

SUMMARY
Background:
Pro-inflammatory fibroblasts are critical for pathogenesis in rheumatoid arthritis, inflammatory bowel disease, interstitial lung disease, and Sjögren’s syndrome and represent a novel therapeutic target for chronic inflammatory disease. However, the heterogeneity of fibroblast phenotypes, exacerbated by the lack of a common cross-tissue taxonomy, has limited our understanding of which pathways are shared by multiple diseases.
Methods:
We profiled fibroblasts derived from inflamed and non-inflamed synovium, intestine, lungs, and salivary glands from affected individuals with single-cell RNA sequencing. We integrated all fibroblasts into a multi-tissue atlas to characterize shared and tissue-specific phenotypes.
Findings:
Two shared clusters, CXCL10+CCL19+ immune-interacting and SPARC+COL3A1+ vascular-interacting fibroblasts, were expanded in all inflamed tissues and mapped to dermal analogs in a public atopic dermatitis atlas. We confirmed these human pro-inflammatory fibroblasts in animal models of lung, joint, and intestinal inflammation.
Conclusions:
This work represents a thorough investigation into fibroblasts across organ systems, individual donors, and disease states that reveals shared pathogenic activation states across four chronic inflammatory diseases.
Funding:
Grant from F. Hoffmann-La Roche (Roche) AG.
Graphical Abstract
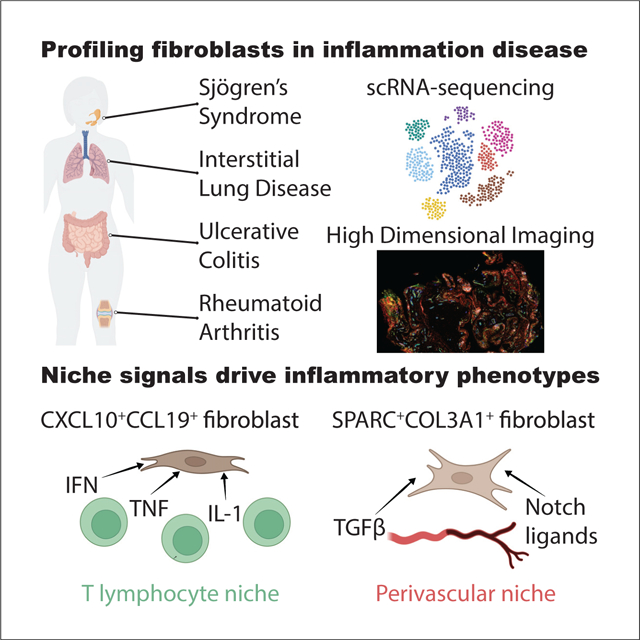
Fibroblasts support tissue re-organization and immunoregulation in inflammatory diseases. Korsunsky et al. construct a single-cell atlas of human fibroblasts from four diseases (Sjögren’s syndrome, interstitial lung disease, ulcerative colitis, and rheumatoid arthritis), define two functionally distinct inflammatory fibroblast phenotypes shared across diseases, and confirm their presence in independent datasets.
INTRODUCTION
Fibroblasts are present in all tissues and adopt specialized phenotypes and activation states to perform essential functions in development, wound healing, and maintenance of tissue architecture as well as pathological functions such as tissue inflammation, fibrosis, and cancer responses.1 Recent studies of chronic inflammatory disease have leveraged advances in high-throughput single-cell genomics, particularly single-cell RNA sequencing (scRNA-seq) to identify molecularly distinct fibroblast populations associated with pathological inflammation in different anatomical sites.2–9 A study of the large intestine from individuals with ulcerative colitis (UC) identified stromal cells expressing the Oncostatin-M receptor (OSMR) and Podoplanin (PDPN) enriched in biopsies tracking with failure to respond to anti-tumor necrosis factor (TNF) therapy.10 Other studies suggested immunomodulatory roles of OSMR+PDPN+ intestinal fibroblasts through interactions with inflammatory monocytes3 and neutrophils.11 Lung investigations identified that COL3A1+ACTA2+ myofibroblasts, PLIN2+ lipofibroblast-like cells, and FBN1+HAS1+ fibroblasts are expanded in lung biopsies from individuals with idiopathic pulmonary fibrosis (IPF).4,5 In the salivary gland, chronic destructive inflammation in primary Sjögren’s syndrome (pSS) with tertiary lymphoid structures is linked to expansion of PDPN+CD34− fibroblasts.12 In synovial tissue, FAPα+CD90+ fibroblasts are expanded in individuals with rheumatoid arthritis (RA)2,13 and drive leukocyte recruitment and activation in an animal model of arthritis.14
In each study, inflammation-associated fibroblasts are characterized by their ability to produce and respond to inflammatory cytokines. These cytokines are often members of conserved families that signal through similar downstream pathways and result in similar effector functions.15 For instance, the inflammatory cytokines interleukin-6 (IL-6), Oncostatin M (OSM), leukemia inhibitory factor (LIF), and IL-11 belong to the gp130 family, whose cognate receptor molecules, including IL-6R, OSMR, LIFR, and IL-11R, contain the glycoprotein 130 (gp130) subunit. In UC, OSMR+ fibroblasts express high levels of the IL-11-encoding gene.3 In RA, a subset of FAPα+CD90+ synovial fibroblasts produce high levels of IL-62 through an autocrine loop involving LIF and LIFR.16,17 In a mouse model for human IPF, IL-11-producing fibroblasts drive fibrosis and chronic pulmonary inflammation.18 These examples of gp130-family cytokines associated with pro-inflammatory fibroblasts highlight that, although individual factors may be tissue specific, their downstream effects may be shared across diseases. This pattern underlines an important question with clinical implications: are inflammation-associated fibroblasts tissue specific, or do they represent shared activation states that manifest a common phenotype across different diseases? A drug that targets a shared pathogenic phenotype can potentially be used to treat multiple inflammatory diseases. Identifying such shared fibroblast programs is a major challenge because these programs are likely to be transient and reversible activation states that vary over the course of a disease rather than representing a static, committed cell lineage.13
There is growing evidence in recent studies of the existence of shared fibroblast transcriptional states across tissues. In particular, single-cell atlas projects that profile tissue samples from multiple organs from the same postmortem individuals provide a unique opportunity to compare fibroblast profiles across tissues while accounting for shared donor effects. When we analyzed fibroblast profiles from two such atlas datasets, the Adult Human Cell Atlas (AHCA)19 and Tabula Sapiens (TS),20 we found that fibroblasts from different tissues group together (Figures S1A and S1B), suggesting that lineage contributes more to transcriptional identity than tissue of origin. When we performed cluster marker analysis on these fibroblast from different tissues (Table S1), we found that of the 2,602 (AHCA) and 2,321 (TS) genes upregulated in fibroblasts in at least one tissue, 1,545 (AHCA) and 1,385 (TS) genes were shared by at least two tissues, and 256 (AHCA) and 357 (TS) genes were universal markers of fibroblasts in all tissues (Figures S1C and S1D). A second line of evidence for universal fibroblasts is presented by Buechler et al.,21 who analyzed mouse fibroblast from 17 distinct tissues and identified shared fibroblast clusters in healthy and perturbed tissues. They experimentally validated the existence of Dpt+ pluripotent universal fibroblasts, present in healthy and perturbed states, that may be able to give rise to functionally distinct fibroblasts in tissue.
Identification of shared cell states across tissues with scRNA-seq has recently become possible with advances in statistical methods for integrative clustering22–24 and reference mapping.25–27 Integrative clustering identifies similar cell states across a range of scRNA-seq datasets even when the datasets come from different donors, species, or tissues. For example, using integrative clustering, Zhang et al.28 identified shared macrophage activation states across five tissues, and Butler et al.24 identified shared pancreatic islet cells between mouse and human datasets. Reference mapping allows rapid comparison of data from a new study to a well-annotated reference even when the study represents a tissue, disease, or species not present in the reference atlas. For instance, Andreatta et al.26 mapped T cell subtypes to a scRNA-seq atlas of annotated tumor-infiltrating T cells, and Lotfollahi et al.25 found disease-related immune states by mapping PBMCs from individuals with coronavirus disease 2019 (COVID-19) to a healthy reference library of immune cells.
In this study, we generated scRNA-seq profiles of CD45−EPCAM− stromal cells derived from affected individuals and then characterized fibroblasts across multiple inflammatory diseases involving lung, intestine, salivary gland, and synovium. After confirming known fibroblast subtypes in our data, we built a de novo, integrated fibroblast atlas and identified five shared phenotypes, two of which are consistently expanded in all four inflammatory diseases. Using reference mapping, we map these to human dermal fibroblasts from inflamed and healthy skin and to fibroblasts from mouse models of lung, synovial, and intestinal inflammation to demonstrate the generalizability of our findings. Our integrated resource represents an important systematic examination of fibroblast subsets and activation states in inflamed tissues. Our identification of two pathogenic fibroblast phenotypes that are shared among four inflammatory diseases suggests potential novel avenues for fibroblast therapeutic targeting. By making available the necessary computational tools to map new datasets to our annotated fibroblast atlas, we provide a common reference for future studies of fibroblasts in tissues and diseases.
RESULTS
Single-cell transcriptional profiles of fibroblasts in human lung, salivary gland, synovium, and intestine
We used droplet-based scRNA-seq to profile individual fibroblasts from a total of 74 high-quality samples in lung, large intestine, lip salivary glands, and joint synovium, selecting donors with inflammatory diseases and controls (Figure 1A). For synovium, we collected arthroplasties and biopsies from 15 individuals with RA and 6 with osteoarthritis (OA) (Table S2). For the lung analysis, we acquired lung biopsies samples from 8 individuals with earlier-stage interstitial lung disease (ILD) who underwent video-assisted thoracoscopic surgical (VATS) biopsy for ILD diagnosis, characterized by inflammatory pathology, and comparator lung transplant samples of explanted lung from 4 control donors and 11 individuals with end-stage IPF and RA-ILD (STAR Methods; Table S3). All individuals were assessed for clinical requirement for supplemental O2 at the time of enrollment (Figure S1E) and assigned to the earlier-stage subcohort when they did not require supplemental O2 treatment. None of the individuals in the earlier-stage subcohort were under consideration for pre-transplantation work-up. To examine salivary glands, we used lip biopsy tissue from 7 individuals with pSS and 6 individuals with non-Sjögren’s Sicca syndrome, characterized as non-autoimmune dryness, as control comparators (Table S4). For the intestine, we collected large intestinal biopsies from individuals with UC (n =7) and control donors (n = 5) (Table S5). Included in the 7 UC samples were 4 individuals for whom we had paired inflamed and adjacent non-inflamed tissue biopsies. To enrich for stromal cells in the synovium and intestinal samples, we used flow cytometry to sort live, CD45−EpCAM− cells (Figure 1A), depleting CD45+ immune and EpCAM+ epithelial populations (Figure S1F). We did not flow sort cells in samples from the salivary gland or lung. For the salivary gland, we avoided flow sorting to optimize cell numbers in small biopsies, and in the lung, we found that flow cytometry compromised fibroblast cell yields. We performed droplet-based scRNA-seq (10X Genomics) on all samples, applied stringent QC to remove low-quality libraries and cells (STAR Methods), and combined all data samples to analyze 221,296 high-quality cells. Using clustering analysis (STAR Methods), we identified 7 major cell types (Figure 1B) across multiple donors (Figure 1C) using canonical markers (Figure 1D): CDH5+ endothelial cells, COL1A1+ fibroblasts, EPCAM+ epithelial cells, GFRA3+ glial cells, JCHAIN+ plasma cells, MCAM+ perivascular murals, and PTPRC+ leukocytes. Consistent with our flow sorting strategy, non-stromal cells (epithelial, glial, and immune) were more abundant in the salivary gland and lung (Figure S1G). We identified stromal (endothelial, mural, and fibroblast) populations in all four tissues, allowing us to carry out a focused analysis of fibroblasts across tissues.
Figure 1. scRNA-seq profiles of and fibroblast heterogeneity within intestine, lung, salivary gland, and synovium.
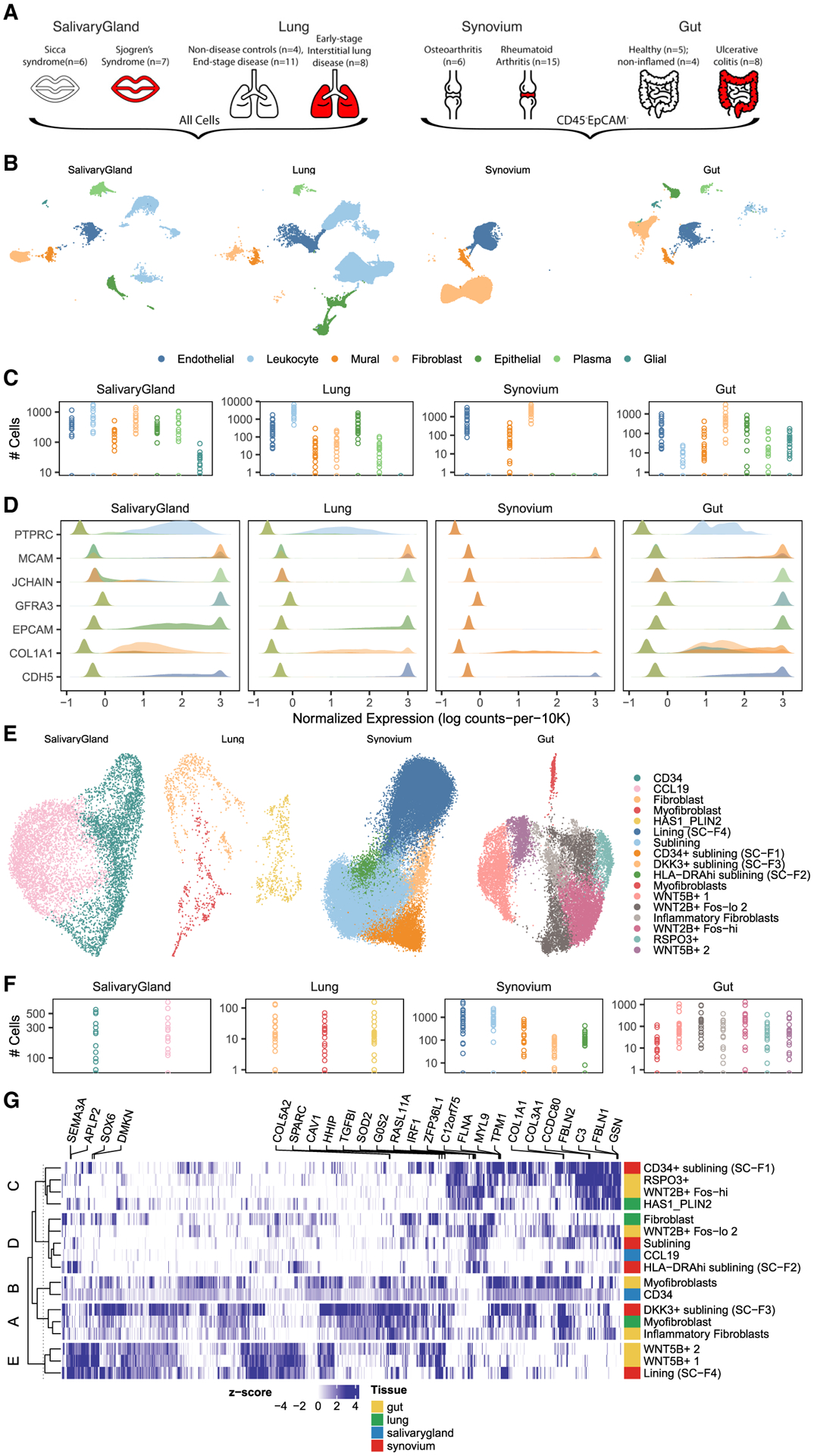
(A and B) Surgical samples were collected from intestine, lung, salivary gland, and synovium from individuals with inflammatory disease and appropriate controls (A). After tissue disaggregation, all cells from lung and salivary gland and CD45−EpCAM− cells from synovium and intestine were profiled with scRNA-seq and (B) analyzed to identify fibroblasts and other major cell types.
(C) Total cell numbers per donor per major cell type in log scale.
(D) Cell type annotation was performed with known markers for each major population.
(E) Fine-grained clustering within fibroblasts was performed for each tissue and plotted with tissue-specific UMAP projections.
(F) Total cell numbers per donor per fibroblast cluster in log scale.
(G) All (n = 894) genes upregulated in a group and shared among tissue clusters in that group were plotted in a heatmap. Color denotes the log fold change, normalized by estimated standard deviation, of a gene in a cluster (versus other clusters in that tissue). The top five genes for each cluster are named above the heatmap. Each row denotes a fibroblast cluster, colored according to the tissue in which it was identified. Rows are clustered into five groups to highlight the similarity of tissue-defined clusters across tissues.
Fibroblast heterogeneity within tissues
We next examined the heterogeneity of fibroblast cell states within individual tissues. We performed a separate fine-grained clustering analysis for fibroblasts within each of the four tissues and annotated clusters with previously identified states (Figure 1E) across individual donors (Figure 1F) by comparing published marker genes with cluster markers in our data (Table S6). In the intestine, we were able to recapitulate 7 of 8 populations identified by Smillie et al.:3 crypt-associated WNT2B+Foshi and WNT2B+Foslo, epithelial-supportive WNT5B+−1 and WNT5B+−2, stem cell niche-supporting RSPO3+, inflammatory, and myofibroblasts. Our data did not support the 2 subtypes of WNT2B+Foslo fibroblasts identified originally by Smillie et al.3 In the lung, Habermann et al.4 described 4 states: HAS1+, PLIN2+, fibroblasts, and myofibroblasts. However, in their analysis, HAS1+ cells were identified in only 1 of 30 donors. When we re-analyzed their data to identify clusters shared by multiple donors, we could not distinguish the HAS1+ from the PLIN2+ population and, thus, merged these two in our annotation. In the salivary gland, the only single-cell study of fibroblasts to date was performed with multi-channel flow cytometry,12 not scRNA-seq. The findings here represent the first set of scRNA-seq data in this context. In our single-cell clusters, we identified the two populations described previously (CD34+ and CCL19+) and confirmed the expression of key distinguishing cytokines and morphogens they measured by qPCR. In the synovium, we clustered 55,143 fibroblasts into 5 major states described in three scRNA-seq studies.2,6,14 These states are largely correlated with anatomical position: THY1−PRG4+ cells in the synovial boundary lining layer and THY1+, DKK3+, HLA-DRA+, and CD34+ cells within the sublining. In total, we labeled 17 fibroblast clusters defined across all four individual tissues.
Next we wanted to determine whether fibroblast states defined within one tissue shared similar expression profiles with states defined in other tissues. To look for these similarities, we selected genes that were significantly (p < 0.01, logFC ≥ 0.5) associated with at least one cluster and computed the correlation of relative gene expression for every pair of clusters (Figure S1H). The clusters naturally grouped across tissues. Using hierarchical partitioning of this correlation matrix, we grouped the 17 tissue-defined clusters into 5 meta-clusters. We then found that 894 marker genes were upregulated in a meta-cluster and shared by all tissue clusters in that meta-cluster (Figure 1G). This heatmap demonstrates shared gene expression profiles across clusters from different tissues and suggests shared functions for these tissue-defined clusters. For instance, COL3A1 shared by group A, with inflammatory fibroblasts in the gut, myofibroblasts in the lung, and DKK3+ sublining fibroblasts in the synovium, may reflect a common extracellular matrix (ECM) modulatory function. Marker genes that are not shared across clusters in the same meta-cluster can arise in two different ways: from a technical artifact, such as different clustering parameters in tissue-specific analyses, or from a true biological signal, such as a tissue-specific microenvironment. To distinguish between the two possibilities, we decided to perform a single integrative clustering analysis with fibroblasts from all tissues. Just as integrative clustering within tissue allowed us to identify clusters shared by multiple donors (Figure S1I), we anticipated that integrative clustering across tissues would highlight shared transcriptional signatures missed in the within-tissue analyses.
Integrative clustering of fibroblasts across tissues
To construct a cross-tissue taxonomy of fibroblast states, we pooled 55,143 synovial, 15,089 intestinal, 7,474 salivary gland, and 1,442 pulmonary fibroblasts and performed integrative clustering analysis. The different numbers of fibroblasts from each tissue, arising because we enriched for stromal cells in intestine and synovium but not in lung and salivary gland, presented a technical challenge. The results of many analyses, including principal-component analysis (PCA), are biased toward tissues with more cells rather than treating each tissue equally. The second major analytical challenge arises because gene expression depends on a complex interplay of tissue, donor, and cell state. As we have described in previous work,22 such confounding variation is particularly challenging to model in scRNA-seq data because the confounder can have global and cell-type-specific effects on gene expression.
We designed an analytical pipeline for integrative clustering to address the two concerns described above (Figure 2A). In this pipeline, we select genes that were informative in the tissue-specific analyses (STAR Methods), associated with cluster identity (Table S6; n = 7,123) or inflammatory status (Table S7; n = 6,476) within tissue, for a total of 9,521 unique genes. To minimize the effect of different cell numbers, we performed weighted PCA, giving less weight to cells from over-represented tissues (e.g., synovium) and more to cells from under-represented tissues (e.g., lung) so that the sum of weights from each tissue is equivalent (STAR Methods). Compared with unweighted PCA, this approach results in principal components whose variation is more evenly distributed among tissues (Figure S2A). As expected, in this PCA space, cells group largely by donor and tissue (Figures S2B and S2C). To appropriately align cell types, we removed the effect of donor and tissue from the cells’ PCA embedding coordinates with a novel, weighted implementation of the Harmony algorithm we developed for this specific application (STAR Methods). Uniform manifold approximation and projection (UMAP) visualization of the harmonized embeddings shows that cells from different tissues are well mixed (Figure 2B). In contrast, fibroblast states identified in tissue-specific analyses are well separated (Figure S2D), suggesting that the integrated embedding faithfully preserves cellular composition. In this integrated space, we performed standard graph-based clustering to partition the cells into 14 fibroblast states (Figure 2C) with representation across multiple donors from all 4 tissues (Figure 2D). These 14 integrated clusters represent putative shared fibroblast states, each of which may be driven by a combination of shared and tissue-specific gene programs.
Figure 2. Integrative clustering and differential expression across tissues.
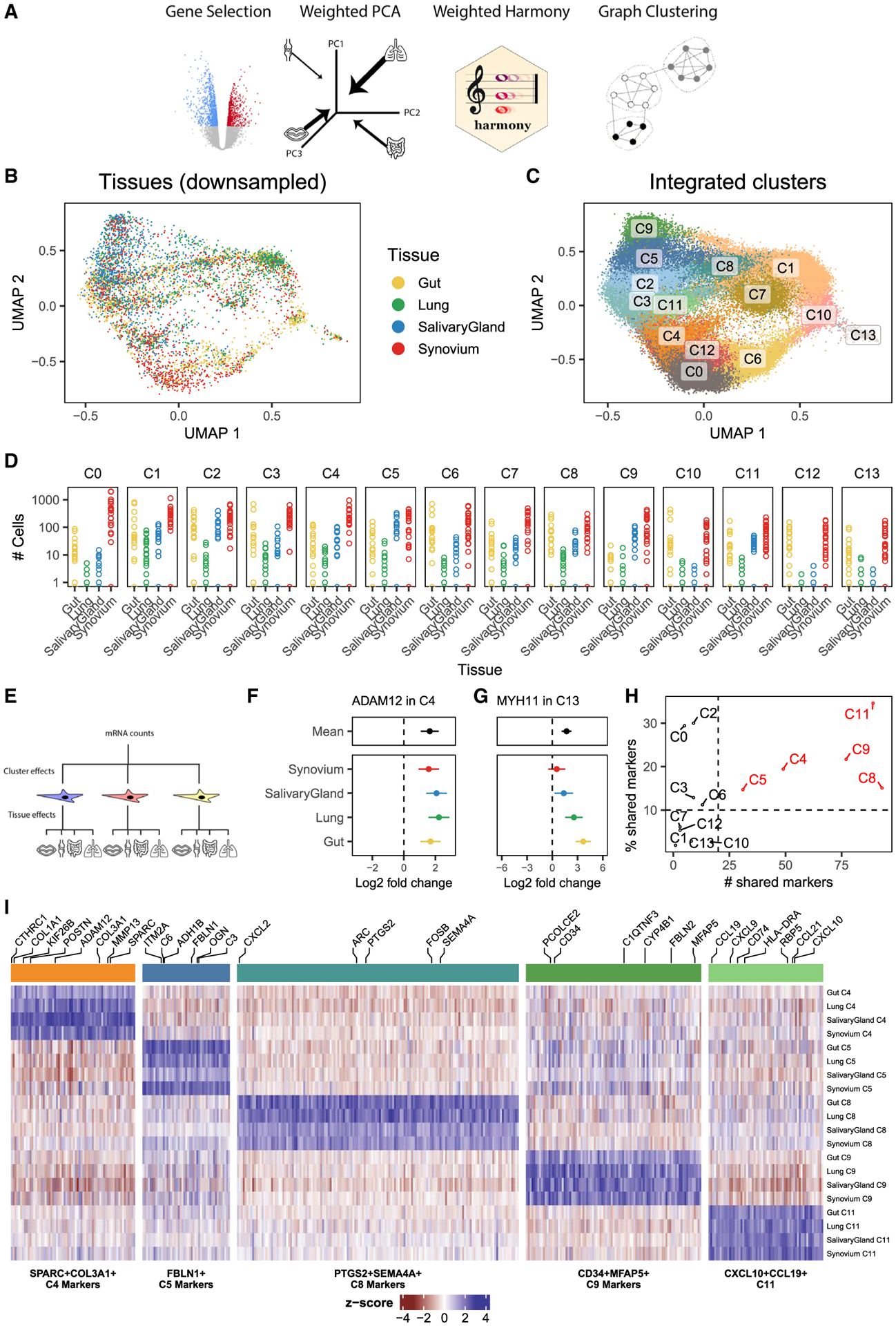
(A) We developed a pipeline to integrate samples from multiple donors and multiple tissues with unbalanced cell numbers. The pipeline starts with gene selection, pooling genes that were informative in single-tissue analyses. With these genes, we performed weighted PCA, reweighting cells to computationally account for the unbalanced dataset sizes among the tissues. These principal components are adjusted with a novel formulation of the Harmony integration algorithm and used to perform graph-based clustering. We applied this pipeline to all fibroblasts across tissues.
