

Summary
Barrier epithelia depend upon resident stem cells for homeostasis, defense and repair. Epithelial stem cells of small and large intestines (ISCs) respond to their local microenvironments (niches) to fulfill a continuous demand for tissue turnover. The complexity of these niches and underlying communication pathways are not fully known. Here, we report a lymphatic network at the intestinal crypt base that intimately associates with ISCs. Employing in vivo loss-of-function and lymphatic:organoid cocultures, we show that crypt lymphatics maintain ISCs and inhibit their precocious differentiation. Pairing single-cell and spatial transcriptomics, we apply BayesPrism to deconvolve expression within spatial features and develop SpaceFold to robustly map the niche at high resolution, exposing lymphatics as a central signaling hub for the crypt in general and ISCs in particular. We identify WNT-signaling factors (WNT2, R-SPONDIN-3) and a hitherto unappreciated extracellular matrix protein, REELIN, as crypt lymphatic signals that directly govern the regenerative potential of ISCs.
Graphical Abstract
ETOC
Niec et al. integrate spatial and single-cell transcriptomics data and develop computational approaches to finely map the cellular and transcriptional landscape of the intestinal crypt-villus axis. Combining these results with functional experiments, the authors expose lymphatics as a central signaling hub that promotes stem cell maintenance in the intestinal niche.
Introduction
At the interface between our body and the external environment, barrier epithelial tissues such as skin and intestine rely on resident tissue stem cells to proliferate and self-renew and repopulate their differentiated progeny. Self-renewal and differentiation must be tightly controlled to prevent excessive growth or poor regeneration and healing. To tailor their activity, stem cells reside in niches, where they communicate with their local microenvironment to receive signals that regulate their maintenance and differentiation (Gehart and Clevers, 2019; Gonzales and Fuchs, 2017; Hsu et al., 2014).
The multifarious nature of barrier epithelia and resident tissue stem cells is exemplified by the intestine. Intestinal stem cells (ISCs) reside within pocket-like crypts of the small intestinal epithelium that include neighboring specialized epithelial cells (Paneth cells), sub-epithelial mesenchymal cells (pericryptal fibroblasts, myofibroblasts, stromal telocytes, trophocytes) and immune cells (Aoki et al., 2016; Baghdadi et al., 2021; Biton et al., 2018; Degirmenci et al., 2018; Gregorieff et al., 2005; Greicius et al., 2018; He et al., 2004; Lindemans et al., 2015; McCarthy et al., 2020; Roulis and Flavell, 2016; Sato et al., 2011; Shoshkes-Carmel et al., 2018; Stzepourginski et al., 2017). ISCs continually produce committed short-lived proliferative offspring that proceed upward along the crypt-villus axis, and differentiate into secretory and absorptive lineage options according to microenvironment and nutrient availability (Beumer and Clevers, 2021). The intestine is subject to dynamic and systemic regulation, indicating a requirement for ISCs to integrate cues from their crypt niches and coordinate their behavior on a tissue-wide scale. A major hurdle in tackling how stem cells balance self-renewal with differentiation and respond to dynamic cues is the incomplete picture of crypt niches, both with regards to the constituents and the signaling molecules they provide.
Improvements in imaging techniques and spatial transcriptomics provide new opportunities to map the stem cell niche and identify features with high resolution. Deep three-dimensional (3D) imaging has provided insights into how tissue stem cells respond to their vascular microenvironment (Comazzetto et al., 2021; Gur-Cohen et al., 2019). Applying these methods to crypt niches, we uncovered lymphatic capillaries as hitherto unappreciated components at ISC niche.
Lymphatics have been well-studied in the small intestine (SI) in the context of lacteal-mediated absorption of lipids and nutrients, immune surveillance and fluid balance (Bernier-Latmani and Petrova, 2017). The submucosal lymphatic capillaries underlying both SI and large intestine (LI) have been largely viewed as conduits for fluids and immune cells. Pathologic changes in intestinal lymphatics have been described in inflammatory bowel disease, a disease characterized by intestinal inflammation and epithelial perturbations (Van Kruiningen and Colombel, 2008; Steven Alexander et al., 2010). However, possible homeostatic roles for lymphatics in governing ISC behavior have been ignored. Here, we make the surprising finding that lymphatics at the base of intestinal crypts function as a signaling hub for the crypt in general and ISCs in particular. Guided by our spatial transcriptomics, we devise new computational methods to inform our in vivo and in vitro studies that unearth physiologically significant interactions between crypt lymphatics and ISCs.
Results
Lymphatic capillaries neighbor stem cells at the intestinal crypt base
To visualize crypt niche diversity that may have been overlooked in the absence of a 3D view, we applied tissue clearing (Richardson and Lichtman, 2015) and whole-mount imaging of the SI. This revealed the well-known capillary plexus, composed of a blood vascular network that surrounds a central lymphatic lacteal along the villus axis (Figures 1A, S1A-B and Video S1) (Bernier-Latmani and Petrova, 2017). As prominent as the lacteals was a rich network of lymphatic capillaries residing just beneath the crypt. Co-immunofluorescence (IMF) of the lymphatic marker LYVE1 and ISC marker LGR5 exposed an association between lymphatics and ISCs, with lymphatic capillaries nesting crypt bases (Figure 1A, S1A-S1B). Ultrastructural analysis confirmed the intimate proximity between lymphatics and crypt base ISCs (Figure 1B). In some cases, lymphatic capillary protrusions were found in contact with the crypt base (Figure S1C). Markers against previously identified mesenchymal and immune cells in the crypt revealed that the lymphatic network was interwoven with these components of the niche (Figures S1D-G).
Figure 1. Lymphatic capillaries nest crypt-based intestinal stem cells.

(A) 3D IMF of SI crypts and villi, revealing LYVE1+ lymphatic capillaries (yellow) nesting LGR5+ crypt-based ISCs (purple). Representative of n > 5 mice. Eye icon indicates plane of view of visualization; horizontal eye = side view, downward facing eye = top view with tissue lying flat. (B) Pseudo-colored ultrastructural images of SI crypt base with lymphatic capillaries (yellow, Lymph.), Paneth cells (P), and ISC (purple, ISCs). (C) Schematic of SI crypt niche, showing ISCs (purple) intermingled with Paneth cells (orange) and the lymphatic vasculature (yellow) (legend in Figure S1A). (D) Top: 3D IMF images of 5 μm z-stacks of the mouse SI lymphatic vasculature (LYVE1+) and differentiated epithelial (ECAD+, aqua) cells or ISCs (LGR5+, green) taken from z planes as illustrated in the schematic (dotted lines) in C. Bottom:Lymphatic vasculature is color-coded according to the distance [μm] to the nearest epithelial/ISC (white outlines). Quantification of the distance between lymphatic surfaces and the epithelium (n = 9 mice with ≥ 3 images/region/mouse) was done using nearest-distance-to-surface analysis in Imaris (Bitplane) and is summarized in the box-plot on the right. **** indicates a p-value of <0.0001 (One-way analysis of variance (ANOVA), Tukey’s multiple comparisons) (E) 3D IMF of LI crypts revealing LYVE1+ lymphatics (yellow) nesting LGR5+ ISCs (purple). Representative of n > 5 mice. (F) 3D IMF images of cleared SI and LI, demonstrating the percentage of LGR5+ crypts (green) that are associated with lymphatic capillaries (LYVE1+, red) (n = 9 mice with ≥ 4 images/region/mouse). Boxed images are magnified. **** indicates a p-value of <0.0001 (Unpaired two-tailed Student’s t-test). (G) Representative 3D IMF images from human SI (terminal ileum) and LI (distal) biopsies, demonstrating conserved association between lymphatics (LYVE1+, red) and the intestinal crypt base (ECAD+, green) (representative of n = 4 individual patients). LGR5-GFP staining in panels (A), (E) and (F) refers to endogenous Lgr5-EGFP expression in Lgr5-EGFP-IRES-CreERT2 mice. Scale bars appear on each image.
In the villus, lacteals were separated from the epithelium by a network of blood vessels, immune cells and abundant stromal cells. By contrast, lymphatic capillaries at the crypt base were more closely associated with resident Paneth and LGR5+ stem and progenitor cells (Figures 1C-1D). This was not the case for the blood vasculature (Figure S1H).
Given the critical function of SI lymphatics in nutrient absorption, we considered that lymphatic endothelial cell proximity to the crypt base might be merely a necessary component of their path from the lacteals to the collecting vessels which flow into mesenteric lymph nodes to ultimately deliver nutrients into the systemic circulation (Bernier-Latmani and Petrova, 2017). If lymphatic:ISC proximity in SI is simply a consequence of lacteal collection then we would not expect to observe this in the LI, as the lymphatic vascular system of the LI is devoid of lacteals (Bernier-Latmani and Petrova, 2017).
Despite lacking a villus and associated lacteals, the LI also displayed LGR5+ ISCs intimately associated with lymphatic capillaries (Figures 1E and 1F, S1I-S1J and Video S2). Although LI lymphatics are known to function in intestinal immunity (Esterházy et al., 2019; Houston et al., 2015), their close proximity to ISCs suggested an additional role. Furthermore, the lymphatic-ISC association was also conserved in humans across the small and large intestine (Figure 1G and Video S3).
Crypt lymphatic endothelial cells directly maintain intestinal stem cells
Prior studies have shown that acute loss of lymphatic lacteals distorts villus architecture and, in combination with the failure of immune surveillance, leads to sepsis (Jang et al., 2013). To determine whether crypt lymphatics are essential for ISC behavior, we used the targeting scheme shown in Figure 2A to induce diphtheria toxin receptor-mediated death in lymphatics. Within 5 days of DT, lymphatics were ablated in the SI, providing a means to interrogate the consequences for the crypt ISCs (Figures 2B and S2A). As judged by OLFM4 IMF, ISCs were significantly reduced upon loss of lymphatic vessels (Figure 2C). This occurred at a time when apoptosis (cleaved caspase 3) and/or DNA damage (TUNEL) were minimal in the crypt (Figure S2B).
Figure 2. In vivo and in vitro, lymphatic endothelial cells maintain ISCs and restrict lineage progression.

(A) Schematic for lymphatic ablation. (B) Representative images of lymphatic ablation in SI upon treatment with Diphtheria toxin (DT) as per schematic in (A). (C) Representative IMF images and quantification of OLFM4+ cells in control and DT-treated mice. (D) EdU pulse-chase: Top: EdU+ cells in control and DT-treated mice 30 minutes after EdU (quantified on the right). Bottom: EdU+ cells 48 hours after EdU. The distance between the top of the crypt and the nearest EdU+ cell [μm] is quantified on the bottom right. n = 3 experiments (E) LEC:organoid coculture schematic (left) and a representative IMF image of organoids (ECAD+, green) and LECs (CD31+, white) (right). (F) Organoid growth from single cells harvested from SI (left) and LI (right). The number of single cell-derived organoids under control and coculture conditions is quantified on the right. (n = 2 experiments). (G) Representative images and boxplot showing the percentage of EdU (magenta)-positive cells after a 10 minute-pulse in crypt-derived SI organoids cultured in the absence or presence of LECs (n = 2 experiments). (H) Quantification of organoid size (area) and number of crypt domains (n = 3 experiments). (I) mTomato+ (magenta) organoids grown with or without LECs. Images show SOX9+ progenitors (light blue) and ALDOB+ differentiated enterocytes (yellow). Quantification of ALDOB maximum fluorescence signal intensity is on the right (n = 3 experiments). * p-value 0.01 to 0.05, ** p-value of 0.001 to 0.01, *** p-value of 0.0001 to 0.001, **** p < 0.0001 (Unpaired two-tailed Student’s t-test).
Probing the roots of ISC reduction, we injected mice with the 5-ethynyl-2’-deoxyuridine (EdU) and then analyzed intestinal tissue at 30 minutes or 48 hours. IMF and FACS quantifications of pulse-chase labeling indicated that lymphatic loss caused elevated proliferation within the crypt, which accelerated the rate of differentiating cells moving up the villus (Figures 2D and S2C). Differentiated cell types were still detected at comparable levels (Figure S2D), suggesting that the consequences of lymphatic loss on the intestinal epithelium were rooted in the flux of ISC-mediated lineage production, rather than lineage choice per se.
We posited that crypt lymphatics might be functioning by maintaining ISC/progenitor cells and restricting the rate of lineage commitment and differentiation. To test this hypothesis and also evaluate the direct action of lymphatic endothelial cells (LECs) on ISCs, we established a murine coculture system to generate 3D intestinal organoids (Sato et al., 2009) under culture conditions that were favorable to LECs, but did not alter organoid formation (Figures 2E, S2E and S2F, Video S4).
Dermal and intestinal lymphatics share many commonalities (Figure S2G) and due to their established purity, faithful identity and ability to be cultured for multiple passages, primary dermal lymphatic endothelial cells (LECs) were chosen for coculture studies. In coculture conditions, LECs formed tubes and maintained expression of common vascular markers (CD31) and key lymphatic-specific markers (PROX1, LYVE1 and Lyve1, Flt4/Vegfr3) (Figures S2F and S2H). We therefore turned to addressing whether, as predicted from our studies in vivo, LECs would enhance secondary organoid formation efficiency of single cells organoids derived from mouse SI and LI crypts.
We seeded single cells atop preformed LEC tubes (Figure 2F). Whether from small or large intestine, organoid forming efficiency of single cells more than doubled when LECs were present (Figure 2F). Organoids derived from crypt:LEC cocultures grew over time, underscoring the viability of the ISCs. Despite the ability of LECs to expand the organoid-forming ISC pool, LEC-cocultured organoids remained in an ‘immature’ state (Lukonin et al., 2020): they were more circular in shape and contained fewer protrusions (crypt domains) than those derived in the absence of LECs (Figure 2H). Such effects were not observed when crypts were cocultured with blood vascular endothelial cells (BECs) (Figure S2I). Moreover, consistent with a reduction in the rapidly dividing, committed transit-amplifying (TA) cells, crypts cultured in the presence of LECs showed less proliferation (Figure 2G). The cultures also displayed fewer differentiated cells, as judged by their fewer crypt domains, less mature shape and reduction in the enterocyte marker Aldolase B (ALDOB) (Figures 2H and 2I). Intriguingly, human LECs induced a similar phenotype in human colonic organoids (Figure S2J).
The effects of LECs on single ISCs and/or crypts in vitro were complementary to and opposite of the effects of lymphatic ablation on ISCs in vivo. The ability to recapitulate in vitro a role for LECs in enhancing ISC fitness and maintenance, while restricting lineage progression, further suggested that LEC-to-ISC communication, rather than lymphatic drainage properties or immune cell trafficking, were at the root of the effects of lymphatics on the crypt.
To determine the nature of the direct effects of LECs, we performed single cell RNA-sequencing (scRNA-seq) on intestinal organoids grown with or without LECs. We focused on SI organoids since these have a greater diversity of lineage options and form more complex organoids. Clustering (Levine et al., 2015) of the pooled epithelial cells from co- and mono-cultures showed that cell types clustered independently of the presence of LECs and segregated into 11 clusters (Figures 3A and S2K-S2L).
Figure 3: scRNA-sequencing supports a role for LECs in enhancing ISCs and reducing terminally differentiated enterocytes within organoids.

(A) Force-directed layout generated from scRNA-seq of organoids cultured with or without LECs. Cells are colored by cell type. (B) Bar plot shows the relative abundance of cell types per condition. (C) Differential abundance testing of cell states in scRNA-seq data using Milo (Dann et al., 2021). Nodes represent Milo neighborhoods, colored by their log2 fold change of cell abundance in LEC-cocultured organoids vs. organoids alone. Node size corresponds to the minus log10 false discovery rate (FDR). Stem/TA cells and differentiated enterocytes are outlined (dotted lines). Mki67+ population from (D) is outlined in red. (D) Force-directed layout colored by the log-transformed normalized gene expression values of Mki67 (cycling cells). (E) Log2 fold change of the expression of villus enterocyte zone genes (Moor et al., 2018) in cocultured and control organoids. Each dot represents a zone gene. Only significantly differentially expressed genes were visualized (absolute value of log2 fold change > 0.1 and FDR < 0.01).
Two clusters expressed markers for Goblet/Paneth cells (e.g. Muc2, Lyz1) and enteroendocrine (e.g. Chga, Chgb) and were assigned unambiguously (Haber et al., 2017) (Figure S2L). Four clusters expressed markers for both stem/progenitor and TA cells (e.g. Olfm4, Ascl2, Tubb5, Top2a) and were labeled as stem/TA (Haber et al., 2017; Nikolaev et al., 2020) (Figure 3A and S2L). The other five clusters expressed enterocyte markers (e.g. Alpi, Fabp1) and correlated with enterocyte differentiation programs along the axis of villus zone positions (Figure S2M) (Moor et al., 2018). Accordingly, we labeled these cell populations as villus bottom-like 1 and 2, mid-like 1 and 2 and top-like. Overall, while many intestinal epithelial populations were identified in this culture system, a few such as Tuft cells, which are rare in vivo, were not in sufficiently high numbers to cluster.
