

Summary
RAF protein kinases are effectors of the GTP-bound form of the small guanosine triphosphatase RAS and function by phosphorylating MEK. We showed here that expression of ARAF activated RAS in a kinase-independent manner. Binding of ARAF to RAS displaced the GTPase-activating protein NF1 and antagonized NF1-mediated inhibition of RAS. This reduced ERK-dependent inhibition of RAS and increased RAS-GTP. By this mechanism, ARAF regulated the duration and consequences of RTK-induced RAS activation and supported the RAS output of RTK-dependent tumor cells. In human lung cancers with EGFR mutation, amplification of ARAF was associated with acquired resistance to EGFR inhibitors, which was overcome by combining EGFR inhibitors with an inhibitor of the protein tyrosine phosphatase SHP2 to enhance inhibition of nucleotide exchange and RAS activation.
Keywords: ARAF, RAS-GTP, NF1, ERK signaling, Receptor tyrosine Kinase inhibitor, Drug Sensitivity
In Brief
Su et al. reveal that ARAF activates RAS by antagonizing its binding to the NF1 RAS-GAP. By this mechanism, the abundance of ARAF determines RAS-GTP levels and the duration and biologic consequences of RTK-activation of RAS signaling. Moreover, ARAF amplification causes resistance of lung cancers to EGFR inhibition.
Graphical Abstract
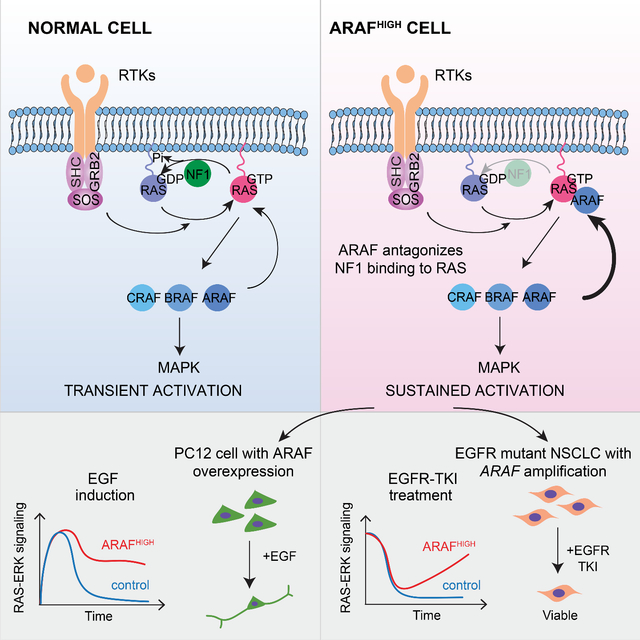
Introduction
The small guanosine triphosphatase RAS signaling pathway regulates multiple cellular functions including proliferation and its dysregulation is a key feature of about half of human tumors (Simanshu et al., 2017). The three RAS family members are 21kd proteins that are converted to their activated GTP-bound state by GDP-GTP exchange factors that are stimulated by upstream receptors. In the GTP-bound state, RAS binds to and activates multiple effector proteins. Ras proteins have endogenous GTPase activity and physiologic regulation of the signal thus involves activation of exchange factors and termination of the signal by hydrolysis of GTP. Endogenous RAS GTPase activity is not itself sufficient for physiologic inactivation and RAS also binds members of a family of GTPase activating proteins (GAPs) that enhance GTP hydrolysis and are required for normal regulation (Simanshu et al., 2017). RAS activity is also modulated by a complex array of ERK protein kinase-dependent negative feedback loops, a subset of which down-regulate upstream activation of nucleotide-exchange (Dong et al., 1996).
The RAS effectors include the three members of the RAF protein kinase family, activation of which promotes cell proliferation (Drosten et al., 2010). RAS-GTP binds to RAF family members and causes their homo- and hetero-dimerization and activation of their kinase activity (Freeman et al., 2013). This constitutes the first step of a kinase cascade: RAF phosphorylates and activates mitogen-activated protein kinase kinases (MEK), which in turn phosphorylates and activates extracellular signal regulated protein kinases 1 and 2 (ERK and ERK2). Phosphorylation of multiple substrates by ERK is required for RAS-induced proliferation in many settings. Activating mutations of members of the RAF, MEK and ERK families have all been identified in cancers with BRAF mutations being by far the most common (Davies et al., 2002; Gao et al., 2018; Goetz et al., 2014). CRAF and ARAF mutations also occur in human cancer but are much rarer. BRAF and CRAF both encode active kinases, knockout of which significantly decreases ERK signaling (Wojnowski et al., 2000). Kinase-independent effects of CRAF have been associated with various cellular phenotypes, including regulation of mitochondrial-dependent apoptosis by blocking the MST2/Hippo pathway (Alavi et al., 2003; Chen et al., 2001; O’Neill et al., 2004; Romano et al., 2014). Less is known about ARAF. It has lower endogenous kinase activity than B- or C-RAF (Marais et al., 1997) and reducing its expression does not impair ERK signaling in mouse embryonic fibroblasts (Mercer et al., 2002). Whereas knockouts of either BRAF or CRAF in mice are embryonic lethal (Mikula et al., 2001; Wojnowski et al., 1997), ARAF knockouts survive to term, although they die soon thereafter with neurologic and intestinal disorders. (Pritchard et al., 1996).
Recurrent ARAF mutations occur in and probably drive human tumors, most notably in the malignant histiocytoses and in adenocarcinomas of the lung (Diamond et al., 2016; Imielinski et al., 2014). We found that these mutants function as constitutively activated RAS-independent dimers, analogous to Class 2 BRAF mutants in all but one respect. ERK activation by activated BRAF mutants causes feedback inhibition of RAS activity, whereas ERK activation by ARAF mutants does not. Moreover, ectopic expression of mutant or wildtype (WT) ARAF increased RAS-GTP in cells. Investigation of these phenomena revealed a mechanism for activating RAS that is mediated by a kinase-independent function of ARAF.
Results
ARAF S214 mutants signal as RAS-independent dimers but do not cause feedback inhibition of cellular RAS-GTP
Recurrent mutations in ARAF have been detected in human tumors (Figure S1A). Somatic mutations of ARAF S214 that activate ERK signaling are oncogenic drivers in lung adenocarcinoma (Imielinski et al., 2014) and in eight other tumor types, including colorectal adenocarcinoma, melanoma and Langerhans cell histiocytosis (http://www.cbioportal.org/and http://cancerhotspots.org/) (Cerami et al., 2012; Chang et al., 2018; Diamond et al., 2016; Sia et al., 2015). Two classes of activated BRAF mutants have been identified: those that can activate ERK signaling as active, RAS-independent monomers and those that function as RAS-independent constitutive dimers. These mutants are thus unaffected by ERK-dependent feedback inhibition of RAS and drive high levels of ERK output (Pratilas et al., 2009; Yao et al., 2015; Yao et al., 2017). To determine the RAS-dependence of ARAF mutants, we used conditional mouse embryonic fibroblasts (MEFs) in which tamoxifen induced the knockout of Kras, the only RAS gene in these cells, and abrogated ERK signaling (Drosten et al., 2014). ARAF S214-mutant alleles supported ERK signaling in these cells after tamoxifen-induced RAS knockout, but WT ARAF did not (Figure 1A). The in vitro kinase activity of ARAF S214 mutant protein purified from these cells is similar to that of mutant enzyme isolated from control MEF cells (Figure S1B). Moreover, ARAF S214 mutants, but not WT ARAF, supported cell proliferation in MEF cells lacking RAS (Figure S1C). Thus, the catalytic activity of ARAF S214 is RAS-independent. In addition, the R362H mutation, which is homologous to the R509H and R401H mutations that abrogate dimerization in BRAF and CRAF (Mooz et al., 2014; Poulikakos et al., 2010), abolished activation of ERK by ARAF S214 in MEF cells lacking RAS (Figure 1B). Thus, ARAF S214 functions in cells as an activated RAS-independent dimer. Moreover, BRAF S365 and CRAF S259 are homologs of ARAF S214, and their mutant alleles also function as RAS-independent dimers in human tumors (Figures S1D and S1E).
Figure 1. ARAF S214 mutants signal as RAS-independent dimers but do not result in feedback inhibition of cellular RAS-GTP. See also Figure S1.
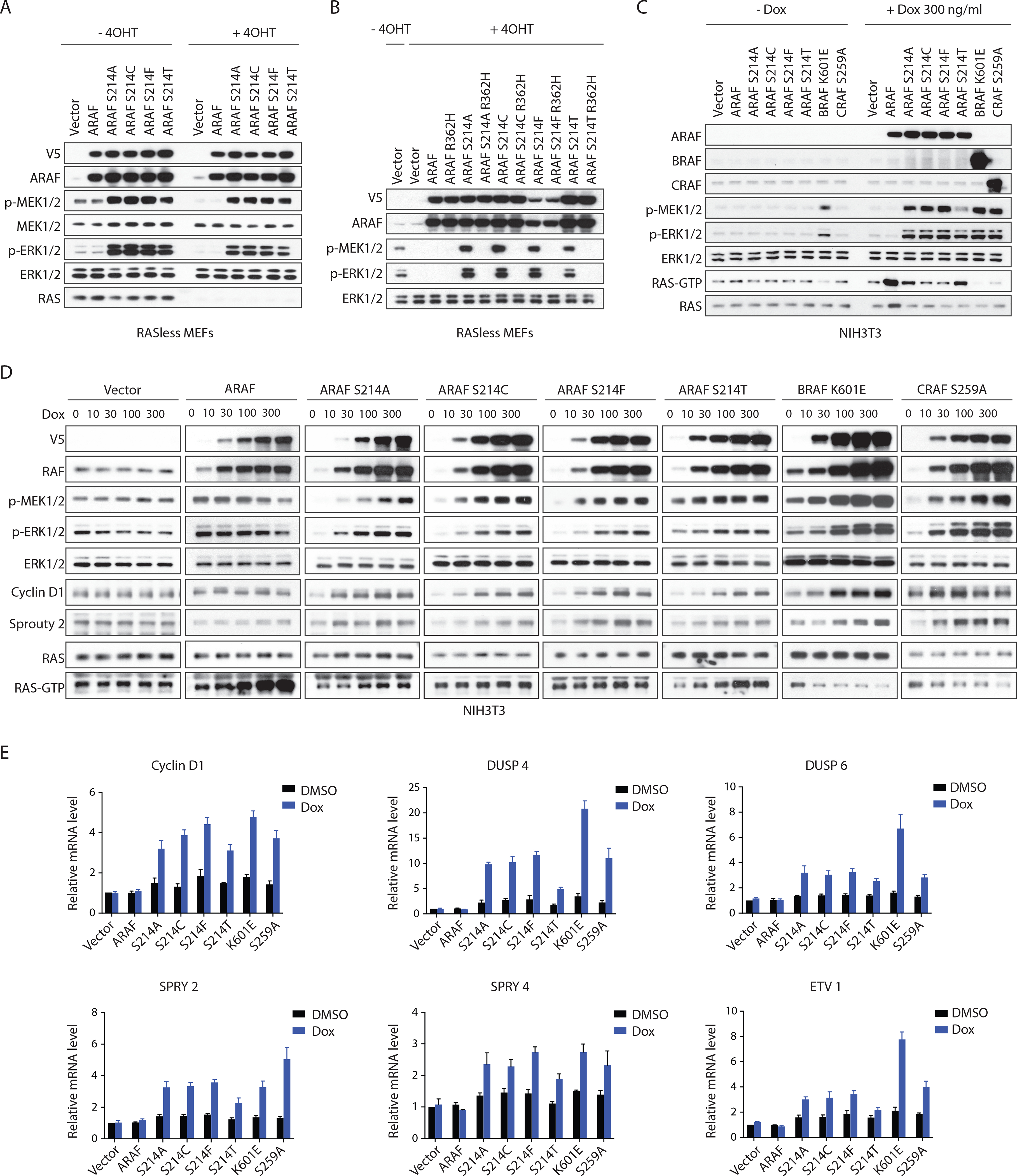
(A) K-Raslox MEF cells transduced with retrovirus carrying doxycycline-inducible WT ARAF or the indicated mutants were grown in medium without or with 1 μM 4-OHT for a week to generate isogenic cells expressing or lacking K-RAS. Cells were then treated with 100 ng/ml doxycycline for 24 hours and subjected to western blot.
(B) K-Raslox MEF cells lacking K-RAS expression were transfected with indicated constructs and subjected to western blot.
(C and D) NIH3T3 cells expressing doxycycline-inducible RAF variants were treated with doxycycline for 48 hr. Signaling activity and RAS-GTP levels were then examined by western blot.
(E) NIH3T3 cells expressing doxycycline-inducible RAF variants were treated with 100 ng/ml doxycycline for 24 hours. Relative expression levels of ERK target genes were examined by qRT-PCR. Error bars, mean±SD triplicate of experiments.
Activating mutants of BRAF, including those that function as constitutive dimers such as BRAF K601E, activate ERK signaling, which, in turn, causes feedback inhibition of RAS activation by inducing the transcription of negative regulators of upstream signaling and by direct inhibitory phosphorylation of upstream regulators such as Son of Sevenless (SOS) (Kamioka et al., 2010; Pratilas et al., 2009; Yao et al., 2015). Thus, in cells expressing activated BRAF mutants, levels of GTP-bound RAS are low (Figures 1C and 1D). By contrast, expression of ARAF S214 mutants in NIH3T3 cells increased ERK signaling (Figures 1C and 1D) and its transcriptional output (Figure 1E) but did not reduce RAS-GTP levels. In fact, some of them increase GTP-bound RAS (Figures 1C and 1D). Induction of RAS-GTP is not a property of activating mutants of BRAF or CRAF, including CRAF S259A, a hotspot mutant similar to ARAF S214 that is activated but reduces RAS-GTP (Figures 1C and 1D). Expression of WT ARAF also increased RAS-GTP but slightly suppressed ERK signaling when expressed at high concentrations (Figures 1C and 1D). ARAF overexpression was associated with a decrease in formation of BRAF/CRAF heterodimers and an increase in BRAF/ARAF heterodimers (Figure S1F). This shift in heterodimer abundance may explain the decrease in ERK phosphorylation, as ARAF has lower kinase activity than BRAF or CRAF and BRAF/CRAF dimers are more active than ARAF dimers (Baljuls et al., 2007; Freeman et al., 2013). Thus, overexpression of WT ARAF can induce RAS activation, and none of the ARAF S214 mutant alleles inhibits RAS activity.
