

Summary
The voltage-gated ion channel activity depends on both activation (transition from the resting state to the open state) and inactivation. Inactivation is a self-restraint mechanism to limit ion conduction and is as crucial to membrane excitability as activation. Inactivation can occur when the channel is open or closed. While open-state inactivation is well understood, the molecular basis of closed-state inactivation has remained elusive. We report cryo-EM structures of human KV4.2 channel complexes in inactivated, open, and closed states. Closed-state inactivation of KV4 involves an unprecedented symmetry breakdown for pore closure by only two of the four S4-S5 linkers, distinct from known mechanisms of open-state inactivation. We further capture KV4 in a putative resting state, revealing how voltage sensor movements activate the pore. Moreover, our structures provide insights regarding channel modulation by KChIP2 and DPP6 auxiliary subunits. Our findings elucidate mechanisms of closed-state inactivation and voltage-dependent activation of the KV4 channel.
In Brief
KV4.2 cryo-EM structures elucidates the molecular basis of state transitions during channel gating, providing a framework to understand the channel’s activation and inactivation. The KV4 channel closed-state inactivation uses a mechanism that is distinct from other voltage-gated ion channels.
Graphical Abstract
Introduction
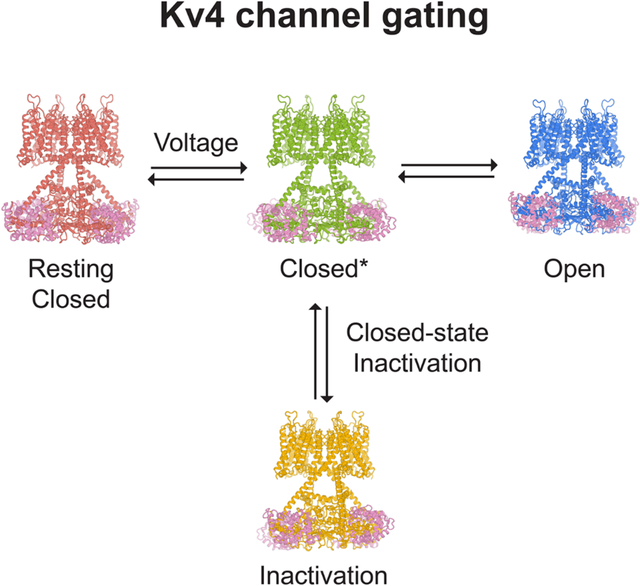
Voltage-gated ion channels activate in response to changes in cell membrane potential and open a pore to allow ion flux. However, upon sustained stimulus, many channels inactivate and are no longer able to conduct ions. This inactivation process acts like a brake to terminate ion flow and its interplay with channel activation is critical for action potential generation and neuron communications (Hille, 2001). Inactivation in voltage-gated ion channels fall into two categories: open-state inactivation and closed-state inactivation (Aldrich and Stevens, 1983). In open-state inactivation (Figure 1A), only after a resting closed channel (C state in Figure 1A) is first activated by voltage and opens (O state), can it inactivate (I state). In closed-state inactivation, however, a channel can inactivate directly from a closed state before it opens (Figure 1B). The two pathways are not mutually exclusive, though many channels inactivate mainly through one of these two pathways. The underlying mechanisms for open-state inactivation have been studied in molecular detail, such as ball-and-chain (N-type) inactivation (Armstrong and Hollingworth, 2018) and C-type inactivation (Hoshi and Armstrong, 2013), with several inactivated structures reported thus far (Cuello et al., 2010; Fan et al., 2020; Pan et al., 2018; Payandeh et al., 2012; Reddi et al., 2021; Tan et al., 2021). In contrast, the mechanism of closed-state inactivation has remained mysterious.
Figure 1. Functions and structures of KV4.2 channel complexes.
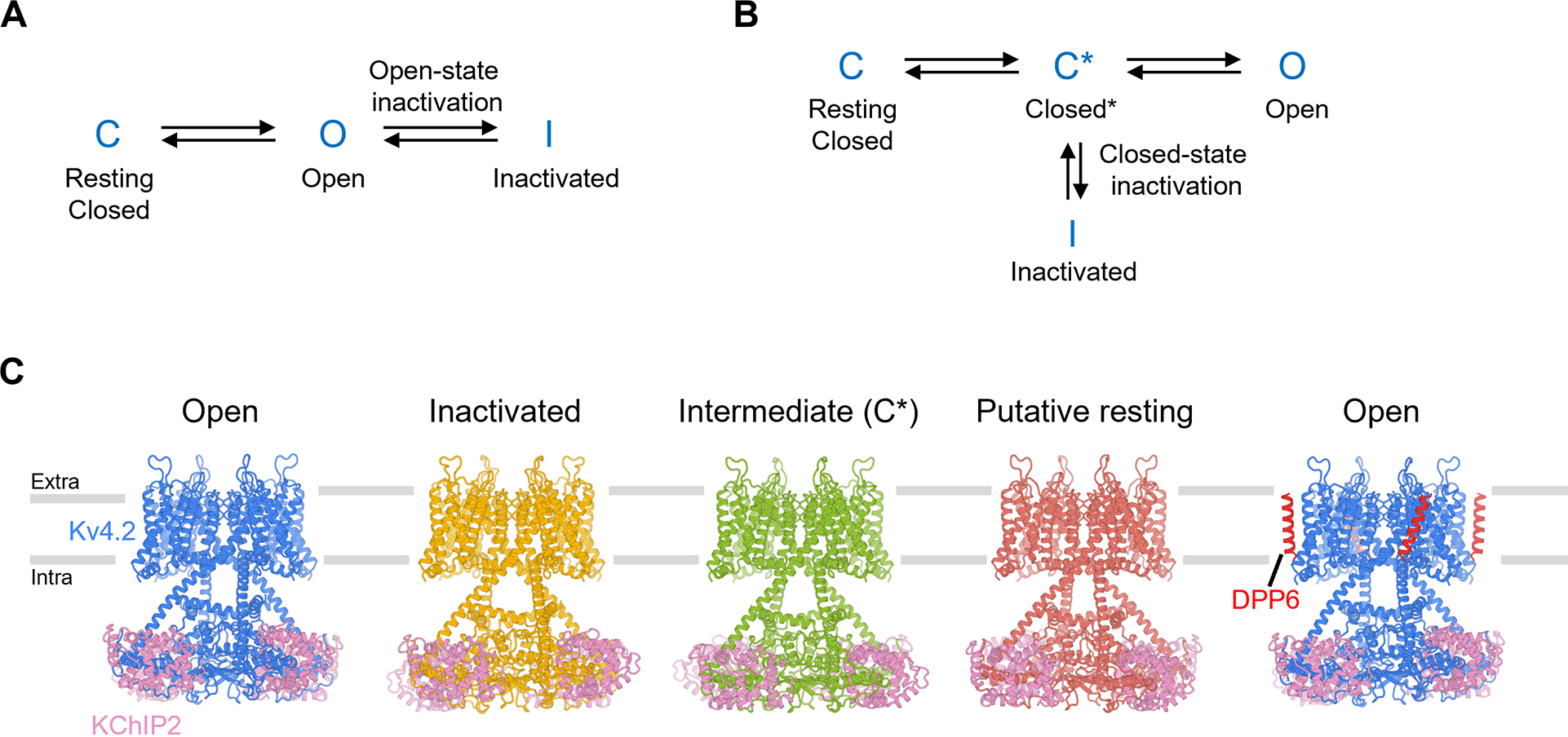
(A) A simple gating model of open-state inactivation.
(B) A simple gating model of closed-state inactivation.
(C) Structures of KV4.2 channel complexes in different states. The gray bars represent approximate boundaries of the membrane.
Here we study the KV4 channel, a voltage-gated K+ channel that undergoes closed-state inactivation (Bahring and Covarrubias, 2011; Bahring et al., 2012). KV4 channels mediate rapidly activating and inactivating potassium currents (Baldwin et al., 1991), often referred to as the transient A-type current (IA) in neurobiology or the transient outward current (Ito) in cardiology. Widely expressed in the brain and heart, KV4 channels regulate neuronal and muscular excitability, as well as orchestrate the firing rhythms (Jerng et al., 2004). KV4 can interact with two types of auxiliary subunits (An et al., 2000; Jerng and Pfaffinger, 2014; Nadal et al., 2003): potassium channel interacting proteins (KChIP) and Dipeptidyl aminopeptidase-like proteins (DPP), both types of auxiliary subunits modulate channel membrane expression as well as activation and inactivation kinetics. Human mutations in KV4 channels or their auxiliary subunits are linked to ataxia, autism, epilepsy, global developmental delay, and cardiac arrhythmia (Alders et al., 2009; Duarri et al., 2015; Hu et al., 2020; Lee et al., 2014; Smets et al., 2015; Zhang et al., 2021).
Previous structural studies have described the overall architecture of KV4 complexes (Kim et al., 2004; Kise et al., 2021; Ma et al., 2022; Pioletti et al., 2006; Wang et al., 2007). As a member of the voltage-gated K+ channel superfamily, KV4 forms a tetrameric channel in which each subunit is composed of a voltage sensor domain (S1 to S4 helices), a pore domain (S5 to S6 helices) and an amphipathic helix running parallel to the membrane plane (S4-S5 linker) that connects these two domains. Although many channels with the same topology inactivate from the open state (e.g. KV1 (Hoshi et al., 1991)), KV4 mostly undergoes closed-state inactivation (Figure 1B). Open-state inactivation, whether N-type or C-type, does not play major roles in KV4 (Shahidullah and Covarrubias, 2003). In a simplified gating scheme of KV4, upon depolarization the channel goes from a resting closed state (C state in Figure 1B) to an activated but non-conducting state (C* state). From there, the channel may either inactivate (I state) or open (O state).
Closed-state inactivation allows a KV4 channel to bypass the open state and become inactivated if the cell membrane is maintained at prolonged subthreshold potentials, and hence it controls the availability of resting A-type K+ channels and regulates cell excitability. During repolarization, an inactivated channel also bypasses the open state and directly return to the resting state, avoiding the resurgent K+ current. With these electrophysiological features, KV4 channels at somatodendritic regions of hippocampal CA1 pyramidal neurons limit action potential back-propagation and regulates synaptic plasticity (Cai et al., 2004; Chen et al., 2006; Kim et al., 2005). However, how KV4 channels activate and inactivate at the molecular level is not fully understood.
We investigate the molecular determinants for closed-state inactivation in human KV4 channels, as well as the structural basis of voltage-dependent activation. Only the open state was captured in recently reported structures of KV4 channel complexes (Kise et al., 2021; Ma et al., 2022). Here we present cryo-electron microscopy (cryo-EM) structures of the human KV4.2 channel in four distinct conformational states — open, inactivated, intermediate, and putative resting states (corresponding to the O, I, C* and C states in Figure 1B). These structures show that closed-state inactivation is achieved by an unprecedented symmetry breakdown in a K+ channel and demonstrate how voltage sensor movements are coupled to the channel gate. Furthermore, our structures of KV4 in complex with KChIP2 and DPP6 elucidate how these auxiliary subunits modulate channel function. Thus, our study reveals the long-sought-after structural mechanism of closed-state inactivation that governs the distinct transient, fast-inactivating currents of Kv4 channels, and provides insights into the electromechanical coupling of voltage-gated ion channels.
Results
Characterization of a KV4.2 channel for structural study
To increase levels of human KV4.2 expressed in mammalian HEK293 cells, we deleted 106 residues from its C-terminus to yield KV4.21–524, hereafter referred to as KV4.2EM. In the presence of auxiliary subunits KChIP2 and DPP6, both full-length KV4.2 and KV4.2EM channels activate upon depolarization and rapidly inactivate, resembling the characteristic A-type K+ currents seen in whole-cell recordings (Figure S1A). The activation and inactivation curves of KV4.2EM are indistinguishable from that of the full-length KV4.2 under the same conditions, with almost the same half-activation and half-inactivation voltages (Figure S1A–C). The time-courses of recovery from inactivation for these two channel constructs are also similar (Figure S1D–F). Hence, KV4.2 activation and inactivation are not altered by the C-terminal truncation.
To examine if the channel truncation affects modulation by KChIP and DPP6, we recorded full-length KV4.2 and KV4.2EM currents from cells coexpressing KChIP2 or DPP6, or both. For both full-length and KV4.2EM channels, KChIP2 shifts half-inactivation potentials to more positive voltages (Figure S1G–I, S1M–N) and accelerates recovery from inactivation (Figure S1J–L, S1O–P), whereas DPP6 shifts half-activation and half-inactivation potentials to more negative voltages (Figure S1G–I, S1M–N), without substantially altering the recovery rates (Figure S1J–L, S1O–P). These experiments demonstrate that KV4.2EM recapitulates the electrophysiology and modulation of the full-length channel.
