

Summary
Activated CD8+ T lymphocytes differentiate into heterogeneous subsets. Using super-resolution imaging, we found that prior to the first division, dynein-dependent vesicular transport polarized active TORC1 towards the microtubule-organizing center (MTOC) at the proximal pole. This active TORC1 was physically associated with active eIF4F, required for the translation of c-myc mRNA. As a consequence, c-myc translating polysomes polarized toward the cellular pole proximal to the immune synapse, resulting in localized c-myc translation. Upon division, the TORC1-eIF4A complex preferentially sorted to the proximal daughter cell, facilitating asymmetric c-Myc synthesis. Transient disruption of eIF4A activity at first division skewed long-term cell fate trajectories to memory-like function. Using a genetic barcoding approach, we found that first-division sister cells often displayed differences in transcriptional profiles that largely correlated with c-Myc and TORC1 target genes. Our findings provide mechanistic insights as to how distinct T cell fate trajectories can be established during the first division.
Keywords: CD8+ T cells, cell fate, c-Myc, translation, eIF4A, STORM, STED, scRNAseq, asymmetric cell division, polarization
In brief
Liedmann et al. found that upon T cell activation, active TORC1-eIF4F complexes on vesicles polarize to the microtubule organizing center and therefore can assort asymmetrically to daughter cells. As a result, differential eIF4F-dependent c-Myc synthesis can produce distinct transcriptional profiles in first-division daughter cells, resulting in distinct cell fates.
Graphical Abstract
Introduction
In response to intracellular microbial infections or cancer mutations, naïve CD8+ T cells expand and develop as a heterogeneous pool of differentiated cells, composed of phenotypically and functionally distinct subsets, each characterized by defining transcriptional profiles (Best et al., 2013; Chang et al., 2014; Kaech et al., 2002). Single-cell transcriptomics have indicated that transcriptional diversity occurs as early as the first division (Arsenio et al., 2015; Kakaradov et al., 2017; Metz et al., 2015). However, the mechanisms by which distinct transcriptional profiles are established during a single cell division to regulate T cell differentiation and function remain unknown. One proposed contributing factor is asymmetric cell division (ACD), an evolutionarily conserved mechanism that describes the genesis of two daughter cells which are distinct in fate and function. CD8+ T cells, once activated by antigen-presenting cells (APCs), frequently undergo ACD such that the daughter cell proximal to the APC displays an effector-like phenotype, and the other one, distal to the APC, is characterized by a memory-like phenotype (Arsenio et al., 2015; Chang et al., 2007; Pollizzi et al., 2016; Verbist et al., 2016). Daughter cells can be distinguished from each other by asymmetric distribution of certain marker proteins, one example being higher c-Myc protein levels in the cell derived from the proximal pole (c-Mychigh sister cell), and lower c-Myc protein levels in the cell derived from the distal pole (c-Myclow sister cell). Since c-Myc is a nuclear protein with a short half-life (Hann and Eisenman, 1984), we asked how the asymmetric distribution of c-Myc might be established.
During T cell activation, the formation of the immunological synapse (IS) at the site of contact between the APC and the T cell provides the cue that dictates the axis of polarity within the undivided T cell. The microtubule organization center (MTOC) is relocated towards the IS within minutes of activation, where it remains until mitosis. During mitosis, Pins/G protein signaling aligns the mitotic spindle with the axis of polarity, with one new nucleus forming close to the proximal pole and the second nucleus forming at the distal pole (Oliaro et al., 2010).
Previously, we determined that ACD in murine CD8+ T cells is associated with asymmetric segregation of c-Myc during telophase and sustained in first-division daughter cells to affect proliferation, metabolism, and differentiation. In addition, we found that brief inhibition of translation (using cycloheximide) was sufficient to rapidly equilibrate c-Myc levels in first-division sister cells (Verbist et al., 2016). However, the molecular events that take place between TCR activation and c-Myc asymmetric distribution observed upon division remained unclear.
The c-Myc protein is generated either by 5’cap-dependent or IRES-dependent translation (Stoneley et al., 2000). The mRNA of c-Myc is characterized by a long and highly structured 5’UTR, rendering its translation dependent on the activity of eIF4A (Wolfe et al., 2014). At the initiation of 5’cap-dependent c-myc translation, the recruitment of ribosomes to mRNAs is mediated by the eukaryotic translation initiation factor 4F (eIF4F), a protein complex consisting of three subunits: (i) the cap-binding protein eIF4E; (ii) the RNA helicase eIF4A, needed to unwind local RNA structures to allow for ribosomal scanning of mRNA templates that possess long and highly structured 5’UTRs (Rubio et al., 2014; Svitkin et al., 2001; Wolfe et al., 2014); and (iii) the scaffold protein eIF4G, which binds eIF4E and eIF4A, and recruits the 43S pre-initiation complex to the mRNA (as reviewed (Pelletier and Sonenberg, 2019)).
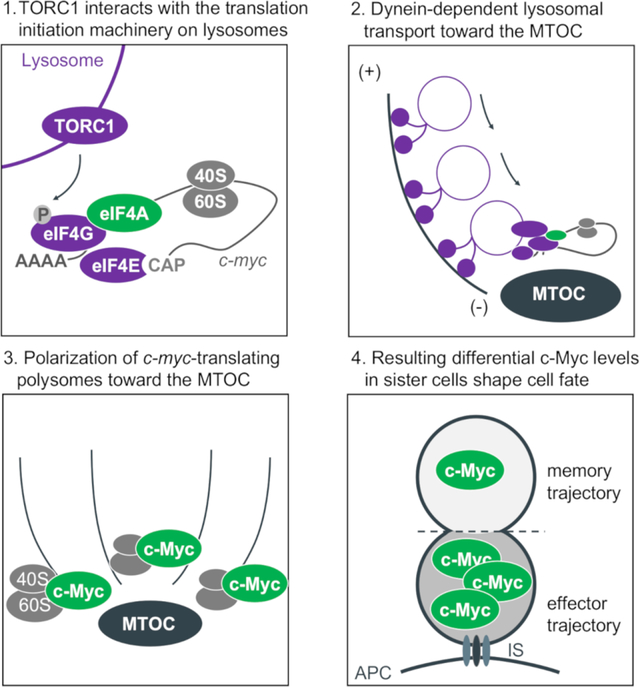
Here, we describe our observations that upon T cell activation, active TORC1, associated with small vesicles (lysosomes or late endosomes with V-ATPase), polarizes towards the microtubule organizing center (MTOC) near the immune synapse. We found that this TORC1 is physically associated with the eIF4F complex, resulting in localized synthesis of c-Myc. Upon division, the TORC1 (Pollizzi et al., 2016) and eIF4F (this paper) therefore often preferentially assort to the proximal daughter, and as a consequence, this daughter expresses more c-Myc protein than does the distal daughter (Verbist et al., 2016). Brief inhibition of eIF4A activity around the time of the first division equilibrates c-Myc levels in the daughter cells, altering the eventual fate of c-Mychigh first-division cells. Using an endogenous barcoding approach, we found that bona fide first-division sister cells often have distinct expression profiles that correlate with TORC1 and c-Myc target genes. Altogether, our findings provide one mechanism by which CD8+ T cell fate trajectories can diverge as early as the first division.
Results
TORC1 signaling is localized at the proximal pole of activated, undivided CD8+ T cells regulating asymmetric c-Myc synthesis
Translation initiation is regulated by multiple signaling pathways, including the phosphoinositide 3-kinase (PI3K)-mechanistic target of rapamycin (mTOR) complex 1 (TORC1) pathway (as reviewed (Roux and Topisirovic, 2018)). Active TORC1 often preferentially assorts to the proximal daughter in first-division CD8+ T cells (Pollizzi et al., 2016), and therefore we asked whether TORC1 activity is polarized prior to division. We analyzed the spatial distribution of TORC1 activity in activated murine OT-I CD8+ T cells prior to the first division in lymph node sections from immunized animals. Active mTOR, as measured by phosphorylation of serine 2481, was polarized towards the proximal pole of the cell (Figure 1A). Similarly, we observed polarization of TORC1 activity to the MTOC in undivided OT-I CD8+ T cells that were activated with plate-bound anti-CD3, anti-CD28 and ICAM1 in vitro (Figure 1B, C). In contrast, non-phosphorylated mTOR showed no polarization towards the proximal pole in activated T cells (Figure S1A, B). The polarization of TORC1 activity appeared to be dependent on TCR engagement, as activation of OT-I CD8+ T cells with phorbol myristate acetate (PMA) plus ionomycin, although resulting in a high level of phospho-mTOR, failed to induce its polarization towards the proximal pole (Figures 1B, D and S1 A, C). This finding is consistent with previous studies in which activation of CD8+ T cells with PMA plus Ionomycin was associated with symmetric distribution of TORC1 activity in dividing sister cells, and also recapitulated the phenotype of CD8high and c-Mychigh cell populations in vivo; that is, such cells showed a reduced potential to respond to a secondary infection (Pollizzi et al., 2016).
Figure 1. TORC1 signaling is localized at the proximal pole of activated, undivided OT-I CD8+ T cells regulating asymmetric c-Myc synthesis.
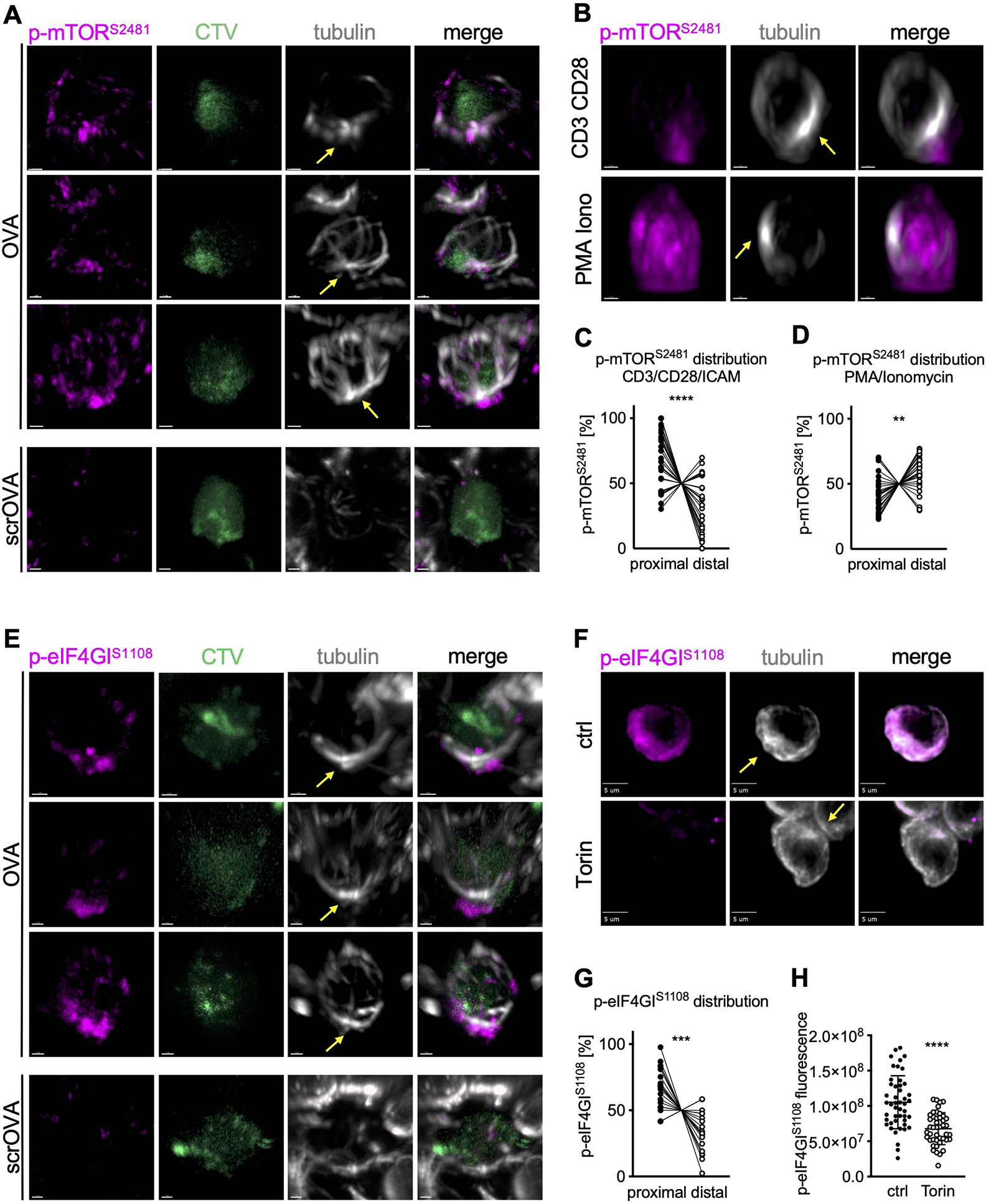
(A) Representative images of undivided OT-I CD8+ T cells in lymph node sections of animals immunized with peptide or scrambled peptide. Scale bar: 1 μm. (B) Representative images of undivided OT-I CD8+ T cells activated on slides coated with anti-CD3, anti-CD28, and ICAM1 or with PMA plus Ionomycin for 2 h. Scale bar: 2 μm. (C, D) Quantification of the distribution of p-mTORS2481 within the cell. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell. (E) Representative images of undivided OT-I CD8+ T cells in lymph node sections from animals immunized with peptide or scramble peptide. Scale bar: 1 μm. (F) Representative images of undivided OT-I CD8+ T cells activated on peptide-pulsed APCs and treated with Torin for the last hour of activation. Scale bar: 5 μm. (G) Quantification of the distribution of p-eIF4GIS1108 signal within the cell. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell. (H) Quantification of sum fluorescence intensity of p-eIF4GIS1108. Each dot represents one cell; mean +/− SD. Arrows mark MTOCs. Compiled from three (C, D, G, H) independent experiments. Significance was calculated with paired (C, D, G) or unpaired (H) Student’s t-test; **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S1.
TORC1 phosphorylates the scaffold protein eIF4GI at serine 1108, although the functional consequences of this modification remain unclear (Raught et al., 2000). We determined that TORC1-mediated phosphorylation of eIF4GI was polarized in undivided cells activated in vitro or in vivo (Figure 1E, F, G). As expected, the phosphorylation of eIF4G1 in activated OT-I CD8+ T cells was prevented by treatment with the TORC1 inhibitor, Torin (Figure 1 F, H). In contrast, unphosphorylated eIF4GI was not polarized in activated cells (Figure S1D, E). Unlike p-eIF4GI, Torin-inhibitable phosphorylation of 4EBP1, a well characterized TORC1-mediated modification of the translation initiation machinery, was not polarized (Figure S1F–H). We expect that this was because phosphorylation of 4EBP1 dissociates it from eIF4F, permitting the binding of eIF4G1 (Gingras et al., 1999). The absence of a significant positive correlation between the distribution of phosphorylated eIF4GI and RPL26 argues against a significant contribution of uneven cytosolic volumes to the observed proximal polarization of p-eIF4GI (Figure S1I).
