

Abstract
Developmental abnormalities of the cerebellum are among the most recognized structural brain malformations in human prenatal imaging. Yet reliable information regarding their cause in humans is sparse, and few outcome studies are available to inform prognosis. We know very little about human cerebellar development, in stark contrast to the wealth of knowledge from decades of research on cerebellar developmental biology of model organisms, especially mice. Recent studies show that multiple aspects of human cerebellar development significantly differ from mice and even rhesus macaques, a nonhuman primate. These discoveries challenge many current mouse-centric models of normal human cerebellar development and models regarding the pathogenesis of several neurodevelopmental phenotypes affecting the cerebellum, including Dandy-Walker malformation and medulloblastoma. Since we cannot model what we do not know, additional normative and pathological human developmental data are essential, and new models are needed.
Keywords: neurogenesis, rhombic lip, ventricular zone, malformation, genetics, histology
1. INTRODUCTION
The human cerebellum contains approximately 80% of all brain neurons yet has long been understudied relative to the cerebral cortex. Although typecast as simply a modulator of motor control, there is longstanding and growing evidence that the cerebellum has much broader roles in brain function (Schmahmann 2019). Animal models provide clear evidence for direct cerebellar modulation of dopaminergic reward circuits regulating language and social behavior, while anatomical studies in mice and nonhuman primates demonstrate reciprocal cerebellar-cerebral circuits in areas primarily regulating higher-order function (Carta et al. 2019, Fiez & Petersen 1998, Hatten 2020, Pisano et al. 2021). Functional MRI studies have shown that just 20% of the human cerebellum is dedicated to areas involved in physical motion. The other 80% is broadly connected to brain regions regulating abstract thinking, planning, emotion, and memory (Marek et al. 2018). Notably, the cerebellum has scaled in size with the cerebral cortex across primate evolution. In macaques, the cerebellum represents just 33% of the surface area of the cerebral cortex. In humans, it represents almost 80% of the cerebral cortex surface area (Sereno et al. 2020), further supporting a prominent role for the cerebellum in the evolution of distinctively human behaviors and cognition. Several human neurodevelopmental disorders display significant cerebellar abnormalities, primarily diagnosed by neuroimaging, since disruptions of the highly stereotypic foliation and lamination of the cerebellum are readily apparent (Figure 1). The existence of these neurodevelopmental disorders and the evolved adaptations of the human cerebellum provide significant impetus to study basic human cerebellar biology in both normal and abnormal development.
Figure 1.
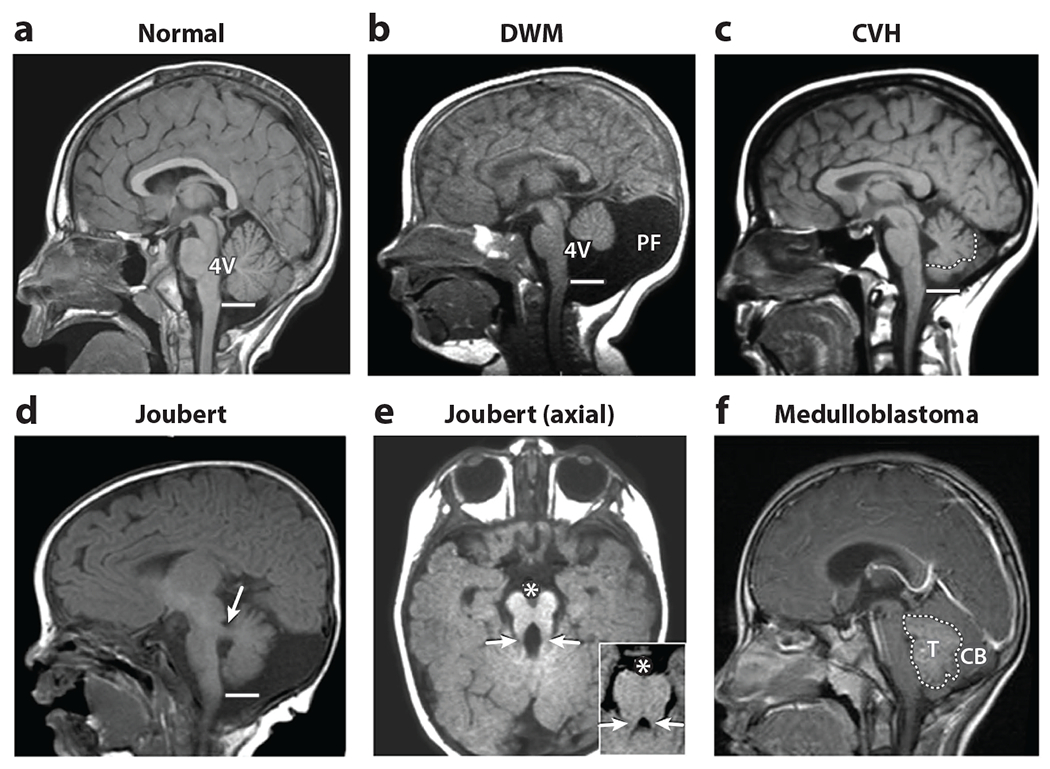
Neuroimaging findings define human cerebellar malformations and anomalies. (a) A normal midline sagittal T1-weighted image demonstrates the exquisite structure of the cerebellar vermis extending to the obex of the brainstem (line) adjacent to the fourth ventricle (4V). (b) Dandy-Walker malformation (DWM) is characterized by a small cerebellar vermis, which is rotated away from the brainstem, resulting in an enlarged 4V and space at the back of the skull [posterior fossa (PF)]. (c) In cerebellar vermis hypoplasia (CVH), the cerebellar vermis is hypoplastic without an enlarged 4V or PF (the vermis is outlined by a dotted line to distinguish it from the lateral cerebellar hemispheres, which collapsed to occupy medial space). (d) Joubert syndrome is characterized by a small cerebellar vermis, which also fails to extend to the obex, together with thickened and abnormally positioned cerebellar peduncles (white arrow) in this paramedial sagittal image. (e) In axial images, these abnormal peduncles (white arrows), together with a large interpeduncular fissure (denoted by *), result in a molar tooth sign (inset), never seen in normal individuals. (f) In this example of medulloblastoma, a tumor mass (T) obscures the normal cerebellar (CB) structure. Figure adapted from Chizhikov & Millen (2013).
Studies, particularly in mice, continue to reveal the cellular and molecular drivers of the developmental programs that generate the cerebellum (Haldipur & Millen 2019, Leto et al. 2016). The basic structure and organization of the cerebellum is largely conserved across vertebrates, yet features such as differential scaling of neurons, extent of foliation, and expansion of the cerebellar hemispheres versus the medial vermis are critical for human cerebellar development and function (Haldipur & Millen 2019, Kebschull et al. 2020, Smaers et al. 2018). Surprisingly little is known about human cerebellar development. Recent studies have begun to identify human-specific developmental programs (Aldinger et al. 2019, 2021; Haldipur et al. 2019, 2021). Some share features with mechanisms of human cerebral cortex neurogenesis and highlight a role for abnormal cerebellar development in more broadly defined neurodevelopmental disorders. Yet there are also unique human cerebellar developmental programs. Both genetic and fetal pathological analyses challenge some of our previously mouse-centric models regarding (a) normal cerebellar development; (b) the pathogenesis of Dandy-Walker malformation (DWM), a well-recognized cerebellar malformation; and (c) the cells of origin of medulloblastoma, one of the most common childhood brain tumors.
In this review we describe what we know about the developing human cerebellum, discuss how this impacts our understanding of neurodevelopmental disorders, and identify the next key areas of focus.