Using cryo-EM, we determined structures of KV4.2EM in complex with KChIP2 or with KChIP2 and DPP6, to 2.2–3 Å resolution (Figure 1C, Figures S2 and S3). The high quality of the cryo-EM density maps allowed the de novo building of atomic models of KV4.2 channel complexes. We resolved structures of the KV4.2EM/KChIP2 channel complex in open, inactivated, intermediate and putative resting states. The transmembrane region of the KV4 channel complex is similar to that of other domain-swapped voltage-gated ion channels, such as KV1-KV7, NaV and CaV channels (Figure 1C). C-terminal helices of the transmembrane domain extend deeper into the cytoplasm. These atypically long helices, together with the cytoplasmic N-terminal region of the channel, assemble with KChIP2 to form a teepee-like structure (Figure 1C). In addition, we determined a structure of the KV4.2EM/KChIP2/DPP6 channel complex in its open state (Figure 1C, Figures S2 and S3). Comparison among these structures reveals remarkable conformational transitions during channel inactivation and activation, and facilitates our understanding as to how KV4.2 is modulated by the two auxiliary subunits.
Channel structures in the inactivated, open, and intermediate states
Cryo-EM analyses of KV4.2EM/KChIP2 channel complexes revealed four high-resolution classes. Two classes are structurally similar (Figure S4A), and together they accounted for half of the particles. We therefore refer to this conformation as the major state (Figure 2A, yellow). The other two classes account for ~25% of the particles each and thus represent two minor states (Figure 2A, blue and green).
Figure 2. Channel pore conformations in the open, inactivated and intermediate states.
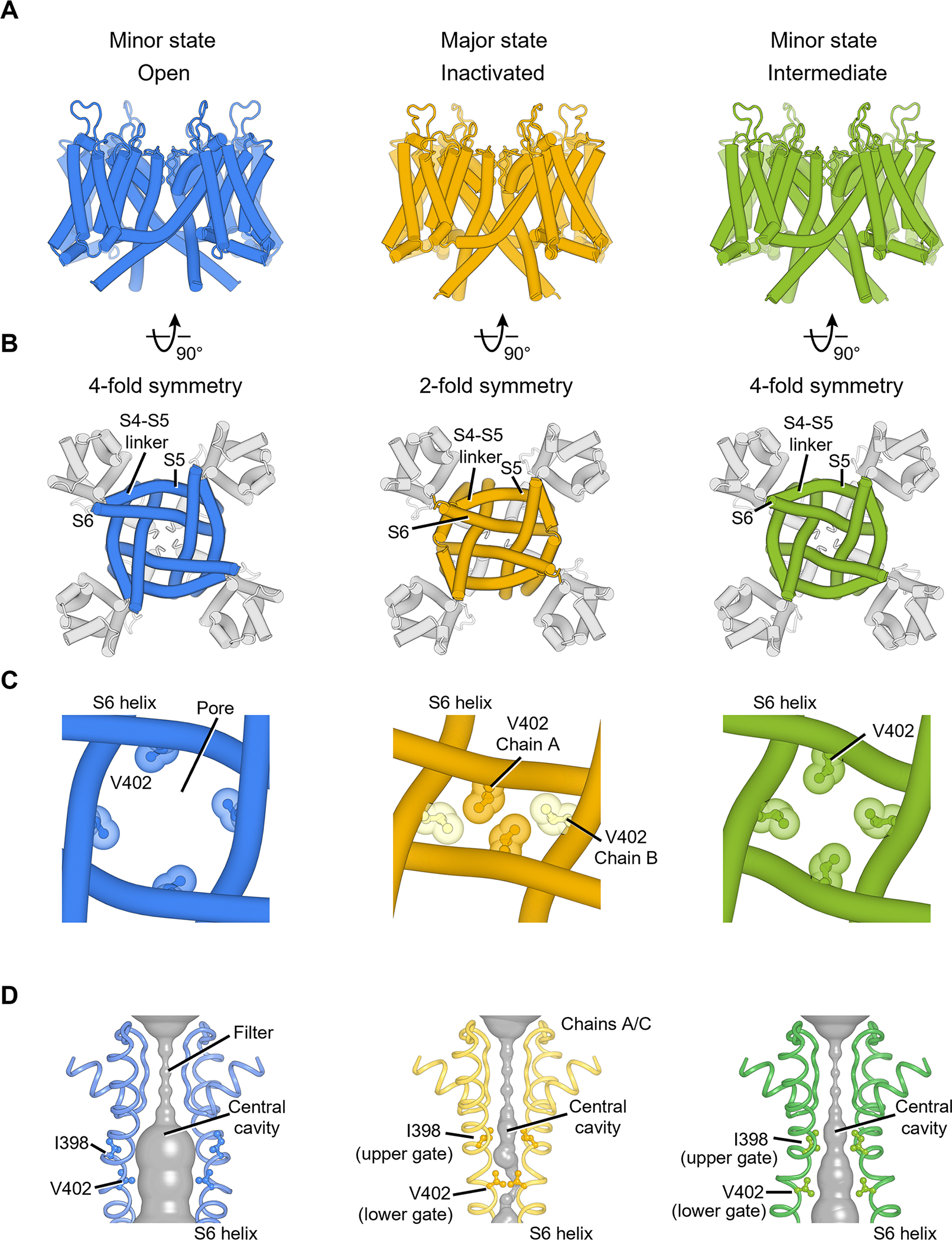
(A) KV4 transmembrane region in different states, viewed parallel to the membrane.
(B) KV4 channel pore domain viewed from the intracellular side. The pore in the inactivated state adopts a 2-fold symmetric conformation.
(C) The pore-forming S6 helices and channel gate V402 residues in different states.
(D) The channel pore with two of four subunits shown for clarity. The minimal radial distance from the center axis to the protein surface is colored in gray. Upper gate and lower gate residues are shown as ball-and-stick representation.
The major state has a very narrow pore (Figure 2C, D, yellow), and we propose that it represents an inactivated conformation (I state in Figure 1B). In this state, the voltage sensor is activated (Figure 3A) but the channel pore is tightly sealed (Figure 2C, D, yellow) with a pore radius small than 0.5 Å (Figure S4B), consistent with the expectations of an inactivated voltage-gated ion channel. This hypothesis is further supported by our electrophysiological studies showing that most of KV4.2 channels inactivate within seconds at 0 mV membrane potential (Figure S1G).
Figure 3. Conformational changes of KV4 during closed-state inactivation.
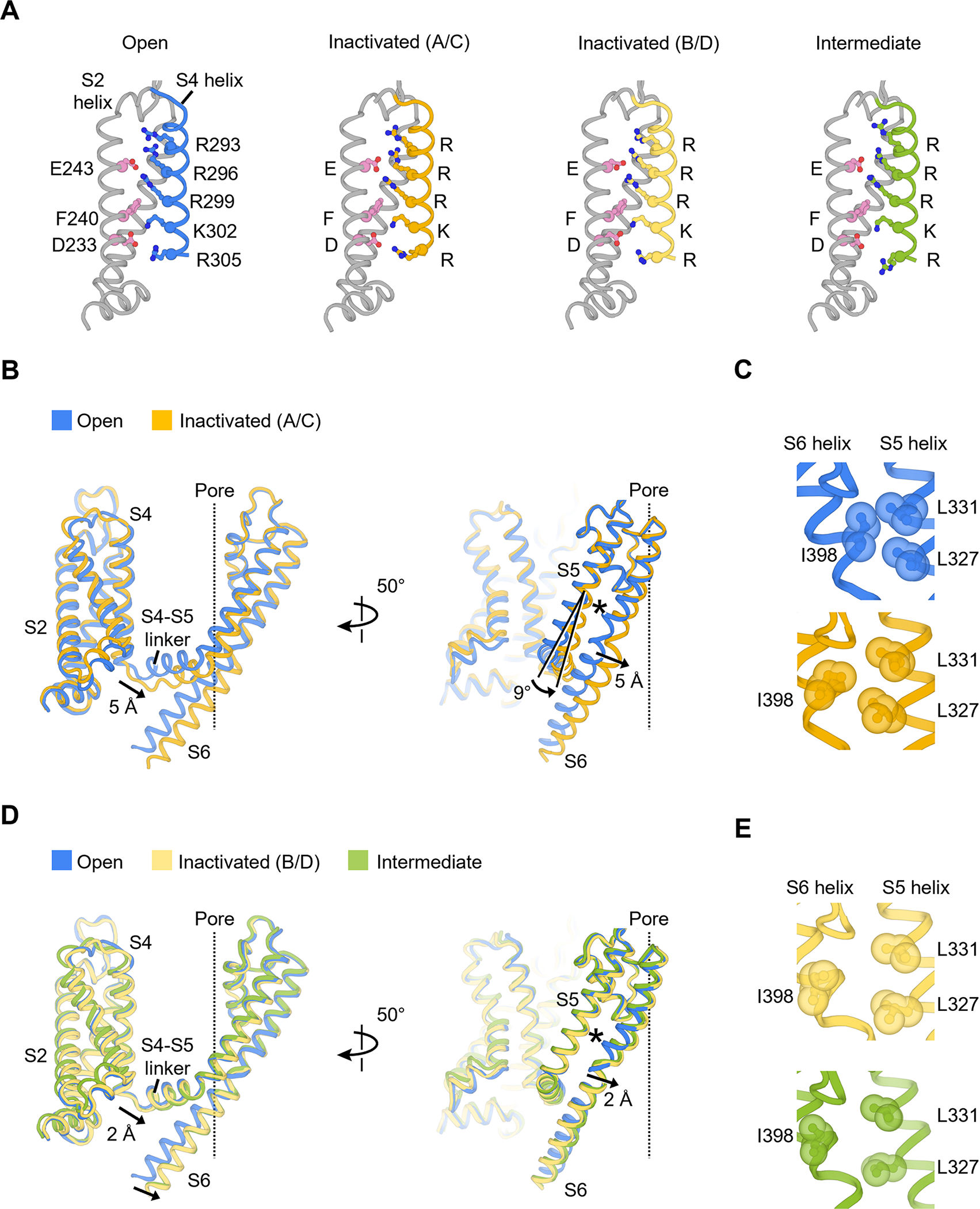
(A) Structures of voltage sensor during inactivation. Residues composing the gating charge transfer center (E243, F240 and D233) and positively charged residues on the S4 helix are shown as ball-and-stick representation.
(B) Conformational changes of the S4-S5 linker and S5-S6 helices in the inactivated state (chain A/C). The central ion pathway is represented by a dash line. The asterisk symbol highlights the location of the interaction shown in (C).
(C) Hydrophobic patch between S5 and S6 helices that controls the upper gate I398 (open state, blue; inactivated state, yellow).
(D) Conformational changes of the S4-S5 linker and S5-S6 helices in the inactivated state (chain B/D) and intermediate state. The central ion pathway is represented by a dash line. The asterisk symbol highlights the location of the interaction shown I).
(E) Hydrophobic patch in the inactivated state (chain B/D, light yellow) and intermediate state (green).
One of the minor states has a wide pore (Figure 2C, D blue), dilated to a radius of 4.1 Å at the constriction site (Figure S4B), which is enough to conduct hydrated K+ ions (radius of ~3.5 Å) (Moldenhauer et al., 2016). This state thus represents the open conformation (O state in Figure 1B). This conformation is similar to what has been observed in previous KV4 structures (Figure S4C) (Kise et al., 2021). In the other minor state (Figure 2C, D, green), the pore adopts an intermediate size (radius of ~2.8 Å) between the inactivated state and the open state, but remains too small to conduct K+ ions (Figure S4B, D), suggesting that it represents an intermediate state that correspond to the closed* state (Figure 1B).
Symmetry breakdown in the inactivated pore
The inactivated state and the open state show striking difference in the pore architecture. Upon inactivation, the homo-tetrameric channel pore loses its four-fold rotational symmetry and becomes two-fold symmetric (Figure 2B, C). Collapse of the symmetry of the pore has been observed in the bacterial NaVAb Na+ channel and TRP channels (Payandeh et al., 2012; Yin et al., 2019; Zubcevic et al., 2018a, 2018b, 2019), but such a symmetry breakdown is unprecedented in voltage-gated K+ channels, and a connection between symmetry breakdown and closed-state inactivation has never been established before. These findings highlight that the mechanism of closed-state inactivation of KV4 channels is distinct from the mechanisms of open-state inactivation of other voltage-gated K+ channels.
The two-fold symmetric pore in the inactivated state allows the formation of tight constrictions along the ion conduction pathway. Specifically, side chains of Val402 from one pair of S6 helices (chains A/C) are packed much closer than Val402 from the other S6 pair (chains B/D) (Figure 2C, yellow), and the four valine residues interlock like a zipper to seal the pore. In fact, Val402 is part of the highly conserved Pro-Val-Pro motif that is critical to gating in many K+ channels (Long et al., 2005a; Webster et al., 2004). Mutations around this motif causing slowed inactivation are associated with autism and epilepsy (Lin et al., 2018) as well as early-onset global developmental delay (Zhang et al., 2021). Ile398 also occupies the central cavity and forms another constriction of the pore (Figure 2D, yellow). We thus define Ile398 as the upper gate and Val402 as the lower gate (Figure S4B).
These two gates adopt different configurations in the open state and the intermediate state. In the open state, the movement of S6 helices dilates the pore and the side chains of both Ile398 and Val402 swing away from the ion pathway (Figure 2D, blue), opening the upper and lower gates and allowing ions to permeate. In the intermediate state, the upper gate remains closed (radius of 1.1 Å) and the lower gate is not as widely open as in the open state (radius of 2.8 Å versus 4.1 Å) (Figure 2D, green and Figure S4B, D), rendering the channel non-conducting. Notably, in this intermediate state, the channel pore is still four-fold symmetric (Figure 2B, C, green), suggesting that the breakdown of symmetry from four-fold to two-fold occurs during the transition from the intermediate to the inactivated state.