Dynein-dependent vesicular transport mediates polarization of the TORC1-eIF4F complex towards the proximal pole of activated, undivided CD8+ T cells
The mTOR signaling pathway integrates environmental cues to direct the production of proteins, lipids, nucleotides, and ATP during cell growth (as reviewed (Liu and Sabatini, 2020)). The activation of TORC1 involves the recruitment of mTOR from the cytoplasm to the lysosomes. In general, lysosomes are transported through the cell along microtubules (Matteoni and Kreis, 1987). In CD8+ T cells, major rearrangements of the cytoskeleton occur in response to activation, including the relocalization of the MTOC to the immune synapse, to allow for directed transport of cargo, such as lysosomes, throughout the cell (Geiger et al., 1982; Ryser et al., 1982; Stinchcombe et al., 2006). The Dynein motor proteins mediate minus-end-directed transport toward the MTOC, whereas Kinesins mediate plus-end-directed transport, away from the MTOC (as reviewed (Sweeney and Holzbaur, 2018)).
To explore the possibility that upon activation, vesicular transport towards the MTOC at the proximal pole of the cell is involved in the polarization of active TORC1 and eIF4F, we employed high resolution Stochastic Optical Reconstruction Microscopy (STORM). Shortly after activation, we observed phosphorylated eIF4GI co-localized with small vATPase+ structures that arrayed along microtubules at the proximal pole of the cell (Figure 2A–C). This observation was confirmed by nanoscale resolution expansion microscopy (Zhang et al., 2020) (Figure S2A–B, Movie S1). Further, we found in immunoprecipitation experiments that p-eIF4GI and eIF4A interacted with the TORC1 components Raptor and RagC (Figures 2D, E, and S2C) and were associated with ribosomal proteins and c-myc mRNA (Figure S2D, E). Brief inhibition of mTOR activity during activation with Torin was sufficient to reduce the interaction of eIF4A both with RagC and eIF4GI (Figure 2D, E). Treatment of activated, undivided cells with the dynein inhibitor Dynarrestin (Hoing et al., 2018) abrogated the polarization of p-eIF4GI and Raptor (Figure 2F–J and S2F, G). Importantly, TORC1 activity in general, as measured by phosphorylation of ribosomal protein S6, remained unaffected (Figure S2H, I).
Figure 2. Dynein-dependent vesicular transport mediates polarization of the TORC1-eIF4F complex towards the proximal pole of activated, undivided OT-I CD8+ T cells.
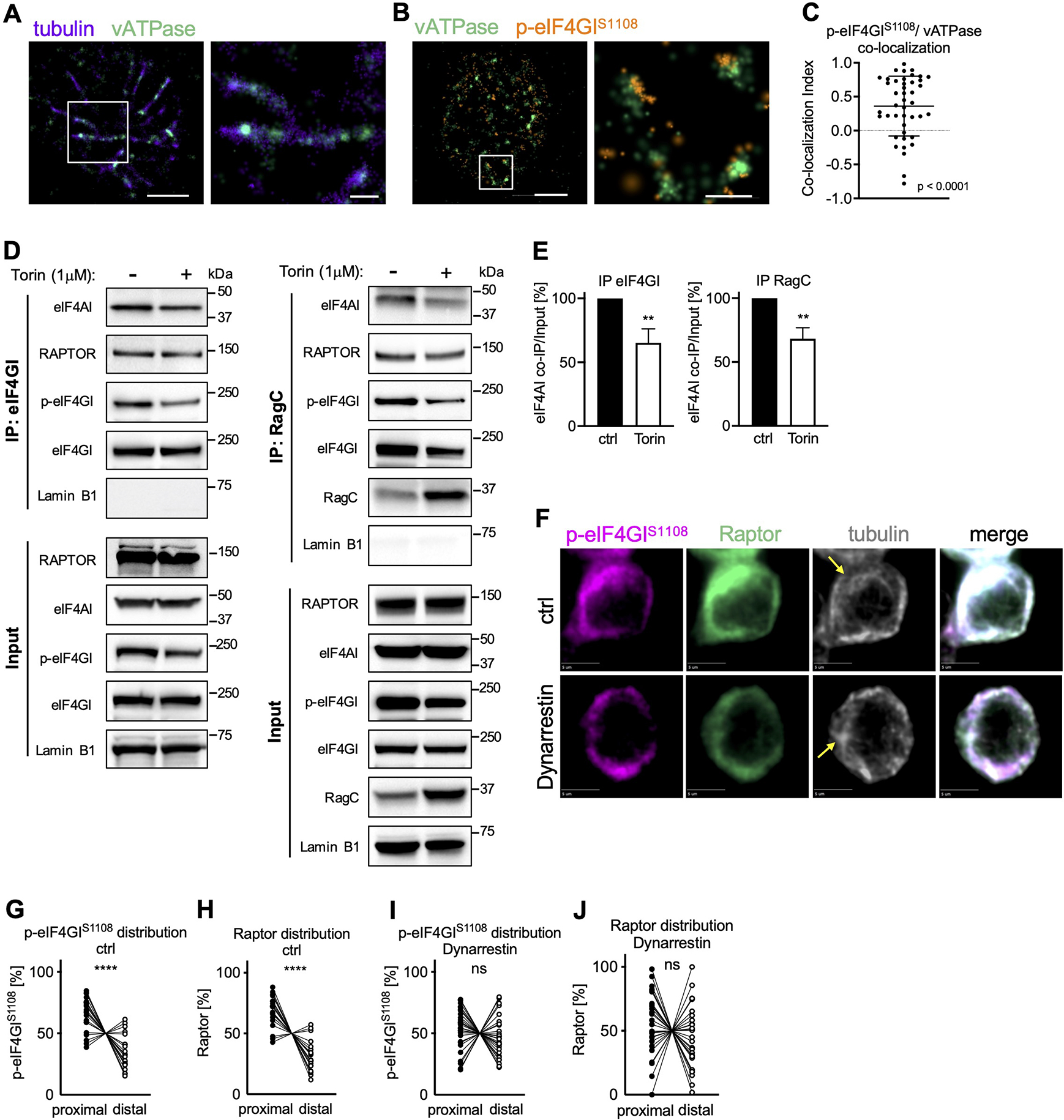
OT-I CD8+ T cells activated on antibody-coated slides as in Figure 1A (A-C) or peptide-pulsed APCs (D-J). (A-C) Association of p-eIF4GIS1108 with vATPase+ vesicular structures along microtubules visualized by STORM. Scale bar wide field (left): 2 μm, zoom (right): 0.5 μm. Representative images (A, B) and quantification (C). (D, E) Co-Immunoprecipitation of eIF4GI (left) and RagC (right) in OT-I CD8+ T cells activated on APCs for 24 h and treated with Torin for the last hour of activation. Representative Western blot (D) and densitometric quantification (E). (F) Representative images of undivided OT-I CD8+ T cells activated on peptide-pulsed APCs and treated with Dynarrestin for the last hour of activation. Scale bar: 5 μm. (G-J) Quantification of the distribution of p-eIF4GIS1108 (G, I) and Raptor (H, J) in control (G, H) and treated (I, J) cells. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell. Arrows mark MTOCs. Compiled from three (C, G-J) or four (E) independent experiments. Significance was calculated with Wilcoxon signed-rank test against 0 (C) or paired (E, G-J) Student’s t-test; **p<0.01, ****p<0.0001, ns=not statistically significant. See also Figure S2.
Taken together, these findings provide evidence that upon T cell activation components of the eIF4F translation initiation machinery associate with and are modified by TORC1 on vATPase+ structures, which are then transported by Dynein along microtubules towards the MTOC.
c-Myc is synthesized at the proximal pole of activated, undivided CD8+ T cells
Our observation that TORC1 and associated eIF4F polarize towards the MTOC suggested that protein synthesis that depends on eIF4F might be similarly polarized. We reasoned that if this complex was active at the proximal pole of activated OT-I CD8+ T cells, the translation of c-myc mRNA would preferentially localize to this region. If so, this would be evidence that the polarized eIF4F complex is functionally active.
Once translated, c-Myc is imported into the nucleus, suggesting that cytosolic c-Myc signal comprises the nascent c-Myc peptide or recently synthesized c-Myc protein. To assess the spatial distribution of c-Myc synthesis in undivided T cells, we therefore visualized cytosolic c-Myc using SoRa (Super-Resolution via Optical Reassignment) imaging. We then measured the distance of cytosolic c-Myc signal to the proximal pole of the cell, using the MTOC as the point of reference. We found that early during T cell activation, cytosolic c-Myc polarized towards the proximal pole of the cell (Figure 3A, B, Movie S2). Similar to c-myc, the Bcl-2 family member mcl1 is an mRNA harboring a long 5’UTR, the translation of which is dependent upon eIF4A (Gandin et al., 2016). We observed that cytosolic Mcl-1 is similarly polarized in activated, undivided cells (Figure S3A, B). In contrast, sirt1 mRNA is characterized by a short, unstructured 5’UTR, the translation of which is less reliant on eIF4A (Elfakess et al., 2011; Sinvani et al., 2015). A similar anfalysis of cytosolic Sirt1 protein, which like c-Myc translocates to the nucleus and thus is likely to be newly synthesized, showed no such polarization with respect to the MTOC (Figure S3C, D, Movie S3). As expected, ribosomes, assessed by staining of RPL26 (Viero et al., 2015), were equally distributed throughout the cell (Figure S3E). The absence of a significant positive correlation between the distribution of cytosolic c-Myc and RPL26 argues against a significant contribution of uneven cytosolic volumes to the observed proximal polarization of cytosolic c-Myc (Figure S3F). Using expansion microscopy, we observed cytosolic c-Myc and p-eIF4GI in the vicinity of the MTOC (Figures S3G and S2B, Movie S4), consistent with these results.
Figure 3. C-Myc synthesis localizes to the proximal pole of activated OT-I CD8+ T cells prior to the first division.
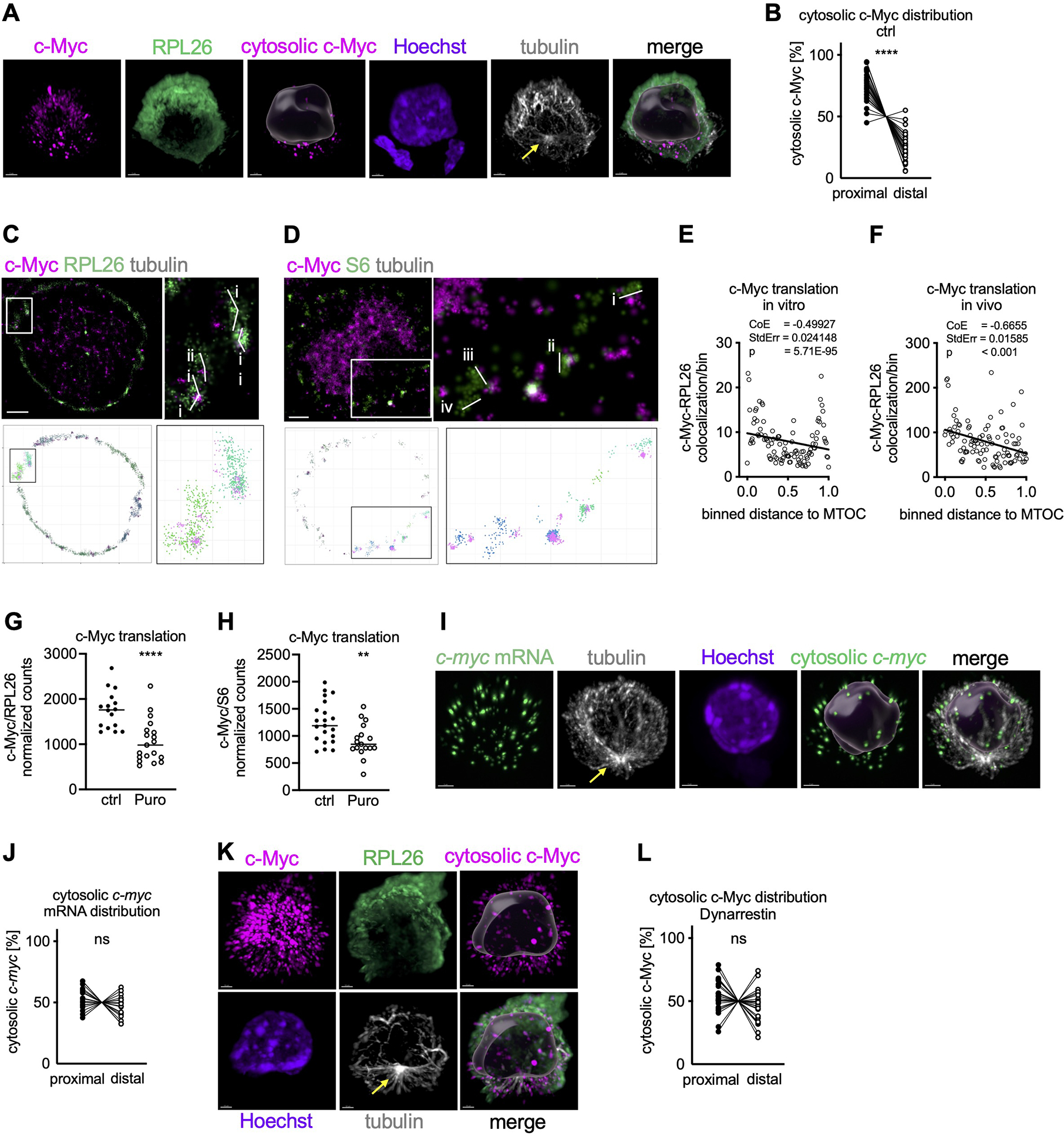
(A, B) Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs using SoRa imaging. Representative image, cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (A). Scale bar: 2 μm. Quantification of the distribution of cytosolic c-Myc within the cell. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell (B). (C, D) Polysomes were identified in STORM images by visual inspection based on size (RPL26: i, 0.52 μm; ii, 0.32 μm; iii, 0.35 μm; iv, 0.45 μm; S6: i, 0.38 μm; ii, 0.32 μm; iii, 0.36 μm; iv, 0.28 μm ) and shape using RPL26 (C) or S6 (D) staining (upper). Polysomes were then identified using DBSCAN in STORM images reconstructed in R (lower). Scale bar: 1 μm. (E, F) Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs (E) or in lymph node sections of peptide-immunized animals (F) using STORM imaging. Relation between number of c-Myc/RPL26 co-localization events and distance towards the MTOC was investigated by Poisson regression. Each dot represents the average number of co-localization events across all analyzed cells. (G, H) Quantification of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs by STORM and treated with Puromycin for the last 10 min of activation. Each dot represents one cell. (I) Representative images of undivided OT-I CD8+ T cells activated on APCs, cytosolic c-myc mRNA is visualized by masking nuclear c-myc mRNA based on Hoechst. Scale bar: 2 μm. (J) Quantification of the distribution of c-myc mRNA within the cell. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell. (K, L) Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs and treated with Dynarrestin for the last hour of activation. Representative SoRa images, cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (K). Scale bar: 2 μm. Quantification of the distribution of cytosolic c-Myc within the cell. Each dot represents signal in the proximal or distal half of a cell. A line connects data points from two halves of the same cell (L). Arrows mark MTOCs. Compiled from two (E, F, G, H, J) or three (B, L) independent experiments. Significance was calculated with paired (B, J, L) or unpaired (G, H) Student’s t-test. **p<0.01, ****p<0.0001, ns=not statistically significant. See also Figure S3.