Organoids grown in the presence of LECs displayed fewer terminally differentiated, top-like enterocytes than stem and progenitor cells (Figure 3B). These findings were also concordant with differential abundance testing (Milo) (Dann et al., 2021), which provided finer resolution (Figure 3C). This analysis revealed that organoids grown in the presence of LECs displayed altered distributions of cells within the stem/progenitor cluster and a skewing of enterocyte progenitors (bottom-like) at the expense of fully differentiated enterocytes (top-like). Consistent with our EdU experiments in organoids as well as pulse-chase experiments after lymphatic ablation in vivo, the transcriptome of LEC-cocultured organoids showed fewer actively cycling (Mki67+) cells (Figures 3C and 3D). As we showed for normal SI tissue (see Figure 2D), these highly proliferative cells are mostly committed TA cells located in the upper portion of the crypt. Additionally, cocultures showed increased transcription levels of enterocyte markers defining bottom zones compared to those defining more mature (top) zones (Figure 3E). Despite an overall decrease in terminal differentiation, we did not observe significant differences across differentiated cell populations (Figure 3B and S2N).
The in vivo spatial maps of cell types in the small and large intestines
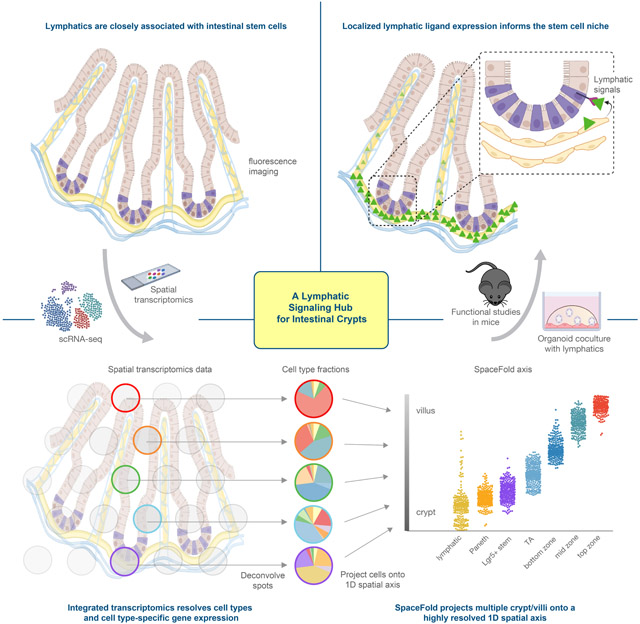
We addressed whether crypt lymphatics might signal to niche neighbors and if so, how. Since lacteal lymphatics might produce a different set of factors than crypt lymphatics, meaningful results necessitated an in vivo spatial transcriptomic approach. Therefore, we sought to cartograph intestinal cell types and their gene expression profiles along the spatial dimensions of the crypt-villus axis (for SI) and crypt axis (for LI) by computationally integrating scRNA-seq and spatial transcriptomic data (Figure 4A). This approach allowed us to (1) identify genes highly and/or uniquely expressed by intestinal LECs and (2) correlate intestinal LEC gene expression patterns according to their spatial proximity to ISCs (and/or other potential receiver cells) within the intestinal epithelium.
Figure 4. Integrated transcriptomics reconstructs cellular and molecular intestinal landscapes.

A) Bayesprism workflow of single-cell:spatial transcriptomic data integration and deconvolution based on BayesPrism to infer joint gene expression and cell type fraction per spot of the 10X Visium gene expression slide. (B and C) Uniform Manifold Approximation and Projection (UMAP) plots of murine scRNA-seq data of SI (B) and LI (C) tissue, containing 2,239 and 5,163 cells respectively. Each cell is colored according to cell type annotation. EE: enteroendocrine cell, pDC: plasmacytoid dendritic cell, cDC: classical dendritic cell, str: stromal cell and TA: transit-amplifying cell. Data were enriched for LECs and LGR5+ ISCs. (D and E) Left: Representative IMF images from SI and LI tissue sections, used for spatial transriptomics. Gray or color-outlined dots reflect the 55 μm (diameter) capture areas. Images show EPCAM+ epithelial cells (white), LYVE1+ lymphatic vasculature (red), DAPI+ nuclei (blue) and, in the SI, OLFM4+ ISCs (green). Right: Pie charts represent the fractions of cell types in each correspondingly colored and numbered spot along the crypt-villus axis deconvolved by BayesPrism. For visualization, multiple crypt-based goblet subtypes were grouped (base goblet).
We collected comprehensive, separate scRNA-seq profiles of intestinal cells from both SI and LI, covering the major cell types from immune, stromal and epithelial lineages within the intestine, while enriching for rare populations of LECs and LGR5+ ISCs (Figures 4B, 4C and S3). Immune cells of SI and LI each encompassed the major lymphoid and myeloid cell populations. Analogously, stromal cells clustered according to glial (e.g. S100b, Gfap), blood endothelial (e.g. Cd31, Cdh5) and lymphatic endothelial (e.g. Lyve1, Prox1), myofibroblast (e.g. Acta2, Myh11) and fibroblast (e.g. Col6a2, Dpt) marker genes (Kinchen et al., 2018). Our clustering further revealed heterogeneity in mesenchymal cell populations as defined by markers for trophocytes (e.g. Cd81), telocytes (e.g. Foxl1) and pericryptal stromal cells (e.g. Cd34, Pdpn) (Figures 4B, 4C and S3A-D). Finally, epithelial cells of SI and LI each reflected all of the main differentiated lineages as highlighted by markers for enterocytes (e.g. Alpi, Fabp1), goblet cells (e.g. Muc2), enteroendocrine cells (e.g. Chga, Chgb), tuft cells (e.g. Dclk1) and Paneth cells (e.g. Lyz1, Defa17), with the latter absent in the LI (Figures 4B, 4C and S3A, S3B). Goblet cells exhibited a high degree of heterogeneity and accounted for a large proportion of epithelial cells in the LI.
Immature epithelial cells segregated into secretory precursors (e.g. Atoh1, Dll1), TA cells (e.g. Stmn1, Tubb5) and LGR5+ progenitors. Bona fide ISCs marked by Lgr5 were found in both scRNA-seq datasets (Barker et al., 2012). Overall, we produced an unprecedented comprehensive atlas of mouse LI epithelium, and a valuable resource of mouse transcriptomes for SI and LI cell populations in general and ISCs in particular.
We next performed ST (10X Visium platform) on matched tissue samples that were immunolabeled for epithelial cells (EpCAM), ISCs (OLFM4, exclusive to the SI) and LECs (LYVE1) (Figures 4D, 4E and S4A, S4B). Commercially available platforms for ST are a valuable tool, yet limited in resolution. To obtain possible signaling routes within the crypt niche required methods to determine (1) the cell types within each spot and (2) the expression level of each gene from each cell type at each spatial position.
We used BayesPrism, a Bayesian statistical model that jointly infers cell type composition and cell type-specific gene expression profiles within each spatial spot by using our scRNA-seq reference from matched tissue as prior information (Figure 4A) (Chu et al., 2022). Our deconvolution assignments per 10X Visium spot were in agreement with the expected cell type distribution and their relative abundance along the crypt-villus or crypt axes, and with IMF profiles of the sequenced tissue (Figures 4D, 4E and S4A, S4B). Benchmarking BayesPrism against other deconvolution tools developed for spatial transcriptomic data (Biancalani et al., 2021; Cable et al., 2021; Elosua-Bayes et al., 2021; Kleshchevnikov et al., 2022; Lopez et al., 2022), we found that BayesPrism showed the highest concordance with IMF signals for ISCs (OLFM4) and lymphatics (LYVE1) (Figure S4C), the two populations of major focus here.
In addition to cell type fractions, BayesPrism provided cell-type specific gene expression. We confirmed the accuracy of our transcript level inference using both IMF and fluorescence in situ hybridization (FISH) to measure SOX9/Sox9, LYZ and LGR5 in LGR5+ ISCs and LYZ1+ Paneth cells (Figures S5A-C). These data were in agreement, validating exclusive differential expression of Lgr5 and Lyz1, as well as higher Sox9 expression in ISCs versus Paneth cells. Similarly, we confirmed expected expression levels of Lyve1, Prox1, and Pecam1 in the blood endothelial cells and LECs (Figures S5D-E).
Cell type and expression cartography reveals in vivo lymphatic:ISC interactome
While 10X Visium allows charting transcripts within a spatial spot, detecting candidates for lymphatic:ISC interactions requires higher-resolution. For this purpose, we developed SpaceFold, a non-linear dimensionality reduction method that projects each spot in our data onto a 1D pseudo-space axis (Figure 5A).
Figure 5. SpaceFold cartography reconstructs transcriptomes along crypt-villus and crypt axis.

(A) SpaceFold schematic. Each spatial spot was deconvolved using BayesPrism (step 1) to infer cell type fractions. Vectors of cell type fractions were reduced to a 1D projection that approximates its physical position along the crypt or crypt-villus axis (step 2). BayesPrism’s cell type-specific gene expression was used to generate trends along this axis for spots containing the cell type(s) of interest (step 3). (B-C) SpaceFold reveals the relative spatial coordinates of cell types, as identified from individual spots, along the SI crypt-villus (B) and LI crypt axis (C). Cell type annotation follows the cluster nomenclature in figures 4B-4C. Each dot represents a Visium spot containing the indicated cell type, plotted along its SpaceFold projection. Black arrow in B denotes computationally reconstructed lymphatic lacteals in the SI, absent in the LI in (C). (D) SpaceFold maps cell type-specific expression of known cell type markers onto the SI crypt-villus axis. X-axes mark the relative SpaceFold spatial coordinate. Histograms show the frequency of spots containing the selected cell types along the crypt-villus axis with corresponding Y-axes on the left. Lines mark smoothed mean values and shaded areas represent the mean ± 2 standard error of total (groups of cell types in left and center panels) or normalized (individual cell types in right panel) gene expression levels inferred by BayesPrism, corresponding to Y-axes on the right. Top panels show the predicted expression in groups of cell types or cell types expected to express the indicated marker genes, while bottom panels show the predicted expression in cell types not expected to express those genes. Black arrowheads indicate the spatial position of crypt-base lymphatics. (E) Intestinal villus epithelial cell zone gene expression distributed spatially along the projected SpaceFold crypt-villus axis. Similar to D, mean z-scores of the normalized expression of each group of zone markers in enterocytes are shown. Each dot represents the mean z-scores averaged over an interval of spatial spots binned by the SpaceFold coordinate. The x-coordinate of each dot represents the mean of the SpaceFold spatial coordinates in each bin. Lines mark the mean values fitted using local polynomial regression. Shaded areas represent the mean ± 2 standard error.
SpaceFold took advantage of the hundreds of highly stereotyped structures of the intestinal epithelium, each comprised of repeating crypt-villus units in SI, or crypt units in LI (Figures S4A, S4B). We hypothesized that, as spatial spots were randomly sampled along the stereotypical axis, the cell type composition of each spatial spot, as inferred by BayesPrism, would inform the relative physical coordinates of cells along a 1D artificial crypt-villus unit.
1D pseudo-space inferred by SpaceFold recapitulated the expected cell type distributions along SI crypt-villus and LI crypt axes and accurately aligned with tissue-specific spatial distribution patterns (Figures 5B, 5C and S5F). Absorptive enterocyte clusters mapped along the villus axis as predicted by villus zone marker genes (Figure 5B). Interestingly, the projected axis distinguished the spots containing the crypt-based lymphatic capillaries from the lacteal lymphatics in SI (Figure 5B, arrow). Lacteal protrusions were not present in the LI data, consistent with the absence of lacteals (Figure 5C, arrow). This cartography also recapitulated the known distribution of ISC niche cells along this axis, with the stromal 3-like population containing trophocytes below the crypt base in SI and LI (Figures 5B, 5C) (Kinchen et al., 2018; McCarthy et al., 2020).
We next cartographed gene expression profiles for each cell type using deconvolved cell type-specific gene expression profiles in each spatial spot. We mapped transcript levels of established markers for ISCs (Lgr5), Paneth cells (Defa5) and stromal cells (e.g. Grem1 as a marker of crypt-based stromal cells/trophocytes) along our projected crypt-villus axis (Figure 5D). A high concordance was found between marker gene expression and the expected spatial distribution of their associated cell types, while unrelated cell types lacked these markers. Intestinal villus epithelial cell zone markers also distributed predictably along the projected villus axis (Figure 5E) (Moor et al., 2018). This was independently validated with IMF for SOX9, distributed differently among crypt cell types (Figure S5G). Taken together, SpaceFold generated an unprecedented high-resolution cartograph of cell types and cell type-specific gene expression profiles in the murine SI and LI.
A lymphatic secretome maintains ISCs and restrains lineage commitment within the crypt niche
To exploit our spatial transcriptomes to unveil potential LEC:ISC interactions in the crypt niche, we began by addressing whether the effects of LECs on ISC behavior were mediated by direct intercellular contact or through LEC-secreted (lymphangiocrine) factors. To this end, we examined the potential of LEC-conditioned medium (LEC-CM) to recapitulate the phenotype of LEC-cocultured organoids. Organoids grown in LEC-CM displayed an increased percentage of cells with organoid forming potential (Figure 6A).
Figure 6. Lymphatics form a signaling hub of secreted factors that localize to the crypt.

(A) Single cells derived from organoids grown in control or LEC-conditioned media and re-seeded in 100% organoid (ENR) media. Quantification was done for organoid number (one value/well) and size of organoids (one value/organoid) on day 10 (n = 2 experiments), revealing that LEC effects on ISCs are reversible. (B) Crypt-derived organoids were cultured alone in 50:50 control or LEC-conditioned media. Violin plots of area and number of crypt domains per organoid following culture for 4 days (n = 3 experiments). (C) Representative IMF images of mTomato+ (magenta) organoids grown in control or LEC-conditioned medium. SOX9+ progenitors (light blue) and ALDOB+ differentiated enterocytes (yellow) are shown. (D) Violin plots of candidate lymphatic-secreted factors in selected cell types from mouse SI scRNA-seq. (E) SpaceFold cartographs of candidate lymphatic-secreted factors normalized by the total gene expression of lymphatics over the crypt-villus axis as in figure 5D. X-axes mark the relative spatial coordinate. Histograms with related y-axes on the left refer to spot frequency. Y-axes on the right show normalized expression of the indicated genes (lines). (F) Whole-mount IMF image of SI crypts harboring LGR5+ ISCs (cyan) that are embedded in a network of LYVE1+ lymphatic capillaries (purple) expressing REELIN (yellow). Eye icon indicates the angle of view. LGR5 staining refers to endogenous Lgr5-EGFP expression in Lgr5-EGFP-IRES-CreERT2 mice. (G) Left panel: 3D-reconstructed IMF whole-mount image of the lymphatic capillary network (LYVE1+, purple) along the crypt-villus axis in the SI. Dotted lines outline individual crypts. REELIN expression in lymphatics is color-coded by fluorescence signal intensity, demonstrating highest intensity in the crypt vs lacteals. Right panel: quantification of the signal intensity of REELIN IMF panel plotted against the y-position along the crypt-villus axis. Each dot represents a surface spot of REELIN fluorescence, generated in Imaris 9.5, color-coded by its’ REELIN signal intensity. (H) Immunofluorescent imaging of REELIN (green) in intestinal lymphatics (purple) of control (ctrl, Relnfl/fl) and VE-Cadherin-CreERT2 Relnfl/delta conditional null (Reln c-null) mice showing loss of REELIN in lymphatics. (I) Incorporation of EdU after a 30-minute pulse in mice of the indicated genotype. Representative immunofluorescent images (left) and quantification of EdU+ cells per crypt (right) are shown. (J) Number of SOX9+ cells out of the crypt (TA cells) in control and lymphatic-ablated (left) and control and Reln c-null mice (center). Representative images showing the distribution of LYZ1+ Paneth cells and SOX9+ cells (stem, Paneth and TA cells) are on the right. Panels H-J, showing control and Reln c-null mice, are representative of n = 2 mice per group. (K) Left: schematic of organoid experiments with recombinant murine REELIN. Middle: violin plots of the area and number of crypt domains per organoid cultured in the presence or absence of murine recombinant REELIN for 4 days. Right: representative IMF images showing SOX9 (green) and mTomato (magenta). Asterisk marks the autofluorescent organoid lumen (n = 3 independent experiments). (L) Western blot of murine SI organoids showing phosphorylation of DAB1 upon stimulation with recombinant REELIN (rRELN) for 10 or 20 minutes +/− inhibitors against REELIN receptors (VLDLRi, ITGB1i). Western blot image is representative of n = 3 independent experiments. SF = serum-free media, * on the right indicate non-specific protein bands. For statistics * indicates a p-value 0.01 to 0.05, ** p-value of 0.001 to 0.01, *** p-value of 0.0001 to 0.001, **** p < 0.0001 (Unpaired two-tailed Student’s t-test).
When re-plated in normal organoid medium, the organoids adopted normal size and morphology, consistent with a reversal in the differentiation block imposed by LEC-secreted factors (Figure 6A). Exposure to LEC-CM, in comparison, yielded less differentiated organoids, recapitulating the phenotype of organoids cocultured with LECs (Figures 6B, 6C). This effect was not observed with BEC-CM (Figure S6A). Together, these data underscored the potent and direct effect of lymphatic-secreted factors on ISC maintenance.