Mechanism of closed state inactivation
To understand the structural basis of closed state inactivation, we first compared the voltage sensor conformations of KV4 channels in the open, intermediate and inactivated states (Figure 3A). The voltage-sensing S4 helices in all three states are in the activated (or depolarized) conformation, as expected for the physiological state at 0 mV membrane potential. Specifically, gating charges Arg293, Arg296 and Arg299 stay above the gating charge transfer center formed by Phe240 and several negatively charged residues including Asp233 (Figure 3A). This observation indicates that S4 helices do not undergo major displacements during inactivation.
The two-fold symmetric inactivation is accomplished by the coupling of the S4-S5 linker to both upper and lower gates (Figure 3B–E). In the inactivated state, one diagonal pair of the S4-S5 linker (chains A/C) moves downward by 5 Å relative to their position in the open state (Figure 3B). This movement of the S4-S5 linker pushes the S6 helix toward the pore axis and narrows the ion conducting pathway to close the lower gate (Figure 2C). Furthermore, the S4-S5 linker movement also triggers a 9° rotation of the S5 helix (Figure 3B). This rotation, together with the S6 helix shift, rearranges the hydrophobic patch involving the upper gate Ile398 (Figure 3C and the asterisk in Figure 3B), leading to the reorientation of the side chain of Ile398 (chains A/C), collapse of the central cavity and closing of the upper gate (Figure 2D, yellow). In this way, both upper and lower gates are tightly shut in the inactivated state.
The asymmetric nature of inactivation is further demonstrated by the observation that, in contrast to chains A/C, the other pair of S4-S5 linker (chains B/D) moves downward only by 2 Å (Figure 3E). Additionally, the upper gate Ile398 in chains B/D reorients more drastically than that in chains A/C (Figure 3E, light yellow and Figure 3C, yellow). The overall conformation of chains B/D resembles the channel conformation in the intermediate state (Figure 3D, E, green).
With structures of KV4 channels in open, intermediate, and inactivated states, we may now understand better the structural transitions that take place during closed-state inactivation (Figure 1B). We propose that the open channel first transits into an intermediate state in which all four S4-S5 linkers tilt towards the intracellular side to narrow the pore. Upon inactivation, an unusual symmetry breakdown occurs, with only two of the S4-S5 linkers (from diagonal subunits) pushing down further to trigger additional conformational changes in the pore-forming S6 helices and to firmly tighten the lower channel gate, preventing ion permeation.
Our structures suggest that, during channel inactivation, only two of the four KV4 subunits undergo the conformation change for inactivation as seen in the A/C chains. We tested this inactivation stoichiometry using a KV4 tandem construct composed of a Val404Met subunit and a wild-type (WT) subunit (V404M-WT). The V404M mutation was initially identified in individuals with autism and epilepsy, and the mutant channel is slower to inactivate and to recover from inactivation compared to the WT channel (Lin et al., 2018), a phenotype that we confirmed here (Figure 4A–C). Our tandem construct (V404M-WT) produces KV4 channels with two diagonally positioned V404M subunits and two diagonally positioned WT subunits. If our hypothesis that only two subunits adopt an inactivated conformation is correct, in most of our V404M-WT channels, only the WT subunits will adopt the inactivated conformation and the recovery speed should be solely determined by the WT subunits. This is because of the slower inactivation of V404M subunits: once WT subunits inactivate, they would prevent V404M subunits from adopting the conformation for inactivation. On the other hand, if more than two subunits can adopt the inactivated conformation, V404M subunits will also take on the inactivated conformation, leading to a significant delay in recovery. To make sure concatenating two subunits does not interfere with channel gating, we also made WT-WT and V404M-V404M concatemers as controls.
Figure 4. Two-fold inactivation stoichiometry probed by KV4 V404M.
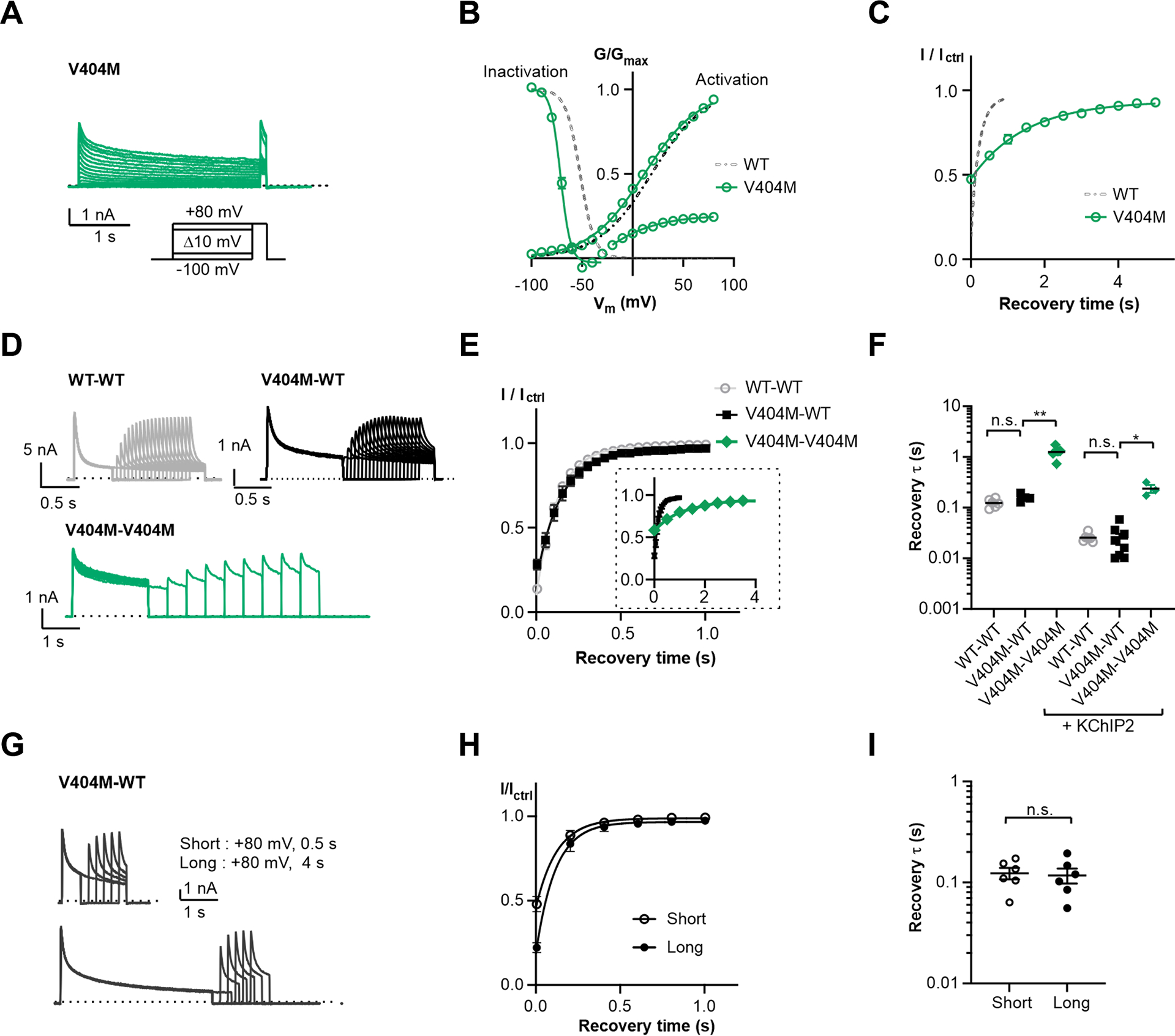
(A) Representative whole-cell recording traces of KV4 V404M.
(B) Steady-state activation and inactivation curves of KV4 V404M. Data shown are mean ± SEM.
(C) Time course of V404M channels recovering from inactivation.
(D) Representative whole-cell recording traces of channels formed by concatenated subunits, showing the channel recovery from inactivation. The time constants of current decay are: WT-WT, τfast = 34±2 ms, τslow = 173±11 ms; V404M-WT, τfast = 104±10 ms, τslow = 1141±115 ms; V404M-V404M, τfast = 204±22 ms, τslow = 2641±343 ms.
(E) Time course of channels formed by concatenated subunits recovering from inactivation. For clarity, V404M-V404M is only included in the insert, displayed on a larger time scale. Data shown are mean ± SEM.
(F) Scatter plot showing recovery time constants (τ) of concatenated channels without or with KChIP2. Individual data points and mean values (horizontal black lines) are shown. *p < 0.05, **p < 0.01, n.s., not significant, as assessed by Dunnett’s test following Kruskal-Wallis test.
(G) Representative whole-cell recording traces with a long depolarizing step on V404M-WT channels. The depolarizing step here is either 0.5 s (short) or 4 s (long), whereas in (D) the step is 1 s for V404M-WT and 2 s for V404M-V404M.
(H) Time courses of V404M-WT channels recovering from inactivation shown in (G). Data shown are mean ± SEM.
(I) Scatter plot showing recovery time constants (τ) from recordings as in (G). Mean values (black lines) and SEM (whiskers) Are shown. n. s., not significant, as assessed by Welch’s t-test.
We found that our V404M-WT channels recover from inactivation with a time constant comparable to WT-WT channels and much faster than V404M-V404M channels (Figure 4D–F). These observations support our hypothesis that only two subunits adopt the inactivated conformation in KV4 channels. We also performed experiments doubling the duration of the depolarizing step (to 4 seconds) to further promote inactivation, but the V404M subunits in V404M-WT channels still did not contribute to inactivation and no further delay in recovery was observed. (Figure 4G–I). Experiments with or without KChIP2 yielded similar results (Figure 4F), demonstrating that this inactivation stoichiometry is an intrinsic property of the KV4 channel and not induced by KChIP2.
We performed control experiments to rule out the possibility that when expressing the tandem constructs, four WT subunits form the tetrameric channel while the four mutated subunits are floating around. We mutated Val406 in the channel pore domain to cystine (Val406Cys, V406C). In contrast to WT KV4.2, which is relatively insensitive to the membrane-permeable bifunctional cysteine crosslinker MTS-4-MTS (1,4-butanediyl bismethanethiosulfonate), KV4.2 V406C mutant is readily inhibited by the MTS reagent (Figure S6A). We then made a V406C-WT tandem construct and examined its response to MTS crosslinkers. If the four WT subunits would make the tetrameric channel, this construct would not respond to the MTS reagent. But that is not the case, as the MTS treatment readily inhibits V406C-WT currents (Figure S5A–E). We also made the V404M/V406C-WT concatemer and found that its current was inhibited by the MTS reagent as well (Figure S5F–H). These data confirm that the currents we recorded were from channels composed of both mutated and WT subunits, rather than those composed of only the WT subunits. Together with the structural information, the electrophysiological recordings strongly support the 2-fold symmetry for the conformation underlying KV4 channel inactivation.
Chemical stabilization to obtain a resting conformation
Having established the transition between the open state and inactivated state in KV4, we sought to understand the transition from either the inactivated or from the open state to the resting closed state. The resting closed state (C state in Figure 1B) has its voltage sensor in a resting (also known as deactivated or hyperpolarized) conformation, which is fundamentally different from the depolarized conformation adopted in the intermediate state (Figure 3A). We hereafter refer to the resting closed state as “resting” state for simplicity. We note that structural determination of this state is difficult, because the channel does not adopt a true resting conformation without an externally applied hyperpolarizing electric field.
To overcome this difficulty, we stabilized KV4 in its resting state using chemical crosslinking. We hypothesized that, in the resting conformation, the channel pore is closed and the pore-forming S6 helices should come close to each other. We therefore substituted different residues located C-terminally to the S6 Pro-Val-Pro motif with cysteine (Figure 5A) and monitored channel currents in response to the crosslinker MTS-1-MTS (1,1-methanediyl bismethanethiosulfonate). MTS-1-MTS inhibits currents of several of the Cys mutants (Figure 5B–D), with strongest inhibition seen for V406C (Figure 5D). To rule out cross-reactions with endogenous cysteines, we mutated the only cysteine within 10 Å of V406, Cys320, to serine (C320S). We did not observe large changes in the inhibition by MTS-1-MTS (Figure 5D), demonstrating that the reagent crosslinks V406C in an inter-subunit fashion to prevent channel activation.
Figure 5. Crosslinking of Kv4 V406C in the resting state.