To further assess whether eIF4F function, as indicated by synthesis of c-Myc, is localized towards the MTOC, we performed STORM imaging (Figure S3H). Using Density-Based Spatial Clustering of Applications with Noise (DBSCAN) (Ester et al., 1996), we identified polysomes using antibodies for the small (RPS6) and the large (RPL26) ribosomal subunits and confirmed visually that these were of the appropriate size and shape (Viero et al., 2015) (Figures 3C, D and S3I). Simulated Emission Depletion (STED) microscopy of RPL26 and S6 confirmed a high degree of colocalization, suggesting the assembly of 80S ribosomes, mirroring our STORM data in size and shape (Figure S3J–M). Next, we found that those polysomes associated with c-Myc N-termini were polarized towards the proximal pole of cells activated in vivo or in vitro, as represented by a negative relation between c-Myc and RPL26 co-association and distance to the MTOC (Figure 3E, F). Brief treatment with the protein synthesis inhibitor puromycin significantly reduced the association of c-Myc with RPL26 and S6, providing further evidence that the identified structures are indeed enriched for functional polysomes engaged in active c-myc translation (Figure 3G, H). In contrast, and in agreement with our SoRa results, Sirt1-associated polysomes and polysomes in general were equally distributed throughout the cytosol (Figure S3N–P). Importantly, c-myc mRNA was equally distributed throughout the cell (Figure 3I, J), supporting the idea that synthesis of c-Myc, rather than location of c-myc mRNA, determines the localization of cytosolic c-Myc.
Together, our observations strongly suggest that the Dynein-mediated polarization of the active TORC1-eIF4F complex to the region of the MTOC results in localized c-Myc synthesis in activated T cells to this region, consistent with the idea that this complex is translationally active. Our results argue against a general polarization of cellular content or c-myc mRNA polarization as an explanation for this localized synthesis of c-Myc. To further test these ideas, we disrupted dynein function with Dynarrestin or Ciliobrevin D, another dynein inhibitor (Firestone et al., 2012). As we had observed for p-eIF4GI and Raptor (Figures 2F–J and S2F, G), brief inhibition of Dynein abrogated the polarization of cytosolic c-Myc (Figures 3K, L and S3Q, R; corresponding untreated control Figure 3A, B; Movies S5, S6).
Inhibition of eIF4A at the time of first division promotes memory-like function
Upon the first division of active CD8+ T cells, TORC1 (Pollizzi et al., 2016) and c-Myc (Verbist et al., 2016) are often asymmetrically distributed in the two daughter cells. Since c-Myc is a short-lived protein, we reasoned that its differential synthesis might be due to an asymmetric assortment of active eIF4F. We therefore examined p-eIF4GI and c-Myc in dividing cells (Figure 4A–C). We found that p-eIF4GI was frequently higher in one daughter, and that this correlated with the distribution of c-Myc. In contrast, total eIF4G was not polarized (Figure S1D, E) and similarly did not assort asymmetrically upon division (Figure S4A, B). Transient treatment with the TORC1 inhibitor Torin was sufficient to equilibrate both p-eIF4GI and c-Myc in dividing sister cells (Figure 4A–C).
Figure 4. Inhibition of eIF4A at the time of first division promotes memory-like function.
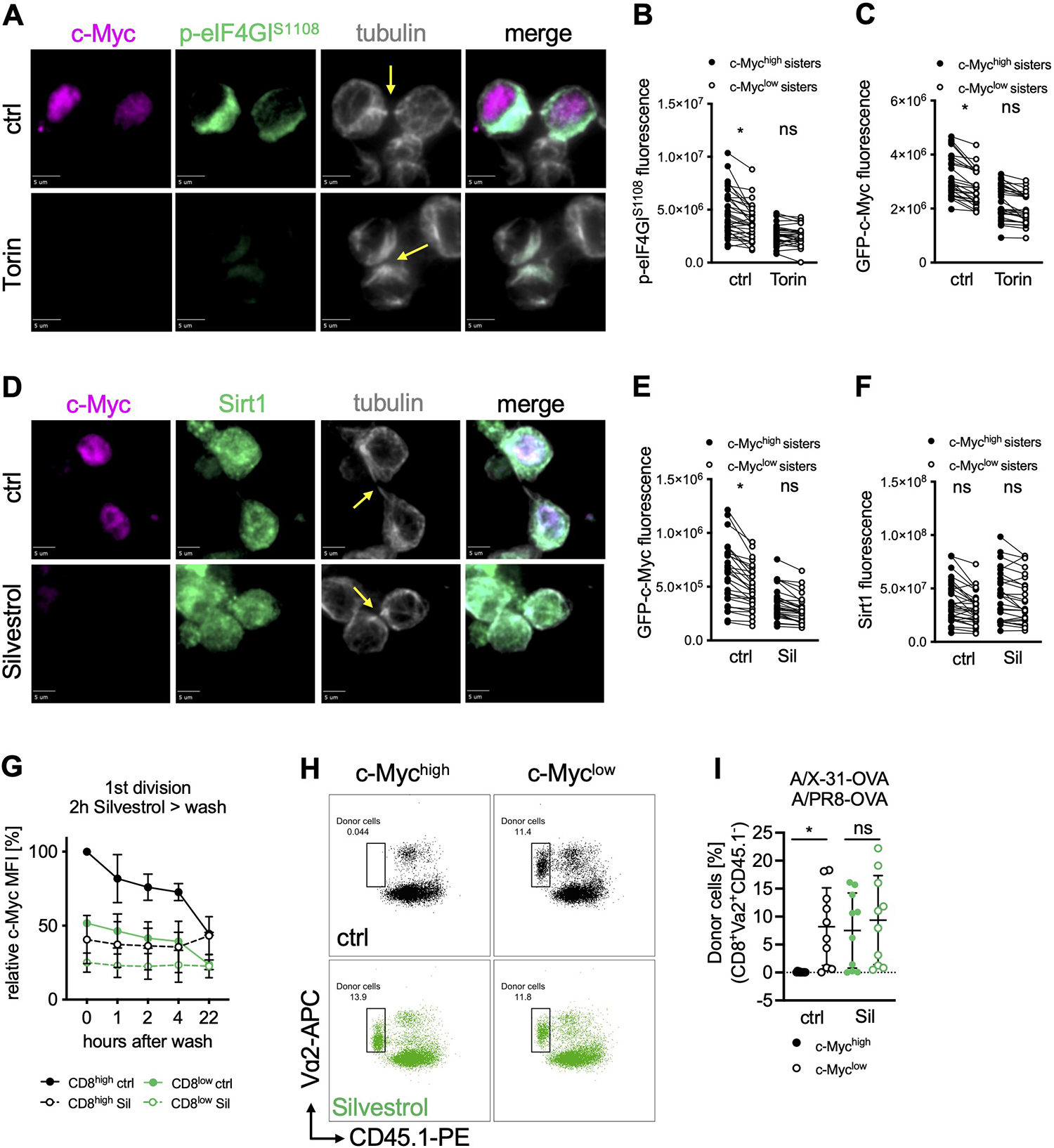
(A) Representative images of conjoined daughter OT-I GFP-c-Myc CD8+ T cells activated for 28 h on peptide-pulsed APCs and treated with Torin for the last hour of activation. Scale bar: 5 μm. (B, C) Quantification of sum fluorescence intensity of p-eIF4GIS1108 (B) or GFP-c-Myc (C) in conjoined sister cells. Each dot represents one cell, data points of sister cells are connected by a line. (D) Representative images of conjoined daughter OT-I GFP-c-Myc CD8+ T cells activated for 28 h on peptide-pulsed APCs and treated with Silvestrol for the last hour of activation. Scale bar: 5 μm. (E, F) Quantification of sum fluorescence intensity of GFP-c-Myc (E) or Sirt1 (F) in conjoined sister cells. Each dot represents one cell, data points of sister cells are connected by a line. (G) MFI of GFP-c-Myc by FACS analysis. OT-I GFP-c-Myc CD8+ T cells were activated for 36 h on peptide-pulsed APCs, treated with Silvestrol for 2 hours, washed and cultured for the indicated time; mean +/− SD, relative to CD8high ctrl. (H, I) Frequency of OT-I GFP-c-Myc CD8+ donor cells (congenic wild type recipients) on day 9 after A/PR/8-OVA challenge. First-division c-Mychigh or c-Myclow cells, treated with Silvestrol for 2 h after sorting, were adoptively transferred into recipient animals before infection with A/X-31-OVA (day 0) and challenge with A/PR/8-OVA (day 30). Representative FACS plots (H) and quantification (I). Each dot represents a recipient animal, n=10; mean +/− SD. Arrows mark tubulin bridges. Compiled from two (G, I) or three (B, C, E, F) independent experiments. Significance was calculated with ANOVA (B, C, E, F, I); *p<0.05, ns=not statistically significant. See also Figure S4 and Table S1.
Silvestrol is a potent inhibitor of eIF4A (Bordeleau et al., 2008; Cencic et al., 2009). Brief treatment of activated OT-I CD8+ T cells with Silvestrol greatly reduced c-Myc bound to chromatin at c-Myc binding sites based on chromatin immunoprecipitation (Figure S4C), as expected (Rubio et al., 2014; Wolfe et al., 2014). Among the affected genes are genes relevant to T cell differentiation and function, including klf4, klf10, bcl6, stat3, and gzmb (Table S1). To test if the asymmetry of c-Myc in first-division sister cells is regulated by eIF4A activity, we analyzed c-Myc protein distribution in cells treated with Silvestrol or Hippuristanol, another eIF4A inhibitor (Bordeleau et al., 2006). We found that inhibition of eIF4A immediately prior to the first division resulted in the rapid equilibration of c-Myc levels in first-division sister cells (Figure 4D, E and S4D–F). Similar effects were observed for Mcl-1, which we found also assorts asymmetrically and is equilibrated by Silvestrol (Figure S4G, H). Since Sirt1 synthesis is less dependent upon eIF4A (Elfakess et al., 2011; Sinvani et al., 2015), we examined Sirt1 protein and found it to be equally distributed between dividing sister cells and unaffected by either Silvestrol or Hippuristanol treatment (Figures 4D, F and S4D, F). The protein distribution of eIF4A, itself, showed no polarization prior to, or asymmetry following first division in activated OT-I CD8+ T cells (Figure S4I–L). Therefore, it is the functional eIF4F complex, rather than eIF4A that appears to be polarized following activation and asymmetrically distributed during cell division. Together, our results demonstrate the importance of eIF4A function for the asymmetric distribution of c-Myc synthesis in first division OT-I CD8+ daughter cells.
We next explored the functional consequences of eIF4A-mediated c-Myc asymmetry. Sorted, first-division c-Mychigh and c-Myclow OT-I CD8+ T cells were treated with Silvestrol for 2 h, sufficient time to reduce c-Myc levels in c-Mychigh cells to those observed in c-Myclow cells (Figure 4G). Next, cells were washed and transferred into congenically distinct wild type recipients. Host animals were infected with a sub-lethal dose of influenza A virus (A/X-31-OVA, H3N2). Nine days post-infection, all populations were present in the peripheral blood at approximately equal levels (Figure S4M). Four weeks post-primary infection, all animals were rechallenged with a heterosubtypic influenza A virus (A/PR/8-OVA, H1N1) and assessed 9 days later. Recovery and analysis of splenic CD8+ T cells revealed that the sorted, untreated c-Mychigh cells failed to respond to the secondary infection, as we had observed previously (Verbist et al., 2016). However, c-Mychigh cells, briefly treated with Silvestrol, were detected at numbers comparable to those found for sorted c-Myclow cells (Figures 4H, I and S4N). These findings suggest that when a naïve CD8+ T cell divides for the first time following activation, the level of c-Myc expression (and potentially other eIF4A-dependent factors) dictates distinct trajectories of cellular function and furthermore, brief inhibition of eIF4A-dependent translation immediately after the first division is sufficient to alter this cell fate trajectory.
After a single cell division, sister cells are often transcriptionally distinct
We next tested the idea that asymmetric synthesis of c-Myc results in transcriptomic differences between sister cells after a single cell division, indicative of distinct cell fate trajectories. Due to technological limitations, strict tracing of sister cell pairs has previously been restricted to small-scale imaging-based methodologies (Chang et al., 2011; Chang et al., 2007; Metz et al., 2015; Oliaro et al., 2010; Pollizzi et al., 2016; Verbist et al., 2016), while larger-scale transcriptomic approaches were limited to population-based analysis, which are unable to identify sister cell pairs (Arsenio et al., 2015; Kakaradov et al., 2017; Metz et al., 2015). Therefore, whole-transcriptome analysis that links ACD to the subsequent function of each sister cell is lacking, culminating in an ongoing debate on the role of ACD in generating transcriptional diversity following the first division of T cells.
We employed an endogenous barcode transgenic mouse model (BCM) (Gerlach et al., 2013) to determine the transcriptional profiles of sister OT-I CD8+ T cells after a single division and assess the contribution of ACD to early transcriptional diversity. In this system, transient V(D)J recombinase expression during T cell development results in recombination and nucleotide diversification of a pseudo-V(D)J substrate. Successful recombination drives GFP expression and provides the cell with a unique genetic barcode, which is inherited by the daughter cells upon division. This allowed the identification of individual sister cell pairs and analysis of their transcriptional profiles.
Using single-cell sequencing of barcode and gene expression libraries (Figure S5A–E, see Methods), we identified 62 barcode pairs from first-division OT-I CD8+ T cells. Once visualized with UMAP plots, it became clear that many putative sister cells could be characterized by transcriptional similarity within the pair, as depicted by two connected cells in close proximity in UMAP space (Figures 5A and S5F). However, within another subset of sister cells, the two cells of a pair were transcriptionally distinct, as might be expected had ACD occurred.