The reversibility on impairing ISC differentiation, coupled with the reliance on sustained exposure to lymphangiocrine factors suggested that in the context of the tissue, LEC effects should be manifested predominantly within the crypt niche, where they co-reside with ISCs. We identified 12 genes encoding intestinal lymphatic ligands that were not expressed in blood vessels and displayed a high probability of being secreted extracellularly based on the presence of N-terminal signal peptide sequences indicative of protein secretion and extracellular localization (Figure S6B) (Emanuelsson et al., 2000; Horton et al., 2007). We excluded genes encoding proteins with either broad expression across cell types, known interactions/functions with non-crypt residents (e.g. platelets or pathogens), and/or established roles in seemingly unrelated pathways such as clotting cascades or copper metabolism. This narrowed our list to six candidate factors that may form a lymphangiocrine signaling hub for the crypt (Figure 6D). Of these six factors, all but IL33 had receptors that were expressed by ISCs (Figure S6C).
All six of these factors were significantly more highly expressed in LECs than in any other crypt cell type, including blood endothelial cells, which we had already shown did not impact ISCs (Figures 6D, S6B and S6D). To further narrow this list, we used our high-resolution cartograph to plot the spatial expression of each of these genes along the crypt-villus axis in a cell type-specific manner (Figure 6E). We used SI data so that we could also highlight lymphatic genes that showed spatial heterogeneity in expression between the crypt and villus. From these analyses, we learned that although some of these genes, e.g. Ntn1 and Il33, were enriched in crypt lymphatics, they were not exclusive to this cell population nor the crypt. By contrast, Reln, Ccl21a, Rspo3 and Wnt2 showed elevated expression at the base where the crypt lymphatics reside (arrow) rather than within the villus, where the lacteals are located.
R-SPONDIN-3, encoded by Rspo3, binds to LGR5 and is known to enhance WNT signaling in ISCs (Mah et al., 2016; Yan et al., 2017). Although Rspo3 has been widely viewed as a factor produced by mesenchymal cells (Greicius et al., 2018), it was recently reported to be expressed by lymphatics (Ogasawara et al., 2018). Our spatial transcriptomic data now identify crypt-based lymphatics as the major source of this essential ISC ligand (Figures 6D-E, S6B and S6D). While WNT2 has not been studied in the context of the crypt, its expression added to the list of canonical WNTs expressed in the niche (David et al., 2020; Mah et al., 2016). Given the importance of WNT-Frizzled signaling in ISC maintenance, the concentration of these factors in crypt-based LECs heightened the importance of lymphatics as a component of the ISC niche.
While the roles of R-SPONDINs and WNTs in ISC function are well established, REELIN has mostly been viewed as a large ECM protein involved in neuronal migration (D’Arcangelo et al., 1995), smooth muscle:lymphatic signaling (Lutter et al., 2012) and cardiac remodeling during development and following injury (Liu et al., 2020). Reln’s exclusive expression and concentration in intestinal crypt lymphatics was striking and although REELIN’s purported receptors (Vldlr/Lrp8 and Itgb1) (D’Arcangelo et al., 1999; Dulabon et al., 2000), were not exclusive to ISCs, they were all expressed by them (Figures 6D-6G and S6E-S6F). This suggested that REELIN may mediate at least some of the effects of LECs and LEC-conditioned medium on ISC maintenance and differentiation. Whole-mount IMF imaging coupled with fluorescence intensity quantifications verified REELIN’s protein expression and concentration in crypt lymphatics, as compared to lacteals (Figures 6F-6G and S6G-S6H, Video S5). Moreover, upon analyzing human scRNA-seq data (Elmentaite et al., 2021), we observed Reln in human intestinal LECs (Figure S6I).
To assess REELIN’s functional importance to ISC behavior, we next examined intestinal tissue from tamoxifen-treated VE-Cadherin-CreERT2 Relnfl/delta mice, in which Reelin is conditionally targeted in the lymphatics (Liu X et al., 2020). Though VE-Cadherin is expressed in all vascular cells, Reelin expression was restricted to lymphatics, effectively providing a conditionally null Reelin model (Reln c-null) for interrogating Reelin’s function shortly after its removal in the lymphatics. Our spatial localization data suggested that the primary early consequences should be in the crypt. Consistent with this, an increase in proliferation was observed within Reln c-null crypt niches, as judged by a pulse of EdU 30 minutes prior to harvesting (Figures 6H and 6I). Immunolabeling for REELIN, LYVE1, OLFM4 and LYSOZYME further suggested that, although REELIN was absent, crypt lymphatics, ISCs and Paneth cells remained (Figures 6H, 6J and S6J). Rather, based upon additional immunolabeling and quantifications for SOX9, which marks ISCs, progenitor cells, TA cells and to a lesser extent Paneth cells within the crypt, the data favored the view that the proliferation differences resulting from REELIN loss reflected a skewing in the ISC:TA populations, similar to what we observed in our lymphatic ablation studies (Figure 6J). Given the complexity and broader distribution of REELIN’s receptors within the niche, we returned to our co-culture system to determine if REELIN directly governs ISC:LEC interactions.
Importantly, the genes encoding all six secreted intestinal lymphatic factors listed in Figure 6 are also expressed by dermal lymphatics (Figure S6K). By ELISA, we found that REELIN levels in the interstitial fluids of SI, LI and skin were similar, with reduced, but appreciable levels in our dermal LEC-CM (Figure S6L). By IMF, it was clear that REELIN was also expressed by cultured dermal LECs (Figure S6M).
The addition of recombinant REELIN to standard organoid cultures recapitulated the immature phenotype of organoids cocultured with LECs and/or cultured in LEC-CM (Figure 6K). Moreover, recombinant REELIN activated the most reliable marker downstream of REELIN-mediated signaling, pDAB1, in organoids (Figure 6L). DAB1’s activation was significantly reduced by blocking antibodies against REELIN’s receptors. Taken together, these data add REELIN as a new component of the intestinal crypt niche that can directly impact ISC behavior and maintain stemness.
Our study illuminated the power of integrating transcriptomic data and high-resolution spatial analysis with functional analyses to dissect the complexity of the intestinal stem cell niche, identify a novel signaling hub within it and unravel its physiologic relevance.
Discussion
The Power and Universality of Our Computational Strategy
The high-resolution spatial map we charted and the associated deep single-cell profiling of small and large intestinal cells offers a rich dataset to query insights into spatially defined cell types, gene expression programs, and complex cellular interactions occurring within the ISC niche. While most previous studies have focused on identifying cell types from ST, our study has highlighted the importance of also inferring spatially resolved cell type-specific gene expression. Two features of BayesPrism make it highly generalizable: (1) It does not rely on the cell type proportions observed in the scRNA-seq data, making it particularly effective for datasets with enriched rare cell types and (2) it is robust to technical and biological variation and so does not require perfectly matched samples from scRNA-seq and ST.
Taking advantage of the stereotypical crypt-villus and crypt units, SpaceFold concentrated transcriptomic data onto a 1D pseudo-space axis, facilitating analysis of spatially restricted gene expression at high-resolution and pooling information across many crypts to elucidate robust shared biological features. This becomes particularly powerful in dissecting the complexities within a stem cell niche that involves cell types and structures (such as lymphatics), which can and often do span diverse tissue regions. SpaceFold can be applied to other tissues with stereotyped structures and extended to project each spot onto two dimensions, to construct a 2D cartography of more complex tissue units (e.g. liver lobules).
Lymphatics as a Signaling Hub for Tissue Stem Cells
Our multifaceted approach exposed niche lymphatic capillaries as a signaling hub for the intestinal crypt. Although ISCs are not the exclusive recipients of lymphatic-secreted factors, our coculture studies showed that lymphatic-derived ligands act directly on ISCs and maintain their self-renewal ability and fitness, while concurrently suppressing differentiation. We were also able to dissect spatial heterogeneity in gene expression within the lymphatic endothelial network across the intestinal crypt-villus axis. This unveiled factors produced specifically by lymphatic capillaries at the crypt base.
We identified lymphangiocrine signals that instruct ISC maintenance and lineage restriction. Lymphatics act as a stationary niche component for ISCs, whose available niche interaction sites are restricted. Thus, as ISC progeny become displaced from their niche signals, these cells execute a program of differentiation. This model was supported by our in vitro studies, showing that LEC-conditioned media sufficed in enhancing stemness and repressing differentiation in organoids.
We learned that crypt lymphatics are the major source of Rspo3, the key WNT-signaling factor for LGR5+ ISCs. Although lymphatics had been reported to express Rspo3, the result had been puzzling given the lacteal-centric focus on intestinal lymphatics, and hence mesenchymal cells seemed a better source for this key ligand in the crypt (Greicius et al., 2018; Ogasawara et al., 2018). Our spatial mapping beautifully showed that, in fact, Rspo3 is predominantly expressed in the crypt lymphatics.
We also discovered that Reln transcripts and REELIN protein concentrate preferentially in the crypt lymphatics. Our spatial transcriptomics revealed that putative receptors for RSPO3, WNT2 and REELIN, three crypt-based lymphangiocrine factors, are expressed by crypt ISCs. While WNTs and RSPONDINs are well known ISC regulators, our in vivo and in vitro studies exposed REELIN, exclusively expressed by lymphatics, as a key balancer of ISC maintenance and tissue regeneration.
Lymphatics and the Intestinal Stem Cell Niche
We brought to light lymphatic capillaries as a key and integral constituent of the crypt niche for both SI and LI. ISCs operate continuously in tissue regeneration, and lymphatic loss caused them to shift even further from self-renewal to tissue regeneration, increasing differentiation and reducing ISC numbers. Interestingly in the hair follicle, conditional lymphatic ablation similarly causes normally quiescent hair follicle stem cells (HFSCs) to undergo precocious proliferation and enter the hair cycle, indicative of lineage commitment and differentiation (Gur-Cohen et al., 2019). However, in contrast to the intestine, lymphatic ablation in the skin did not cause immediate stem cell exhaustion. It is notable that while ISCs cycle continuously, HFSCs undergo protracted bouts of quiescence. When hairs are repetitively plucked, thereby perturbing the niche and placing HFSCs in continual motion, stem cell exhaustion occurs as well (Lay et al., 2016). These comparisons yield two general insights. First, despite very different tissue demands, intestinal and hair follicle stem cells rely upon lymphatics to maintain their stemness and restrict tissue regeneration. Second, when lymphatics malfunction and other niche constraints fail to curb accelerated tissue production, stem cell exhaustion can occur.
Broader Implications for Lymphatic-ISC Interactions
Although we focused on direct communication between LECs and ISCs here, lymphatics are also likely to mediate other crypt dynamics, including crosstalk with other niche cells, as suggested by the myriad of additional putative secreted factors, such as IL33, expressed by lymphatic capillaries. As REELIN’s receptors are also expressed by other niche cell types, the action of this ligand may extend beyond lymphatic: ISC signaling.
Lymphatic capillaries could also be important in trafficking of niche-specific signaling molecules, fluid drainage and immune cell crosstalk. Indeed, while immune cells can directly signal to ISCs to orchestrate pathogen responses (Biton et al., 2018; von Moltke et al., 2016), immune-mediated effects on ISCs also occur in graft-versus-host disease (Fu et al., 2019), hinting at an important role for immune cell drainage from the niche via lymphatics.
With the diversity of roles that lymphatics can play, their association with ISCs is likely to have important consequences in disease states. In this regard, inflammatory diseases of the intestine have been associated with lymphatic abnormalities and dysfunction (Alexander et al., 2010; D’Alessio et al., 2014; Jang et al., 2013; Van Kruiningen and Colombel, 2008). Additionally, obesity alters LEC density, proliferation, and permeability (Zhang et al., 2018). Any or all of these perturbations in lymphatics could impact ISCs directly and/or through other niche components. To guide future efforts, it will be important to interrogate how the lymphatic transcriptome is altered in disease states, how it interprets inflammatory and metabolic cues, and how it relays these to the ISC niche.
Limitations of the Study
Our new algorithms have enabled us to discover that lymphatic capillaries form a signaling hub for ISC maintenance and intestinal homeostasis. However, the field of spatial transcriptomics is still unfolding and resolution will continue to improve as bar-coded spot sizes are reduced. In addition, although our coculture studies were ideal for documenting direct effects of lymphatics on intestinal organoids, they do not preclude multi-cellular communication networks, e.g. between lymphatics, ISCs and mesenchymal or immune cells. As we delve deeper into lymphatic:stem cell communication, we’ll also want to turn to disease states, such as inflammatory bowel disease, to unearth whether and how lymphatic-to-ISC communication is altered in disease states.
Regarding functional studies, the use of systemic lymphatic ablation to evaluate the function of lymphatic signaling in ISC maintenance has caveats, including the collapse of the lymphatic network at the base of the crypts and resultant lack of drainage. We chose an early time point for analysis to mitigate this. However, genetic models, such as Reelin c-null mice used here or conditional ablation of factors not uniquely expressed by lymphatics, will be needed to probe deeper into the features of crypt lymphatic signaling within the ISC niche.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Elaine Fuchs (fuchslb@rockefeller.edu).