(B) The integrated UMAP projection shows cells from all tissues mixed in one space. For clarity, we down-sampled each tissue to the smallest tissue, the lung, choosing 1,442 random fibroblasts from intestine, synovium, and salivary gland.
(C) Graph-based clustering proposed 14 fibroblast clusters in the integrated embedding.
(D) Total cell numbers per donor per integrated cluster in log scale.
(E–G) Gene-level analysis to find upregulated marker genes for clusters was done with hierarchical regression to model complex interactions between clusters and tissues (E). This strategy distinguishes cluster marker genes that are (F) shared among tissues, such as ADAM12 in C4, from those that are (G) tissue-specific, such as MYH11 in C13. Points denote log fold change (cluster versus other fibroblast), and error bars mark the 95% CI for the fold change estimate.
(H) The number of shared genes (x axis) as well as the percentage of shared over total marker genes (y axis) for each cluster, ranked from most to least, prioritizes clusters with large evidence of shared gene expression (red) from those with little evidence (black).
(I) Marker genes for the 5 shared clusters plotted in a heatmap. Each block represents the (differential) gene expression of a gene (column) in a cluster for a tissue (row).
Identification of shared and tissue-specific marker genes in integrated clusters
Next we modeled gene expression to define active gene programs in the 14 integrative fibroblast clusters. We wanted to distinguish between two types of cluster markers: tissue shared and tissue specific. Tissue-shared markers are highly expressed in the cluster for all four tissues. Tissue-specific markers are highly expressed in the cluster for at least one tissue but not highly expressed in at least one other tissue. In our expression modeling analysis, we needed to allow for the possibility that tissue gene expression will be consistent in clusters and variable in others (Figure 2E). As we explain in our approach below, we will use ADAM12 expression in cluster C4 as an example of a tissue-shared gene and MYH11 expression in cluster C13 as an example of a tissue-specific gene.
Typically, cluster marker analysis is done with regression to associate gene expression with cluster identity. To address the complex interaction between cluster and tissue identity in our data, we used mixed-effects regression to perform hierarchical cluster marker analysis (STAR Methods). This analysis estimated two sets of differential expression statistics for each gene: mean log2 fold change (e.g., cluster 0 versus all other clusters) and tissue-specific log2 fold change (e.g., cluster 0 in lung versus all other clusters in lung). This approach distinguishes shared marker genes, defined by minimal tissue-specific contributions, from tissue-specific marker genes, defined by large tissue-specific fold changes, relative to the mean fold change. To demonstrate this, we plotted the estimated log2 fold changes, with a 95% confidence interval, for one shared (Figure 2F) and one tissue-specific (Figure 2G) cluster marker. ADAM12, a shared marker for cluster C4, has significant (log2 fold change = 1.6, p = 6.5 × 10−9) mean differential expression in C4, whereas the tissue-specific effects (in color) are not significantly different for any one tissue (Figure 2F). In contrast, MYH11 is differentially overexpressed in cluster C13 for intestinal (log2 fold change = 3.7, p = 8.5 × 10−16) and lung fibroblasts (log2 fold change = 2.6, p = 5.9 × 10−7) but not for synovial or salivary gland cells (Figure 2G). Because MYH11 is so strongly overexpressed in intestinal and lung fibroblasts, the mean log2 fold change is also significant (log2 fold change = 1.7, p = 5.7 × 10−9) and, therefore, is not a good metric alone to determine whether a marker is shared or tissue specific.
We defined tissue-shared cluster markers conservatively by requiring a marker gene to be significantly overexpressed in all four tissues, such as ADAM12 above. With this criterion, we quantified the number of shared marker genes per cluster (Figure 2H). Clusters C0, C1, C2, C3, C6, C7, C10, C12, and C13 each had fewer than 20 shared markers. Based on this cutoff, we decided that these clusters had too little evidence of shared marker genes to be reliably called shared clusters. We assigned names for the remaining clusters based on their shared gene markers: SPARC+COL3A1+ C4, FBLN1+ C5, PTGS2+SEMA4A+ C8, CD34+MFAP5+ C9, and CXCL10+CCL19+ C11. We then plotted the log2 fold change values of all 1,524 shared markers for these clusters in Figure 2I and report the results of the full differential expression analysis in Table S8.
Testing for overintegration
Harmony integration of tissues and donors is necessary to find reproducible fibroblast clusters. Without Harmony, most clusters would be specific not only to each tissue but to a single donor (Figures S2B and S2C). We were concerned about the possibility of overintegration. We therefore performed rigorous analyses to address the potential for overintegration in our study.
Some algorithms are more prone to overintegration than others. We performed integration with three alternative algorithms, BBKNN,29 scVI,30 and Scanorama,31 recommended by a benchmarking study32 that ranked algorithms by their ability to removal technical noise and preserve biological variability. Unlike Harmony, these algorithms can only integrate over one variable at a time. Thus, we first tested the ability of each algorithm to integrate donors within each tissue separately. Scanorama introduced many outlier clusters that did not exist in the original data, suggestive of overfitting (Figure S3A). BBKNN failed to run altogether in the lung because of insufficient cell numbers and barely integrated donors in the remaining tissues (Figure S3B). Only scVI was able to adequately integrate donors within tissues (Figure S3C). Based on these results, we moved forward with scVI to integrate our full dataset, first integrating over donor (scVI-donor) and then over tissue (scVI-tissue). scVI-donor merged donors within tissue but kept each tissue separate (Figure S3D). Conversely, scVI-tissue merged cells across tissues but failed to merge donors within tissue (Figure S3E). Although scVI is sufficient to analyze datasets with only one major confounder (e.g., donor), it is insufficient to integrate cells in our multi-donor, multi-tissue dataset.
Another concern of integration lies in the ability of Harmony to integrate explicitly over two variables. If Harmony can model the effects of donor and tissue, then will Harmony always find shared clusters, even when none exist? To explore this, we first re-analyzed our datasets with Harmony integration over donor only. Integrating over donor yielded results similar to two-level integration in terms of the degree of mixing among tissues (Figure S3F) and separation among clusters (Figure S3G). The only difference is that we needed a more aggressive cluster diversity penalty in the donor-only integration (θdonor = 2), whereas we used very mild penalties in the donor and tissue integration (θdonor = 0.25, θtissue = 0.25). This reflects the fact that, if we correctly specify the sources of variation in our dataset, then we do not require strong statistical priors to enforce mixing. Next we wanted to determine whether Harmony would always find shared clusters across donors and tissues. We first devised an extreme where we know the ground truth by attempting to integrate 10,000 randomly selected fibroblasts from synovium with 10,000 epithelial cells from lung. Using the same pipeline we used in the cross-tissue analysis in Figure 3, Harmony correctly failed to integrate synovium with lung cells here, keeping the biologically distinct fibroblasts and epithelial cells in two separate clusters (Figure S3H). Next we took a less stark example and integrated lung, salivary gland, and gut epithelial cells, which we expect to have more tissue-specific types than stromal or immune populations. Although we found some overlapping cells between lung and salivary gland fibroblasts, most cells failed to mix among tissues (Figure S3I). Thus, the cross-tissue integration we achieved with fibroblasts is not a priori guaranteed by Harmony and reflects a greater degree of shared transcriptional profiles than what we found in epithelial cells.
Figure 3. Identification of inflammation-associated clusters.
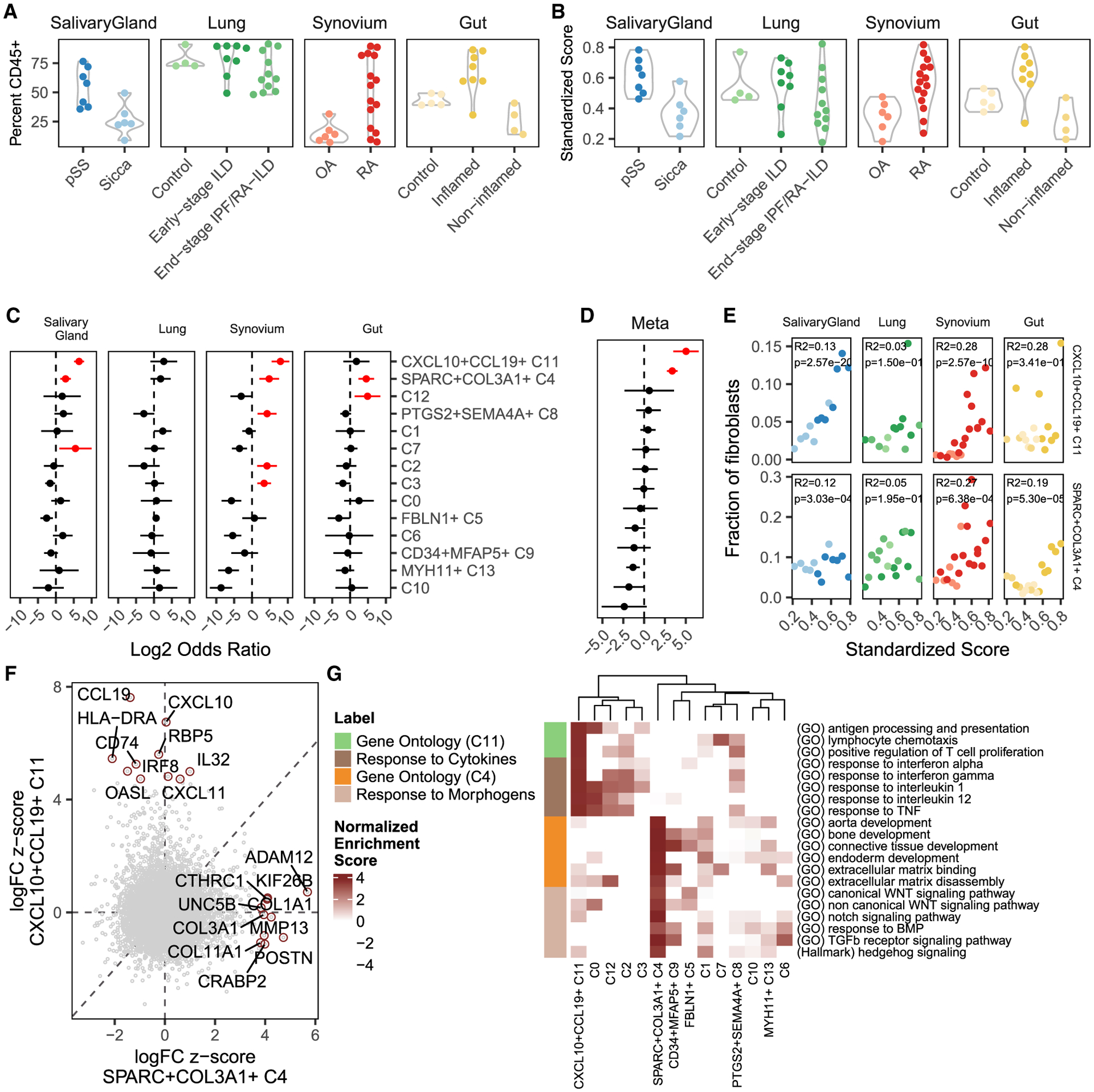
(A) We computed the relative abundance of CD45+ immune cells among all cells in each sample.
(B) We standardized these frequencies across tissues into an inflammation score that ranges from 0–1 and removes distributional differences.
(C) Association analysis results between fibroblast cluster abundance and standardized inflammation scores. Each point represents the log fold change in fibroblast cluster abundance with increasing inflammation, and the line represents that point’s 95% CI. Red denotes estimates with one-tailed FDR < 5%.
(D) The tissue-specific results were summarized using meta-analysis.
(E) For CXCL10+CCL19+ (C11) and SPARC+COL3A1+ (C4) fibroblasts, scatterplots relating to standardized inflammation scores (x axis) to relative fibroblast frequency (y axis). The colors in each panel refer to the clinical status of each donor, as denoted in (A) and (B). Reported p values were computed from logistic mixed-effects regression test and R2 statistics using McKelvey’s method.
(F) Comparison of differential gene expression between CXCL10+CCL19+ and SPARC+COL3A1+ fibroblasts shows that these two inflammation-expanded clusters are characterized by distinct genes. The top 10 markers for each cluster are named.
(G) Gene set enrichment analysis (GSEA) with Gene Ontology and MSigDB hallmark pathways shows distinct functions for the C4 (orange) and C11 (lime) states. These states may be explained by response to distinct sets of signaling molecules: inflammatory cytokines for C4 (brown) and tissue modeling morphogens for C11 (tan). The heatmap shows normalized enrichment scores from GSEA, focusing only on positive enrichment for clarity.
Finally, we evaluated the ability of Harmony to identify dataset-specific clusters in our study. This is critical for interpretation of our fibroblast atlas, particularly when we want to identify disease-specific and tissue-specific clusters. Because it is difficult to know when a cluster is truly dataset specific in real data, we performed this analysis by artificially removing pre-labeled clusters from our fibroblasts datasets, establishing a ground truth for evaluation. We chose six donors with UC from the intestinal datasets, artificially split the donors into two groups, and removed all WNT5B+ fibroblasts from group A (Figure S3J). We then integrated the down-sampled dataset with Harmony, which successfully mixed cells from the 6 donors (Figure S3K) while correctly separating the WNT5+ fibroblasts from group B from group A fibroblasts (Figure S3I). To further quantify these results, we performed de novo clustering of the down-sampled, integrated dataset to test whether we can find a cluster specific to group B (Figure S3M). Our unsupervised clustering results identified one cluster (denovo1) that is substantially over-represented in group B (Figure S3N). In group A, only 39 of 1,928 cells were assigned to cluster denovo1. Differential abundance testing confirmed that denovo1 as the only cluster significantly (adjusted p = 0.003) differentially abundant between groups A and B. This analysis demonstrates the behavior of Harmony with condition-specific fibroblasts and shows that we can identify condition-specific fibroblasts with differential abundance testing.
Correspondence between fibroblast clusters defined in integrative analysis and single-tissue analyses
We determined how the clusters labeled in the single-tissue analyses (Figure 1E) correspond to our new shared cross-tissue taxonomy. Because we used the same cells for within-tissue and cross-tissue analyses, we were able to directly observe the overlap of cross-tissue clusters with tissue-defined clusters in the tissue-defined UMAP projections (Figure S4A) and conditional co-occurrence bar plots (Figure S4B). For a more formal approach, we used a statistical test to quantify the enrichment of cross-tissue membership within each of the tissue-defined clusters (Figure S4C).
The CXCL10+CCL19+ C11 cluster overlapped significantly (FDR < 5%) with THY1+ sublining (OR = 3.8, 95% CI[2.2, 6.7]) and HLA-DRAhi synovial fibroblasts (OR = 39.2, 95% CI[22.2, 69.0]), with CCL19+ fibroblasts in the salivary gland (OR = 9.1, 95% CI[6.3, 13.0]), with RSPO3+ (OR = 16.1, 95% CI[12.0, 21.7]) and WNT2B+Foshi (OR = 2.3 95% CI[1.7, 3.1]) fibroblasts in the intestine and did not overlap significantly with any one cluster in the lung. Here, odds ratio (OR) refers to the probability of a cell being in a cross-tissue cluster (versus not), given that the cell belongs to some within-tissue clusters. The SPARC+COL3A1+ C4 cluster was split between DKK3+ and THY1+ sublining fibroblasts in the synovium, corresponded exclusively to myofibroblasts in the lung, split between inflammatory fibroblasts and myofibroblasts in the intestine, and corresponded to CD34+ fibroblasts in the salivary gland. None of these associations was one to one. HLA-DRA+ synovial fibroblasts, CCL19+ salivary gland fibroblasts, and RSPO3+ and WNT2B+Foshi intestinal fibroblasts corresponded to multiple clusters that were expanded in one or more tissues: C3 (lung and synovium), C2 (synovium), C12 (intestine), and C8 (salivary gland and synovium). Similarly, the myofibroblasts in the lung and intestine as well as DKK3+ synovial fibroblasts corresponded to C13 and vascular fibroblasts (C4).
Cluster C13 aligned strikingly with intestinal and pulmonary myofibroblasts. Although C13 contained cells from all tissues, it only expressed the canonical myofibroblast genes MYH11, MYL9, and ACTA2 in intestinal and pulmonary cells (Figure S4D). Although myofibroblasts are absent in synovium, synovial C13 cells may reflect an activated phenotype involved in tissue repair. This is supported by synovium-specific upregulation of the bone and cartilage repair genes TFF3, BMP6, HTRA1, and HBEGF (Figure S4E).
In the synovium and intestine, several clusters have been shown previously to be associated with distinct anatomical locations:2,3,6 PRG4+ synovial lining fibroblasts, THY1+ sublining synovial fibroblasts, WNT5B+ villus-associated fibroblasts, and WNT2B+ crypt-associated fibroblasts. Many of the integrated clusters we identified grouped along these anatomically defined lines. Clusters C0, C6, C10, and C12 were most associated with PRG4+ lining-associated synovial and WNT5B+ villus-associated gut fibroblasts, whereas clusters C1, C2, C3, and C8 mapped to THY1+ sublining-associated synovial and WNT2B+ crypt-associated gut fibroblasts. Except for cluster C8, clusters that were strongly associated with anatomical locations in gut and synovium had fewer numbers of shared marker genes across tissues, potentially reflecting tissue-specific functions dictated by the specific anatomical constraints and physiological functions of the tissue.
FBLN1+ C5 and CD34+MFAP5+ C9 states mapped strongly to RSPO3+ intestinal, HAS1+PLIN2+ pulmonary, and CD34+THY1+ synovial fibroblasts. The remaining cluster, C7, did not correspond well to intestinal or synovial clusters. Subsequent analysis of marker genes within tissues suggested enrichment in doublets: the epithelial markers KRT7 and ADGRF5 in lung and the macrophage markers C1QB, C1QA, and SPP1 in the salivary gland. This suggests that, despite our best efforts to filter doublets during QC preprocessing, some contaminating doublets were retained. This makes further inference about cluster C7 less reliable.
Comparison of cross-tissue clusters with independent fibroblast annotations
We next compared our cross-tissue clusters with cross-tissue fibroblast annotations defined in a similar study performed with publicly available mouse scRNA-seq datasets.21 Here the authors used Harmony to integrate public datasets into two study-integrated fibroblast atlases: one with fibroblasts from healthy mice and one from perturbed tissues (i.e., disease models). Using gene set enrichment analysis with their published cluster marker sets, we found strong correspondence between our cluster definitions (Figure S2E). We observed the strongest correspondence with our C5, C9, C11, C4, C10, C0, and C12. The C5 and C9 clusters corresponded specifically to the Col15a1+ and Pi16+ clusters, respectively, both of which were confirmed experimentally to have the plasticity to give rise to multiple other fibroblast clusters in vivo in multiple organ systems. The SPARC+COL3A1+ (C4) cluster corresponded mostly to the Comp5+ cluster in healthy and perturbed tissue and the Lrrc15+ cluster only observed in perturbed tissue, which Buechler et al.21 associated with functions involved in fibrosis, wound repair, and muscle injury. The CXCL10+CCL19+ (C11) cluster corresponded to the healthy and perturbed CCL19+ clusters, which Buechler et al.21 labeled as specific to the lymph node and spleen. The C0 cluster corresponded to their Cxcl5+ cluster, only identified in perturbed tissue and associated with muscle injury. The C10 cluster corresponded to their Bmp4+ cluster, identified only in healthy large intestine samples. Finally, C12 fibroblasts corresponded to Adamdec1+ fibroblasts, identified only in perturbed gut tissue.
Identification of fibroblast states expanded in inflamed tissue
We next addressed which cross-tissue fibroblast states were expanded in inflamed tissues. To perform this association across tissues, we first needed to define a common measure of tissue inflammation. Although histology is often the gold standard to assess inflammation, histological features are inherently biased to tissue-specific pathology. Instead, we decided to define inflammation in a tissue-agnostic way, as the relative abundance of immune cells in each sample. Although immune cell abundance alone oversimplifies complex pathological processes, it is a ubiquitous and quantifiable measure of chronic inflammation. We quantified the fraction of immune cells based on previously labeled scRNA-seq clusters (Figure 1B) for salivary gland and lung samples and based on the proportion of CD45+ cells by flow cytometry (Figure S1F) for synovium and intestine (Figure 3A). These estimates are quantified with dissociated cells from cryopreserved tissue (STAR Methods) and thus lack granulocytes, such as neutrophils, which constitute an important part of tissue inflammation. To obtain comparable results across tissues, we standardized the raw tissue-specific immune cell frequencies to a common scale from 0 (not inflamed) to 1 (inflamed) (Figure 3B). Importantly, this transformation (STAR Methods) removes the effect of distributional differences among tissues and preserves the order of scores within each tissue.
Using these standardized inflammation scores, we performed a separate association analysis with mixed-effects logistic regression for each tissue. This analysis provided, for each tissue and fibroblast state, the effect of increased inflammation on cluster abundance (Figure 3C). Positive log ORs denote expansion with inflammation, whereas negative ratios denote a diminishing population. Some clusters, such as C2, C3, C7, PTGS2+SEMA4A+ C8, and C12, were significantly (false discovery rate [FDR] < 5%, red) expanded in only one tissue. Others, such as CXCL10+CCL19+ C11 and SPARC+COL3A1+ C4, were significantly expanded in multiple tissues. We confirmed that association with normalized inflammation scores did not change the qualitative results within tissue but did make the results more interpretable across tissues (Figure S5A). Within tissue, information about and individual’s treatment status (Tables S2, S3, S4, and S5) did not systematically explain the range of inflammation scores (Figure S5B). We then performed a meta-analysis of these tissue-specific effects (STAR Methods) to prioritize clusters expanded consistently across all tissues (Figure 3D). This meta-analysis identified two fibroblast states significantly expanded in inflamed samples from all 4 tissues (Figure 3E): SPARC+COL3A1+ (C4) (OR = 10.4, 95% CI[6.6, 16.2], p = 9.4 × 10−25) and CXCL10+CCL19+ (C11) fibroblasts (log OR = 32.7, 95% CI [11.4, 94.0], p = 9.6 × 10−11). The reported OR values denote the odds of a cell being in a cluster (versus not), given that it came from an inflamed sample. Because the effects for these clusters were similar across tissues, pooling in the meta-analysis increased the power to detect these abundance changes.
We noted that the associations of C4 and C11 clusters in the lung alone were not statistically significant. We hypothesized that this could arise from our overly simplistic inflammation score. For instance, the number of alveolar macrophages in lung can vary by anatomical region, and this anatomical variation could confound our scores based on the total percentage of CD45 cells. Thus, we quantified an alternative inflammation score for lung samples based on the proportion of lymphoid cells (Figure S5C), which was weakly correlated (r = 0.38, p = 0.07) to the scores based on CD45+ cells. We defined lymphocytes in our dataset as the aggregate of CD3+ T, CD20+ B, CD56+ natural killer (NK), and JCHAIN+ plasma cells, identified in a fine-clustering analysis of lung cells (Figures S5D and S5E). We then associated fibroblast cluster abundance with this targeted inflammation score and compared the results with association with the percentage of CD45+ cells (Figure S5F). Overall, the association of fibroblast cluster frequency with the two scores is correlated (r = 0.60, p = 0.02), and, in particular, the log ORs for the C4 and C11 clusters are consistent (βC4|%CD45 = 1.90 ± 1.43 versus βC4|%lymphocytes = 1.29 ± 0.86; βC11|%CD45 = 2.75 ± 1.90 versus βC11|%lymphocytes = 3.14 ± 1.41). Based on this analysis, the lack of statistical significance in the lung-only association of C4 and C11 clusters is not due to the coarse nature of the inflammation score and more likely due to the smaller number of fibroblasts profiled in the lung.
Distinct immune-interacting and vascular-interacting fibroblast states expanded in tissue inflammation
The two fibroblast states consistently expanded in inflamed tissue are characterized by distinct gene programs (Figure 3F) that reflect putative distinct functions during tissue inflammation. To explore these potential roles, we performed gene set enrichment analysis with 6,369 Gene Ontology (GO)33 and 50 MSigDB hallmark pathways (Table S9; Figure 3G).34 Marker genes for CXCL10+CCL19+ fibroblasts were enriched for pathways involved in direct interaction with immune cells, including lymphocyte chemotaxis (GO:0048247, adjusted p < 0.005; includes CCL19, CCL2, and CCL13), antigen presentation (GO:0019882, adjusted p < 0.005; includes CD74, HLA-DRA, and HLA-DRB1), and positive regulation of T cell proliferation (GO:0042102, adjusted p < 0.005; includes TNFSF13B, VCAM1, and CCL5). CXCL10+CCL19+ fibroblasts show broad evidence of response to the key pro-inflammatory cytokines interferon (IFN) γ (GO:0034341, adjusted p = 0.005), IFN α (GO:0035455, adjusted p = 0.02), TNF-α (GO:0034612, adjusted p < 0.005), IL-1 (GO:0070555, adjusted p < 0.005), and IL-12 (GO:0070671, adjusted p < 0.005). Although TNF-α, IL-1, and IL-12 responses are broadly enriched in several fibroblast populations, an IFN response (IFN α) is more specific to CXCL10+CCL19+ fibroblasts. In contrast to these cytokine signaling pathways, SPARC+COL3A1+ fibroblast marker genes were enriched in pathways centered around ECM binding (GO:0050840, adjusted p < 0.005; includes COL11A1, SPARC, and LRRC15) and disassembly (GO:0022617, adjusted p = 0.005; includes MMP13, MMP11, and FAP) and numerous developmental pathways (GO:0035904, GO:0060348, GO:0061448, and GO:0007492; adjusted p < 0.005; includes COL3A1, COL1A1, COL5A1, and TGFB1).