Figure 5. Transcriptional heterogeneity in sister cells is established as early as the first division.
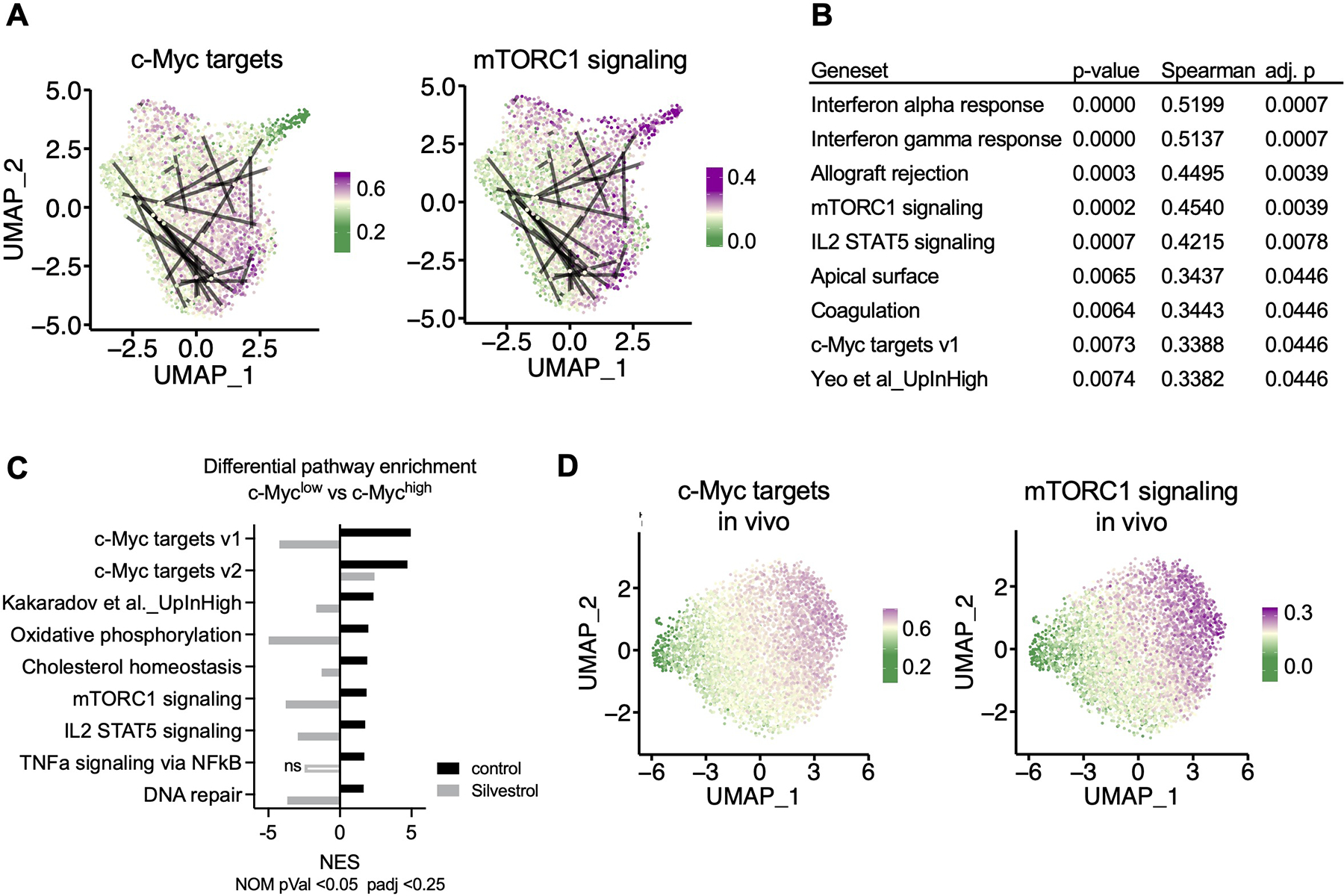
OT-I BCM CD8+ T cells were active for 36h on peptide-pulsed APCs and subjected to single cell RNA-Seq (A, B, D) or microarray (C). (A) Uniform Manifold Approximation and Projection (UMAP) plots representing single-cell gene expression data of murine first division OT-I BCM CD8+ T cells obtained from barcode experiment replicate A. Lines connect putative sister cells. Color gradient corresponds to specific gene set module scores, where the gradient midpoint (light yellow) is set to the median module score. (B) Statistical summary of Spearman correlations between pairwise Euclidian PCA distances and absolute differences in gene set module scores for putative sister cell pairs. P-values were adjusted to an FDR <0.05, and only correlations with an adjusted p-value of <0.05 were included. (C) Gene Set Enrichment Analysis comparing first-division c-Mychigh and c-Myclow sorted cells, control or treated with Silvestrol for the last hour of activation. Normalized Enrichment Score (NES) is presented for each treatment for all gene sets that were significantly (p < 0.05, FDR < 0.25) enriched in the c-Myc high control condition. Positive NES corresponds to enrichment in the c-Myc high condition, negative NES corresponds to enrichment in the c-Myc low condition. (D) UMAP plots representing single-cell gene expression of first-division OT-I GFP-c-Myc CD8+ T cells obtained from replicate A of three independent in vivo experiments, otherwise presented as described in (A). See also Figure S5.
To investigate potential regulators of transcriptional similarity and dissimilarity between confirmed sister cell pairs in an unbiased manner, we asked if variation in known gene sets (see Methods) correlated with broadly defined signals of general transcriptional variation. Using the first two principal components from PCA analysis, we calculated Euclidean distance between putative sisters. We use distances calculated from PCA space because distances in UMAP space can be warped. We looked for correlations between those distances and absolute differences in gene set expression modules. Of the 54 gene sets tested, we found significant correlations between sister-pair distances and gene set differences for 9 sets after adjustment for multiple comparison (Figure 5B). These significant correlations included gene sets associated with c-Myc target gene expression, mTORC1 signaling, and a gene set previously associated with asymmetric division and cell fate determination (Kakaradov et al., 2017). The enrichment of these gene sets was lost upon transient inhibition of eIF4A with Silvestrol, as assessed by microarray analysis comparing first-division c-Mychigh and c-Myclow cells (Figure 5C).
Using a probabilistic model of V(D)J rearrangement specifically for this BCM system (Marcou et al., 2018), we were able to estimate probabilities of generation (Pgen) for observed barcodes. By comparing the barcode Pgen for each putative sister pair to their pairwise Euclidean PCA distance, we found that the barcodes for transcriptionally distinct sister pairs were no more or less likely to arise in the system than those from transcriptionally similar sisters (Figure S5G), ruling out the possibility that these analyses were biased due to potential convergence in barcode recombination. In order to further contextualize the extent of transcriptional differences between putative sisters, we also related sister-pair distances to the general transcriptional heterogeneity within each experiment, dividing each distance by the median distance from all randomly selected cell-cell pairs within the experiment. With this Median Random Pair Distance (MRPD) ratio, a value of 0 therefore indicates identical cell pairs, while a value of 1 indicates sisters that exhibit the median transcriptional differences between randomly selected cell pairs in the experiment. Distances between putative sister-cells were significantly greater than would be expected of identical cell pairs (p = 7.766e-12, two-sided Wilcoxon Rank Sum test with a null hypothesis of 0), and on average reflected 1.03x (SD = 0.75) the median transcriptional distance within an experiment (Figure S5H). Thirty putative sister cell pairs exhibited distances greater than 1x (relatively asymmetric), while distances of 32 sister cell pairs were shorter than 1x (relatively symmetric), which reflects frequencies of asymmetric cell division observed in other studies (Chang et al., 2007; Verbist et al., 2016).
To ensure that differences between sister pairs were not specific to our in vitro barcode model, we also generated single-cell expression datasets from first-division T cells without barcodes, stimulated in vivo. These data exhibited similar patterns to those described for our in vitro experiments (Figures 5D and S5I).
Taken together, our data suggest that a major source of transcriptional variation across populations of first-division T cells correlates with gradations in expression of c-Myc targets and mTORC1 signaling and, furthermore, that distinct transcriptional profiles between sister cells are established as early as the first division.
Discussion
Our results provide mechanistic insight into the early molecular events that occur during the first division of an activated T cell promoting transcriptional and functional diversity. As part of the early events during T cell activation and mediated by Dynein-dependent lysosomal transport, active TORC1 localizes towards the MTOC at the proximal pole of the cell. The interaction of TORC1 with components of the eIF4F translation initiation complex results in the polarization of polysomes translating c-myc. As cells enter mitosis, the MTOC divides to form two centrioles, with the original MTOC representing the “mother centriole”. The localization of the active TORC1-eIF4F complex near this centriole results in its asymmetric distribution upon division, predisposing the two daughter cells to differentially synthesize c-Myc, where it contributes to the initiation of distinct transcriptional profiles.
Previous studies have shown that the affinity of the T cell receptor for its cognate ligand influences whether an activated T cell will undergo asymmetric or symmetric division, with higher affinities corresponding to the former (King et al., 2012). It will be interesting to determine if affinity affects the localization of the MTOC and/or Dynein-dependent transport of the TORC1-eIF4F complex to the proximal pole. If so, this may help to explain the association between higher affinity interactions and ACD.
Our data suggest that at the time of the first division, cell fate trajectories can be manipulated by targeting c-Myc expression (and potentially other proteins that rely on eIF4F-dependent translation). Brief treatment of c-Mychigh, first division cells with Silvestrol was sufficient to induce long-term effects on their memory-like potential. In addition to c-Myc, Silvestrol inhibits the translation of components of the cell cycle machinery, including Cyclin D1 (Rubio et al., 2014; Wolfe et al., 2014). Consistent with this, we have observed that c-Myclow first division daughter cells cycle more slowly than do their c-Mychigh counterparts (Verbist et al., 2016). Nevertheless, reduction of c-Myc levels in c-Myc+/− CD8+ T cells promotes memory-like T cell fate (Verbist et al., 2016), suggesting that the effects we observed with transient Silvestrol treatment were due to the reduction in c-Myc levels in the period following first division.
In addition to c-Myc, we found that the synthesis of Mcl-1 was polarized in activated, undivided CD8+ T cells, and asymmetrically assorted to first division daughter cells. Like c-Myc, Mcl-1 synthesis is dependent upon the function of the eIF4F complex and TORC1 activity (Gandin et al., 2016). Our observations are therefore consistent with the idea that the localization of active TORC1-eIF4F complex leads to its asymmetric distribution upon cell division, and thus unequal expression of eIF4F-dependent proteins. Mcl-1 is an anti-apoptotic member of the Bcl-2 family, with a short half-life and important roles in development and homeostasis (Perciavalle and Opferman, 2013). While Mcl-1 is required for the survival of naïve and activated T cells (Dzhagalov et al., 2008), we do not know if such asymmetric expression of Mcl-1 protein plays any role in cell fate.
The principles we have elucidated in dividing CD8+ T cells may not apply to other cell types that undergo ACD. We previously found that levels of the protein Numb positively correlate with c-Myc in T cells that had divided asymmetrically (Verbist et al., 2016). In contrast, in asymmetrically dividing hematopoietic stem cells (HSC), Numb and c-Myc were observed to inversely correlate (Loeffler et al., 2019). Further, in this study, while lysosomes were found to asymmetrically assort in daughter cells, those with fewer lysosomes were associated with increased expression of c-Myc. The authors concluded that higher levels of protein degradative machinery, including lysosomes and autophagosomes, was associated with the maintenance of stemness in the daughters. Since autophagy is negatively regulated by TORC1, these results suggest that TORC1 activity is not associated with lysosomes in dividing HSC. Indeed, unlike activated CD8+ T cells, HSC did not begin to express elevated c-Myc until after cell division had completed (Loeffler et al., 2019).
Compartmentalized translation as a mechanism of spatiotemporal regulation of cellular processes has been described in various models including axonal translation in neurons, local translation at cell protrusions, the outer mitochondria membrane or the endoplasmic reticulum, chromosomal translation during meiotic progression of mammalian oocytes, and asymmetric protein translation in embryogenesis. Most commonly, compartmentalized protein translation is achieved by mRNA localization. The process of mRNA localization in general is regulated by a target sequence within the mRNA molecule, which is recognized by RNA-binding proteins that facilitate interaction with the intracellular transport machinery, as well as transport and anchoring to the final destination. Less commonly, mRNA localization is mediated by localized protection from degradation or passive diffusion and local entrapment (as reviewed (Blower, 2013; Buxbaum et al., 2015)). The coding region of c-myc mRNA is targeted by c-myc coding region determinant-binding protein (CRB-BP), a member of an RNA-binding protein family implicated in the regulation of intracellular RNA localization (Doyle et al., 1998). However, while binding of CRB-BP to c-myc mRNA affected its stability (Doyle et al., 1998), a potential impact on c-myc mRNA localization within a cell has not been reported. In this study, we found no evidence of localization of c-myc mRNA. Instead, we provide evidence that the polarization of c-Myc synthesis in activated T cells is due to polarization of the translational machinery required to express this protein. It will be interesting to determine if other specialized translational machinery can also be polarized under some conditions to localize synthesis of specific proteins.
Previous studies have shown that CD8+ T cells can display differences in gene expression indicative of cell fate as early as the first cell division (Chang et al., 2011; Chang et al., 2007; Metz et al., 2015; Oliaro et al., 2010; Pollizzi et al., 2016; Verbist et al., 2016). Combining a genetic barcode with single-cell sequencing technology, our data provide a quantitative assessment of the occurrence of asymmetrically divided first division T cells as well as an unbiased analysis of transcriptional variation between genuine sister cells. Our findings provide a demonstration that ACD can contribute to differential cell fate trajectories. This idea, however, is controversial for several reasons, in part due to the demonstration that memory T cells show evidence of having expressed markers of effector T cells, such as Granzyme B (Jacob and Baltimore, 1999). At this point, we do not know if the c-Myclow first-division cells that contribute to the recall response similarly have expressed Granzyme B at some point in their fate trajectory. Nevertheless, while we cannot assert that ACD is necessary for the generation of T cell memory, as suggested by others (Borsa et al., 2021), we suggest that ACD can contribute to the generation of T cells with divergent cell fate trajectories that can be manipulated through interventions (such as Silvestrol treatment) shortly after the first division. A recent study has similarly shown that inhibition of TORC1 can promote ACD and the generation of cells with increased memory potential and improved recall responses following viral infection (Borsa et al., 2019). The ability to shape T cell function through minimal pharmacological intervention opens potential therapeutic avenues to improve vaccines and advance T cell immune therapies.