Materials availability
No unique reagents or materials were generated in this study. Materials used in this study are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Olfm4 (D6Y5A) XP® Rabbit mAb (Mouse Specific) | Cell Signaling Technology | Cat#39141S; RRID: AB_2650511 |
| Mouse Reelin Antibody AF3820 | R&D systems | Cat#AF3820; RRID: AB_2253745 |
| Human LYVE-1 Antibody (goat) | R&D systems | Cat#AF2089; RRID: AB_355144 |
| Human/Mouse E-Cadherin Antibody | R&D systems | Cat#AF748; RRID: AB_355568 |
| E-Cadherin (24E10) Rabbit mAb #3195 | Cell Signaling Technology | Cat#3195S; RRID: AB_2291471 |
| Anti-Aldolase B Antibody | Abcam | Cat#ab75751; RRID: AB_2226682 |
| CD140a (PDGFRA) Monoclonal Antibody (APA5), APC, eBioscience™ | ThermoFisher | Cat#17-1401-81; RRID: AB_529482 |
| APC/Cyanine7 anti-mouse CD45 Antibody | Biolegend | Cat#103116; RRID: AB_312981 |
| LYVE1 Monoclonal Antibody (ALY7), eFluor 660, eBioscience™ | ThermoFisher | Cat#50-0443-82; RRID: AB_10597449 |
| LYVE1 Monoclonal Antibody (ALY7), eBioscience | ThermoFisher | Cat#14-0443-82; RRID: AB_1633414 |
| anti-mouse LYVE-1 | Angiobio | Cat#11-034; RRID: AB_2813732 |
| PE anti-mouse Podoplanin Antibody | Biolegend | Cat#127408; RRID: AB_2161928 |
| PerCP/Cyanine5.5 anti-mouse Podoplanin Antibody | Biolegend | Cat#127422; RRID: AB_2814016 |
| E-Cadherin (4A2) Mouse mAb #14472 | Cell Signaling Technology | Cat#14472S; RRID: AB_2728770 |
| Purified anti-mouse CD326 (Ep-CAM) Antibody | Biolegend | Cat#118202; RRID: AB_1089027 |
| FITC anti-mouse CD31 Antibody | Biolegend | Cat#102506; RRID: AB_312913 |
| PE/Cyanine7 anti-mouse CD326 (Ep-CAM) Antibody | Biolegend | Cat#118216; RRID: AB_1236471 |
| Anti-Prox1 Ab (rabbit, 100 ug) | Angiobio | Cat#11-002P |
| PE/Cyanine7 anti-mouse CD31 Antibody | Biolegend | Cat#102524; RRID: AB_10013720 |
| Endomucin Antibody (V.7C7): sc-65495 | SantaCruz | Cat#SC65495; RRID: AB_2100037 |
| Polyclonal Rabbit Anti-Human Lysozyme | Dako | Cat#EC 3.2.1.17; RRID: AB_2341231 |
| Anti-Reelin (CR-50) mAb (Monoclonal Antibody) | MBL International | Cat# D223-3; RRID: AB_843523 |
| TruStain FcX™ (anti-mouse CD16/32) Antibody | Biolegend | Cat#101320; RRID: AB_1574975 |
| Donkey anti-Sheep IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | ThermoFisher | Cat#A-21448; RRID: AB_2535865 |
| APC anti-mouse CD170 (Siglec-F) Antibody | Biolegend | Cat#155508; RRID: AB_2750237 |
| Phospho-Dab1 (Tyr220) Antibody #3327 | Cell Signaling Technology | Cat#3327S; RRID: AB_2267690 |
| DAB1 Antibody (PA5-85453) | ThermoFisher | Cat#PA5-85453; RRID: AB_2792593 |
| Anti-GFP Polyclonal Chicken | Abcam | Cat#ab13970; RRID: AB_300798 |
| Guineapig anti Sox9 | Elaine Fuchs’ Lab | N/A |
| Recombinant Anti-SOX9 antibody [EPR14335-78] (ab185966) | Abcam | Cat#ab185966; RRID: AB_2728660 |
| Cleaved Caspase-3 (Asp175) (5A1E) Rabbit mAb #9664 | Cell Signaling Technology | Cat#9664; RRID: AB_2070042 |
| Anti-PECAM-1 Antibody, clone 2H8, Azide Free | MilliporeSigma | Cat#MAB1398Z; RRID: AB_94207 |
| Purified NA/LE Hamster Anti-Rat CD29 | BD bioscience | Cat#555002; RRID: AB_395636 |
| Purified Mouse Anti-CD29 | BD bioscience | Cat#610467; RRID: AB_2128060 |
| α-Smooth Muscle Actin (D4K9N) XP® | Cell Signaling Technology | Cat#19245; RRID: AB_2734735 |
| β-Actin (8H10D10) Mouse mAb #3700 | Cell Signaling Technology | Cat#3700S; RRID: AB_2242334 |
| Biological samples | ||
| Human organoids | Memorial Sloan Kettering Cancer Center | N/A |
| Human intestinal tissue | Jill Roberts Center for Inflammatory Bowel Disease | N/A |
| Mouse intestinal tissue from VE-Cadherin-CreERT2 Reelinflox/Δ mice and Reelinflox/flox mice | Gift from Guillermo Oliver | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant Mouse Reelin Protein | R&D Systems | Cat#3820-MR-025 |
| Recombinant Human R-Spondin 1 Protein | R&D Systems | Cat#4645-RS-250 |
| Recombinant Murine Noggin | Prepotech | Cat#250-38 |
| EGF Recombinant Mouse Protein | ThermoFisher | Cat#PMG8041 |
| Recombinant Murine Wnt-3a | Prepotech | Cat#315-20 |
| Recombinant Mouse LRPAP Protein, CF | R&D Systems | Cat#4480-LR-050 |
| N-Acetyl-L-cysteine | MilliporeSigma | Cat#A7250-50G |
| EDTA (0.5 M), pH 8.0, RNase-free | ThermoFisher | Cat#AM9260G |
| Gelatin-Based Coating Solution | Cell Biologics | Cat#6950 |
| Ethyl cinnamate | MilliporeSigma | Cat#112372-100G |
| L-Glutamine (200 mM) | ThermoFisher | Cat#25030081 |
| GlutaMAX™ Supplement | ThermoFisher | Cat#35050061 |
| TRIzol™ Reagent | ThermoFisher | Cat#15596026 |
| Diphtheria Toxin from Corynebacterium diphtheriae | MilliporeSigma | Cat#D0564 |
| Tamoxifen | MilliporeSigma | Cat#T5648 |
| Gibco™ B-27™ Supplement (50X), serum free | Fisher Scientific | Cat#17-504-044 |
| HEPES (1 M) | ThermoFisher | Cat#15630080 |
| PRIMOCIN | Fisher Scientific | Cat#NC9141851 |
| NORMOCIN | Fisher Scientific | Cat#NC9273499 |
| Gentle Cell Dissociation Reagent | Stem Cell Technology | Cat#100-0485 |
| Corning® Matrigel® Matrix for Organoid Culture, Phenol Red-free, LDEV-free | Corning | Cat#356231 |
| Cultrex UltiMatrix Reduced Growth Factor Basement Membrane Extract | R&D systems | Cat#BME001-10 |
| Gibco™ TrypLE™ Express Enzyme (1X), no phenol red | Fisher Scientific | Cat#12-604-021 |
| COMPLETE MOUSE ENDOTHELIAL CELL MEDIUM /W KIT | Cell Biologics | Cat# M1168 |
| Advanced DMEM/F-12 | Thermo Fisher | Cat#12634028 |
| Endothelial Cell Growth Medium MV 2 | PromoCell | Cat#C-22121 |
| ProLong™ Gold Antifade Mountant with DAPI | Thermo Fisher | Cat#P36935 |
| Normal Donkey Serum | JacksonImmunoResearch Lab | Cat#017-000-121 |
| RIPA Lysis and Extraction Buffer (Roche) | ThermoFisher | Cat#89901 |
| PhosSTOP™ (Roche) | MilliporeSigma | Cat#4906837001 |
| cOmplete™ Protease Inhibitor Cocktail (Roche) | MilliporeSigma | Cat#11836145001 |
| NuPAGE™ LDS Sample Buffer (4X) | ThermoFisher | Cat#NP0008 |
| NuPAGE™ Sample Reducing Agent (10X) | ThermoFisher | Cat#NP0009 |
| Precision Plus Protein™ Dual Color Standards | Biorad | Cat#1610374EDU |
| NuPAGE™ 4 to 12%, Bis-Tris, 1.0–1.5 mm | ThermoFisher | Cat#:NP0321BOX |
| NuPAGE™ MOPS SDS Running Buffer (20X) | ThermoFisher | Cat#NP0001 |
| NuPAGE™ Transfer Buffer (20X) | ThermoFisher | Cat#NP00061 |
| 1x Tris Buffered Saline (TBS) | Biorad | Cat#1610782 |
| Pierce™ ECL Plus Western Blotting Substrate | ThermoFisher | Cat#32132 |
| SuperSignal™ West Femto Maximum Sensitivity Substrate | ThermoFisher | Cat#34095 |
| NGS-WNT | Immunoprecise | Cat#N001 |
| EGF | Peprotech | Cat#AF-100-15 |
| A83-01 | Tociris | Cat#2939 |
| FGF2 | Peprotech | Cat#100-18B |
| IGF-1 | Peprotech | Cat#100-11 |
| Noggin Condition Media | Immunoprecise | Cat#N002 |
| R-Spondin 1 Conditioned Media | Made in Ganesh lab with R-Spondin1-Fc 293T line | |
| Alexa Fluor™ 488 Phalloidin | ThermoFisher | Cat#A12379 |
| Critical commercial assays | ||
| Visium Spatial gene Expression Starter Kit | 10X Genomics | Cat#1000200 |
| Mix-n-Stain 568/591 nm | Biotium | Cat#92336 |
| Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 647 dye | ThermoFisher | Cat#C10340 |
| LIVE/DEAD™ Fixable Aqua Dead Cell Stain Kit, for 405 nm excitation | ThermoFisher | Cat#L34957 |
| Mouse Reelin ELISA Kit (Colorimetric) | Novus Biologic | Cat#NBP2-82424; RRID: |
| In Situ Cell Death Detection Kit, TMR red | Roche | Cat#12156792910 |
| Direct-zol RNA Microprep | Zymo Research | Cat#R2062 |
| Direct-zol RNA Miniprep | Zymo Research | Cat#R2050 |
| SuperScript™ VILO™ cDNA Synthesis Kit | ThermoFisher | Cat#11754050 |
| Power SYBR™ Green PCR Master Mix | ThermoFisher | Cat#4368577 |
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| Deposited data | ||
| Raw single and spatial transcriptomic data (mouse small and large intestine) | This paper | GEO: GSE190037 |
| Raw bulk RNA-sequencing data (mouse large intestine) | This paper | GEO: GSE199082 |
| Re-analyzed bulk RNA-sequencing data (mouse skin) | (Gur-Cohen et al., 2019) | GEO: GSE130976 |
| Re-analyzed single cell RNA-sequencing data (human) | (Elmentaite et al., 2021) | EMBL-EBI Array Express: E-MTAB-9532, E-MTAB-8901 |
| Experimental models: Cell lines | ||
| C57BL/6 MOUSE PRIMARY DERMAL LYMPHATIC ENDOTHELIAL CELLS | Cell Biologics | Cat#C57-6064L |
| B129 Mouse Primary Dermal Microvascular Endothelial Cells | Cell Biologics | Cat#B129-7064 |
| Human Dermal Lymphatic Endothelial Cells (HDLEC) - adult, cryopreserved | PromoCell | Cat#C-12217 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | 000664 |
| Mouse: Lgr5-EGFP-IRES-CreERT2 (B6.129P2-Lgr5tm1(cre/ERT2)Cle/J) | The Jackson Laboratory | 008875 |
| Mouse: PDGFRa-H2B-EGFP (B6.129S4-Pdgfratm11(EGFP)Sor/J) | The Jackson Laboratory | 007669 |
| Mouse: ROSAmT/mG (Gt(ROSA)26Sortm4 (ACTB-tdTomato,-EGFP)Luo/J)) | The Jackson Laboratory | 007676 |
| Mouse: B6.Cg- Tg(Grem1-cre/ERT)3Tcw/J | The Jackson Laboratory | 027039 |
| Mouse: Vegfr3-CreERT2 iDTRflox/+ mice and Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice | (Gardenier et al., 2016) | N/A |
| Oligonucleotides | ||
| Quantitative real-time PCR primers (see table S1) | Eurofins Genomics | N/A |
| RNAscope® 2.5 LS Probe - Mm-Sox9-C2 - Mus musculus SRY-box containing gene 9 (Sox9), mRNA | ACD | 401058-C2 |
| RNAscope® 2.5 LS Probe - Mm-Lyz1-C3 - Mus musculus lysozyme 1 (Lyz1) mRNA | ACD | 415138-C3 |
| RNAscope® LS 2.5 Probe - Mm-Lgr5 - Mus musculus leucine rich repeat containing G protein coupled receptor 5 (Lgr5), mRNA | ACD | 312178-C1 |
| Software and algorithms | ||
| Prism | https://www.graphpad.com/scientific-software/prism/ | N/A |
| Imaris | https://imaris.oxinst.com/ | N/A |
| ImageJ | https://fiji.sc/ | N/A |
| FlowJo | https://www.flowjo.com | N/A |
| Biorender | https://biorender.com/ | N/A |
| Cellranger (version 6.0.1) | https://10xgenomics.com | N/A |
| Spaceranger (version 1.2.1) | https://10xgenomics.com | N/A |
| BayesPrism (R package) (version 1.3) | (Chu et al., 2022) | N/A |
| SpaceFold (R package) (version 1.0) | This paper: DOI 10.6084/m9.figshare.19715029 | N/A |
| phateR (R package) (version 1.0.7) | (Moon et al., 2019) | N/A |
| MAST (R package) (version 1.18.0) | (Finak et al., 2015) | N/A |
| biomaRt (R package) (version 2.48.3) | (Durinck et al., 2005, 2009) | N/A |
| miloR (R package) (version 1.0.0) | (Dann et al., 2021) | N/A |
| RCTD (R package) (version 1.2.0) | (Cable et al., 2021) | N/A |
| SPOTlight (R package) (version 0.99.8) | (Elosua-Bayes et al., 2021) | N/A |
| slalom (python package) (version 1.0.0.dev11) | (Buettner et al., 2017) | N/A |
| ForceAtlas2 (python package) (version 0.3.5) | (Jacomy et al., 2014) | N/A |
| scanpy (python package) (version 1.8.1) | (Wolf et al., 2018) | N/A |
| Phenograph (python package) (version 1.5.7) | (Levine et al., 2015) | N/A |
| scran (python package) (version 1.20.1) | (Lun et al., 2016) | N/A |
| mclust (python package) (version 5.4.7) | (Scrucca et al., 2016) | N/A |
| mixtools (python package) (version 1.2.0) | (Benaglia et al., 2010) | N/A |
| CellBender (python package) (version 1.0) | (Fleming et al., 2019) | N/A |
| DoubletDetection (python package) (version 3.0) | (Gayoso and Shor, 2020) | N/A |
| Scrublet (python package) (version 0.2.3) | (Wolock et al., 2019) | N/A |
| Stereoscope (python package) (version 0.3.1) | (Andersson et al., 2020) | N/A |
| Cell2location (python package) (version 0.6a0) | (Kleshchevnikov et al., 2022) | N/A |
| tangram-sc (python package) (version 1.0.1) | (Biancalani et al., 2021) | N/A |
| Other | ||
| BD FACSAria Cell Sorter | BD Bioscience | N/A |
| BD LSRII Analyzer | BD Bioscience | N/A |
| Synergy Neo2 Multi-Mode Reader | BioTek | |
Data and code availability
All data that support the findings of this study are available within the paper and its supplementary files. Single-cell and spatial transcriptomic data have been deposited in the Gene Expression Omnibus (GEO) repository with the accession code GSE190037. Newly generated bulk RNA sequencing data are associated with the accession code GSE199082. All original code used to analyze the data is available at https://github.com/dpeerlab/SpaceFold_paper.git. The SpaceFold R package along with tutorials is available at https://github.com/dpeerlab/SpaceFold.git as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6 mice, Lgr5-EGFP-IRES-CreERT2 (B6.129P2-Lgr5tm1(cre/ERT2)Cle/J), PDGFRα-H2B-EGFP (B6.129S4-Pdgfrαtm11(EGFP)Sor/J) and ROSAmT/mG (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J)) mice were purchased from the Jackson Laboratories. Both male and female mice were used. Mice were maintained in the Association for Assessment and Accreditation of Laboratory Animal Care-accredited animal facility of The Rockefeller University (RU), and procedures were performed with Institutional Animal Care and Use Committee (IACUC)-approved protocols. Mice of all strains were housed in an environment with controlled temperature and humidity, on a 12-hour light cycle, and fed regular rodent’s chow. The numbers of animals used for each experiment is specified in the figure legends or methods as n = x mice per group, per timepoint analyzed. Animals were used at 6-12 weeks of age for experiments unless indicated otherwise. (B6.Cg-Tg(Grem1-cre/ERT)3Tcw/J) mice were a gift from Dr. Timothy C. Wang (Columbia University, New York). ROSAmT/mG (Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J) mice were used for the majority of crypt isolation (organoid) experiments. Intestinal tissue from VE-Cadherin-CreERT2 Reelinflox/Δ mice were a gift from Dr. Guillermo Oliver (Northwestern University). For lymphatic ablation experiments Vegfr3-CreERT2 iDTRflox/+ mice (originally a gift from Dr. Babak Mehrara (MSKCC)) or Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice were used.
Human tissues
De-identified intestinal biopsy specimens were obtained from the Jill Roberts Center for Inflammatory Bowel Disease Research at Weill Cornell Medical College, Department of Gastroenterology (New York, NY) in compliance with federal and state laws, and National Institute of Health guidelines.
Primary Cell Culture
Mouse small intestinal organoid culture
To grow small intestinal organoids crypts were isolated from the small intestine of either C57BL6/J or ROSAmT/mG mice at an age of 8-12 weeks. See method details.
Mouse large intestinal organoid culture
To grow large intestinal organoids crypts were isolated from the colon of 1-2 ROSAmT/mG mice at an age of 8-12 weeks. See method details.
Mouse lymphatic endothelial cell culture
C57BL/6 mouse primary dermal lymphatic endothelial cells (LECs) (Cell Biologics) were expanded on 100 mm dishes, pre-coated with gelatin (Sigma), in Complete Mouse Endothelial Cell Medium (Cell Biologics). LECs were utilized at passages 4-5 in all experiments.
Mouse blood endothelial cell culture
Blood endothelial cells (BECs) were derived by sorting alive, CD31+, LYVE1−, PDPN− single cells from C57BL/6 mouse primary dermal microvascular endothelial cells (Cell Biologics) after in vitro expansion. Sorted BECs were used at passages 4-5 for all experiments.
Human lymphatic endothelial cell culture
Human dermal lymphatic endothelial cells (adult donor, PromoCell) were cultured in Endothelial Cell Media MV/MV2 (PromoCell) on gelatin (Sigma)-coated dishes. Cells were used at passages 4-5 for experiments.
Human colonic organoid culture
Experiments involving human tissue from normal colon biopsies were carried out under the MSKCC Institutional Review Board (IRB) protocol # 14-244. Details on human organoid derivation can be found in the detailed methods.
METHOD DETAILS
Animal treatments
Tamoxifen treatments
To generate conditional Reelin-knockout mice from VE-Cadherin-CreERT2 Reelinflox/Δ mice, animals were intraperitoneally injected with tamoxifen (5 mg/40 g body weight) 2-3 times a week for 2 consecutive weeks at an age of 8-10 weeks. (B6.Cg-Tg(Grem1-cre/ERT)3Tcw/J) mice were intraperitoneally injected with 100 μl tamoxifen (2% w/v) once at an age of 7 weeks and sacrificed 1 week later. Vegfr3-CreERT2 iDTRflox/+ mice and Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice were intraperitoneally injected with tamoxifen (75 mg/kg body weight) (Sigma) dissolved in corn oil (Sigma) 3 times every second day at 6-8 weeks of age to activate the expression of diphtheria toxin receptor.
Acute lymphatic ablation experiments
For acute lymphatic ablation experiments tamoxifen-treated Vegfr3-CreERT2 iDTRflox/+ mice and Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice were intraperitoneally injected with 200 ng diphtheria toxin (DT) (Sigma Aldrich) diluted in sterile PBS for 3 times every second day (day 0-2-4) at 8-10 weeks of age. Animals were euthanized on day 5 for experiments. Mice undergoing lymphatic ablation and controls were sex-matched and co-housed. Acute ablation experiments were repeated 3 times (n = 7 mice total per experimental group) for quantification of EdU signal on tissue sections and qPCRs, while 2 additional repeats (n = 10 mice total per experimental group) were performed for quantification of EdU signal via flow cytometry.