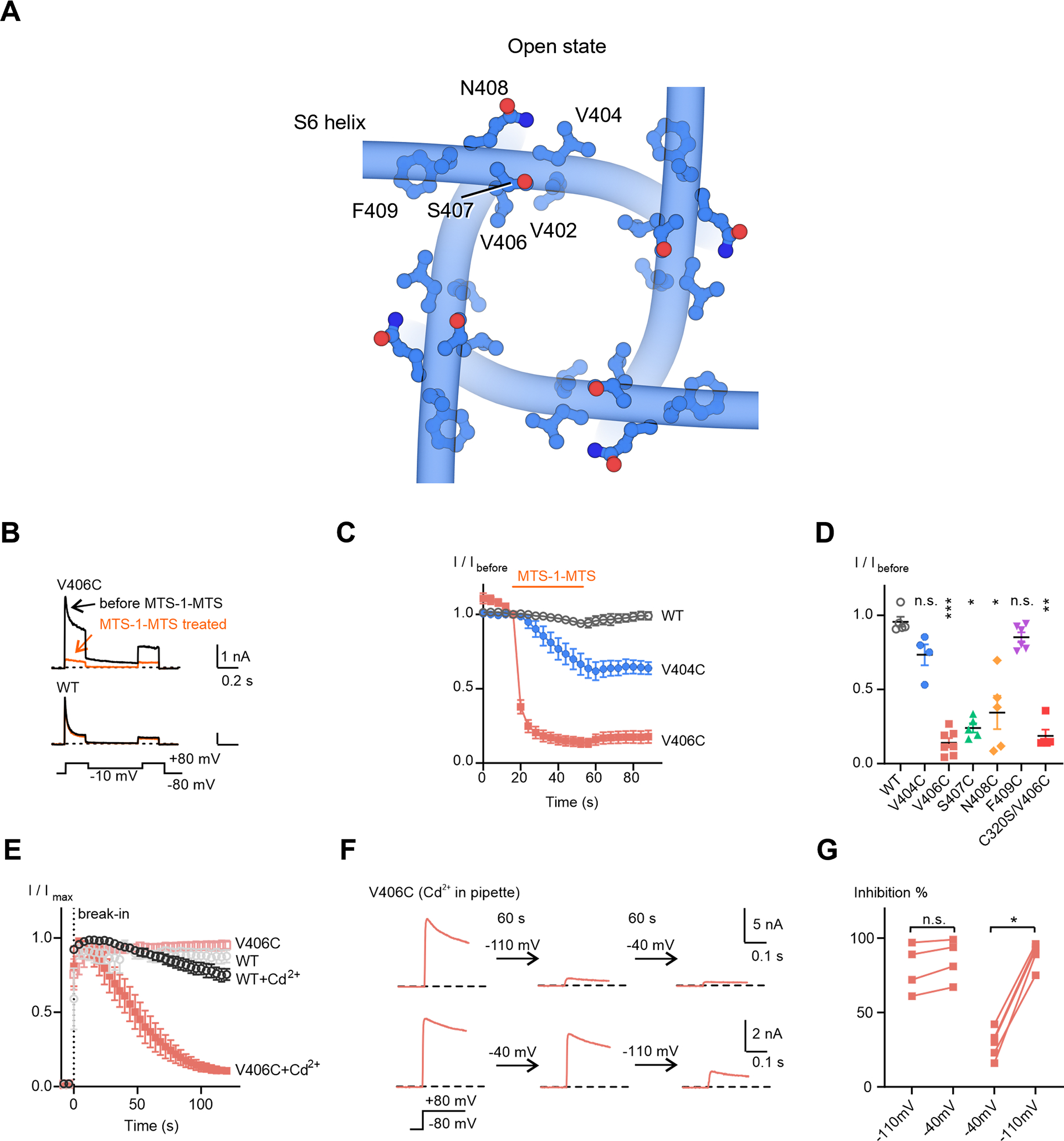
(A) Residues located C-terminally to the channel lower gate V402.
(B) Representative whole-cell recording traces of KV4 channels before and after a 40-second MTS-1-MTS treatment.
(C) Time courses of current responses to MTS-1-MTS treatment. The current magnitudes were normalized to that at the sweep before treatment (I/Ibefore) for each cell. Data shown are mean ± SEM.
(D) Scatter plot showing current responses to MTS-1-MTS treatment for each mutant. Mean values (horizontal black lines) and SEM (colored whiskers) are shown. *p < 0.05, **p < 0.01, ***p < 0.001, n.s., not significant, as assessed by Dunnett’s test following Kruskal-Wallis test.
(E) Time courses of changes in KV4 peak currents in the absence or presence Cd2+. The recordings start before breaking into the cells (before the 0 s time point). Data shown are mean ± SEM.
(F) Representative traces of KV4 V406C in the presence of Cd2+, showing the state-dependence of Cd2+ inhibition. Currents were evoked by a depolarizing step to +80 mV from −80 mV. After recording for 2 to 3 sweeps, the membrane potential was held at the indicated potentials for 60 seconds, followed by subsequent recordings with the same protocol.
(G) Summary of Cd2+ inhibition. Two sequential recordings from the same cell are connected by a line. *p < 0.05, n.s., not significant, as assessed by Dunnett’s test following Kruskal-Wallis test.
We next tested crosslinkers that were shorter than MTS-1-MTS, to reduce potential conformational heterogeneity and facilitate structural determination. Knowing that Cd2+ ions can crosslink cysteines within ~5 Å (Linsdell, 2015), we included Cd2+ in the pipette solution within the patch electrode for whole-cell recording and found that the V406C channel current gradually reduced in size after breaking into the cell with the patch pipette (Figure 5E), indicative of the formation of Cd2+ crosslinks in V406C.
We then determined whether V406C crosslinking stabilizes the resting state or the inactivated state. When the cell membrane potential was held at −110 mV (a voltage that promotes the resting state) for 60 seconds after breaking in, Cd2+ crosslinks rapidly formed and the majority of channels (80 ± 8 %) were prevented from activation within 60 seconds (Figure 5F upper traces, and Figure 5G). Stepping the membrane potential to −40 mV (a voltage that promotes the inactivated state) for another 60 seconds did not induce further inhibition of channel activation (Figure 5F, G). In contrast, when the membrane potential was held at −40 mV first, only about 30% of the channel were inhibited (Figure 5F lower traces, and Figure 5G) while further inhibition could be achieved by stepping the membrane potential to −110 mV (Figure 5F, G). A similar state-dependence was also observed using a different crosslinker MTS-4-MTS (Figure S6A–C). These results indicate that the crosslinking of V406C occurs when the channel is in the resting state.
Structure of KV4 in a putative resting state
We determined the structure of the KV4.2EM V406C channel in the presence of the crosslinker Hg2+ (Figure 6 and Figures S2, S3, S6D, E). We used Hg2+ instead of Cd2+ because of its higher affinity toward cysteine (Ajsuvakova et al., 2020). Compared to the open state structure, the voltage sensor of the crosslinked channel adopts a different conformation where the S4 helix is displaced toward the intracellular side (Figure 6A) and the channel pore is constricted (Figure 6C, D), consistent with a putative resting state. The conformational changes involving the S4 helix are distant from the cysteine introduced in the pore-forming S6 helical bundle. These findings are consistent with the notion that the S4 helix is strongly coupled to the pore in the resting state, allowing motions of voltage sensors to be transmitted efficiently to the pore upon channel activation.
Figure 6. Structure of KV4 in the putative resting state.
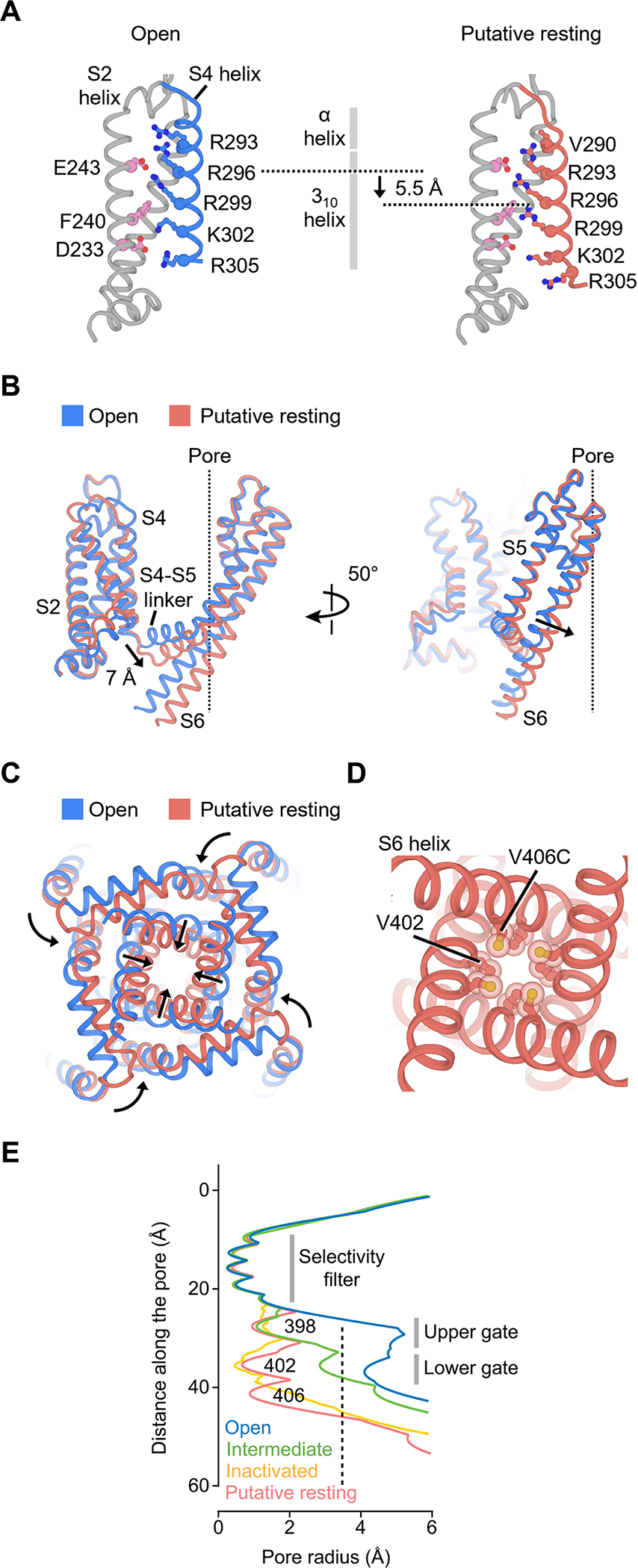
(A) Movement of the S4 helix in the putative resting voltage sensor.
(B) Conformational changes of the S4-S5 linker and S5-S6 helices in the putative resting state. The central ion pathway is represented by a dash line
(C) Conformational changes of the pore in the putative resting state.
(D) Arrangement of V406C and the lower gate V402 residues in the putative resting state.
(E) Radius of the pore in different states. The radius is plotted as a function of the distance along the pore axis. The minimal pore radius for hydrated K+ ions to pass through (3.5 Å) is highlighted by a dash line.
In the putative resting conformation, the S4 helix is shifted by one helical turn (~5.5 Å) (Figure 6A), but the 310 helical region within S4 spans a similar range in the membrane in the putative resting and open states (Figure 6A, gray vertical bar). Thus, the S4 movement must be accompanied by a reorganization in its secondary structure, with residues in the α-helical segment near the extracellular side rearranging into a 310 helix. This helical transition or “concertina effect” (Lee and MacKinnon, 2019; Long et al., 2007) allows the S4 helix to translate without rotating along its long axis.
In the putative resting voltage sensor, the gating residue Arg299 moves into the gating charge transfer center (below Phe240) while Lys302 moves out of the center. The repositioning of Arg299 is a key step along the activation pathway and correlates with the closing or opening of the channel pore. With Arg299 in the transfer center, the shifted S4 helix displaces the S4-S5 linker by 7 Å toward the intracellular side (Figure 6B). The linker helices in turn compress the S6 helices to close the channel gate (Figure 6C, D). In contrast to the observed movement of the KV4 channel, in the resting structures of other voltage gated channels, including the HCN, NaV and TPC1 channels (Guo et al., 2016; Kintzer and Stroud, 2016; Lee and MacKinnon, 2019; Wisedchaisri et al., 2019), the S4 helices are shifted toward the intracellular side by about two helical turns (~10–11 Å), resulting in a displacement of two gating charges. To our knowledge, this KV4 structure represents the first visualization of any voltage-gated K+ channel in a putative resting state, and the direct comparison of the putative resting and open structures reveals the mechanism of voltage-dependent gating of KV4 channels.
Channel modulation by auxiliary subunits
Finally, we investigated how the two types of auxiliary subunits, KChIP2 and DPP6, interact with the channel and modulate properties of activation and inactivation. In our structure, four KChIP2 subunits bind to a channel tetramer to form an octamer (Figure 7A), similar to the arrangement of the KV4.2/KChIP1 complex (Kise et al., 2021). KChIP2 and the KV4.2 channel form an extensive interaction network: every KChIP2 molecule communicating with two KV4.2 channel subunits. KChIP2 binds not only an N-terminal helix from one KV4.2 subunit (Figure 7A zoomed-in view, light blue), but also two C-terminal helices from an adjacent KV4.2 subunit (Figure 7A zoomed-in view, blue) (Callsen et al., 2005). To validate these interactions, we truncated these helices in KV4.2 and measured KChIP2 modulation on inactivation recovery. KChIP2 modulation is completely abolished with the N-terminal helix deletion (Figure 7B, D, KV4.2 Δ7–21), while its modulation is compromised with C-terminal helix deletion (Figure 7C, D, KV4.21–473). These results support our structural findings and suggest that interactions of KChIP2 with the KV4.2 N-terminal helix play a more prominent role in modulating inactivation than those interactions with the KV4.2 C-terminal helices.
Figure 7. KV4 modulation by auxiliary subunits KChIP2 and DPP6.