We performed transcription factor (TF) analysis to infer which TFs may be active in the C4 and C11 states. Following the recommend standards for TF analysis,35 we used the Viper algorithm36 and the TRRUST37 and DoRothEA38 databases. Examining the top 10 TFs assigned to each of the C4 and C11 clusters, we found consistent results with the gene set analysis above (Figure S5G). C11 is most associated with TFs in inflammatory signaling pathways, such as RELA and NFKB1 in nuclear factor κB (NF-κ B) signaling; STAT1, STAT2, IRF1, IRF2, and IRF9 in IFN signaling; and RXFAP and RFXANK in major histocompatibility complex (MHC) class II signaling. C4 is most associated with TFs in morphogen signaling and developmental pathways, such as CREB3L1 and RUNX2, key TFs in bone development and homeostasis; HMBOX1 in developmental tissue patterning; MAZ in the MYC pathway; SMAD3 in transforming growth factor β (TGF-β) signaling, and HES1 in NOTCH signaling.
The pathway and TF analyses suggests that SPARC+COL3A1+ fibroblasts may be driven by conserved developmental pathways during tissue remodeling in chronically inflamed diseases. Given the extensive enrichment in developmental pathways in these fibroblasts, we hypothesized that this state could be driven by morphogens within the tissue microenvironment. Indeed, we observed enrichment in the key morphogen signaling pathways hedgehog (adjusted p = 0.005), TGF-β (GO:0007179, adjusted p < 0.005), WNT (canonical [GO:0060070, adjusted p = 0.007] and non-canonical [GO:0035567, adjusted p = 0.005]), BMP (GO:0071772, adjusted p = 0.01), and Notch (GO:0007219, adjusted p < 0.005). Of these pathways, Notch signaling was the most specific to SPARC+COL3A1+ fibroblasts (Figure 3G), with non-significant (raw p > 0.20) enrichment in all other clusters. Because we previously identified Notch3 signaling as a key driver in differentiation of disease-associated perivascular fibroblasts in RA synovium,13 we predict that this cluster may represent a similar endothelium-driven, activated fibroblast state across inflammatory diseases involving other organ tissues. We explored this hypothesis with ligand receptor analysis (STAR Methods). We started with manually curated cognate ligand and receptor pairs39 and, for each pair, looked for high expression of one gene in endothelial cells within our libraries (Figure 1B) and its partner in each fibroblast state. Filtering for only differentially expressed genes, we found a total of 63 putative signaling interactions (Figure S5H). Notably, 19 of these interactions were between SPARC+COL3A1+ fibroblasts and endothelial cells, including Notch activation through the DLL4:NOTCH3 interaction, as described earlier for the synovium,13 as well as the morphogen TGF-β, the growth factor platelet-derived growth factor β (PDGF β), the angiogenic factors Ephrin-α and Ephrin-β,40 and the angiogenic and mitogenic factors MDK and PTN.41 This large variety of putative signaling interactions (Figure S5H) from and to endothelial cells suggests that SPARC+COL3A1+ fibroblasts participate in signaling cross-talk with endothelial cells. These pathway and cross-talk analyses suggest two independent, conserved populations that support tissue inflammation: immune cell-interacting CXCL10+CCL19+ immunofibroblasts and endothelium-interacting SPARC+COL3A1+ vascular-associated fibroblasts.
We next explored the possibility that disease-related genes may be upregulated within the C4 and C11 clusters and missed by using cluster marker analysis alone. Within each cross-tissue cluster, we correlated gene expression with normalized inflammation score to find intra-cluster inflammation-associated signatures (Table S10). We found that the number of significantly associated genes largely depended on the number of cells in a cluster, reflecting increased power to find statistically significant associations in larger clusters (Figure S5I). When we examined the genes most associated with inflammation score within C4 and C11, they were markers for that cluster or associated with inflammation in multiple clusters. For instance, in synovium, among the top inflammation-associated genes were the cluster C11 marker genes CCL19, CXCL9, and CD74 and DNAJB1, HSPH1, and MAFF, which were associated with inflammation in all synovial clusters. Thus, although our focus on cluster markers misses some inflammation-induced genes, these genes may represent a generic inflammatory response and do not help characterize distinct functional roles for distinct transcriptional states.
In situ localization of vascular and immuno-fibroblasts
The gene enrichment and ligand-receptor analyses suggest that the CXCL10+CCL19+ (C11) immuno-fibroblast and SPARC+COL3A1+ (C4) vascular fibroblast phenotypes are driven by distinct T lymphocyte- and vascular-derived signals in their microenvironments. We used high-dimensional imaging (STAR Methods) to determine the spatial co-localization of CCL19+ and SPARC+ fibroblasts with T lymphocytes and vascular endothelial cells, respectively, in inflamed synovium, lip, and gut tissue. We performed segmentation analysis, marker intensity quantification, and image-based quality control filtering (STAR Methods) to identify high-dimensional molecular profiles for 355,227 high-quality cells (Table S11): 58,471 cells from 2 synovial samples, 195,617 cells from 1 lip sample, and 101,139 cells from 3 gut tissue samples (Figure 4A). Within these cells, we identified fibroblasts based on expression of PDPN and/or PDGFRA, as in a previous study,21 and used clustering and gating strategies to identify SPARC+ and CCL19+ fibroblast subgroups. To facilitate statistical quantification of co-localization between fibroblasts and elements of their microenvironment, we analytically partitioned each tissue into anatomical niches. Here we consider a niche to be a spatially connected region with a well-defined cellular composition profile that reflects the function of the anatomical region (STAR Methods). When we identify such niches, we quantify co-localization as the frequency with which each fibroblast subtype is located inside versus outside of that niche.
Figure 4. Quantitative co-localization of inflammation-expanded fibroblast phenotypes in vascular and lymphoid niches.
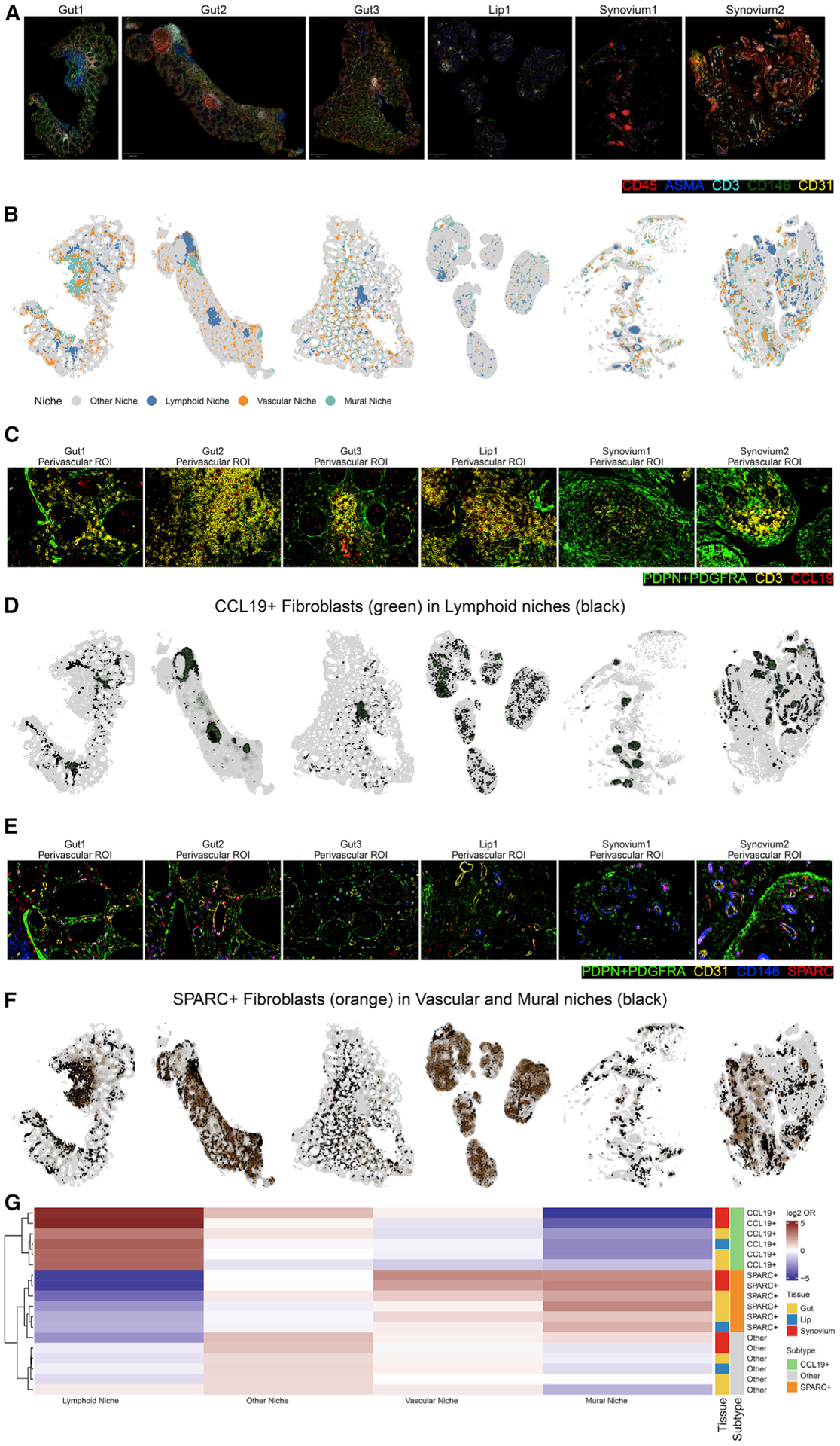
(A) Normalized intensities of the representative markers CD45, ASMA, CD3, CD146, and CD31 in segmented cells from surgical tissues samples of UC gut, pSS lip, and RA synovium.
(B) Visualization of molecularly distinct anatomical niches based on the 5 markers in (A).
(C) Manually selected regions of interest from the images in (A) highlight a region with abundance of CD3+ T lymphocytes next to (PDPN/PDGFRA)+CCL19+ fibroblasts.
(D) The same tissues from (B), colored to highlight lymphoid regions (black) and CCL19+ fibroblasts (green).
(E) Manually selected regions of interest from the images in (A) highlight a region with abundance of CD31+ vascular and CD146+ perivascular mural cells near (PDPN/PDGFRA)+SPARC+ fibroblasts.
(F) The same tissues from (B), colored to highlight vascular regions (black) and SPARC+ fibroblasts (orange).
(G) Heatmap depicting results of co-localization analysis between niches (columns) defined in (B) and three fibroblast subtypes (rows). Color in the heatmap denotes the log2 OR from the logistic regression test. Color bars for rows specify the tissue and fibroblast subtype of each test.
We used spatial clustering analysis (STAR Methods) to identify 4 anatomical niches present in all samples (Figure S6A; Figure 4B). We then used differential expression analysis to associate each niche with its predominant cell types based on lineage markers (Figure S6B). We labeled the lymphoid niche based on the abundance of CD45+CD3+ T lymphocytes, the vascular niche based on the abundance of CD31+CD146+ endothelial cells, and the mural niches based on abundance of CD146+ASMA+ mural cells. CD146+ mural cells, which include pericytes and vascular smooth muscle cells, are usually considered to be perivascular cells that play important roles in modulating vascular structure and growth.42 This localization is consistent with our data, where each CD146+ASMA+ mural niche is usually adjacent to a CD31+ vascular niche, particularly in the synovial tissue samples (Figure 4B). However, ASMA also marks highly contractile cells, such as submucosal smooth muscle cells in the intestine, visible as large, expanded ASMA+ regions in the Gut1 and Gut2 samples but not in the Gut3 sample, which lacks submucosal tissue (Figure 4B). All anatomical regions that did not fit into the lymphoid, vascular, or mural categories was labeled “other.” Although these regions likely contain functionally important niches, we chose to not label them to keep the focus of co-localization analyses based on lymphoid, vascular, and perivascular regions.
We next sought to identify CCL19+ and SPARC+ fibroblasts and test their co-localization within the lymphoid, vascular, and mural niches defined above. With manual inspection of the niches, we identified representative regions of interest in which (PDPN/PDGFRA)+CCL19+ cells localized next to CD3+ T cell-enriched regions (Figure 4C). To quantify this relationship across all datasets, we identified (PDPN/PDGFRA)+CCL19+ cells using a two-step clustering analysis (Figure S6C): coarse-grained clustering to identify (PDPN/PDGFRA)+ cells and then fine-grained clustering to identify CCL19+ fibroblasts (Figure S6D). The output of this analysis allowed us to map the location of all CCL19+ fibroblasts in the niche-annotated images (Figure 4D). We repeated the procedure to identify SPARC+ fibroblasts, which we manually identified near CD31+ vasculature in representative regions (Figure 4E). The same two-step clustering analysis described above also identified SPARC+ fibroblasts and mapped them into the niche-annotated images (Figure 4F). Finally, we quantified the statistical enrichment of co-localization between our fibroblast subsets and niches (Figure 4G). The results show that CCL19+ fibroblasts are significantly enriched in the lymphoid niche (log2 OR = 2.7 ± 0.53, p = 2.81 × 10−7). Although SPARC+ fibroblasts are nominally enriched in the vascular niche in some tissues (log2 OR = 0.88 ± 0.66, p = 0.09), the association between SPARC+ fibroblasts and the predominantly perivascular ASMA+CD146+ mural niche is considerably stronger (log2 OR = 1.5 ± 0.42, p = 1.54 × 10−4). These co-localization analyses confirm that CCL19+ fibroblasts localize to T lymphocyte-enriched anatomical regions, whereas SPARC+ fibroblasts localize to mural cell-enriched regions, which includes perivascular zones in all tissues as well as tissue-specific regions enriched for ASMA+ contractile cells.
We also performed these high-dimensional in situ experiments in inflamed lung tissue but were not able to robustly identify fibroblasts because of lack of PDGFRA staining. Qualitatively, these data show co-localization of SPARC and ASMA in the same regions in the lung (Figure S6E), consistent with the quantitative co-localization results above. These images also qualitatively confirmed that CCL19 is expressed by non-epithelial (CK8−), non-leukocytes (CD45−), non-perivascular (CD146−, CD31−) cells, suggesting the presence of CCL19+ fibroblasts around CD3+ T cells (Figure S6F).
T cells and vascular endothelium induce convergence of fibroblast states
The co-localization data above show that immuno- and vascular fibroblasts co-localize with T cells and endothelial cells in inflamed tissue, respectively. However, physical proximity alone does not prove that signals from T cells and vascular cells are sufficient to polarize fibroblasts into the divergent phenotypes observed in our atlas. To test this hypothesis, we obtained fibroblasts from IPF/ILD lungs and RA synovia and stimulated them with supernatant from in-vitro-activated T cells or cultured in the presence of endothelial cells to mimic the tissue microenvironment in inflammatory diseases (Figure 5A). For consistency, we plated cells on a 2D hard surface under each condition and performed scRNA-seq profiling on 18,000 fibroblasts. To avoid confounding effects from experimental batches, we profiled fibroblasts from one tissue in a single 10X library, pooling cells from multiple donors (n = 3) and conditions within each tissue using cell-hashing-based multiplexing (STAR Methods). After demultiplexing and standard QC, we recovered a total of 22,473 scRNA-seq profiles of cultured fibroblasts with more than 1,000 cells in most replicates (Figure 5B). Separate UMAP analyses of synovial and lung fibroblasts (Figure S7A) show that multiplexing successfully grouped cells primarily by culture condition and then by donor ID.
Figure 5. Convergence of fibroblasts from distinct tissues with in vitro activation.
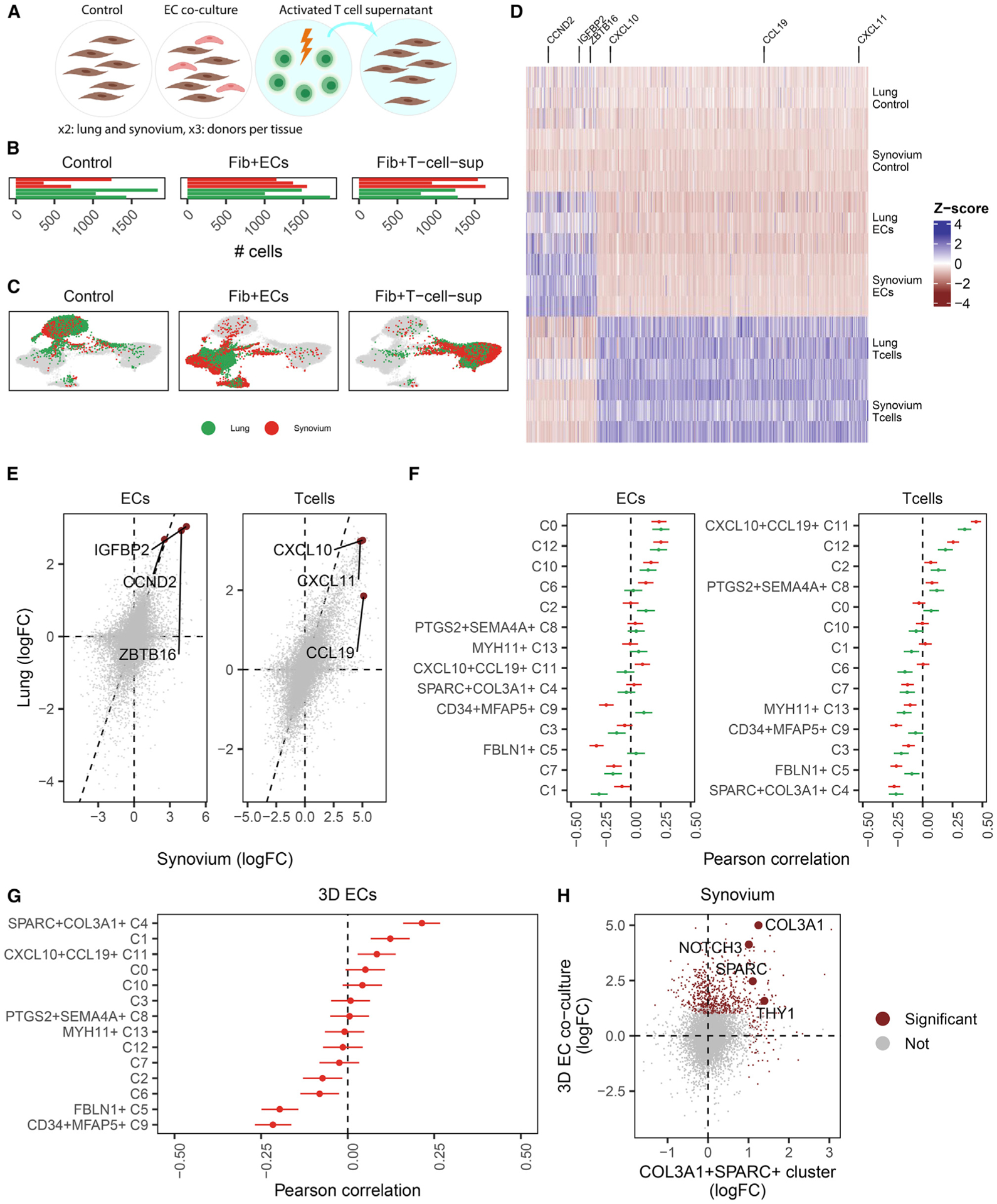
(A) Study design with three conditions: fibroblasts cultured alone (control), with an equal mixture of endothelial cells (ECs), and in supernatant extracted from activated T cells (T cells). Each condition was repeated in three lung-derived and three synovium-derived fibroblast cell lines.
(B) Total cell numbers per donor per condition in log scale.
(C) scRNA-seq profiles of cultured cells were visualized in 3D with integrative analysis and UMAP projection. Each subpanel highlights the location of fibroblasts from the control, T cell, and EC conditions and colors fibroblasts by tissue.
(D) Within each tissue, activation signatures were derived for the EC and T cell conditions and plotted in a heatmap of pseudobulk samples (rows) by genes (columns), colored by centered and scaled log2 fold change (versus control). Three representative genes were selected for each activation signature.
(E) For each condition, we plotted the per-gene changes for synovial fibroblasts (x axis) against lung fibroblasts (y axis) and highlighted the three representative genes from (D).
(F) We compared the in vitro activation changes with cluster marker signatures from the cross-tissue atlas with correlation analysis. Error bars denote 99% CI for the Pearson correlation statistic.
(G) Correlation analysis of fibroblasts cultured with ECs in a 3D culture system.
(H) Magnification of the correlation of SPARC+COL3A1+ (C4) cluster markers (x axis) with the 3D EC synovial activation signature (y axis). Genes significantly (p < 0.01, log2 FC > 1) upregulated on either axis are colored red, and canonical markers of the C4 cluster are highlighted with text.
We first wanted to determine whether the effect of activation condition was similar across tissues or whether lung and synovium-derived fibroblasts responded with unique gene expression programs to the same conditions. Harmony and UMAP analyses of fibroblasts from lung and synovium together (STAR Methods) groups fibroblasts largely by culture condition (Figure 5C), suggesting that fibroblasts from different tissues share transcriptional profiles driven by experimental perturbations. To identify which genes are driven by shared responses to culture conditions and which are tissue specific, we performed differential expression analysis within each tissue to find response signatures to each activation condition (Figure 5D; Table S12). The immune-activated signature contains key IFN-responsive genes, such as CXCL10, CXCL11, and CCL19, whereas the endothelial-activated signature contains genes related to cell cycle and differentiation pathways, such as IGFBP2, ZBTB16, and CCND2. To look for tissue versus condition specificity of signature, we directly compared these signatures across tissues. We found that genes upregulated in synovial fibroblasts were highly correlated with those upregulated in lung fibroblasts in response to both endothelial cell (EC) co-culture (ρPearson = 0.55, p < 10−16) and T cell supernatant culture (ρPearson = 0.79, p < 10−16) (Figure 5E). In contrast, within tissue, the response to different conditions induced less correlated (synovial fibroblasts ρ = 0.25, lung ρ = 0.07) gene expression programs (Figure S7B). These results suggest that responses to T cells or vascular ECs induce fibroblasts from different tissue sources to converge on shared phenotypes.
Next we wanted to determine whether in vitro activation by T cells or vascular ECs was sufficient to reproduce gene expression programs that define the immune-interacting and vascular-interacting phenotypes in our cross-tissue atlas. More concretely, we wanted to find out which atlas cluster markers are most enriched in each of the activation response signatures. Using correlation analysis on differentially expressed genes, we correlated the relative gene expression profiles in the fibroblast atlas clusters to those in the culture experiments (Figure 5F). Genes that responded to T cell-derived signals under T cell culture conditions were specifically correlated with the immune-interacting CXCL10+CCL19+ cluster in lung (ρ = 0.35, 95% confidence interval [CI] = [0.31, 0.39]) and synovium (ρ = 0.45, 95% CI = [0.41, 0.48]). Marker genes for immune-interacting fibroblasts were upregulated in response to secreted signals from activated T cells (Figure S7C). This correlation plot also shows an important asymmetry; although most CXCL10+CCL19+ cluster markers are upregulated in response to activated T cell signals, the opposite is not true. That is, many genes upregulated by supernatant from activated T cells are not associated with CXCL10+CCL19+ fibroblasts, suggesting that the supernatant contains signals required for CXCL10+CCL19+ fibroblast activation in addition to those that are not. The gene signature for EC co-culture non-specifically and weakly (ρ ≤ 0.25) matched multiple clusters (C0, C10, and C12) but not the vascular SPARC+COL3A1+ cluster. Deeper pathway analysis with Gene Ontology shows upregulation of transcriptional and translational pathways in EC co-cultured fibroblasts (Table S13), suggesting that overall activation of gene and protein production, not a specific response to endothelial-derived signals, drives the similarity of these fibroblasts in 2D culture. The lack of enrichment of vascular signature in fibroblast co-cultured with ECs in a 2D system could reflect a requirement of vascular endothelial tubes to fully elicit a vascular fibroblast phenotype. Fibroblasts from a 3D synovial organoid system, in which spontaneously assembled into vascular tubules, exhibited enrichment of the SPARC+COL3A1+ vascular marker gene signature (Figure 5G), as reflected by the key signature genes SPARC, COL3A1, NOTCH3, and THY1 (Figure 5H).