ARAF increases GTP-bound WT RAS in a kinase-independent manner
We tested whether induction of RAS activation is a specific property of ARAF proteins. Overexpression of either WT BRAF or WT CRAF (8~ 15-fold increases over endogenous concentrations) in NIH3T3 cells, caused small increases in ERK signaling but had little or no effect on cellular RAS-GTP (Figure 2A) By contrast, increasing WT ARAF caused a concentration-dependent increase in RAS-GTP accompanied by a decrease in ERK signaling (Figure 2A and 2B). Multiple ARAF S214 mutant alleles also increased RAS-GTP in a concentration-dependent manner (Figure S2A). The binding of WT and mutant ARAF to RAS increased with increasing levels of ARAF expression, but WT ARAF caused a greater increase in RAS-GTP than any of the activating mutants did (Figures 1C and S2A).
Figure 2. ARAF increases GTP-bound WT RAS in a kinase-independent manner. See also Figure S2.
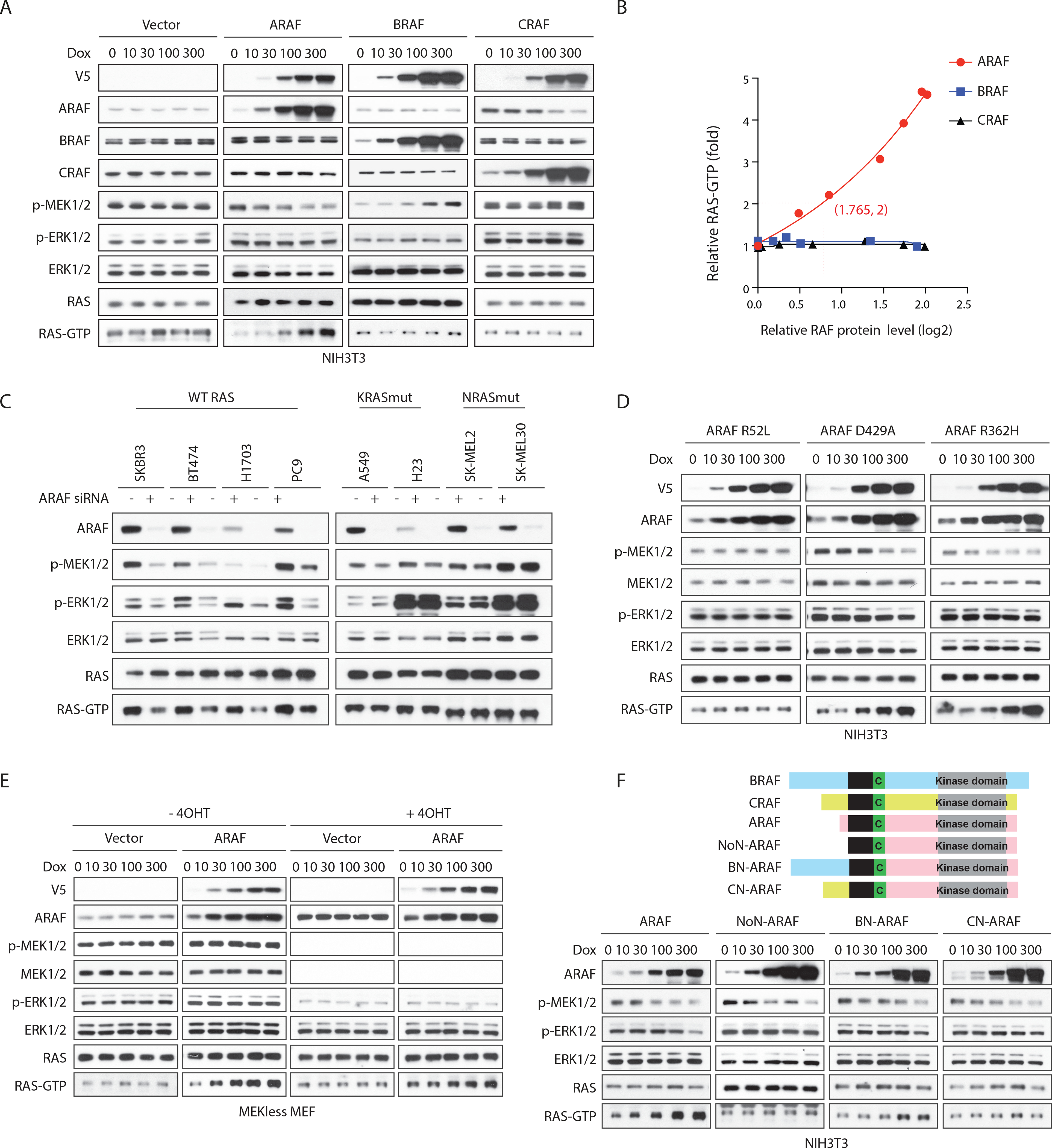
(A and B) NIH3T3 cells expressing doxycycline-inducible A-, B- or C-RAF were treated with doxycycline for 48 hours and subjected to western blot (A). RAS-GTP levels were quantified (B).
(C) SKBR3, BT474, H1703, PC9, A549, H23, SK-MEL-2 and SK-MEL30 cells were transfected with scramble or ARAF siRNA. Cells were collected 48 hours after transfection.
(D) NIH3T3 cells expressing doxycycline-inducible ARAF mutants were treated with doxycycline for 48 hours.
(E) MEK-less MEF cells expressing vector or doxycycline-inducible WT ARAF were grown in medium without or with Adeno-Cre particles plus 1 μM 4-OHT for a week to generate isogenic cells expressing or lacking MEK1. Cells were then treated with doxycycline for 48 hours.
(F) Upper panel, schematic representation of functional domains of RAF proteins and engineered ARAF fusion proteins with different N-terminus: NoN-ARAF, 18 amino acids of ARAF N-terminus is deleted; BN-ARAF, 18 amino acids of ARAF N-terminus is replaced by 154 amino acids from BRAF N-terminus; CN-ARAF, 18 amino acids of ARAF N-terminus is replaced by 55 amino acids from CRAF N-terminus. Lower panel, NIH3T3 cells expressing doxycycline-inducible WT or engineered ARAF variants were treated with doxycycline for 48 hours.
ARAF overexpression increased RAS-GTP in cells with WT RAS, but had no effect in SK-MEL2 cells, which harbor an activated NRAS Q61R mutant (Figure S2B). Thus, ARAF may regulate the activation of RAS by a mechanism that does not apply to mutant RAS proteins. In support of this idea, depletion of ARAF decreased RAS-GTP and the phosphorylation of MEK and ERK in tumor cell lines with WT RAS (SKBR3 and BT474, human breast cancer cells; H1703 and PC9, human lung cancer cells), but not in cells expressing mutant K- or N-RAS (KRAS: A549 and H23, human lung cancer cells; NRAS: SK-MEL-2 and SK-MEL-30, human melanoma cells) (Figure 2C). Depletion of BRAF or CRAF had little effect on RAS-GTP in any of these cells, whether or not they contained mutant RAS (Figure S2C). Thus, whereas increasing ARAF expression increases RAS-GTP without increasing MEK and ERK phosphorylation, decreasing ARAF reduces both. This occurs in tumor cells driven by upstream RTKs and suggests that, in such tumors, ARAF expression enhances receptor-driven signaling.
We explored the mechanism whereby ARAF activates WT RAS. Three ARAF mutants were utilized: ARAF R52L, which does not bind to RAS, ARAF R362H, which does not dimerize, and ARAF D429A, which is catalytically inactive (Imielinski et al., 2014; Mooz et al., 2014; Rebocho and Marais, 2013). Neither D429A nor R362H affected the ability of ARAF to increase RAS-GTP, so neither the kinase activity of ARAF nor its homo- or heterodimerization is required (Figure 2D). However, binding of ARAF to RAS is required for RAS activation, as the ARAF R52L mutant failed to increase RAS-GTP or to decrease ERK signaling (Figure 2D). Furthermore, whereas oncogenic ARAF S214 mutants do not inhibit RAS activation (Figure 1C), ARAF S214mut/R52L did (Figure S2D). RAS activation by ARAF does not require changes in ERK signaling, overexpression of ARAF in MEF cells that lack both MEK1 and MEK2 induces RAS activation without affecting ERK phosphorylation (Drosten et al., 2014; Gao et al., 2018) (Figure 2E).
Because the three RAF proteins share a highly conserved RAS binding domain (RBD) and all three RAF isoforms can bind to RAS-GTP, we sought to understand why only binding of ARAF causes RAS activation. The lengths and the sequences of the amino-termini of the three RAF proteins diverge upstream of the RBD (Lavoie and Therrien, 2015) and ARAF has by far the shortest N-terminal region (Figure 2F). We tested whether this region plays a role in mediating its activation of RAS. Overexpression of N-terminal-truncated ARAF failed to increase the cellular RAS-GTP in NIH3T3 cells (Figure 2F).
Neither B- nor C-RAF caused accumulation of RAS-GTP when their N-terminus was deleted (Figures S2E–S2G), showing that neither of their N-termini suppresses RAS activation. We generated chimeric proteins in which the N-terminal region of BRAF or the N-terminal of CRAF were joined to the N-terminal truncated ARAF (BN-ARAF and CN-ARAF), respectively (Figure 2F). In NIH3T3 cells, overexpression of BN-ARAF partially restored ARAF’s ability to promote accumulation of RAS-GTP. CN-ARAF had no effect (Figure 2F). Thus, the ARAF N-terminus appears to play an essential role in its promotion of GTP loading of RAS.
To test whether this N-terminal region of ARAF is sufficient to confer activation of RAS by BRAF or CRAF, we generated BRAF and CRAF proteins AN-BRAF and AN-CRAF, from which the B- or C-RAF N-terminal regions were replaced by the ARAF region. The AN-CRAF had no effect on RAS-GTP, whereas overexpression of AN-BRAF did cause an accumulation of RAS-GTP (Figures S2E–S2G). Thus, the ARAF amino terminus is necessary for induction of RAS activation but not sufficient. There may be sequences downstream of the amino terminus play a role as well. Thus, effect of ARAF on GTP-bound WT RAS requires its unique amino terminus and its direct binding to RAS, but not its dimerization or kinase activity.
ARAF increases RAS-GTP by competing with RASGAP NF1 for binding to RAS
Levels of RAS activation in cells with WT RAS are determined by dynamic changes in guanine nucleotide exchange factor (GEF)-mediated GTP-exchange rates and in GAP activation of RAS-mediated GTP-hydrolysis. ARAF binds to GTP-loaded RAS (Lavoie and Therrien, 2015). We assayed in vitro RAS GTPase activity in the presence or absence of ARAF using the GTPase-Glo assay, which measures the amount of free GTP remaining as a function of time during a RAS-dependent GTP hydrolysis reaction. The rate of intrinsic GTP hydrolysis by RAS is low (Figure S3A). Incubation of any of the three RAS proteins with any of the three RAF proteins had almost no effect on basal GTP-hydrolysis (Figure S3B). Thus, none of the RAF proteins alter intrinsic RAS GTPase activity.
GAP activity is required for physiologic regulation of WT RAS(Scheffzek et al., 1997). We tested whether activation of RAS-GTP hydrolysis by the Neurofibromatosis 1 (NF1) GAP was affected by ARAF. The GTP hydrolysis rate of WT RAS is increased by approximately 105-fold by catalytic domains of GAPs (Gideon et al., 1992; Nixon et al., 1995). NF1 promoted RAS-mediated GTP hydrolysis with an EC50 of approximately 2 μg/ml for N- and H-RAS, and 20 μg/ml for KRAS (Figures S3C–S3E). We examined the effect of ARAF on the kinetics of NF1 activated GTP-hydrolysis in vitro. When incubated with RAS and a catalytic fragment of the GAP-related domain of NF1 (NF1-333, comprising residues 1198–1530 of human NF1) (Wiesmuller and Wittinghofer, 1992) that stimulates GTP-hydrolysis by WT RAS, ARAF decreased the rate and extent of GTP hydrolysis over two hours in a concentration-dependent manner (Figure 3A). Neither BRAF, CRAF nor the ARAF R52L mutant that doesn’t bind to RAS had this effect (Figures 3A and S3F). GTP hydrolysis was enhanced by NF1-333 and both the initial velocity of the reaction and the total amount of GTP hydrolyzed was decreased by ARAF. When NF1-333 was incubated with recombinant N-, H- or K-RAS for 30 minutes, only ARAF, not BRAF or CRAF, antagonized NF1-dependent RAS GTPase activity (Figures S3C–S3E), and its effect was concentration-dependent (Figure S3G). Given that ARAF must bind RAS to activate its function, ARAF and NF1 might compete for binding to RAS.
Figure 3. ARAF increases RAS-GTP by competing with RASGAP NF1 for binding to RAS. See also Figure S3.