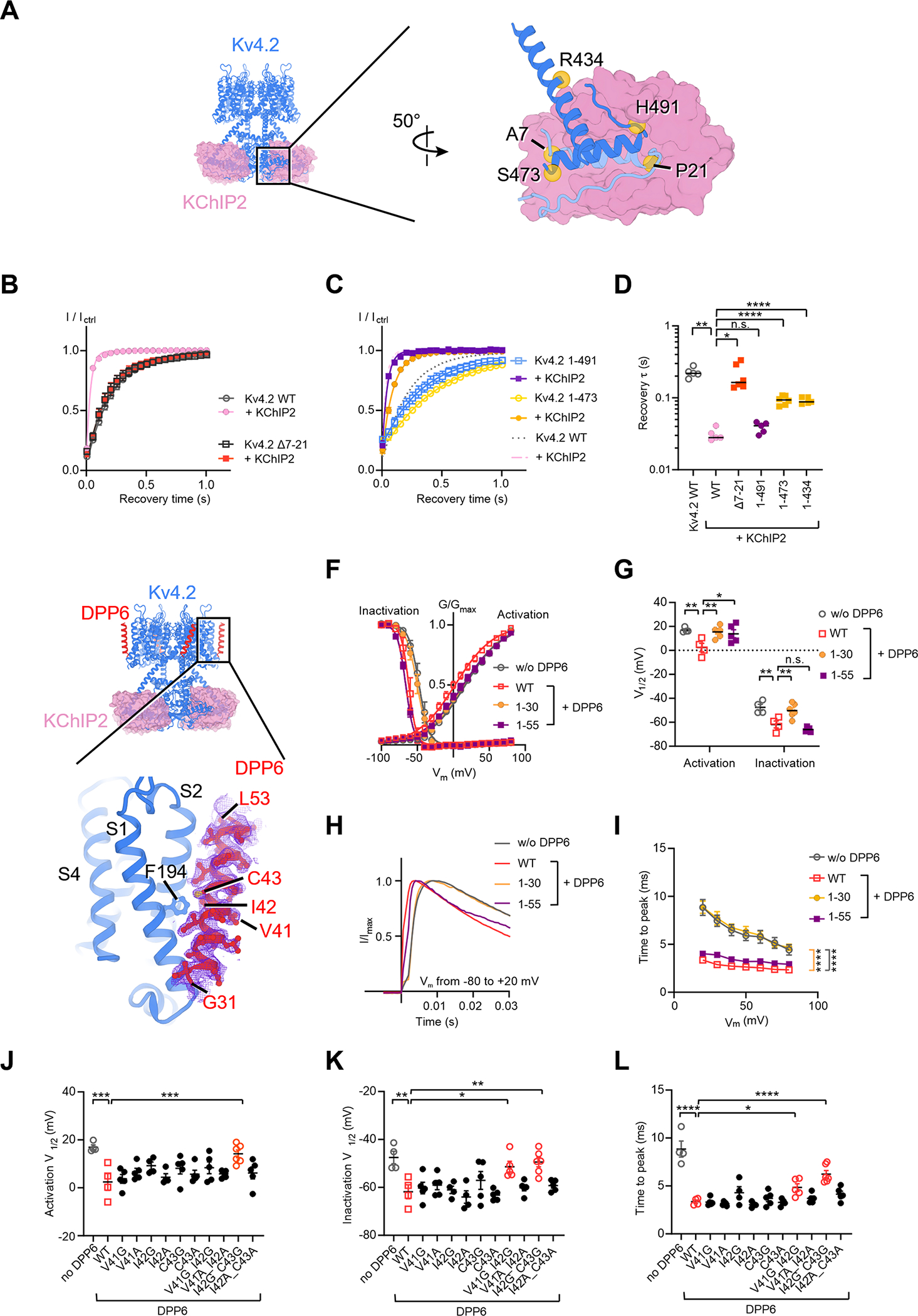
(A) Interfaces between KV4 and KChIP2. In the zoomed-in view, one KV4 subunit is colored in light blue and an adjacent KV4 subunit is in blue.
(B) and (C) Time courses of KV4 deletion mutants recovering from inactivation in absence or presence of KChIP2. Data shown are mean ± SEM.
(D) Scatter plot showing recovery time constants (τ) of KV4 deletion mutants. Individual data points and mean values (horizontal black lines) are shown. *p < 0.05, **p < 0.01, ****p < 0.0001, n.s., not significant, as assessed by Dunnett’s test following Brown-Forsythe ANOVA test.
(E) Interfaces between KV4 and DPP6. The cryo-EM density of DPP6 is shown as purple mesh.
(F) Steady-state activation and inactivation curves of KV4 in the absence or presence of DPP6 variants. Data shown are mean ± SEM.
(G) Scatter plot showing half-activation and half-inactivation voltage (V1/2) from recordings as in (F). Individual data points and mean values (horizontal black lines) are shown. *p < 0.05, **p < 0.01, n.s., not significant, as assessed by Welch’s t-test.
(H) Representative normalized traces showing the time to peak Kv4 current.
(I) Time to peak current at different depolarizing voltages. Data shown are mean ± SEM. ****p < 0.0001, as assessed by Dunnett’s test following two-way ANOVA.
(J) (K) and (L) Scatter plots showing half-activation voltages (J), half-inactivation voltages (K), and the time to peak current (L) of KV4 in the presence of DPP6 variants. In (L), the time to peak current was measured in recordings where membrane potential was depolarized to +20 mV from −80 mV. Mean values (horizontal black lines) and SEM (colored whiskers) are shown. For clarity, only comparisons with p < 0.05 are labeled. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, as assessed by Dunnett’s test following one-way ANOVA (against KV4 with wild-type DPP6).
To understand the modulation of DPP6, we determined the structure of the KV4.2EM/KChIP2/DPP6 channel complex (Figure 7E). DPP6 is a single-span transmembrane protein with a large C-terminal extracellular domain. In our cryo-EM reconstruction, the transmembrane region of DPP6 is clearly resolved, while the extracellular domain is poorly resolved probably due to its high flexibility. Nevertheless, the extracellular domain appears to be largely dispensable for DPP6 modulation because (1) a previous study showed that the extracellular domain of DPP6 barely interacts with the channel (Kise et al., 2021). (2) Deletion of the DPP6 extracellular domain (DPP61–55) does not alter DPP6 modulation on channel inactivation or the time course of channel activation (Figure 7F–I, purple). In contrast, deletion of the DPP6 transmembrane helix (DPP61–30) eliminates the modulation (Figure 7F–I, yellow), indicating that the transmembrane domain of DPP6 is essential to its function.
The structure of the transmembrane domain of the KV4.2EM/KChIP2/DPP6 channel complex was determined to a resolution of 2.2 Å and the well-defined density of the DPP6 transmembrane segment allows us to unambiguously assign the amino acid residues in our model (Figure 7E). Our assignment is different by one residue from a recent structure with a lower resolution (Kise et al., 2021). DPP6 interacts with the voltage-sensing domain of KV4.2 but does not directly contact the S4 helix. The extracellular half of the tilted DPP6 helix leans against the S2 helix and its intracellular half contacts the S1 helix (Figure 7E). In the middle of the membrane Ile42 and Cys43 of DPP6 sit next to Phe194 of the S2 helix in KV4.2. However, alanine substitutions of these residues do not affect DPP6 modulation of channel activity (Figure 7J–L). We hypothesize that the helix rigidity of DPP6 may be important for its interaction with the S1 and S2 helices. In support of this hypothesis, mutants with double-glycine substitutions in the middle of the helix, V41G/I42G and I42G/C43G, show compromised modulation of KV4.2 currents, while mutants with double-alanine substitutions or with a single-glycine substitution have no significant effects (Figure 7J–L).
Discussion
In this work we resolved the conformational changes that take place during closed-state inactivation, revealing that KV4 channels potentially inactivate through an intermediate state involving breakdown of its four-fold symmetry. Furthermore, using chemical crosslinking, we captured the channel in a putative resting state, providing a mechanism for voltage-dependent gating. Comparisons of channels in different conformations allow us to correlate structural states to functional states, and to define the molecular rearrangements that underlie major channel gating pathways. We propose that during depolarization, the resting S4 helices move toward the extracellular side, pulling along the S4-S5 linkers. This displacement widens the lower gate of the channel while the pore remains closed (C* state, Figure 1B). Further movements of the S4-S5 linkers in the same outward direction propagate to S5 and S6 helices to fully open the upper and lower gates (O state, Figure 1B). Alternatively, from the intermediate state, one diagonal pair of the S4-S5 linkers can disengage from the voltage sensors in the activated conformation due to depolarization and fall back to the intracellular side — such movements trigger channel inactivation and squeeze the square-shaped pore into a rhombic one (I state, Figure 1B).
We have revealed for the first time the potential structural mechanism of closed-state inactivation. Inactivation is essential to electrical signaling of voltage-gated ion channels, but much less is known about closed-state inactivation as compared to open-state inactivation. The ball-and-chain type of open-state inactivation was initially discovered in sodium channels of squid axons (Armstrong et al., 1973). Later studies in Shaker-related K+ channels show that the N-terminus of channel subunits could be the inactivation “ball” that blocks the channel pore, giving the name N-type inactivation (Hoshi et al., 1990; Zagotta et al., 1990). This type of inactivation has been structurally characterized in the prokaryotic MthK channel (Fan et al., 2020). The molecular basis of fast inactivation in NaV channels has also been revealed: the fast inactivation motif binds to a pocket adjacent to the S6 intracellular gate to inactivate the channel (Clairfeuille et al., 2019; Jiang et al., 2021a; Pan et al., 2018). Another type of open-state inactivation, C-type inactivation (Hoshi et al., 1991), involves the rearrangement of the selectivity filter, and the underlying structural mechanisms have been explored (Cuello et al., 2010; Pau et al., 2017; Reddi et al., 2021; Tan et al., 2021; Wang and MacKinnon, 2017). Compared to open-state inactivation, close-state inactivation is arguably equally important for channel function, but its structural principles have remained enigmatic. Here we show that closed-state inactivation of KV4 channels involves a completely different mechanism that directly affects the channel gate through the S4-S5 linker and includes a breakdown of channel symmetry. In addition to providing key insights into the major form of closed-state inactivation, our work builds a framework for future studies of other forms of closed-state inactivation (Bahring and Covarrubias, 2011; Bahring et al., 2012; Yan et al., 2016).
Asymmetric pore collapse has been shown to be a mechanism of slow inactivation in the bacterial NaVAb channel (Payandeh et al., 2012). Such an inactivation process is fundamentally distinct from the closed-state inactivation of KV4, both functionally and structurally. While both channels show 2-fold symmetry in the pore region in their inactivated states, there are important differences. In the inactivated state of Kv4, the bottoms of S4 helices and the S4-S5 linkers undergo large conformational changes in only two out of the four subunits (Fig S7A). These regions are important channel elements that couple the voltage sensors to the gating of the pore. This is in sharp contrast to NaVAb, where the equivalent regions are in a same conformation within a tetramer (i.e., retain roughly 4-fold symmetry) (Fig S7A). Moreover, the pores of Kv4 and NaVAb collapse in different ways (Fig S7B), potentially due to the distinct movements of the S4-S5 linkers. Finally, the selectivity filter of KV4 does not change in the inactivated state, whereas the selectivity filter of NaVAb becomes asymmetric in its inactivated states (Payandeh et al., 2012). Ion channels have developed different gating mechanisms and alternative ways for inactivation, in order to fulfill different biological needs of cells, tissues and organisms. Nevertheless, the observation of symmetry transitions in inactivation of distantly related channels suggests that symmetry breakdown may be one common mechanism for voltage-gated channel inactivation, first uncovered by the seminal study of NaVAb (Payandeh et al., 2012).
Symmetry transition has also been observed in TRPV and TRPM channels (Yin et al., 2019; Zubcevic et al., 2018a, 2018b, 2019). However, the transition in TRP channels occurs in intermediate states during channel activation, in contrast to our findings with KV4 channels or the previous study of NaVAb. We speculate that a transition from four to two-fold symmetry may be a general feature for many tetrameric channels, but it could occur during different steps of channel gating.
Structures of several voltage-gated K+ channels have been reported, such as KV 1.2, KV1.2–2.1 chimera, KV7, KV11, KV12, and Ca2+-activated K+ channel (Hite et al., 2017; Long et al., 2005b, 2007; Matthies et al., 2018; Sun and MacKinnon, 2017; Tao et al., 2017; Wang and MacKinnon, 2017; Whicher and MacKinnon, 2016). These structures are determined in various conditions that populated different functional states, but in all of them, the voltage sensors are in the activated, “up” conformation characteristic of channels at depolarized membrane potential. For other voltage-gated ion channels, several approaches have been applied to stabilize the voltage sensors in hyperpolarized/deactivated/resting states for structural study, including the use of toxins (Clairfeuille et al., 2019; Xu et al., 2019), disulfide bridges (Wisedchaisri et al., 2019), divalent ligands (Guo et al., 2016), or metal bridges (Lee and MacKinnon, 2019). All of those methods directly target the voltage sensor. Here we develop an alternative approach and target the pore domain to stabilize the channel in a resting state. Guided by structural information, we engineered cysteine-mediated coordination of metal ion that takes place preferably in a resting state, and we used IIB-group metal ions to stabilize the channel in the resting state. With this approach, we succeeded in determining the first putative resting state structure of a voltage-gated K+ channel and anticipate that our approach can be applied to other voltage-gated ion channels or other channels in the superfamily, such as TRP channels, which involve coupling of the pore gating to the movement of S1-S4 helices.