Validation of lung results with independent cohorts
Given the small number of lung fibroblasts represented in our study, we were concerned about how our results would generalize to independent cohorts. In the following analyses, we compared our results with two independent studies, one of healthy lung fibroblasts43 and one in a study of late-stage IPF.5
The authors of the human healthy lung atlas identified 9 distinct non-endothelial stromal cells, of which 5 are fibroblasts (adventitial, alveolar, lipofibroblasts, fibromyocytes, and myofibroblasts), 2 are muscle (vascular smooth muscle and airway smooth muscle), 1 is mesothelial, and 1 is a pericyte population. Using the authors’ published marker gene profiles (Figure S8A), we re-analyzed the lung mesenchymal cells in our dataset and were able to label clusters (Figure S8B) that accounted for 97.5% of cells in the healthy atlas (Figure S8C). In our analysis, we did not discern two rare (≤ 2%) populations the authors had annotated: mesothelial cells and fibromyocytes. These annotations may not have been robust because both populations were present in only a single donor in the original publication (Figure S8C). Next we wanted to determine how the four fibroblast clusters defined in the healthy atlas compared with our cross-tissue clusters (Figure S8D). We found strong correspondence between the atlas-derived labels and the cross-tissue labels (Figure S8E); alveolar fibroblasts map to C1 and C3, adventitial fibroblasts map to FBLN1+C5 and CD34+MFAP+ C9, lipofibroblasts map to C2 and PTGS2+SEM4A+ C8, and myofibroblasts map to SPARC+COL3A1+ C4, C10, and MYH11+ C13. These results demonstrate that our lung dataset is sufficiently rich to capture reliably annotated states present in healthy lung.
We next wanted to determine whether our cross-tissue clusters are informative in an independent lung cohort with healthy and diseased donors. Our dataset is the first scRNA-seq study of lung tissue to describe early-stage, inflammatory ILD. Previous studies4,5,44 compared non-diseased lungs with lungs from individuals with late-stage IPF, a disease defined more by fibrosis than active inflammation. We downloaded the data from one such study5 and mapped it into our cross-tissue atlas (Figure S8F). Among 5,380 fibroblasts from 58 donors, we recovered proportions of clusters comparable with those in our lung dataset (Figure S8G). We then looked for fibroblast clusters expanded in IPF-derived samples compared with non-IPF controls (Figure S8H). MYH11+ (C13) myofibroblasts and SPARC+COL3A1+ (C4) vascular fibroblasts were most expanded in IPF samples, whereas CXCL10+CCL19+ (C11) immuno-fibroblasts were not significantly expanded (Figure S8I). The expansion of fibrosis-associated (C13) myofibroblasts and collagen-enriched (C4) vascular fibroblasts and the absence of (C11) lymphocyte interacting immuno-fibroblasts are consistent with the non-inflammatory fibrotic pathology of the late-stage IPF individuals in this cohort.
Finally, we tested the reproducibility of our cluster marker results for lung fibroblasts in the cross-tissue atlas. We performed differential expression analysis in the IPF dataset described above and compared the cluster markers profiles between our data and this independent cohort. Using correlation analysis, we confirmed that the cluster marker profiles between the two cohorts are concordant (Pearson ρ ∈ [0.34, 0.83]) (Figure S8J). To illustrate these results, we focused on cluster markers for the two clusters expanded in individuals with IPF: MYH11+ (C13) myofibroblasts (Figure S8K) and SPARC+COL3A1+ (C4) vascular fibroblasts (Figure S8I). We found significant concordance between differentially expressed genes in the two cohorts (C4 ρ = 0.76, p = 1.06 × 10−139, C13 ρ = 0.69, p = 3.74 × 10−78), particularly among canonical genes we used to label the clusters: ACTA2 (βadams = 1.60 ± 0.23, βAtlas = 2.05 ± 0.49), MYH11 (βadams = 2.60 ± 0.24, βAtlas = 2.57 ± 0.53), and MYL9 (βadams = 0.78 ± 0.26, βAtlas = 0.60 ± 0.40) in myofibroblasts and THY1 (βadams = 0.54 ± 0.18, βAtlas = 0.71 ± 0.29), SPARC (βadams = 1.37 ± 0.19, βAtlas = 1.24 ± 0.30), and COL3A1 (βadams = 1.52 ± 0.24, βAtlas = 1.38 ± 0.30) in vascular fibroblasts. These results confirm that, even with small cell numbers, the disease-related clusters we identified in our lung disease cohort are defined by the same genes as the disease-related clusters in the independent IPF cohort.
Comparison of gut clusters with independent healthy adult atlas
We also performed a comparison with a non-diseased atlas within our gut cells, leveraging the recently published gut cell atlas by Elmentaite et al.45 The authors identified 8 types of fibroblasts with sufficient representation (>25 total cells) in large intestine tissues sampled from healthy adult donors: myofibroblast, myofibroblast (RSPO2+), stromal 1 (ADAMDEC1+), stromal 1 (CCL11+), stromal 2 (NPY+), stromal 3 (C7+), T reticular, and transitional stromal 3 (C3+). We re-analyzed our gut fibroblasts and were able to identify all 8 phenotypes described by Elmentaite et al.45 (Figure S9A). We next compared these cluster labels with our integrated fibroblast phenotypes. We found a strong correspondence between the two labels (Figure S9B): MYH11+ C13 maps to both myofibroblast clusters; C12, C0, C10, and C6 map to stromal 2 (NPY+); CXCL10+CCL19+ C11 maps to T reticular cells; SPARC+COL3A1+ C4, C3, C2, and C1 map mostly to stromal 1 (CCL11+); CD34+MFAP5+ C9 and FBLN1+ C5 map to transitional stromal (C3+); and PTGS2+SEM4A1+ C8 maps to stromal 1 (ADAMDEC1+) and stromal 3 (C7+). Finally, we confirmed our labels by using the top 50 marker genes associated with the authors’ phenotypes (Figure S9C). These results confirm that our gut fibroblast dataset captures the heterogeneity identified in healthy tissue and that the healthy-gut classification system agrees with our cross-tissue clusters, although some cross-tissue clusters are coarser than the healthy-gut atlas phenotypes.
Validation in an alternative tissue: Dermal fibroblasts in atopic dermatitis
As a proof of principle, we next explored whether the fibroblast states discovered in the four tissues could generalize to a tissue not explored in this study by examining cells from an independent dataset. We analyzed data from a study by He et al.46 of atopic dermatitis (AD), a chronic inflammatory condition of the skin (Figure 6A). The authors performed droplet-based scRNA-seq on all cells from cryopreserved skin biopsies of 5 individuals with AD (4 samples from skin lesions and 5 samples from skin outside of lesions) and 7 healthy donors. After removing low-quality (STAR Methods) cells and 3 samples with fewer than 500 high-quality cells, we clustered 29,625 cells from 13 samples to identify the following major cell types (Figures S10A and S10B): MLANA+ melanocytes, KRT15+ epithelial cells, CD3G+ T cells, C1QB+ myeloid cells, PROX1+ lymphatic ECs, ACKR1+ vascular ECs, ACTA2+ mural cells, and COL1A1+ fibroblasts. As before, we used immune cell abundance to quantify a relative inflammation score in each sample (Figure 6B). Immune cell abundance correlated with histological classification, highest in samples from skin lesions and lowest in samples from non-diseased controls (Figure 6B).
Figure 6. Dermal fibroblast scRNA-seq profiles mapped to the cross-tissue fibroblast atlas.
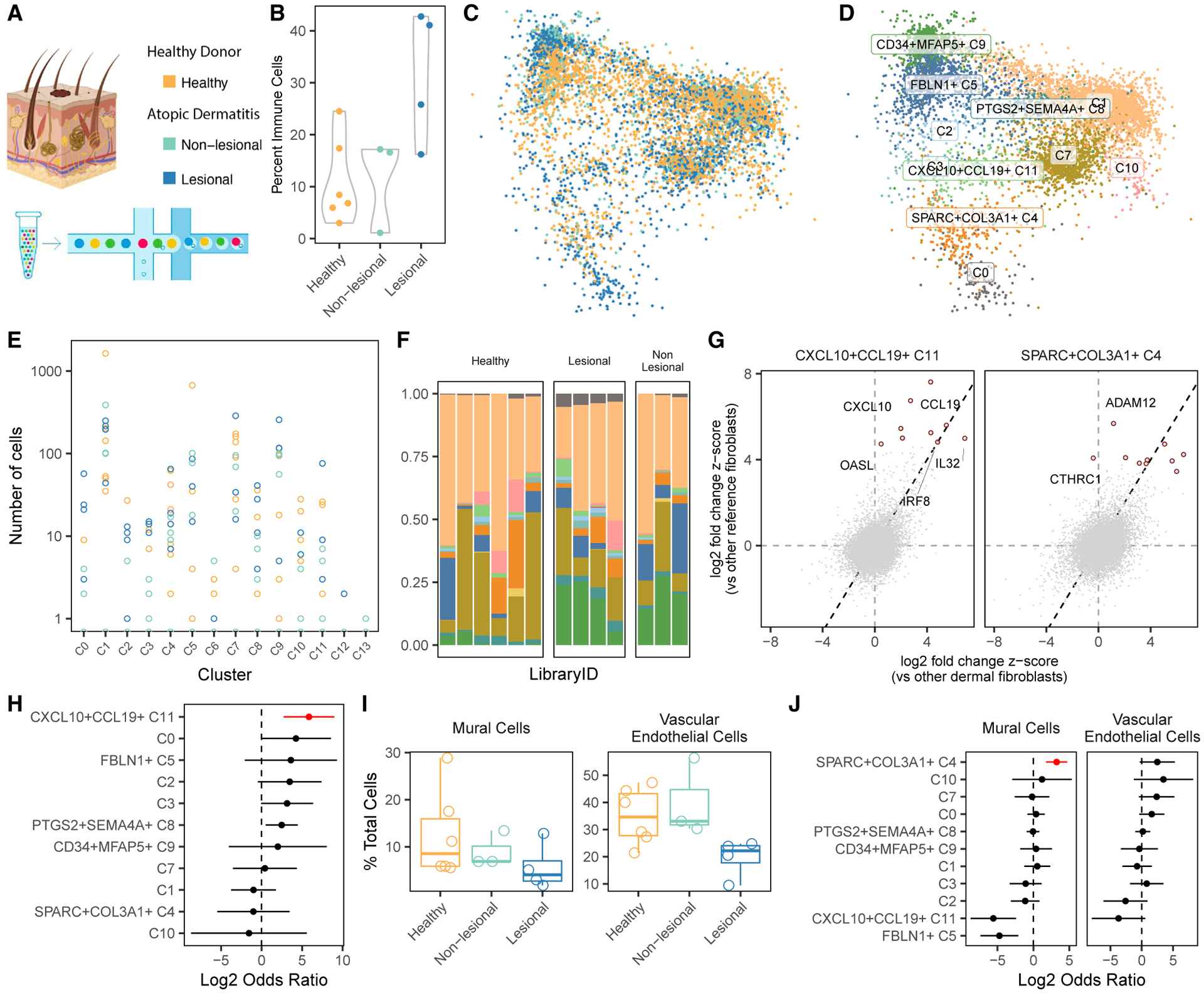
(A) To validate our results, we mapped scRNA-seq profiles of dermal fibroblasts from lesion biopsies from individuals with atopic dermatitis (AD), non-lesional biopsies from individuals with AD, and control skin biopsies from healthy donors.
(B) Based on the relative frequency of immune cells in each biopsy, we computed standardized inflammation scores from 0–1.
(C–F) We mapped dermal fibroblasts to our fibroblast atlas (C) and labeled dermal fibroblasts according to their most similar atlas cluster (D). Shown are per-donor (E) absolute and (F) relative frequencies of all reference-mapped inferred clusters. Clusters are colored according to the names in (D).
(G) We confirmed that the gene expression profiles of inferred dermal fibroblast clusters correlated with expression profiles of their reference fibroblast clusters. This is demonstrated for clusters C4 and C11 by plotting the (differential) gene expression in dermal (x axis) versus reference (y axis) clusters and calling out the top marker genes identified in the reference clusters.
(H) Only CXCL10+CCL19+ (C11) fibroblast frequency was significantly (FDR < 5%) associated with dermal inflammation.
(I) Cells from skin with lesions (blue) had considerably less evidence of vasculature, measured by the abundance of perivascular mural cells and vascular ECs.
(J) Relative abundance of mural cells and ECs was most strongly associated with cluster C4. Red denotes one-tailed FDR < 5%.
We wanted to compare dermal fibroblasts directly with clusters defined in our fibroblast atlas. To do this, we leveraged a novel algorithm, Symphony27 (STAR Methods), designed to quickly and accurately map new scRNA-seq profiles into a harmonized atlas to compare them with annotated reference cells. Using Symphony, we mapped dermal fibroblasts into our multi-tissue fibroblast atlas and projected them into the reference UMAP space for visual comparison (Figure 6C). For quantitative comparison of fibroblast subtypes, we labeled individual dermal fibroblasts by their most similar reference clusters (Figure 6D). Dermal fibroblasts from all donors (Figures 6E and 6F) mapped primarily to all clusters except C6, C12, and C13, three clusters we identified as more tissue specific (Figure 2G). We computed marker genes for these clusters in skin (Table S14) and compared them with the markers we computed in the cross-tissue analysis. The gene expression profile of each dermal fibroblast cluster most closely resembled that of its corresponding reference cluster (Figure S10C). As two examples of this expression concordance, we plotted gene expression of immune (C11) and vascular (C4) fibroblasts inferred in the skin dataset versus those labeled in the reference (Figure 6G), highlighting the top 10 marker genes upregulated in each of the fibroblast clusters in the reference.
We associated the abundance of inferred dermal fibroblast clusters with the sample-level inflammation score (Figure 6H). CXCL10+CCL19+ (C11) fibroblasts were most significantly expanded in inflamed skin samples (OR = 57, 95% CI [6.5, 503], p = 2 × 10−4), even when performing the association within histological groups (OR > 1000, p = 1.8 × 10−11) (Figure S10D). SPARC+COL3A1+ fibroblasts, expanded in the original four tissues, were less abundant in inflamed skin. Given the previous association of SPARC+COL3A1+ fibroblasts with vasculature, we explored the relative degree of vascular cell types in each skin sample. Lesional samples had significantly fewer vascular ECs (one-tailed t test, p = 0.004) and perivascular mural cells (one-tailed t test, p = 0.07) (Figure 6I), compared with non-lesional and healthy samples together. The lack of vascular fibroblast expansion in inflamed samples from skin lesions is consistent with this decreased vascularization. In fact, the abundance of vascular fibroblasts is associated nominally with the abundance of vascular ECs (log OR = 2.5, p = 0.04) and strongly with perivascular mural cells (log OR = 3.2, p = 1.8 × 10−5) when taking into account the histological status (Figure 6J).
The original analysis of dermal fibroblasts by He et al.46 identified a novel COL6A5+ COL18A1+ population expanded in lesional skin biopsies. This population contained inflammatory (e.g., CCL19, CCL2, IL32) and ECM remodeling (e.g., POSTN, COL3A1, TWIST2) genes and likely represents two distinct subpopulations, as reflected by the different anatomical localization of CCL19 and POSTN. We next wanted to determine where the signature for these COL6A5+COL18A1+ fibroblasts appears in our shared clusters. With gene set enrichment analysis, we found that clusters C0, C4, and C11 were significantly enriched in COL6A5+COL18A1+ marker genes (Figure S10E). Genes that contributed to enrichment in C0 and C11 were more related to inflammation (e.g., CCL19, CCL2, IL32, and IFI27), whereas genes that contributed to enrichment in C4 were more ECM modulatory (e.g., COL3A1, POSTN, and TWIST2) (Figure S10F). With further gene expression and pathway analysis, we found that C0 and C11 represent distinct inflammatory activation programs; C0-associated genes were more enriched in NF-κB signaling, whereas C11-associated genes were more enriched in IFNγ signaling (Figure S10G). Our analysis deciphered subtle heterogeneity of three potentially inflammation-associated dermal fibroblast states that was previously described as one cluster.
Cross-species mapping identifies shared fibroblast activation states in disease animal models of pulmonary, synovial, and intestinal inflammation
Next we tested whether our two shared inflammation-associated fibroblast subtypes were identifiable in single-cell datasets from mouse models of tissue inflammation. By defining which aspects of fibroblast-driven pathology are reproduced in mouse models, it may be possible to elucidate which pathological processes in murine models best parallel human fibroblast cell states. We found three publicly available single-cell RNA-seq datasets that included inflamed and non-inflamed samples in matched mouse tissues, which we could use to analyze the conservation of cluster markers and the expansion of inflammation-associated immuno-fibroblasts and vascular fibroblasts (Figure 7A). Kinchen et al.8 profiled 8,113 cells, CD45− gated to enrich for stroma, from 3 healthy and 3 mice with dextran sulfate sodium (DSS)-induced colitis. Tsukui et al.47 profiled 15,095 cells, Col1a1+ gated to enrich for fibroblasts, from 2 healthy and 2 bleomycin-induced lung injury mouse lungs, profiled 14 days after treatment. Wei et al.13 profiled 8,738 total synovial cells from mice with K/BxN serum transfer (ST)-induced arthritis, half with active inflammation and half with abated disease by inhibition of Notch3 signaling, by genetic knockout (Notch3−/−) and blocking antibody (anti-Notch3 monoclonal antibody [mAB]). Although the K/BxN transgenic model generates autoreactive antibodies through a lymphocyte-mediated etiology, mice receiving those autoreactive antibodies through ST develop arthritis through a lymphocyte-independent etiology.48 Therefore, we did not expect to see changes in the frequency of T cell-interacting immunofibroblasts with this model. For this reason, we also generated a novel scRNA-seq dataset (STAR Methods) of collagen induced arthritis (CIA), an antigen-based model of arthritis that involves T cells49 and, thus, is more likely to involve immunofibroblasts.
Figure 7. Replication in disease models of pulmonary, intestinal, and synovial inflammation.
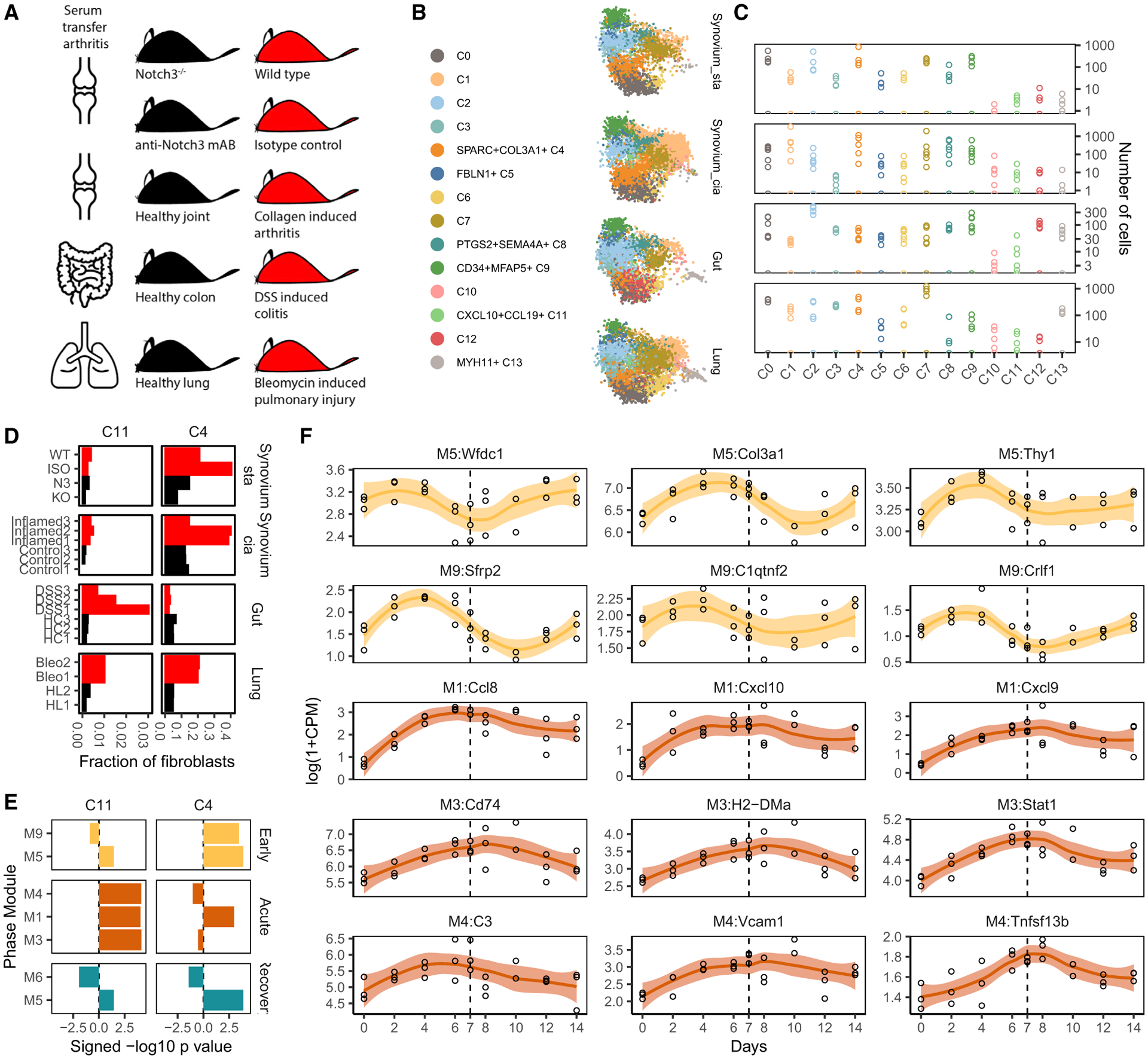
(A) We collected studies of inflammation in mouse models of human disease: bleomycin-induced ILD, DSS-induced colitis, ST arthritis, and CIA.
(B) Fibroblasts from each study were mapped to the human fibroblast atlas and labeled with their most closely mapped clusters.
(C) Total cell numbers per replicate per integrated cluster in log scale. Each panel corresponds to the aligned tissue in (B).
(D) Frequencies of the human inflammatory states C4 and C11 in each study sample, colored to denote samples from animals with high (red) and low (black) inflammation.
(E) GSEA with modules associated with early, acute, and recovery phases of DSS-induced colitis shows that C4 and C11 gene signatures are activated at distinct stages of inflammation.
(F) Time course expression profiles of key C4 and C11 marker genes that overlap with the early (yellow) and acute (orange) phase-associated modules. A dotted line denotes the time point (day 7) when DSS was removed from mice.
Within each dataset, we identified fibroblasts (6,979 intestinal, 10,320 pulmonary, 5,704 K/BxN ST synovial, and 15,965 CIA synovial) with clustering and marker analyses (Figures S11A and S11B). We then mapped these fibroblasts to our human cross-tissue reference with the Symphony pipeline (STAR Methods) and labeled mouse cells with the most similar reference fibroblast subtypes (Figure 7B). Although most clusters were well represented across tissues (Figure 7C), two appeared to be more tissue specific (Figure S11C). Myofibroblast-enriched C13 was mostly absent in both datasets for synovium, which is known to lack myofibroblasts. Cluster C12, which mapped well to the intestinal WNT5B+ 2 cluster in our initial analyses (Figure S4B), was enriched in intestinal fibroblasts in this mouse analysis. To test the degree to which gene markers are conserved between mouse and human, we performed cluster marker analysis in the mouse fibroblasts (Table S15) and compared cluster expression profiles between mouse genes and human orthologs (Figure S11D). Importantly, the most similar gene expression profiles were between corresponding clusters in mouse and human. For most clusters, expression profiles were even more similar between matched tissues.
We next wanted to determine whether the same fibroblast subtypes were expanded in inflamed tissues in human disease and mouse models. Thus, we performed differential abundance analysis within each mouse dataset, comparing inflamed cases with matched controls (STAR Methods) to determine which populations expanded in human tissues were also expanded in mouse models (Figure S11E), focusing particularly on the inflammation-associated SPARC+COL3A1+ and CXCL10+CCL19+ populations (Figure 7D). Overall, we found a high degree of concordance between expanded clusters in human and mouse tissues (Figure S11E). In bleomycin-treated lungs, the most expanded populations were SPARC+COL3A1+ (OR = 5.2, 95% CI [4.5, 6.0], p < 10−8) and CXCL10+CCL19+ (OR = 3.8, 95% CI[2.2, 6.6], p = 2.5 × 10−6) fibroblasts. The expansion of both populations is consistent with the known pathology of the bleomycin model,50 which is characterized by lymphocyte infiltration and fibrosis on day 14. In particular, the expansion of SPARC+COL3A1+ fibroblasts in the fibrotic mouse lungs is consistent with our results of fibrotic human disease in individuals with late-stage IPF (Figure S8I). In contrast to the C4 and C11 phenotypes, clusters C1 and C10, which are among the most expanded in inflamed human lungs, were not expanded in the mouse data (ORs 0.43 for C1 and 0.24 for C10). Given the low statistical confidence in associations in the human lung dataset (Figure 3C), we were less confident which clusters that were nominally expanded in inflamed human lung tissue would generalize to mice. In the K/BxN ST arthritis model, the Notch signaling-enriched (Figure 7D) SPARC+COL3A1+ cluster was greatly diminished with therapeutic Notch3 inhibition (OR = 3.8, 95% CI[1.5, 9.4], p = 4.1 × 10−3). On the other hand, the frequency of lymphocyte-interacting CXCL10+CCL19+ fibroblasts was not associated with disease activity (OR = 1.2, 95% CI[0.47, 3.3], p = 0.6). This result is consistent with the known lymphocyte independence of the ST model etiology.48 In contrast, in the CIA model of arthritis, which requires T cells, the SPARC+COL3A1+ (OR = 1.40, 95% CI[1.31, 5.29], p = 0.003) and CXCL10+CCL19+ (OR = 1.34, 95% CI[0.79, 8.12], p = 0.05) fibroblast clusters were expanded. The two transcriptionally related clusters C2 and C8 were expanded (OR = 18.4, % CI[2.80, 121], p = 8.5 × 10−3) in human RA but not in either mouse model. In contrast, C0 fibroblasts were found to be depleted (OR = 0.18, % CI[0.002, 0.13], p = 4.22 × 10−5) in human inflammatory arthritis but significantly expanded (OR = 2.52, % CI[1.89, 3.36], p = 1.08 × 10−10) in the mouse model. In DSS-induced colitis, CXCL10+CCL19+ fibroblasts were significantly expanded (OR = 6.1, 95% CI[1.9, 19.3], p = 2.3 × 10−3), as reported previously,8 whereas SPARC+COL3A1+ fibroblasts were actually diminished (OR = 0.5, 95% CI[0.4, 0.7], p = 9.2 × 10−7) in frequency.