2. HUMAN NEURODEVELOPMENTAL DISORDERS AFFECTING THE CEREBELLUM
Many cerebellar malformations and tumors are recognized as disorders of cerebellar development. Cerebellar malformations are heterogeneous developmental anomalies that are frequently associated with a broad range of prognoses and associated medical concerns (Aldinger & Doherty 2016, Bolduc et al. 2011, Bolduc & Limperopoulos 2009). They are diagnosed by recognizing a specific pattern in neuroimaging (Figure 1) and through histopathology at autopsy. Diagnosis commonly occurs during pregnancy due to the widespread use of ultrasound and fetal MRI. A malformed cerebellum can be unusually small, dysplastic, or abnormally large. The prevalence of cerebellar malformations is unknown because few population-based cohorts capture these phenotypes.
Collectively cerebellar malformations are classified as primarily affecting the cerebellum or both the cerebellum and brainstem. They encompass a wide range of morphological presentations, including nonspecific diagnoses that are idiopathic. Joubert syndrome, which is diagnosed based on the presence of the pathognomonic molar tooth sign in the brainstem (Bachmann-Gagescu et al. 2020, Parisi & Glass 1993), is a notable exception. Though cerebellar malformations have historically been used as primary diagnoses, cerebellar abnormality diagnosis alone rarely provides prognostic information for families. Although they can occur in isolation, cerebellar anomalies often occur as part of syndromes involving multiple brain regions and organ systems and have variable expressivity (Aldinger et al. 2019, Stambolliu et al. 2017). Cerebellar abnormalities have been linked to both genetic and nongenetic mechanisms, including rare chromosomal abnormalities and monogenic syndromes, infection, stroke, prenatal hemorrhage, and prematurity (Aldinger et al. 2019, Limperopoulos et al. 2005b, Murray et al. 1985, Reeder et al. 2015). Cerebellar hypoplasia is often a nonspecific feature associated with distinct chromosomal abnormalities that impact many genes. It is also observed in a subset of individuals diagnosed with autism spectrum disorder, along with fewer Purkinje cells (PCs) (Bauman & Kemper 2005, Skefos et al. 2014), and an impaired eye blink conditioning response (Oristaglio et al. 2013), all of which implicate cerebellar dysfunction in this disorder.
Most monogenic syndromes featuring a cerebellar anomaly are inherited in an autosomal-recessive manner. These conditions include congenital disorders of glycosylation, caused by mutations in over 100 genes (Paprocka et al. 2021); Joubert syndrome, caused by mutations in about 50 genes (Parisi & Glass 1993); and pontocerebellar hypoplasia, with 11 genetically defined subtypes (van Dijk et al. 2018). Gene identification has converged on critical pathways and cellular processes that include cilia formation and function in Joubert syndrome and RNA metabolism in pontocerebellar hypoplasia. Rhombencephalosynapsis, characterized by fusion of the cerebellar hemispheres with partial or complete absence of a recognizable vermis, has only been associated with a single, rare genetic cause so far despite being a highly stereotyped anomaly (Aldinger et al. 2018, Mak et al. 2020). DWM, in the absence of a congenital disorder of glycosylation, has been associated with a rare genetic cause only when additional syndromic features are present (Aldinger et al. 2019). Interestingly, case reports with serial neuroimaging document prenatal cerebellar hypoplasia, followed by prenatal cerebellar hemorrhage, that ultimately presents as DWM (Pichiecchio et al. 2016, Shiohama et al. 2017). Further investigation is required to evaluate whether a genetic risk factor and a prenatal injury such as hemorrhage interact to cause DWM or other cerebellar abnormalities.
Although not specifically a neurodevelopmental disorder, medulloblastoma is an embryonal tumor of the cerebellum and is among the most common brain tumors in children. Diagnosis is based on clinical symptoms, neuroimaging, and integrated histopathological and molecular analysis. Four distinct subgroups are recognized based on transcriptional profiling studies: wingless (WNT), sonic hedgehog (SHH), group 3, and group 4. Each subgroup is characterized by a unique set of demographic and clinical features, genetics, and gene expression, with molecular subclassification correlating well with outcome (Garcia-Lopez et al. 2021, Hovestadt et al. 2020, Northcott et al. 2019).
Collectively, numerous diverse disorders that include developmental abnormalities of the human cerebellum are recognized. Since pediatric cerebellar phenotypes are primarily defined by neuroimaging, ascertainment bias has clouded the interpretation of these disorders. Many cerebellar disorders are associated with other central nervous system (CNS) deficits, which may or may not be readily identified via neuroimaging studies. Cerebellar-specific injury can also lead to secondary deficiencies elsewhere (Limperopoulos et al. 2014). Further, many fundamental neurodevelopmental mechanisms such as neuronal migration (Allen & Lyons 2018, Hatten 1999) are shared throughout the developing CNS. To delve deeper into the causes and pathogenesis of human cerebellar abnormalities, it is first helpful to review what we know about normal cerebellar development.
3. MOUSE CEREBELLAR DEVELOPMENT
A rich knowledge base on cerebellar development is available from decades of research in model vertebrates, particularly mice. Here we review the major developmental programs known to drive cerebellar neurogenesis (Figure 2) and to define the spatial relationships of progenitor zones and primary derivatives, as this information is particularly relevant to human cerebellar developmental disorders.
Figure 2.
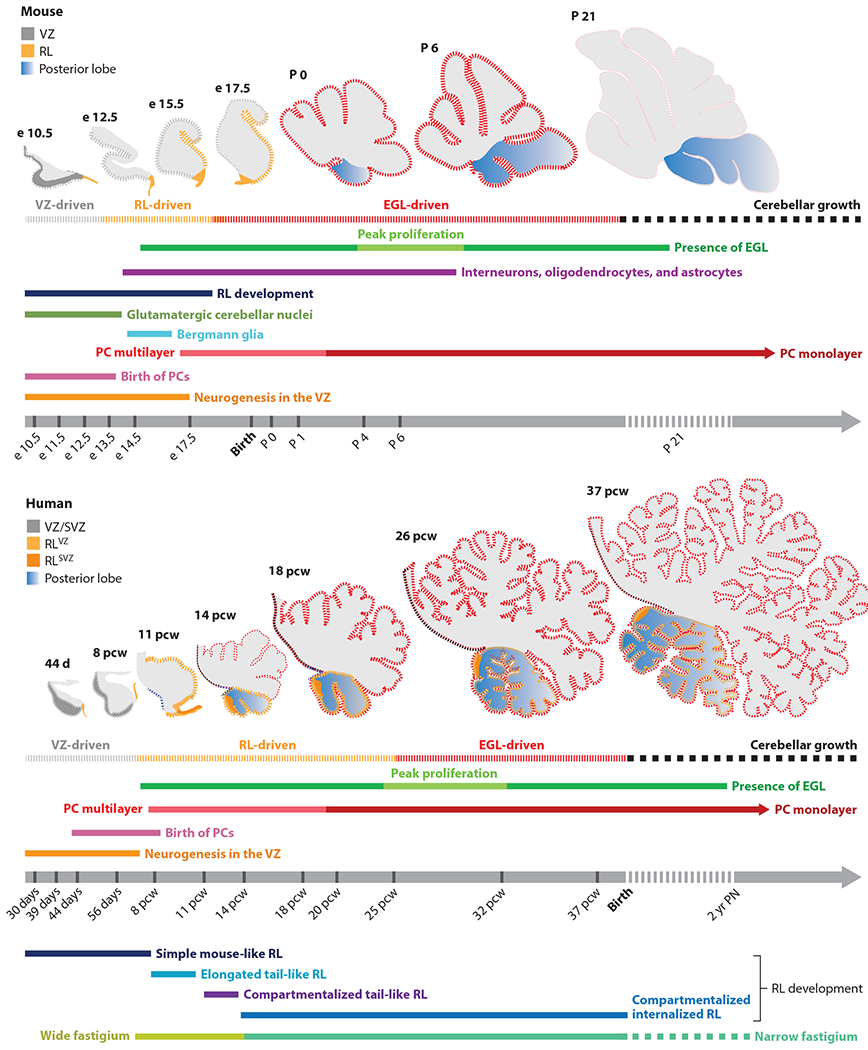
Timeline of mouse and human cerebellar development. Cerebellar neurogenesis in the mouse and human is driven by three progenitor zones: two primary zones, namely the VZ and RL, and one secondary zone, the EGL. Development of each cell type is indicated by a unique colored line. In mice, cerebellar development takes place over a period of approximately 30–35 days, with a significant portion occurring in the postnatal period. In humans, cerebellar development is protracted, taking place over 2–3 years, beginning approximately 30 days post-conception, with all major developmental events taking place in utero. Peak progenitor proliferation is indicated by thickened dashed lines. In both species, VZ- and RL-driven growth precedes an EGL-driven increase in cerebellar volume and foliation. However, unlike in the mouse, where RL presence is transient, in humans, the RL is spatiotemporally expanded and promotes growth and maintenance of the posterior lobe throughout gestation. Abbreviations: e, embryonic day; EGL, external granule layer; P, postnatal day; PC, Purkinje cell; pcw, postconceptional week; PN, postnatal; RL, rhombic lip; SVZ, subventricular zone; VZ, ventricular zone.