The fact that we observed a conformational change on the S4 sensor when crosslinking the pore domain suggests that there is strong coupling between the pore and voltage sensor in the KV4 channel. We thus expect that under such crosslinked conditions, the S4 sensor is locked and the gating current of KV4 is largely reduced. This is distinct from the HCN channel, where one could lock the pore but the voltage sensor could still move (Ryu and Yellen, 2012). In HCN channels, the coupling between the voltage sensors and the channel gate is weak, so crosslinking at the pore did not immobilize the gating charge. Consistently, in the hyperpolarized HCN1 channel structure (Lee and MacKinnon, 2019), the voltage sensor is in a hyperpolarized conformation, but the pore (S6 helices) does not undergo larger conformational changes (a sign of weak coupling).
From the putative resting state to the open state, the S4 helix of KV4.2 is shifted by one helical turn toward the extracellular side and Arg299 is translocated across the gating charge transfer center (Figure 6A). This is different from voltage sensors in other voltage-gated ion channels, where the S4 helix moves by two-helical turns and two positive-charged residues are translocated (Clairfeuille et al., 2019; Guo et al., 2016; Jiang et al., 2021b; Lee and MacKinnon, 2019; Wisedchaisri et al., 2019; Xu et al., 2019). Since positively charged gating residues are aligned into a single file on the S4 helix, one would expect them to move across the center sequentially; thus, the structural transition we observed in KV4 may be the final step towards the open conformation. It is also possible the movement of the S4 helix and its gating charges during activation might differ across voltage-gated ion channels, despite their overall conserved architecture. For example, while the voltage sensors of KAT1 and HCN1 channels show very similar architecture (Clark et al., 2020; Lee and MacKinnon, 2017), the S4 sensor of KAT1 is suggested to move only ~5–7 Å (one gating charge translocated) (Clark et al., 2020), compared to the ~10 Å in HCN1 (Lee and MacKinnon, 2019).
Limitations of the study
Our work provides a general framework that captures the essence of KV4 gating. We reported four distinct channel conformations that likely represent the resting, inactivated, open, and intermediate, and open states of the channel gating cycle. Our assignment of channel structures to functional states are based on cryo-EM structural analyses and on the electrophysiology experiments, and the results from those orthogonal approaches are consistent with each other. However, we cannot rule out other alternative interpretations of state assignments. For example, the inactivated conformation we observed may reflect another intermediate state that is on the pathway toward the open state, although the existence of such a transient state with a breakdown of symmetry had not been reported in KV4 before.
We used a crosslinker to stabilize a resting state of the KV4.2 channel and are reasonably confident that the conformation stabilized is similar to a conformation favored by the channel in the resting membrane potential. However, it is possible that the crosslinker could slightly alter the nature of the resting structure. We also note that voltage-gated ion channels can have multiple resting states (DeCaen et al., 2008, 2009, 2011; Yarov-Yarovoy et al., 2012). Our electrophysiology and structural data suggest that our structure represent a bona fide physiologically relevant resting state, but we do not claim this is the only resting state. We did not observe that the KV4 sensor further moves downward (i.e., two gating charges translocated) to another resting state, but we could not exclude the possibility that the KV4 sensor in detergent may still have some mobility even after crosslinking, and the sensor gradually shifts its conformation to the resting conformation that we captured.
Finally, unlike the KV4.2EM/KChIP2 channel complex, only open conformation is observed for the KV4.2EM/KChIP2/DPP6 channel complex. Given that in electrophysiological experiments most channels inactivate at 0 mV (Figure 1D), it is likely that the detergent glyco-diosgenin used for purifying this particular complex, although beneficial for biochemical stability, inadvertently traps the channel in the open state. In line with this hypothesis, the recently reported KV4 complex structures, which are also determined in glyco-diosgenin, adopt the same open conformation (Kise et al., 2021; Ma et al., 2022). Despite these limitations, our study significantly expands our knowledge of channel inactivation and reveals previously unknown aspects of voltage-dependent activation in a K+ channel.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Chia-Hsueh Lee (ChiaHsueh.Lee@stjude.org)
Materials availability
Reagents generated in this study are available from the lead contact with a completed Materials Transfer Agreement.
Data and code availability
The structural models have been deposited in the Protein Data Bank under the accession codes: 7UK5, 7UKC, 7UKD, 7UKE, 7UKF, 7UKG and 7UKH. The density maps have been deposited in the Electron Microscopy Data Bank under the accession codes: EMD-26575, EMD-26576, EMD-26577, EMD-26578, EMD-26579, EMD-26580, and EMD-26581.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293 cells were cultured in DMEM with 4.5 g/L glucose, L-glutamine, and sodium pyruvate containing 10% fetal bovine serum and 1% penicillin-streptomycin, at 37 °C with 5% CO2. Sf9 cells were cultured in ESF 921 medium at 28 °C. HEK293S GnTI–cells were cultured in Freestyle 293 medium supplemented with 2% fetal bovine serum at 37 °C.
METHOD DETAILS
Construct design
The complementary DNA (cDNA) encoding human KV4.2 (NP_036413.1), KChIP2 (NP_775284), or DPP6 (AAI50305.1) was cloned into the pEG BacMam vector (Goehring et al., 2014). For KV4.2EM, the coding sequence was followed by a 3C protease cleavage site and a C-terminal green fluorescent protein (GFP) tag. For DPP6, a N-terminal mCherry tag was inserted before the coding sequence.
Site-directed mutagenesis were performed using Phusion or Q5 polymerase (New England Biolabs). For concatemer experiments, the cDNA encoding the second subunit was amplified, with EcoRI restriction site introduced to both ends, digested with EcoRI-HF (NEB BioLabs), and ligated into EcoRI-HF-digested, CIP (calf intestinal alkaline phosphatase, NEB BioLabs)-treated vector of the first subunit, using T4 DNA ligase (NEB BioLabs). The two subunits were connected by a SNSGGGGGGGGS linker.
For structural experiments, the expression cassette containing each gene was amplified and assembled into the pBIG1a vector using biGBac method (Weissmann et al., 2016). The multigene expression construct containing KV4.2EM/KChIP2 or KV4.2EM/KChIP2/DPP6 was used for large-scale protein expression. All constructs were verified with Sanger sequencing.
Cell culture for electrophysiology
HEK293 cells were lifted with trypsin-EDTA (Life Technologies) and plated in 6-well places (Warner Instruments) 3–4 d before recording. Transient transfection was performed with Lipofectamine 2000 (Thermo Fisher Scientific) 1 day before recording. For co-transfection, the vector ratio of KV4.2 to KChIP2, or of KV4.2 to DPP6 was 1:10. The ratio of concatemeric KV4.2 vector to KChIP2 vector was 1:20. Before recording, cells were lifted with trypsin-EDTA and kept in modified Tyrode’s saline (140 mM NaCl, 5 mM KCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, pH 7.2–7.3 with HCl), and were used within 8 hours. For recording, an aliquot of cells was transferred to a recording chamber on a Nikon-TE2000 Inverted Scope (Nikon Instruments), and transfection was confirmed with fluorescent microscopy. Cells were washed for 3 times with Tyrode’s saline. Once the recoding from a cell was completed, cells in the same chamber were discarded, and new aliquots of cells were replated for the next recording. All recordings were performed at room temperature (22–24 °C). All chemicals without notes were purchased from Sigma-Aldrich.
Electrophysiology
Patch borosilicate pipets (Sutter Instrument) were pulled from a Sutter P-97 puller with resistances of 2–3 MΩ. Data were acquired using an Axopatch 200B amplifier controlled by Clampex 10.2 via Digidata 1550A (Axon Instruments), sampled at 10 kHz, filtered at 2 kHz. Membrane capacitance was around 15 pF. Access resistance was around 5 MΩ. In most experiments, pipette solution contained 150 mM KCl, 10 mM HEPES, 5 mM EGTA, 2.5 mM CaCl2 (~100 nM free Ca2+). For experiments involving Hg2+ or Cd2+, 100 μM Hg2+ or 500 μM Cd2+ were added to pipette solution without EGTA and Ca2+. In most experiments, the bath was modified Tyrode’s solution as previously stated. For each solution, final pH was adjusted to 7.2–7.3 with KOH, NaOH or HCl. The osmolality of each solution was 290–310 mOsm/kg. MTS-1-MTS or MTS-4-MTS (Santa Cruz Biotechnology) were dissolved in glucose-free Tyrode’s solution, puffed with VC3–8xP pressurized perfusion system (ALA Science).
Steady-state activation and inactivation were recorded with a hybrid protocol consisting of a voltage family and a depolarizing step to +80 mV at the end of each sweep. In brief, membrane potential was held at −80 mV or −100 mV, and steady-state current was evoked by a voltage family protocol with 10-mV increments. In most experiments, each step lasted 0.5 s, unless for V404M, in which case the step was elongated to 3 s. A depolarizing step to +80 mV was applied at the end of each sweep for measuring steady-state inactivation.
For recordings of recovery from inactivation, membrane potential was held at −80 mV, and the current was first evoked by membrane depolarization to +80 mV, followed by hyperpolarization to −80 mV for a duration with appropriate increments for every sweep, and then the current was evoked again by membrane depolarization to +80 mV. Here, the first pulse was 0.5 s for most experiments, unless for channels including V404M subunit, in which case the pulse was elongated to 1 s, 2 s or otherwise stated. The increment per sweep of deactivation length varied from 5 ms to 500 ms, depending on whether reasonable data points can be obtained to fit with a single-exponential plateau growth curve. For measuring the time course of current decay, the currents were fitted with a two-exponential curve. For recordings of MTS reagents, Cd2+, and Hg2+ inhibitions, membrane potential was held at −80 mV, and the current was evoked by a depolarizing step to +80 mV for 0.2 s.
Statistical analyses for electrophysiology
All data were first visualized with pClamp10 (Molecular Devices). Data were then imported to Jupyter notebook with pyabf, analyzed and organized with open-source python packages: numpy, pandas and scipy. Data were saved as csv-format tables and imported into GraphPad Prism (GraphPad Software), with which graphs were generated. For activation curves, the peak conductance at every voltage was calculated from the respective peak current magnitude (see below). For each cell, peak conductances were then fit with the Boltzmann equation against voltages, yielding midpoint (activation V1/2), slope factor and maximal conductance (Gmax). For inactivation curves, the conductance was calculated by dividing the peak current magnitude at +80 mV following every conditioning voltage by the same driving force at +80 mV. Similarly, for each cell, peak conductances were then fit with the Boltzmann equation against conditioning voltages, yielding midpoint (inactivation V1/2), slope factor and maximal conductance (Gmax). For KV4 V404M mutant which manifest U-shaped inactivation, the ascending phase was not included in fitting. For both activation and inactivation curves, G/Gmax was calculated by normalizing the conductances of each cell to its respective Gmax, and was averaged by group at the end.
The equation for K+ equilibrium potential, which is needed to calculate conductance, is as follows: , where R is the universal gas constant (8.314 J K−1 mol−1), T is temperature (295 K), F is the Faraday’s constant (96485 C mol−1), [K]o and [K]i are extracellular and intracellular K+ concentrations. Conductance is calculated with the Ohm’s law: G = I / (Vm - EK).
For curves showing the time course of recovery, the current magnitude at each sweep evoked by the second depolarization pulse was normalized to that by the first pulse (I/Ictrl), and for each cell, the normalized current magnitudes (I/Ictrl) were fit with a single-exponential plateau growth equation against deactivation duration, yielding time constant (τ). I/Ictrl was averaged by group at the end.
Data represents mean ± SEM. For all channel intrinsic kinetic parameters, such as half-activation and half-inactivation voltages (V1/2), recovery time constants (τ) and the time to peak current, we assume normal distribution within each group and perform t-test or ANOVA for comparison. For recovery time constants (τ), we do not assume equal variations due to their several orders of magnitude difference. For experiments such as the change of current magnitude in response to treatments, we used non-parametric comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, n.s. p > 0.05.
Protein expression and purification
The KV4.2EM/KChIP2 complex or KV4.2EM/KChIP2/DPP6 complex was expressed in HEK293S GnTI- cells using the BacMam method (Goehring et al., 2014). Baculoviruses were produced by transfecting Sf9 cells with the bacmids using TransIT. After one rounds of amplification, viruses were used for cell transduction. When HEK293S GnTI–suspension cultures were grown at to the density of 3.5 × 106 cells/ml at 37 °C, baculoviruses (10 % v/v) were added to initiate transduction. After 14 hours, 10 mM sodium butyrate was supplemented to the cultures and the culture temperature was shifted to 30 °C. Cells were collected at 60 h post-transduction.