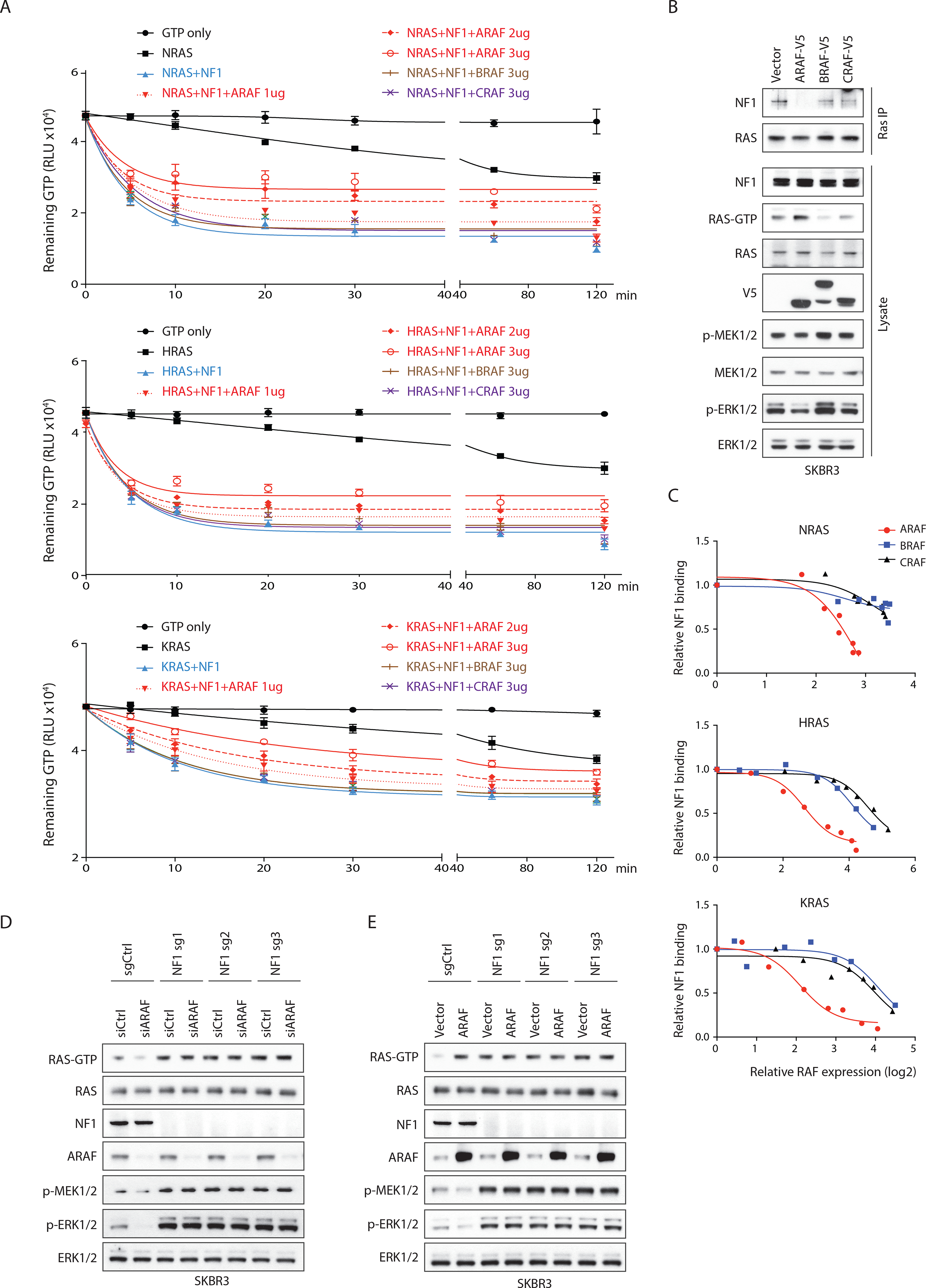
(A) GTP hydrolysis reactions were assembled with purified 0.5 μg (1 μM) N-, H-, or K-RAS and NF1-333 (50 ng/50 nM for N- or H-RAS, 500 ng/500 nM for KRAS) in the presence/absence of RAF proteins (60 nM to 1.8 μM). Reactions were incubated for the indicated time points and luminescence was recorded. Bars, mean±SD biological triplicate of experiments.
(B) SKBR3 cells transfected with indicated constructs were collected 24 hours after transfection. Endogenous RAS was immunoprecipitated.
(C) 293FT cells co-transfected with FLAG tagged N-, H- or K-RAS, NF1 and different amounts of RAF constructs were collected 24 hours after transfection. NF1 was co-immunoprecipitated with RAS and the NF1-RAS binding was quantified and normalized to basal levels.
(D and E) Cas9 expressing SKBR3 cells were transfected with sgRNAs targeting NF1. After 48 hours, cells were transfected with indicated siRNAs (D), or constructs (E).
To test this hypothesis, we expressed ARAF with various amounts of NF1 in 293FT cells. NF1 reduced the increase in RAS-GTP caused by ARAF overexpression (Figure S3H). In SKBR3 (WT RAS, amplified HER2-driven human breast cancer) cells, overexpression of ARAF but not B- or C-RAF caused accumulation of RAS-GTP and decreased the amount of NF1 bound to RAS, as measured in RAS-pulldowns (Figure 3B and S3I). ARAF did not affect the binding of other RASGAPs (p120, RASA3, RASA4, RASAL1, RASAL3, DAB2IP), to RAS (Data not shown). We assessed the degree of overexpression of ARAF required to exert these effects. ARAF bound with similar affinity to all three RAS family members, as tested in 293FT cells (Figures S3I–S3K) (Terrell et al., 2019). When N-, H- or K-RAS were co-expressed with NF1 and expression of each isoform was increased a 4 to 6-fold increase of ARAF expression compared to control was sufficient to decrease NF1 binding to RAS by 50%. Much higher amounts of CRAF or BRAF expression (15 to 30-fold over the endogenous expression levels) were required to cause a similar reduction (Figures 3C, S3I–S3K).
We assessed the effects of manipulating ARAF expression in SKBR3 cells from which NF1 was depleted by CRISPR-Cas9 gene editing. In control cells expressing a non-targeting sgRNA, the overexpression of ARAF increased and the knockdown of ARAF decreased RAS activation, while both manipulations decreased MEK and ERK phosphorylation. Levels of RAS-GTP in the SKBR3 cells lacking NF1 were twice those observed in the NF1 expressing parent. Neither overexpressing nor decreasing ARAF expression had any effect on cellular RAS-GTP or p-MEK or p-ERK in the cells lacking NF1 (Figures 3D and 3E). These data support the idea that ARAF modulation of RAS activation results from altered binding of RAS to NF1. Changes in ARAF expression also had no effect on RAS-GTP in a mouse melanoma cell line Yumm 4.1 (Meeth et al., 2016) (WT RAS, WT RAF, PTEN null, CDKN1A null) in which we depleted NF1 (Figure S3L).
Thus, increased ARAF activates RAS by antagonizing its inhibition by NF1.
The level of ARAF expression is a determinant of the duration of RAS activation in cells
Physiologic activation of ERK signaling is limited by ERK-dependent feedback inhibition of RAS and of pathway components downstream of RAS signaling. NF1 is important in mediating the former (Hennig et al., 2016; Nissan et al., 2014). The prevention of ERK-dependent feedback inhibition of RAS by activated ARAF mutants (Figure 1D) and the antagonism of NF1 binding to RAS by WT ARAF (Figures 3A and 3B) suggests that it may modulate upstream feedback and thus determine the duration of the ERK signal. We used genetically modified MEFs to assess the duration of ERK pathway activation after growth factor stimulation. Epidermal growth factor (EGF) stimulation caused a rapid (within one minute) increase in EGFR phosphorylation, RAS activation and phosphorylation of MEK and ERK in control MEF cells (Figure 4A). Phosphorylation of EGFR was increased for 10 minutes and then declined slowly but was still increased 6 hours later. Downstream signaling declined even faster, suggesting a role for feedback inhibition of other components of the pathway. Levels of GTP-bound RAS fell to baseline between 10 and 30 minutes after ligand stimulation. ERK phosphorylation was slightly more prolonged, it was still higher than its basal level 30 minutes after stimulation and fell to baseline 90 minutes later (Figure 4A). In MEF cells expressing only ARAF, basal levels of RAS-GTP were higher than those in control cells or in MEFs expressing either BRAF or CRAF alone (Figure S4A). The kinetics of ligand-induced EGFR phosphorylation were similar in all four cell lines but the increase in RAS activation and in the phosphorylation of MEK and ERK were prolonged in the MEFs expressing only ARAF. Levels of RAS-GTP declined to baseline two hours after EGF stimulation and ERK signaling only began to decline at this time. In contrast, in MEFs expressing only BRAF or only CRAF, the duration of RAS activation and ERK phosphorylation was shorter than that observed in control MEFs. RAS-GTP declined to basal levels within 5 to 10 minutes after stimulation and ERK signaling declined within 10 to 30 minutes (Figure 4A). Thus, in cells expressing only ARAF, RTK-activated RAS signaling is prolonged. Consistent with this observation, in MEFs lacking ARAF expression, EGF stimulation of RAS activation was more transient than that observed in control MEFs (Figure 4B). Activation of RAS-GTP, p-MEK and p-ERK fell to baseline rapidly, within 10–30 minutes of stimulation. In contrast, in MEFs in which BRAF or CRAF were depleted, EGF-induced signaling declined to basal level at about 60 minutes (Figure 4B). Similar results were obtained in LIM1215, an EGFR-dependent human colorectal cancer cell line with WT RAS and RAF. Depletion of ARAF in this cell line reduced RAS-GTP, p-MEK and p-ERK, whereas overexpression of ARAF increased RAS-GTP but had little effect on MEK and ERK phosphorylation (Figure S4A). Activation of RAS, p-MEK and p-ERK by EGF was more transient in LIM1215 cells lacking ARAF than in controls, and ARAF overexpression prolonged activation (Figures S4B and S4C). WT ARAF expression supports RAS activation and signaling in this EGFR-dependent model as well. Neither BRAF or CRAF knockdown nor their overexpression altered the duration of EGF-stimulated RAS-ERK signaling (Figures S4B and S4C). Thus, the expression level of ARAF in these systems is a determinant of the duration of EGF-stimulated RAS/ERK signaling.
Figure 4. The level of ARAF expression is a determinant of the duration of RAS activation in cells. See also Figure S4.
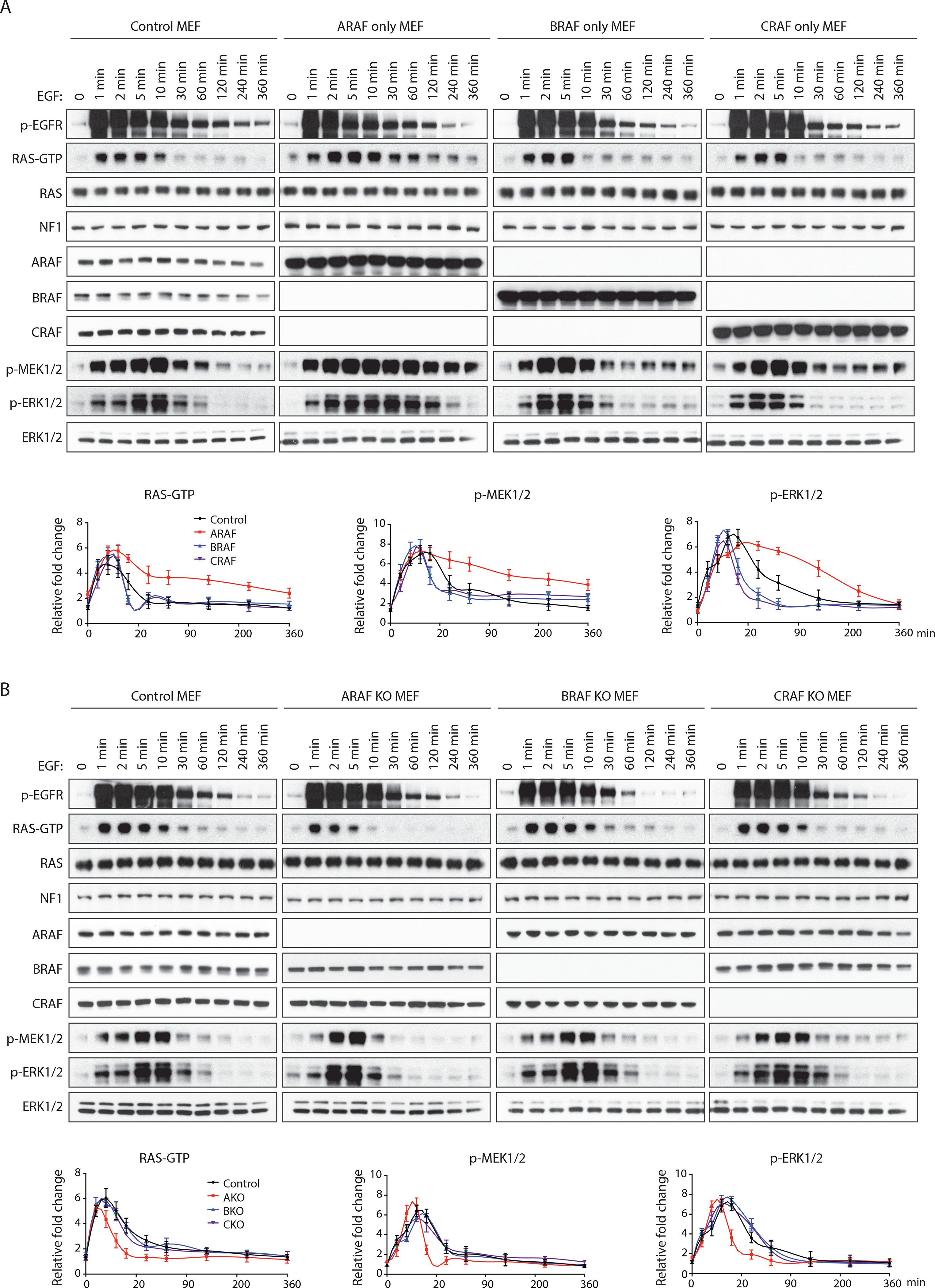
RAF-less MEF cells expressing only A-, B-, or C-RAF were treated with 100 ng/ml recombinant EGF for the indicated time points. Signaling activity and RAS-GTP levels were examined by western blot (top). RAS-GTP, p-MEK and p-ERK levels were quantified and normalized to basal levels (bottom). Bars, mean±SD biological triplicate of experiments. of experiments.
MEF cells derived from A-RAF, B-RAF or C-RAF knockout mice were treated with 100 ng/ml recombinant EGF for the indicated time points. Bars, mean±SD biological triplicateof experiments.
Expression of ARAF influences the biological consequences of ligand-activated RAS signaling
The duration of activation of ligand-induced RAS-ERK signaling can determine its cellular effects. In PC12 cells, prolonged activation of ERK by nerve growth factor (NGF) causes neural differentiation, whereas EGF induces transient ERK activation and causes proliferation (Traverse et al., 1992). We tested whether modulating ARAF expression changes the duration and consequences of pathway activation in these cells. NGF induced similar levels of TRK-A phosphorylation in control PC12 cells and in those lacking ARAF, but the duration of the RAS-ERK signal was shortened in the latter (Figure S5A).
In these cells, induction of neurite formation by NGF was decreased and fewer than 10% of the cells differentiated at 24 hours compared 60% of control cells. Fewer than 20% of the cells without ARAF had neurite-like structures even after 72 hours NGF treatment, whereas more than 80% of the control cells differentiated at this time (Figure 5A). A short prolongation (~30%) of NGF-induced ERK signaling in PC12 cells in which ARAF was overexpressed did not cause an increase in differentiation compared to that observed in control cells (Figures S5B and S5C).