The cerebellum is a derivative of the anterior dorsal hindbrain. Cerebellar development is initiated by the emergence of the isthmic organizer at the mid-/hindbrain boundary by a signaling cascade involving fibroblast growth factor (FGF) and WNT signaling (Li et al. 2002). Two primary zones of neurogenesis are induced to generate all cerebellar neurons: the ventricular zone (VZ) and the rhombic lip (RL) (Leto et al. 2016). The VZ generates all cerebellar GABAergic neurons and most glial cells, including radial glial cells, which serve as scaffolds for migration of neurons into the cerebellar anlage, and a subset transform into Bergmann glial cells. The dorsally located RL, induced by dorsal bone morphogenetic protein (BMP) signaling, produces all cerebellar glutamatergic neurons and contributes multiple populations to the developing brainstem, including the pontine nuclei.
Cerebellar GABAergic neurons are produced in overlapping waves (Sudarov et al. 2011). Initially, GABAergic cerebellar nuclei neurons are generated, although the mechanisms driving their neurogenesis remain unclear. Subsequently, PCs and PAX2+ interneuronal progenitors (PIPs) are generated and migrate into the cerebellar anlage. Both are produced in distinct domains and at different time periods. The PTF1A+ VZ is subdivided into two main zones: an OLIG2+ zone that produces PCs and a GSX1+ zone that produces PIPs. During the birth of PCs [embryonic day (e)10.5–13.5], a major portion of the VZ expresses OLIG2, while GSX1 expression is confined to the most ventral region. However, as the production of PCs begins to wane in favor of PIPs, the OLIG2+ domain shrinks at the expense of GSX1, which spreads dorsally and by the end of embryonic development covers almost the entire VZ (Seto et al. 2014). Early-born PIPs differentiate into inhibitory neurons of the cerebellar nuclei, while those born later generate Golgi cells and precursors of basket and stellate cells, with cell fate determined by laminar location (Leto et al. 2012).
Glutamatergic neurons are produced in waves of neurogenesis from the RL. From e10 to e13.5, glutamatergic neurons of the cerebellar nuclei, plus multiple brainstem populations, are generated. This is followed by the production of granule cell precursors that form the external granule layer (EGL) (Machold & Fishell 2005) and, unlike other derivatives, remain proliferative. Between e16.5 and birth, when the mouse RL disappears, the mouse RL also produces unipolar brush cells (UBCs) (Englund et al. 2006, Yeung et al. 2014). The RL, though short lived in the mouse, produces granule cell precursors that differentiate into granule neurons, which account for nearly 60% of all neurons in the mouse brain (Lange 1975). The mouse RL is small and lacks structural compartmentalization. However, like the VZ, it exhibits molecular compartmentalization. The early RL is composed almost entirely of WLS+ cells. However, as development proceeds, it expands into molecularly distinct zones that contain a population of cells that express LMX1A, PAX6, and TBR2 in isolation or in combination (Yeung et al. 2014). While cell fates are assigned within the RL, mechanisms remain unclear, with the exception of an important role for LMX1A (Chizhikov et al. 2010).
As granule cell progenitors exit the RL to migrate over the anlage to form the EGL, they express ATOH1+ and proliferate, populating the presumptive anterior lobes followed by those building the posterior lobe (Machold & Fishell 2005). Extensive proliferation within the EGL occurs during the postnatal period in mice, with proliferation peaking around postnatal day (P)6–8 induced by SHH and other mitogens secreted by underlying PCs. Blocking SHH results in a striking reduction in granule cell progenitors and reduced cerebellar volume (Corrales et al. 2004, Dahmane & Ruiz i Altaba 1999). ATOH1, along with other players such as MYCN and JAG1, maintains granule cell precursors in a proliferative state. Following clonal expansion, these cells exit the cell cycle associated with NEUROD1 upregulation and cell polarity changes (Ong et al. 2020) and enter the inner compartment of the EGL preparing for their eventual inward migration along Bergmann glial fibers (Solecki et al 2009, Wilson et al. 2010). Postmitotic granule neurons migrate radially inward along Bergmann glia fibers that differentiated from ventricular radial glia. Simultaneously, they extend parallel fibers that run along the molecular layer. Eventually, they settle in the internal granule layer beneath the PC layer, finalizing the mature laminar structure of the cerebellum.
Although numerous intrinsic molecular programs drive cerebellar development, complex interactions between cerebellar cell types plus extracerebellar influences from the choroid plexus and posterior fossa mesenchyme are also essential. For example, the choroid plexus secretes mitogens, including SHH, which through transventricular signaling aids expansion of the ventricular zone neuroepithelium (Huang et al. 2010). Additionally, the posterior fossa mesenchyme overlaying the developing cerebellum secretes factors, including SDF1a, that regulate proliferation and migration of VZ- and RL-derived populations. Disrupting mesenchyme function in mouse cerebellum causes aberrant migration of PCs and granule cell precursors that build the posterior vermis, leading to a hypoplastic cerebellum with ectopic populations, including heterotopia (Haldipur et al. 2014, 2017).
While mice have been instrumental for understanding cerebellar development and disease, mice lack key transient human developmental structures (Haldipur et al. 2019), a key limitation of this model.
4. HUMAN CEREBELLAR DEVELOPMENT
The human cerebellum shares foundational similarities in lamination, circuitry, neuronal subtypes, and basic patterns of foliation with mice. However, there are several significant differences between the two species. The human cerebellum has 750-fold greater surface area and contains about 80% of all neurons compared to that of mice, which only contains 60%. Neuronal subtype ratios differ between species, suggesting variations in developmental neurogenesis. For example, in mice, the granule cell to PC ratio is 200:1, while in humans this ratio is 3,000:1 (Lange 1975). Additionally, the human cerebellum also exhibits greater folial complexity, with massively enlarged cerebellar hemispheres relative to the medial cerebellar vermis.
Studies detailing human cerebellar development are scant. The earliest reports were before publication of photographic plates or the advent of high-resolution microscopes but included tracings and diagrams of cerebellar foliation patterns (Larsell & Jansen 1972, Larsell & Stotler 1947). In their landmark paper, Rakic & Sidman (1970) described histogenesis in the developing human cerebellum. Documented in these early studies, and verified in recent work, human cerebellar development is protracted, beginning prior to 30 days post conception (dpc) and ending during the second postnatal year (Haldipur et al. 2011, 2019; Rakic & Sidman 1970) (Figure 2).
Human data prior to 30 dpc are unavailable. By 30 dpc, the nascent cerebellar anlage is specified and composed almost entirely of dividing progenitors in a simple neuroepithelium. By 40–45 dpc, the human cerebellar VZ is conspicuously split into a VZ and a subventricular zone (SVZ) (Haldipur et al. 2019), which is never seen in mice or other models. Early data are unavailable for other primates. Elaboration of the human cerebellar VZ is reminiscent of the subdivision of the VZ in the developing human cerebral cortex that occurs later, at approximately 8 post-conceptional weeks (pcw) (Zecevic et al. 2005). Notably, the cerebellar SVZ, like its cerebral counterpart, is composed of basally mitotic outer radial glia-like progenitors. However, unlike in the cerebral cortex, these basally located progenitors are not confined to a defined domain but are scattered throughout the cerebellar SVZ. Differentiation of GABAergic neurons, including PCs, begins by 47 dpc in the outer SVZ (Haldipur et al. 2019). VZ neurogenesis is largely complete by 56 dpc, corresponding to the end of the embryonic period and beginning of fetal development in humans. By 56 dpc, the SVZ is occupied by differentiated immature PCs, while the VZ is thin and lined by residual progenitors, a subset of which are dividing and likely giving rise to PIPs.
