immigration from other hosts in the microbiome metacommunity (i.e., other local communities). Briefly, local communities have a constant size N, and individual microbes within each local community die at a constant rate. These deaths create vacancies that can be occupied, either by individuals immigrating from the microbiome metacommunity (with probability m), or by daughter cells from any taxon within the local community (i.e., from reproduction within the same host, with probability 1-m). Taxa that are common in the metacommunity have a higher chance of occupying vacancies than rare taxa. Without immigration from the microbiome metacommunity, ecological drift leads each host’s microbial diversity to reduce to a single taxon. Thus, the migration probability, m, represents the metacommunity-wide probability that any taxon, randomly lost from a given host/local community, will be replaced by dispersal from the microbiome metacommunity, as opposed to reproduction within hosts40, 41. Following Burns et al.57, m can be interpreted as a measure of dispersal limitation, such that low migration probabilities signify high dispersal limitation. We estimated season and hydrological year-specific values for m by defining the microbiome metacommunity as either the hosts’ social group or the whole host population. We fit neutral models using nonlinear least-squares regression as implemented in the R package tyRa58.
To quantify the relative magnitude of idiosyncratic versus synchronized dynamics for community metrics and common families and phyla, we used generalized additive models (GAMs) to capture the non-linear, longitudinal dynamics of 52 features, including the first three principal components of ASV-level composition, three indices of alpha diversity (ASV richness, the exponent of ASV-level Shannon’s H, and the inverse Simpson index for ASVs, as computed by the function reyni from the R package vegan59), and the clr-transformed abundances of 12 phyla and 34 families present in >20% of samples. We analyzed phyla and families (as opposed to genera or ASVs) because phyla and families are highly prevalent across samples (mean prevalence=85.6% for the 12 phyla and 73.7% for the 34 families), offering excellent power to compare their dynamics between different baboons. However, phyla and families might exhibit stronger synchrony than lower-level taxa because, compared to species or strains, the dynamics of families and phyla reflect multiple microbial processes and interactions, which are expected to buffer them against large fluctuations in abundance. Further, the processes and interactions that a given phylum or family collectively encompasses may be more consistent across hosts than those carried out by an individual species or strain (although this consistency will vary depending on the phylum, family, or process in question18, 60).
Our GAMs allowed us to calculate the percent deviance in each feature’s dynamics attributable to factors that could contribute to synchronized dynamics at different scales (percent deviance is a measure of goodness-of-fit for nonlinear models and is analogous to the unadjusted R2 for linear models). We considered deviance explained by factors at three scales: those experienced by the whole host population (e.g., rainfall and temperature), those differentiated by social groups (e.g., group identity, group home range location, and diet), and those differentiated at the level of individual hosts (e.g., host identity, sex, age, and social dominance rank; see below for complete model structures). If microbiome community dynamics are largely idiosyncratic, then population- and group-level factors will not explain considerable deviance in microbiota change over time, and instead, a large fraction of the deviance will be attributable to host identity, controlling for shared environments, behaviors, and traits. Alternatively, if shared environments and behaviors across the population and within social groups synchronize gut microbiota, then population- and group-level factors should explain substantial deviance in community dynamics. To ensure sufficiently dense sampling for identifying host- and group-level dynamics, all three GAMs were run on a subset of the full data set, consisting of 4,277 16S rRNA gene sequencing profiles from the 56 best-sampled baboons living in the 5 social groups sampled the longest (between 2002 and 2010; median=72.5 samples per host; minimum=48 samples; maximum=164 samples; Supplementary Fig. 9). GAMs were fit using the R package mgcv61, 62, 63.
To test whether host genetic effects contribute to gut microbial idiosyncrasy, we performed a post hoc analysis of the relationship between the deviance explained in the GAMs for each microbial taxon and the heritability of that taxon’s relative abundance32. If host effects on microbiome dynamics are in part explained by host genotype, we predicted that taxon heritability should be positively correlated with deviance explained at the host level (i.e., model P+G+H), but not at the group or population level (i.e., model P and model P+G).