Figure 5. Expression of ARAF influences the biological consequences of ligand activated RAS signaling. See also Figure S5.
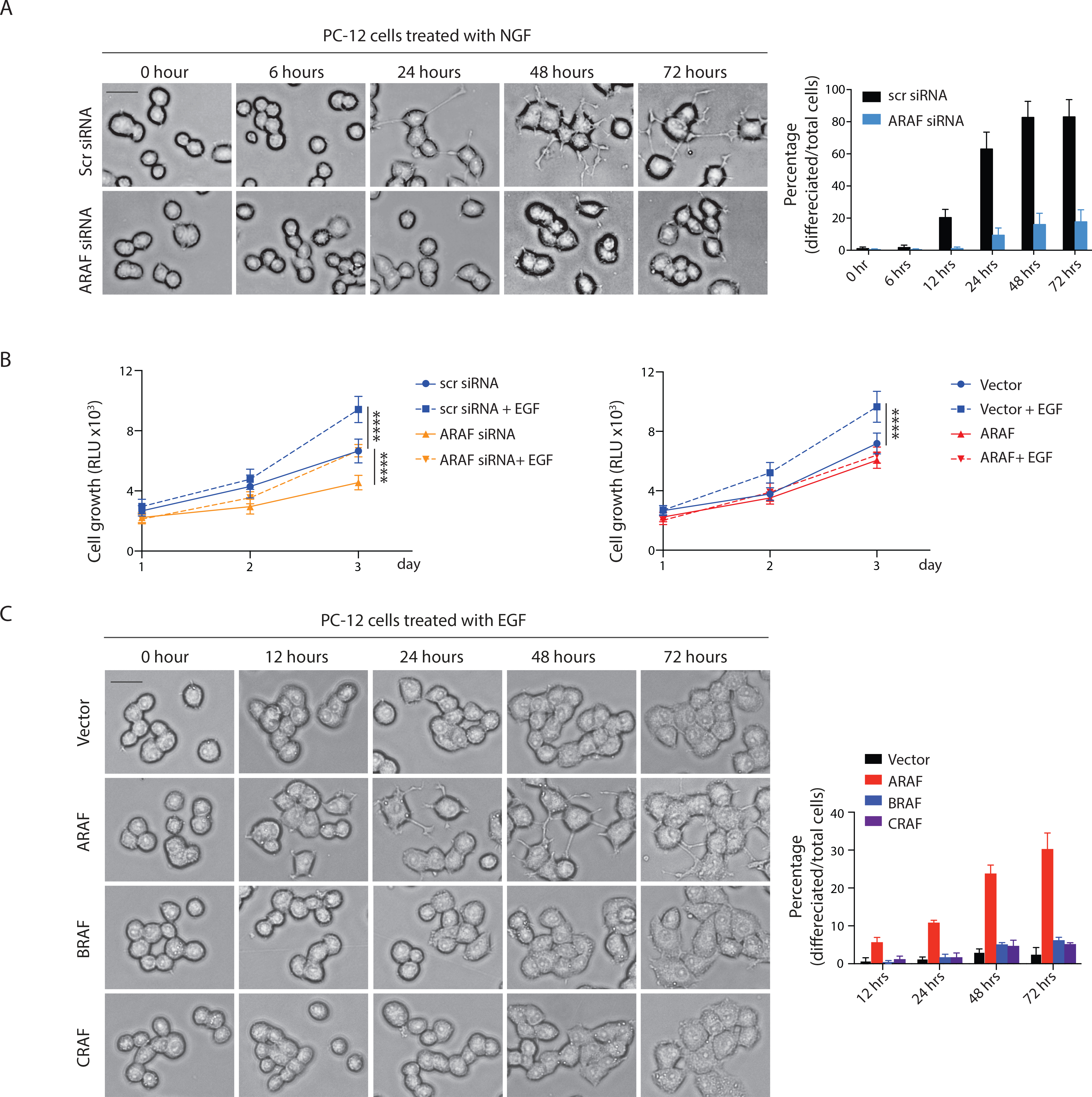
(A) PC12 cells were transfected with indicated siRNAs for 48 hours and then treated with 50 ng/ml recombinant NGF. Images were taken at the indicated time points (left). Percentage of differentiated cells was quantified (right). Scale bar, 20 μM. Bars, mean±SD biological triplicate of experiments.
(B) PC12 cells transfected with indicated siRNAs (left) or expressing indicated constructs (right) were seeded in 96 well plates at a density of 2,000 cells/well and supplemented with or without 100 ng/ml EGF. Cell growth was measured. Bars, mean±SD biological triplicate of experiments. **** p<0.0001, two-tailed Student’s t test.
(C) PC12 cells expressing doxycycline-inducible A-, B- or C-RAF were treated with 100 ng/ml doxycycline for 24 hours. Cells were then treated with 100 ng/ml EGF. Scale bar, 20 μM. Bars, mean±SD biological triplicate of experiments.
In contrast, EGF stimulation caused a rapid but transient activation of RAS signaling in PC12 cells and enhanced their proliferation without inducing cellular differentiation (Figures S5D, 5B and 5C). EGF-stimulated activation of RAS-ERK signaling was shortened (30min to 10min) in cells with reduced ARAF expression and prolonged (30min to 2hrs) when ARAF was overexpressed (Figures S5D and S5E). Both manipulations impaired the proliferative response to EGF stimulation (Figure 5B). However, only increased ARAF expression caused EGF to induce the differentiation of PC12 cells. After 72 hours treatment, about 30% of the cells with ARAF overexpression underwent differentiation compared to fewer than 10% of the control cells or of cells with either BRAF or CRAF overexpression (Figure 5C). Overexpression of mutants that increase RAS-GTP (dimerization incompetent ARAF R362H and kinase dead D429A) increased EGF induced differentiation whereas the ARAF R52L mutant that does not bind to RAS or increase RAS GTP failed to do so (Figure S5F). Together, the results confirm the previous work of Marshall’s laboratory that the duration of ERK signaling influences the effects of activating the pathway and shows that duration of signaling induced by a given ligand and its cellular effects can be altered by manipulating ARAF expression. Neither ARAF nor NF1 levels are altered by either ligand (Figures S5A and S5D), so, in this system, the level of ARAF expression is a necessary but not sufficient determinant of the duration of the signal.
ARAF overexpression causes acquired resistance of EGFR mutant lung cancers to EGFR inhibitors
We identified ARAF gene amplification (5 to 10 copies gain) in 49 patients whose tumors were characterized by MSK-IMPACT (Zehir et al., 2017). Of those, 17 were non-small cell lung cancers (NSCLC), and 12 of them also had activating EGFR mutations (Figure S6A). These EGFR mutant NSCLC tumors are typically sensitive to EGFR tyrosine kinase inhibitors (TKI) but resistance develops in almost all cases. One of these patients had serial tumor samples collected before and after initiation of EGFR inhibitor therapy. This patient developed a bony metastasis after 6 months treatment (Figure 6A). DNA sequencing revealed focal ARAF amplification in the resistant tumor, but not in the specimen obtained before treatment (Table S1 and Figure S6B). An average of 6-fold ARAF amplification in the resistant tumor was validated by fluorescence in-situ hybridization (Figure 6B). Immunohistochemical staining confirmed increases in ARAF protein expression in this tumor compared to the sample collected prior to treatment. In contrast, BRAF staining was essentially unchanged (Figure S6B). The erlotinib-resistant tumor from the patient did not harbor an EGFRT790M mutation or MET amplification (Table S1), which account for the majority of cases of acquired resistance to erlotinib (Kobayashi et al., 2005; Pao et al., 2005; Sequist et al., 2011). Upon making this observation we searched for further evidence for association of ARAF amplification with resistance to EGFR inhibitors. In a Japanese cohort of 97 TKI resistant EGFR mutant lung cancer patients, ARAF amplification (5–7 copies) was acquired in 5 of the 55 patient tumors that had none of the known resistance mechanisms (Figure S6C).
Figure 6. ARAF overexpression causes acquired resistance of EGFR mutant lung cancers to EGFR inhibitors. See also Figure S6 and Table S1–S3.
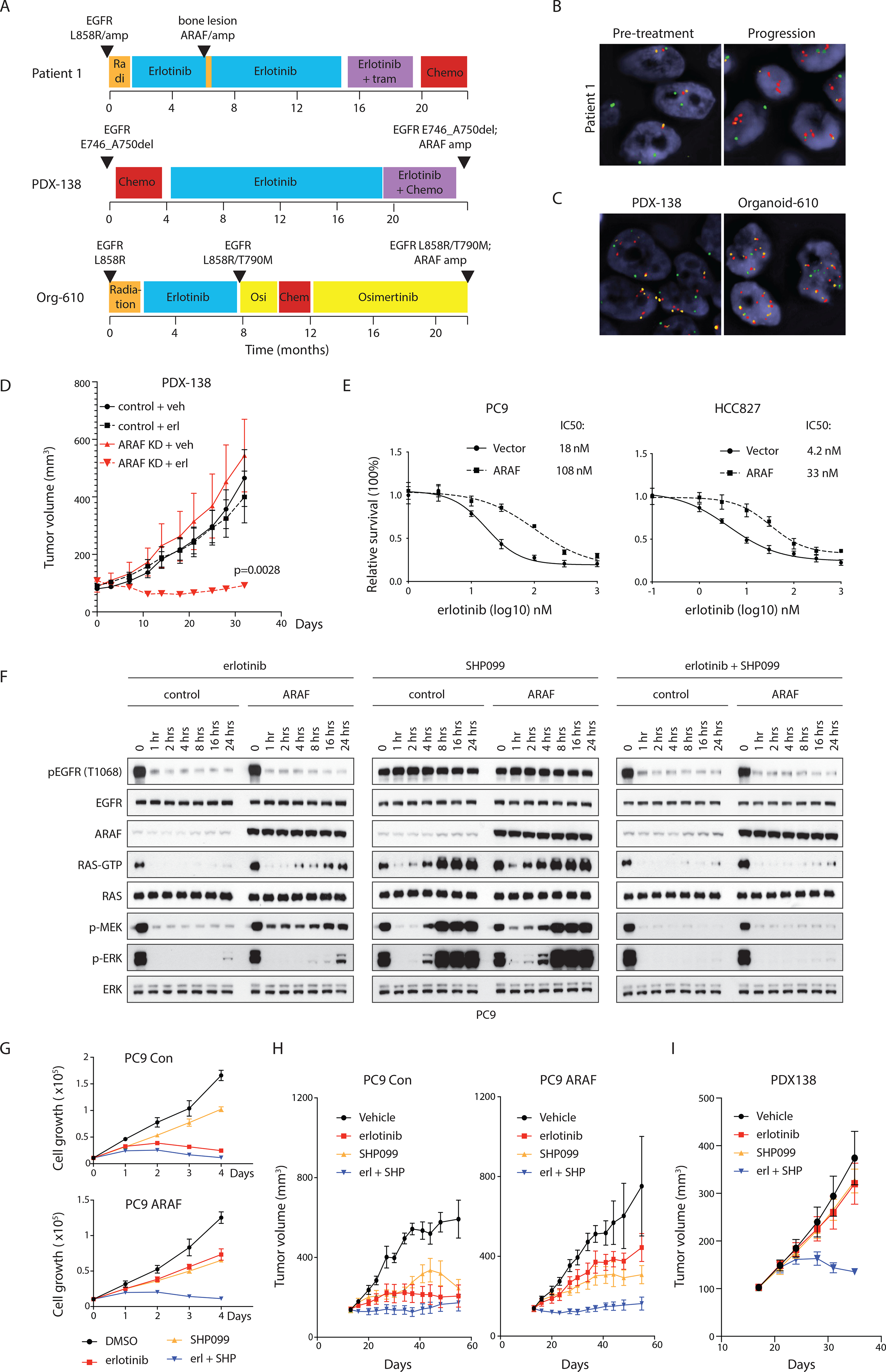
(A) Treatment timeline for three NSCLC patients with tumor ARAF amplification. (B and C) Fluorescence in situ hybridization for ARAF and BRAF in the patient specimens. Red, ARAF; green, BRAF; orange, centromeric of chromosome X. Scale bar, 5 μM.
(D) Control or ARAF knockdown PDX-138 cells were subcutaneously injected into NSG mice. Erlotinib was given at 25 mg/kg once a day. Bars, mean±SEM, n=5, two-tailed Student’s t test.
(E) PC9 or HCC827 cells expressing vector or doxycycline-inducible ARAF were seeded in 96 well plates at a density of 2,000 cells/well and supplemented with 100 ng/ml doxycycline. After 24 hours induction, cells were treated with erlotinib for 72 hours. Cell survival was normalized to untreated controls. Drug concentrations inducing 50% inhibition in survival (IC50 nmol/L) are indicated. Bars, mean±SD triplicate of experiments.
(F) PC9 cells expressing vector or doxycycline-inducible ARAF were treated with 100 ng/ml doxycycline for 24 hours. Cells were then treated with 100 nM erlotinib, 3 μM SHP099 or in combination.
(G) PC9 cells expressing vector or doxycycline-inducible ARAF were seeded in 96 well plates at a density of 2,000 cells/well and supplemented with 100 ng/ml doxycycline. Cells were then treated with 100 nM erlotinib, 3 μM SHP099, or in combination. Bars, mean±SD triplicate of experiments.
(H) PC9 cells expressing doxycycline-inducible ARAF were subcutaneously injected into nude mice. Mice were fed either normal or doxycycline containing food. Erlotinib was given at 12.5 mg/kg once a day, SHP099 at 75 mg/kg once a day. Bars, mean±SEM, n=5.
(I) PDX-138 cells were subcutaneously injected into NSG mice. Erlotinib was given at 25 mg/kg once a day, SHP099 at 150 mg/kg once a day. Bars, mean±SEM, n=5.