The pellet of cells expressing KV4.2EM/KChIP2 complex was resuspended using hypotonic buffer (10 mM KCl, 1 mM MgCl2, 20 mM Tris pH 8, benzonase, and protease inhibitors) for 30 min. The cell lysate was then spun at 39,800g for 30 min to sediment crude membranes. The membrane pellet was mechanically homogenized and solubilized in extraction buffer (10 mM LMNG, 2mM CHS, 150 mM KCl, 20 mM Tris pH 8, and protease inhibitors) for 1.5 hours. Solubilized membranes were clarified by centrifugation at 39,800g for 45 min. The supernatant was applied to the GFP nanobody-coupled Sepharose resin, which was subsequently washed with 10 column volumes of wash buffer (0.05 mM DDM, 0.01 mM CHS, 150 mM KCl, and 20 mM Tris pH 8). The washed resin was incubated with 3C protease overnight at a target protein to protease ratio of 40:1 (w/w) to cleave off GFP and release the protein from the resin. The protein was eluted with wash buffer, concentrated, and further purified by gel-filtration chromatography using a Superose 6 increase column equilibrated with SEC buffer (0.05 mM DDM, 0.01 mM CHS, 150 mM KCl, and 20 mM Tris pH 8). For KV4.2EM/KChIP2/DPP6 complex, wash buffer and SEC buffer contain 0.005% GDN instead of DDM and CHS. Peak fractions were pooled and concentrated to desired concentrations for cryo-EM experiments.
EM data acquisition
Prior to EM grid preparation, protein samples were concentrated to 6–7 mg/ml; 200 μM HgCl2 was added to the protein sample when required. Aliquots of 3.5-μl protein samples were applied to plasma-cleaned UltrAuFoil R1.2/1.3 300 mesh grids. After 15 s, the grids were blotted for 4 s and plunged into liquid ethane using a Vitrobot Mark IV (FEI) operated at 10 °C and 100% humidity. The grids were loaded onto a 300 kV Titan Krios transmission electron microscope with a K3 detector. Micrographs were recorded in super-resolution mode using SerialEM (Mastronarde, 2005) with a physical pixel size of 0.826 Å per pixel and a nominal defocus range of 0.6–1.6 μm. The exposure time for each micrograph was 2.4–3 s, dose-fractionated into 60–70 frames with a dose rate of 1.18–1.27 e−/pixel/s. Image acquisition parameters are summarized in Table S1.
EM data processing
The image stacks were gain-normalized, binned by 2, and corrected for beam-induced motion using MotionCor2 (Zheng et al., 2017). Defocus parameters were estimated from motion-corrected images using cryoSPARC3 (Punjani et al., 2017). Micrographs not suitable for further analysis were removed by manual inspection. Particle picking and 2D classifications were done in cryoSPARC3. The initial 3D models of KV4.2EM/KChIP2 and KV4.2EM/KChIP2/DPP6 complex were generated by cryoSPARC3 ab initio reconstruction. For the dataset of KV4.2EM/KChIP2, a consensus 3D refinement on selected particles was run in cryoSPARC3. A 3D classification focused on the transmembrane region was then performed without particle alignment using RELION 3 (Zivanov et al., 2018), which resulted in two inactivated classes, one open class and one intermediate class. The selected classes of particles were then refined in cryoSPARC3. Nonuniform refinements were performed (Punjani et al., 2019), followed by local refinements focusing on either the transmembrane region or the intracellular region with appropriate symmetry imposed. For the datasets of V406C mutant and KV4.2EM/KChIP2/DPP6, refinements were done in a similar manner in cryoSPARC3 without performing focused 3D classification in RELION. The mask-corrected FSC curves were calculated in cryoSPARC3, and reported resolutions were based on the 0.143 criterion. A summary of reconstructions is shown in Table S1.
Model building
Homology models of KChIP2 and cytosolic domains of KV4.2 were generated by the SWISS-MODEL server (Biasini et al., 2014). These models were docked into the density maps using Chimera (Pettersen et al., 2004). For the transmembrane helices of KV4.2 and DPP6, de novo models were built manually in Coot (Emsley et al., 2010). All models were then refined iteratively using Coot and Phenix (Afonine et al., 2018). The KV4.2 structural model includes KV4.2 residues 4–153, residues 166–451, and residues 472–497. The KChIP2 model includes residues 72–252. The DPP6 model includes residues 31–53. Structural model validation was done using Phenix and MolProbity (Chen et al., 2010). A summary of model refinement and validation is shown in Table S1. Figures were prepared using PyMOL and Chimera.
Quantification and Statistical Analysis
Resolution estimations of all cryo-EM density maps are based on the 0.143 criterion. Electrophysiological data in all figures are shown as mean ± SEM. Statistical significance is evaluated using the method indicated in the figure legends.
Supplementary Material
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial Strains | ||
| Turbo competent cells | New England Biolabs | Cat#C2984H |
| DH10Bac competent cells | Thermo Fisher Scientific | Cat#10361012 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Protease inhibitor cocktail | MedChemExpress | Cat#HY-K0010 |
| Lauryl maltose neopentyl glycol (LMNG) | Anatrace | Cat#NG310 |
| n-Dodecyl-β-D-Maltopyranoside (DDM) | Anatrace | Cat#D310S |
| Glyco-Diosgenin (GDN) | Anatrace | Cat#GDN101 |
| Cholesteryl hemisuccinate tris salt | Anatrace | Cat#CH210 |
| ESF 921 Medium | Expression Systems | Cat#96–001–01 |
| Freestyle 293 expression medium | Gibco | Cat#12338018 |
| DMEM/F-12 medium | Gibco | Cat#11330–032 |
| Penicillin-streptomycin solution, 100x | Corning | Cat#30–002-CI |
| Fetal bovine serum | Gibco | Cat#16140089 |
| Lipofectamine 2000 reagent | Invitrogen | Cat#11668019 |
| TransIT-Insect transfection reagent | Mirus | Cat#MIR6100 |
| 1,1-Methanediyl Bismethanethiosulfonate (MTS-1-MTS) | Santa Cruz Biotech | Cat#SC-208706 |
| 1,4-Butanediyl Bismethanethiosulfonate (MTS-4-MTS) | Santa Cruz Biotech | Cat#SC-208789 |
| Deposited Data | ||
| KV4.2/KChIP2 channel complex, open state | This study | PDB: 7UK5; EMDB: EMD-26575 |
| KV4.2/KChIP2 channel complex, inactivated state, class 1 | This study | PDB: 7UKC; EMDB: EMD-26576 |
| KV4.2/KChIP2 channel complex, inactivated state, class 2 | This study | PDB: 7UKD; EMDB: EMD-26577 |
| KV4.2/KChIP2 channel complex, intermediate state | This study | PDB: 7UKE; EMDB: EMD-26578 |
| KV4.2/KChIP2 channel complex, putative resting state | This study | PDB: 7UKF; EMDB: EMD-26579 |
| KV4.2/KChIP2/DPP6 channel complex, open state, transmembrane region | This study | PDB: 7UKG; EMDB: EMD-26580 |
| KV4.2/KChIP2/DPP6 channel complex, open state, intracellular region | This study | PDB: 7UKH; EMDB: EMD-26581 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat#CRL-1573 |
| Sf9 | ATCC | Cat#CRL-1711 |
| HEK293S GnTI– | ATCC | Cat#CRL-3022 |
| Recombinant DNA | ||
| pEG-BacMam Human Kv4.2EM | This study | N/A |
| pEG-BacMam Human KChIP2 | This study | N/A |
| pEG-BacMam Human DPP6 | This study | N/A |
| Software and Algorithms | ||
| Serial EM | Mastronarde, 2005 | http://bio3d.colorado.edu/SerialEM |
| CTFFIND4 | Rohou and Grigorieff, 2015 | http://grigoriefflab.janelia.org/ctffind4 |
| RELION | Scheres, 2012 | http://www2.mrclmb.cam.ac.uk/relion |
| cryoSPARC | Punjani et al., 2017 | https://cryosparc.com/ |
| Coot | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| MolProbity | Williams et al., 2018 | http://molprobity.biochem.duke.edu |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera |
| PyMOL | Schrodinger | http://www.pymol.org |
| pCLAMP | Molecular Devices | https://www.moleculardevices.com/ |
| Anaconda | Anaconda | https://www.anaconda.com/ |
| Other | ||
| CNBR-activated Sepharose beads | Cytiva | Cat#17043001 |
| Superose 6 Increase 10/300 GL SEC column | Cytiva | Cat#29091596 |
| R1.2/1.3 400 mesh UltrAuFoil gold grids | Quantifoil | Cat#Q350AR13A |
Highlights.
KV4 channel structures in closed, intermediate, inactivated, and open states are solved
Upon closed-state inactivation, the pore collapses into a 2-fold symmetric conformation
Only two of the four S4-S5 linkers move to seal the pore during inactivation
Comparison of resting and open states reveals the activation mechanism of the channel
Acknowledgments
We thank M. Grabe, I. Chen, W. Lü, C. Zhao, J. Payandeh, Y. Niu and F. Liu for helpful discussions. We thank A. Myasnikov for assistance in cryo-EM data collection and staffs at the Cryo-EM Center of St. Jude Children’s Research Hospital for support with computer infrastructure. W.Y. was partially supported by Sandler program for breakthrough biomedical research, through Postdoctoral Independent Research Program of University of California, San Francisco. L.Y.J. is an investigator of the Howard Hughes Medical Institute. This study was supported by NIH (R01MH065334, L.Y.J.; R01GM143282, C.-H.L.) and by ALSAC (C.-H.L.).
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, and Adams PD (2018). Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74, 531–544. 10.1107/S2059798318006551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajsuvakova OP, Tinkov AA, Aschner M, Rocha JBT, Michalke B, Skalnaya MG, Skalny AV, Butnariu M, Dadar M, Sarac I, et al. (2020). Sulfhydryl groups as targets of mercury toxicity. Coord Chem Rev 417, 213343. 10.1016/j.ccr.2020.213343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alders M, Koopmann TT, Christiaans I, Postema PG, Beekman L, Tanck MWT, Zeppenfeld K, Loh P, Koch KT, Demolombe S, et al. (2009). Haplotype-Sharing Analysis Implicates Chromosome 7q36 Harboring DPP6 in Familial Idiopathic Ventricular Fibrillation. The American Journal of Human Genetics 84, 468–476. 10.1016/j.ajhg.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW, and Stevens CF (1983). Inactivation of Open and Closed Sodium Channels Determined Separately. Cold Spring Harb Symp Quant Biol 48, 147–153. 10.1101/SQB.1983.048.01.017. [DOI] [PubMed] [Google Scholar]
- An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, et al. (2000). Modulation of A-type potassium channels by a family of calcium sensors. Nature 403, 553–556. 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, and Hollingworth S (2018). A perspective on Na and K channel inactivation. J Gen Physiol 150, 7–18. 10.1085/jgp.201711835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F, and Rojas E (1973). Destruction of Sodium Conductance Inactivation in Squid Axons Perfused with Pronase. Journal of General Physiology 62, 375–391. 10.1085/jgp.62.4.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahring R, and Covarrubias M (2011). Mechanisms of closed-state inactivation in voltage-gated ion channels. J Physiol 589, 461–479. 10.1113/jphysiol.2010.191965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahring R, Barghaan J, Westermeier R, and Wollberg J (2012). Voltage sensor inactivation in potassium channels. Front Pharmacol 3, 100. 10.3389/fphar.2012.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin TJ, Tsaur ML, Lopez GA, Jan YN, and Jan LY (1991). Characterization of a mammalian cDNA for an inactivating voltage-sensitive K+ channel. Neuron 7, 471–483. 10.1016/0896-6273(91)90299-f. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42, W252–W258. 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Liang CW, Muralidharan S, Muralidharan S, Kao JPY, Tang C-M, and Thompson SM (2004). Unique roles of SK and Kv4.2 potassium channels in dendritic integration. Neuron 44, 351–364. 10.1016/j.neuron.2004.09.026. [DOI] [PubMed] [Google Scholar]
- Callsen B, Isbrandt D, Sauter K, Hartmann LS, Pongs O, and Bahring R (2005). Contribution of N- and C-terminal Kv4.2 channel domains to KChIP interaction [corrected]. J Physiol 568, 397–412. 10.1113/jphysiol.2005.094359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan L-L, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, and Johnston D (2006). Deletion of Kv4.2 Gene Eliminates Dendritic A-Type K+ Current and Enhances Induction of Long-Term Potentiation in Hippocampal CA1 Pyramidal Neurons. J. Neurosci. 26, 12143–12151. 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clairfeuille T, Cloake A, Infield DT, Llongueras JP, Arthur CP, Li ZR, Jian Y, Martin-Eauclaire MF, Bougis PE, Ciferri C, et al. (2019). Structural basis of alpha-scorpion toxin action on Nav channels. Science 363. 10.1126/science.aav8573. [DOI] [PubMed] [Google Scholar]
- Clark MD, Contreras GF, Shen R, and Perozo E (2020). Electromechanical coupling in the hyperpolarization-activated K+ channel KAT1. Nature 583, 145–149. 10.1038/s41586-020-2335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, and Perozo E (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature 466, 203–208. 10.1038/nature09153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Zhao Y, Scheuer T, and Catterall WA (2008). Disulfide locking a sodium channel voltage sensor reveals ion pair formation during activation. Proc Natl Acad Sci U S A 105, 15142–15147. 10.1073/pnas.0806486105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Sharp EM, Scheuer T, and Catterall WA (2009). Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc Natl Acad Sci U S A 106, 22498–22503. 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCaen PG, Yarov-Yarovoy V, Scheuer T, and Catterall WA (2011). Gating charge interactions with the S1 segment during activation of a Na+ channel voltage sensor. Proc Natl Acad Sci U S A 108, 18825–18830. 10.1073/pnas.1116449108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarri A, Lin MC, Fokkens MR, Meijer M, Smeets CJ, Nibbeling EA, Boddeke E, Sinke RJ, Kampinga HH, Papazian DM, et al. (2015). Spinocerebellar ataxia type 19/22 mutations alter heterocomplex Kv4.3 channel function and gating in a dominant manner. Cell Mol Life Sci 72, 3387–3399. 10.1007/s00018-015-1894-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Sukomon N, Flood E, Rheinberger J, Allen TW, and Nimigean CM (2020). Ball-and-chain inactivation in a calcium-gated potassium channel. Nature 580, 288–293. 10.1038/s41586-020-2116-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring A, Lee C-H, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, and Gouaux E (2014). Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585. 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Zeng W, Chen Q, Lee C, Chen L, Yang Y, Cang C, Ren D, and Jiang Y (2016). Structure of the voltage-gated two-pore channel TPC1 from Arabidopsis thaliana. Nature 531, 196–201. 10.1038/nature16446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ion Channels of Excitable Membranes (Sinauer Associates, Inc.). [Google Scholar]