Supplementary Material
Acknowledgments:
We thank Jeanne Altmann for her essential role in stewarding the Amboseli Baboon Project, and in collecting and maintaining the fecal samples used in this manuscript. This work was supported by the National Science Foundation and the National Institutes of Health, especially NSF Rules of Life Award DEB1840223 (EAA, JAG), and the National Institute on Aging R21AG055777 (EAA, RB) and NIH R01AG053330 (EAA), NIH R01AG071684 (EAA), and NIHR35 GM128716 (RB), the Duke University Population Research Institute P2C-HD065563 (pilot to JT), the University of Notre Dame’s Eck Institute for Global Health (EAA), and the Notre Dame Environmental Change Initiative (EAA). Since 2000, long-term data collection in Amboseli has been supported by NSF and NIH, including IOS1456832 (SCA), IOS1053461 (EAA), DEB1405308 (JT), IOS0919200 (SCA), DEB0846286 (SCA), DEB0846532 (SCA), IBN0322781 (SCA), IBN0322613 (SCA), BCS0323553 (SCA), BCS0323596 (SCA), P01AG031719 (SCA), R21AG049936 (JT, SCA), R03AG045459 (JT, SCA), R01AG034513 (SCA), R01HD088558 (JT), and P30AG024361 (SCA). We also especially thank the members of the Maasai pastoralist communities in the Amboseli-Longido areas. We thank the Kenya Wildlife Service, Kenya’s Wildlife Research & Training Institute, the National Council for Science, Technology, and Innovation, and the National Environment Management Authority for permission to conduct research and collect biological samples in Kenya. We also thank the University of Nairobi, Institute of Primate Research, National Museums of Kenya, the Enduimet Wildlife Management Area, Ker & Downey Safaris, Air Kenya, and Safarilink for their cooperation and assistance in the field. We thank Karl Pinc for managing and designing the database. We also thank Tawni Voyles, Anne Dumaine, Yingying Zhang, Meghana Rao, Tauras Vilgalys, Amanda Lea, Noah Snyder-Mackler, Paul Durst, Jay Zussman, Garrett Chavez, and Reena Debray for contributing to fecal sample processing. Complete acknowledgments for the ABRP can be found online at https://amboselibaboons.nd.edu/acknowledgements/.
Footnotes
Competing interests: The authors declare no competing interests.
Code availability:
Analyzed data and code is available on the JRB’s Open Science Framework / GitHub repository at https://doi.org/10.17605/OSF.IO/ERDXA
Data availability:
16S rRNA gene sequences are deposited on EBI-ENA (project ERP119849) and Qiita [study 1294964]. Note that our research permission from Kenya Wildlife Service prohibits third party sharing of the biological samples themselves.
References
- 1.Kolodny O, et al. Coordinated change at the colony level in fruit bat fur microbiomes through time. Nature Ecology & Evolution 3, 116–124 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Schlomann BH, Parthasarathy R. Timescales of gut microbiome dynamics. Curr Opin Microbiol 50, 56–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koch H, Schmid-Hempel P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proceedings of the National Academy of Sciences 108, 19288–19292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finnicum CT, et al. Cohabitation is associated with a greater resemblance in gut microbiota which can impact cardiometabolic and inflammatory risk. BMC Microbiology 19, 230 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bashan A, et al. Universality of human microbial dynamics. Nature 534, 259–262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science 336, 1255–1262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miller ET, Svanback R, Bohannan BJ. Microbiomes as metacommunities: understanding host-associated microbes through metacommunity ecology. Trends in Ecology & Evolution, (2018). [DOI] [PubMed] [Google Scholar]
- 8.Bjork J, Díez-Vives C, Astudillo-García C, Archie EA, Montoya JM. Vertical transmission of sponge microbiota is inconsistent and unfaithful. Nature Ecology & Evolution 3, 1172–1183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieber M, et al. Neutrality in the Metaorganism. PLoS Biol 17, e3000298 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tredennick AT, dr Mazancourt C, Loreau M, Adler PB. Environmental responses, not species interactions, determine synchrony of dominant species in semiarid grasslands. Ecology 98, 971–981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loreau M, de Mazancourt C. Species synchrony and its drivers: neutral and nonneutral community dynamics in fluctuating environments. American Naturalist 172, E48–66 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Isbell FI, Polley HW, Wilsey BJ. Biodiversity, productivity and the temporal stability of productivity: patterns and processes. Ecol Lett 12, 443–451 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Hector A, et al. General stabilizing effects of plant diversity on grassland productivity through population asynchrony and overyielding. Ecology 91, 2213–2220 (2010). [DOI] [PubMed] [Google Scholar]