Temporal ordering of C4 and C11 activation in DSS-induced colitis
We were surprised that SPARC+COL3A1+ fibroblasts were not significantly expanded in a DSS-induced colitis model despite their significance in the human cohorts. Further analysis of vascular ECs, lymphatic ECs, and mural cells in the same mice shows a lack of evidence of vascular expansion in this dataset (Figure S12A). The lack of vascular fibroblast signal in the diseased mice could mean that DSS-induced colitis utilizes an alternative inflammatory process. However, the difference may also reflect the kinetics of disease. Because DSS-induced inflammation is an acute process, reversible with removal of the chemical irritant, cross-sectional cellular compositions in that model may differ from compositions of chronically inflamed UC intestine. Specifically, if SPARC+COL3A1+ fibroblasts are responsible for tissue remodeling to enable leukocyte infiltration, then genes associated with SPARC+COL3A1+ fibroblasts should precede those associated with CXCL10+CCL19+ fibroblasts. To test this hypothesis, we used recently published time course transcriptional profiles of DSS-induced colitis, which tracks gene expression changes with the induction and resolution of inflammation.51 The authors induced intestinal inflammation in female 8- to 12-week-old C57BL/6J mice by putting DSS in their drinking water for 7 days and allowed resolution of inflammation by removing DSS for another 7 days. Measuring gene expression profiles with RNA-seq approximately every 2 days, the authors defined gene modules M5 and M9, associated with early inflammation (2–4 days); M1, M3, and M4, associated with acute inflammation (6–8 days); and M5 and M6, associated with resolution (10–14 days). We analyzed the enrichment of these phase-associated modules in our fibroblast marker profiles to associate the expansion of fibroblast subtypes with distinct phases of DSS-induced inflammation and resolution (Table S16). Strikingly, CXCL10+CCL19+ fibroblasts exclusively mapped to the three acute-phase modules M1, M3, and M4, whereas SPARC+COL3A1+ fibroblasts mapped to two early-phase modules, M5 and M9, and only the M1 acute-phase module (Figure 7E). Time course profiles of representative genes demonstrate the early- and resolution-phase activation of SPARC+COL3A1+-associated genes and acute phase activation of CXCL10+CCL19+-associated genes (Figure 7F). Importantly, this temporal pattern was obscured in the single-cell DSS-induced colitis dataset analyzed above because all mice in that study were euthanized on day 7, at the height of the acute inflammation phase. Given our hypothesis that SPARC+COL3A1+ fibroblasts are involved in vascular remodeling, whereas CXCL10+CCL19+ fibroblasts interact with infiltrating immune cells, the early upregulation of SPARC+COL3A1+-association gene suggests that vascular remodeling precedes leukocyte infiltration in the DSS colitis model.
We next wanted to determine whether these dynamic signatures were driven specifically by changes in fibroblasts or in another cell type because the analysis above was done with whole-tissue RNA-seq profiles. For instance, genes associated with CXCL10+CCL19+ immuno-fibroblasts include IFN response genes, such as Cxcl9 and Cxcl10, which are upregulated in stromal and immune cells downstream of IFNγ signaling. To look for changes in fibroblast-specific transcriptional profiles in the DSS model, we generated a novel time course RNA seq dataset of DSS mice (STAR Methods), profiling RNA from flow-sorted fibroblasts rather than from whole tissue. On days 2, 4, 7, 9, 11, and 14 of the model, we sacrificed 6 mice with DSS-induced colitis and 1 healthy control mouse, flow-sorted Epcam−Cd45−Cd31−Pdpn+Pdgfra+ disaggregated colon cells, and profiled 1,000 sorted fibroblasts from each mouse with RNA seq. This novel fibroblast-specific dataset allowed us to identify dynamic patterns arising from changes in fibroblast states and rule out changes because of fluctuations in cell type abundance.
Dynamic expression analysis across time points (STAR Methods; Table S17) identified 52 time-related genes that were also upregulated at least 1 time point versus healthy controls (Figure S12B). Expression levels for most of these genes peaked on days 7 and 9, at the height of leukocyte infiltration (Figure S12C), and on day 14, the onset of resolution. We compared these inflammation-phase and resolution-phase gene sets with our cluster markers using gene set enrichment analysis and found significant (FDR < 1%) enrichment with marker genes for clusters C2, C12, and C11 (Table S18), with the strongest association (p = 1.3 × 10−20) for cluster C11 (Figure S12D), driven by inflammation-associated genes such as Ccl19, Cxcl9, and Gbp4 (Figure S12E). This enrichment supports our hypothesis that C11 immuno-fibroblast abundance expands with the peak of leukocyte infiltration. We found no significant association at any time points with marker genes from the C4 cluster, which, as we know from our analysis of single-cell data, should be depleted on day 7 (Figure 7D). This lack of signal suggests that these data do not capture transcriptional changes arising from expansion or depletion of vascular fibroblasts, potentially because we excluded these cells with the double-positive sort for Pdpn+Pdgfra+ cells.
DISCUSSION
In this study, we sought to define whether shared fibroblast states exist across four diverse tissues affected by clinically distinct inflammatory diseases. We postulated that defining shared pathogenic, inflammation-associated fibroblast states across diseases will help inform common therapeutic strategies targeting fibroblasts across different inflammatory diseases. Comparison of pathogenic fibroblast phenotypes across diseases that manifest in different tissues is hampered by the lack of an accepted, tissue-independent taxonomy by immune and vascular cells. We thus approached this question by generating novel scRNA-seq profiles of fibroblasts and analyzing the fibroblasts together to identify shared phenotypes across diseases. Cross-tissue analysis of gene expression is a challenging task, as evidenced by the plethora of statistical methods introduced to analyze even non-single-cell, multi-tissue data generated by the Genotype-Tissue Expression (GTEx) project.52 Using sophisticated statistical methods for cross-tissue analysis, we were able to identify fibroblast phenotypes that were shared by all tissues as well as fibroblast adaptations unique to a subset of tissues.
The lack of universal definitions for key concepts, such as fibroblast identity and inflammation scoring, that apply equally well to all tissues presented a major challenge to our effort to associate fibroblast phenotypes with inflammation. In particular, the lack of a universal, pan-fibroblast surface marker that is uniformly expressed on all fibroblasts prevented us from directly isolating fibroblasts with flow cytometry. We addressed this problem with negative selection, using specific markers to filter out non-fibroblast populations, thus defining fibroblasts based on high-dimensional scRNA-seq data as non-epithelial, non-immune, non-endothelial, and non-mural cells with some known tissue-specific fibroblast markers, such as PDPN, PDGFRA, and COL1A1. The lack of a quantifiable score for inflammation prevented us from directly using standard tools from meta-analysis, which assume a standardized phenotype that can be measured equally well across all organ tissues. Inflammation in each disease is defined by disease-specific pathological processes, reflected in tissue-specific histological scores, such as the Krenn inflammation score in RA53 and the Nancy index in UC.54 We approached this challenge by intentionally selecting four chronic inflammatory diseases with distinct pathological and inflammatory processes. By analyzing fibroblasts from a range of diverse pathologies, we maximized the chances of identifying fibroblast phenotypes common to inflammation in four tissues. We chose the simplest aspect of inflammation that can be measured in all tissues: the proportion of immune cells infiltrating each tissue sample. Despite this simplicity, our definition robustly identified two shared fibroblast states, CXCL10+CCL19+ (C11) and SPARC+COL3A1+ (C4), associated with inflammation across tissues.
The presence of multiple inflammation-related fibroblast states suggests multiple distinct functions for fibroblasts during inflammation. Understanding this functional specialization is critical for accurate therapeutic targeting and is missed by studies that look for a single inflammatory fibroblast state. For instance, 47% of inflammation-associated fibroblasts (IAFs) identified by Smillie et al.3 to be expanded in UC map to two distinct clusters in our study: vascular-associated (C4) fibroblasts and the C12 cluster, which is uniquely expanded in the gut in our study. In the gut, C4 and C12 fibroblasts may be divided along anatomical lines because WNT2B (higher in C4) and WNT5B (higher in C12) have been associated with crypt-associated and villus-associated fibroblasts, respectively. C12 has higher expression of IL11 and IL13RA2, two canonical markers of IAFs suggested by Smillie et al.3 The remaining 22% of IAFs map to our C2 and C3 clusters, which are enriched for pro-inflammatory cytokine signaling pathways (Figure 3G). IAFs have been localized to the ulcer bed in a subset of individuals with inflammatory bowel disease (IBD)11 and might exert multiple functions depending on their proximity to different niches; one highlighted by our C4 cluster is shared across multiple inflammatory tissue and involved remodeling of the ECM around vessels to facilitate immune cells recruitment. In individuals with IBD with ulcers, vessels are expanded, and some IAFs are found in close proximity to them.11 IAFs do not map to the immune-interacting (C11) cluster, which is better resembled by a CCL19+ subset of the RSPO3+ cluster defined in Smillie et al.3 and CCL19+CD74high fibroblasts identified by Kinchen et al.8 to be the primary stromal cluster associated with UC in their cohort. A caveat of our definition of inflammation is that the other fibroblast clusters may be associated with distinct aspects of inflammation. For instance, PTGS2+SEM4A+ (C8) fibroblasts express the neutrophil-recruiting genes CXCL1 and CXCL2, are critical in a subset of IBD patient with ulceration,11 and are likely associated with neutrophil infiltration. In the same study, CCL19 was associated with another subset of individuals characterized by the presence of lymphoid aggregates in IBD but not with neutrophil infiltration and ulceration.11 Future studies with more nuanced definitions of inflammation could address the heterogeneous nature of inflammatory chronic diseases and may find additional pathological associations among our fibroblast clusters.
The complexity of our study design, with cells measured from multiple donors, tissues, and diseases, presented a second major challenge to our study. Algorithms to identify shared clusters in scRNA-seq datasets from multiple donors and tissues do not address key issues such as data imbalance or downstream analysis of gene expression in multi-tissue studies of human disease. Analyses that do not account for these factors in this complex setting may result in diminished power and spurious associations. Here we use weighted PCA and weighted Harmony to account for imbalanced datasets and mixed-effects Poisson regression to account for the effect of complex interactions between covariates on gene expression. Our analytical approach to decipher tissue-shared and tissue-specific gene expression serves as a template for well-powered and robust analysis of single-cell cluster markers, which is particularly relevant to the growing number of studies designed to identify shared etiology across tissues and diseases.28,55,56
Based on marker gene profiles, we believe that some of the clusters in our analysis have been previously described in single-cell and functional studies of individual tissues, potentially with the exception of pSS, in which a scRNA-seq atlas has not been described to date. For the first time, we provide a common frame of reference to cross-compare these diverse populations objectively across tissues. As a powerful corollary, we can draw upon functional studies performed in individual tissues to interpret the biological significance of our clusters.
CXCL10+CCL19+ (C11) fibroblasts closely resemble functionally well-characterized CCL19+PDPN+ immunofibroblasts in the salivary gland. These CCL19+ fibroblasts co-localize with CD3+ T cells and underlie the formation of salivary gland tertiary lymphoid structures in human tissue and in an animal model.12 This putative interaction with T cells is suggested by the expression of HLA genes in the synovial fibroblasts expanded in individuals with RA.2 Here, HLA-DRA+ fibroblasts show strong evidence of response to IFN γ, and functional work demonstrated that IFN γ is mostly produced by CD8+ T cells in inflamed synovium. Kinchen et al.8 also identified CCL19+ fibroblasts in the inflamed UC intestine, and numerous studies57,58 have identified T cells as the primary source of IFN γ in intestinal inflammation. CXCL10+CCL19+ (C11) fibroblasts expressed IRF8, and TF enrichment score analysis (Figure S5G) was enriched for STAT2-regulated genes, particularly in this cluster. Those are downstream targets of IFNα, and GO analysis highlights a potential role of IFNα in driving this phenotype. This suggests that T cell recruitment driven by CCL19+ fibroblasts and IFN-activated fibroblasts is a shared feature of inflammation across multiple diseases, and further studies are required to distinguish the activity of type I and type III IFN signaling in fibroblasts. Additional functional studies are required to investigate the complex interactions between T cells and fibroblasts in individual inflammatory diseases. Our integrative results provide generalizable markers that may identify such T cell-interacting fibroblasts across tissues.
SPARC+COL3A1+ (C4) fibroblasts closely resemble the CD90hi NOTCH3-activated synovial fibroblasts that are located near arterial blood vessels and pericytes and expanded in RA.13 Despite their perivascular location, NOTCH3+ fibroblasts, like our SPARC+COL3A1+ fibroblasts, are distinct from pericytes, as evidenced by their lack of the canonical pericyte genes ACTA2 and MCAM.59 Our cross-tissue analysis suggests that these vascular fibroblasts, which clustered separately from MCAM+ pericytes (Figure 1B), may also play a role in vascular remodeling in the lung, intestine, and salivary gland. In the time-series analysis of acute inflammation in the mouse intestine, we found that expansion of vascular fibroblasts preceded expansion of CXCL10+CCL19+ immune-interacting fibroblasts. If this temporal ordering holds tissues, then it suggests a two-stage mechanism for fibroblast-mediated regulation of inflammation, initiated by vascular remodeling that enables greater leukocyte infiltration into the tissue. Further mechanistic studies are needed to elucidate the additional endothelium-derived or angiocrine factors60 that mediate perivascular fibroblast differentiation and the mechanistic relationship between vascular and immune-interacting fibroblasts.
We focused on the histological characterization of the C4 and C11 cell clusters because we found them to be consistently expanded with inflammation across tissues. As a result, we focused less on three remaining clusters with a preponderance of shared genes: C8, C5, and C9. These clusters may be related to shared homeo-static functions or shared pathological functions not captured by our coarse-grained inflammation score based only on the percentage of CD45+ cells. Indeed, when we compared these shared clusters with previously defined fibroblast clusters in a cross-tissue study of mouse fibroblasts,21 we found a significant (p < 10−10) enrichment of genes from two experimentally validated universal progenitor states, Pi16+ and Col15a1+ fibroblasts, in our C5 and C9 clusters (Figure S2E). This comparison suggests a role of C5 and C9 fibroblasts as pluripotent progenitor states shared among our tissues. In contrast, our C8 cluster mapped well to multiple tissue-specific clusters: (perturbed and healthy) Cxcl12+ fibroblasts from joint tissue and Adamdec1+ and Fbln1+ fibroblasts from the intestine. Pathways analysis for cluster C8 suggested an inflammation-induced phenotype with evidence of response to TNF-α, IL-1, and IFNγ activation (Figure 3G). Upregulated genes included those associated with granulocyte recruitment, such as IL6, CXCL2, CXCL3, and ICAM1. Our study did not measure granulocyte abundance in tissue because neutrophils are poorly captured in scRNA studies. Thus, as mentioned earlier, if C8 is related to granulocyte trafficking, then our inflammation association test would not have picked up an expansion of this cluster. Finally, our C4 and C11 clusters were also described in the Buechler et al.21 fibroblast atlas and associated with perturbed tissue states. However, their experimental characterization of fibroblast phenotypes focused on the Pi16+ and Col15a1+ phenotypes, which are not associated with tissue pathology. Thus, our study provides a complementary view of fibroblasts in human tissues, with a particular focus on states universally expanded in inflammatory conditions as opposed to homeostasis.
When interpreting clusters with more tissue-specific than tissue-shared genes, we noticed that tissue-specific programs often express genes with tissue repair functions. This observation may reflect the tissue-specific needs for maintenance and repair, defined by that tissue’s unique anatomical structures.61 In contrast, clusters with more tissue-shared genes were enriched in biological processes, such as immune cell recruitment (C11 and C8), processes that are independent of tissue architecture, and interaction with blood vessels (C4), structures that are present in all tissues. This dichotomy between functions tailored to a tissue’s structural composition versus functions common to all tissues explains why some fibroblast phenotypes in scRNA-seq appear to be more tissue specific and others more tissue shared.
Although our analyses were focused on associations between tissue inflammation and fibroblast subtypes, we also found evidence to support the potential role of the inflammation-expanded C4 fibroblast cluster in fibrosis. Comparison of late-stage fibrotic disease in the lung with non-fibrotic lung found expansion of C4 but not C11 fibroblasts (Figure S8H). The C4 signature is enriched in the DSS-induced colitis model on day 14, which has been proposed as a model of intestinal fibrosis. Inflammation and fibrosis are tightly linked processes, but the cellular mechanisms that connect the two pathological processes are not well understood62 and may vary across diseases. For instance, although expansion of C4 and C11 fibroblasts may occur at different times in DSS-induced colitis (Figure 7E), our data on the mouse model of human IPF show concurrent expansion of C4 and C11 fibroblasts on day 14 (Figure 7D), consistent with the presence of fibrosis and lymphocyte infiltration at this time point. Finally, our C4 cluster shows evidence of perivascular localization and fibrosis-related genes (e.g., COL3A1). This combination makes C4 fibroblasts an attractive cellular phenotype to study the connection between vascular pathology and fibrosis. Multiple studies have noted that fibrosis occurs near vasculature63 and have suggested perivascular mesenchymal cells as precursors to profibrotic myofibroblasts.64,65 We also found that C4 cluster and SPARC expression was enriched around smooth muscle cell in the lung and in the intestine, two tissues prone to develop fibrosis. In future studies, we will investigate the role of C4 fibroblasts as a potential stromal mediator between pathological vascular processes and tissue fibrosis.
Our results suggest that distinct local microenvironments, some enriched for vascular cells and others for lymphocytes, are key to determining the fibroblast state. This would be impossible to determine from global clinical characteristics of individuals or even from non-anatomically matched molecular measurements. For instance, the proportion of CD45+ cells in several individuals with the more inflammatory RA diagnosis was lower than in those with the comparator diagnosis OA. The separation of CD45+ cells in inflamed versus control and non-inflamed gut samples was more concordant because inflamed samples were selected for evidence of local pathology. The frequency of immune cells in our lung cohort was the most diverse. Although we selected individuals with early-stage ILD to enrich for inflammatory disease, we found a wide variation in the frequency of CD45+ cells and lymphocytes among early-stage and end-stage disease. In the same tissue samples derived from affected individuals, we performed additional histopathological assays to obtain absolute quantification of lymphocytes (Table S19) identified by histological examination of formalin-fixed and paraffin-embedded (FFPE) lung tissue sections stained with hematoxylin and eosin (STAR Methods). Because of the destructive nature of tissue disaggregation for scRNA-seq, these histology scores profile different anatomical regions and, thus, different local microenvironments. The number of lymphocytes counted in these is not correlated with the number of lymphocytes profiled by scRNA-seq (p = 0.34). Thus, if we had measured fibroblast profiles in one region and inflammatory status in a separate region of the lung, then we would have captured distinct microenvironments and missed the correlation between fibroblast and immune cell abundance. This point highlights the importance of paired molecular and spatial profiling to understand the functional roles of fibroblasts and motivates use of emerging spatial transcriptomics technologies in future studies. With these emergent technologies, we will be able to measure more clinically relevant local inflammation states that were not available to us during this study.
Our in vitro co-culture experiments with lung and synovial fibroblasts derived from affected individuals highlight key limitations of inducing fibroblast phenotypes ex vivo. We found that fibroblasts co-cultured with disaggregated ECs failed to reproduce the COL3A1+SPARC+ phenotype, whereas fibroblasts co-cultured with ECs in a 3D organoid were induced toward the COL3A1+SPARC+ phenotype. This discrepancy highlights the need for more realistic physical culture settings to faithfully reproduce some fibroblast phenotypes ex vivo. In contrast, the co-culture experiment with supernatant from stimulated T cells was able to polarize fibroblasts toward the CXCL10+CCL19+ (C11) phenotype without the need for a 3D system. This co-culture condition captures the response of fibroblasts to secreted inflammatory T cell-derived signals. We found enrichment of genes associated with response to inflammatory cytokine pathways in multiple clusters: C0, C2, C3, C8, C11, and C12 (Figure 3G). However, this co-culture condition specifically upregulated genes associated with cluster C11. Thus, although multiple fibroblast clusters show evidence of response to inflammatory signals, likely originating from different sources, only CXCL10+CCL19+ (C11) fibroblasts are specifically enriched for signals derived from stimulated T lymphocytes.
We used a novel type of analysis from single-cell analysis, Symphony reference mapping,27 to compare human dermal fibroblasts and mouse lung, synovial, and lung fibroblasts with our annotated cross-tissue atlas. Reference mapping let us avoid intensive and error-prone manual interpretation steps in de novo analysis of the external datasets. We anticipate that this strategy can improve reproducibility in single-cell analysis in general and particularly in fibroblasts, whose phenotypes are often difficult to identify with one or two canonical marker genes. To promote reproducible research and cross-disease insights into fibroblast biology, we made the fibroblast atlas (github.com/immunogenomics/fibroblastlas) and the tools needed to map data (github.com/immunogenomics/symphony) publicly available.
Fibroblasts are essential players in inflammatory disease, fibrotic disease, and cancer. The potential to target fibroblasts therapeutically is growing with the number of single-cell and functional studies on fibroblast heterogeneity.66,67 Although early studies of fibroblast heterogeneity focused on positional identity, more recent studies have focused on functional states that mediate pathological processes. Our study provides the first cross-tissue analysis that rigorously distinguishes tissue-specific from tissue-shared identity in fibroblasts. We described two fibroblast states that may be universal to inflammatory disease across tissues. We created the first single-cell reference atlas of fibroblast heterogeneity to unify fibroblast research and prevent a confusing sprawl of fibroblast names across disciplines. The next critical step is to define how these fibroblast states behave in different clinical contexts and how they respond to the wide range of therapeutic agents available for immune-mediated inflammatory diseases. In our study, although many individuals were actively receiving immune-modulatory therapeutic agents (Tables S2, S3, S4, and S5), we did not have sufficient power in the design to look for systematic patterns with these therapeutic agents. Future studies that control for therapeutic intervention can use our atlas to identify which fibroblast states are associated with particular interventions and potentially find a stromal basis to explain the heterogeneous clinical response to immune-modulatory drugs. Moreover, our finding that C4 vascular fibroblasts are expanded in inflammation (Figure 3D) in fibrosis (Figure S8I) suggests that the same fibroblast states in our atlas can be relevant to multiple pathological processes and targeted to treat multiple, diverse indications. Finally, we proposed an analytical pipeline for studying shared pathological processes across diseases that can readily be applied to all cell types and tissues.
Limitations of the study
We would like to emphasize several limitations of our study. First, as detailed above, our definition of inflammation score is based on total leukocyte frequency within each sample. Although this definition allowed us to create a tissue-independent score to perform cross-tissue meta-analyses, it also limits our ability to correlate fibroblast phenotypes with more subtle and functionally specific aspects of inflammation. The second limitation of our study arises from the inability of scRNA-seq to capture certain key populations of cells, such as neutrophils, mast cells, and adipocytes, which do not survive the tissue disaggregation and encapsulation procedures of the droplet-based scRNA-seq pipeline. It is also well known that large cells that cannot effectively fit into a droplet, such as muscle cells and nerves, are not captured by droplet-based scRNA-seq. It is possible that certain fibroblast states may fall into this category and are thus not represented in our fibroblast atlas. The third limitation of our study is in the sample size of our cohort. Although 74 donors represent a sizeable scRNA-seq resource, this sample size is not powered to correlate molecular results with demographic and clinical features, such as sex, age, and medication status.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Soumya Raychaudhuri soumya@broadinstitute.org.