Human RL development is protracted. It is readily evident at 30 dpc, but, unlike the VZ, it does not disappear by 56 dpc. Instead, the RL undergoes extensive morphological and substructural transformations, only disappearing near the end of gestation (Haldipur et al. 2019). The embryonic (<56 dpc) RL is slender and cellularly homogeneous and composed of progenitors. By analogy to the mouse, this simple embryonic RL likely produces glutamatergic cerebellar nuclei neurons and brainstem-destined populations. By 8 pcw, the human RL becomes strikingly more elaborate than that of the mouse or even rhesus macaque, a nonhuman primate. It expands into a tail-like structure, trailing from the dorsal posterior end of the cerebellum. This tail-like RL develops significant substructure by 11 pcw, expanding and splitting into a molecularly distinct VZ and SVZ, separated by a conspicuous vascular bed. The RLVZ contains SOX2+ self-renewing progenitors, while the RLSVZ contains progenitors destined to differentiate into either TBR2+ UBCs that migrate into the core of various cerebellar lobes or ATOH1+ granule cell precursors that migrate onto the surface of the anlage to form the EGL where they extensively divide, driving foliation and increased cerebellar volume. As the posterior cerebellum grows, the RL becomes embedded in the most posterior lobe of the cerebellum, retaining substructure throughout gestation, with a few residual progenitors present at birth (Haldipur et al. 2019).
The human EGL first appears at the beginning of 8 pcw, growing as PCs coalesce beneath the nascent EGL, secreting SHH, which induces proliferation of granule cell precursors. Growth then induces foliation and the formation of lobes and lobules (Aguilar et al. 2012, Haldipur et al. 2012). The principal lobes are first visible between 11 and 13 pcw. As proliferation continues, each lobe foliates further, increasing the overall area and volume of the cerebellum. The three main lobes do not grow concurrently. The anterior lobe expands much earlier compared to the central and posterior lobes. Posterior lobe growth lags, and the rate of proliferation in the posterior EGL is significantly slower compared to the anterior and central lobes (Haldipur et al. 2021). While mechanisms leading to this difference are unknown, it correlates with the graded maturation of PCs in the cerebellum. Peak EGL proliferation and thickness occur between 26 and 32 pcw, which causes the area and volume of the cerebellum to increase 30- and fivefold, respectively, between 26 pcw and birth (Abraham et al. 2001, Haldipur et al. 2011, Rakic & Sidman 1970, Volpe 2009). By 40 pcw, the EGL thins but remains present until the end of the first postnatal year.
Since human cerebellar development is protracted, peak proliferation and expansion of each progenitor zone is largely nonoverlapping, making it difficult to directly compare timing between mouse and human development (Figure 2) but making it easier to divide human cerebellar neurogenesis into distinct epochs. During the embryonic period (<56 dpc), VZ neurogenesis occurs. Thus, GABAergic neurons such as PCs and PAX2+ interneuron progenitors and glutamatergic neurons of the cerebellar nuclei are produced considerably ahead of neuronal production within the cerebral cortex. Concomitantly, the simply embryonic RL produces glutamatergic neurons of the cerebellar nuclei and multiple brainstem populations. As VZ neurogenesis concludes, there is dramatic expansion and internalization of the human RL. From 8 to 11 pcw, the early RL tail (preinternalized) likely produces granule cell precursors that establish the cardinal lobes. Following internalization around 14 pcw, the RL primarily produces UBCs and drives growth of the posterior lobe (Haldipur et al. 2019, 2021). Finally, an extensive growth phase occurs in the third trimester driven by extensive EGL proliferation. The protracted time course provides substantial opportunities for genetic and environmental insults to derail development. Yet it also offers significant opportunity to define key molecular regulators, since intermediate/transitional cell states are more likely to be found in humans versus other vertebrates.
5. TRANSCRIPTIONAL ARCHITECTURE OF HUMAN CEREBELLAR DEVELOPMENT
Transcriptomics provides an unbiased means to study the genetic architecture of the human developing cerebellum that can be applied across areas, layers, and cell types. Large-scale atlases that include spatiotemporal coverage of the cerebellum have been produced for the adult mouse (Lein et al. 2007) and the developing mouse (Carter et al. 2018, Sarropoulos et al. 2021, Thompson et al. 2014, Vladoiu et al. 2019, Wizeman et al. 2019). Histological analyses have distinguished mouse and human cerebellar developmental programs, yet transcriptomic analyses of human brain development have largely focused on neocortical regions, with little representation of cerebellum (Kang et al. 2011, Miller et al. 2014). To address this significant knowledge gap, we generated the first Developmental Cell Atlas of the Human Cerebellum profiling bulk and three laser-microdissected developmental compartments and ~70,000 single nuclei of human cerebellum across 13 weeks of early and mid-fetal development (Aldinger et al. 2021). Profiling gene expression of progenitor zones beyond midgestation is challenging due to their waning size and abundance relative to the exponential growth of the cerebellum and the scarcity of late-stage tissue.
Since developmental compartments are spatially demarcated within the prenatal human cerebellum, they are readily isolated using various microdissection methods then profiled to obtain complete transcriptomic signatures. These compartments include progenitor zones (RL and EGL) and migrating neurons (EGL and PC layer), each with transcriptional signatures that reflect their cellular composition and biological activity (Aldinger et al. 2021). We confirmed that glutamatergic progenitors of the RL and EGL are dividing cells with transcriptional profiles enriched in genes controlling cell cycle and DNA replication. These gene expression signatures also reflect glial and endothelial cells residing in these compartments and likely contamination with cells abundant in tissues adjacent to the RL and EGL, especially mesenchyme and choroid plexus. Within the RL, progenitors in the RLVZ and RLSVZ subcompartments are transcriptionally distinct, with RLVZ gene expression profiles enriched in cell morphology and morphogenesis and those of the RLSVZ enriched in cell signaling (Haldipur et al. 2019). Not only do the RL and EGL transcriptional profiles demonstrate similarities due to their cellular makeup but they also show unique biological activity. Genes comprising the Hippo signaling pathway are highly expressed in the RL, while genes constituting the MAPK signaling pathway are highly expressed in the EGL. In mice, the Hippo signaling pathway is essential for maintaining glutamatergic progenitors by establishing the radial glia scaffold and junctional stability of the cerebellar neuroepithelium (Hughes et al. 2020, Yang & Joyner 2019), while the MAPK signaling pathway is critical for maintaining the proliferation of granule cell progenitors (Guldal et al. 2012). The precise role of these pathways in human cerebellar development requires further investigation.
Two additional developmental compartments bear mentioning: the VZ and the PC layer. The VZ is short lived, existing prior to 56 dpc. Data sets defining the VZ transcriptional signature have yet to be published, but they are critical to elucidate the molecular specification and developmental trajectories of VZ lineages. The PC layer evolves throughout fetal development (Haldipur et al. 2011, Zecevic & Rakic 1976). Initially, composed of clusters of nondividing newborn PCs migrating radially away from the VZ toward the outer surface of the cerebellum. Immature PCs initially aggregate in multiple layers, known as the PC plate, that occupy most of the early developing cerebellar anlage. By 30 pcw, PCs arrange into their final monocellular alignment whereby they undergo axon growth and dendritic tree expansion, which continues after birth. During early to mid-fetal development (9–21 pcw), well before PCs establish a monolayer, the transcriptional profile of the PC layer is enriched in genes that are involved in synapse organization and signaling, but no specific pathways emerge. We conclude that PCs are not dynamic during mid-fetal development. Investigating the molecular diversity and developmental trajectories among clusters of PCs requires additional sampling from earlier time points that capture VZ progenitors and from later time points that capture multiple stages of PC maturation.