- 14.de Mazancourt C, et al. Predicting ecosystem stability from community composition and biodiversity. Ecology Letters 16, 617–625 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Gross K, et al. Species richness and the temporal stability of biomass production: a new analysis of recent biodiversity experiments. Am Nat 183, 1–12 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Louca S, et al. Function and functional redundancy in microbial systems. Nat Ecol Evol 2, 936–943 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Rainey PB, Quistad SD. Toward a dynamical understanding of microbial communities. Philos Trans R Soc Lond B Biol Sci 375, 20190248 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martiny JB, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: A phylogenetic perspective. Science 350, aac9323 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Debray R, Herbert RA, Jaffe AL, Crits-Christoph A, Power ME, Koskella B. Priority effects in microbiome assembly. Nat Rev Microbiol 20, 109–121 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Risely A, Wilhelm K, Clutton-Brock T, Manser MB, Sommer S. Diurnal oscillations in gut bacterial load and composition eclipse seasonal and lifetime dynamics in wild meerkats. Nat Commun 12, 6017 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Franzosa EA, et al. Identifying personal microbiomes using metagenomic codes. Proceedings of the National Academy of Sciences 112, E2930–E2938 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faith JJ, et al. The long-term stability of the human gut microbiota. Science 341, 1237439 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bik EM, et al. Marine mammals harbor unique microbiotas shaped by and yet distinct from the sea. Nat Commun 7, 10516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caporaso JG, et al. Moving pictures of the human microbiome. Genome Biology 12, R50 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Flores GE, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biology 15, 531 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson AJ, et al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans. Cell Host & Microbe 25, 789–802 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Smits SA, Marcobal A, Higginbottom S, Sonnenburg JL, Kashyap PC. Individualized Responses of Gut Microbiota to Dietary Intervention Modeled in Humanized Mice. mSystems, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothschild D, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 555, 210–215 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Falony G, et al. Population-level analysis of gut microbiome variation. Science 352, 560–564 (2016). [DOI] [PubMed] [Google Scholar]
- 31.Zhernakova A, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grieneisen L, et al. Gut microbiome heritability is nearly universal but environmentally contingent. Science 373, 181–186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alberts SC, Altmann J. The Amboseli Baboon Research Project: Themes of continuity and change. In: Long-term field studies of primates (eds Kappeler P, Watts DP). Springer Verlag; (2012). [Google Scholar]
- 34.Ren T, Grieneisen L, Alberts SC, Archie EA, Wu M. Development, diet, and dynamism: longitudinal and cross-sectional predictors of gut microbial communities in wild baboons. Environmental Microbiology 18, 1312–1325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grieneisen L, et al. Genes, geology, and germs: gut microbiota across a primate hybrid zone are explained by site soil properties, not host species. Proceedings of the Royal Society 286, 20190431 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hicks AL, et al. Gut microbiomes of wild great apes fluctuate seasonally in response to diet. Nat Commun 9, 1786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orkin JD, Campos FA, Myers MS, Cheves Hernandez SE, Guadamuz A, Melin AD. Seasonality of the gut microbiota of free-ranging white-faced capuchins in a tropical dry forest. ISME J 13, 183–196 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baniel A, et al. Seasonal shifts in the gut microbiome indicate plastic responses to diet in wild