Limitations of the Study
While we provide several lines of evidence that the function of the eIF4F complex is localized towards the MTOC in activated CD8+ T cells and can asymmetrically assort to first-division daughter cells, direct visualization of translation of specific proteins in single cells (Biswas et al., 2019) is lacking for primary T cells. Such direct visualization would require extensive genetic engineering, for example, of the c-myc locus. Similarly, assessment of the effects of removing the requirement for eIF4A for c-myc translation, e.g., by replacing the 5’ UTR of c-myc may prove interesting. Another limitation of our study concerns the small numbers of confirmed sister cell pairs we could assess by single cell RNA-Seq. This was due to the small numbers of primary cells that expressed the endogenously generated barcode and the stringent analysis we employed to confirm unique barcodes, and the diversity of barcodes generated by the system. Finally, we note that our studies relied on the use of the OVA-specific OT-I transgene, and it will be important to extend our results to other T cell receptors.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Douglas Green (Douglas.Green@stjude.org).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Single-cell RNA-seq data have been deposited at BioProject, accession PRJNA641918. CUT&RUN data has been deposited at GEO accession GSE196967.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 and OT-I Tg mice (C57BL/6-Tg (TcraTcrb)1100Mjb/J) were acquired from The Jackson Laboratory (Bar Harbour, Maine). GFP-c-Myc fusion knock-in mice were generated and provided by B. Sleckman (Huang et al., 2008) and bred with OT-I Tg mice. BCM mice were generated and provided by T. Schumacher (Gerlach et al., 2013). All animal experiments were performed with both female and male sex- and age-matched littermate controls (6–10 weeks old). All mice were bred and housed in specific pathogen-free facilities, in a 12-hour light/dark cycle in ventilated cages, with chow and water supply ad libitum, at the Animal Resources Center at St. Jude Children’s Research Hospital.
Mouse studies were conducted in accordance with protocols approved by the St. Jude Children’s Research Hospital Committee on Care and Use of Animals and in compliance with all relevant ethical guidelines.
Murine OT-I CD8+ T lymphocytes
For OT-I CD8+ T cell stimulation, splenocytes obtained from C57BL/6 mice were enriched for antigen presenting cells (APCs) using CD11c MicroBeads (130–108-338, Miltenyi Biotec) and pulsed with 5 nM of SIINFEKL peptide (AS-60193–1, AnaSpec Inc., not specified as free of endotoxin) for 1 h at 37°C. OT-I CD8+ T cells were isolated from lymph nodes of either OT-I Tg or GFP-c-Myc OT-I using the Dynabeads Untouched Mouse CD8 Cells Kit (11417D, Invitrogen) and overlain onto the peptide-pulsed APCs. Alternatively, OT-I CD8+ T cells were stimulated on plate-bound anti-CD3ε (1 μg ml−1, BE0001–1, Bio X Cell), anti-CD28 (1 μg ml−1, BE0015–1, Bio X Cell), and recombinant human ICAM (0.5 μg ml−1) produced in insect cells (Huppa et al., 2010); or by incubation with PMA (10 ng/ml, HY-18739, MedChemExpress) and Ionomycin (1 μM, HY-13434, MedChemExpress). Whenever assessment of cell division was required, OT-I CD8+ T cells were labeled using the CellTrace Violet (CTV) Cell Proliferation Kit (C34557, Invitrogen) at 5 μM per 1 × 107 cells for 10 min at 37°C. For in vivo stimulation, CTV labeled OT-I CD8+ T cells were transferred by tail vein injection of wild-type recipients. 24 h after transfer, recipients were immunized by subcutaneous injection of 50 μg of SIINFEKL peptide into the neck region. Donor cells were obtained from axillary and brachial lymph nodes 8 h after immunization. OT-I CD8+ T cells were cultured in RPMI 1640 (11875093, Gibco) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS), 1x MEM non-essential amino acids (11140050, Gibco), 1mM Sodium pyruvate (11360070, Gibco), 2 mM L-glutamine (25030081, Gibco), 55 μM 2-mercaptoethanol (21985023, Gibco), 50 U ml−1 penicillin and 50 μg ml−1 streptomycin (30001-Cl, Corning) at 37°C in 5% CO2. For all imaging, CD8+ T cells were cultured in 4-well μ-slides (80426, ibidi). Where indicated, OT-I CD8+ T cells were treated with Silvestrol (200 nM, HY-13251, MedChemExpress), Hippuristanol (500 nM, gifted by J. Pelletier), Torin 1 (1 μM, S2827, Selleckchem), Dynarrestin (25 μM, SML2332, Sigma), or Ciliobrevin D (50 μM, HY-122632, MedChemExpress) for the last hour of activation, or Puromycin (91 μM, 4098, Tocris).
METHOD DETAILS
Single-cell RNA sequencing (scRNA-Seq)
OT-I BCM CD8+ T cells labeled with CTV were stimulated on peptide-pulsed APCs for 36 h. Cells were stained with anti-CD8-APC (17–0081-82, Invitrogen) and anti-CD44-PE/Cy7 (103030, BioLegend) for sorting on a MoFlow (Beckman-Coulter). First-division OT-I BCM CD8+ T cells expressing a barcode (CD8+, CD44+, CTV 2nd peak, GFP-bcm+) were then sorted into culture medium, washed once with PBS + 0.04% BSA, and re-suspended in 32 μl PBS + 0.04% BSA. Single-cell suspensions were loaded onto the Chromium Controller to generate up to 10000 single-cell gel beads in emulsion (GEMs) per sample. Single-cell gene expression libraries were prepared using the Chromium Single Cell 5’ v2 Library and Gel Bead Kit (10x Genomics) for BCM experiments and the 3’ v2 kit for non-BCM experiments. Resulting libraries were sequenced on Illumina HiSeq or NovaSeq platforms at 26×98bp.
For in vivo stimulation, CTV labeled OT-I CD8+ T cells were transferred by tail vein injection of wild-type recipients. 24 h after transfer, recipients were immunized by subcutaneous injection of 50 μg of SIINFEKL peptide into the neck region. Donor cells were obtained from spleens and peripheral lymph nodes 24 h after immunization and labeled, sorted, and Barcode-containing transcripts were enriched from amplified cDNA via target enrichment PCR amplification (1st fwd-AAT GAT ACG GCG ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT C, 1st rev-GCT GAA CTT GTG GCC GTT TA, 2nd fwd-AAT GAT ACG GCG ACC GAG ATC T, 2nd rev-CGT CCA GCT CGA CCA GGA T) in order to create a separate single-cell barcode library (Kapa HyperPrep Kit), which was sequenced at 250×250bp. Quality controls and quantifications were performed using High Sensitivity D5000 Screen Tape (5067–5588, Agilent Technologies) with a 4200 TapeStation (Agilent Technologies).
Analysis for scRNA-Seq data
The single-cell data can be divided into two broad experiments, each with independent replicates. For the barcode experiments, single-cell gene expression data were processed with CellRanger (v3.1.0, 10xGenomics) using the accompanying mouse transcriptomic reference (v3.0.0), and the processed libraries were aggregated after normalizing for the number of confidently mapped reads per cell (BCM and non-BCM libraries were aggregated separately due to the distinct molecular chemistry used to generate each). Filtered aggregation outputs were subsequently analyzed using Seurat (v4.0.2) (Hao et al., 2021) and following standard procedures. For BCM libraries, the aggregated data were used to create a Seurat object with 10,149 putative cells (3,511 and 6,638 for each replicate, respectively) and 12,038 genes (only retaining genes found in a minimum of 0.3% of cells, i.e., ~30 cells). Independent replicates exhibited similar distributions of typical QC features, including number of genes per cell, number of RNA molecules per cells, and percent of mitochondrial expression per cell (Figure S5B); however, the replicate with the larger number of cells exhibited slightly more cells that fell outside the general range of genes per cell and RNA molecules per cell, as expected given the increasing frequency of cell multiplets as more cells are recovered from a 10x reaction. Filtering thresholds were decided based on the distributions of these QC metrics. To exclude putative multiplets, we filtered out cells with more than 6,000 genes or more than 42,000 UMIs. We also filtered out cells with >7.5% of expression owed to mitochondrial genes to exclude dead or dying cells (Figure S5C). We subsequently identified a population of cells with relatively few genes and very low mitochondrial expression (a feature commonly observed in some versions of the CellRanger cell calling algorithm); to exclude these transcriptionally inactive cells and false cell calls, we filtered out cells with <0.1% of expression owed to mitochondrial genes and cells with fewer than 300 genes. After filtering, we were left with 2,192 and 3,771 cells per replicate, respectively; importantly, these filtered data exhibited expected relationships between the observed number of genes and mitochondrial expression (Figure S5D) and between the number of UMIs and number of genes (Figure S5E).
Data were then LogNormalized within Seurat using a scale.factor of 1e4, and cell cycle phase was inferred using the CellCycleScoring function with markers obtained from Tirosh et al. (Tirosh et al., 2016). After filtering the two independent replicates together to ensure no biases in these processing steps, the two independent replicates were then split from the filtered, normalized Seurat object to be analyzed independently. This was done to demonstrate independent replication of the patterns we observed. For each independent replicate, we separately scaled the data, regressing out the number of UMIs and the percent of expression owed to mitochondrial genes in each cell. We then used the ‘vst’ method to find 2,000 variable features (again, separately for each replicate), after excluding any possible variable features owing to V(D)J gene segments, which are known to map poorly. We used those variable features for Principal Component Analysis (PCA) and UMAP analysis (with min.dist = 0.5) for each replicate.
In order to broadly survey variation in major functional pathways, we utilized expression modules from a number of curated gene sets, including the entire Hallmark collection (obtained from the msigdbr package in R; (Subramanian et al., 2005)), two gene sets previously described in the context of asymmetrical T cell division (Kakaradov et al., 2017), and a gene set comprised of c-Myc target genes involved in metabolic processes (Wang et al., 2011). To do this, we used the AddModuleScore function in Seurat for each gene set within each independent Seurat object. To match the maximum number of genes between the gene sets and the mouse reference, we utilized the UpdateSymbolList function directly rather than using the search = TRUE parameter in the AddModuleScore function.
Once each cell was assigned a module score for each gene set, we were able to look for differences between cell-cell pairs. In the barcode experiments, for putative sister cells specifically, we calculated absolute differences in each module between the two sister cells. We also calculated distances between cell pairs using Euclidean distances between PC1 and PC2 (using the pointDistance function in the raster R package). Correlations were assessed using Spearman correlations (specifically using the cor.test function with method = “Spearman” and use = “complete.obs”), and p-values were adjusted for multiple testing with False Discovery Rate (FDR).
Barcode-containing transcripts were enriched from amplified cDNA via target enrichment PCR to create single-cell barcode libraries, which were sequenced at 250×250bp and processed with CellRanger VDJ (v3.0.2) using a custom reference based on the potential barcode segments and arrangements present in the endogenous BCM system (Gerlach et al., 2013). Filtered contigs from the VDJ assemblies were aligned to the genomic sequence corresponding to the region of barcode rearrangement using MUSCLE (v3.8.31) (Edgar, 2004). The resulting alignments were parsed with custom python scripts to trim the contigs to only span between the conserved regions immediately upstream (TTACCTCCTCGAGGTCA) and downstream (CATGGTGAGCAAGGGC) of the rearrangements.
Because of the known limitations of this BCM system, we set out to characterize patterns of convergent recombination among cells within each experiment by identifying barcodes that appeared more than once in an undivided population, more than twice in a first-division population, or three or more times in undivided and first-division populations from the same experiment. To this end, we activated BCM OT-I transgenic CD8+ T cells on APCs pulsed with the OT-I T cell cognate antigen SIINFEKL for 36 h. OT-I CD8+ T cells expressing a barcode (CD8+, CD44+, GFP-bcm+) were sorted by flow cytometry into undivided, first and second division cell populations based on CellTrace Violet intensity followed by preparation and sequencing of single-cell barcode libraries using 10x genomics technology. In total, we sequenced 7,975 cell-specific barcodes across 11 barcode libraries derived from either undivided, first-division, or second division cells. Overall, barcodes were assembled from a median of 51,891 sequencing reads derived from a median of 56 unique transcripts (range: 1–557 UMIs) per cell. Convergent rearrangements were responsible for approximately half of the barcodes we sequenced, but the majority of these problematic barcodes were derived from a limited number of rearrangements; for instance, more than 43% of the problematic barcodes we identified were derived from a single common rearrangement that was only observed in a single experiment. The 62 barcodes that were each shared between a single pair of first-division cells were assembled from a median of 52,116 reads (range: 6,321–178,130) derived from a median of 39.5 unique transcripts (range: 8–198 UMIs) per cell and exhibited variation in length ranging from 92 to 132bp.
To address if putative sisters that were transcriptionally different may have arisen from distinct lineages and were only sharing a barcode as a result of instances of convergent barcode rearrangement that we were unable to exclude, we generated a probabilistic model of V(D)J rearrangement specifically for this BCM system. Probabilities of generation (Pgen) for barcode sequences were estimated with IGoR (Marcou et al., 2018), using the BCM segments and potential arrangements as a custom mouse TCRβ reference. The recombination model was inferred using the total set of barcode sequences (including duplicates), with a probability ratio threshold of 1e-20, a sequence likelihood threshold of 1e-100, and 10 expectation-maximization algorithm iterations. We were able to generate finite estimates of Pgen for >99% of unique barcode sequences using this model.
Whereas barcodes we identified as problematic exhibited a median probability of generation of approximately 1.8 in 100,000, all other barcodes had much lower Pgen estimates, with a median probability of approximately 6 in 100 million. In contrast, we estimated a Pgen of 7 in 10,000 for the most frequently observed convergent rearrangement across experiments.
Single-cell data from first-division cells activated in vivo were generated using 3’ v2 10x kits. The data were processed in the same manner as described above. Filtering thresholds were changed slightly to reflect the distinct distributions of the QC metrics for each library.