In vitro cell culture
Mouse small intestinal organoid culture
To grow small intestinal organoids crypts of the mouse small intestine were isolated as described with some alterations (O’Rourke et al., 2016). Briefly, three-quarters of the small intestine were flushed with ice-cold phosphate-buffered saline (PBS), opened longitudinally and scraped twice with a glass slide to remove villus epithelium. The tissue was cut into ~ 5-10 mm pieces, which were washed 5-8 times in ice-cold PBS with 1.5 mM Dithiothreitol (DTT) (Sigma). Remnant villi were removed by shaking tissue pieces at 4°C in an initial 5 minute incubation in PBS with 10 mM RNase-free Ethylenediaminetetraacetic acid (EDTA) pH 8.0 (Invitrogen). Crypts were collected and filtered through 70 μm cell strainers in tubes pre-coated with PBS and 0.2% bovine serum albumin (BSA) or fetal bovine serum (FBS) in subsequent fractions, for which the pieces were incubated in PBS + 5 mM EDTA for 10 minutes shaking at 4°C. The best fractions were pooled and centrifuged at 4°C in subsequent spins at 290 g and 200 g for 6 minutes each. 300-500 crypts were plated per dome. Per well of a 48-well plate 20 μl organoid droplets were plated and incubated at 37°C for 15 minutes before adding 300 μl of organoid media.
Organoids were maintained in Advanced DMEM/F12 medium (Thermo Fisher) with L-glutamine (2 mM), Penicillin-Streptomycin (100 μg/ml), HEPES buffer (10 mM) (Fisher Scientific) and N-Acetylcysteine (1 mM) (Sigma Aldrich). Growth factors facilitating organoid growth were added and included recombinant murine EGF (50 ng/ml) (Thermo Fisher), recombinant murine Noggin (50 ng/ml) (Peprotech) and recombinant human R-Spondin-1 (500 ng/ml) (R&D systems or Peprotech) to make ENR medium. To avoid fungal contamination Normocin (InvivoGen) antimicrobial reagent was added at 1:200 to ENR organoid medium. Indicated numbers of crypts or single cells were plated in growth-factor-reduced Matrigel (Corning, R&D) mixed 1:1 with ENR medium. The media was replaced every 2 days and for the first two days in culture after crypt isolation ROCK inhibitor (1:5000) was added to ENR medium.
To passage organoids, the media was aspirated and replaced with Gentle Cell Dissociation Reagent (Stemcell Technologies) on days 6-7. Matrigel domes were collected in a precoated 15 ml falcon tube and incubated in Gentle Cell Dissociation Reagent for 10 minutes on a rocker at room temperature. Organoids were spun down in subsequent centrifugation steps at 290 g, 200 g and 150 g for 6 minutes and resuspended in ENR organoid medium mixed 1:1 with Matrigel to plate new domes.
Mouse large intestinal organoid culture
To isolate crypts from the murine colon, the large intestine of 1-2 mice was cut, flushed with ice-cold PBS, opened longitudinally and cut into ~5 mm pieces. The tissue pieces were washed in ice-cold PBS for at least 8 times by vigorous shaking. The tissue was incubated in PBS with 5 mM EDTA, shaken vigorously before replacing the solution with fresh PBS and 2.5 mM EDTA. Tissue pieces were incubated on a horizontal shaker at 4°C for 30 minutes, shaken vigorously and the solution was passed through a 70 μm strainer to collect the first fraction. The tissue pieces were incubated again for 10 minutes in PBS with 2.5 mM EDTA and up to 3 additional fractions were collected. The best fractions were pooled and spun down at 300 g for 5 min at 4°C. The pellet was resuspended in PBS with 0.2% BSA or FBS and filtered through a 70 μm strainer again. 400-500 crypts were plated per dome. Large intestinal organoids were grown and maintained in IntestiCult media (Stemcell technologies) with Normocin (1:200) and ROCK inhibitor (1:5000), which was added for the first two days.
Human colonic organoid culture
Normal colon biopsies were transferred in a wash buffer (DPBS supplemented with 2% dialyzed FBS, 10 mM EDTA, and 2 mM GlutaMAX) containing antibiotics (Pen/Strep 1x, Primocin 100 μg/ml, Primocin 25 μg/ml, Amphotericin B 1.25 μg/ml, Nystatin 50 U/ml) and washed until the solution was clear. Specimens were sliced into 2-mm pieces using sharp blades, resuspended into 15 mL dissociation buffer (DPBS supplement with 8 mM EDTA, and 10 mM DTT) and incubated at 4°C for 30 min with gentle agitation. The tissue fragments were allowed to settle under normal gravity for 1 minute, the supernatant was removed and replaced with 15 mL of wash buffer. The suspension was shaken vigorously by hand to promote the detachment of intestinal crypts from the colonic mucosa. The remaining tissue fragments were allowed to settle under normal gravity for 1 minute, then the supernatant was transferred into a fresh tube upon filtering through a 1 mm strainer (PluriStrainer, cat. # 43-51000-03), and visually inspected under the microscope for the presence of crypts. This step was repeated until no crypts were found in supernatants. The supernatants containing crypts were centrifuged at 300 g for 5 minutes, embedded in growth factor-reduced basement membrane (Corning, Matrigel, cat. # 356231) and cultured in a base media (Advanced DMEM/F12 supplemented with HEPES 10 mM, GlutaMAX 2mM, N-Acetyl-L-cysteine 1mM, Primocin 100 μg/ml, and B-27 supplement with Vit. A 1x) with the addition of the following growth factors and inhibitors (NGS-WNT 0.5 nM, EGF 50 ng/ml, A83-01 500 nM, FGF2 50 ng/mL, IGF-I 100 ng/mL, Noggin conditioned media 1% v/v, R-Spondin 1 conditioned media 2% v/v).
Lymphatic endothelial cell-intestinal organoid coculture
For LEC:intestinal organoid coculture experiments, 70,000-100,000 LECs per well were plated on Matrigel-coated 48-well-plates or 8-well glass slides (Millipore, LabTek). LECs were allowed to adhere and form tubes for 4-6 hours before medium was aspirated and ~100-120 small intestinal crypts from previously cultured organoids (P0-1) were plated atop in 20 μl Matrigel domes mixed with organoid media. The same number of crypts was plated across conditions per experiments. LEC:intestinal organoid cocultures were maintained in a medium containing 50% Complete Mouse Endothelial Cell Medium and 50% ENR small intestinal organoid medium for coculture experiments with crypts derived from small intestinal organoids or IntestiCult medium for experiments with crypts derived from large intestinal organoids (50:50 medium). Cocultures initiated from intestinal crypts were maintained for up to 4 days.
For experiments with REELIN, intestinal organoids were grown in ENR organoid medium supplemented daily with 10 nM mouse recombinant REELIN (R&D) following what has been previously done in vitro in other studies (Telese et al., 2015). For Western blot experiments purified NA/LE Hamster anti-rat CD29 (BD Biosciences), a blocking antibody for REELIN receptor ITGB1, at 20 μg/ml or recombinant mouse LRPAP protein (R&D systems), a chaperone blocking REELIN receptor VLDLR, at 200 ng/ml were added for 3 hours prior to adding recombinant REELIN.
Generation of LEC- and BEC-conditioned media
For experiments with conditioned media, LECs or BECs at 90% confluence were washed 3x in PBS before replacing with 7 ml Complete Mouse Endothelial Cell Medium per 100 mm dish of cells. The medium was collected after 16 or 24 hours as indicated and filtered through a 0.22 μm MBS/Polypropylene vacuum filter unit (MilliporeSigma™ Steriflip™). For experiments a mixture of 50% ENR small intestinal organoid medium and 50% LEC- or BEC-conditioned media was used.
Organoid replate assay
For the organoid replate assay, as shown in figure 6, organoids grown in 50:50 medium with normal LEC or LEC-conditioned medium on day 4 were collected in Gentle Cell Dissociation Reagent as described earlier. To get a single-cell-suspension crypt pellets were washed twice in PBS, resuspended in 200 μl TrypLE Express (Gibco) with DNase I (0.1 mg/ml) (Roche) and incubated at 37°C for up to 20 minutes with intermittent mixing every 5 minutes. Once the majority of the suspension consisted of single cells, PBS with 2% fetal bovine serum (FBS) was added 1:1 and the cells were centrifuged at 1400 rpm for 5 minutes. Cells were counted and plated at 5-10 x 104 cells per dome. Cells were grown in ENR organoid medium with ROCK inhibitor (1:5000) for up to 10 days and phenotypic analyses were done on organoids on day 10.
Single-cell-derived organoid tracking experiments
For time-course experiments tracking the development of small and large intestinal organoids starting from single cells (as shown in Figure 2) mTmG organoids were utilized to facilitate tracking of organoid structures. Single cells were derived by dissociating organoids that have been cultured for 6 days (small intestine) or 4 days (large intestine) via the TrypLE Express-based approach as described. Cells were plated at a density of 5-10 x 104 cells per dome alone or atop 8 x 104 LECs/well that were seeded on a thin layer of Matrigel 4-6 hours in advance. Cells were grown for 10 days in LEC (25%) : organoid (75%) medium, using ENR medium for small intestinal organoid-derived and IntestiCult medium for large intestine organoid-derived single cells. ROCK inhibitor was added (1:5000) for the first 4 days. Media was exchanged every second day. The growth and development of organoids was monitored by taking images on a 10x objective on the indicated days using the phase contrast and RFP channel for mTomato signal from organoids. The number of organoids across conditions was counted on day 10.
Human lymphatic endothelial cell-intestinal organoid coculture
For human lymphatic endothelial cell (LEC):intestinal organoid coculture experiments, 80,000 LECs (P4-5) per well of a 48-well plate were seeded on Matrigel and left to adhere and form tubes for 4-6 hours. Human colonic organoids were dissociated to organoid fragments and seeded alone or on top of LECs in 20 μl Matrigel:human organoid media droplets and incubated at 37°C for 10 minutes before adding 300 μl of 100% human organoid media per well. Media was exchanged every second day and organoids were imaged on day 4 with the Synergy Neo2 Multi-Mode Reader (BioTek). Organoids were dissociated in Cell Recovery Solution (Corning) at 4°C for 45 minutes. Organoids were collected, spun down and fixed in 4% PFA for 50 minutes at 4°C for subsequent staining. Five biological replicates, i.e. human crypts derived from n = 5 individual patient samples, were used for human lymphatic endothelial cell-intestinal organoid coculture experiments. Patient samples included tissue specimens from healthy individuals or healthy adjacent sections from inflammatory bowel disease patients.
Immunofluorescence staining
Primary antibody labeling
Anti-lysozyme antibody (EC3.2.1.17, Dako) was labelled with CF570 (RRX equivalent) fluorophore using a labelling kit, Mix-N-Stain (Biotium), according to the manufacturer’s instructions and as previously described (Lukonin et al., 2020). The conjugated LYZ1 antibody was used for staining in Figure 6J and S5G.
Primary cell immunofluorescence staining
Primary murine lymphatic endothelial cells (Cell Biologics) at passages 4-5 were cultured on Millicell EZ 8-well glass slides (Millipore) pre-coated with gelatin (Sigma) in Complete Mouse Endothelial Medium (Cell Biologics). At 80-90% confluence cells were washed with PBS and fixed in 4% paraformaldehyde (PFA) for 30 minutes at room temperature (RT). Cells were washed twice in PBS and incubated in blocking solution (PBS with 0.3% Triton-X-100 (PBST), 2.5% normal donkey serum, 1% gelatin and 1% bovine serum albumin (BSA)) for 1 hour at RT. Primary antibodies were added in blocking solution for 1 hour at RT. Cells were washed 3 times in PBST, secondary antibodies (1:500) were added in blocking buffer at RT for 2 hours. Cells were washed 3 times in PBST and mounted in 4′,6-diamidino-2-phenylindole (DAPI)-containing ProLong Anti-fade Diamond mountant (Thermo Fisher Scientific).
Lymphatic endothelial cell:organoid coculture immunofluorescence staining
For stainings of LEC:organoid cocultures in Matrigel, cocultures were washed once in PBS, fixed in 2% or 4% PFA for 30-60 minutes at RT on day 4, washed 3 times in PBS and permeabilized in PBS with 0.3% Triton-X-100 (PBST) for at least 15 minutes. Cells were blocked for 1 hour at RT in PBST with normal donkey serum (NDS, 2.5%), gelatin (1%) and bovine serum albumin (BSA) (1%) before adding primary antibodies in blocking buffer overnight at 4°C. On the next day the samples were allowed to reach RT for 1 hour before washing 3 times in PBST with 2% NDS and incubating with secondary antibodies (1:200) at RT for 2 hours. Samples were washed at least 3 times in PBST with 2 μg/ml DAPI (1:500). LEC:organoid cocultures that were grown on 8-well glass slides were subsequently mounted with DAPI-containing mountant and a glass coverslip. Cocultures fixed in 48-well-plates were imaged as is in PBS.
Staining of mouse and human organoids out of Matrigel were performed as previously described. Briefly, organoids were washed once in PBS before adding 300 μl of ice-cold cell recovery solution (Corning) per well. Organoids were incubated for 30-60 minutes at 4°C until Matrigel was completely dissolved. Well contents were gently resuspended 5-10 times, transferred to a 15 ml falcon tube and centrifuged in a volume of 10 ml for 3 minutes at 70 g and 4°C. The pellet was resuspended in 1 ml of ice-cold PFA (4%) and organoids were fixed at 4°C for 45 min. 1 ml of ice-cold PBS with 0.1% Tween-20 was added, organoids were mixed, incubated for 10 minutes and spun down at 70 g. The pellet was resuspended in organoid wash buffer (OWB; PBS + 0.1% Triton-X-100 + 10% SDS + 0.1% BSA). Organoids were incubated on a rocker (40 rpm) at 4°C for at least 15 minutes before primary antibodies were added overnight at 4°C. On the next day organoids were washed in OWB in 3 x 2 hour steps. Secondary antibodies (1:500) were added in OWB with 1 μg/ml DAPI (1:1000) and organoids were incubated overnight at 4°C. Washing was repeated on the following day before organoids were spun at 70 g and gently resuspended in mountant as described by van Ineveld et al. Organoids were mounted onto 35 mm glass-bottom dishes (Cellvis) and imaged via confocal microscopy.
Intestinal tissue section immunofluorescence staining
For immunofluorescence analysis, the mouse intestine was dissected, flushed with ice-cold PBS and fixed with 4% PFA in PBS for 1 - 2 hours at 4°C, washed three times with PBS and incubated with 30% sucrose overnight at 4°C. The tissue was embedded in OCT (Tissue Tek) the following day. Frozen tissue blocks were sectioned at 18-30 μm on a cryostat (Leica), and mounted on SuperFrost Plus slides (Fisher). The tissue sections were blocked for 30-60 minutes at room temperature in blocking solution (PBST with 2.5% NDS, 1% BSA, 2.5% gelatin). Sections were incubated with primary antibodies diluted in blocking solution at 4°C overnight or 1 hour at RT. Sections were washed three times with PBS and incubated with secondary antibodies conjugated with Alexa-488, RRX or Alexa-647 (1:800, Life Technologies) in blocking solution at room temperature for 2 hours. Finally, sections were washed three times with PBS and mounted with ProLong Gold or Diamond Anti-fade Mountant (Thermo Fisher Scientific).
Whole-mount immunofluorescence staining
Isolated small and large intestinal tissues from mice were flushed with ice-cold PBS. Tissues were opened longitudinally and spread with the epithelial (luminal) side up on Whatman filter paper or left as tubes. If left as tubes the intestinal tissue was cut under a dissection microscope (Leica) into ~2 mm thick strips or rings containing 1-2 rows of intact crypt-villus axes to image a side view of the intestine. Mouse and human biopsy tissues were fixed with 4% PFA in PBS for 1-2 hours at 4°C, washed three times in PBS and then permeabilized for at least 3 hours in PBST followed by 1 hour in blocking solution (PBST with 2.5% NDS, 1% BSA, 2.5% gelatin). For immunolabeling, samples were incubated with primary antibodies in PBST for 24 hours at room temperature followed by 8-10 washes in PBST. Samples were incubated overnight at room temperature with secondary antibodies conjugated with Alexa-488, RRX, or Alexa-647 (1:200 Life Technologies) in blocking solution with DAPI (2 μg/ml, 1:500). Samples were washed at least 8 times with PBST containing DAPI (1:500) for 4 hours at room temperature prior to tissue clearing.
Tissue clearing
Ethyl-cinnamate-based tissue clearing was performed as previously described with some modifications (Gur-Cohen et al., 2019). Stained intestinal tissue was transferred through 30%, 50% and 70% ethanol diluted in UltraPure water and adjusted to a pH of 9.0 for 1 hour each, all on a rocker at room temperature (RT). Dehydrated tissues were then incubated twice in 100% ethanol for at least 1 hour each at RT. Tissues were next transferred into 500 μl of ethyl cinnamate (Sigma) in Eppendorf tubes (Polypropylene) for clearing. Cleared intestinal tissue was mounted with ethyl cinnamate drops between 2 glass cover slips size 22x40 mm, #0 (Electron Microscopy Science), and placed in the microscope slide holder to acquire images.