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-sequencing data have been deposited in GEO, Broad’s Single Cell Portal, and NIAID ImmPort. High dimensional images for Cell Dive experiments have been deposited in NIAID ImmPort. Accession numbers are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EPCAM/CD326, clone 9C4, Mouse IgG2b | Biolegend | Cat# 324233 |
| CD45, clone HI30, mouse IgG1 | Biolegend | Cat# 304005 |
| CD31, clone WM59, mouse IgG1 | Biolegend | Cat# 303133 |
| CD146, clone P1H12, Mouse IgG1 | Biolegend | Cat# 361003 |
| PDPN, clone NZ-1.3, rat IgG2a | eBioscience/ThermoFisher | Cat #46-4321-82 |
| CD90, clone 5E10, mouse IgG1 | Biolegend | Cat# 328109 |
| Alexa Fluor® 647 - Mouse monoclonal (D2-40) anti-Podoplanin (Lymphatic Endothelial Marker) antibody | Biolegend | Cat# 916610 RRID : AB_2810816 |
| BSA and Azide free -Recombinant Rabbit monoclonal [EPR5480] Anti PDGFR alpha | Abcam | Cat# ab248689 RRID : N/A |
| Alexa Fluor® 488 - Recombinant Rabbit monoclonal [EPR3208] Anti-CD146 antibody | Abcam | Cat# ab196448; RRID:AB_2868591 |
| BSA and Azide free – Recombinant Rabbit monoclonal [EPR3132] Anti CD90 / Thy1 antibody | Abcam | Cat# ab181885 RRID : N/A |
| Alexa Fluor® 555 – Recombinant Rabbit monoclonal [EPR20545] Anti-CD68 antibody | Abcam | Cat# ab280860 RRID : N/A |
| BSA and Azide free - Recombinant Rabbit monoclonal [SP162] Anti-CD3 antibody | Abcam | Cat# ab245731 RRID : N/A |
| Alexa Fluor® 647 – Mouse monoclonal (JC/70A) anti CD31/PECAM-1 antibody | Novus | Cat# NB600-562AF647 RRID: N/A |
| Alexa Fluor® 647 – Mouse monoclonal (C31.3) anti CD31/PECAM-1 antibody | Novus | Cat# NB P2-33154AF647 RRID: N/A |
| BSA and Azide free – Recombinant Rabbit monoclonal [SP205] Anti-SPARC antibody | Abcam | Cat# ab245733 RRID: N/A |
| Unconjugated - Goat Polyclonal Anti-Human Ccl19 / mip-3 beta antibody | R and D systems | Cat# AF361; RRID:AB_355323 |
| Alexa Fluor® 488 – Mouse monoclonal [1A4] Anti-alpha smooth muscle Actin antibody | Abcam | Cat# ab184675; RRID:AB_2832195 |
| Alexa Fluor® 647 Mouse monoclonal (2D1) anti-human CD45 antibody | BioLegend | Cat# 368538; RRID:AB_2716028 |
| Alexa Fluor® 647 Mouse monoclonal (C8/144B) anti-human CD8a antibody | BioLegend | Cat# 372906; RRID:AB_2650712 |
| Alexa Fluor® 488 Recombinant Rabbit monoclonal [EP1628Y] Anti-Cytokeratin 8 antibody | Abcam | Cat# ab192467; RRID:AB_2864346 |
| Alexa Fluor® 555 Recombinant Rabbit monoclonal [EPR3776] Anti-Vimentin antibody - Cytoskeleton Marker | Abcam | Cat# ab203428 RRID : N/A |
| Alexa Fluor® 488 Mouse monoclonal (TAL 1B5) Anti-HLA-DR Antibody | Santacruz | Cat# sc-53319AF488 RRID N/A |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447; RRID:AB_2535864 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572; RRID:AB_162543 |
| CD16/CD32 Monoclonal Antibody (93) (FcR block) 1:200 | ThermoFisher Scientific | 14-0161-86 |
| FITC EpCAM (G8.8) 1:200 | Biolegend | 118208 |
| PE-Cy7 Podoplanin (8.1.1) 1:200 | Biolegend | 127412 |
| APC CD140a (APA5)1:200 | Biolegend | 135907 |
| BV605 CD31 (390) 1:200 | Biolegend | 102427 |
| BV711 CD45 (30-F11) 1:200 | Biolegend | 103147 |
| BV711 CD45 (30-F11) 1:200 | Biolegend | 103147 |
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis H37Ra | BD Difco | BD 231141 |
| Chemicals, peptides, and recombinant proteins | ||
| Glycerol | Sigma-Aldrich | Cat# G5516 RRID N/A |
| Trizma®base | Sigma-Aldrich | Cat# T6066 RRID N/A |
| Sodium bicarbonate | Sigma-Aldrich | Cat#S6297 RRID N/A |
| Tween20 | Sigma-Aldrich | Cat#P9416 RRID N/A |
| Triton X-100 | Sigma-Aldrich | Cat#T9284 RRID N/A |
| Propyl gallate | Sigma-Aldrich | Cat#02370 RRID N/A |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A7906 RRID N/A |
| Target Retrieval Solution 10X Concentrate | Dako | Cat#S1699 |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | Cat#E9884 |
| eBioscience Fixable Viability Dye eFluor 780 1:1000 | ThermoFisher Scientific | 65-0865-18 |
| DAPI Solution 50 pg/mL | ThermoFisher Scientific | 62248 |
| 2.5% w/v dextran sulfate sodium | MP Biomedicals | 216011080 |
| Liberase TL | Roche | 5401020001 |
| DNase 1 | Sigma | 11284932001 |
| Quick-RNA 96 Kit | Zymo Research | R1052 |
| Donkey Serum | Biorad | Cat# C06SB RRID N/A |
| Hydrogen Peroxide Solution | Sigma-Aldrich | Cat#216763 RRID N/A |
| FcR Blocking Reagent | Miltenyi | Cat# 130-059-901; RRID:AB_2892112 |
| Bovine Tyope II Collagen | Richard Williams https://doi.org/10.1385/1-59259-771-8:207 |
N/A |
| Critical commercial assays | ||
| Dynabeads Human T-activator CD3/CE28 for T cell expansion and activation | ThermoFisher | Cat# 11132D |
| 10× Genomics Chromium Single Cell 3’ (v2 and v3 chemistry) | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/technical-note-assay-scheme-and-configuration-of-chromium-single-cell-3-v2-libraries |
| Cell Dive | General Electric | https://www.ge.com/research/project/multiplexed-tissue-imaging-platform |
| CD45RA MicroBeads (human) | Miltenyi | Cat# 130-045-901 |
| Pan T Cell Isolation Kit (human) | Miltenyi | Cat# 130-096-535 |
| LS columns | Miltenyi | Cat# 130-042-401 |
| Deposited data | ||
| CellDive imaging data: https://www.immport.org/shared/study/SDY1765 | This paper | ImmPort:SDY1765 |
| Human fibroblast atlas: https://sandbox.zenodo.org/record/772596#.YGsi6BRKg-Q | This paper | https://doi.org/10.5072/zenodo.772596 |
| Atopic dermatitis scRNAseq | He et al., 2020a | GSE147424 |
| Serum transfer arthritis scRNAseq | Wei et al., 2020 | GSE145286 |
| DSS induced colitis scRNAseq | Kinchen et al., 2018 | GSE114374 |
| bleomycin induced lung injury scRNAseq | Tsukui et al., 2020 | GSE132771 |
| Adult human cell atlas | He et al., 2020b | GSE159929 |
| Tabula sapiens: https://figshare.com/projects/Tabula_Sapiens/100973 | The Tabula Sapiens Consortium and Quake, 2021 | N/A |
| Healthy lung atlas scRNAseq | Travaglini et al. | Synapse:syn21041850 |
| IPF lung cohort scRNAseq | Adams et al. | GSE136831 |
| Healthy gut atlas scRNAseq | Elmentaite et al., 2021 | E-MTAB-9536 |
| Raw human sequencing data: https://www.immport.org/shared/study/SDY1765 | This paper | ImmPort:SDY1765 |
| Processed human sequencing data:https://singlecell.broadinstitute.org/single_cell/study/SCP738 | This paper | BroadSingleCellPortal:SCP738 |
| Raw and processed mouse sequencing data | This paper | GSE185711 |
| Experimental models: Cell lines | ||
| Primary lung fibroblast cell lines from earlier-stage fibrotic interstitial lung disease | This paper | Lung fibroblast cell lines named V3, V6, V7, V11. |
| Primary Synovial fibroblasts | This paper | synovial fibroblast cell lines named: STB-009, RA200212, AMP-005 |
| Primary human T cells isolated from leukopak mononuclear cells | This paper | N/A |
| Collagen Induced Arthritis | https://doi.org/10.1385/1-59259-771-8:207 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000,664 |
| Mouse: DBA/1J | MRC Harwell | N/A |
| Software and algorithms | ||
| Code to Reproduce Analyses and Figures for Fibroblast Atlas 2022 | This Paper | https://doi.org/10.5281/zenodo.6510339 |
| Symphony v1.0 | Kang et al., 2021 | https://github.com/immunogenomics/symphony |
| DeepCell | Greenwald et al., 2021 | https://github.com/vanvalenlab/deepcell-tf |
| uwot v0.1.10 | Melville et al., 202068 | https://github.com/jlmelville/uwot |
| arm v1.11.2 | Gelman and Su, 202093 | https://github.com/gelman/arm |
| lme4 v1.1.27.1 | Bates et al., 2015 | https://github.com/lme4/lme4 |
| presto | This paper | https://github.com/immunogenomics/presto/tree/glmm |
| fgsea v1.18.0 | Sergushichev, 2016 | https://github.com/ctlab/fgsea |
| msigdbr v7.4.1 | Dolgalev, 201894 | https://github.com/igordot/msigdbr |
| scanorama v1.7.1 | Hie et al., 2019 | https://github.com/brianhie/scanorama |
| bbknn v1.5.1 | Polański et al., 2020 | https://github.com/Teichlab/bbknn |
| scvi-tools v0.6.8 | Lopez et al., 2018 | https://github.com/scverse/scvi-tools |
| scanpy v1.7.1 | Wolf et al., 2018 | https://github.com/scverse/scanpy |
| VGAM v1.1.5 | Yee, 2010 | https://github.com/cran/VGAM |
| limma v3.48.3 | Ritchie et al., 2015 | https://github.com/cran/limma |
| kallisto | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| bustools | Melsted et al., 2019 | https://github.com/BUStools/bustools |
| harmony v1.0 | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony/tree/weights |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human research and sample acquisition
Synovial study samples for transcriptomic and imaging studies were obtained from Brigham and Women’s Hospital, Hospital for Special Surgery, and the University of Birmingham under IRB-approved protocols. Synovial tissue from patients with clinically diagnosed rheumatoid arthritis were obtained from ultrasound-guided joint biopsy (University of Birmingham, BEACON cohort ethics number 07/H1203/57) or arthroplasty or synovectomy procedures (Brigham and Women’s Hospital, Hospital for Special Surgery, and The Royal Orthopaedic Hospital (Birmingham)). For arthroplasty and synovectomy tissue samples, the diagnosis of rheumatoid arthritis was confirmed clinically through clinical chart review. Synovial tissue from patients with osteoarthritis were obtained from arthroplasty procedures. Synovial tissues were cryopreserved on-site in Cryostor CS10, then shipped to BWH under a BWH IRB-approved protocol PROSET for tissue dissociation and single-cell transcriptomic analysis.
Intestinal samples were obtained from Ulcerative colitis (UC) or from healthy individuals by endoscopic biopsy. Healthy patients were recruited as a part of the research tissue bank ethics 16/YH/0247 and Inflammatory Bowel Diseases (IBD) patients among the Inflammatory Bowel Cohort 09/H1204/30 by the Translational Gastroenterology Unit Biobank at the John Radcliffe Hospital in Oxford. All patients gave informed consent and collection was approved by NHS National Research Ethics Service. Samples were immediately placed on ice (RPMI1640 medium) and processed within 3 h.
Labial minor salivary gland samples were obtained from patients recruited in the Optimising Assessment in Sjögren’s Syndrome (OASIS) cohort69 which recruits new patients attending the multidisciplinary Sjögren’s clinic at the Queen Elizabeth Hospital Birmingham, UK for assessment. Sjögren’s syndrome patients had a physician diagnosis of primary Sjögren’s syndrome and fulfilled the 2016 ACR/EULAR classification criteria. Participants with non-Sjögren’s sicca syndrome had signs and/or symptoms of dryness but did not have a physician diagnosis of SS or fulfill 2016 classification criteria. Salivary gland biopsy samples were divided in two: one for the scRNAseq study and the second for histological analysis to confirm diagnosis. Histological diagnosis is summarized in Supplemental Data: Table S4 and reported as presence of focal lymphocytic sialadenitis (FLS, suggestive of Primary Sjögren’s Syndrome, PSS) or non-specific chronic sialadenitis (NSCS), in the case of non-Sjögren’s sicca syndrome. Focus score (FSC, number of inflammatory foci/4mm2 of tissue) is also reported in Table S1. All OASIS participants provided written informed consent and the study was approved by the Wales Research Ethics Committee 7 (WREC 7) formerly Dyfed Powys REC; 13/WA/0392.
Lung samples were obtained from patients recruited at the Brigham and Women’s Hospital with informed consent under MGB IRB protocols 2014P002558 and 2019P003592 approved by the Mass General Brigham IRB (PROSET). As enumerated in Supplemental Data: Table S2, samples coded Lung1–15, which included control donor lung and later-stage ILD diagnoses (IPF, Rheumatoid Arthritis [RA]-ILD) were explants from lung transplant surgery. Samples coded Lung 16–23 (unclassifiable (u)ILD, IPF, NSIP), which constitute the earlier-stage ILD subcohort, were from Video-assisted thoracoscopic surgical (VATS) lung biopsies for diagnosis of ILD. No patients with earlier-stage ILD (n = 8) had a clinical requirement for supplemental oxygen, whereas all patients with late-stage ILD (n = 11) required supplemental oxygen at time of enrollment. Earlier disease defined as requirement of VATS for diagnosis of ILD and no requirement for supplemental O2. Later disease is defined as lung explanted for lung transplantation. For patients with pulmonary function testing within six months prior to enrollment, TLC and DLCO statistics were collected. The patient condition is the diagnosis determined by clinical providers after their inter-disciplinary review of patient history, exam, clinical laboratory testing (e.g., serologies), imaging and histopathology of the explanted or biopsied lung tissue. The presence or absence of anti-CCP antibodies is noted.
Where possible, patient data on sex, gender, and age were gathered from clinical records. Data on socioeconomic status, race/ancestry, and ethnicity were not reported in this study.
METHOD DETAILS
Cell isolation for single-cell RNA-sequencing
Synovial tissues were cryopreserved on site, thawed and disaggregated into single-cell suspension as previously described.70 Four pairs of intestinal biopsies were pooled, minced and frozen in 1 mL of CryoStor® CS10 (StemCell Technologies) at −80°C then transferred in LN2 within 24 h. Single-cell suspensions from these endoscopic biopsies were then prepared by thawing, washing and subsequent mincing of the tissue using surgical scissors. Minced tissue was then subjected to rounds of digestion in RPM-1640 medium (Sigma) containing 5% Fetal Bovine Serum (FBS, Life Technologies), 5 mM HEPES (Sigma), antibiotics as above, and Liberase TL (Sigma), with DNAse I. After 30 min, digestion supernatant was taken off, filtered through a cell strainer, spun down, and resuspended in 10 mL of PBS containing 5% BSA and 5 mM EDTA. Remaining tissue was then topped up with fresh digestion medium until no more cells were liberated from the tissue. Cells were then stained and FACS-sorted for live EPCAM−CD45− cells, before being taken for microfluidic partitioning.
Lung tissues were cryopreserved on site, thawed and disaggregated into single-cell suspension. Each lung tissue was frozen in 1 mL of CryoStor CS10 in −80 °C with a controlled rate of freezing and then transferred to LN2 within two weeks. On the day of single-cell analysis, the cryopreserved lung tissue was rapidly thawed, serially rinsed with DMEM (GIBCO) supplemented with 10% FBS and then DMEM with 2% FBS on ice. Lung tissue was minced using surgical scissors and then transferred to a polypropylene tube with digestion media containing Liberase TL, hyaluronidase (Worthington Biochemical Corporation), Elastase (Worthington Biochemical Corporation), DNAse (Sigma) and 1% FBS. The addition of FBS improved cell viability without reducing yield of viable stromal cells. After 20 min of incubation at 37° C warm room with agitation by stir bar, the supernatant containing single cells was collected, and fresh digestion media was added. After 20 min of addition digestion, the tissue and supernatant were filtered through a 70 micron cell strainer and washed in DMEM with 2% FBS twice. Dead cells were removed using a magnetic column based method per manufacturers protocols (Dead Cell Removal kit, Miltenyi Biotec). Then single cells were taken for microfluidic partitioning.
Minor salivary gland biopsies were taken surgically from the lip and frozen in 1 mL of CryoStor® CS10 (StemCell Technologies) at −80°C. For preparation of single-cell suspension, firstly the frozen tissue sample in Cryotube were quickly thawed in water bath at 37° C and washed twice in pre-warmed 5%FBS RPMI media. The salivary gland biopsies were then enzymatically digested as previously described (PMID: 31213547). Dead cells were removed using the EasySepTM Dead Cell Removal (Annexin V) kit from the digested samples following manufacturer’s instructions before proceeding for the scRNA sequencing using the 10× platform.
RNA-sequencing
Single-cell RNA-sequencing experiments for lung, intestine, and synovium samples were performed through the Brigham and Women’s Hospital Single Cell Genomics Core. Viable cells in single-cell suspension were resuspended in 0.4% BSA in PBS at a concentration of 1,000 cells per ul. 7,000 cells were loaded onto a single lane (Chromium chip, 10X Genomics) followed by encapsulation in lipid droplet, with the 10× Genomics Single-Cell 3′ kit (Version 2 for synovium and intestine, Version 3 for lung) followed by cDNA and library generation per manufacturer protocol. cDNA libraries were sequenced to an average of 50,000 reads per cell using Illumina Nextseq 500. Single-cell RNA-sequencing experiments for salivary gland samples were performed at Oxford University. For each library, 10,000 cells were counted using the automated cell counter Bio-Rad TC20 and loaded onto a single 10× lane and processed with the 10× Genomics Single Cell 3′ kit (Version 3). Sequencing was done using Illumina NovaSeq 6000 and libraries were sequenced to a minimum of 50000 reads/cell.
In vitro fibroblast studies
Early passage synovial and lung fibroblast cell lines (passage 3 to 5) derived from rheumatoid arthritis and ILD patients, respectively, were used for in vitro functional experiments. Human Umbilical Vein Endothelial Cells (HUVECs) were purchased from Lonza and expanded in the presence of growth factors (EGM-2, Lonza). Unmanipulated non-naïve human T cells were isolated using a Miltenyi Pan T Cell Isolation Kit supplemented with CD45RA MicroBeads, yielding >95% CD45RO+ T cells. T cells were resuspended in DMEM media containing 1% fetal calf serum and transferred to multiple wells of a 96-well flat-bottom plate at 100,000 cells/well. The cells were then stimulated for 16 h with Dynabeads Human T-Activator CD3/CD28 beads (ThermoFisher) at a 1:1 ratio of beads to cells. Supernatants were harvested and pooled, and any remaining T cells were removed by centrifugation. For fibroblast-endothelial co-culture experiments, 3 synovial fibroblasts cell lines and 3 ILD fibroblast cell lines were co-cultured with HUVECs at a 2:1 ratio for 7 days. Fibroblasts were serum-starved for 24 h followed by incubation with undiluted supernatants from activated T cells for 24 h prior to harvest for sorting and single-cell RNA-seq.
Single cell RNA-seq experiments were performed by the Brigham and Women’s Hospital Single Cell Genomics Core. Cells were trypsinized and stained with Fixable Viability dye (ThermoFisher), anti-CD31 (clone wm59, Biolegend), anti-CD3 (clone UCHT1, Biolegend), and a unique barcoded antibody (Cell-hashing antibody, TotalSeq-A, Biolegend) as previously described. For scRNAseq analysis of the fibroblasts, viable, CD31−, and CD3− cells from organoids were isolated by FACs. Next, 3,000 CD31−CD3− cells from each condition were resuspended in 0.4% BSA in PBS at a concentration of 1,000 cells per μL, pooled together, then loaded onto a single lane (Chromium chip, 10X Genomics) followed by encapsulation in a lipid droplet (Single Cell 3′kit V3.1, 10X Genomics) followed by cDNA and library generation according to the manufacturer’s protocol. mRNA libraries were sequenced to an average of 50,000 reads per cell and HTO (Cell Hashing antibodies) libraries sequenced to an average of 5,000 reads per cell, both using Illumina Novaseq. ADT reads from scRNA-seq reads were processed with Cell Ranger v3.1, which demultiplexed cells from different samples. Gene quantification was performed using the kallisto and bustools pipeline, as described above.
Collagen-induced arthritis mouse model
For collagen induced arthritis, bovine type II collagen (CII; generously provided by Prof. Richard Williams, Kennedy Institute Oxford) was dissolved in 0.1 M acetic acid at 4 mg/mL. Complete Freund’s adjuvant (CFA) was generated using incomplete Fruend’s adjuvant (IFA; BD Difco, #BD263910) containing Mycobacterium tuberculosis H37Ra (4 mg/mL; BD Difco, #BD231141). Male DBA/1 mice were immunised with 200 μg of CII emulsified 1:1 in CFA and were boosted 21 days later with 200 μg CII in IFA. Onset of arthritis was determined by a paw score ≥2. 5–7 days following onset, mice were culled and inflamed rear limbs were harvested. Mouse rear limb were dissected and bones (including femur, tibia, fibular; and rear foot bones calcaneus, tarsals, metatarsals and phalanges) with intact joint tissue were transferred into RPMI-1640 media (+2% FCS) containing 0.1 g/mL Collagenase D (Roche), 0.01 g/mL of DNase I (Sigma-Aldrich). Samples were incubated at 37° C, 45 min, followed by a second incubation with RPMI-1640 media (+2% FCS) containing 0.1 g/mL Collagenase Dispase (Roche) and 0.01 g/mL DNase I at 37° C for 30 min. Cells were labelled with anti-mouse CD45 APC-cy7 (1/500, 30-F11; BioLegend, #103116) and 7-AAD (7-Aminoactinomycin D, 1/1000; ThermoFisher, #A1310) viability dye. From this, live, CD45 negative cells were sorted using a MoFlow Astrios EQ (100 μM nozzle size). Cells were counted and loaded into 10X chromium controller for a 5000 cell target recovery.
DSS colitis time course
Animal experiments were carried out under the relevant Home Office license at the University of Oxford (PPL: P508FFA1F). Female 7-week–old C57BL/6J mice were obtained from The Jackson Laboratory and housed in ventilated cages with 12-h light cycles under specific pathogen-free conditions (SPF). They received food and water ad libitum. For induction of colitis, 2.5% w/v dextran sulfate sodium (DSS; MP Biomedicals) was supplemented in drinking water and given to mice for six consecutive days, with renewal on day three. After the treatment was ceased, mice returned to receiving standard water for the remaining time. Mice were monitored every day for alterations in body weight and clinical disease scores, until euthanised with carbon dioxide. Colons were resected, cleaned and washed in RPMI1640 supplemented with 5% fetal calf serum (FCS), 1% Penicillin-Streptomycin and 5 mM EDTA for two total washes of 30 and 20 min respectively at 37° C to remove bulk epithelium. This was followed by tissue digestion using 100 μg/mL Liberase TL and 40 μg/mL DNAse for 3 × 1-h cycles to dissociate all tissue into single-cell suspension. These were then stained with relevant reagents and antibodies (below) and FACS sorted (BD FACSAria III and BD FACSDiva 8.0.1) for live fibroblasts on CD45 −ve, EpCAM −ve, CD31 −ve, Podoplanin +ve, Pdgfra +ve cells at the time points described. Cells were directly sorted into RNA lysis buffer and RNA was isolated using Zymo Quick RNA 96 kits as per the manufacturer’s instructions. Libraries were prepared using NEBNext ultra-low input RNA library prep with 1 ng of RNA per sample and sequenced on a NovaSeq6000 (150 paired-end).
High-dimensional proteomics imaging using cell DIVE
Slide clearing and blocking.
Formalin-Fixed Paraffin Embedded (FFPE) tissues slides from synovium, intestine, salivary glandy, and lung were deparaffinised and rehydrated. The slides were then permeabilised for 10 min in 0.3% Triton X-100 and washed further in 1× PBS for 5 min. Antigen retrieval was performed using the NxGen decloaking chamber (Biocare Medical, Pacheco, CA, USA) in boiling pH6 Citrate (Agilent, S1699) and pH9 Tris-based antigen retrieval solutions for 20 min each. Tissue slides were blocked in 1xPBS with a 3% BSA (Merck, A7906), 10% Donkey serum (Bio-Rad, C06SB) and FcR Blocking Reagent, human (Miltenyi, 130-059-901, 1:200 dilution) solution for 1 h at room temperature. Slides were washed in 1xPBS for 10 min and then stained with DAPI (Thermo, D3571) for 15 min. Slides were washed in 1xPBS for 5 min and coverslipped with mounting media (50% glycerol – Sigma, G5516 and 4% propyl gallate – Sigma, 2370).
Scan plan and background acquisition.
The GE Cell DIVE system71 was used to image all FFPE slides. A scan plan was acquired at 10X magnification to select regions of interest followed by imaging at 20X to acquire background autofluorescence and generate virtual H&E images. Background imaging is used to subtract autofluorescence from all subsequent rounds of staining. Slides are decoverslipped in 1xPBS prior to staining.
Staining and bleaching.
Each staining round consisted of a mix of 3 antibodies prepared in blocking buffer (PBS, 3% BSA, 10% donkey serum, FcR blocking Re-agent). The initial round used primary antibodies which were incubated overnight at 4C° followed by 3× washes in 1xPBS and 0.05% Tween20 (Sigma P9416). Secondary antibodies raised in Donkey were then incubated for an additional hour at room temperature which were either conjugated to Alexa Fluorophore 488, 555 or 647 (Invitrogen). Each subsequent staining round used directly conjugated antibodies to either of these dyes (Antibodies list in table below) and were incubated overnight at 4C° or for an hour at room temperature. Antibodies manually conjugated were purchased in a BSA-AZIDE free format and conjugated using antibody labelling kit (Invitrogen).
Fluorophores were bleached between each staining round using NaHCO3 (0.1 M, pH 11.2. Sigma - S6297) and 3% H2O2 (Merck – 216763) (Gerdes, Sevinsky et al. 2013). Fresh bleaching solutions were prepared and slides were bleached 2 times (15 min each) with a 1 min 1xPBS wash in between bleaching rounds. Slides were re-stained for DAPI for 2 min and washed in 1xPBS for 5 min before imaging the dye-inactivated round as the new background round (for subsequent background subtraction). DAPI staining between imaging rounds assists in image registration and alignment. Slides were multiplexed with the next panel of three markers with iterative staining, bleaching and imaging.