Complementing bulk transcriptional profiling, we applied single-cell transcriptomics to nuclei isolated from the prenatal cerebellum, identifying 21 cell types among ~70,000 nuclei (Aldinger et al. 2021). The proportions of major cerebellar cell types, including PCs, RL progenitors, granule cell progenitors, and granule neurons, mirror well-known changes in their cellular composition across developmental time, with concordance between humans and mice. PCs comprise nearly all nuclei in the cerebellar anlage at 9 pcw and only a third of nuclei at 20 pcw, while granule neurons are present in inverse proportions. RL lineage derivatives, including granule cell progenitors, granule neurons, excitatory cerebellar neurons, and UBCs, are among the identified cell types, as are subcompartments of the RL. Although excitatory cerebellar neurons and UBCs emerge from the RL at different times, these cell types could not be distinguished in the current data set. More refined single-cell studies of enriched cell populations, using microdissection or fluorescence-activated nuclei sorting, for example, that are aimed at distinguishing excitatory cerebellar neurons from UBCs and resolving the dynamics of progenitor cell types are required.
In its current form, transcriptional signatures from the Developmental Cell Atlas of the Human Cerebellum can be used to deconvolute existing bulk transcriptome data to reveal the cell types captured in bulk samples. Furthermore, this atlas provides an important resource to investigate comparative evolution, benchmark model systems, and investigate the cell type origins of human diseases.
6. REEVALUATING NEURODEVELOPMENTAL DISORDERS INVOLVING THE CEREBELLUM
A convergence of recent human genetic studies and data from human fetal tissue analyses considerably shifts our paradigms regarding the causes of several neurodevelopmental disorders involving the cerebellum. Genetics demonstrates that cerebellar developmental abnormalities are often a part of broader neurodevelopmental disorders wherein there is likely a significant interplay of genetic and environmental risk factors that contribute. Histological and molecular analyses of human developing cerebellar tissue are facilitating new hypotheses regarding causes and timing of insults not previously possible from mouse data.
The only exome sequencing study of individuals with cerebellar malformations found cerebellar malformations occur as a comorbidity of a monogenic neurodevelopmental disorder in about one third of patients included in the study. Aldinger et al. (2019) studied 100 samples from individuals with cerebellar malformations from mostly trio-based studies totaling 282 exomes. They identified rare, mostly de novo variants in 27 neurodevelopmental disorder–associated genes with variable expressivity of any cerebellar malformation largely unique to a single family. Some of these genetic disorders, including BCL11A and FOXP1, were highly penetrant for cerebellar malformations, though a cerebellar phenotype had not previously been described for these syndromes. Individuals who received a molecular diagnosis had intellectual disability and other developmental comorbidities, including autism and/or epilepsy, and often a cerebellar anomaly other than DWM. Indicative of these findings, expression of the impacted genes in human development is correlated between cerebellum and neocortex. Thus, individuals diagnosed with a cerebellar malformation and additional neurodevelopmental comorbidities are most likely to receive a genetic diagnosis. These individuals are also more likely to have an unfavorable long-term neurodevelopmental outcome (Bolduc et al. 2011). This relationship underscores the value of prenatal genetic testing to inform prenatal counseling of a cerebellar abnormality diagnosis, since the presence of additional neurodevelopmental features is often not detectable by prenatal imaging, and they only become visible as clinical symptoms as the child grows. Categorical diagnoses that have long been used by both clinicians and scientists fail to capture the variability of symptoms within categories and the co-occurrence of diagnoses within and among individuals (Astle et al. 2021). Cerebellar malformations are often categorical diagnoses that provide little prognostic information on their own. Further, the extent to which cerebellar dysfunction contributes to the impact and prognosis of neurodevelopmental disorders is not well known and warrants further study.
Twinning, prenatal hemorrhage, premature birth, and select infertility treatments are risk factors for cerebellar anomalies, especially DWM (Aldinger et al. 2019, Limperopoulos et al. 2005a, Pichiecchio et al. 2016, Reeder et al. 2015, Reefhuis et al. 2011). Among the two-thirds of individuals in the cerebellar malformation exome study who did not receive a molecular diagnosis, half had evidence of a prenatal injury, while the remaining individuals lacked both a molecular diagnosis and evidence of a prenatal injury (Aldinger et al. 2019) (Figure 3). Interestingly, exome sequencing studies of individuals with cerebral palsy, long presumed to be caused by environmental risk factors or prenatal injury, are revealing an impact to genes important for early brain development that are associated with known neurodevelopmental disorders or predisposition to known environmental risk factors (Jin et al. 2020, May et al. 2021, Moreno-De-Luca et al. 2021). A complex relationship between genetic and environmental risk factors may similarly underlie cerebellar anomalies. Furthermore, while copy number variants are rarely associated with cerebellar malformations (Sajan et al. 2013), additional genetic variation not captured by exome sequencing, including noncoding variants and repeat polymorphisms, warrants further study.
Figure 3.
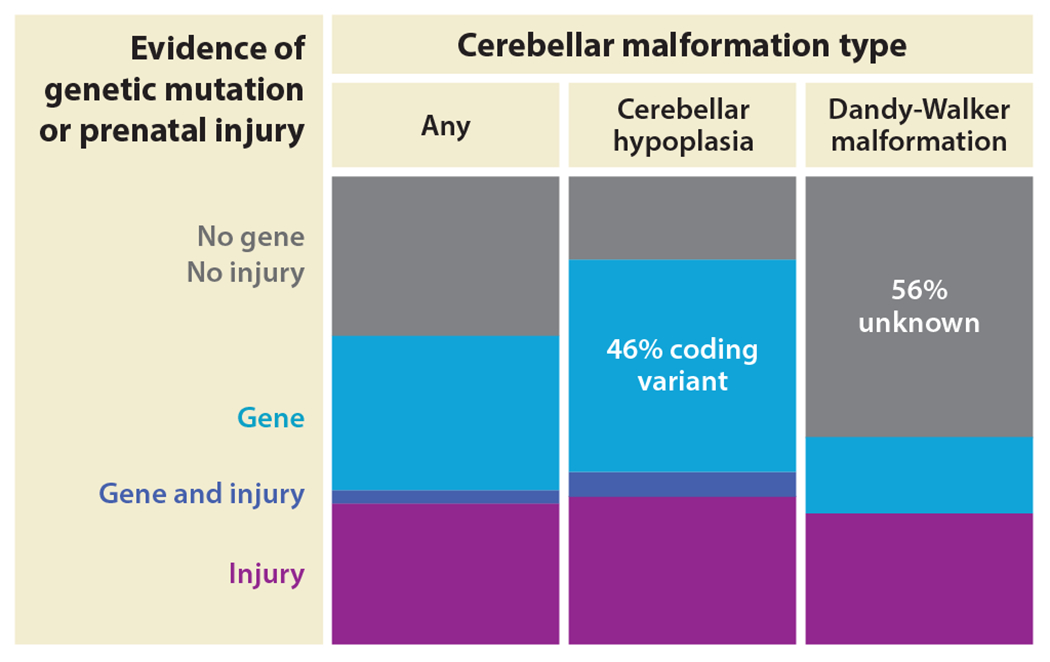
Genetic and prenatal factors cause cerebellar malformations. The cumulative genetic and prenatal injury rates associated with cerebellar malformations are depicted as stacked bar graphs. The prenatal injury rate is constant among cerebellar malformation types, but genetic rates vary among cerebellar malformation diagnoses. Exome sequencing is nondiagnostic for Dandy-Walker malformation among the patients tested. Here, cerebellar hypoplasia is used as a catchall term for cerebellar malformation types that are not Dandy-Walker malformation. Data are based on published exome sequencing, review of prenatal history, and review of neuroimaging (Aldinger et al. 2019).