geladas. Microbiome 9, 26 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellard JP, Audoye P, Loreau M. Seasonal patterns in species diversity across biomes. Ecology 100, e02627 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environmental Microbiology 8, 732–740 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Sloan WT, Woodcock S, Lunn M, Head IM, Curtis TP. Modeling Taxa-Abundance Distributions in Microbial Communities using Environmental Sequence Data. Microbial Ecology 53, 443–455 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Tung J, et al. Social networks predict gut microbiome composition in wild baboons. eLife 4, e05224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moeller AH, Foerster S, Wilson ML, Pusey AE, Hahn BH, Ochman H. Social behavior shapes the chimpanzee pan-microbiome. Science Advances 2, e1500997 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lax S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amato KR, et al. Patterns in gut microbiota similarity associated with degree of sociality among sex classes of a neotropical primate. Microbial Ecology 74, 250–258 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Amato KR, et al. The role of gut microbes in satisfying the nutritional demands of adult and juvenile wild, black howler monkeys (Alouatta pigra). American Journal of Physical Anthropology 155, 652–664 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Perofsky AC, Leriw RJ, Abondano LA, Di Fiore A, Meyers LA. Hierarchical social networks shape gut microbial composition in wild Verreaux’s sifaka. Proceedings of the Royal Society 284, 20172274 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Silk JB. Activities and feeding behavior of free-ranging pregnant baboons. International Journal of Primatology 8, 593–613 (1987). [Google Scholar]
- 49.Altmann SA. Foraging for Survival: Yearling Baboons in Africa. University of Chicago Press; (1998). [Google Scholar]
- 50.Bronikowski AM, Altmann J. Foraging in a variable environment: Weather patterns and the behavioral ecology of baboons. Behavioral Ecology and Sociobiology 39, 11–25 (1996). [Google Scholar]
- 51.Muruthi P, Altmann J, Altmann S. Resource base, parity and reproductive condition affect females’ feeding time and nutrient intake within and between groups of a baboon population. Oecologia 87, 467–472 (1991). [DOI] [PubMed] [Google Scholar]
- 52.Shopland JM. Food quality, spatial deployment, and the intensity of feeding interference in yellow baboons (Papio cynocephalus). Behav Ecol Sociobiol 21, 149–156 (1987). [Google Scholar]
- 53.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morton JT, et al. Establishing microbial composition measurement standards with reference frames. Nat Commun 10, 2719 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front Microbiol 8, 2224 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.R Core Team R. R: A language and environment for statistical computing. (available at http://www.R-project.org/). In: R Foundation for Statistical Computing; ) (2020). [Google Scholar]
- 57.Burns AR, et al. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME Journal 10, 655–664 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sprockett D tyRa: Build Models for Microbiome Data. GitHub repository; (available at https://danielsprockett.github.io/tyRa/articles/tyRa.html). (2020). [Google Scholar]
- 59.Oksanen J, et al. vegan: Community Ecology Package. R package version 2.5–7. (2020). [Google Scholar]
- 60.Vieira-Silva S, et al. Species-function relationships shape ecological properties of the human gut microbiome. Nature Microbiology 1, 16088 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Wood SN. Stable and Efficient Multiple Smoothing Parameter Estimation for Generalized Additive Models. Journal of the American Statistical Association 99, 673–686 (2004). [Google Scholar]
- 62.Wood SN. Fast stable restricted maximum likelihood and marginal likelihood estimation of semiparametric generalized linear models. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 73, 3–36 (2011). [Google Scholar]
- 63.Wood SN. Generalized Additive Models: An Introduction with R, Second Edition CRC Press; (2017). [Google Scholar]
- 64.Gonzalez A, et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat Methods 15, 796–798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
16S rRNA gene sequences are deposited on EBI-ENA (project ERP119849) and Qiita [study 1294964]. Note that our research permission from Kenya Wildlife Service prohibits third party sharing of the biological samples themselves.