Staining for proliferation via EdU incorporation
Staining for proliferation in tissue sections and organoids was performed according to manufacturer’s instructions using the Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 647 dye (Invitrogen). To label proliferative cells in small intestinal organoids, 10 μM 5-ethynyl-2’-deoxyuridine (EdU) (Thermo Fisher) was added to ENR medium in vitro for 10 minutes before dissociating organoid samples in cell recovery solution as described. For pulse-chase experiments in vivo mice were intraperitoneally injected with 500 ng of EdU diluted in sterile PBS 30 minutes (short timepoint) or 48 hours (long timepoint) before sacrifice.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed using the Click-iT Alexa Fluor 647 Imaging kit (Thermofisher) according to manufacturer’s instructions on tissue sections from control and DT-treated Vegfr3-CreERT2 iDTRflox/+ mice and Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice to label cells exhibiting DNA fragmentation. Briefly, fixed tissue sections were washed and permeabilized with Proteinase K solution for 15 minutes, washed and immersed in 4% paraformaldehyde (PFA) for 5 minutes at 37°C. After that slides were washed again, rinsed in deionized water and TdT Reaction buffer was added to each slide. Slides were incubated at 37°C for 10 minutes, TdT reaction buffer was removed and TdT reaction mixture was added to each slide for a 60 minutes long incubation at 37°C. Slides were rinsed in deionized water, washed with 3% BSA and 0.1% Triton X-100 in PBS for 5 minutes and rinsed in PBS. This was followed by the TUNEL Click-iT reaction i.e. Click-iT Plus TUNEL reaction cocktail was added and slides were incubated at 37°C for 30 minutes, washed with 3% BSA in PBS and rinsed in PBS. For additional immunofluorescence stainings slides were processed as described i.e. the TUNEL Click-iT reaction was followed by blocking and incubation in primary antibodies.
RNAScope
RNAScope probes for Sox9, Lgr5 and Lysozyme were purchased from ACD and RNA fluorescence in situ hybridization as well as imaging was performed at the Molecular Cytology Core at Memorial Sloan Kettering Cancer Center (MSKCC) according to manufacturer’s instructions. RNAScope was performed on paraffin sections of a healthy small intestine from a C57BL/6 male mouse, which was sacrificed at 10 weeks of age.
Imaging and confocal microscopy
Tissue sections, primary cells and organoid samples were imaged with the 20x objective of an AxioObserver.Z1 epifluorescence microscope equipped with a Hamamatsu ORCA-ER camera and ApoTome.2 (Carl Zeiss) slider. Additionally, organoid samples were imaged at 20x on an inverted LSM 780 laser scanning confocal microscope (Zeiss) or an inverted spinning disk confocal system (Andor Technology Inc). The LSM 780 confocal microscope is equipped with an inverted Zeiss Axio Observer Z1 microscope with definite focus, 405, 440, 488, 514, 561, 594 and 633 laser lines. The system was driven by the Zen software (Zeiss). Cleared whole-mount tissue specimens were exclusively imaged using the 20x objective of an inverted Dragonfly 202 spinning disk confocal system (Andor Technology Inc) equipped with a 40 μm pinhole and a Leica DMi8 camera with AFC (20x air objective). Four laser lines (405, 488, 561 and 625 nm) were used for near simultaneous excitation of DAPI, Alexa-448, RRX and Alexa-647 fluorophores. The system was driven by the Andor Fusion software. For the majority of experiments z-stack images of 1-3 μm steps were collected. For each experiment images were acquired at identical settings.
Electron microscopy
Intestinal tissues were flushed with ice-cold PBS and fixed in 2% glutaraldehyde, 4% PFA, and 2 mM CaCl2 in 0.1 M sodium cacodylate buffer (pH 7.2) for >1 h at room temperature, post-fixed in 1% osmium tetroxide, and processed for Epon embedding. Ulltrathin sections (60–65 nm) were counterstained with uranyl acetate and lead citrate. Images were taken with a transmission electron microscope (Tecnai G2-12; FEI) equipped with a digital camera (AMT BioSprint29).
Image processing and analysis
Processing and rendering of imaging data was performed in Imaris 9.5 (Bitplane) and included initial image processing, 3D-surface rendering, cell-spotting analysis as well as mean signal intensity, volumetric, distance measurements and statistical color-coding of objects. 3D surface rendering and spots analysis were based on signal intensity per channel and a cell diameter estimate of 5-7 μm. Color-coding of objects was performed according to mean fluorescence intensity (for quantification of REELIN by intensity in figure 6 and quantification of Sox9 in Supplemental Figure 5) or distance to another object (for epithelial-vascular distance measurements in figure 1 and supplemental figure 1) as specified in the respective experiments. For the epithelial-vascular distance analyses in Figure 1 and its supplemental figures, surfaces were created for lymphatic and blood vessels as well as E-Cadherin-positive epithelial and Lgr5-positive stem cells. Surfaces of vascular structures were subsequently color-coded according to their distance from the epithelial surfaces (see the respective color bars), for which the values were calculated in Imaris by the nearest-distance-to-surface analysis. All image analyses were done at pre-defined identical settings across images and experimental replicates.
To quantify the number of EdU+ or OLFM4+ cells in control (n = 7) and DT-treated Vegfr3-CreERT2 iDTRflox/+ mice (n = 7), control (n = 2) and VE-Cadherin-CreERT2 Relnflox/Δ (n = 2) mice only well-oriented and fully shown crypts were taken into consideration. Cells were counted across at least 4 images per mouse in a blinded manner and independently by 2 researchers.
Analysis of RNAScope images, i.e. adjusting color and brightness parameters, was carried out using the CaseViewer (3D Histech Ltd.) software.
Phenotypic characterization of organoids
Images of intestinal organoids for morphological characterization were acquired using a ZEISS Axio Observer Life Science Inverted Microscope (10x objective). Organoids in brightfield images were outlined and hence defined as individual objects in ImageJ (NIH). Subsequent phenotypic analysis was done in ImageJ (analyze – set measurements) using the statistics parameters area, perimeter and circularity. Object circularity in ImageJ is calculated according to the formula circularity = 4pi (area/perimeter^2), whereby a circularity value of 1 indicates a perfect circle. The number of crypt domains, i.e. clearly defined budding protrusions, per organoids was counted manually in a blinded manner. To quantify total organoid numbers, only fully developed structures which exhibited a morphology as expected after x days of culture were counted. The number of EdU+ cells per organoid was calculated in Imaris 9.5, plotting the number of EdU+ spots out of total DAPI+ spots (% EdU+ out of total) per organoid structure.
Western blotting
Bradford assay
Protein was collected from murine small intestinal organoids at days 4-5 that had been starved in serum-free DMEM media (Thermo Fisher) for at least 4 hours, treated for 3 hours with inhibitors against REELIN receptors (purified NA/LE Hamster anti-rat CD29 (BD Biosciences) (20 μ/ml), recombinant mouse LRPAP protein (R&D systems) (200 ng/ml)) and stimulated with recombinant REELIN (mouse recombinant REELIN (R&D) (10 nM)) for 10-20 minutes as indicated. After stimulation organoids were washed in PBS, dissociated with the Gentle Cell Dissociation Reagent (Stemcell Technologies), washed twice in PBS with 0.1% BSA and lysed in ~50 μl RIPA Lysis and Extraction Buffer (Thermo Fisher) with added PhosSTOP™ (Roche) and cOmplete Protease Inhibitor Cocktail (Roche). After lysis, samples were spun down at 4°C and 16000g for 15 minutes and the supernatant was transferred to a new tube. We used the Pierce™ BCA Protein Assay Kit (Thermo Fisher) according to manufacturer’s instructions to measure protein amount at 562 nm on a Synergy Neo2 Multi-Mode Reader (BioTek) relative to a BSA standard curve. 10-15 μg of total protein were loaded for all experiments.
Western blot
Protein samples were mixed with NuPAGE™ LDS Sample Buffer (4X) and NuPAGE™ Sample Reducing Agent (10X) (Thermo Fisher), denatured at 95°C for 5 minutes and loaded together with the Precision Plus Protein Dual Standard ladder (Bio-Rad) in a NuPAGE™ 4-12%, Bis-Tris Protein Gel (Invitrogen). NuPAGE™ MOPS SDS Running Buffer (20X) was used for running the gel at 80V (10 minutes) and 120V in subsequent steps. Afterwards the gel was transferred to a Nitrocellulose Membrane (0.45 μm) (Thermo Scientific) in Transfer Buffer with NuPAGE™ Transfer Buffer (20X) (Thermo Fisher) and 2% Methanol on ice at 90V for 2 hours. Membranes were blocked in 1X Tris-Buffered Saline Solution (Bio-Rad) with 0.1% Tween-20 (Sigma) (TBST) and 5% BSA (Sigma) for 1 hour at room temperature before being incubated in TBST with 3% BSA and primary antibody at 4°C overnight. The following primary antibodies were used: p-DAB1 (Cell Signaling 3327S, 1:300), total DAB1 (Invitrogen PA5-85453, 1:500) and β-ACTIN (Cell Signaling 3700S, 1:5000). Membranes were washed 3 times in TBST, incubated with HRP-conjugated secondary antibody (Jackson ImmunoResearch, 1:10 000) and washed 3 times in TBST. Membranes were incubated with Pierce™ ECL Plus Western Blotting Substrate (Thermo Scientific) for 4 minutes to visualize β-ACTIN and SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific) for the rest. Chemiluminescent signal was evaluated using CL-XPosure Film (Thermo Fisher).
Enzyme-linked immunoassay (ELISA)
To measure REELIN concentration in vivo and in vitro we used the Mouse Reelin ELISA colorimetric Kit (Novus Biologicals) according to manufacturer’s instructions. To collect interstitial fluid from the intestine of n = 4 C57BL/J male mice (10 weeks of age), the small and large bowels were dissected, flushed twice with ice-cold PBS and cut into ~1 cm long pieces. The pieces were opened longitudinally and with the luminal side down were placed on a 40 μm mesh filter. From the same mouse we took the backskin after shaving, scraped off residual fat, cut ~1 x 1 cm2 pieces and placed them with the dermal side down onto mesh filter. Samples were spun down at 4°C and 140g for 20 minutes and interstitial fluid was collected into 1.5 ml Eppendorf tubes. Samples were pooled per tissue, spun down again at 4°C and 1000g for 20 minutes and the supernatant was transferred to a new tube. Samples were diluted 1:2–1:5. In terms of the sample dilution ranges for the ELISA, LEC-conditioned media (24h) as well as small, large intestinal and dermal interstitial fluid were used at 1 – 1:2 – 1:4 – 1:8 serial dilutions. Protein concentration was measured at 450 nm on a Synergy Neo2 Multi-Mode Reader (BioTek). The protein concentrations (pg/ml) within range of the standard curve are visualized in the graph in Figure S6.
Quantitative real-time PCR (qPCR)
RNA isolation
RNA was isolated from whole tissue that had been snap-frozen in liquid nitrogen. 10–20 mg of frozen distal small intestinal tissue from control and DT-treated Vegfr3-CreERT2 iDTRflox/+ mice or Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice were manually homogenized and the resulting tissue powder was dissolved in 1-2 ml of TRIzol Reagent (Thermo Fisher). RNA was isolated using the Direct-zol RNA MiniPrep Kit (Zymo Research) according to manufacturer’s instructions including treatment with DNase I. The final elution volume was 25-30 μl and RNA quantity and quality were measured on a NanoDrop 8000 spectrophotometer (Thermo Fisher). The same amount of RNA (up to 1.5 mg) was loaded per sample for subsequent cDNA synthesis. For RNA isolation from sorted small intestinal blood endothelial, lymphatic endothelial and fibroblast cells 105 cells were sorted into 300 μl of TRIzol Reagent using a 70 μm nozzle. RNA was isolated using the Direct-zol RNA MicroPrep Kit (Zymo Research) including treatment with DNase I and a final elution volume of 10 μl. RNA from cultured dermal lymphatic endothelial cells was collected by lysing 90% confluent cells seeded in a 6-well plate in 0.5 – 1 ml TRIzol Reagent. RNA isolation was performed with the Direct-zol RNA MiniPrep Kit (Zymo Research).
cDNA synthesis
At least 100 ng of RNA were loaded per sample and cDNA was synthesized using the SuperScript™ VILO™ cDNA Synthesis Kit (Thermo Fisher). Briefly, in a final volume of 20 μl we mixed VILO Reaction mix (5X), SuperScript Enzyme (10X), RNA template and DEPC-treated water (Thermo Fisher). cDNA was synthesized using the following PCR program: 10 minutes at 25°C – 60 minutes at 42°C – 5 minutes at 85°C – 12°C. For all quantitative real-time PCR (qPCR) reactions cDNA was diluted to get a final concentration of 5 ng/μl.
Quantitative real-time PCR (qPCR)
For qPCR equal amounts of cDNA template were mixed with validated gene-specific primers, DEPC-treated water and Power SYBR green PCR Master Mix (Thermo Fisher). qPCR was performed with technical replicates using an Applied Biosystems 7900HT Fast Real-Time PCR machine and relative expression (RE) was calculated as RE = 2−ΔΔCt. Primer sequences can be found in supplementary table S1.
Flow cytometric analysis
EdU flow cytometric analysis of small intestinal epithelial cells
Vegfr3-CreERT2 iDTRflox/+ or Vegfr3-CreERT2 iDTRflox/+ tdTomatoflox/+ mice were treated with tamoxifen and diphtheria toxin as described above. For pulse-chase experiments in vivo mice were intraperitoneally injected with 500 ng of EdU diluted in sterile PBS 30 minutes before sacrifice. To obtain small intestinal epithelial cells enriched for intestinal stem cells, we isolated crypts from the entire small intestine as described. The best fractions were pooled and spun down at 1300 rpm for 5 minutes, resuspended in 1 ml TrypLE Express (Gibco) and incubated at 37°C for 20 minutes. PBS with FBS was added up to 10 ml and the cells were spun down at 1300 rpm, resuspended in PBS, filtered through a 40 μm filter and stained for 30 minutes at 4°C in PBS with LIVE/DEAD Fixable Aqua Dead Cell dye (1:1000) (Invitrogen) in the dark. Samples were washed in PBS, spun down for 5 minutes and 4°C at 1300 rpm and resuspended in PBS + 2% BSA and 2.5 mM EDTA (FACS buffer) with TruStain FcX™ (anti-mouse CD16/32) antibody (1:500). After a 2 minutes long incubation on ice, samples were spun down and resuspended in FACS buffer containing the following antibodies: CD45-APC-Cy7 (BioLegend, 1:250), CD326-PE-Cy7 (Biolegend, 1:200), CD31 (MEC13.3)-FITC (Biolegend, 1:200) and Podoplanin (PDPN)-PerCP-Cy5.5 (Biolegend, 1:100). Samples were stained in the dark on ice for 20-30 minutes and subsequently washed in FACS buffer before cells were fixed for 15 minutes at 4°C in EdU Click-iT fixative (Thermo Fisher). EdU Click-iT staining (with AF647) was performed according to manufacturer’s instructions. Briefly, cells after fixation were washed in FACS buffer, resuspended in 100 μl of 1X Click-iT permeabilization buffer for 15 minutes at room temperature. 200 μl of 1X Click-iT EdU reaction cocktail per sample were added on top and cells were stained for 30 minutes in the dark at room temperature. After that cells were washed once in Click-iT permeabilization and wash reagent, resuspended in FACS buffer and analyzed on an LSR-II flow cytometer (BD Biosciences). Data from flow cytometric analysis were exported as FCS 3.0 files and analyzed with the FlowJo software. EdU was quantified by plotting the percentage of EdU+ CD326+ cells out of the total population of CD45− CD31− alive single cells or alternatively, EdU signal in a population of CD326+ CD45− CD31− alive single cells was plotted as a histogram.
Fluorescence-activated cell sorting (FACS)
Cell sorting for all experiments was performed at the Rockefeller Flow Cytometry Resource Center on a BD FACSAria Cell Sorter. Cells for RNA isolation in TRIzol were sorted with a 70 μm, while cells for subsequent single-cell RNA-sequencing analyses were sorted using a 100 or 85 μm nozzle.
Intestinal epithelial cell isolation from cultured organoids for single-cell RNA-sequencing (scRNA-seq)
Wild-type organoids (P1) from n = 16 wells per condition were pooled and collected in Gentle Cell Dissociation Reagent and subsequently dissociated into single cells using the TrypLE Express solution (Gibco) with DNase I (0.1 mg/ml) as described. Single cells were resuspended in FACS buffer (PBS with 2% BSA or FBS), filtered and counted. 105 - 106 cells were resuspended in FACS buffer with 1 μg/ml TruStain FcX™ anti-mouse CD16/32 antibody (BioLegend) to block for 5 minutes before adding conjugated surface antibodies (CD326-PE-Cy7 (1:200) (BioLegend)) in FACS buffer. Cells were stained for 20-30 minutes at 4°C in the dark and washed twice in FACS buffer. Cells were resuspended in FACS buffer with DAPI (33 ng/ml) at a final concentration of 5-6 million cells/ml for subsequent fluorescence-activated cell sorting into a pre-coated tube filled with FACS buffer. Cells were then spun down at 1400 rpm for 5 minutes and resuspended to reach a final concentration of 1200 cells/μl. 1.5 x 105 cells from the organoids only condition and 105 cells from co-cultured organoids were collected.
Intestinal stem cell (ISC) isolation from mouse small and large intestine for scRNA-seq
To enrich for intestinal stem cells (ISCs) in subsequent 10X-based single-cell RNA-sequencing experiments, male Lgr5-EGFP-IRES-CreERT2 mice were utilized at 8-10 weeks of age. Mice were age- and sex-matched between single-cell and spatial transcriptomic sequencing experiments. Isolation of ISCs was performed in separate experiments for the small and large intestine.