The complex and protracted development of the human cerebellum leaves it vulnerable to genetic and environmental insults, whereby timing of insult differentially impacts the extent of anatomical damage. The human RL is a highly vascular and fragile tissue as it transitions from simple embryonic structure to a long tail and then internalized stem cell population from 8 to 14 pcw, which underscores its vulnerability to vascular or other insults. Pathologic studies of fetal DWM suggest early insults to the RL during this period are correlated with more severe hypoplasia that can alter anterior cerebellar development. This contrasts with later insults to the RL after internalization that likely lead to disproportionate underdevelopment of only the posteriormost lobule (Haldipur et al. 2021). The highly proliferative EGL is also vulnerable to insults related to premature birth such as hemorrhage and hypoxia, which can cause generalized cerebellar hypoplasia, although mechanisms of damage to the granule cell progenitors remain underexplored(Limperopoulos et al. 2005c, Nguyen et al. 2018, Volpe 2009, Yoo et al. 2014). Since development and maturation of each cerebellar neuronal population is highly interdependent, disruptions of any one neuronal type can have broader impacts on cerebellar neuron scaling and circuit function (Beckinghausen & Sillitoe 2019, van der Heijden et al. 2021, Willett et al. 2019). Defining the impacts of these insults within the developing cerebellum and impact on the cerebellum’s connections to cortical regions is essential to understand the functional implications for any disruption of cerebellar development (Boswinkel et al. 2019, Dijkshoorn et al. 2020, Limperopoulos et al. 2005c, van der Heijden et al. 2021).
The complexity and longevity of progenitors in both human RL and EGL make them exceptional candidates for cells of origin for medulloblastoma. While granule cell progenitors have long been implicated in several medulloblastoma subtypes, the cells of origin for groups 3 and 4 have remained elusive, based on a lack of clear similarity to mouse progenitors (Hovestadt et al. 2019, Luo et al. 2021, Ocasio et al. 2019, Vladoiu et al. 2019). We and others are currently testing the hypothesis that human-specific RL developmental programs and progenitors will provide new clues regarding cells of origin for these problematic tumors and lead to new approaches for much-needed targeted therapeutics.
7. NEW MODELS ARE NEEDED
Much remains to be discovered about human cerebellar development. What are the genetic programs driving the dramatic cerebellar hemisphere expansion across primate evolution? Are the SVZs found in the human cerebellar VZ and RL uniquely human innovations? What are the programs driving these innovations, and how do they compare to other brain regions? How do perturbations of human cerebellar neurogenesis relate to human neurodevelopmental disorders? These questions and many others require access to cerebellar tissue from not just humans but other nonhuman primates and nontraditional mammalian models across developmental time, which are scarce. New models are required. There are ongoing efforts to develop cerebellar 2D and 3D differentiation cultures from human pluripotent stem cells (Ballabio et al. 2020; Behesti et al. 2021; Buchholz et al. 2020; Hua et al. 2021; Muguruma et al. 2015; Nayler et al. 2021; Silva et al. 2020a,b). Yet, cerebellar differentiation methods are in their infancy compared to those of human cerebral cortex. Extensive additional normative human developmental data are required to validate cellular models, particularly from stages prior to 8–9 pcw, when VZ and early RL progenitors are specified and initiate differentiation. Since in vivo human cerebellar development is a 2-year process, it will also be essential to develop strategies to facilitate more rapid maturation in vitro. One solution may involve modeling the complexity of human progenitor zones in mice, once molecular mechanisms are defined.
8. CONCLUSION
Our understanding of neurodevelopmental disorders affecting the cerebellum is growing. Studies detailing the development of the human cerebellum are imperative to understand the role of the cerebellum in higher-order functions and disease and to benchmark in vitro models. Insights from human transcriptomics and histological analyses emphasize the importance of continuing human studies of the developing cerebellum. We cannot model what we do not know, and there remains so much more to discover.
ACKNOWLEDGMENTS
This work was supported by the following funding sources: National Institutes of Health (NIH) R37NS095733 to K.J.M., NIH R21MH126244 to K.A.A., and NIH R21NS117848 and Brain & Behavior Research Foundation Young Investigator Award to P.H. We would also like to acknowledge Jake Millman and Kathleen Tran Bach for their assistance with the manuscript.
Footnotes
The Annual Review of Neuroscience is online at neuro.annualreviews.org
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- Abraham H, Tornoczky T, Kosztolanyi G, Seress L. 2001. Cell formation in the cortical layers of the developing human cerebellum. Int. J. Dev. Neurosci 19:53–62 [DOI] [PubMed] [Google Scholar]
- Aguilar A, Meunier A, Strehl L, Martinovic J, Bonniere M, et al. 2012. Analysis of human samples reveals impaired SHH-dependent cerebellar development in Joubert syndrome/Meckel syndrome. PNAS 109:16951–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinger KA, Dempsey JC,Tully HM, Grout ME, Mehaffey MG, et al. 2018. Rhombencephalosynapsis: fused cerebellum, confused geneticists. Am. J. Med. Genet. C Semin. Med. Genet 178:432–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinger KA, Doherty D. 2016. The genetics of cerebellar malformations. Semin. Fetal Neonatal. Med 21:321–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinger KA, Thomson Z, Phelps IG, Haldipur P, Deng M, et al. 2021. Spatial and cell type transcriptional landscape of human cerebellar development. Nat. Neurosci 24:1163–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldinger KA, Timms AE, Thomson Z, Mirzaa GM, Bennett JT, et al. 2019. Redefining the etiologic landscape of cerebellar malformations. Am. J. Hum. Genet 105:606–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NJ, Lyons DA. 2018. Glia as architects of central nervous system formation and function. Science 362:181–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astle DE, Holmes J, Kievit R, Gathereole SE. 2021. Annual research review: The transdiagnostic revolution in neurodevelopmental disorders. J. Child Psychol. Psychiatry In press [DOI] [PubMed] [Google Scholar]
- Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, Chen ML, D’Arrigo S, et al. 2020. Healthcare recommendations for Joubert syndrome. Am. J. Med. Genet. A 182:229–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballabio C, Anderle M, Gianesello M, Lago C, Miele E, et al. 2020. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun 11:583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. 2005. Neuroanatomic observations of the brain in autism: a review and future directions. Int. J. Dev. Neurosci 23:183–87 [DOI] [PubMed] [Google Scholar]
- Beckinghausen J, Sillitoe RV. 2019. Insights into cerebellar development and connectivity. Neurosci. Lett 688:2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behesti H, Kocabas A, Buchholz DE, Carroll TS, Hatten ME. 2021. Altered temporal sequence of transcriptional regulators in the generation of human cerebellar granule cells. eLife 10:e67074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc ME, Du Plessis AJ, Sullivan N, Khwaja OS, Zhang X, et al. 2011. Spectrum of neurodevelopmental disabilities in children with cerebellar malformations. Dev. Med. Child Neurol 53:409–16 [DOI] [PubMed] [Google Scholar]
- Bolduc ME, Limperopoulos C. 2009. Neurodevelopmental outcomes in children with cerebellar malformations: a systematic review. Dev. Med. Child Neurol 51:256–67 [DOI] [PubMed] [Google Scholar]
- Boswinkel V, Steggerda SJ, Fumagalli M, Parodi A, Ramenghi LA, et al. 2019. The CHOPIn study: a multi-center study on cerebellar hemorrhage and outcome in preterm infants. Cerebellum 18:989–98 [DOI] [PubMed] [Google Scholar]