These results and the finding that increased ARAF expression increases RAS activation suggest that amplification of ARAF might mediate resistance to EGFR inhibitors. To test this possibility, we utilized a patient-derived xenograft (PDX) model derived from a patient with an EGFR exon19 deletion NSCLC tumor with acquired resistance to erlotinib (Figure 6A). Copy number analysis and sequencing data showed that this PDX had focal amplification of ARAF (>=6 copies) and no EGFR T790M mutation (Figure 6C and Table S2). When ARAF was depleted with shRNA in the PDX, no tumor growth inhibition was observed. However, these tumors were now sensitive to erlotinib treatment, as opposed to control tumors expressing non-targeted shRNA which remained resistant (Figure 6D). These data suggest that acquired resistance of this tumor to erlotinib was dependent on amplification of ARAF. To examine whether overexpression of ARAF is sufficient to cause resistance, we used two human mutant EGFR-driven NSCLC cell lines, PC9 and HCC827. In PC9, a 5-fold increase of ARAF expression led to a 4 to 5-fold increase in GTP-bound RAS, whereas AKT and MEK phosphorylation and cyclin D1 expression were unchanged. Accumulation of RAS-GTP was not accompanied by a change in EGFR phosphorylation (Figure S6D). Inhibition of EGFR with erlotinib reduced RAS-GTP in both control and ARAF-overexpressed cells, however, in the latter, RAS-GTP after treatment was 5- to 6-fold higher than that in the treated control cells. Similarly, after treatment, the downstream targets of RAS signaling, p-ERK, p-AKT, and cyclin D1, were higher in the cells with ARAF-overexpression compared to the control cells (Figure S6D). The proliferation of cells that overexpressed ARAF was less sensitive to erlotinib than that of the controls (IC50: 18 nM versus 108 nM) (Figure 6E). Similar results were obtained in HCC827 (4.2 nM versus 33 nM) (Figure 6E). When increasing amounts of ARAF were expressed in PC9 and HCC827, a 5-fold increase in ARAF expression caused a 4 to 5-fold increase in the IC50 of erlotinib in both cell lines (Figure S6E). Moreover, in both cells, overexpression of the ARAF R362H and D429A mutants decreased their sensitivity to erlotinib treatment, but ARAF R52L mutant had no effect (Figure S6F). Thus, increased ARAF expression in tumors may decrease the sensitivity of RAS signaling to EGFR inhibition and cause resistance by that mechanism.
Because ARAF blocks NF1-GAP function, increased RAS activation is due to unopposed GEF activation. To test whether increased RAS inhibition could restore erlotinib sensitivity, we combined erlotinib with SHP099, a protein tyrosine phosphatase 2 (SHP2) inhibitor that suppresses activation of its downstream target, the RAS-GEF SOS and thus RAS activation (Chen et al., 2016; Dardaei et al., 2018; Fedele et al., 2018; Nichols et al., 2018). Increased inhibition of exchange factor activation by combining EGFR and SHP2 inhibition could counteract the decrease in GAP-activated GTPase activation caused by ARAF overexpression. In PC9 and HCC827 cells with ARAF overexpression, RAS-GTP levels were 6-fold higher than those found in control cells. Erlotinib alone caused a rapid inhibition of RAS-GTP and ERK signaling in the controls and the ARAF cells. This inhibition persisted in control cells up to 24 hours after EGFR inhibition. In contrast, in ARAF-overexpressing cells, levels of RAS-GTP began to rebound 4 to 8 hours after EGFR inhibition and this was accompanied by induction of phosphorylation of MEK and ERK, consistent with amplification of signaling reactivation in tumors with high amounts of ARAF. SHP099 alone caused transient inhibition of RAS-GTP and MAPK signaling in control PC9 cells and cells with ARAF overexpression, both of which rebounded 2 hours after drug inhibition and reached higher than basal levels after 8 hours in both control and ARAF-overexpressing PC9 cells (Figure 6F). In HCC827 cells, the rebounds of RAS-GTP and MEK/ERK phosphorylation occurred about 4 hours after treatment, but to a lesser extent than that observed in PC9 cells (Figure S6G). By 8 hours after treatment, in control or ARAF-overexpressing HCC827 cells, both RAS-GTP and MAPK signaling were reactivated but remained lower than the pre-treatment level (Figure S6G).
By contrast, combined inhibition of EGFR and SHP2 overcame reactivation of RAS signaling in both control and ARAF-overexpressed cells (Figures 6F and S6G). In control PC9 and HCC827 cells, 3 μM SHP099 had small inhibitory effects, whereas erlotinib alone or combination treatment arrested the growth of cells. In contrast, in the cells with ARAF overexpression, only the combination treatment prevented cell growth (Figures 6G and S6H). These effects of treatment were observed in PC9 and HCC827 xenograft models in vivo as well (Figure 6H and S6I). Moreover, in the PDX138 model, only the combination treatment inhibited tumor growth (Figure 6I), suggesting that SHP2 inhibitor amplifies EGFR inhibition and that there is synergistic effect of the two inhibitors in a tumor model with ARAF-mediated resistance to EGFR-TKI. This combination effect was also observed in a patient-derived organoid model, which was derived from an EGFR L858R/T790M NSCLC patient with ARAF amplification (~7 copies) and is resistant to the 3rd generation of EGFRi osimertinib (Figures 6A, 6C and Table S3). 3 μM SHP099 alone weakly inhibited growth of the organoid but sensitized it to osimertinib (IC50 of osimertinib decreased 5-fold when combined with SHP099) (Figure S6J). Thus, increased expression of ARAF decreases the sensitivity of the EGFR mutant tumors to EGFR inhibitors. This is attributable to the increased RAS activation in these tumors, treatment of which requires more effective inhibition of nucleotide exchange than is afforded by EGFR inhibitors. Effective inhibition of RAS can be accomplished by combining EGFR inhibitors with drugs that inhibit nucleotide exchange by other mechanisms, such as SHP2 inhibition.
Discussion
Binding of RAF protein kinases to GTP-bound RAS activates their catalytic activity and drives ERK signaling. We show that binding of ARAF to RAS also antagonizes its binding to RASGAP NF1 and increases levels of the activated GTP-bound form of RAS. Thus, ARAF is a RAS effector that also affects RAS activation. This is a mechanism for controlling RAS activation that is not shared by other RAS effectors including the other two members of the RAF family, BRAF and CRAF. As predicted by this model, increasing ARAF expression promotes RAS activation. Since NF1 mediates, in part, ERK dependent feedback inhibition of RAS (Hennig et al., 2016), ARAF expression is expected to decrease the sensitivity of the pathway to feedback. Consistent with this hypothesis, alteration of ARAF expression was shown to influence the duration of growth factor induced RAS-RAF-MEK-ERK pathway activation (Figures 4 and S4). Moreover, overexpressing activated oncogenic mutants of ARAF failed to cause feedback inhibition of RAS activation.
Previous work has focused on the role of RAF protein kinases in transducing the RAS signal by phosphorylating MEK1 and MEK2. Kinase-independent effects of CRAF have been associated with a variety of cellular phenotypes, including regulation of mitochondrial dependent apoptosis (Alavi et al., 2003). However, none of these involves activation of RAS. Some early work reported that RAF family proteins may work together to influence the activation and duration of ERK signaling (Mercer et al., 2002; Mercer et al., 2005), but the mechanism was not defined. ARAF has significantly lower kinase activity than BRAF and CRAF (Baljuls et al., 2007; Marais et al., 1997; Mercer et al., 2002), although it can support ERK activation in cells that do not express B- or CRAF. We showed here that overexpression of ARAF in cells with all three RAF family members decreased ERK signaling by increasing ARAF containing RAF dimers, at the expense of the more active BRAF and CRAF homo- and heterodimers (Figures 1D and S1F). ARAF overexpression did exert positive phenotypes, including increased duration of RAS-ERK activation and induction of resistance of tumors to the receptor tyrosine kinase inhibitors. Reduction of ARAF expression reduced signaling duration and reduced RAS activation and output in receptor tyrosine kinase driven tumor cell lines (Figure 2C).
It has been demonstrated that the biologic consequences of ERK activation can vary depending on the duration of the signal. This was first demonstrated by Traverse, Marshall and colleagues who showed in PC12 cells that EGF stimulation caused transient activation of RAS-ERK signaling that resulted in stimulation of proliferation, whereas NGF stimulation caused more sustained pathway activation and resulted in neural differentiation (Marshall, 1995; Traverse et al., 1992). We showed here that manipulating ARAF expression in these cells altered the duration of the signal stimulated by both ligands and their biologic effects. Thus, overexpression of ARAF resulted in prolongation of the EGF-induced signal and caused differentiation, not proliferation (Figures 5C and S5E). By contrast, depletion of ARAF in these cells shortened the signal induced by NGF and abrogated its ability to cause differentiation (Figures 5A and S5A).
Glucocorticoid receptor binding sites have been identified in the ARAF promoter (Lee et al., 1996) but, whether steroid hormones regulate ARAF expression and, in doing so, alter RAS or ERK signaling is unknown. Induction of ARAF expression has also been observed during adipose differentiation of 3T3-L1 in cell culture and after partial hepatectomy during liver regeneration in rodents (Silverman et al., 1989; Zmuidzinas et al., 1989). Little is known about how ARAF expression is regulated in these systems, whether it has functional consequences, and whether those consequences require its catalytic activity.
ARAF and NF1 compete for binding to RAS and have opposing effects on its activation. Previous research suggests that reduced NF1 expression confers resistance to EGFR inhibitors (de Bruin et al., 2014). We showed that ARAF amplification in a small cohort of EGFR mutant-driven lung cancers was associated with acquired resistance to EGFR inhibitors. None of the common mechanisms for acquired resistance were present in these tumors. In EGFR mutant lung cancer cells, inhibition of RAS-GTP by EGFR inhibitor treatment was attenuated when ARAF was over-expressed and these cells were 6 to 8-fold less sensitive to EGFR inhibitors (Figure 6E). We generated a single PDX model from a tumor with clinically acquired resistance to EGFR inhibitors and ARAF amplification. In the model, depletion of ARAF had no effect on tumor growth in vivo but did sensitize it to EGFR inhibition (Figure 6D). Taken together, the data suggest that amplification of ARAF may cause acquired resistance to EGFR inhibitors.
A balance of nucleotide exchange factor activation and GAP activity functions to regulate RAS activation. We hypothesized that more effective exchange factor inhibition might reverse the resistance to EGFR inhibitors due to ARAF amplification. We did find that the resistant PDX model was sensitive to the combined inhibition of SHP2 and EGFR (Figure 6I). Further research will determine whether more potent inhibition of nucleotide exchange by combining RTK inhibitors with either SHP2 or SOS1 inhibitors could be an effective therapy for RTK driven tumors in which resistance is mediated by ARAF amplification, NF1 loss, or other mechanisms for activating wild type RAS.
Limitations of the study:
Several experiments in this study were carried out in models in which ARAF expression was manipulated in cells. Significant overexpression might lead to artifactual results. We thus used an inducible expression system to maintain a moderate level of ARAF expression at levels consistent with those seen in cell lines and tissues. and validated the key finding in various models. We believe these experiments were necessary for understanding ARAF function. Similarly, the activity, functions, and biochemical mechanism of action of the oncogenic ARAF S214 mutants were examined by ectopically expressing the mutants in cellular models. There are currently no tumor cell lines that endogenously express ARAF S214 mutant and it will be useful to confirm these experiments in such model when they are available. Finally, we show that ARAF antagonized NF1 function, but not that of other RASGAP proteins in the tested cell line models. Studies in other cellular and lineage contexts will extend and confirm our model.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Neal Rosen (rosenn@mskcc.org).
Materials availability
Plasmids and cell lines generated in this study will be available upon request. A Material Transfer Agreement will be needed for sharing patient-derived xenograft and organoid cells.