Microarray
CTV-labeled GFP-c-Myc OT-I CD8+ T cells were activated on peptide-pulsed APCs for 36 h. First-division GFP-c-Mychigh and GFP-c-Myclow cell populations (CD8+, CTV 2nd peak, highest and lowest 20% GFP-c-Myc) were sorted into medium with or without 200 nM Silvestrol and cultured for additional 2 h. RNA was isolated as described below. RNA quality was assessed on the Agilent 2100 Bioanalyzer with the nano reagent kit, and concentration was determined by nanodrop. 125 ng of intact high-quality RNA was processed using the Affymetrix Whole Transcript (WT) Plus assay (Thermo Fisher). Resulting cDNA was then incubated on the Clariom S mouse array (Thermo Fisher) for 16 hours at 45°C while rotating at 60 rpm. The cartridges were stained and washed on a Gene Chip FS450 fluidics station and scanned with the Gene Chip Scanner 3000 7G (Thermo Fisher). Data were characterized using Gene Set Enrichment Analysis (GSEA, v4.2.2; (Subramanian et al., 2005)) with the hallmark gene set collection from MSigDB (Liberzon et al., 2015) as well as two gene sets previously described in the context of asymmetrical T cell division (Kakaradov et al., 2017). Probes were mapped using the Clariom_S_Mouse.r1_MSigDB.v7.1_ REMAPPED_PATCH chip from MSigDB, and enrichment was detected with gene set permutation. P-values were adjusted for multiple comparisons using the False Discovery Rate (FDR) over the entire set of gene sets tested, with significant enrichment denoted by a nominal p-value < 0.05 and an FDR q-value < 0.25 as recommended by GSEA (Subramanian et al., 2005).
Immunofluorescence staining and imaging
OT-I CD8+ T cells were stimulated on peptide-pulsed APCs for 28 h followed by fixation for 10 min at room temperature (RT) with paraformaldehyde (PFA) (15710, Electron Microscopy Science) directly added to the culture medium at 4% (v/v) final concentration. Cells were rinsed with TBS (50 mM Tris pH 8 (T3038, Sigma-Aldrich), 100 mM Sodium Chloride (S5150, Sigma-Aldrich) in ddH2O) and permeabilized with TBS + 0.1% (v/v) Triton X-100 (93443, Sigma-Aldrich) for 3 min at RT. Non-specific binding was blocked with TBS + 2% BSA (A7030, Sigma-Aldrich) for 30 min at RT. Samples were incubated overnight at 4°C with the following primary antibodies: anti-c-Myc (1:500, 5605, Cell Signaling Technology), anti-Sirt1 (1:500, ab110304, Abcam), anti-eIF4A1 (1:500, ab31217, Abcam), anti-phospho-eIF4GIS1108 (1:500, 2441, Cell Signaling Technology), anti-eIF4GI (1:500, 2858, Cell Signaling Technology), anti-Mcl1 (1:500, 24296, Cell Signaling Technology), anti-phospho-mTORS2481 (1:500, 2974, Cell Signaling Technology), anti-mTOR (1:500, 4517, Cell Signaling Technology), anti-CD11c (1:500, 117301, BioLegend), anti-tubulin (1:1000, MA1–80189, Invitrogen), anti-S6 (1:500, 2317, Cell Signaling Technology), anti-tubulin (1:1000, PA5–19489, Invitrogen), anti-tubulin (1:1000, 13–8000, Invitrogen). Samples were washed with TBS and incubated for 1 h at RT with the following secondary antibodies: Donkey anti-mouse AFplus488 (1:1000, A32766, Invitrogen), donkey anti-rabbit AFplus555 (1:1000, A32794, Invitrogen), donkey anti-rat AF647plus (1:1000, 712–605-153, Jackson ImmunoResearch), and goat anti-hamster AF488 (1:500, 127–545-160, Jackson ImmunoResearch).
For imaging of in vivo stimulated OT-I CD8+ T cells, axillary and brachial lymph nodes were fixed in PBS containing 2% PFA, 0.1% T-100, and 1% DMSO overnight at 4°C. Tissues were subsequently cryosectioned onto charged glass slides. Sections were blocked in PBS containing 1% BSA prior to incubation with primary antibodies mentioned above at 1:250 dilution overnight at 4°C. Slides were washed in PBS and incubated for 1 h at RT with secondary antibodies mentioned above at 1:500 dilution. Slides were mounted with Prolong Glass Antifade Mountant (P36980, Invitrogen).
Detection of c-myc mRNA was facilitated using reagents from Advanced Cell Diagnostics, per the manufacturer’s protocol. In brief, antibody-activated cells were fixed with 4% PFA in PBS for 15 min, washed, permeabilized with 0.1% Triton-100 for 3 min, and blocked with 1% BSA in PBS for 10 min at RT. Cells were subsequently stained with rat anti-tubulin antibody (clone YOL1/34) for 15 min at RT prior to detection with AF647-conjugated donkey anti-rat secondary (Jackson Immuno Research) for 15 min at RT. Samples were post-fixed in 1% PFA in PBS prior to dehydration in an ethanol series. Cells were rehydrated to PBS and protease digested for 15 min, followed by hybridization with a c-Myc RNA probe (413451, ACD) and detection utilizing a fluorescent reagent (320851, ACD) per the manufacturer’s direction.
For imaging, a Marianas confocal (Intelligent Imaging Innovations) comprised of a CSU-X spinning disk, Prime95B sCMOS camera, and 405, 488, 561 and 640 nm laser lines was used. Alternatively, a CSU-W (Yokogawa) spinning disk to facilitate super resolution via optical reassignment (SoRA) imaging in combination with a 100X 1.45 NA oil objective and Prime 95B camera was used.
Diving sister cells were identified by the presence of an intercellular cytokinetic bridge based on tubulin staining. Using Slidebook 6 imaging software (Intelligent Imaging Innovations), regions for each sister cell were created and converted to masks. Masks statistics of the sum fluorescent intensities were analyzed.
The distribution of proteins in undivided cells was analyzed using Imaris 9.5 software (Oxford Instruments). A surface object and spots were created to segment the microtubule-organizing center (MTOC) and the indicated proteins, respectively. The distance of protein to the MTOC was measured via intensity to distance transformation and the proportion of protein in the proximal vs. distal half of the cell was calculated.
Stochastic Optical Reconstruction Microscopy (STORM)
For STORM analysis of translating polysomes in vitro, OT-I CD8+ T cells were stimulated on peptide-pulsed APCs in a 96-well round bottom plate. PFA was added to the culture at a final concentration of 4% (v/v), the cell suspension was transferred into 1.5 μl eppendorf tubes, and fixation continued for 10 min at room temperature (RT). During all sample preparation, cells were pelleted for 3 min at 2000 rpm between each buffer. Cells were permeabilized in TBS + 0.1% (v/v) Triton X-100 for 3 min at RT. Reactive groups were quenched in 20 mM glycine, and non-specific antibody binding was blocked in TBS + 2% BSA. Samples were incubated overnight at 4°C with the following antibodies: anti-c-Myc (1:500, 5605, Cell Signaling Technology), anti-RPL26 (1:1000, PLA0299, Sigma-Aldrich), anti-Sirt1 (1:500, ab110304, Abcam), anti-tubulin (1:1000, MA1–80189, Invitrogen). Cells were washed with TBS and incubated with the following secondary antibodies for 1 h at RT: CF488-labeled donkey anti rat (1:1000, 20027, Biotium), CF568-labeled donkey anti Goat (1:1000, 20106, Biotium), and AF647-labeled donkey anti rabbit (1:1000, 711–605-152, Jackson ImmunoResearch). Following two washes with TBS, cells were post-fixed in 1% (v/v) PFA for 5 min at RT, pelleted and resuspended in 20 μl Geltrex (A1413201, Gibco). A 10 μl drop of the cell suspension was placed into a cryomold, overlaid with Tissue Freezing Medium (15–183-40, Fisher Scientific Company) and snap frozen on dry ice. 10 μm thick cryosections were placed on poly-L-lysine coated #1.5 18 mm coverslips (72294–12, Electron Microscopy Sciences) and affixed to 35 mm dishes (P35.G-1.5–14-C, Mattek) using epoxy resin. Stochastic optical reconstruction microscopy was performed using an N-STORM system (Nikon Instruments) as previously reported (Liedmann et al., 2014). Co-ordinate (x,y) positions of identified single molecules were exported for analysis. At least 25 cells from two independent experiments were analyzed.
For STORM analysis of translating polysomes in vivo, 24 h after immunization axillary and brachial lymph nodes were harvested and fixed in PBS containing 2% PFA, 0.1% Triton X-100 and 1% DMSO overnight at 4°C. 10 um thick cryosections were placed on poly-L-lysine coated coverslips and affixed to MatTek dishes as described above. Sections were blocked in PBS containing 1% BSA prior to incubation with primary and secondary antibodies as described above. 6 cells from two independent experiments were analyzed.
Analysis for STORM data
After the removal of noise signals from outside the cell borders and the nucleus the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) (Ester et al., 1996) algorithm was employed to identify RPL26 and S6 clusters, defined by a proximity size of 100 nm and minimum number of signals of 10 within 100 nm. RPL26 and S6 clusters with c-Myc or Sirt1 molecules in close vicinity of 100 nm were defined as c-Myc or Sirt1 translating polysomes. Next, the physical distances of c-Myc or Sirt1 molecules within RPL26 and S6 clusters from the MTOC were calculated and binned between 0 and the largest distance from the MTOC, where the largest distance from the MTOC is normalized to be 1. The counts of c-Myc/Sirt1 molecules in binned groups were regressed against average distances by generalized linear mixed-effect models with Poisson link across all images analyzed. The estimated slopes and the standard errors in the regressions for the effects of distances from the MTOC onto the counts of c-Myc/Sirt1 molecules were used to test the polarization of c-myc or sirt1 translating ribosomes towards the MTOC.
Rigorous statistical analysis of single molecule data, allowing statistical comparison across groups, required the development of a new algorithm denoted ‘normalized spatial intensity correlation (NSInC) (Liu et al., 2022). Briefly, co-ordinate data of identified single molecules were analyzed for the degree of bi-directional co-localization using the NSInC algorithm, which incorporates terms for normalization and correction of edge effect, and which is proven to be unbiased under conditions of complete spatial randomness. According to the algorithm, a value of 0 corresponds to complete spatial randomness between the molecules in question, while a value of 1 corresponds to complete co-localization and a value of −1 represents complete exclusion of molecules within the two-dimensional or three-dimensional study region. The NSInC algorithm was applied to STORM data sets containing coordinate data for vATPase and p-eIF4GI wherein multiple regions of interest (ROI) were analyzed per cell, with ROI defined by the inclusion of at least 3 vATPase+ cytoplasmic lysosomal structures.
Simulated Emission Depletion (STED) microscopy
For super resolved confocal imaging, cells were imaged with a 100× 1.4NA oil objective on a Leica TCS SP8 STED 3X microscope equipped with a 405 nm diode laser, a tunable (470–670 nm tunability range) white light laser and three STED depletion lasers with wavelength of 592 nm, 660 nm, and 775 nm. Hoechst channel was excited with 405 nm diode laser and detected with a PMT detector in the 415–478 nm range. Alexa-488, Alexa-555, Alexa-594, and Atto-647N dye-labeled probe channels were excited at 494 nm, 552 nm, 590 nm, and 650 nm wavelengths, respectively, using the white light laser and detected with Leica GaAsP HyD detectors in 498–536 nm, 556–584 nm, 595–645 nm, and 655–740 nm ranges, respectively. Alexa-594 and Atto-647N dye-labeled probe channels were imaged in 3-D STED mode using 775 nm depletion laser with pinhole diameter set to 0.8 Airy unit calculated for the lowest wavelength of the two dyes. Alexa-488 and Alexa-555 dye-labeled probe channels were imaged in confocal mode with pinhole diameter set to 0.4 Airy unit calculated for the lowest wavelength of the two dyes. Hoechst channel was imaged in confocal mode with pinhole diameter set to 0.8 Airy unit. All images were acquired with LAS X software (version 3.5) as 3-D stacks with 22 nm XY pixel size and either 120 or 150 nm Z-step size. Post-acquisition, the images were processed with built-in Lightning adaptive deconvolution module with optimized settings for each channel.
Expansion microscopy
Expansion microscopy was performed as recently described (Zhang et al., 2020). Briefly, cells were cultured for 2 h on anti-CD3/CD28-coated 12 mm glass coverslips, prior to fixation with 4% PFA in PBS for 15 min. Cells were subsequently permeabilized with 0.1% Triton X-100 for 3 min and blocked with 1% BSA in PBS for 30 min prior to incubation with primary antibodies overnight at 4°C. Samples were washed in PBS prior to detection with the following secondary antibodies: anti-mouse 488plus (A32766, ThermoFisher), anti-rabbit 488plus (A32790, ThermoFisher), CF568 conjugated anti-rat (20092, Biotium), or Atto647N conjugated anti-rat (Rockland). Following incubation with secondary antibodies, samples were washed in PBS prior to incubation with 0.1 mg/ml acryloyl-X-SE (A20770, ThermoFisher;) overnight at 4°C. Samples were subsequently washed in PBS prior to gelation with an acrylate/acrylamide solution, followed by proteinase K digestion as described previously (Zhang et al., 2020). Samples were finally washed three times in water to facilitate expansion and were visualized using SoRa.
Adoptive transfer and in vivo infection
To study the functional consequence of brief eIF4A inhibition in vivo, CTV-labeled GFP-c-Myc, OT-I+ CD45.2+ CD8+ T cells were activated on peptide-pulsed APCs for 36 h. First-division GFP-c-Mychigh and GFP-c-Myclow cell populations (CD8+, CTV 2nd peak, highest and lowest 20% GFP-c-Myc) were sorted into medium with or without 200 nM Silvestrol and cultured for additional 2 h. Next, 5×105 cells were transferred i.v. into CD45.1+ recipient animals, which were infected with 4000 EID50 influenza A virus A/X-31 (H3N2) expressing SIINFEKL peptide (P. Thomas, St. Jude Children’s Research Hospital). 30 days later animals were challenged with 105 EID50 influenza A virus A/PR/8 (H1N1) expressing SIINFEKL peptide (P. Thomas, St. Jude Children’s Research Hospital). Nine days after the secondary infection spleens were collected and analyzed by Flow cytometry for the presence of donor cells.
Flow cytometry
Cells were stained for 20 min at 4°C in PBS + 5% BSA with the following antibodies from eBioscience at 1:300 dilutions: Anti-CD8-PerCP5.5 (45–0081-82), anti-CD45.1-PE (12–0453-82), anti-CD45.2-FITC (11–0454-82), anti-TCR-Va2-APC (17–5812-82), anti-CD44-PE-Cy7 (25–0441-82), and anti-CD62L-BV605 (104437, BioLegend). Samples were acquired on a SP6800 Spectral Cell Analyzer (Sony Biotechnology) and analyzed using FlowJo 10.1r7 software (FlowJo, LLC).