- Buchholz DE, Carroll TS, Kocabas A, Zhu X, Behesti H, et al. 2020. Novel genetic features of human and mouse Purkinje cell differentiation defined by comparative transcriptomics. PNAS 117:15085–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carta I, Chen CH, Schott AL, Dorizan S, Khodakhah K. 2019. Cerebellar modulation of the reward circuitry and social behavior. Science 363:aav0581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RA, Bihannic L, Rosencrance C, Hadley JL, Tong Y, et al. 2018. A single-cell transcriptional atlas of the developing murine cerebellum. Curr. Biol 28:2910–20.e2 [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Lindgren AG, Mishima Y, Roberts RW, Aldinger KA, et al. 2010. Lmx1a regulates fates and location of cells originating from the cerebellar rhombic lip and telencephalic cortical hem. PNAS 107:10725–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov VV, Millen KJ. 2013. Chapter 22: Neurogenesis in the cerebellum. In Comprehensive Developmental Neuroscience: Patterning and Cell Type Specification in the Developing CNS and PNS, ed. Rubenstein JL, Rakic P, pp. 417–34. London: Academic [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. 2004. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development 131:5581–90 [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. 1999. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 126:3089–100 [DOI] [PubMed] [Google Scholar]
- Dijkshoorn ABC, Turk E, Hortensius LM, van der Aa NE, Hoebeek FE, et al. 2020. Preterm infants with isolated cerebellar hemorrhage show bilateral cortical alterations at term equivalent age. Sci. Rep 10:5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Kowalczyk T, Daza RA, Dagan A, Lau C, et al. 2006. Unipolar brush cells of the cerebellum are produced in the rhombic lip and migrate through developing white matter. J. Neurosci 26:9184–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiez JA, Petersen SE. 1998. Neuroimaging studies of word reading. PNAS 95:914–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez J, Kumar R, Smith KS, Northcott PA. 2021. Deconstructing sonic hedgehog medulloblastoma: molecular subtypes, drivers, and beyond. Trends Genet. 37:235–50 [DOI] [PubMed] [Google Scholar]
- Guldal CG, Ahmad A, Korshunov A, Squatrito M, Awan A, et al. 2012. An essential role for p38 MARK in cerebellar granule neuron precursor proliferation. Acta Neuropathol. 123:573–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Aldinger KA, Bernardo S, Deng M, Timms AE, et al. 2019. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science 366:454–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Bernardo S, Aldinger KA, Sivakumar T, Millman J, et al. 2021. Evidence of disrupted rhombic lip development in the pathogenesis of Dandy-Walker malformation. Acta Neuropathol. 142:761–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Bharti U, Alberti C, Sarkar C, Gulati G, et al. 2011. Preterm delivery disrupts the developmental program of the cerebellum. PLOS ONE 6:e23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Bharti U, Govindan S, Sarkar C, Iyengar S, et al. 2012. Expression of Sonic hedgehog during cell proliferation in the human cerebellum. Stem Cells Dev. 21:1059–68 [DOI] [PubMed] [Google Scholar]
- Haldipur P, Dang D, Aldinger KA,Janson OK, Guimiot F, et al. 2017. Phenotypic outcomes in mouse and human Foxc1 dependent Dandy-Walker cerebellar malformation suggest shared mechanisms. eLife 6:e20898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Gillies GS, Janson OK, Chizhikov VV, Mithal DS, et al. 2014. Foxc1 dependent mesenchymal signalling drives embryonic cerebellar growth. eLife 3:e03962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldipur P, Millen KJ. 2019. What cerebellar malformations tell us about cerebellar development. Neurosci. Lett 688:14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME. 1999. Central nervous system neuronal migration. Annu. Rev. Neurosci 22:511–39 [DOI] [PubMed] [Google Scholar]
- Hatten ME. 2020. Adding cognitive connections to the cerebellum. Science 370:1411–12 [DOI] [PubMed] [Google Scholar]
- Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. 2020. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat. Rev. Cancer 20:42–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw ML, et al. 2019. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature 572:74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua TT, Bejoy J, Song L, Wang Z, Zeng Z, et al. 2021. Cerebellar differentiation from human stem cells through retinoid, Wnt, and Sonic hedgehog pathways. Tissue Eng. Part A 27:881–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Liu J, Ketova T, Fleming JT, Grover VK, et al. 2010. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. PNAS 107:8422–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LJ, Park R, Lee MJ, Terry BK, Lee DJ, et al. 2020. Yap/Taz are required for establishing the cerebellar radial glia scaffold and proper foliation. Dev. Biol 457:150–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SC, Lewis SA, Bakhtiari S, Zeng X, Sierant MC, et al. 2020. Mutations disrupting neuritogenesis genes confer risk for cerebral palsy. Nat. Genet 52:1046–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, et al. 2011. Spatio-temporal transcriptome of the human brain. Nature 478:483–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebschull JM, Richman EB, Ringach N, Friedmann D, Albarran E, et al. 2020. Cerebellar nuclei evolved by repeatedly duplicating a conserved cell-type set. Science 370:abd5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange W 1975. Cell number and cell density in the cerebellar cortex of man and some other mammals. Cell Tissue Res. 157:115–24 [DOI] [PubMed] [Google Scholar]
- Larsell O, Jansen J. 1972. The Comparative Anatomy and Histology of the Cerebellum: The Human Cerebellum, Cerebellar Connections and Cerebellar Conex. Minneapolis: Univ. Minn. Press [Google Scholar]
- Larsell O, Stotler WA. 1947. Some morphological features of the human cerebellum. Anat. Rec 97:352. [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, et al. 2007. Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–76 [DOI] [PubMed] [Google Scholar]
- Leto K, Arancillo M, Becker EB, Buffo A, Chiang C, et al. 2016. Consensus paper: cerebellar development. Cerebellum 15:789–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto K, Rolando C, Rossi F. 2012. The genesis of cerebellar GABAergic neurons: fate potential and specification mechanisms. Front. Neuroanat 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JYH, Lao Z, Joyner AL. 2002. Changing requirements for Gbx2 in development of the cerebellum and maintenance of the mid/hindbrain organizer. Neuron 36:31–43 [DOI] [PubMed] [Google Scholar]
- Limperopoulos C, Benson CB, Bassan H, Disalvo DN, Kinnamon DD, et al. 2005a. Cerebellar hemorrhage in the preterm infant: ultrasonographic findings and risk factors. Pediatrics 116:717–24 [DOI] [PubMed] [Google Scholar]
- Limperopoulos C, Chilingaryan G, Sullivan N, Guizard N, Robertson RL, du Plessis AJ. 2014. Injury to the premature cerebellum: Outcome is related to remote cortical development. Cereb. Cortex 24:728–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C, Soul JS, Gauvreau K, Huppi PS, Warfield SK, et al. 2005b. Late gestation cerebellar growth is rapid and impeded by premature birth. Pediatrics 115:688–95 [DOI] [PubMed] [Google Scholar]
- Limperopoulos C, Soul JS, Haidar H, Huppi PS, Bassan H, et al. 2005c. Impaired trophic interactions between the cerebellum and the cerebrum among preterm infants. Pediatrics 116:844–50 [DOI] [PubMed] [Google Scholar]
- Luo W, Lin GN, Song W, Zhang Y, Lai H, et al. 2021. Single-cell spatial transcriptomic analysis reveals common and divergent features of developing postnatal granule cerebellar cells and medulloblastoma. BMC Biol. 19:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machold R, Fishell G. 2005. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron 48:17–24 [DOI] [PubMed] [Google Scholar]
- Mak CCY, Doherty D, Lin AE, Vegas N, Cho MT, et al. 2020. MN1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis. Brain 143:55–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek S, Siegel JS, Gordon EM, Raut RV, Gratton C, et al. 2018. Spatial and temporal organization of the individual human cerebellum. Neuron 100:977–93.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May HJ, Fasheun JA, Bain JM, Baugh EH, Bier LE, et al. 2021. Genetic testing in individuals with cerebral palsy. Dev. Med. Child Neurol 63 (12): 1448–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, et al. 2014. Transcriptional landscape of the prenatal human brain. Nature 508:199–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-De-Luca A, Millan F, Pesacreta DR, Elloumi HZ, Oetjens MT, et al. 2021. Molecular diagnostic yield of exome sequencing in patients with cerebral palsy. JAMA 325:467–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. 2015. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 10:537–50 [DOI] [PubMed] [Google Scholar]