Data and Code Availability
Original western data and microscopy data for figures in this paper have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ARAF | Santa Cruz | sc-408 (AB_630882) |
| BRAF | Santa Cruz | sc-5284 (AB_626760) |
| BRAF | Sigma | HPA001328 (AB_1078296) |
| CRAF | BD | 610152 (AB_397553) |
| FLAG | Sigma | F1804 (AB_262044) |
| V5 | Thermo Fisher | R960-25 (AB_2556564) |
| GAPDH | Santa Cruz | sc-32233 (AB_627679) |
| RAS | Thermo Fisher | MA1-012 (AB_2536664) |
| RAS | Santa Cruz | sc-14022 (not available) |
| RAS | Millipore | 05-516 (AB_11211664) |
| NF1 | Santa Cruz | sc-67 (AB_2149681) |
| Cyclin D1 | Santa Cruz | sc-718 (AB_2070436) |
| Sprouty2 | Abcam | ab85670 (not available) |
| p-MEK1/2 (Ser217/221)) | Cell Signaling | 9154 (AB_2138017) |
| MEK1/2 | Cell Signaling | 4694 (AB_10695868) |
| p-p44/42 MAPK (Erk1/2) | Cell Signaling | 9101 (AB_331646) |
| p44/42 MAPK (Erk1/2) | Cell Signaling | 4696 (AB_390780) |
| p-EGFR (Tyr1068) | Cell Signaling | 3777 (AB_2096270) |
| EGFR | Cell Signaling | 4267 (AB_2246311) |
| p-TrkA | Cell Signaling | 4619 (AB_10235585) |
| AKT | Cell Signaling | 9272 (AB_329827) |
| p-AKT (Ser473) | Cell Signaling | 4060 (AB_2315049) |
| p-AKT (Ser308) | Cell Signaling | 4056 (AB_331163) |
| Chemicals, peptides and recombinant proteins | ||
| DMEM | ThermoFisher Scientific | 11965-092 |
| RPMI 1640 | ThermoFisher Scientific | 61870-036 |
| DMEM/F12 | ThermoFisher Scientific | 12634-010 |
| Renaissance Essential Tumor Medium | Cellaria | CM-0001 |
| B27 supplement | ThermoFisher Scientific | 17504044 |
| Matrigel | Corning | 354234 |
| L-glutamine | Corning | 25005CI |
| penicillin G-streptomycin | Corning | 30004CI |
| Trypsin-EDTA (0.05%) | Invitrogen | 25300054 |
| TrypLE Express | Invitrogen | 12604013 |
| CD45 MicroBeads, human | Miltenyl Biotex | 130-045-801 |
| Doxycycline | Sigma | D3072 |
| 4-OHT | Sigma | H6278 |
| Erlotinib | MedChem Express | HY-50896 |
| Osimertinib | Selleckchem | S7297 |
| Puromycin | Invitrogen | A1113802 |
| hygromycin | Invitrogen | 10687010 |
| Blasticidin | Invitrogen | A1113903 |
| SHP099 | Novartis | N/A |
| Recombinant human ARAF | Origene | TP300737 |
| Recombinant human BRAF | Origene | TP311013 |
| Recombinant human CRAF | Origene | TP301983 |
| Recombinant human NF1-333 | Creative Biomart | NF1-334H |
| Recombinant human EGF | Invitrogen | PHG0311 |
| Recombinant rat NGF | R & D | 7185-NG-025 |
| Recombinant human KRAS | Origene | TP301958 |
| Recombinant human NRAS | Cell Biolabs | STA-749 |
| Recombinant human HRAS | Cell Biolabs | STA-747 |
| Recombinant K97R MEK | Millipore | 14-737 |
| Critical commercial assays | ||
| CellTiter-Glow | Promega | G7573 |
| CellTiter-Glow 3D | Promega | G9682 |
| Active RAS pull-down assay | ThermoFisher Scientific | 16117 |
| GTPase-Glow assay | Promega | V7681 |
| Experimental models: Organisms/strains | ||
| Athymic nude mice (female) | Envigo | Hsd:Athymic Nude-Foxn1nu |
| Nod SCID gamma mice (female) | The Jackson Laboratory | 005557 |
| Experimental models: cell lines | ||
| 293FT (human) | ThermoFisher Scientific | R70007 (CVCL_6911) |
| NIH 3T3 (mouse) | ATCC | CRL-1658 (CVCL-0594) |
| SKBR3 (human) | ATCC | HTB-30 (CVCL_0033) |
| PC9 (human) | Sigma | 90071810 (CVCL_B260) |
| HCC827 (human) | ATCC | CRL-2868 (CVCL_2063) |
| LIM1215 (human) | Sigma | 10092301 (CVCL_2574) |
| BT474 (human) | ATCC | CRL-3247 (CVCL_AQ07) |
| H1703 (human) | ATCC | CRL-5889 (CVCL_1490) |
| A549 (human) | ATCC | CCL-185 (CVCL_0023) |
| H23 (human) | ATCC | CRL-5800 (CVCL_1547) |
| PC12 Tet-On (rat) | Clonetech | 631137 (CVCL_V333) |
| SK-Mel2 (human) | MSKCC | (CVCL_0069) |
| SK-Mel30 (human) | MSKCC | (CVCL_0039) |
| K-Raslox MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| RAF-less MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| MEK-less MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| ARAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| BRAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| CRAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| YUMM 4.1 (mouse) | Marcus Bosenberg | doi: 10.1111/pcmr.12498. |
| Oligonucleotides | ||
| Human ARAF siRNA | Dharmacon | L-003563-00-0020 |
| Human BRAF siRNA | Dharmacon | L-003460-00-0020 |
| Human CRAF siRNA | Dharmacon | L-003601-00-0020 |
| Rat ARAF siRNA | Dharmacon | L-088540-00-0020 |
| Mouse ARAF siRNA | Dharmacon | L-042948-00-0020 |
| Mouse BRAF siRNA | Dharmacon | L-040325-00-0020 |
| Mouse CRAF siRNA | Dharmacon | L-040149-00-0020 |
| Human ARAF shRNA1 | Sigma | TRCN0000000571 |
| Human ARAF shRNA2 | Sigma | TRCN0000199465 |
| NF1 sgRNA1 | Cellecta | CATGAGACCACTGTCTACGT |
| NF1 sgRNA2 | Cellecta | CCACCACCTAGAATCGAAAG |
| NF1 sgRNA3 | Cellecta | GAGGAAGCAGATATCCGGTG |
| Cyclin D1 qPCR probe | ThermoFisher Scientific | Mm00432359_m1 |
| DUSP4 qPCR probe | ThermoFisher Scientific | Mm00732761_m1 |
| DUSP6 qPCR probe | ThermoFisher Scientific | Mm00518185_m1 |
| SPRY2 qPCR probe | ThermoFisher Scientific | Mm00442344_m1 |
| SPRY4 qPCR probe | ThermoFisher Scientific | Mm00442345_m1 |
| ETV1 qPCR probe | ThermoFisher Scientific | Mm00514804_m1 |
| GAPDH qPCR probe | ThermoFisher Scientific | Mm99999915_g1 |
| Recombinant DNA | ||
| pcDNA3.1-hARAF-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214A-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214C-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214F-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214T-V5 | This paper | N/A |
| pcDNA3.1-hARAF-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-R52L-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214A-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214C-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214F-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214T-R362H-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365A-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365C-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-R509H-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365C-R509H-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-R401A-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-S259A-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-S259A-R401H-V5 | This paper | N/A |
| pTTIGFP-hARAF-V5 | This paper | N/A |
| pTTIGFP-hBRAF-V5 | This paper | N/A |
| pTTIGFP-hCRAF-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214A-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214C-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214F-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214T-V5 | This paper | N/A |
| pTTIGFP-hBRAF-K601E-V5 | This paper | N/A |
| pTTIGFP-hCRAF-S259A-V5 | This paper | N/A |
| pTTIGFP-hARAF-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-D429A-V5 | This paper | N/A |
| pTTIGFP-hARAF-R362H-V5 | This paper | N/A |
| pTTIGFP-BN-ARAF-V5 | This paper | N/A |
| pTTIGFP-CN-ARAF-V5 | This paper | N/A |
| pTTIGFP-NoN-ARAF-V5 | This paper | N/A |
| pTTIGFP-NoN-BRAF-V5 | This paper | N/A |
| pTTIGFP-AN-BRAF-V5 | This paper | N/A |
| pTTIGFP-NoN-CRAF-V5 | This paper | N/A |
| pTTIGFP-AN-CRAF-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214A-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214C-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214F-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214T-R52L-V5 | This paper | N/A |
| pXretro-FLAG-NRAS | This paper | N/A |
| pXretro-FLAG-HRAS | This paper | N/A |
| pXretro-FLAG-KRAS | This paper | N/A |
| pMSCV-rtTA3-PGK-Hygro | Scott Lowe | N/A |
| TTIGFP-MLUEX | Scott Lowe | N/A |
| pEF-DEST51-NF1-V5-His | Frank McCormick | N/A |
| Deposited data | ||
| Raw western blot and microscopy data | Mendeley | doi: 10.17632/yx7s86mcrn.1 |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com |
Experimental MODEL AND SUBJECT DETAILS
Cell lines
293FT cells were obtained from Invitrogen. PC12 (tet on) cells were from Clontech. LIM1215 and PC9 cells were from Sigma. The conditional RAS knockout MEF, conditional RAF knockout cells and conditional MEK knockout MEF cell lines were kindly provided by Mariano Barbacid, the Spanish National Cancer Research Center, Spain. The ARAF knockout MEF, BRAF knockout MEF and CRAF knockout MEF cell lines were kindly provided by Dr. Manuela Baccarini, University of Vienna, Austria. The ARAF only, BRAF only and CRAF only MEF cell lines were generated from the conditional RAF knockout MEF cells. The YUMM 4.1 cells were kindly provided by Marcus Bosenberg, Yale University, USA. All other cell lines were obtained from the Memorial Sloan Kettering Cancer Center (MSKCC) cell collection and the American Type Culture Collection. SKBR3 cells were cultured in DMEM/F12 supplemented with 10% Fetal Bovine Serum, 2 mM L-Glutamine (Glu), 100 IU/ml Penicillin/Streptomycin. LIM1215, PC12, H1703, PC9, HCC827, A549 and H23 cells were cultured in RPMI supplemented with 10% Fetal Bovine Serum, 2 mM Glu, 100 IU/ml Penicillin/Streptomycin. All other cell lines were cultured in DMEM high Glucose supplemented with 10% Fetal Bovine Serum, 2 mM Glu, 100 IU/ml Penicillin/Streptomycin
Mouse studies
All mouse studies were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee of Memorial Sloan Kettering Cancer Center (MSKCC). Animal treatments were conducted by the MSKCC core facility. Staff was not aware of each treatment when delivering drugs or assessing the outcomes.
Cell xenograft models
For localized tumor growth assays, 5 million cells were resuspended in 100 μl PBS with Matrigel (Corning) in 1:1 ratio and subcutaneously injected into rear flanks of 6–8 weeks old female athymic mice (Envigo). Animals were excluded from the study if not properly injected. When tumor volumes reached 100–150 mm3, mice were randomized (n=5 mice per group) and dosed with a control vehicle, erlotinib, SHP099, or their combination for 6 consecutive weeks. Mice were observed daily throughout the treatment period for signs of morbidity/mortality, and body weights were recorded twice weekly. Tumors were measured twice weekly using calipers, and volume was calculated using the formula length × width2 × 0.52.
PDX model
Tumor tissue from a patient with a NSCLC EGFR E746_A750del tumor, that had been treated with erlotinib and standard chemotherapy, was collected under MSKCC IRB protocol 14–091 (informed consent was obtained from the patient) and implanted subcutaneously in 6–8 weeks old female NSG mice (Jackson Laboratory) as previously described (Mattar et al., 2018). The generated PDX was subjected to high coverage next generation sequencing with the MSK-IMPACT assay (Cheng et al., 2015). For ARAF depletion, PDX tumor tissue was dissociated into single cells in vitro. Expanded cells infected with control or ARAF shRNAs were selected with puromycin for 48 hours and then implanted in the flank of 6- to 8-week-old female NSG mice. When tumor volumes reached 100–150 mm3, mice were randomized (n = 5 mice per group) and dosed with a control vehicle, erlotinib, SHP099, or their combination for 4 consecutive weeks. Tumor volumes and clinical signs were monitored as described above.
Patient-derived organoid model
A pleural effusion specimen was obtained under IRB 02–180 at the Dana Farber Cancer Institute as part of clinical care from a patient who progressed on osimertinib. Informed consent was obtained from the patient. After RBC lysis and CD45 depletion (Miltenyl Biotex), ice-cold Matrigel (Corning) was added to the cells and incubated on pre-warmed 6 cm plate to solidify. To generate primary cancer organoids, cells were cultured in Renaissance Essential Tumor Medium (RETM; Cellaria) supplemented with B27 (Thermo Fisher Scientific). Organoids passaged over three times were used for experiments or cryopreserved.
METHOD DETAILS
Compounds
erlotinib was obtained from MedChem Express. osimertinib was obtained from SelleckChem Express. SHP099 was provided by Novartis. Doxycycline and 4-OHT were from Sigma Aldrich. Puromycin and hygromycin stock solution were from Invitrogen. Drugs were dissolved in DMSO to yield 10mM stock and store at −20°C.
Generation of RAS-less, RAF-less and MEK-less cells
Hras−/−; Nras−/−; Kraslox/lox; RERTert/ert (K-Raslox) MEFs, A-RAFlox/lox; B-RAFlox/lox; C-RAFlox/lox; RERTert/ert (RAF-less) MEFs and Mek2−/−; Mek1lox/lox; RERTert/ert (MEK-less) MEFs were generously provide by Mariano Barbacid. MEF cells were isolated from embryonic day 13.5 embryos and immortalized using standard methods. The protocol for the generation of Ras-less cells was followed as previously reported (Drosten et al., 2010).
Gene inducible expression cell system
Retrovirus encoding rtTA or the RAF genes was packaged in 293FT cells obtained from Invitrogen. The medium containing virus was filtered with 0.45 PVDF filters followed by incubation with the target cells for 12 hr. Cells were then maintained in virus free media for 2 days. Cells were selected using Hygromycin (250 μg/ml) or Puromycin (2 μg/ml) for 3 days. The positive infected cell populations were further sorted using GFP as a marker after overnight exposing to 1 μg/ml doxycycline. GFP positive cells were then cultured and expanded in medium without doxycycline but with antibiotics.
Generation of ARAF only, BRAF only and CRAF only MEF cells
A-RAFlox/lox; B-RAFlox/lox; C-RAFlox/lox; RERTert/ert MEF cells were stably transduced with retrovirus carrying doxycycline-inducible ARAF, BRAF or CRAF. Cells were then grown in medium containing Adeno-Cre particles and 1 μM 4-OHT for a week to generate isogenic cells lacking endogenous RAFs. 100 ng/ml doxycycline was added to the medium to maintain the expression of exogenous RAF proteins.
Analysis of protein and mRNA expression
For immunoblotting, cells were washed with PBS and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors (Calbiochem) and phosphatase inhibitors (PhosSTOP, Roche Life Science). Protein concentrations were measured by using the DC Protein Assay (Bio-Rad). Total RNA was extracted using the RNeasy Mini kit coupled with RNase-free DNase set (Qiagen) and reverse transcribed with SuperScript III First-Strand Synthesis SuperMix (Invitrogen). cDNA corresponding to approximately 10 ng of starting RNA was used for one reaction. Q-PCR was performed with Taqman Gene Expression Assay (Applied Biosystems). All quantifications were normalized to endogenous GAPDH.
Active RAS pull-down assay
Cells were cultured in 10cm dishes until 70–80% confluence. GTP-bound RAS was quantitated using the RAF1 RAS-binding domain (RBD) pull-down from Detection Kit (Thermo Fisher Scientific), as instructed by the manufacturers.
In vitro kinase assay
Cells were collected and stored on ice and lysed in lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% NP40, 1 mM EDTA) supplemented with protease inhibitors (Calbiochem) and phosphatase inhibitors (PhosSTOP, Roche Life Science). Immunoprecipitations were carried out at 4 °C for 2 h, followed by five washes with lysis buffer. When the in vitro kinase assay was performed after immunoprecipitation, 0.02% SDS was added to the wash buffer and one extra wash with kinase buffer (25 mM Tris, pH 7.5, 10 mM MgCl2) was required. RAF kinase assays were conducted in the presence of 200 μM ATP, at 30 °C for 20 min. Recombinant inactive K97R MEK (Millipore) was used as a substrate and the reaction was terminated with 1 × SDS loading buffer and boiling. Kinase activity was estimated by immunoblotting for p-MEK.
Cell growth assay
Cells were seeded into 96 well plates at 2000–3000 cells per well. Cell growth was quantitated using the CellTiter-Glo assay (Promega) every 24 hr. For each condition, 8 replicates of each concentration were measured.
Organoid growth assay
The cells in 3D matrices were harvested and dissociated using TrypLE Express (Invitrogen) at 37°C. The single cells were resuspended in cold RETM/10% Matrigel and plated into 384-well ultra-low attachment microplates (Corning) at 1000 cells per well. On the next day, cells were treated with DMSO, osimertinib, SHP099, or in combinations as indicated for 4 days. Each dose had more than three replicates. The viability assay was performed using CellTiter-Glo 3D (Promega) according to the manufacturer’s protocol.