To obtain small intestinal ISCs the last ⅔ of the small intestine of each mouse (n = 6 Lgr5-EGFP-IRES-CreERT2 mice) were used to isolate crypts as described. All fractions were spun down at 1300 rpm for 5 minutes, resuspended in 1 ml TrypLE Express (Gibco) each and incubated at 37°C for 5-10 minutes. PBS was added up to 10 ml and the cells were spun down at 1400 rpm, resuspended in FACS buffer (PBS + 2% FBS), filtered and stained for 20 minutes at 4°C in the dark with the following antibodies: CD45-APC-Cy7 (1:250) (BioLegend), CD31-PE-Cy7 (1:200) (BioLegend). Samples were washed twice in FACS buffer and resuspended at 5-6 million cells/ml in FACS buffer with DAPI (33 ng/ml). For single-cell RNA-sequencing of the small intestine 50000 Lgr5-positive ISCs (alive, CD45−, CD31−, Lgr5-EGFP+ singlets) were spiked into 50000 bulk intestinal cells, which were generated from the lymphatic endothelial cell isolation of the small intestine that was done in parallel.
ISC isolation from the large intestine was performed in a similar way with modifications. The entire large intestine was taken per mouse (n = 6 Lgr5-EGFP-IRES-CreERT2 mice), cleaned and processed as described. For the first incubation large intestinal tissue pieces were placed into PBS with 5 mM EDTA pH 8.0. Pieces were pipetted vigorously and allowed to settle by gravity before discarding the supernatant. Tissue pieces were resuspended in PBS with 2 mM EDTA pH 8.0 and put on a rocker at 4°C for 30 min. Pieces were shaken vigorously and the supernatant was discarded. After that serial fractions containing released crypts were collected by vigorous shaking. Fractions were spun at 1300 rpm and resuspended in TrypLE analogously to the protocol for the small intestine. For the large intestine 70,000 LGR5-positive ISCs were spiked into 70,000 bulk intestinal cells. For all single-cell sequencing experiments cells were resuspended at a final concentration of 1200 cells/μl.
Intestinal lymphatic isolation from mouse small and large intestine for scRNA-seq
For bulk intestinal and lymphatic endothelial cell sorting experiments male C57BL6/J mice, age- and sex-matched with mice from ISC sorting experiments, were utilized.
To isolate lymphatic endothelial cells (LECs) from the small intestine, the entire small intestine per mouse (n = 6 C57BL6/J mice) was dissected, washed and cut into ~5 mm fragments into a 50 ml conical tube filled with PBS for additional washes. Tissue pieces were transferred into 25 ml of wash solution (RPMI (Gibco) with 2% FBS, 100 μg/ml Penicillin-Streptomycin, 25 mM HEPES buffer and 2 mM L-Glutamine) with 1 mM EDTA pH 8.0 and 1 mM DTT for shaking at 225 rpm and 37°C for 15 minutes. The supernatant was discarded and tissue fragments were resuspended in 8 ml advanced DMEMF12 media with 100 μg/ml Penicillin-Streptomycin, 25 mM HEPES and 2 mM L-glutamine as well as DNase I (0.1 mg/ml) (Roche), 0.25% collagenase (Sigma) and 2 U/ml Dispase (Gibco). 2 ceramic beads were added per tube to mince the tissue during a 20 minute incubation at 37°C and 225 rpm. The solution was filtered through a 70 μm cell strainer, spun at 1300 rpm and the cell pellet was resuspended in FACS buffer. Cells were stained for 20 minutes at 4°C in the dark with the following antibodies: CD45-APC-Cy7 (1:250) (BioLegend), CD31-PE-Cy7 (1:200) (BioLegend), PDPN-PE (1:300) (BioLegend), LYVE1-ef660 (1:100, BioLegend). Cells were washed twice and resuspended at a final concentration of 5-6 million cells/ml for sorting. For single-cell RNA-sequencing of the small intestine 25000 lymphatic endothelial cells (alive CD45−, CD31+, PDPN+, LYVE1+ singlets) were spiked into 50000 bulk intestinal cells from the same sample prep.
LEC isolation from the large intestine from n = 10 C57BL6/J mice was done without the addition of ceramic beads for tissue fractionation. For single-cell RNA-sequencing of the large intestine 18000 LECs were spiked into 70000 bulk intestinal cells. For all single-cell sequencing experiments cells were resuspended at a final concentration of 1200 cells/μl.
Spatial transcriptomic analyses (10X Visium) of mouse small and large intestine
Spatial transcriptomics with immunofluorescence staining was performed following 10X Genomics guidelines. Immunofluorescence staining and imaging of intestinal tissue sections was optimized using the 10X Genomics tissue optimization slide. For spatial gene expression analysis, the distal parts of the small and large intestine from a C57BL6/J male mouse at 8 weeks of age were dissected, cleaned and cut into ~7 mm long tubes. Tubes were directly embedded in OCT (TissueTek) in 7 mm x 7 mm cryomolds. Four tubes were embedded per block. 10 μm thick sections were taken and immediately placed onto the 10X Visium gene expression. Serial sections were used as technical replicates for small and large intestine. Tissue sections were incubated at 37°C for 1 minute followed by fixation in pre-chilled methanol at −20°C for 30 minutes. Tissue sections were blocked for 5 minutes at room temperature (RT), before adding primary antibodies (1:50) and incubating at RT for 45 minutes. Sections were quickly washed 5 times before adding Alexa-488, RRX- and Alexa-647-conjugated secondary antibodies (1:1000) with DAPI (1:300) and incubating at RT for 30 minutes. Sections were washed 5 times and mounted in glycerol-based mounting medium. Recipes for mountant, blocking and wash buffers can be found in the 10X Visium spatial transcriptomics manual. Gene expression slides were immediately imaged on a 3DHistech Panoramic Scanner (20x/0.8NA air objective) followed by tissue permeabilization for RNA release and cDNA synthesis. Spatial gene expression sequencing was done in collaboration with the Molecular Cytology Core and Integrated Genomics Operation at Memorial Sloan Kettering Cancer Center (MSKCC). Image processing was done using the CaseViewer Software (3DHistech). Images were manually aligned with the spot layout per capture area of the gene expression slide in Loupe Browser (10X Genomics) and exported as .json files for subsequent data processing in Space Ranger. FASTQ files from sequencing (NovaSeq) were processed via Space Ranger (version 1.2.1) count to align reads to the GRCm38 (mm10) reference genome and generate count matrices for subsequent bioinformatic analyses. We obtained 2,097 and 2,574 spots under tissue with median UMIs of 10,199 and 9,009 per spot for two replicates of small intestines. For the two replicates of large intestines, we obtained 2,096 and 2,180 spots under tissue with median UMIs of 19,320 and 19,814 per spot.
Intestinal fibroblast, lymphatic and blood endothelial cell isolation from mouse small intestine for quantitative real-time PCR
To compare the relative expression of candidate lymphatic ligands via quantitative real-time PCR (qPCR) in cell types of the intestinal stem cell niche we sorted mouse small intestinal lymphatic endothelial, blood endothelial cells and fibroblasts directly into TRIzol Reagent (Thermo Fisher). Per tube and population we sorted 105 cells into 300 ul TRIzol in a ratio of 1:3 TRIzol : cells in n = 2-3 technical replicates per population, which were immediately placed on dry ice and stored at −80°C until RNA isolation. Lymphatic and blood endothelial cells as well as fibroblasts were isolated from the small intestines of n = 3 C57BL/J male 2-months-old mice in 2 independent experiments using our lymphatic endothelial cell isolation protocol as described. Cells after tissue isolation were stained with CD45-APC-Cy7 (1:250), CD326-PE-Cy7 (1:200), PDPN-PE (1:300), CD31 (MEC13.3)-FITC (1:200) and DAPI (33 ng/ml). From alive single CD45− cells our populations of interest were defined and sorted as follows: CD31+ PDPN+ lymphatic endothelial cells, CD31+ PDPN− blood endothelial cells and CD31− PDPN+ fibroblasts. Subsequent RNA isolation, cDNA synthesis and qPCR were performed as previously described.
Intestinal lymphatic isolation from mouse large intestine for bulk RNA-seq
Isolation of lymphatic endothelial cells from the murine large intestine for subsequent bulk RNA-seq was performed with some alterations. Briefly, the large intestine of n = 3 C57BL/J male mice at 7 weeks of age was removed and flushed with ice-cold PBS before it was opened longitudinally. The tissue was cut into ~5 mm pieces, washed and incubated in RPMI with 1 mM DTT and 5 mM EDTA as described. Afterwards the remaining tissue was incubated with ceramic beads in Liver Digest Media (Thermo Fisher) with DNase I (0.1 mg/ml) (Roche) and 0.25% collagenase (Sigma) at 37°C for 30 minutes and subsequently passed through 70 μm and 40 μm cell strainers. Cells were stained in FACS buffer with the following antibodies: CD45-FITC (1:500) (BioLegend), CD90.2-PerCP-Cy5.5 (1:300) (BioLegend), PDPN-PE (1:300) (BioLegend), CD31-PE-Cy7 (1:300) (BioLegend) and LYVE1 (rabbit, 1:300) (AngioBio) followed by staining with a secondary anti-rabbit AF647 conjugated antibody (1:500) (Life Technologies). Dead cells were separated via DAPI (33 ng/ml) staining. For bulk RNA-sequencing of large intestinal lymphatic endothelial cells 2-3 x 104 single alive CD45−, CD31+, PDPN+, LYVE1+ cells per mouse (n = 3) were sorted directly into TRIzol Reagent (Thermo Fisher).
Protein secretion prediction
Genes expressed at significant levels in LECs (expressed in >20% of LECs, 2055 genes), but not expressed at a significant level in >20% of blood endothelial were analyzed to identify those likely to encode lymphatic-secreted proteins. Proteins with a high probability of being secreted extracellularly were identified using TargetP 1.1 (Emanuelsson et al., 2000) with a cutoff of 0.8 (80% chance of being secreted) and WoLF PSORT (Horton et al., 2007) with a cutoff score of 20 for extracellular location. This generated a list of 12 genes: Reln, CCl21a, Sned1, Wnt2, Cp, Rnase4, Lbp, Mmrn1, Ntn1, Thy1, Rspo3, Il33. For downstream analysis, we excluded Sned1 (encodes extracellular matrix protein), Cp (encodes copper binding glycoprotein, involved in ion transport), Rnase4 (encodes ribonuclease), Lbp (encodes lipopolysaccharide binding protein, acute phase protein), Mmrn1 (encodes carrier protein for platelet factor V), Thy1 (encodes a cell surface protein in immunoglobulin family) based on a low probability of being directly relevant to ISC function (Bateman et al., 2021).
Sequencing analyses
scRNA-seq of bulk intestinal tissue and small intestinal organoids
All single-cell RNA-seq experiments were run on an Illumina NovaSeq instrument, and FASTQ files were generated via cellranger mkfastq (version 6.0.1.). Cell Ranger counts were used to align reads to the GRCm38 (mm10) reference genome for intestinal organoid samples and a modified version of GRCm38 (mm10) that includes the sequence of the Lgr5-EGFP-ires-CreERT2 knock-in cassette for bulk intestinal tissue samples, given that intestinal stem cells for those samples were derived from Lgr5-EGFP-ires-CreERT2 mice. Finally, Cell Ranger counts were used to generate count matrices for subsequent bioinformatic analyses
scRNA-seq data analyses
scRNA-seq data from the murine small and large intestine were analyzed separately due to inherent differences in tissue structure (e.g. crypts and villi exist in small intestine, whereas only crypts exist in the large intestine) and composition (e.g. presence of some non-overlapping cell types and differing cell type abundance).
Preprocessing.
We first used CellBender (Fleming et al., 2019) to remove ambient RNA molecules from the raw count matrices with the parameter --expected-cells 5000 for the small intestine dataset, 4000 for large intestine, 800 for organoids co-cultured with LECs, and 400 for organoids alone. These parameters were chosen by inspecting the barcode rank plot generated by Cell Ranger, following author recommendations. Defaults were used for all other parameters.
We next removed low quality cells, unexpressed genes and potential doublets from the CellBender output using scanpy. For the in vivo data, we performed filtering on each dataset separately. For the organoid data, we pooled datasets from two conditions and filtered them together. For each dataset, we filtered to exclude i) genes detected in fewer than 3 cells, ii) cells with under 200 genes or 1000 UMIs, and iii) cells with a mitochondrial fraction above 15%. Mitochondrial and protein-coding ribosomal protein coding genes were also omitted from downstream analyses, as these are often a large source of spurious variance that can dominate clustering and confound deconvolution (Chu et al., 2022).
To remove doublets, we assessed the performance of DoubletDetection (Gayoso and Shor, 2020) and Scrublet (Wolock et al., 2019) on filtered count matrices. For organoid datasets, we first split matrices by condition. In the co-cultured organoid dataset, which has more cells, we observed a 57-cell cluster in which both methods detected an enrichment of doublets. DoubletDetection called more doublets in this cluster, corresponding to ~2-fold higher sensitivity (33.3% of cells in this cluster called as doublets, compared to 17.5% by Scrublet), at similar specificity (98.4% of cells outside this cluster called as negative, compared to 99.4% by Scrublet); thus, we chose to use DoubletDetection for the organoid datasets. Since Scrublet yields a more restricted set of doublets, we used it on the in vivo tissue data, which is more continuous in nature and therefore contains more cells that are intermediate between two cell types (DoubletDetection is known to remove transitional states at times).
We used Scrublet with sim_doublet_ratio = 20 to prevent excessive removal of transitional cell types that would be critical for downstream lineage trajectory analysis. After Scrublet, 2239, 5193, 896, and 430 cells were retained for downstream analyses, with a median of 17,789, 24,316, 90,373 and 84,660 UMIs per cell in small intestine, large intestine, organoids with LECs, and organoids alone datasets, respectively. Raw counts were normalized by the library size of each cell divided by 104, followed by log2 (X+1) transformation.
Dimension reduction, clustering and annotation – organoid datasets.
We performed a single round of dimension reduction and clustering on filtered cells that we pooled across organoid conditions. The pooling strategy is supported by high mixing across conditions in the KNN graph (Figure S2K): we performed dimension reduction on log-transformed UMI counts (see preprocessing section) using PCA with 30 PCs, explaining 71% of total variance, then computed the KNN graph with k = 10 in this PC space, and found that the majority of neighborhoods (84.1%) contain cells from both conditions.
Next, we used Slalom (Buettner et al., 2017) to factor out cell cycle effects that could obscure signals from differentiation processes in downstream clustering and trajectory analyses. Specifically, cell cycle markers from Kowalczyk et al. and Tirosh et al. (Kowalczyk et al., 2015; Tirosh et al., 2016) were converted to mouse homologs using the biomaRt package (Durinck et al., 2005, 2009). Normalized and log-transformed data were used as input to Slalom. G2M and S phase genes were used for initialization, employing the Gaussian noise model with pruneGenes enabled, and treating condition (LEC:organoid cocultures vs organoids alone) as a covariate. We regressed out the difference between the G2M and S phase factor scores from log-normalized gene expression, and we used residuals from this regression to perform dimension reduction, clustering and trajectory analysis.
For downstream analysis, we first selected the top 5000 highly variable genes in each dataset using the scanpy function pp.highly_variable_genes, with raw counts as input, n_top_genes = 5000, flavor = ‘seurat_v3’ and span = 1. For these genes, we performed dimension reduction over the residuals of the cell cycle regression, using the scanpy.tl.pca function with the arpack solver. We selected 30 PCs, which explain 71% of variance. The nearest neighbor graph was computed over the 30 PCs, which were used as the input for computing the force-directed layout with the ForceAtlas2 package (Jacomy et al., 2014) and scanpy function scanpy.tl.draw_graph. For clustering, we used Phenograph (Levine et al., 2015) with k = 30 and clustering_algo = 'leiden', which generated 11 clusters in total.
We used known cell type markers to annotate the clusters. Five clusters expressed strong enterocyte markers (e.g. Alpi, Ephx, Apoa1), 4 expressed stem cell and transient amplifying cell (TA) markers, (e.g. Lgr5, Olfm4, Axin2, Top2a, Tubb5) and the remaining 2 expressed goblet/Paneth (e.g. Lyz, Muc2) and enteroendocrine (e.g. Chga, Chgb) markers respectively (Figure S2L). To assign enterocyte clusters, we isolated these cells for analysis, and computed the z scores of markers of each enterocyte zone reported in Moor et al. (Moor et al., 2018) then averaged z scores across markers of each zone (Figure S2M). Two clusters exhibited high scores for zone 2 marker genes and were labeled bottom-like 1 and bottom-like 2; two clusters with highest zone 3 marker scores were labeled mid-like 1 and mid-like 2; and the cluster with highest zone 5 marker scores was labeled top-like.
Dimension reduction, clustering and cell type annotation – in vivo datasets.