- Murray JC, Johnson JA, Bird TD. 1985. Dandy-Walker malformation: etiologic heterogeneity and empiric recurrence risks. Clin. Genet 28:272–83 [DOI] [PubMed] [Google Scholar]
- Nayler S, Agarwal D, Curion F, Bowden R, Becker EBE. 2021. High-resolution transcriptional landscape of xeno-free human induced pluripotent stem cell-derived cerebellar organoids. Sci. Rep 11:12959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen V, Sabeur K, Maltepe E, Ameri K, Bayraktar O, Rowitch DH. 2018. Sonic hedgehog agonist protects against complex neonatal cerebellar injury. Cerebellum 17:213–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, et al. 2019. Medulloblastoma. Nat. Rev. Dis. Primers 5:11. [DOI] [PubMed] [Google Scholar]
- Ocasio J, Babcock B, Malawsky D, Weir SJ, Loo L, et al. 2019. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat. Commun 10:5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong T, Trivedi N, Wakefield R, Frase S, Solecki DJ. 2020. Siah2 integrates mitogenic and extracellular matrix signals linking neuronal progenitor ciliogenesis with germinal zone occupancy. Nat. Commun 11:5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oristaglio J, Hyman West S, Ghaffari M, Lech MS, Verma BR, et al. 2013. Children with autism spectrum disorders show abnormal conditioned response timing on delay, but not trace, eyeblink conditioning. Neuroscience 248:708–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paprocka J, Jezela-Stanek A, Tylki-Szymanska A, Grunewald S. 2021. Congenital disorders of glycosylation from a neurological perspective. Brain Sci. 11:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Glass I. 1993. Joubert syndrome. In GeneReviews, ed. Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, et al. Seattle: Univ. Wash. [PubMed] [Google Scholar]
- Pichieechio A, Decio A, Di Perri C, Parazzini C, Rossi A, Signorini S. 2016. “Acquired” Dandy-Walker malformation and cerebellar hemorrhage: usefulness of serial MRI. Eur. J. Paediatr. Neurol 20:188–91 [DOI] [PubMed] [Google Scholar]
- Pisano TJ, Dhanerawala ZM, Kislin M, Bakshinskaya D, Engel EA, et al. 2021. Homologous organization of cerebellar pathways to sensory, motor, and associative forebrain. Cell Rep. 36:109721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Sidman RL. 1970. Histogenesis of cortical layers in human cerebellum, particularly the lamina dissecans. J. Comp. Neurol 139:473–500 [DOI] [PubMed] [Google Scholar]
- Reeder MR, Botto LD, Keppler-Noreuil KM, Carey JC, Byrne JL, et al. 2015. Risk factors for Dandy-Walker malformation: a population-based assessment. Am. J. Med. Genet. A 167A:2009–16 [DOI] [PubMed] [Google Scholar]
- Reefhuis J, Honein MA, Schieve LA, Rasmussen SA, Natl. Birth Defects Prev. Study. 2011. Use of clomiphene citrate and birth defects, National Birth Defects Prevention Study, 1997–2005. Hum. Reprod 26:451–57 [DOI] [PubMed] [Google Scholar]
- Sajan SA, Fernandez L, Nieh SE, Rider E, Bukshpun P, et al. 2013. Both rare and de novo copy number variants are prevalent in agenesis of the corpus callosum but not in cerebellar hypoplasia or polymicrogyria. PLOS Genet. 9:e1003823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarropoulos I, Sepp M, Fromel R, Leiss K, Trost N, et al. 2021. Developmental and evolutionary dynamics of cis-regulatory elements in mouse cerebellar cells. Science 373:abg4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmahmann JD. 2019. The cerebellum and cognition. Neurosci. Lett 688:62–75 [DOI] [PubMed] [Google Scholar]
- Sereno MI, Diedrichsen J, Tachrount M, Testa-Silva G, d’Arceuil H, De Zeeuw C. 2020. The human cerebellum has almost 80% of the surface area of the neocortex. PNAS 117:19538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto Y, Nakatani T, Masuyama N, Taya S, Kumai M, et al. 2014. Temporal identity transition from Purkinje cell progenitors to GABAergic interneuron progenitors in the cerebellum. Nat. Commun 5:3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiohama T, Ando R, Fujii K, Mukai H, Naruke Y, et al. 2017. An acquired form of Dandy-Walker malformation with enveloping hemosiderin deposits. Case Rep. Pediatr 2017:3861608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TP, Bekman EP, Fernandes TG, Vaz SH, Rodrigues CAV, et al. 2020a. Maturation of human pluripotent stem cell-derived cerebellar neurons in the absence of co-culture. Front. Bioeng. Biotechnol 8:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva TP, Fernandes TG, Nogueira DES, Rodrigues CAV, Bekman EP, et al. 2020b. Scalable generation of mature cerebellar organoids from human pluripotent stem cells and characterization by immunostaining. J. Vis. Exp 160:e61143. [DOI] [PubMed] [Google Scholar]
- Skefos J, Cummings C, Enzer K, Holiday J, Weed K, et al. 2014. Regional alterations in Purkinje cell density in patients with autism. PLOS ONE 9:e81255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smaers JB, Turner AH, Gomez-Robles A, Sherwood CC. 2018. A cerebellar substrate for cognition evolved multiple times independently in mammals. eLife 7:e35696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki DJ, Trivedi N, Govek EE, Kerekes RA, Gleason SS, Hatten ME. 2009. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron 63:63–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolliu E, Ioakeim-Ioannidou M, Kontokostas K, Dakoutrou M, Kousoulis AA. 2017. The most common comorbidities in Dandy-Walker syndrome patients: a systematic review of case reports. J. Child Neurol 32:886–902 [DOI] [PubMed] [Google Scholar]
- Sudarov A, Turnbull RK, Kim EJ, Lebel-Potter M, Guillemot F, Joyner AL. 2011. Ascl1 genetics reveals insights into cerebellum local circuit assembly. J. Neurosci 31:11055–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CL, Ng L, Menon V, Martinez S, Lee CK, et al. 2014. A high-resolution spatiotemporal atlas of gene expression of the developing mouse brain. Neuron 83:309–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Heijden ME, Lackey EP, Perez R, Isleyen FS, Brown AM, et al. 2021. Maturation of Purkinje cell firing properties relies on neurogenesis of excitatory neurons. eLife 10:e68045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk T, Baas F, Barth PG, Poll-The BT. 2018. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J. Rare Dis 13:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, et al. 2019. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature 572:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. 2009. Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J. Child Neurol 24:1085–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett RT, Bayin NS, Lee AS, Krishnamurthy A, Wojcinski A, et al. 2019. Cerebellar nuclei excitatory neurons regulate developmental scaling of presynaptic Purkinje cell number and organ growth. eLife 8 :e50617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson PM, Fryer RH, Fang Y, Hatten ME. 2010. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J. Neurosci 30:8529–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wizeman JW, Guo Q, Willon EM, Li JYH. 2019. Specification of diverse cell types during early neurogenesis of the mouse cerebellum. eLife 8:e42388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Joyner AL. 2019. YAP1 is involved in replenishment of granule cell precursors following injury to the neonatal cerebellum. Dev. Biol 455:458–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung J, Ha TJ, Swanson DJ, Choi K, Tong Y, Goldowitz D. 2014. Wls provides a new compartmental view of the rhombic lip in mouse cerebellar development. J. Neurosci 34:12527–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo JY, Mak GK, Goldowitz D. 2014. The effect of hemorrhage on the development of the postnatal mouse cerebellum. Exp. Neurol 252:85–94 [DOI] [PubMed] [Google Scholar]
- Zecevic N, Chen Y, Filipovic R. 2005. Contributions of cortical subventricular zone to the development of the human cerebral cortex. J. Comp. Neurol 491:109–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecevic N, Rakic P. 1976. Differentiation of Purkinje cells and their relationship to other components of developing cerebellar cortex in man. J. Comp. Neurol 167:27–47 [DOI] [PubMed] [Google Scholar]