GTPase-Glo assay
Recombinant RAS proteins were obtained from Cell Biolabs or Origene. Recombinant RAF proteins were obtained from Origene. Recombinant human NF1-333 was from Creative Biomart. RAS GTPase activity was quantitated using the GTPase-Glo Kit (Promega), GTPase/GAP buffer was used as instructed by the manufacturers. Concentrations of reaction components were indicated in figure legends.
CRISPR-Cas9 mediated NF1 knockout
Two-vector CRISPR-Cas9 system was obtained from Cellecta. Lentivirus encoding Cas9 or the NF1 sgRNAs was packaged in 293FT cells obtained from Invitrogen. The medium containing virus was filtered with 0.45 PVDF filters followed by incubation with the SKBR3 cells for 12 hr. Cells were then maintained in virus free media and selected using Blasticidin (5 μg/ml) or Puromycin (2 μg/ml) for 2 days. Knockout efficiency was determined by western blot.
The YUMM 4.1 cells stably expressing Cas9 proteins were infected with virus containing NF1 sgRNAs and single cell colonies were isolated. NF1 knockout clones were verified through western blot.
PC12 cell differentiation
PC12 cells were either transfected with RAF siRNAs or treated with doxycycline to induce RAF protein expression. Cells were then treated with 50 ng/ml NGF or 100 ng/ml EGF. Images were taken using light microscope over time.
Gene copy number analysis
Ninety-seven EGFR-mutant NSCLC patients with acquired resistance to either gefitinib or erlotinib undergoing standard post-resistance biopsy of their tumor from 2012 to 2016 at Aichi Cancer Center were included in the analysis. Gene copy number of ARAF was analyzed using TaqMan gene copy number assay (Assay ID: Hs05691600_cn, Applied Biosystems), according to the manufacturer’s instructions. TaqMan Copy Number Reference Assay, human RNase P was used as the endogenous reference gene. Fold increase in copy number was calculated as the ratio of the ARAF signal in each tumor to that obtained in the normal ARAF gene in human genomic DNA (Promega). Gene amplification was defined as tumors harboring ARAF copy number control ratio 5.0 or higher. The study was conducted in accordance with ethical guidelines in the Declaration of Helsinki and approved by Institutional Review Board (protocol 2019–1-224 and 2019–1-418) at Aichi Cancer Center. Informed consent was obtained from all patients.
ARAF immunohistochemistry
Progression biopsies and collection of patient samples were conducted under appropriate Institutional Review Board/Privacy Board protocols and waivers (protocols 06–107, 12–245, 14–019). Participating patients signed written informed consent for these biospecimen protocols. This study was conducted in accordance with ethical guidelines in the Declaration of Helsinki. Immunohistochemistry was performed by the Molecular Cytology Core Facility in MSKCC. The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems), antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 minutes with Background Buster solution (Innovex), followed by avidin-biotin blocking for 8 minutes (Ventana Medical Systems). Sections were incubated with anti-ARAF (Santa Cruz sc-408) or anti-BRAF (Sigma HPA001328) antibodies for 5 hours, followed by 60 minutes incubation with biotinylated horse anti- rabbit (Vector Labs, cat# PK6101) at 1:200 dilution. The detection was performed with DAB detection kit (Ventana Medical Systems) according to manufacturer instruction. Slides were counterstained with hematoxylin and coverslipped with Permount (Fisher Scientific).
FISH
FISH analysis was performed on formalin fixed paraffin embedded (FFPE) sections using a 3-color BRAF/ARAF/Cen X Probe. The probe mix consisted of BAC clones containing the full length BRAF gene (clones RP11-788O6, RP11-1065D4, and RP11-133N19; labeled with Green dUTP), the full length ARAF gene (clones RP11-466C12 and RP11-404P16; labeled with Red dUTP) and a centromeric repeat plasmid for chromosome X served as the control for ARAF (clone pSV2×5; labeled with Orange dUTP). Probe labeling, tissue processing, hybridization, post-hybridization washing, and fluorescence detection were performed according to standard laboratory procedures. Slides were scanned using a Zeiss Axioplan 2i epifluorescence microscope equipped with a megapixel CCD camera (CV-M4+CL, JAI) controlled by Isis 5.5.9 imaging software (MetaSystems Group Inc, Waltham, MA).
The entire section was scanned under 63X or 100X objective, intra-tumoral heterogeneity assessed, and representative regions imaged through the depth of the tissue (compressed/merged stack of 12 z-section images taken at 0.5 micron intervals). At least 5 images per representative region were captured and a minimum of 50 discrete nuclei analysed within each region. Amplification was defined as ARAF:Cen X ratio of ≥2.0, ≥6 copies of ARAF/BRAF (independent of control locus) or at least one small cluster of ARAF/BRAF(≥4 signals resulting from tandem repeat/duplication). In cells with high-level amplification, signals ≥20 cannot be accurately counted and therefore given a score of 20. Additionally, amplification was categorized as homogeneously staining region (HSR)when observed as small to large clustered signal. HSRs correspond to linearly integrated chromosomal/DNA segments that appear homogeneous and stain uniformly by conventional cytogenetic methods. Cells with 3–5 copies of ARAF/BRAF were considered to be polysomic.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses used GraphPad Prism 8 software, with a minimum of three biologically independent samples for significance. For animal experiments, each mouse was counted as biologically independent sample. Results are reported as mean+SD or mean+SEM. Comparisons between two groups were performed using an unpaired two-sided Student’s t test. P values of less than 0.05 were considered significant. All experiments were reproduced at least three times, unless otherwise indicated.
Supplementary Material
Highlights:
ARAF activates RAS by antagonizing its interaction with NF1
ARAF controls the duration and consequences of receptor activated ERK signaling
Increased ARAF expression in lung cancer causes acquired resistance to EGFR inhibitors.
Acknowledgements
We are grateful to Mariano Barbacid (Centro Nacional de Investigaciones Oncológicas) for sharing the conditional RAS/RAF/MEK knockout cells. We are grateful to Manuela Baccarini (University of Vienna, Austria) for sharing the A-, B-, and C-RAF knockout MEF cells. We thank Sandra Misale (MSKCC) for providing the NF1 knockout Yumm4.1 cell lines, which were modified from mouse melanoma Yumm4.1 cells originally generated by Marcus W. Bosenberg from Yale University.
Funding:
This research was supported by grants (to N.R.) from the National Institutes of Health (NIH) (P01 CA094060; R35 CA210085); the Geoffrey Beene Cancer Research Center and the Emerson Collective Research Grant. Support was also received from LCRF Pilot Grant (2019) (to Z. Y. and W.S.), the NIH MSKCC Cancer Center Core Grant P30 CA008748, R01 CA233736 (to R.Y.), R01 CA201247 (to O.A.W.), R01 CA204749 (to B.S.T.), R01 CA207244 (to B.S.T.), U01 CA199215 (to C.M.R.), R35CA220497 (to P.A.J.) and U54 OD020355–01 (to E.S.). Additional funding was provided by grants from the Histiocyte Society, the Erdheim-Chester Global Alliance, the Functional Genomics Initiative of MSKCC, the Histiocytosis Association and the Leukemia & Lymphoma Society (to O.A.W.), the Fund for the Promotion of Joint International Research from JSPS (15KK0303) and P-CREATE from the AMED under grant number 19cm0106513h0004 (to H. E.) We would like to acknowledge the support of the Arlene and Joseph Taub Foundation, Paula and Thomas Mclnerney and Debra Black, without whom this work would not have been possible.
Footnotes
Declaration of Interests
N.R. is on the scientific advisory board (SAB) and owns equity in Beigene, Zai Labs, MapKure, Ribon and Effector. N.R. is also on the SAB of Astra-Zeneca and Chugai and a past SAB member of Novartis, Millennium-Takeda, Kura, and Araxes. N.R. is a consultant to RevMed, Tarveda, Array-Pfizer, Boeringher-Ingelheim and Eli Lilly. He receives research funding from Revmed, AstraZeneca, Array Pfizer and Boerhinger-Ingelheim and owns equity in Kura Oncology and Fortress. Z. Y. is a past SAB member of MapKure and currently an employee of Loxo oncology at Lilly. R.Y. has received research support from Array BioPharma/Pfizer, Novartis, and Boehringer-Ingelheim; and served as an advisor for Array BioPharma/Pfizer, Mirati Therapeutics, and Natera. O.A.W. received research funding from H3B Biomedicine and personal fees from H3B Biomedicine, Foundation Medicine Inc, Merck and Jansen. C.M.R has consulted regarding cancer drug development with AbbVie, Amgen, Ascentage, Bicycle, Celgene, Daiichi Sankyo, Genentech/Roche, Ipsen, Loxo, and PharmaMar, and serves on the SAB of Bridge Medicines and Harpoon Therapeutics. P.A.J. has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Roche/Genentech, Merrimack Pharmaceuticals, Chugai Pharmaceuticals, Ariad Pharmaceuticals, Eli Lilly and Company, Araxes Pharama, Ignyta, Novartis, Mirati Therapeutics, Takeda Oncology, Daiichi Sankyo, Biocartis, Voronoi, SFJ Pharmaceuticals, and LOXO Oncology; receives post-marketing royalties from DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp; has sponsored research agreements with AstraZeneca, Daichi Sankyo, Boehringer Ingelheim, PUMA, Eli Lilly, Astellas Pharmaceuticals, and Takeda Oncology; and has stock ownership in Gatekeeper Pharmaceuticals and LOXO Oncology. F.M. is shareholder in Olema, Opna, Kura, BridgeBio, Avidity, Quartz, Quadriga and Wellspring and is a consultant for Amgen, Pfizer, Daiichi Sankyo, Ideaya, BridgeBio, PMV, Caris, and Aduro.
Inclusion and Diversity
We worked to ensure gender balance and ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alavi A, Hood JD, Frausto R, Stupack DG, and Cheresh DA (2003). Role of Raf in vascular protection from distinct apoptotic stimuli. Science 301, 94–96. [DOI] [PubMed] [Google Scholar]
- Baljuls A, Mueller T, Drexler HC, Hekman M, and Rapp UR (2007). Unique N-region determines low basal activity and limited inducibility of A-RAF kinase: the role of N-region in the evolutionary divergence of RAF kinase function in vertebrates. J Biol Chem 282, 26575–26590. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, Chakravarty D, Phillips S, Kandoth C, Penson A, et al. (2018). Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer discovery 8, 174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Fujii K, Zhang L, Roberts T, and Fu H (2001). Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci U S A 98, 7783–7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, Chen Z, Cooke VG, et al. (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. (2015). Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD 17, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, Kerr G, Yu K, Liang J, Cao Y, et al. (2018). SHP2 inhibition restores sensitivity in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nature medicine 24, 512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. (2002). Mutations of the BRAF gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, Gettinger S, Walther Z, Wurtz A, Heynen GJ, et al. (2014). Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer discovery 4, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond EL, Durham BH, Haroche J, Yao Z, Ma J, Parikh SA, Wang Z, Choi J, Kim E, Cohen-Aubart F, et al. (2016). Diverse and Targetable Kinase Alterations Drive Histiocytic Neoplasms. Cancer discovery 6, 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Waters SB, Holt KH, and Pessin JE (1996). SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J Biol Chem 271, 6328–6332. [DOI] [PubMed] [Google Scholar]
- Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, and Barbacid M (2010). Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J 29, 1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosten M, Sum EY, Lechuga CG, Simon-Carrasco L, Jacob HK, Garcia-Medina R, Huang S, Beijersbergen RL, Bernards R, and Barbacid M (2014). Loss of p53 induces cell proliferation via Ras-independent activation of the Raf/Mek/Erk signaling pathway. Proceedings of the National Academy of Sciences of the United States of America 111, 15155–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, Araki K, Ozerdem U, Simeone DM, Miller G, et al. (2018). SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer discovery 8, 1237–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman AK, Ritt DA, and Morrison DK (2013). Effects of Raf dimerization and its inhibition on normal and disease-associated Raf signaling. Mol Cell 49, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Chang MT, McKay D, Na N, Zhou B, Yaeger R, Torres NM, Muniz K, Drosten M, Barbacid M, et al. (2018). Allele-Specific Mechanisms of Activation of MEK1 Mutants Determine Their Properties. Cancer Discov 8, 648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon P, John J, Frech M, Lautwein A, Clark R, Scheffler JE, and Wittinghofer A (1992). Mutational and kinetic analyses of the GTPase-activating protein (GAP)-p21 interaction: the C-terminal domain of GAP is not sufficient for full activity. Mol Cell Biol 12, 2050–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz EM, Ghandi M, Treacy DJ, Wagle N, and Garraway LA (2014). ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res 74, 7079–7089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig A, Markwart R, Wolff K, Schubert K, Cui Y, Prior IA, Esparza-Franco MA, Ladds G, and Rubio I (2016). Feedback activation of neurofibromin terminates growth factor-induced Ras activation. Cell Commun Signal 14, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imielinski M, Greulich H, Kaplan B, Araujo L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M, and Carbone DP (2014). Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J Clin Invest 124, 1582–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamioka Y, Yasuda S, Fujita Y, Aoki K, and Matsuda M (2010). Multiple decisive phosphorylation sites for the negative feedback regulation of SOS1 via ERK. J Biol Chem 285, 33540–33548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, Janne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, and Halmos B (2005). EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. The New England journal of medicine 352, 786–792. [DOI] [PubMed] [Google Scholar]
- Lavoie H, and Therrien M (2015). Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16, 281–298. [DOI] [PubMed] [Google Scholar]