Lung slides histopathology scoring
The presence and quantity of interstitial lymphocytes were assessed using a standard Olympus Bx50 microscope on 4 uM hematoxylin and eosin-stained sections prepared from formalin-fixed paraffin-embedded tissue. Interstitial lymphocytes only were counted manually using a cell counter in 50 high power fields (HPF; 400× magnification), by a pathologist with expertise in pulmonary pathology and idiopathic interstitial lung diseases. Total lymphocytes in 50 HPF were averaged to number of lymphocytes per 2mm2 for comparison between samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
scRNAseq gene quantification
For all scRNAseq datasets analyzed in this manuscript, we quantified gene expression ab initio from FASTQ files. Human reads were mapped to the GRCh3872 reference and genes annotated with Gencode73 v33. Mouse reads were mapped to mm10 reference and genes annotated with Gencode v25. For both human and mouse data, we filtered transcripts for the annotation “protein_coding” and ignored the rest. Reads from distinct transcripts of the same gene were collapsed by summation. We used kallisto74 v0.46.0 to map reads to transcriptomes and bustools75 v0.39.3 to collapse duplicate reads by UMI and return gene-cell count matrices. We downloaded read level data for the following publicly available scRNAseq datasets: PRJNA61453946 (atopic dermatitis), PRJNA5423508 (DSS model), and PRJNA54894747 (Bleomycin model). After contacting the authors, the PRJNA542350 data turned out to be BAM files rather than FASTQ. Per their suggestion, we used the 10X Cell Ranger76 bamtofastq utility (version 1.3.2), with default parameters, to convert the BAMs back into FASTQs for remapping. doc. The code to perform all steps of this mapping are implemented as functions in the github repository for this manuscript.
scRNAseq quality control, pre-processing, and normalization
After quantifying gene count matrices with kallisto and bustools (above), we filtered out poor quality cells with three metrics. (1) Cells must have at least 500 unique genes. (2) Cells must have more than 20% of the total UMIs mapped to non-mitochondrial genes. (3) Cells must be inferred as singlets by algorithmic doublet identification. For doublet identification, we used the scDblFinder algorithm, with default parameters, separately within each 10X library. We normalized for read depth with the standard logCP10K normalization procedure for gene g and cell i:
Inflammation score normalization across tissues
Inflammation scores computed within each tissue had ranges and distributions. To be able to compare inflammation associated phenotypes across tissues, we normalized the distributions by performing quantile normalization. Because the number of samples was relatively small, we did not use an empirical distribution. Instead, we normalized to the quantiles of a parametric distribution. We chose the beta distribution (α = 3, β = 3) to map the scores to an interpretable interval, between 0 (low inflammation) and 1 (high inflammation).
Gene selection
For analyses with one tissue, we used the VST method for variable gene selection, reimplemented from the Seurat package24 as a stand along function in our github at immunogenomics/singlecellmethods. We used default parameters and kept the top 2000 genes, ranked by standardized variance. For the multi-tissue integrated analysis, we used genes that we found informative in at least one of the tissue-specific analyses of lung, salivary gland, intestine, and synovium. We defined informative genes with two analyses. The first analysis is differential expression of cluster-markers for tissue-specific fibroblast subtypes (Figure 4A). We kept cluster-informative genes with p < 0.05 and |β| ≥ 0.5. The second analysis found broadly inflammation associated genes by fitting a Poisson log-normal GLMMs to each gene. We kept inflammation associated genes with p < 0.05 and |β| ≥ 0.1.
Weighted PCA
We implemented principle components analysis that gives equal weight to each tissue while preserving the total cell number . The weights given to each cell were determined to meet this equal weight condition. These weights were then used in the scaling and SVD steps. For scaling, we computed weighted means and variance with the following formulas: , . For SVD, we modified the PCA covariance decomposition formula to allow for observation weights with a diagonal matrix W : XWXT = UDUT. This decomposition is achieved by performing SVD on the weighted matrix XW1/2 = UDVT. Because W is diagonal, its square root is the element-wise square root. This SVD solution now represents the original data as X = UDVTW−1/2, with gene loadings U and cell embeddings VTW−1/2. Weighted PCA is implemented on our github at immunogenomics/single-cellmethods with the weighted_pca function.
Weighted Harmony
We modified the Harmony algorithm to include observation weights. To achieve this, we modified the clustering objective function and rederiving the optimization steps for this function. The new objective function modifies the original only by multiply the per-cell cost (inside the summation) by . The rest of the formula is unchanged and described in detail in the original Harmony manuscript.22 This modified Harmony implementation is available on our github at immunogenomics/harmony, under the weights branch.
UMAP visualization
We used the UMAP algorithm to visualize cells in two dimensional embeddings. We used the uwot R package with parameters n_neighbors = 30L, metric = ‘Euclidean’, init = ‘Laplacian’, spread = 0.3, min_dist = 0.05, set_op_mix_ratio = 1.0, local_connectivity = 1L, repulsion_strength = 1, and negative_sample_rate = 1. For all other parameters, we used default values. In the symphony pipeline, we visualized mapped query cells by using the UMAP object learned for the reference analysis. The umap reference projection was done with the umap_transform function in uwot.
Clustering
We performed graph based clustering with the Louvain algorithm,77 implemented in Seurat.24 Instead of constructing the kNN and sNN graphs from scratch, we used the uniform manifold graph estimated in the UMAP algorithm. In the uwot package, this data structure is directly available in the fgraph field when umap is run with option ret_extra = c(‘fgraph’).
Hierarchical gene expression modelling
Statistical model.
We modeled the expression of each gene using Poisson lognormal GLMM regression. This framework allows us to model the hierarchical design in our multi-tissue, multi-donor dataset. We fit the following GLMM for the integrated, multi-tissue analysis, regressing to the frequency of gene g in observation i.
We chose to model the cluster interaction terms with donor and tissue. As many papers have observed,22,78 the effect of biological and technical covariates are often cell type specific. This is why integration algorithms cannot adjust every cell type by the same amount to account for batch, donor, or tissue variability. Unfortunately, the absence of some donors and tissues in some clusters means that interaction terms may be very poorly estimated. To address this issue, we model all terms except for the global intercept (β0) with Gaussian priors, allowing each effect to have a different size, denoted by τ2, the variance of the priors. These priors shrink β s toward zero, stabilizing estimation for terms with little data to draw from.
We performed cluster marker analysis with the estimated β s, estimating both marginal effects and tissue-specific effects. Marginal cluster effects are only concerned with the βCluster term. For instance, the differential expression for cluster 3 is . This comparison can be compactly repre sented with the contrast vector such that the differential expression can be computed with the linear operation . Following the example of significance testing in DESeq2, the standard errors of contrasts are in the diagonal elements of , in which Σ is the covariance matrix of β levels. In our example, Σ is a cluster by cluster covariance matrix and the standard error for cluster 3 would be . There is generally no analytical way to compute Σ for random effects, so we estimate it with simulation, using the arm R package,79 with 1000 simulations. Tissue-specific cluster effects take into account both the cluster and tissue-cluster interaction term. For instance, if we wanted to know how a gene is associated with cluster 3 in the lung, we would compute . The contrast vector now includes terms that represent the β s estimated for lung tissue as well. The statistical procedures to compute and are the same as before. For both marginal and tissue-specific effects, we use a Gaussian approximation to estimate p values for each effect: βDGE ~ N(Δβ, ΔΣClusterΔT).
Implementation.
We fit GLMMs with the glmer function in the lme4 R package80 and estimated random effect covariance with the sim function in the R arm package.79 Initially, we found it difficult to tie model fitting and simulation seamlessly with differential expression analysis. For instance, building contrasts for nested effects and estimating significance for multiple gene queries was difficult to do. Moreover, the memory footprint of lme4 models makes it impractical to fit and save models for 1000 s of genes for downstream inference. To make lme4 and arm more accessible for gene expression analysis, we created the Presto package. Presto extracts the necessary components from lme4 models, saves them in efficient data structures, and has all necessary functions to do efficient contrast analysis for differential expression. We made Presto available as an R package, available on github at immunogenomics/presto under the GLMM branch.
To make the models more numerically stable, we enforced a minimum value for the size of random effects: σ ≥ 0.5. This prevented degenerate solutions with σ = 0, local minima which may arise in GLMM optimization. As a side effect, this Bayesian variance prior also enforces a conservative null model on random effects, effectively setting the null effect size to 0.5 rather than 0. This results in higher estimated uncertainty thus more conservative p values. In developing this software, QQ plot analysis was deflated and resembled post-hoc adjusted (e.g. Bonferroni) p values more than nominal p values from independent tests. Others have noted a similarity between post hoc correction and shrinkage integrated into the model.81 For our analyses, we consider significance with respect to these shrunken p values, estimated with random effects, without doing additional post hoc shrinkage.
We made two decisions to make Presto scale to large datasets. First, we fit the model with pseudobulk, rather than single-cell RNAseq profiles. Note that in the formula above, the cluster, tissue, and donor covariates are not unique to single cells. Therefore, we collapse reads from cells with same cluster, donor, and tissue identity into one observation. This approach has strong precedent.82 It is important to note that in this strategy, the number of parameters to estimate is equal to the number of observations. With fixed effects, this model is under-determined. However, because we shrink estimates to 0 with Gaussian priors, the effective number of independent parameters shrinks too. The second decision is with the choice of generative model. Many RNAseq differential expression tools used the Negative Binomial distribution, which uses Gamma rather than lognormal priors to model over-dispersion. For completeness, we also included negative binomial GLMMs in Presto. In practice, we found that this error model yielded almost identical results but took ten times longer to run.
Tissue heterogeneity.
We took a very simple approach to labeling genes as conserved or heterogeneous cluster makers. Conserved markers were significantly (p < 0.05) overexpressed (β > 0) in all four tissues. If a gene was not upregulated in at least one tissue, we considered it to be a heterogeneous marker. Effect heterogeneity has a rich statistical treatment, especially in meta-analysis. We decided to not use these more sophisticated techniques, although the parameters learned in Presto could be used for such analyses.
Analyses.
To find marker genes for dermal fibroblasts, we fit the same model as above but omitted the Tissue terms: . For the mouse scRNAseq analyses, we used the same hierarchical formula with all Tissue terms.
Pathway analysis
All formal geneset enrichment was done with the GSEA algorithm, implemented in the fgsea R package.83 To enrich pathways for marker analyses (Figure 5D), we used the H (hallmarks) and C5 (Gene Ontology) genesets from MSigDB, accessed with the msigdbr R package. To enrich for different phases of inflammatory response in DSS-induced colitis (Figure 7E), we used the published genesets, provided as supplemental materials in the manuscript.51
Abundance modeling
We associated inflammation score with cluster abundance using logistic regression, following the MASC method,85 with the following formula: . As in MASC, the response variable models the log odds of being in cluster k vs not, to test for which factors contribute to cluster k abundance. This probability is a function of (1) an intercept, which reflects the average abundance of cluster k in the data, (2) fixed effect for Score, the normalized inflammation score for each sample, (3) random effect for 10X library, to account for dependence of cells within a library, and (4) cell quality statistics MT (percent mitochondrial reads) and DS (doublet score), separately within each library. The association between inflammation and cluster abundance is captured in the β statistic. We computed significance for each β with the following Gaussian approximation, using the standard error σ provided by lme4: β ~ N(0, σ2): To combine MASC results from individual tissue analyses, we used inverse variance weighted meta analysis with random effects. The variance from random effects was estimated with the DerSimonian and Laird (DL) method.86,87
Cluster correspondence analysis
To compare the co-occurrence of the fibroblast cluster labels, within-tissue (Figure 3) and integrative (Figure 4), we used a similar framework to abundance modeling above. We used the following formula: . The contrast term of interest is the random effect (1|ClusterTissue), a categorical variable that encodes the within-tissue cluster identity. We chose to model this with a random effect for numerical stability. To estimate significance, we used Wald’s approximation and simulated covariance for the levels of (1|ClusterTissue) with the R arm package.
Symphony projection
The Symphony pipeline is described in detail in a separate manuscript.27 To infer reference cluster identity in query cells, we used a k-NN classifier. K = 10 nearest neighbors were estimated with Symphony projected low dimensional embeddings, based on cosine distance (σ = 0. 1).
Ligand receptor analysis
We started with a curated list of known interacting ligand-receptor pairs, from.39 To predict putative interactions between endothelial cells and fibroblast subsets, we performed differential expression on the pooled dataset of endothelial cells and fibroblasts. We filtered for differentially expressed genes and kept interaction pairs in which the ligand was overexpressed (p < 0.05, β > 0) in endothelial cells and the receptor in a fibroblast subset, or vice versa. For these pairs, we computed the interaction scores (Figure 4E) as the mean of the ligand’s and receptor’s z-scores.
Integration with alternative algorithms
We used the python packages Scanorama v1.7.1, bbknn v1.5.1, and scvi-tools v0.6.8 to integrate over donor effect within each tissue. We imported data and performed appropriate pre-processing with scanpy v1.7.188 and used default parameters for each integration method. Reproducible code for these analyses is available on the github repository associated with this manuscript (github.com/immunogenomics/fibroblastatlas2022).
Dynamic expression analysis of sorted fibroblast DSS time-course dataset
We identified genes with significant associations with time point using spline regression analysis. Specifically, we used the R VGAM package89 to construct a natural spline basis function with 4 degrees of freedom and used this non-linear expanded basis to perform association testing with linear regression using the limma package.90 Significant values were assessed with the F-statistic computed in limma, with a cutoff of FDR<20%. To identify association of gene expression with each time point, we repeated limma regression analysis, using time point as a categorical variable to compute log fold change at each time point versus healthy controls.
Cell Dive analysis
Segmentation and quantification.
We performed cell segmentation using the Deep Cell model, a pre-trained neural network specialized in segmentation of cytoplasm and nucleus of cells in tissue.91 For image-preprocessing, we used the CLAHE histogram normalization method recommended in91 and implemented in the python scikit-image library.92 We then produced two-channel images, one with nucleus intensity and one with cytoplasm and membrane intensity. For nucleus intensity, we used the DAPI intensity at the final round of imaging. For cytoplasm and membrane intensity, we averaged all remaining channels. After segmentation, we used the regionprops function from scikit-image to quantify cell location, cell area, and total intensity per cell. We removed cells whose area was too small (<50 pixels) and cells whose DAPI intensities at the first and last rounds of imaging were >50% discordant.
Spatial niche identification.
To identify spatial niches, we quantified the average intensity of each marker in each cell’s local neighborhood. The local neighborhoods were computed with Delaunay triangulation. These neighborhood-averaged marker intensities were normalized by cell area, z-scored with marker, processed with PCA, and projected into 2D space with UMAP. The neighborhood graph computed in UMAP was then used to perform graph-based clustering with the Louvain algorithm. Area-normalized marker intensities were associated with cluster identity using the Wilcoxon rank sum test. Cells from each tissue were analyzed separately. For tissues with more than 1 dataset (i.e. gut and synovium), we used Harmony to integrate over dataset identity after PCA.
Fibroblast subtype identification.
Cells were clustered with the same pipeline described above, using area-normalized cell marker intensities instead of neighborhood-averaged intensities. The first pass of clustering was used to separate fibroblasts using lineage markers PDGFRA and PDPN. For each tissue, we then isolated fibroblasts and repeated the full clustering procedure (from normalization to Louvain) to find fine-grained fibroblast clusters. We identified fine-grained clusters enriched in CCL19 (and SPARC) and performed additional gating based on CCL19 (and SPARC) to identify CCL19+ (and SPARC+) fibroblasts.
Colocalization enrichment.
With the analyses above, each cell is annotated with a cell type (fibroblast, CCL19+ fibroblast, SPARC+ fibroblast, or other cell) and a spatial niche (lymphoid, vascular, mural, and other). Within each dataset, we tested for the association between cell type and spatial niche using logistic regression, implemented in the R lme4 package80 with the formula: CellType ~ 1 + (1|Niche). Here, the CellType variable is used to test each of the four cell types, one at a time. We decided to model Niche as a random effect to avoid unstable model estimates. P-values were estimated using posterior simulation with the R arm package79 and the Wald test on the simulation-estimated confident intervals.
Supplementary Material
Highlights.
Cross-disease single-cell RNA sequencing study of human inflammatory fibroblasts
CXCL10+CCL19+ inflammatory fibroblasts localize to a T cell-enriched niche
SPARC+COL3A1+ fibroblasts localize to a perivascular niche
Both inflammatory fibroblast phenotypes were confirmed in mouse models
Context and significance.
Tissue-resident cells called fibroblasts orchestrate immune and repair functions in multiple organ systems. Signaling failures between immune cells and fibroblasts cause diverse inflammatory diseases.
Researchers from Brigham and Women’s Hospital, the University of Oxford, and the University of Birmingham conducted a study to determine whether disease-related fibroblasts share common features across diverse diseases.
The authors studied fibroblasts from target tissues from individuals with rheumatoid arthritis, ulcerative colitis, interstitial lung disease, and Sjögren’s syndrome and found two types of fibroblasts associated with inflammation in all diseases. The authors then showed that two shared fibroblast states present across all four organ types play distinct roles: one communicates with immune cells, and the other communicates with blood vessels.
ACKNOWLEDGMENTS
I.K., K.W., and M.P. conceptualized the study and co-wrote manuscript under supervision of S.R., M.B.B., C.D.B., and F.P.I.K. and J.B.K. performed analyses. K.W., M.P., G.F.M.W., E.Y.K., M.F., J.T., S.N., T.M., E.T., R.R., D.W., S.K., A.H.J., Y.J., and H.A. performed experiments. E.Y.K., M.P., M.F., A.F., K.R., F.B., B.A.F., S.J.B., C.D.B., and A.P.C. performed human sample acquisition. All authors agreed to submit the manuscript, have read and approved the final draft, and take full responsibility of its content, including the accuracy of the data and their statistical analysis.
This work was supported by a grant from F. Hoffmann-La Roche (Roche) AG. We thank David Lee for having the vision and organizing this Roche network to study stromal biology across tissues. I.K. is supported by a NIH-NIAMS training award (K01AR078355). K.W. is supported by a National Institute of Arthritis and Musculoskeletal and Skin Diseases award (K08AR077037), a Rheumatology Research Foundation Innovative Research award, and a Burroughs Wellcome Fund Career Award for Medical Scientists. E.Y.K. is supported by an American Lung Association Dalsemer Research Award (DA-827785) in Interstitial Lung Disease. B.A.F and S.J.B. have received support from the National Institute for Health Research (NIHR) Birmingham Biomedical Research Centre and the NIHR/Wellcome Trust Birmingham Clinical Research Facility. J.B.K. is supported by an award (T32GM007753) from the National Institute of General Medical Sciences. S.R. is supported by funding from the National Institutes of Health (U19AI111224, U01 HG009379, and R01AI049313). K.R. is supported by the NIHR Birmingham Biomedical Research Centre. A.P.C. receives funding from a Kennedy Trust for Rheumatology Research Senior Fellowship and Foundation for Rheumatology Research (FOREUM) Career Grant. C.B. is a co-founder of Mestag Therapeutics. H.E. holds grant funding from and has consulted for Hoffman La-Roche, Janssen, GSK, and AstraZeneca.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.medj.2022.05.002.
DECLARATION OF INTERESTS
E.Y.K. is a member of the advisory board for Cell Reports Medicine. In disclosures unrelated to this work, E.Y.K. is a member of the Steering Committees for and receives no financial renumeration from NCT04409834 (Prevention of arteriovenous thrombotic events in critically ill COVID-19 patients, TIMI group) and REMAP-CAP ACE2 renin-angiotensin system (RAS) modulation domain. E.Y.K. receives unrelated research funding from Bayer AG. In the past, E.Y.K. received unrelated research funding from Windtree Therapeutics. T.T. receives unrelated support from the Zoll Foundation. K.W. is a consultant to Mestag and Gilead Sciences and reports grant support from Gilead Sciences. S.R. is a scientific advisor for Rheos Medicines, Janssen, and Pfizer and a founder of Mestag, Inc. Y.J. and H.A. receive unrelated support from Bayer AG. M.B.B. is a consultant to GSK and 4FO Ventures and a founder of Mestag Therapeutics. M.L.S. receives unrelated research funding and institution consulting fees from Genentech, institution consulting fees from Lilly, research funding from Bristol Myers Squibb, and personal consulting fees from GV20 Therapeutics. M.C. is a co-founder of Mestag Therapeutics and obtains grant funding from and has consulted for Hoffman La-Roche. S.B. has provided paid consultancy services regarding Sjögren’s syndrome clinical trial design for the following companies in the past 3 years: Abbvie, AstraZeneca, BMS, Galapagos, Novartis, and Resolve Pharma. A.F. has received personal renumerations from Abbvie, Roche, and Janssen in the last 2 years and institutional research funding from Roche, UCB, Nascient, Mestag, GSK, and Janssen. A.P.F. and K.G.L. reported being employees of F. Hoffmann-La Roche (Roche) AG.