- Hite RK, Tao X, and MacKinnon R (2017). Structural basis for gating the high-conductance Ca(2+)-activated K(+) channel. Nature 541, 52–57. 10.1038/nature20775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, and Armstrong CM (2013). C-type inactivation of voltage-gated K+ channels: Pore constriction or dilation? Journal of General Physiology 141, 151–160. 10.1085/jgp.201210888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, and Aldrich RW (1990). Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250, 533–538. 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, and Aldrich RW (1991). Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron 7, 547–556. 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Hu J-H, Malloy C, Tabor GT, Gutzmann JJ, Liu Y, Abebe D, Karlsson R-M, Durell S, Cameron HA, and Hoffman DA (2020). Activity-dependent isomerization of Kv4.2 by Pin1 regulates cognitive flexibility. Nat Commun 11, 1567. 10.1038/s41467-020-15390-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, and Pfaffinger PJ (2014). Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Front Cell Neurosci 8, 82. 10.3389/fncel.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, and Covarrubias M (2004). Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci 27, 343–369. 10.1016/j.mcn.2004.06.011. [DOI] [PubMed] [Google Scholar]
- Jiang D, Banh R, Gamal El-Din TM, Tonggu L, Lenaeus MJ, Pomes R, Zheng N, and Catterall WA (2021a). Open-state structure and pore gating mechanism of the cardiac sodium channel. Cell 184, 5151–5162 e11. 10.1016/j.cell.2021.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D, Tonggu L, Gamal El-Din TM, Banh R, Pomes R, Zheng N, and Catterall WA (2021b). Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat Commun 12, 128. 10.1038/s41467-020-20078-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Wei D-S, and Hoffman DA (2005). Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurones. J Physiol 569, 41–57. 10.1113/jphysiol.2005.095042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim LA, Furst J, Gutierrez D, Butler MH, Xu S, Goldstein SAN, and Grigorieff N (2004). Three-Dimensional Structure of Ito: Kv4.2-KChIP2 Ion Channels by Electron Microscopy at 21 Å Resolution. Neuron 41, 513–519. 10.1016/S0896-6273(04)00050-9. [DOI] [PubMed] [Google Scholar]
- Kintzer AF, and Stroud RM (2016). Structure, inhibition and regulation of two-pore channel TPC1 from Arabidopsis thaliana. Nature 531, 258–262. 10.1038/nature17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kise Y, Kasuya G, Okamoto HH, Yamanouchi D, Kobayashi K, Kusakizako T, Nishizawa T, Nakajo K, and Nureki O (2021). Structural basis of gating modulation of Kv4 channel complexes. Nature 10.1038/s41586-021-03935-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, and MacKinnon R (2017). Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell 168, 111–120.e11. 10.1016/j.cell.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CH, and MacKinnon R (2019). Voltage Sensor Movements during Hyperpolarization in the HCN Channel. Cell 179, 1582–1589 e7. 10.1016/j.cell.2019.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lin MC, Kornblum HI, Papazian DM, and Nelson SF (2014). Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 23, 3481–3489. 10.1093/hmg/ddu056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MA, Cannon SC, and Papazian DM (2018). Kv4.2 autism and epilepsy mutation enhances inactivation of closed channels but impairs access to inactivated state after opening. Proc Natl Acad Sci U S A 115, E3559–E3568. 10.1073/pnas.1717082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linsdell P (2015). Metal bridges to probe membrane ion channel structure and function. Biomolecular Concepts 6, 191–203. 10.1515/bmc-2015-0013. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, and Mackinnon R (2005a). Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903. 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, and MacKinnon R (2005b). Voltage Sensor of Kv1.2: Structural Basis of Electromechanical Coupling. Science 309, 903–908. 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, and MacKinnon R (2007). Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 450, 376–382. 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Ma D, Zhao C, Wang X, Li X, Zha Y, Zhang Y, Fu G, Liang P, Guo J, and Lai D (2022). Structural basis for the gating modulation of Kv4.3 by auxiliary subunits. Cell Res 32, 411–414. 10.1038/s41422-021-00608-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51. 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Matthies D, Bae C, Toombes GE, Fox T, Bartesaghi A, Subramaniam S, and Swartz KJ (2018). Single-particle cryo-EM structure of a voltage-activated potassium channel in lipid nanodiscs. Elife 7. 10.7554/eLife.37558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldenhauer H, Díaz-Franulic I, González-Nilo F, and Naranjo D (2016). Effective pore size and radius of capture for K+ ions in K-channels. Sci Rep 6, 19893. 10.1038/srep19893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadal MS, Ozaita A, Amarillo Y, de Miera EV-S, Ma Y, Mo W, Goldberg EM, Misumi Y, Ikehara Y, Neubert TA, et al. (2003). The CD26-Related Dipeptidyl Aminopeptidase-like Protein DPPX Is a Critical Component of Neuronal A-Type K+ Channels. Neuron 37, 449–461. 10.1016/S0896-6273(02)01185-6. [DOI] [PubMed] [Google Scholar]
- Pan X, Li Z, Zhou Q, Shen H, Wu K, Huang X, Chen J, Zhang J, Zhu X, Lei J, et al. (2018). Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 362. 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Pau V, Zhou Y, Ramu Y, Xu Y, and Lu Z (2017). Crystal structure of an inactivated mutant mammalian voltage-gated K(+) channel. Nat Struct Mol Biol 24, 857–865. 10.1038/nsmb.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, and Catterall WA (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139. 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pioletti M, Findeisen F, Hura GL, and Minor DL Jr. (2006). Three-dimensional structure of the KChIP1-Kv4.3 T1 complex reveals a cross-shaped octamer. Nat Struct Mol Biol 13, 987–995. 10.1038/nsmb1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14, 290–296. 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Punjani A, Zhang H, and Fleet DJ (2019). Non-uniform refinement: Adaptive regularization improves single particle cryo-EM reconstruction. BioRxiv 2019.12.15.877092. 10.1101/2019.12.15.877092. [DOI] [PubMed] [Google Scholar]
- Reddi R, Matulef K, Riederer EA, Whorton MR, and Valiyaveetil FI (2021). Structural basis for C-type inactivation in a Shaker family voltage gated K+ channel. BioRxiv 2021.10.01.462615. 10.1101/2021.10.01.462615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu S, and Yellen G (2012). Charge movement in gating-locked HCN channels reveals weak coupling of voltage sensors and gate. J Gen Physiol 140, 469–479. 10.1085/jgp.201210850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, and Covarrubias M (2003). The link between ion permeation and inactivation gating of Kv4 potassium channels. Biophys J 84, 928–941. 10.1016/S0006-3495(03)74910-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smets K, Duarri A, Deconinck T, Ceulemans B, van de Warrenburg BP, Züchner S, Gonzalez MA, Schüle R, Synofzik M, Van der Aa N, et al. (2015). First de novo KCND3 mutation causes severe Kv4.3 channel dysfunction leading to early onset cerebellar ataxia, intellectual disability, oral apraxia and epilepsy. BMC Medical Genetics 16, 51. 10.1186/s12881-015-0200-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, and MacKinnon R (2017). Cryo-EM Structure of a KCNQ1/CaM Complex Reveals Insights into Congenital Long QT Syndrome. Cell 169, 1042–1050 e9. 10.1016/j.cell.2017.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X-F, Bae C, Stix R, Fernandez AI, Huffer K, Chang T-H, Jiang J, Faraldo-Gómez JD, and Swartz KJ (2021). Structure of the Shaker Kv channel and mechanism of slow C-type inactivation. BioRxiv 2021.09.21.461258. 10.1101/2021.09.21.461258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Hite RK, and MacKinnon R (2017). Cryo-EM structure of the open high-conductance Ca(2+)-activated K(+) channel. Nature 541, 46–51. 10.1038/nature20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, and MacKinnon R (2017). Cryo-EM Structure of the Open Human Ether-a-go-go-Related K(+) Channel hERG. Cell 169, 422–430 e10. 10.1016/j.cell.2017.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yan Y, Liu Q, Huang Y, Shen Y, Chen L, Chen Y, Yang Q, Hao Q, Wang K, et al. (2007). Structural basis for modulation of Kv4 K+ channels by auxiliary KChIP subunits. Nat Neurosci 10, 32–39. 10.1038/nn1822. [DOI] [PubMed] [Google Scholar]
- Webster SM, Del Camino D, Dekker JP, and Yellen G (2004). Intracellular gate opening in Shaker K+ channels defined by high-affinity metal bridges. Nature 428, 864–868. 10.1038/nature02468. [DOI] [PubMed] [Google Scholar]
- Weissmann F, Petzold G, VanderLinden R, Huis in ‘t Veld PJ, Brown NG, Lampert F, Westermann S, Stark H, Schulman BA, and Peters J-M (2016). biGBac enables rapid gene assembly for the expression of large multisubunit protein complexes. Proc Natl Acad Sci U S A 113, E2564–E2569. 10.1073/pnas.1604935113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher JR, and MacKinnon R (2016). Structure of the voltage-gated K(+) channel Eag1 reveals an alternative voltage sensing mechanism. Science 353, 664–669. 10.1126/science.aaf8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisedchaisri G, Tonggu L, McCord E, Gamal El-Din TM, Wang L, Zheng N, and Catterall WA (2019). Resting-State Structure and Gating Mechanism of a Voltage-Gated Sodium Channel. Cell 178, 993–1003 e12. 10.1016/j.cell.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Li T, Rohou A, Arthur CP, Tzakoniati F, Wong E, Estevez A, Kugel C, Franke Y, Chen J, et al. (2019). Structural Basis of Nav1.7 Inhibition by a Gating-Modifier Spider Toxin. Cell 176, 702–715 e14. 10.1016/j.cell.2018.12.018. [DOI] [PubMed] [Google Scholar]
- Yan J, Li Q, and Aldrich RW (2016). Closed state-coupled C-type inactivation in BK channels. Proc Natl Acad Sci U S A 113, 6991–6996. 10.1073/pnas.1607584113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, DeCaen PG, Westenbroek RE, Pan C-Y, Scheuer T, Baker D, and Catterall WA (2012). Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc Natl Acad Sci U S A 109, E93–102. 10.1073/pnas.1118434109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Wu M, Hsu AL, Borschel WF, Borgnia MJ, Lander GC, and Lee SY (2019). Visualizing structural transitions of ligand-dependent gating of the TRPM2 channel. Nat Commun 10, 3740. 10.1038/s41467-019-11733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, and Aldrich RW (1990). Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science 250, 568–571. 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Tachtsidis G, Schob C, Koko M, Hedrich UBS, Lerche H, Lemke JR, van Haeringen A, Ruivenkamp C, Prescott T, et al. (2021). KCND2 variants associated with global developmental delay differentially impair Kv4.2 channel gating. Human Molecular Genetics 10.1093/hmg/ddab192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J-P, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2 - anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14, 331–332. 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. ELife 7, e42166. 10.7554/eLife.42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubcevic L, Herzik MA, Wu M, Borschel WF, Hirschi M, Song AS, Lander GC, and Lee S-Y (2018a). Conformational ensemble of the human TRPV3 ion channel. Nat Commun 9, 4773. 10.1038/s41467-018-07117-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubcevic L, Le S, Yang H, and Lee S-Y (2018b). Conformational plasticity in the selectivity filter of the TRPV2 ion channel. Nat Struct Mol Biol 25, 405–415. 10.1038/s41594-018-0059-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubcevic L, Hsu AL, Borgnia MJ, and Lee S-Y (2019). Symmetry transitions during gating of the TRPV2 ion channel in lipid membranes. ELife 8, e45779. 10.7554/eLife.45779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The structural models have been deposited in the Protein Data Bank under the accession codes: 7UK5, 7UKC, 7UKD, 7UKE, 7UKF, 7UKG and 7UKH. The density maps have been deposited in the Electron Microscopy Data Bank under the accession codes: EMD-26575, EMD-26576, EMD-26577, EMD-26578, EMD-26579, EMD-26580, and EMD-26581.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.