- Lee JE, Beck TW, Wojnowski L, and Rapp UR (1996). Regulation of A-raf expression. Oncogene 12, 1669–1677. [PubMed] [Google Scholar]
- Marais R, Light Y, Paterson HF, Mason CS, and Marshall CJ (1997). Differential regulation of Raf-1, A-Raf, and B-Raf by oncogenic ras and tyrosine kinases. J Biol Chem 272, 4378–4383. [DOI] [PubMed] [Google Scholar]
- Marshall CJ (1995). Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185. [DOI] [PubMed] [Google Scholar]
- Mattar M, McCarthy CR, Kulick AR, Qeriqi B, Guzman S, and de Stanchina E (2018). Establishing and Maintaining an Extensive Library of Patient-Derived Xenograft Models. Front Oncol 8, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeth K, Wang JX, Micevic G, Damsky W, and Bosenberg MW (2016). The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res 29, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer K, Chiloeches A, Huser M, Kiernan M, Marais R, and Pritchard C (2002). ERK signalling and oncogene transformation are not impaired in cells lacking A-Raf. Oncogene 21, 347–355. [DOI] [PubMed] [Google Scholar]
- Mercer K, Giblett S, Oakden A, Brown J, Marais R, and Pritchard C (2005). A-Raf and Raf-1 work together to influence transient ERK phosphorylation and Gl/S cell cycle progression. Oncogene 24, 5207–5217. [DOI] [PubMed] [Google Scholar]
- Mikula M, Schreiber M, Husak Z, Kucerova L, Ruth J, Wieser R, Zatloukal K, Beug H, Wagner EF, and Baccarini M (2001). Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. EMBO J 20, 1952–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooz J, Oberoi-Khanuja TK, Harms GS, Wang W, Jaiswal BS, Seshagiri S, Tikkanen R, and Rajalingam K (2014). Dimerization of the kinase ARAF promotes MAPK pathway activation and cell migration. Science signaling 7, ra73. [DOI] [PubMed] [Google Scholar]
- Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, Tzitzilonis C, Mordec K, Marquez A, Romero J, et al. (2018). RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell Biol 20, 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissan MH, Pratilas CA, Jones AM, Ramirez R, Won H, Liu C, Tiwari S, Kong L, Hanrahan AJ, Yao Z, et al. (2014). Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res 74, 2340–2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon AE, Brune M, Lowe PN, and Webb MR (1995). Kinetics of inorganic phosphate release during the interaction of p21ras with the GTPase-activating proteins, p120-GAP and neurofibromin. Biochemistry 34, 15592–15598. [DOI] [PubMed] [Google Scholar]
- O’Neill E, Rushworth L, Baccarini M, and Kolch W (2004). Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science 306, 2267–2270. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, and Varmus H (2005). Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS medicine 2, e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, and Rosen N (2010). RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, and Rosen N (2009). (V600E)BRAF is associated with disabled feedback inhibition of RAF-MEK signaling and elevated transcriptional output of the pathway. Proc Natl Acad Sci U S A 106, 4519–4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CA, Bolin L, Slattery R, Murray R, and McMahon M (1996). Post-natal lethality and neurological and gastrointestinal defects in mice with targeted disruption of the A-Raf protein kinase gene. Curr Biol 6, 614–617. [DOI] [PubMed] [Google Scholar]
- Rebocho AP, and Marais R (2013). ARAF acts as a scaffold to stabilize BRAF:CRAF heterodimers. Oncogene 32, 3207–3212. [DOI] [PubMed] [Google Scholar]
- Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, and Kolch W (2014). Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat Cell Biol 16, 673–684. [DOI] [PubMed] [Google Scholar]
- Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F, and Wittinghofer A (1997). The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338. [DOI] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. (2011). Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Science translational medicine 3, 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sia D, Losic B, Moeini A, Cabellos L, Hao K, Revill K, Bonal D, Miltiadous O, Zhang Z, Hoshida Y, et al. (2015). Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat Commun 6, 6087. [DOI] [PubMed] [Google Scholar]
- Silverman JA, Zurlo J, Watson MA, and Yager JD (1989). Expression of c-raf-1 and A-raf-1 during regeneration of rat liver following surgical partial hepatectomy. Mol Carcinog 2, 63–67. [DOI] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, and McCormick F (2017). RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrell EM, Durrant DE, Ritt DA, Sealover NE, Sheffels E, Spencer-Smith R, Esposito D, Zhou Y, Hancock JF, Kortum RL, et al. (2019). Distinct Binding Preferences between Ras and Raf Family Members and the Impact on Oncogenic Ras Signaling. Mol Cell 76, 872–884 e875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traverse S, Gomez N, Paterson H, Marshall C, and Cohen P (1992). Sustained activation of the mitogen-activated protein (MAP) kinase cascade may be required for differentiation of PC12 cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem J 288 ( Pt 2), 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesmuller L, and Wittinghofer A (1992). Expression of the GTPase activating domain of the neurofibromatosis type 1 (NF1) gene in Escherichia coli and role of the conserved lysine residue. J Biol Chem 267, 10207–10210. [PubMed] [Google Scholar]
- Wojnowski L, Stancato LF, Larner AC, Rapp UR, and Zimmer A (2000). Overlapping and specific functions of Braf and Craf-1 proto-oncogenes during mouse embryogenesis. Mech Dev 91, 97–104. [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Zimmer AM, Beck TW, Hahn H, Bernal R, Rapp UR, and Zimmer A (1997). Endothelial apoptosis in Braf-deficient mice. Nat Genet 16, 293–297. [DOI] [PubMed] [Google Scholar]
- Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q, de Stanchina E, Abdel-Wahab O, Solit DB, Poulikakos PI, et al. (2015). BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell 28, 370–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. (2017). Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature 548, 234–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, et al. (2017). Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature medicine 23, 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuidzinas A, Gould GW, and Yager JD (1989). Expression of c-raf-1 and A-raf-1 during differentiation of 3T3-L1 preadipocyte fibroblasts into adipocytes. Biochem Biophys Res Commun 162, 1180–1187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original western data and microscopy data for figures in this paper have been deposited at Mendeley Data and are publicly available as of the date of publication. The DOI is listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ARAF | Santa Cruz | sc-408 (AB_630882) |
| BRAF | Santa Cruz | sc-5284 (AB_626760) |
| BRAF | Sigma | HPA001328 (AB_1078296) |
| CRAF | BD | 610152 (AB_397553) |
| FLAG | Sigma | F1804 (AB_262044) |
| V5 | Thermo Fisher | R960-25 (AB_2556564) |
| GAPDH | Santa Cruz | sc-32233 (AB_627679) |
| RAS | Thermo Fisher | MA1-012 (AB_2536664) |
| RAS | Santa Cruz | sc-14022 (not available) |
| RAS | Millipore | 05-516 (AB_11211664) |
| NF1 | Santa Cruz | sc-67 (AB_2149681) |
| Cyclin D1 | Santa Cruz | sc-718 (AB_2070436) |
| Sprouty2 | Abcam | ab85670 (not available) |
| p-MEK1/2 (Ser217/221)) | Cell Signaling | 9154 (AB_2138017) |
| MEK1/2 | Cell Signaling | 4694 (AB_10695868) |
| p-p44/42 MAPK (Erk1/2) | Cell Signaling | 9101 (AB_331646) |
| p44/42 MAPK (Erk1/2) | Cell Signaling | 4696 (AB_390780) |
| p-EGFR (Tyr1068) | Cell Signaling | 3777 (AB_2096270) |
| EGFR | Cell Signaling | 4267 (AB_2246311) |
| p-TrkA | Cell Signaling | 4619 (AB_10235585) |
| AKT | Cell Signaling | 9272 (AB_329827) |
| p-AKT (Ser473) | Cell Signaling | 4060 (AB_2315049) |
| p-AKT (Ser308) | Cell Signaling | 4056 (AB_331163) |
| Chemicals, peptides and recombinant proteins | ||
| DMEM | ThermoFisher Scientific | 11965-092 |
| RPMI 1640 | ThermoFisher Scientific | 61870-036 |
| DMEM/F12 | ThermoFisher Scientific | 12634-010 |
| Renaissance Essential Tumor Medium | Cellaria | CM-0001 |
| B27 supplement | ThermoFisher Scientific | 17504044 |
| Matrigel | Corning | 354234 |
| L-glutamine | Corning | 25005CI |
| penicillin G-streptomycin | Corning | 30004CI |
| Trypsin-EDTA (0.05%) | Invitrogen | 25300054 |
| TrypLE Express | Invitrogen | 12604013 |
| CD45 MicroBeads, human | Miltenyl Biotex | 130-045-801 |
| Doxycycline | Sigma | D3072 |
| 4-OHT | Sigma | H6278 |
| Erlotinib | MedChem Express | HY-50896 |
| Osimertinib | Selleckchem | S7297 |
| Puromycin | Invitrogen | A1113802 |
| hygromycin | Invitrogen | 10687010 |
| Blasticidin | Invitrogen | A1113903 |
| SHP099 | Novartis | N/A |
| Recombinant human ARAF | Origene | TP300737 |
| Recombinant human BRAF | Origene | TP311013 |
| Recombinant human CRAF | Origene | TP301983 |
| Recombinant human NF1-333 | Creative Biomart | NF1-334H |
| Recombinant human EGF | Invitrogen | PHG0311 |
| Recombinant rat NGF | R & D | 7185-NG-025 |
| Recombinant human KRAS | Origene | TP301958 |
| Recombinant human NRAS | Cell Biolabs | STA-749 |
| Recombinant human HRAS | Cell Biolabs | STA-747 |
| Recombinant K97R MEK | Millipore | 14-737 |
| Critical commercial assays | ||
| CellTiter-Glow | Promega | G7573 |
| CellTiter-Glow 3D | Promega | G9682 |
| Active RAS pull-down assay | ThermoFisher Scientific | 16117 |
| GTPase-Glow assay | Promega | V7681 |
| Experimental models: Organisms/strains | ||
| Athymic nude mice (female) | Envigo | Hsd:Athymic Nude-Foxn1nu |
| Nod SCID gamma mice (female) | The Jackson Laboratory | 005557 |
| Experimental models: cell lines | ||
| 293FT (human) | ThermoFisher Scientific | R70007 (CVCL_6911) |
| NIH 3T3 (mouse) | ATCC | CRL-1658 (CVCL-0594) |
| SKBR3 (human) | ATCC | HTB-30 (CVCL_0033) |
| PC9 (human) | Sigma | 90071810 (CVCL_B260) |
| HCC827 (human) | ATCC | CRL-2868 (CVCL_2063) |
| LIM1215 (human) | Sigma | 10092301 (CVCL_2574) |
| BT474 (human) | ATCC | CRL-3247 (CVCL_AQ07) |
| H1703 (human) | ATCC | CRL-5889 (CVCL_1490) |
| A549 (human) | ATCC | CCL-185 (CVCL_0023) |
| H23 (human) | ATCC | CRL-5800 (CVCL_1547) |
| PC12 Tet-On (rat) | Clonetech | 631137 (CVCL_V333) |
| SK-Mel2 (human) | MSKCC | (CVCL_0069) |
| SK-Mel30 (human) | MSKCC | (CVCL_0039) |
| K-Raslox MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| RAF-less MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| MEK-less MEF (mouse) | Mariano Barbacid | doi: 10.1073/pnas.1417549111. |
| ARAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| BRAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| CRAF knockout MEF (mouse) | Manuela Baccarini | N/A |
| YUMM 4.1 (mouse) | Marcus Bosenberg | doi: 10.1111/pcmr.12498. |
| Oligonucleotides | ||
| Human ARAF siRNA | Dharmacon | L-003563-00-0020 |
| Human BRAF siRNA | Dharmacon | L-003460-00-0020 |
| Human CRAF siRNA | Dharmacon | L-003601-00-0020 |
| Rat ARAF siRNA | Dharmacon | L-088540-00-0020 |
| Mouse ARAF siRNA | Dharmacon | L-042948-00-0020 |
| Mouse BRAF siRNA | Dharmacon | L-040325-00-0020 |
| Mouse CRAF siRNA | Dharmacon | L-040149-00-0020 |
| Human ARAF shRNA1 | Sigma | TRCN0000000571 |
| Human ARAF shRNA2 | Sigma | TRCN0000199465 |
| NF1 sgRNA1 | Cellecta | CATGAGACCACTGTCTACGT |
| NF1 sgRNA2 | Cellecta | CCACCACCTAGAATCGAAAG |
| NF1 sgRNA3 | Cellecta | GAGGAAGCAGATATCCGGTG |
| Cyclin D1 qPCR probe | ThermoFisher Scientific | Mm00432359_m1 |
| DUSP4 qPCR probe | ThermoFisher Scientific | Mm00732761_m1 |
| DUSP6 qPCR probe | ThermoFisher Scientific | Mm00518185_m1 |
| SPRY2 qPCR probe | ThermoFisher Scientific | Mm00442344_m1 |
| SPRY4 qPCR probe | ThermoFisher Scientific | Mm00442345_m1 |
| ETV1 qPCR probe | ThermoFisher Scientific | Mm00514804_m1 |
| GAPDH qPCR probe | ThermoFisher Scientific | Mm99999915_g1 |
| Recombinant DNA | ||
| pcDNA3.1-hARAF-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214A-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214C-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214F-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214T-V5 | This paper | N/A |
| pcDNA3.1-hARAF-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-R52L-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214A-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214C-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214F-R362H-V5 | This paper | N/A |
| pcDNA3.1-hARAF-S214T-R362H-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365A-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365C-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-R509H-V5 | This paper | N/A |
| pcDNA3.1-hBRAF-S365C-R509H-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-R401A-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-S259A-V5 | This paper | N/A |
| pcDNA3.1-hCRAF-S259A-R401H-V5 | This paper | N/A |
| pTTIGFP-hARAF-V5 | This paper | N/A |
| pTTIGFP-hBRAF-V5 | This paper | N/A |
| pTTIGFP-hCRAF-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214A-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214C-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214F-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214T-V5 | This paper | N/A |
| pTTIGFP-hBRAF-K601E-V5 | This paper | N/A |
| pTTIGFP-hCRAF-S259A-V5 | This paper | N/A |
| pTTIGFP-hARAF-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-D429A-V5 | This paper | N/A |
| pTTIGFP-hARAF-R362H-V5 | This paper | N/A |
| pTTIGFP-BN-ARAF-V5 | This paper | N/A |
| pTTIGFP-CN-ARAF-V5 | This paper | N/A |
| pTTIGFP-NoN-ARAF-V5 | This paper | N/A |
| pTTIGFP-NoN-BRAF-V5 | This paper | N/A |
| pTTIGFP-AN-BRAF-V5 | This paper | N/A |
| pTTIGFP-NoN-CRAF-V5 | This paper | N/A |
| pTTIGFP-AN-CRAF-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214A-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214C-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214F-R52L-V5 | This paper | N/A |
| pTTIGFP-hARAF-S214T-R52L-V5 | This paper | N/A |
| pXretro-FLAG-NRAS | This paper | N/A |
| pXretro-FLAG-HRAS | This paper | N/A |
| pXretro-FLAG-KRAS | This paper | N/A |
| pMSCV-rtTA3-PGK-Hygro | Scott Lowe | N/A |
| TTIGFP-MLUEX | Scott Lowe | N/A |
| pEF-DEST51-NF1-V5-His | Frank McCormick | N/A |
| Deposited data | ||
| Raw western blot and microscopy data | Mendeley | doi: 10.17632/yx7s86mcrn.1 |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com |