REFERENCES
- 1.Koliaraki V, Prados A, Armaka M, and Kollias G (2020). The mesenchymal context in inflammation, immunity and cancer. Nat. Immunol 21, 974–982. 10.1038/s41590-020-0741-2. [DOI] [PubMed] [Google Scholar]
- 2.Zhang F, Wei K, Slowikowski K, Fonseka CY, Rao DA, Kelly S, Goodman SM, Tabechian D, Hughes LB, Salomon-Escoto K, et al. (2019). Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol 20, 928–942. 10.1038/s41590-019-0378-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smillie CS, Biton M, Ordovas-Montanes J, Sullivan KM, Burgin G, Graham DB, Herbst RH, Rogel N, Slyper M, Waldman J, et al. (2019). Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 178, 714–730.e22. 10.1016/j.cell.2019.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL, Peter L, Chung MI, Taylor CJ, Jetter C, et al. (2020). Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv 6, eaba1972. 10.1126/sciadv.aba1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, et al. (2020). Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv 6, eaba1983. 10.1126/sciadv.aba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mizoguchi F, Slowikowski K, Wei K, Marshall JL, Rao DA, Chang SK, Nguyen HN, Noss EH, Turner JD, Earp BE, et al. (2018). Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun 9, 789. 10.1038/s41467-018-02892-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang B, Chen Z, Geng L, Wang J, Liang H, Cao Y, Chen H, Huang W, Su M, Wang H, et al. (2019). Mucosal profiling of pediatric-onset colitis and IBD reveals common pathogenics and therapeutic pathways. Cell 179, 1160–1176.e24. 10.1016/j.cell.2019.10.027. [DOI] [PubMed] [Google Scholar]
- 8.Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, Ashley N, Cubitt L, Mellado-Gomez E, Attar M, et al. (2018). Structural remodeling of the human colonic mesenchyme in inflammatory bowel disease. Cell 175, 372–386.e17. 10.1016/j.cell.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martin JC, Chang C, Boschetti G, Ungaro R, Giri M, Grout JA, Gettler K, Chuang L, Nayar S, Greenstein AJ, et al. (2019). Single-cell analysis of Crohn’s disease lesions identifies a pathogenic cellular module associated with resistance to anti-TNF therapy. Cell 178, 1493–1508.e20. 10.1016/j.cell.2019.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, Coccia M, Gortz D, This S, Stockenhuber K, Pott J, Friedrich M, Ryzhakov G, Baribaud F, Brodmerkel C, Cieluch C, Rahman N, Muller-Newen G, Owens RJ, Kuhl AA, Maloy KJ, Plevy SE, Oxford IBD Cohort Investigators, Keshav S, Travis SPL, and Powrie F (2017). Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med 23, 579–589. 10.1038/nm.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedrich M, Pohin M, and Jackson M (2021). IL-1-driven stromal-neutrophil interaction in deep ulcers identifies a pathotype of therapy non-responsive inflammatory bowel disease. Nat Med 27, 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nayar S, Campos J, Smith CG, Iannizzotto V, Gardner DH, Mourcin F, Roulois D, Turner J, Sylvestre M, Asam S, et al. (2019). Immunofibroblasts are pivotal drivers of tertiary lymphoid structure formation and local pathology. Proc. Natl. Acad. Sci. U S A 116, 13490–13497. 10.1073/pnas.1905301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei K, Korsunsky I, Marshall JL, Gao A, Watts GFM, Major T, Croft AP, Watts J, Blazar PE, Lange JK, et al. (2020). Notch signalling drives synovial fibroblast identity and arthritis pathology. Nature 582, 259–264. 10.1038/s41586-020-2222-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor AJ, Kemble S, et al. (2019). Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251. 10.1038/s41586-019-1263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.West NR (2019). Coordination of immune-stroma crosstalk by IL-6 family cytokines. Front. Immunol 10, 1093. 10.3389/fimmu.2019.01093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nguyen HN, Noss EH, Mizoguchi F, Huppertz C, Wei KS, Watts GF, and Brenner MB (2017). Autocrine loop involving IL-6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 46, 220–232. 10.1016/j.immuni.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slowikowski K, Nguyen HN, Noss EH, Simmons DP, Mizoguchi F, Watts GF, Gurish MF, Brenner MB, and Raychaudhuri S (2019). CUX1 and IκBζ mediate the synergistic inflammatory response to TNF and IL-17A in stromal fibroblasts. Preprint at bioRxiv. 10.1101/571315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ng B, Dong J, Viswanathan S, Widjaja AA, Paleja BS, Adami E, Ko NSJ, Wang M, Lim S, Tan J, et al. (2020). Fibroblast-specific IL11 signaling drives chronic inflammation in murine fibrotic lung disease. FASEB J. 34, 11802–11815. 10.1096/fj.202001045rr. [DOI] [PubMed] [Google Scholar]
- 19.He S, Wang LH, Liu Y, Li YQ, Chen HT, Xu JH, Peng W, Lin GW, Wei PP, Li B, et al. (2020). Single-cell transcriptome profiling of an adult human cell atlas of 15 major organs. Genome Biol. 21, 294. 10.1186/s13059-020-02210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.The Tabula Sapiens Consortium, and Quake SR (2021). The Tabula Sapiens: a single cell transcriptomic atlas of multiple organs from individual human donors. Preprint at bioRxiv. 10.1101/2021.07.19.452956. [DOI] [Google Scholar]
- 21.Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, Yang YA, Tam L, Caothien R, Roose-Girma M, et al. (2021). Cross-tissue organization of the fibroblast lineage. Nature 593, 575–579. 10.1038/s41586-021-03549-5. [DOI] [PubMed] [Google Scholar]
- 22.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran HTN, Ang KS, Chevrier M, Zhang X, Lee NYS, Goh M, and Chen J (2020). A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 21, 12. 10.1186/s13059-019-1850-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lotfollahi M, Naghipourfar M, Luecken MD, Khajavi M, Büttner M, Avsec Z, Misharin AV, and Theis FJ (2020). Query to reference single-cell integration with transfer learning. Preprint at bioRxiv. 10.1101/2020.07.16.205997. [DOI] [Google Scholar]
- 26.Andreatta M, Corria-Osorio J, Müller S, Cubas R, Coukos G, Carmona SJ, and Projecting single-cell transcriptomics data onto a reference T cell atlas to interpret immune responses. (2020). Preprint at bioRxiv. 10.1101/2020.06.23.166546. [DOI] [Google Scholar]
- 27.Kang JB, Nathan A, Weinand K, Zhang F, Millard N, Rumker L, Moody DB, Korsunsky I, and Raychaudhuri S (2021). Efficient and precise single-cell reference atlas mapping with Symphony. Nat. Commun 12, 5890. 10.1038/s41467-021-25957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang F, Mears JR, Shakib L, Beynor JI, Shanaj S, Korsunsky I, Nathan A, Donlin LT, and Raychaudhuri S (2020). IFN- γ and TNF- α drive a CXCL10 + CCL2 + macrophage phenotype expanded in severe COVID-19 and other diseases with tissue inflammation. Preprint at bioRxiv. 10.1101/2020.08.05.238360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polański K, Young MD, Miao Z, Meyer KB, Teichmann SA, and Park JE (2020). BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965. 10.1093/bioinformatics/btz625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez R, Regier J, Cole MB, Jordan MI, and Yosef N (2018). Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058. 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hie B, Bryson B, and Berger B (2019). Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat. Biotechnol 37, 685–691. 10.1038/s41587-019-0113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luecken MD, Buttner M, Chaichoompu K, Danese A, Interlandi M, Mueller MF, Strobl DC, Zappia L, Dugas M, Colome-Tatche M, and Theis FJ (2022). Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50. 10.1038/s41592-021-01336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liberzon A, Subramanian A, Pinchback R, Thorvaldsdottir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holland CH, Tanevski J, Perales-Paton J, Gleixner J, Kumar MP, Mereu E, Joughin BA, Stegle O, Lauffenburger DA, Heyn H, et al. (2020). Robustness and applicability of transcription factor and pathway analysis tools on single-cell RNA-seq data. Genome Biol. 21,36. 10.1186/s13059-020-1949-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, and Califano A (2016). Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet 48, 838–847. 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Kim CY, Lee M, Kim E, et al. (2018). TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 46, D380–D386. 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garcia-Alonso L, Holland CH, Ibrahim MM, Turei D, and Saez-Rodriguez. (2019). Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 29, 1363–1375. 10.1101/gr.240663.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramilowski JA, Goldberg T, Harshbarger J, Kloppmann E, Lizio M, Satagopam VP, Itoh M, Kawaji H, Carninci P, Rost B, and Forrest ARR (2015). A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun 6, 7866. 10.1038/ncomms8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudno-Rudzińska J, Kielan W, Frejlich E, Kotulski K, Hap W, Kurnol K, Dzierzek P, Zawadzki M, and Halon A (2017). A review on Eph/ephrin, angiogenesis and lymphangio-genesis in gastric, colorectal and pancreatic cancers. Chin. J. Cancer Res 29, 303–312. 10.21147/j.issn.1000-9604.2017.04.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weckbach LT, Groesser L, Borgolte J, Pagel JI, Pogoda F, Schymeinsky J, Muller-Hocker J, Shakibaei M, Muramatsu T, Deindl E, and Walzog B (2012). Midkine acts as proangiogenic cytokine in hypoxia-induced angiogenesis. Am. J. Physiol. Heart Circ. Physiol 303, H429–H438. 10.1152/ajpheart.00934.2011. [DOI] [PubMed] [Google Scholar]
- 42.von Tell D, Armulik A, and Betsholtz C (2006). Pericytes and vascular stability. Exp. Cell Res 312, 623–629. 10.1016/j.yexcr.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 43.Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, et al. (2020). A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625. 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, et al. (2019). Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis. Am. J. Respir. Crit. Care Med 199, 1517–1536. 10.1164/rccm.201712-2410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, Kleshchevnikov V, Dabrowska M, Pritchard S, Bolt L, et al. (2021). Cells of the human intestinal tract mapped across space and time. Nature 597, 250–255. 10.1038/s41586-021-03852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, Kameyama N, Estrada Y, Der E, Krueger JG, et al. (2020). Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J. Allergy Clin. Immunol 145, 1615–1628. 10.1016/j.jaci.2020.01.042. [DOI] [PubMed] [Google Scholar]
- 47.Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, Adams TS, Schupp JC, Poli SD, Rosas IO, et al. (2020). Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat. Commun 11, 1920. 10.1038/s41467-020-15647-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Monach P, Hattori K, Huang H, Hyatt E, Morse J, Nguyen L, Ortiz-Lopez A, Wu HJ, Mathis D, and Benoist C (2007). The K/BxN mouse model of inflammatory arthritis. Methods Mol. Med 136, 269–282. 10.1007/978-1-59745-402-5_20. [DOI] [PubMed] [Google Scholar]
- 49.Brand DD, Latham KA, and Rosloniec EF (2007). Collagen-induced arthritis. Nat. Protoc 2, 1269–1275. 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 50.Izbicki G, Segel MJ, Christensen TG, Conner MW, and Breuer R (2002). Time course of bleomycin-induced lung fibrosis. Int.J. Exp. Pathol 83, 111–119. 10.1046/j.1365-2613.2002.00220.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Czarnewski P, Parigi SM, Sorini C, Diaz OE, Das S, Gagliani N, and Villablanca EJ (2019). Conserved transcriptomic profile between mouse and human colitis allows unsupervised patient stratification. Nat. Commun 10, 2892. 10.1038/s41467-019-10769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.GTEx Consortium (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Krenn V, Morawietz L, Burmester GR, Kinne RW, Mueller-Ladner U, Muller B, and Haupl T (2006). Synovitis score: discrimination between chronic low-grade and high-grade synovitis. Histopathology 49, 358–364. 10.1111/j.1365-2559.2006.02508.x. [DOI] [PubMed] [Google Scholar]
- 54.Marchal-Bressenot A, Salleron J, Boulagnon-Rombi C, Bastien C, Cahn V, Cadiot G, Diebold MD, Danese S, Reinisch W, Schreiber S, et al. (2017). Development and validation of the Nancy histological index for UC. Gut 66, 43–49. 10.1136/gutjnl-2015-310187. [DOI] [PubMed] [Google Scholar]
- 55.Szabo PA, Levitin HM, Miron M, Snyder ME, Senda T, Yuan J, Cheng YL, Bush EC, Dogra P, Thapa P, et al. (2019). Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun 10, 4706. 10.1038/s41467-019-12464-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieto P, Elosua-Bayes M, Trincado JL, Marchese D, Massoni-Badosa R, Salvany M, Henriques A, Mereu E, Moutinho C, Ruiz S, et al. (2020). A single-cell tumor immune atlas for precision oncology. Cold Spring Harbor Lab. 10, 1913–1926. 10.1101/2020.10.26.354829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Breese E, Braegger CP, Corrigan CJ, Walker-Smith JA, and MacDonald TT (1993). Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology 78, 127–131. [PMC free article] [PubMed] [Google Scholar]
- 58.Bisping G, Lugering N, Lutke-Brintrup S, Pauels HG, Schurmann G, Domschke W, and Kucharzik T (2001). Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon-gamma (IFN-γ) in peripheral CD8+ lymphocytes co-cultured with intestinal epithelial cells. Clin. Exp. Immunol 123, 15–22. 10.1046/j.1365-2249.2001.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armulik A, Genové G, and Betsholtz C (2011). Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 21, 193–215. 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Rafii S, Butler JM, and Ding B-S (2016). Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325. 10.1038/nature17040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, and Brown PO (2002). Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc. Natl. Acad. Sci. U. S. A 99, 12877–12882. 10.1073/pnas.162488599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mack M (2018). Inflammation and fibrosis. Matrix Biol. 68–69, 106–121. 10.1016/j.matbio.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 63.Asano Y, and Sato S (2015). Vasculopathy in scleroderma. Semin. Immunopathol 37, 489–500. 10.1007/s00281-015-0505-5. [DOI] [PubMed] [Google Scholar]
- 64.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, Valerius MT, McMahon AP, and Duffield JS (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol 176, 85–97. 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hung C, Linn G, Chow YH, Kobayashi A, Mittelsteadt K, Altemeier WA, Gharib SA, Schnapp LM, and Duffield JS (2013). Role of lung pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med 188, 820–830. 10.1164/rccm.201212-2297oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dakin SG, Coles M, Sherlock JP, Powrie F, Carr AJ, and Buckley CD (2018). Pathogenic stromal cells as therapeutic targets in joint inflammation. Nat. Rev. Rheumatol 14, 714–726. 10.1038/s41584-018-0112-7. [DOI] [PubMed] [Google Scholar]
- 67.Davidson S, Coles M, Thomas T, Kollias G, Ludewig B, Turley S, Brenner M, and Buckley CD (2021). Fibroblasts as immune regulators in infection, inflammation and cancer. Nat. Rev. Immunol 21, 704–717. 10.1038/s41577-021-00540-z. [DOI] [PubMed] [Google Scholar]
- 68.Melville J, Lun A, Djekidel MN, and Hao Y. uwot: The uniform manifold approximation and projection (UMAP) method for dimensionality reduction. R package version 15. https://cran.r-project.org/web/packages/uwot/index.html.
- 69.Machowicz A, Hall I, De Pablo P, Rauz S, Richards A, Higham J, Poveda-Gallego A, Imamura F, Bowman SJ, Barone F, and Fisher BA (2020). Mediterranean diet and risk of Sjögren’s syndrome. https://www.clinexprheumatol.org/abstract.asp?a=15905. [PubMed]
- 70.Donlin LT, Rao DA, Wei K, Slowikowski K, McGeachy MJ, Turner JD, Meednu N, Mizoguchi F, Gutierrez-Arcelus M, Lieb DJ, et al. (2018). Methods for high-dimensional analysis of cells dissociated from cryopreserved synovial tissue. Arthritis Res. Ther 20, 139. 10.1186/s13075-018-1631-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. U. S. A 110, 11982–11987. 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. (2017). Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 27, 849–864. 10.1101/gr.213611.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773. 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 75.Melsted P, Ntranos V, and Pachter L (2019). The barcode, UMI, set format and BUStools. Bioinformatics 35, 4472–4473. 10.1093/bioinformatics/btz279. [DOI] [PubMed] [Google Scholar]
- 76.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Blondel VD, Guillaume J-L, Lambiotte R, and Lefebvre E (2008). Fast unfolding of communities in large networks. J. Stat. Mech. Theor. Exp 2008, P10008. 10.1088/1742-5468/2008/10/p10008. [DOI] [Google Scholar]
- 78.Haghverdi L, Lun ATL, Morgan MD, and Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol 36, 421–427. 10.1038/nbt.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gelman A, and Su Y-S (2020). Arm: Data Analysis Using Regression and Multilevel/Hierarchical Models.
- 80.Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting linear mixed-effects models using lme4. J. Stat. Softw 67, 1–48. 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 81.Gelman A, Hill J, and Yajima M (2012). Why we (usually) don’t have to worry about multiple comparisons. J. Res. Educ. Eff 5, 189–211. 10.1080/19345747.2011.618213. [DOI] [Google Scholar]
- 82.Lun ATL, and Marioni JC (2017). Overcoming confounding plate effects in differential expression analyses of single-cell RNA-seq data. Biostatistics 18, 451–464. 10.1093/biostatistics/kxw055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sergushichev A (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Preprint at bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- 85.Fonseka CY, Rao DA, Teslovich NC, Korsunsky I, Hannes SK, Slowikowski K, Gurish MF, Donlin LT, Lederer JA, Weinblatt ME, et al. (2018). Mixed-effects association of single cells identifies an expanded effector CD4+ T cell subset in rheumatoid arthritis. Sci. Transl. Med 10, eaaq0305. 10.1126/scitranslmed.aaq0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.DerSimonian R, and Laird N (1986). Meta-analysis in clinical trials. Control Clin. Trials 7, 177–188. 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 87.Veroniki AA, Jackson D, Viechtbauer W, Bender R, Bowden J, Knapp G, Kuss O, Higgins JP, Langan D, and Salanti G (2016). Methods to estimate the between-study variance and its uncertainty in meta-analysis. Res. Synth. Methods 7, 55–79. 10.1002/jrsm.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wolf FA, Angerer P, and Theis, F.J.S.C.A.N.P.Y. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yee TW (2010). TheVGAMPackage for categorical data analysis. J. Stat. Softw 32, 1–34. 10.18637/jss.v032.i10. [DOI] [Google Scholar]
- 90.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Greenwald NF, Miller G, Moen E, Kong A, Kagel A, Dougherty T, Fullaway CC, McIntosh BJ, Leow KX, Schwartz MS, et al. (2021). Whole-cell segmentation of tissue images with human-level performance using large-scale data annotation and deep learning. Nat. Biotechnol 40, 555–565. 10.1038/s41587-021-01094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.van der Walt S, Schonberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, and Yu T (2014). scikit-image: image processing in Python. PeerJ 2, e453. 10.7717/peerj.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gelman A, Su Y-S, Yajima M, Hill J, Pittau MG, Kerman J, et al. arm: Data analysis using regression and multilevel/hierarchical models. R package version 1.10–1. https://cran.r-project.org/web/packages/arm/.
- 94.Dolgalev I. msigdbr: MSigDB Gene Sets for Multiple Organisms in a Tidy Data Format. 2018. R package version 7.5.1. https://cran.r-project.org/web/packages/msigdbr/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-sequencing data have been deposited in GEO, Broad’s Single Cell Portal, and NIAID ImmPort. High dimensional images for Cell Dive experiments have been deposited in NIAID ImmPort. Accession numbers are listed in the key resources table.
All original code has been deposited at Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| EPCAM/CD326, clone 9C4, Mouse IgG2b | Biolegend | Cat# 324233 |
| CD45, clone HI30, mouse IgG1 | Biolegend | Cat# 304005 |
| CD31, clone WM59, mouse IgG1 | Biolegend | Cat# 303133 |
| CD146, clone P1H12, Mouse IgG1 | Biolegend | Cat# 361003 |
| PDPN, clone NZ-1.3, rat IgG2a | eBioscience/ThermoFisher | Cat #46-4321-82 |
| CD90, clone 5E10, mouse IgG1 | Biolegend | Cat# 328109 |
| Alexa Fluor® 647 - Mouse monoclonal (D2-40) anti-Podoplanin (Lymphatic Endothelial Marker) antibody | Biolegend | Cat# 916610 RRID : AB_2810816 |
| BSA and Azide free -Recombinant Rabbit monoclonal [EPR5480] Anti PDGFR alpha | Abcam | Cat# ab248689 RRID : N/A |
| Alexa Fluor® 488 - Recombinant Rabbit monoclonal [EPR3208] Anti-CD146 antibody | Abcam | Cat# ab196448; RRID:AB_2868591 |
| BSA and Azide free – Recombinant Rabbit monoclonal [EPR3132] Anti CD90 / Thy1 antibody | Abcam | Cat# ab181885 RRID : N/A |
| Alexa Fluor® 555 – Recombinant Rabbit monoclonal [EPR20545] Anti-CD68 antibody | Abcam | Cat# ab280860 RRID : N/A |
| BSA and Azide free - Recombinant Rabbit monoclonal [SP162] Anti-CD3 antibody | Abcam | Cat# ab245731 RRID : N/A |
| Alexa Fluor® 647 – Mouse monoclonal (JC/70A) anti CD31/PECAM-1 antibody | Novus | Cat# NB600-562AF647 RRID: N/A |
| Alexa Fluor® 647 – Mouse monoclonal (C31.3) anti CD31/PECAM-1 antibody | Novus | Cat# NB P2-33154AF647 RRID: N/A |
| BSA and Azide free – Recombinant Rabbit monoclonal [SP205] Anti-SPARC antibody | Abcam | Cat# ab245733 RRID: N/A |
| Unconjugated - Goat Polyclonal Anti-Human Ccl19 / mip-3 beta antibody | R and D systems | Cat# AF361; RRID:AB_355323 |
| Alexa Fluor® 488 – Mouse monoclonal [1A4] Anti-alpha smooth muscle Actin antibody | Abcam | Cat# ab184675; RRID:AB_2832195 |
| Alexa Fluor® 647 Mouse monoclonal (2D1) anti-human CD45 antibody | BioLegend | Cat# 368538; RRID:AB_2716028 |
| Alexa Fluor® 647 Mouse monoclonal (C8/144B) anti-human CD8a antibody | BioLegend | Cat# 372906; RRID:AB_2650712 |
| Alexa Fluor® 488 Recombinant Rabbit monoclonal [EP1628Y] Anti-Cytokeratin 8 antibody | Abcam | Cat# ab192467; RRID:AB_2864346 |
| Alexa Fluor® 555 Recombinant Rabbit monoclonal [EPR3776] Anti-Vimentin antibody - Cytoskeleton Marker | Abcam | Cat# ab203428 RRID : N/A |
| Alexa Fluor® 488 Mouse monoclonal (TAL 1B5) Anti-HLA-DR Antibody | Santacruz | Cat# sc-53319AF488 RRID N/A |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447; RRID:AB_2535864 |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572; RRID:AB_162543 |
| CD16/CD32 Monoclonal Antibody (93) (FcR block) 1:200 | ThermoFisher Scientific | 14-0161-86 |
| FITC EpCAM (G8.8) 1:200 | Biolegend | 118208 |
| PE-Cy7 Podoplanin (8.1.1) 1:200 | Biolegend | 127412 |
| APC CD140a (APA5)1:200 | Biolegend | 135907 |
| BV605 CD31 (390) 1:200 | Biolegend | 102427 |
| BV711 CD45 (30-F11) 1:200 | Biolegend | 103147 |
| BV711 CD45 (30-F11) 1:200 | Biolegend | 103147 |
| Bacterial and virus strains | ||
| Mycobacterium tuberculosis H37Ra | BD Difco | BD 231141 |
| Chemicals, peptides, and recombinant proteins | ||
| Glycerol | Sigma-Aldrich | Cat# G5516 RRID N/A |
| Trizma®base | Sigma-Aldrich | Cat# T6066 RRID N/A |
| Sodium bicarbonate | Sigma-Aldrich | Cat#S6297 RRID N/A |
| Tween20 | Sigma-Aldrich | Cat#P9416 RRID N/A |
| Triton X-100 | Sigma-Aldrich | Cat#T9284 RRID N/A |
| Propyl gallate | Sigma-Aldrich | Cat#02370 RRID N/A |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A7906 RRID N/A |
| Target Retrieval Solution 10X Concentrate | Dako | Cat#S1699 |
| Ethylenediaminetetraacetic acid | Sigma-Aldrich | Cat#E9884 |
| eBioscience Fixable Viability Dye eFluor 780 1:1000 | ThermoFisher Scientific | 65-0865-18 |
| DAPI Solution 50 pg/mL | ThermoFisher Scientific | 62248 |
| 2.5% w/v dextran sulfate sodium | MP Biomedicals | 216011080 |
| Liberase TL | Roche | 5401020001 |
| DNase 1 | Sigma | 11284932001 |
| Quick-RNA 96 Kit | Zymo Research | R1052 |
| Donkey Serum | Biorad | Cat# C06SB RRID N/A |
| Hydrogen Peroxide Solution | Sigma-Aldrich | Cat#216763 RRID N/A |
| FcR Blocking Reagent | Miltenyi | Cat# 130-059-901; RRID:AB_2892112 |
| Bovine Tyope II Collagen | Richard Williams https://doi.org/10.1385/1-59259-771-8:207 |
N/A |
| Critical commercial assays | ||
| Dynabeads Human T-activator CD3/CE28 for T cell expansion and activation | ThermoFisher | Cat# 11132D |
| 10× Genomics Chromium Single Cell 3’ (v2 and v3 chemistry) | 10× Genomics | https://support.10xgenomics.com/single-cell-gene-expression/library-prep/doc/technical-note-assay-scheme-and-configuration-of-chromium-single-cell-3-v2-libraries |
| Cell Dive | General Electric | https://www.ge.com/research/project/multiplexed-tissue-imaging-platform |
| CD45RA MicroBeads (human) | Miltenyi | Cat# 130-045-901 |
| Pan T Cell Isolation Kit (human) | Miltenyi | Cat# 130-096-535 |
| LS columns | Miltenyi | Cat# 130-042-401 |
| Deposited data | ||
| CellDive imaging data: https://www.immport.org/shared/study/SDY1765 | This paper | ImmPort:SDY1765 |
| Human fibroblast atlas: https://sandbox.zenodo.org/record/772596#.YGsi6BRKg-Q | This paper | https://doi.org/10.5072/zenodo.772596 |
| Atopic dermatitis scRNAseq | He et al., 2020a | GSE147424 |
| Serum transfer arthritis scRNAseq | Wei et al., 2020 | GSE145286 |
| DSS induced colitis scRNAseq | Kinchen et al., 2018 | GSE114374 |
| bleomycin induced lung injury scRNAseq | Tsukui et al., 2020 | GSE132771 |
| Adult human cell atlas | He et al., 2020b | GSE159929 |
| Tabula sapiens: https://figshare.com/projects/Tabula_Sapiens/100973 | The Tabula Sapiens Consortium and Quake, 2021 | N/A |
| Healthy lung atlas scRNAseq | Travaglini et al. | Synapse:syn21041850 |
| IPF lung cohort scRNAseq | Adams et al. | GSE136831 |
| Healthy gut atlas scRNAseq | Elmentaite et al., 2021 | E-MTAB-9536 |
| Raw human sequencing data: https://www.immport.org/shared/study/SDY1765 | This paper | ImmPort:SDY1765 |
| Processed human sequencing data:https://singlecell.broadinstitute.org/single_cell/study/SCP738 | This paper | BroadSingleCellPortal:SCP738 |
| Raw and processed mouse sequencing data | This paper | GSE185711 |
| Experimental models: Cell lines | ||
| Primary lung fibroblast cell lines from earlier-stage fibrotic interstitial lung disease | This paper | Lung fibroblast cell lines named V3, V6, V7, V11. |
| Primary Synovial fibroblasts | This paper | synovial fibroblast cell lines named: STB-009, RA200212, AMP-005 |
| Primary human T cells isolated from leukopak mononuclear cells | This paper | N/A |
| Collagen Induced Arthritis | https://doi.org/10.1385/1-59259-771-8:207 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | RRID:IMSR_JAX:000,664 |
| Mouse: DBA/1J | MRC Harwell | N/A |
| Software and algorithms | ||
| Code to Reproduce Analyses and Figures for Fibroblast Atlas 2022 | This Paper | https://doi.org/10.5281/zenodo.6510339 |
| Symphony v1.0 | Kang et al., 2021 | https://github.com/immunogenomics/symphony |
| DeepCell | Greenwald et al., 2021 | https://github.com/vanvalenlab/deepcell-tf |
| uwot v0.1.10 | Melville et al., 202068 | https://github.com/jlmelville/uwot |
| arm v1.11.2 | Gelman and Su, 202093 | https://github.com/gelman/arm |
| lme4 v1.1.27.1 | Bates et al., 2015 | https://github.com/lme4/lme4 |
| presto | This paper | https://github.com/immunogenomics/presto/tree/glmm |
| fgsea v1.18.0 | Sergushichev, 2016 | https://github.com/ctlab/fgsea |
| msigdbr v7.4.1 | Dolgalev, 201894 | https://github.com/igordot/msigdbr |
| scanorama v1.7.1 | Hie et al., 2019 | https://github.com/brianhie/scanorama |
| bbknn v1.5.1 | Polański et al., 2020 | https://github.com/Teichlab/bbknn |
| scvi-tools v0.6.8 | Lopez et al., 2018 | https://github.com/scverse/scvi-tools |
| scanpy v1.7.1 | Wolf et al., 2018 | https://github.com/scverse/scanpy |
| VGAM v1.1.5 | Yee, 2010 | https://github.com/cran/VGAM |
| limma v3.48.3 | Ritchie et al., 2015 | https://github.com/cran/limma |
| kallisto | Bray et al., 2016 | https://github.com/pachterlab/kallisto |
| bustools | Melsted et al., 2019 | https://github.com/BUStools/bustools |
| harmony v1.0 | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony/tree/weights |