CUT&RUN
First-division OT-I CD8+ T cells were sorted into medium with or without 200 nM Silvestrol and cultured for additional 2 h. CUT&RUN experiments were performed as previously described (Meers et al., 2019) with slight modifications. Briefly, 3×105 cells were washed with Wash Buffer (20mM HEPES (H3375, Sigma-Aldrich), 150 mM NaCl (AM9760G, Invitrogen), 0.5 mM spermidine (S0266, Sigma-Aldrich), and Protease inhibitor cocktail (5056489001, Sigma-Aldrich) twice. Cells were then resuspended and bound to Concanavalin A-coated magnetic beads (BP531, Bang Laboratories). Samples were placed on the magnet stand to pull off the liquid and beads were resuspended in 100 μl antibody buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM spermidine, 0.01% digitonin (300410, Milipore), 2 mM EDTA (AM9260G, Invitrogen), and Protease inhibitor cocktail). Anti-c-Myc antibody (13987, Cell Signaling Technology) was added to the samples at a final concentration of 1:100 and incubated overnight at 4°C. The next day, samples were washed with cold Dig-wash buffer (20 mM HEPES, 150 mM NaCl, 0.5 mM spermidine, 0.01% digitonin and protease inhibitor) twice. After washing, pAG-MNase (123461, Addgene) was added to tubes and rotated at 4°C for 1h. After two washes with Dig-wash buffer, samples were resuspended in 50 μl Dig-wash buffer, 2 μl of 100 mM CaCl2 (2115, Sigma-Aldrich) was added per sample, briefly vortexed, and immediately placed on ice for 30 min. 50 μl 2xSTOP buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA (E3889, Sigma-Aldrich), 100 μg/mL RNAse A (EN0531, Thermo-Fisher), and 50 μg/mL GlycoBlue (AM9515, Invitrogen)) were added and mixed by gentle vertexing. Then, samples were incubated 30 min at 37°C to release CUT&RUN fragments. Fragmented DNA was purified with the NEB Monarch PCR&DNA Cleanup Kit (T1030S, NEB). DNA libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit (E7645S, NEB) and purified with AMPure SPRI beads (B23318, Beckman-Coulter). Libraries were quantified using Qubit and size distribution was determined by Agilent 4200 TapeStation analysis before 15 million paired-end sequencing was performed.
Analysis for CUT&RUN data
Paired-end sequencing reads were trimmed with Trim Galore (version 0.5.0) (Krueger, 2020) with default parameters. Reads were aligned to the reference mouse mm10 assembly using Bowtie 2 (version 2.3.5.1) (Langmead and Salzberg, 2012) with settings --end-to-end --very-sensitive --no-mixed --no-discordant -q --phred33 -I 10 -X 700. The resulting alignments, recorded in BAM file, were sorted, indexed, and marked for duplicates with Picard MarkDuplicate function (version 2.19.0, Broad Institute). The BAM file was filtered with SAMtools (version 1.9) (Li et al., 2009), BamTools (version 2.5.1) (Barnett et al., 2011), and scripts of nf-core/chipseq (Ewels et al., 2020)to discard reads, mates that were unmapped, PCR/optical duplicates, not primary alignments, mapped to multiple locations, mapped to ENCODE blacklisted regions (Amemiya et al., 2019) or that have more than 4 mismatches (-F 0x004 -F 0x008 -F 0x0100 -f 0x001 -q 1). MACS (version 2.1.2) (Zhang et al., 2008) was used to call peaks from the BAM file with narrowPeak setting, IgG control, and recommended mappable genome size (default value for other parameters). C-Myc occupancy was normalized by scaling to 1 million mapped reads using BEDTools (version 2.27.1) (Quinlan and Hall, 2010) and bedGraphToBigWig (version 377) (Kent et al., 2010) and visualized as heatmaps using deepTools plotHeatmap (version 3.2.1) (Ramirez et al., 2016).
Immunoprecipitation
OT-I CD8+ T cells were stimulated on peptide-pulsed APCs for 24 h, treated with Torin for 1 h, and harvested in cold PBS. Immunoprecipitations were performed using the Dynabeads Co-Immunoprecipitation Kit (14321D, Invitrogen) following manufacturer instructions. In brief, pelleted cells were lysed in lysis buffer supplemented with phosphatases (11836153001, Roche) and proteases inhibitors (04906837001, Roche). Primary antibodies against anti-RagC (5466, Cell Signaling Technology) and anti-eIF4GI (8701, Cell Signaling Technology) were covalently coupled to Dynabeads M-270 Epoxy beads. For detection of co-precipitated proteins by Western blot, membranes were blocked in 5% BSA, and probed against anti-eIF4A (1:1000, Ab31217, Abcam), anti-Raptor (1:1000, 2280, Cell Signaling Technology) or anti-phospho-eIF4G (1:1000, 2441, Cell Signaling Technology). Anti-Lamin B1 (1:1000, Ab16048, Abcam) served as loading control. For quantification, densitometric signals of co-immunoprecipitated proteins were normalized with respective signals in the input samples.
RNA isolation, reverse transcription, and qPCR
Total RNA from IP samples spiked with drosophila mRNA was isolated using RNeasy Mini Kit (74104, Qiagen). First-strand synthesis was performed using M-MLV Reverse Transcriptase (2805013, Thermo Fisher Scientific) and oligo(dT). cDNA was preamplified using PreAmp Master Mix (1005580, Fluidigm) following manufacturer’s instructions. Quantitative amplification was performed using SYBR Green PCR Master Mix in an 7900HT thermocycler (Applied Biosystems). mRNA level were detected using the following primer sequences: c-myc fwd TTT GTC TAT TTG GGG ACA GTG TT, c-myc rev-CAT CGT GGC TGT CTG, dro-rpl32 fwd-ATG CTA AGC TGT CGC ACA AAT G, dro-rpl32 rev-GTT CGA TCC GTA ACC GAT GT. mRNA level were normalized to drosophila rpl32 mRNA level.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical power was calculated to determine sample size. Data were plotted and analyzed with GraphPad Prism 8.0 software (GraphPad Software). Statistical significance was accessed with unpaired or paired two-tailed Student’s t-test or ANOVA, as specified in the figure legends. Poisson regression was used on imaging data to assess co-localization between two proteins. Differences were considered statistically significant when the p-value was less than 0.05. An innovative NSinC algorithm was used for quantification and statistical comparison of protein molecule colocalization across groups. For detailed information on statistical analysis of scRNAseq or STORM data please refer to the respective method section.
Supplementary Material
Movie S1: p-eIF4GIS1108 associates with vATPase+ vesicles along microtubules. Related to Figures 2 and S2.
Association of p-eIF4GIS1108 (orange) with vATPase+ vesicular structures (green) along microtubules (purple) in proximity to the MTOC was visualized by expansion microscopy.
Movie S2: Synthesis of c-Myc is polarized toward the proximal pole of the cell. Related to Figures 3 and S3
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs was visualized using SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S3: Synthesis of Sirt1 is equally distributed throughout the cell. Related to Figures 3 and S3
Spatial distribution of Sirt1 synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs was visualized using SoRa imaging. Tubulin (white), Sirt1 (pink). Cytosolic Sirt1 is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S4: C-Myc associates with vATPase+ vesicles along microtubules. Related to Figures 3 and S3.
Association of c-Myc (orange) with vATPase+ vesicular structures (green) along microtubules (purple) in proximity to the MTOC was visualized by expansion microscopy.
Movie S5: Dynein-inhibitor Dynarrestin disrupts localized c-Myc synthesis. Related to Figures 3 and S3.
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs treated with Dynarrestin for the last hour of activation. SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S6: Dynein-inhibitor Ciliobrevin D disrupts localized c-Myc synthesis. Related to Figures 3 and S3.
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs treated with Ciliobrevin D for the last hour of activation. SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Table S1: Inhibition of eIF4A reduces c-Myc binding to chromatin. Related to Figures 4 and S4.
C-Myc binding to chromatin based on chromatin immunoprecipitation. First-division OT-I CD8+ T cells were treated with Silvestrol for 2 hours before CUT&RUN procedure.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD3ε | Bio X Cell | BE0001-1 |
| anti-CD28 | Bio X Cell | BE0015-5 |
| anti-CD44-PE/Cy7 | BioLegend | 103030 |
| anti-c-Myc | CST | 5605 |
| anti-Sirt1 | Abcam | ab110304 |
| anti-eIF4A1 | Abcam | ab31217 |
| anti-phospho-eIF4G1S1108 | CST | 2441 |
| anti-eIF4G1 | CST | 2858 |
| anti-phospho-mTORS2481 | CST | 2974 |
| anti-mTOR | CST | 4517 |
| anti-CD11c | BioLegend | 117301 |
| anti-tubulin | Invitrogen | MA1-80189 |
| anti-tubulin | Invitrogen | PA5-19489 |
| anti-tubulin | Invitrogen | 13-8000 |
| anti-RPL26 | Sigma-Aldrich | PLA0299 |
| anti- CD8-PerCP5.5 | eBioscience | 45-0081-82 |
| anti-CD45.1-PE | eBioscience | 12-0453-82 |
| anti-CD45.2-FITC | eBioscience | 11-0454-82 |
| anti-TCR-Va2-APC | eBioscience | 17-5812-82 |
| anti-CD62L-BV605 | Biolegend | 104437 |
| anti-RagC | CST | 5466 |
| anti-eIF4G1 | CST | 8701 |
| anti-Raptor | CST | 2280 |
| anti-lamin B1 | abcam | ab16048 |
| anti-S6 | CST | 2317 |
| anti-Mcl1 | CST | 94296 |
| donkey anti-mouse AFplus488 | Invitrogen | A32766 |
| donkey anti-rabbit AFplus555 | Invitrogen | A32794 |
| donkey anti-rat AF647plus | Jackson ImmunoResearch | 712-605-153 |
| goat anti-hamster AF488 | Jackson ImmunoResearch | 127-545-160 |
| donkey anti-rat CF488 | Biotium | 20027 |
| donkey anti-goat CF568 | Biotium | 20106 |
| donkey anti-rabbit AF647 | Jackson ImmunoResearch | 711-605-152 |
| donkey anti-rabbit AF488plus | ThermoFisher | A32790 |
| donkey anti-rat CF568 | Biotium | 20092 |
| Bacterial and Virus Strains | ||
| influenza virus A/X-31-OVA (H3N2) | kindly gifted by P. Thomas | |
| influenza virus A/PR/8-OVA (H1N1) | kindly gifted by P. Thomas | |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| SIINFEKL peptide | AnaSpec Inc. | AS-60193-1 |
| human ICAM | Huppa et al., 2010 | |
| PMA | MedChemExpress | HY-18739 |
| Ionomycin | MedChemExpress | HY-13434 |
| Silvestrol | MedChemExpress | HY-13251 |
| Hippuristanol | gifted by J. Pelletier | N/A |
| Torin 1 | Selleckchem | S2827 |
| Dynarrestin | Sigma | SML2332 |
| Ciliobrevin D | Selleckchem | S2827 |
| Puromycin | Tocris | 4098 |
| PFA | Electron Microscopy Science | 15710 |
| PhosSTOP | Sigma-Aldrich | 04906837001 |
| cOmplete, Mini Protease Inhibitor Cocktail | Roche | 11836153001 |
| RNAse A | ThermoFisher | EN0531 |
| GlycoBlue | Invitrogen | AM9515 |
| M-MLV Reverse Transcriptase | ThermoFisher | 2805013 |
| acryloyl-X-SE | ThermoFisher | A20770 |
| Critical Commercial Assays | ||
| CD11c MicroBeads | Miltenyi Biotec | 130-108-338 |
| Dynabeads Untouched Mouse CD8 Cell Kit | Invitrogen | 11417D |
| CellTrace Violet (CTV) Cell Proliferation Kit | Invitrogen | C34557 |
| High Sensitivity D5000 Screen Tape | Aligent Technologies | 5067-5588 |
| Prolong Glass Antifade Mountant | Invitrogen | P36980 |
| Dynabeads Co-Immunoprecipitation Kit | Invitrogen | 14321D |
| Dynabeads M-270 Epoxy beads | Invitrogen | 14301 |
| Concanavalin A-coated magnetic beads | Bang Laboratories | BP531 |
| NEB Monarch PCR&DNA Cleanup Kit | NEB | T1030S |
| NEBNext Ultra II DNA Library Prep Kit | NEB | E7645S |
| AMPure SPRI beads | Beckman-Coulter | B23318 |
| RNeasy Mini Kit | Qiagen | 74104 |
| PreAmp Master Mix | Fluidigm | 1005580 |
| RNAscope Probe-Mm-Myc | ACD | 413451 |
| RNAscope Fluorescent Miltiplex Detection Kit | ACD | 320851 |
| Deposited Data | ||
| Single Cell RNAseq data | BioProject | PRJNA641918 |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| BCM1+ mice | kindly provided by T. Schumacher (Gerlach et al., 2013) | N/A |
| GFP-c-Myc fusion knock-in mice | kindly provided by B. Sleckman (Huang et al., 2008) | N/A |
| OT-I Tg (C57BL/6-Tg (TcraTcrb)1100Mjb/J) | Jackson Laboratory | 003831 |
| Oligonucleotides | ||
| BCM-amp_1st fwd | this paper | N/A |
| AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT C | ||
| BCM-amp_1st rev | this paper | N/A |
| GCT GAA CTT GTG GCC GTT TA | ||
| BCM-amp_2nd fwd | this paper | N/A |
| AAT GAT ACG GCG ACC ACC GAG ATC T | ||
| BCM-amp_2nd rev | this paper | N/A |
| CGT CCA GCT CGA CCA GGA T | ||
| c-myc fwd | this paper | N/A |
| TTT GTC TAT TTG GGG ACA GTG TT | ||
| c-myc rev | this paper | N/A |
| CAT CGT CGT GGC TGT CTG | ||
| Dro-rpl32-fwd | this paper | N/A |
| ATG CTA AGC TGT CGC ACA AAT G | ||
| Dro-rpl32-fwd | this paper | N/A |
| GTT CGA TCC GTA ACC GAT GT | ||
| Recombinant DNA | ||
| pAG-MNase | Addgene | 123461 |
| Software and Algorithms | ||
| CellRanger VDJ v3.0.2 | 10x Genomics | |
| MUSCLE v3.8.31 | Edgar, 2004 | |
| Seurat v3.1.4 | Stuart et al., 2019 | |
| Uniform Manifold Approximation and Projection (UMAP) | McInnes et al., 2018 | |
| Density-Based Spatial Clustering of Applications with Noise (DBSCAN) | Ester et al., 1996 | |
| normalized spatial intensity correlation (NSInC) | Lui et al. under revision | |
| Trim Galore v0.5.0 | Krueger et al., 2020 | |
| Bowtie 2 v2.3.5.1 | Langmead and Salzberg, 2012 | |
| Picard Mark Duplicate v2.19.0 | Broad Institute | |
| SAMtools v1.9 | Li et al., 2009 | |
| MACS v2.1.2 | Zhang et al., 2008 | |
| BEDTools v2.27.1 | Quinlan and Hall, 2010 | |
| bedGraphToBigWing v377 | Kent et al., 2010 | |
| deepTools plot Heatmap v3.2.1 | Ramirez et al., 2016 | |
| Slidebook 6 | Intelligent Imaging Innovations | |
| Imaris 9.5 | Oxford Instruments | |
| FlowJo 10.1r7 | FlowJo, LLC | |
| GraphPad Prism 8.0 | GraphPad Software | |
| Other | ||
Highlights.
TORC1 physically associates with active eIF4F in activated, undivided CD8+ T cells
c-myc-translating polysomes polarize toward the immune synapse
sister cells have distinct transcriptional profiles after a single cell division
eIF4F inhibition upon the first cell division promotes T cell memory
Acknowledgments
The authors thank Patrick Fitzgerald, Mao Yang, Lacie Harris, Richard Cross, Greig Lennon, John Easton, Xiang Chen, and Khaled Khairy (SJCRH) for technical assistance, and Beiyun Liu, Kaitlynn Allen, Helen Beere (SJCRH), Pradyot Dash (Miltenyi), and Peer Karmaus (NIEHS) for insights and discussion. We also thank Jerry Pelletier (McGill) for providing Hippuristanol, Ton Schumacher (NKI) for providing BCM mice, and Barry Sleckman (UAB) for providing GFP-c-Myc mice.
Funding
Images were acquired at the Cell & Tissue Imaging Center and RNA sequencing was performed at the Hartwell center (supported by SJCRH and NCI P30 CA021765). This work was supported by ALSAC, the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award numbers RO1AI123322, R01AI154470, and R01AI136514, and the German Research Foundation under award number LI 2967/1–1. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing interests. DRG consults for Inzen Therapeutics and Ventus Therapeutics. DR is a shareholder of eFFECTOR Therapeutics. DRG is a member of Molecular Cell’s Advisory Board.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE Blacklist: Identification of Problematic Regions of the Genome. Sci Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenio J, Metz PJ, and Chang JT (2015). Asymmetric Cell Division in T Lymphocyte Fate Diversification. Trends Immunol 36, 670–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett DW, Garrison EK, Quinlan AR, Stromberg MP, and Marth GT (2011). BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best JA, Blair DA, Knell J, Yang E, Mayya V, Doedens A, Dustin ML, Goldrath AW, and Immunological Genome Project C (2013). Transcriptional insights into the CD8(+) T cell response to infection and memory T cell formation. Nat Immunol 14, 404–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas J, Liu Y, Singer RH, and Wu B (2019). Fluorescence Imaging Methods to Investigate Translation in Single Cells. Cold Spring Harb Perspect Biol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD (2013). Molecular insights into intracellular RNA localization. Int Rev Cell Mol Biol 302, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordeleau ME, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, and Pelletier J (2006). Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol 2, 213–220. [DOI] [PubMed] [Google Scholar]
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, Brem B, Greger H, Lowe SW, Porco JA Jr., et al. (2008). Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 118, 2651–2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsa M, Barandun N, Grabnitz F, Barnstorf I, Baumann NS, Pallmer K, Baumann S, Stark D, Balaz M, Oetiker N, et al. (2021). Asymmetric cell division shapes naive and virtual memory T-cell immunity during ageing. Nat Commun 12, 2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsa M, Barnstorf I, Baumann NS, Pallmer K, Yermanos A, Grabnitz F, Barandun N, Hausmann A, Sandu I, Barral Y, et al. (2019). Modulation of asymmetric cell division as a mechanism to boost CD8(+) T cell memory. Sci Immunol 4. [DOI] [PubMed] [Google Scholar]
- Buxbaum AR, Haimovich G, and Singer RH (2015). In the right place at the right time: visualizing and understanding mRNA localization. Nat Rev Mol Cell Biol 16, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cencic R, Carrier M, Galicia-Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, Brem B, Teodoro JG, Greger H, Tremblay ML, et al. (2009). Antitumor activity and mechanism of action of the cyclopenta[b]benzofuran, silvestrol. PLoS One 4, e5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, McClurkin CE, Dejong CS, Mooney EC, Kim JS, Steinel NC, Oliaro J, et al. (2011). Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity 34, 492–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, et al. (2007). Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 315, 1687–1691. [DOI] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15, 1104–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle GA, Betz NA, Leeds PF, Fleisig AJ, Prokipcak RD, and Ross J (1998). The c-myc coding region determinant-binding protein: a member of a family of KH domain RNA-binding proteins. Nucleic Acids Res 26, 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzhagalov I, Dunkle A, and He YW (2008). The anti-apoptotic Bcl-2 family member Mcl-1 promotes T lymphocyte survival at multiple stages. J Immunol 181, 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfakess R, Sinvani H, Haimov O, Svitkin Y, Sonenberg N, and Dikstein R (2011). Unique translation initiation of mRNAs-containing TISU element. Nucleic Acids Res 39, 7598–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M, Kriegel H-P, Sander J, and Xu X (1996). A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (Portland, Oregon, AAAI Press; ), pp. 226–231. [Google Scholar]
- Ewels PA, Peltzer A, Fillinger S, Patel H, Alneberg J, Wilm A, Garcia MU, Di Tommaso P, and Nahnsen S (2020). The nf-core framework for community-curated bioinformatics pipelines. Nat Biotechnol 38, 276–278. [DOI] [PubMed] [Google Scholar]
- Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, and Chen JK (2012). Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Masvidal L, Hulea L, Gravel SP, Cargnello M, McLaughlan S, Cai Y, Balanathan P, Morita M, Rajakumar A, et al. (2016). nanoCAGE reveals 5’ UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res 26, 636–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Rosen D, and Berke G (1982). Spatial relationships of microtubule-organizing centers and the contact area of cytotoxic T lymphocytes and target cells. J Cell Biol 95, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach C, Rohr JC, Perie L, van Rooij N, van Heijst JW, Velds A, Urbanus J, Naik SH, Jacobs H, Beltman JB, et al. (2013). Heterogeneous differentiation patterns of individual CD8+ T cells. Science 340, 635–639. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, and Sonenberg N (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13, 1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR, and Eisenman RN (1984). Proteins encoded by the human c-myc oncogene: differential expression in neoplastic cells. Mol Cell Biol 4, 2486–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. (2021). Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 e3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoing S, Yeh TY, Baumann M, Martinez NE, Habenberger P, Kremer L, Drexler HCA, Kuchler P, Reinhardt P, Choidas A, et al. (2018). Dynarrestin, a Novel Inhibitor of Cytoplasmic Dynein. Cell Chem Biol 25, 357–369 e356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CY, Bredemeyer AL, Walker LM, Bassing CH, and Sleckman BP (2008). Dynamic regulation of c-Myc proto-oncogene expression during lymphocyte development revealed by a GFP-c-Myc knock-in mouse. Eur J Immunol 38, 342–349. [DOI] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, and Davis MM (2010). TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J, and Baltimore D (1999). Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature 399, 593–597. [DOI] [PubMed] [Google Scholar]
- Kaech SM, Hemby S, Kersh E, and Ahmed R (2002). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851. [DOI] [PubMed] [Google Scholar]
- Kakaradov B, Arsenio J, Widjaja CE, He Z, Aigner S, Metz PJ, Yu B, Wehrens EJ, Lopez J, Kim SH, et al. (2017). Early transcriptional and epigenetic regulation of CD8(+) T cell differentiation revealed by single-cell RNA sequencing. Nat Immunol 18, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CG, Koehli S, Hausmann B, Schmaler M, Zehn D, and Palmer E (2012). T cell affinity regulates asymmetric division, effector cell differentiation, and tissue pathology. Immunity 37, 709–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F (2020). FelixKrueger/TrimGalore. htps://github.com/FelixKrueger/TrimGalore.
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedmann S, Hrincius ER, Guy C, Anhlan D, Dierkes R, Carter R, Wu G, Staeheli P, Green DR, Wolff T, et al. (2014). Viral suppressors of the RIG-I-mediated interferon response are pre-packaged in influenza virions. Nat Commun 5, 5645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GY, and Sabatini DM (2020). mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XY, Guy C, Boada-Romero E, Green D, Flanagan M, Cheng C, and Zhang H (2022). Unbiased and robust analysis of co-localization in super-resolution Images. Statistical Methods in Medical Research, accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeffler D, Wehling A, Schneiter F, Zhang Y, Muller-Botticher N, Hoppe PS, Hilsenbeck O, Kokkaliaris KD, Endele M, and Schroeder T (2019). Asymmetric lysosome inheritance predicts activation of haematopoietic stem cells. Nature 573, 426–429. [DOI] [PubMed] [Google Scholar]
- Marcou Q, Mora T, and Walczak AM (2018). High-throughput immune repertoire analysis with IGoR. Nat Commun 9, 561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteoni R, and Kreis TE (1987). Translocation and clustering of endosomes and lysosomes depends on microtubules. J Cell Biol 105, 1253–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L, Healy J, and Melville J (2018). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. In arXiv e-prints, p. arXiv:1802.03426. [Google Scholar]
- Meers MP, Bryson TD, Henikoff JG, and Henikoff S (2019). Improved CUT&RUN chromatin profiling tools. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz PJ, Arsenio J, Kakaradov B, Kim SH, Remedios KA, Oakley K, Akimoto K, Ohno S, Yeo GW, and Chang JT (2015). Regulation of asymmetric division and CD8+ T lymphocyte fate specification by protein kinase Czeta and protein kinase Clambda/iota. J Immunol 194, 2249–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliaro J, Van Ham V, Sacirbegovic F, Pasam A, Bomzon Z, Pham K, Ludford-Menting MJ, Waterhouse NJ, Bots M, Hawkins ED, et al. (2010). Asymmetric cell division of T cells upon antigen presentation uses multiple conserved mechanisms. J Immunol 185, 367–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J, and Sonenberg N (2019). The Organizing Principles of Eukaryotic Ribosome Recruitment. Annu Rev Biochem 88, 307–335. [DOI] [PubMed] [Google Scholar]
- Perciavalle RM, and Opferman JT (2013). Delving deeper: MCL-1’s contributions to normal and cancer biology. Trends Cell Biol 23, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, Tam AJ, Blosser RL, Wen J, Delgoffe GM, et al. (2016). Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol 17, 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raught B, Gingras AC, Gygi SP, Imataka H, Morino S, Gradi A, Aebersold R, and Sonenberg N (2000). Serum-stimulated, rapamycin-sensitive phosphorylation sites in the eukaryotic translation initiation factor 4GI. EMBO J 19, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, and Topisirovic I (2018). Signaling Pathways Involved in the Regulation of mRNA Translation. Mol Cell Biol 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio CA, Weisburd B, Holderfield M, Arias C, Fang E, DeRisi JL, and Fanidi A (2014). Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol 15, 476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryser JE, Rungger-Brandle E, Chaponnier C, Gabbiani G, and Vassalli P (1982). The area of attachment of cytotoxic T lymphocytes to their target cells shows high motility and polarization of actin, but not myosin. J Immunol 128, 1159–1162. [PubMed] [Google Scholar]
- Sinvani H, Haimov O, Svitkin Y, Sonenberg N, Tamarkin-Ben-Harush A, Viollet B, and Dikstein R (2015). Translational tolerance of mitochondrial genes to metabolic energy stress involves TISU and eIF1-eIF4GI cooperation in start codon selection. Cell Metab 21, 479–492. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Majorovits E, Bossi G, Fuller S, and Griffiths GM (2006). Centrosome polarization delivers secretory granules to the immunological synapse. Nature 443, 462–465. [DOI] [PubMed] [Google Scholar]
- Stoneley M, Subkhankulova T, Le Quesne JP, Coldwell MJ, Jopling CL, Belsham GJ, and Willis AE (2000). Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res 28, 687–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, and Sonenberg N (2001). The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5’ secondary structure. RNA 7, 382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney HL, and Holzbaur ELF (2018). Motor Proteins. Cold Spring Harb Perspect Biol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, and Green DR (2016). Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature 532, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viero G, Lunelli L, Passerini A, Bianchini P, Gilbert RJ, Bernabo P, Tebaldi T, Diaspro A, Pederzolli C, and Quattrone A (2015). Three distinct ribosome assemblies modulated by translation are the building blocks of polysomes. J Cell Biol 208, 581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, et al. (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe AL, Singh K, Zhong Y, Drewe P, Rajasekhar VK, Sanghvi VR, Mavrakis KJ, Jiang M, Roderick JE, Van der Meulen J, et al. (2014). RNA G-quadruplexes cause eIF4A-dependent oncogene translation in cancer. Nature 513, 65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Kang JS, Asano SM, Gao R, and Boyden ES (2020). Expansion Microscopy for Beginners: Visualizing Microtubules in Expanded Cultured HeLa Cells. Curr Protoc Neurosci 92, e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1: p-eIF4GIS1108 associates with vATPase+ vesicles along microtubules. Related to Figures 2 and S2.
Association of p-eIF4GIS1108 (orange) with vATPase+ vesicular structures (green) along microtubules (purple) in proximity to the MTOC was visualized by expansion microscopy.
Movie S2: Synthesis of c-Myc is polarized toward the proximal pole of the cell. Related to Figures 3 and S3
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs was visualized using SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S3: Synthesis of Sirt1 is equally distributed throughout the cell. Related to Figures 3 and S3
Spatial distribution of Sirt1 synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs was visualized using SoRa imaging. Tubulin (white), Sirt1 (pink). Cytosolic Sirt1 is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S4: C-Myc associates with vATPase+ vesicles along microtubules. Related to Figures 3 and S3.
Association of c-Myc (orange) with vATPase+ vesicular structures (green) along microtubules (purple) in proximity to the MTOC was visualized by expansion microscopy.
Movie S5: Dynein-inhibitor Dynarrestin disrupts localized c-Myc synthesis. Related to Figures 3 and S3.
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs treated with Dynarrestin for the last hour of activation. SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Movie S6: Dynein-inhibitor Ciliobrevin D disrupts localized c-Myc synthesis. Related to Figures 3 and S3.
Spatial distribution of c-Myc synthesis in undivided OT-I CD8+ T cells activated on peptide-pulsed APCs treated with Ciliobrevin D for the last hour of activation. SoRa imaging. Tubulin (white), c-Myc (pink). Cytosolic c-Myc is visualized by masking nuclear c-Myc based on Hoechst (purple).
Table S1: Inhibition of eIF4A reduces c-Myc binding to chromatin. Related to Figures 4 and S4.
C-Myc binding to chromatin based on chromatin immunoprecipitation. First-division OT-I CD8+ T cells were treated with Silvestrol for 2 hours before CUT&RUN procedure.
Data Availability Statement
Single-cell RNA-seq data have been deposited at BioProject, accession PRJNA641918. CUT&RUN data has been deposited at GEO accession GSE196967.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.