Considering that cell types from multiple lineages are present in the in vivo datasets, we performed clustering in a hierarchical manner. The first round of clustering was optimized to distinguish lineages. Given a good coarse clustering, sub-setting cells by lineage is relatively confident and loses little information. After sub-setting the cells by lineage, highly variable genes were re-calculated within each lineage, resulting in biologically relevant features optimized for each lineage to give rise to more accurate clustering. The recalculated sets of highly variable genes (HVGs) were then used for the additional rounds of dimension reduction and re-clustering. This strategy allowed us to optimize the features selected for each round of clustering and avoid noise arising when using a large number of PCs in a single round of clustering. We performed an initial round of dimension reduction and clustering in the in vivo tissue data, and then zoomed in on heterogeneous clusters for additional rounds of refinement. For initial clustering, we selected the top 5000 HVGs in each dataset using the scanpy function pp.highly_variable_genes with raw counts as input, n_top_genes = 5000, flavor = ‘seurat_v3’ and span = 1. We applied PCA to these genes using the scanpy.tl.pca function with the arpack solver. We selected 30 PCs, which explain 63.6% and 63.1% of variance in small and large intestine datasets, respectively, and were able to capture coarse phenotyping of main cellular compartments. For clustering, we used Phenograph with k = 15 and clustering_algo = 'leiden', which generated 27 and 35 clusters in the small and large intestine datasets, respectively.
The in vivo data cover a broad spectrum of immune, stromal, endothelial and epithelial lineages. Thus, cell types of similar lineage may not cluster accurately with 30 PCs, prompting us to perform a second round of clustering among cell types from similar lineages. During this refinement stage, we selected the top 3000 HVGs for each subset of cells (described below), and performed PCA with 30 PCs. We then re-clustered cells using Phenograph with k = 30. The higher k value in this round was chosen to prevent over-clustering. Over-clustering can cause the reference cell states used for deconvolution (in spatial transcriptomics analysis) to be highly linearly dependent, resulting in a possible identifiability issue. To further reduce the linear dependency between clusters, we also regrouped subclusters that express similar marker genes profiles (see below).
We re-clustered cells for each dataset as follows. For small intestine, we pooled and re-clustered the stem, TA and mature enterocyte clusters (N = 848), then further divided and separately re-clustered mature enterocytes (N = 428) and stem/TA cells (N = 420). For each of the 9 resulting mature enterocyte clusters, we compared mean z scores between bottom and top zone landmark genes (Moor et al., 2018) (Figure S3E). Six clusters with lowest top-to-bottom ratio were labeled bottom zone enterocytes (N = 325); two with marginally higher ratios were labeled mid zone enterocytes (N = 63); and one cluster with substantially higher ratio was labeled top zone enterocytes (N = 40). The stem/TA group (N = 420) was refined into 7 clusters; three lacking Lgr5 expression were labeled TA (N = 191); two with high expression of Lgr5 and other ISC markers were labeled Lgr5+ stem; (N = 121); and two with highest TA marker expression, including Stmn1 and Tubb5, and moderate Lgr5 were labeled Lgr5+ progenitor (N = 108). The goblet cell group (N = 216) was refined into 5 clusters; one expressing Paneth, goblet and secretory progenitor cell markers was labeled secretory progenitor (N = 35); two with high Mki67 expression were labeled goblet_cycling (N = 79); and the remaining two clusters were labeled goblet_1 (N = 46) and goblet_2 (N = 56). Refinement of cells expressing stromal marker genes (N = 84) yielded three sub-clusters; one with high myofibroblast marker expression was labeled as such (N = 14); and two that do not perfectly match known cell types were labeled str1 (N = 38) and str2 (N = 32), which may represent a mixture of stromal cell types.
For the large intestine dataset, we first re-clustered a population expressing high stem and enterocyte and low goblet cell markers (N = 2187), yielding 16 clusters. Four clusters exhibit high enterocyte and low stem and TA marker expression; two of these have high crypt top enterocyte marker expression and were labeled mature enterocyte 1 (N = 49) and 2 (N = 102); and the other two were labeled enterocyte 1 (N = 72) and 2 (N = 78). Eleven clusters exhibit high expression of Lgr5 and other stem cell markers such as Ascl2 and Smoc2; six of these have high cell cycle marker expression and were labeled Lgr5+ stem cycling (N = 815); and the remaining five were labeled Lgr5+ stem (N = 946). The final cluster has higher TA but intermediate stem marker expression, and was labeled TA (N = 125). Re-clustering of cells expressing goblet markers (N = 1577) yielded 10 clusters; four similar clusters that express high cell cycle markers were labeled goblet cycling (N = 486); we combined one small cluster goblet_2 (N = 16) with goblet_8, the cluster with highest Pearson correlation, generating the group goblet_2_8 (N = 246); and the remaining four clusters were labeled by Phenograph cluster ID. Re-clustering of stromal cells (N = 473) generated 8 clusters; three with high glial cell, pericyte and myofibroblast marker expression were labeled with the corresponding cell type (N = 12, 17 and 91, respectively); three clusters express str1, 2, and 3 marker genes from Kinchen et al. (Kinchen et al., 2018) and were labeled accordingly (N = 116, 65 and 172, respectively); and we excluded a small cluster (N = 30) with low library size and few detected genes, which likely reflects low quality cells, from trajectory and deconvolution analyses.
We evaluated the stability of clustering at the final refinement stage using Rand index. Briefly, we clustered the subsetted cells using PhenoGraph with k ranging from 21 to 42 at an interval of 3, and computed the Rand index between each pair of clustering results. For the small intestine dataset, medians of pairwise Rand indices were 0.77, 0.85, 0.87 and 1 for enterocyte, stem cell, goblet and stromal subclusters respectively. For the large intestine dataset, medians of Rand indices were 0.85, 0.78, 0.84 for enterocyte/stem cell, goblet and stromal subclusters respectively. Due to the regrouping step, we expect the actual clustering stability should be substantially higher than the Rand index computed using the raw clusters.
Marker genes used in Figure S2 and Figure S3 for cell type identification.
To identify cell types in the organoid sequencing data, gene sets highlighting differentiated enterocyte subsets along the intestinal villus axis (Moor et al., 2018) and those specifying remaining epithelial populations, i.e. stem/TA, goblet/Paneth and enteroendocrine cells (Biton et al., 2018), were taken from the respective literature. Cell type annotation in the in vivo sequencing data was done based on established marker gene sets to identify cells of the epithelial (Haber et al., 2017), stromal (Kinchen et al., 2018) and immune (Xu et al., 2019) composition present in the mouse intestine.
Differential abundance analysis – organoid dataset.
We adapted Milo (Dann et al., 2021) to test for the differential abundance of cells within defined neighborhoods, between two conditions lacking replicates. The preprocessed, cell-cycle-corrected organoid datasets (above) were used as input by Milo. We first used the buildGraph function to construct a KNN graph with k = 30, using 30 principal components (d = 30). Next, we used the makeNhoods function from Milo function to assign cells into neighborhoods based on their connectivity over the KNN graph. For computational efficiency, we subsampled 10% of representative cells, i.e. the index cells, from the KNN graph to define neighborhoods using Milo's min-max sampling scheme, originally proposed in (Setty et al., 2016). This step generated 95 neighborhoods. Next, we tested for differences in the abundance of cells from each condition in each neighborhood. As our data lacks replicates, we modified the negative binomial test used by the original Milo implementation to a two-sided Fisher’s exact test. Lastly, we corrected for multiple testing for p values outputted by Fisher’s exact tests to account for the overlap between neighborhoods using the Milo function graphSpatialFDR. Specifically, the p value of each neighborhood was corrected using the Benjamini–Hochberg method, but weighted by the reciprocal of the neighborhood connectivity. Connectivity was measured as the Euclidean distance in PC space (d = 30) to the kth neighbor of the index cell that defines each neighborhood, with k = 10. In addition, we computed the log2 fold change of number of cells in LEC co-culture compared to the organoid-only condition. The spatial FDR and log2 fold change of number of cells between two conditions in each neighborhood was used for visualization.
Differential gene expression analyses for the organoid dataset using Mast.
We performed differential expression analysis using Mast (Finak et al., 2015) on organoid data using the preprocessed log normalized expression data described above. We first subsetted the cells from the enterocyte clusters in each condition, and fitted the zero-inflated regression model using the zlm function of the Mast R package. We performed a likelihood ratio test over two linear models to derive the log2 fold change and statistical significance of change between the gene expression from two culture conditions, i.e. LEC coculture vs. organoids alone. The alternative model used two covariates. The first covariate is a categorical variable which represents the culture condition. The second covariate is a continuous variable representing the cellular detection rate which was calculated as the number of cells with zero expression followed by scaling to mean of zero and standard deviation of one. Only the second covariate is included by the null model. Multiple testing correction was performed using false discovery rate. We defined genes significantly changed between two conditions as those with FDR less than 0.01, and log2 fold change greater than 0.1. The log2 fold change of genes significantly differentially expressed were visualized for each zone marker genes in Figure 3E.
BayesPrism algorithm
BayesPrism is a fully Bayesian deconvolution method that jointly models the cell type fractions and cell type-specific gene expression from each mixture using scRNA-seq as the reference (Chu et al., 2022). It was previously developed for deconvolving bulk RNA-seq data, and we show here that it represents a general deconvolution framework that naturally extends to spatial transcriptomic data. For spatial transcriptomic data, BayesPrism takes 3 inputs, the raw count matrix of scRNA-seq, cell type labels of each cell and the raw count of each spot from Visium data (10x Genomics). The first two inputs were derived from the preprocessing and clustering/cell type annotation steps of the in vivo scRNA-seq data described above. A high-level description of the algorithm is as follows. The scRNA-seq count matrix was collapsed to generate the gene expression profile matrix φ, by summing over the cells in each cell type and then renormalizing such that the sum across genes is 1. The matrix φ essentially describes the multinomial parameters for the count matrix of each cell type. It is used as the prior information to infer the posterior distribution of cell type fractions and gene expression conditional on the observed expression in each spatial spot:
Unsg represents the number of counts assigned to the gth gene of sth cell type in the nth spot. μns represents the estimates of fraction of counts from the sth cell type and the nth spot, an estimate assumed to be proportional to the fraction of cells in each spot. Xng represents the counts of the gth gene in the nth spot, measured by spatial transcriptomics. α is a non-informative weak Dirichlet prior hyper-parameter set to the small value of 10−8 by default. BayesPrism implements an efficient Gibbs sampling scheme to sample U and μ from the posterior distribution, followed by marginalizing each other and then summarizing using the posterior mean. We also leveraged the information shared across thousands of spatial spots in each dataset to update φ using U., to construct the updated gene expression profile ψ of the same dimension as φ, by setting the argument first.gibbs.only = FALSE in the run.Ted function provided by BayesPrsim. This procedure allows for better accommodating biological variation between the reference scRNA-seq data and spatial transcriptomics. ψ was then used to re-estimate the cell type fractions using the posterior distribution described above, while marginalizing U:
where θfns is the updated version of μns, which also represents the estimates of fraction of counts from the sth cell type and the nth spot. θf was used for all downstream analysis.
BayesPrism has been mathematically shown to be intrinsically invariant to linear batch effects while being robust to other types of non-linear batch effects, as well as true biological variation between the samples being integrated. All other methods developed for deconvolving spatial transcriptomic data require a large number of spatial spots to learn the linear correction factor for batch effects between scRNA-seq and spatial transcriptomics, but we show for the first time that directly modeling such linear batch effects is not necessary (see benchmarking described below). While most spatial deconvolution methods only supply cell-type fractions, BayesPrism supplies both cell-type fractions and cell-type-specific gene expression for each spatial transcriptomic spot. BayesPrism thus represents a powerful deconvolution tool for spatial transcriptomics.
Cell type deconvolution using BayesPrism
We deconvolved the Visium small and large intestine datasets using the corresponding in vivo scRNA-seq data. Mitochondrial and ribosomal protein coding genes were excluded from deconvolution analysis. To increase the signal-to-noise ratio for deconvolution, we selected marker genes that are differentially upregulated in each cell type. We then took the union of these marker genes and deconvolved over these genes. Specifically, we performed the pairwise t-test using the findMarker function from the scran package (Lun et al., 2016) between each pair of cell types over the log-transformed reads generated by our scRNA-seq pipeline. We then required both the maximum p value to be less than 0.05, and the minimum log2 fold change to be above 0.05. This generated a total of 3,265 genes for the small intestine dataset and 3,588 genes for the large intestine dataset. To deconvolve the expression profiles for known receptor and ligand genes, we also added 1,349 receptor ligand genes compiled from multiple sources (Baccin et al., 2019; Jin et al., 2021), totaling 4,266 and 4,487 genes for the small and large intestine datasets, respectively. In addition, we excluded spots with low UMI counts from deconvolution. Spots with less than 2000 UMIs in the small intestine dataset and 4000 UMIs in the large intestine dataset were removed. Each biological replicate was deconvolved separately. To ensure a fair comparison between deconvolution methods, we used the same set of genes and spatial spots in benchmarking.
Benchmarking deconvolution of spatial transcriptomics
A big challenge in the deconvolution field is the lack of ground truth for benchmarking. We performed benchmarking of the inferred cell type distribution using two strategies.
First, we compared the spatial pattern of cell types imputed by BayesPrism with known cell type distributions. In the small and large intestine, Lgr5+ stem cells reside in the base of the crypt and enterocytes mature along the crypt-villus axis. In the small intestine, Paneth cells intermingle with Lgr5+ ISCs. Lymphatics and myofibroblasts in the large intestine are positioned below the crypt base. Lymphatics in the small intestine are both below the crypt base and protrude into the villi (lacteals). These distributions are consistent with BayesPrism imputation (Figures S4A and S4B).
Second, to benchmark BayesPrism imputation against published methods, we generated ground truth based on immunofluorescence, using markers for lymphatics and intestinal stem cells, measured directly on the Visium slide (note that immunofluorescence was performed prior to sequencing). Since many cells lie on the boundary of Visium spots, computing the exact proportion of cell types can be difficult. To circumvent this, we generated a binary validation set of spots, i.e. spots were labeled 1 - containing the cell type of interest or 0 - not (top row of Figure S4C). We focused on cell types that are most central to our study and built the validation set for stem cells in the small intestine based on the immunofluorescence of the stem cell marker OLFM4, and for lymphatics in both small and large intestine based on lymphatic marker LYVE1 (OLFM4 is not expressed in large intestine). Negative labels were created by first excluding the positive labels followed by selecting spots from the top of villus, where no stem cell and lymphatics are expected. We then computed the area under the ROC curve (AUC) for each method (bottom row of Figure S4C).
To benchmark, we used the same set of marker genes described in Cell type deconvolution using BayesPrism across all methods to ensure a fair comparison. For Stereoscope (Andersson et al., 2020), we trained the scRNA-seq data with 1000 epochs, and the deconvolution model with 10000 epochs until convergence. For Cell2location (Kleshchevnikov et al., 2022), we trained the scRNA-seq data by RegressionModel using batch_size = 2500, train_size = 1, lr = 0.002, max_epochs = 40000 (small intestine), and 30000 (large intestine), followed by estimating the cluster mu by simulating 1000 samples from the posterior distribution using the same batch size. The deconvolution model was trained with the hyper-parameters N_cells_per_location = 30, detection_alpha = 200, max_epochs = 30000, batch_size = None and train_size = 1. The same setup as the scRNA-seq model was used to draw samples from the posterior distribution to estimate the cell abundance score. The 5% quantile of this posterior distribution of the cell abundance scores was used for downstream benchmarking. For destVI (Lopez et al., 2022), we trained the scRNA-seq model using max_epochs = 250, and the deconvolution model using max_epochs = 2500 until convergence. For RCTD (Cable et al., 2021), we used the parameter doublet_mode = ‘full’ to output the fraction of every cell type without forcing a constraint to make a fair comparison with other tools. For Tangram (Biancalani et al., 2021), we used the function map_cells_to_space to map each cell to each spatial spot, followed by computing the probability of the presence of each cell type in each spot using the function project_cell_annotations. Outputs from the project_cell_annotations function was used for downstream benchmarking. For SPOTlight (Elosua-Bayes et al., 2021), as marker genes cannot be directly supplied, we followed author’s recommendation to use the scoreMarkers to define genes with mean.AUC > 0.8 as markers, while using top 5000 HVGs computed in our pre-processing step. The mean.AUC was also used as the weights by SPOTlight. To improve computational efficiency, we randomly selected 100 cells per cell type (or used all cells for cell types with fewer than 100 cells) from the scRNA-seq dataset, following author recommendations. All other parameters were at their default. We mapped each factor to their corresponding cell types by the cell type with the maximum number of cells aligned to each topic profile determined by the function plotTopicProfiles, with the arguments facet = TRUE, min_prop = 0.4. The mat entry of the output was used for downstream benchmarking. For BayesPrism, default values were used for all parameters.
SpaceFold cartography
Projecting spots onto a 1D spatial axis.
The Visium data only comprised ~4 spots for each crypt. To achieve higher spatial resolution, we used the following features of our data: (1) our spatial transcriptomics data consists of hundreds of crypts, (2) each crypt is organized in a highly stereotypical fashion and (3) spots along each crypt are each shifted in a slightly different position (see Figure 5A, left). Given the stereotypical nature of the crypt, we assume that two spots with similar cell type fractions are in a similar spatial position along the crypt, and moreover these cell type fractions change gradually along the crypt axis. Therefore, we describe each spatial transcriptomic spot using a vector representing the cell type abundances as computed by BayesPrism (in this application ~30 cell types). In the next step, we aimed to use non-linear dimensionality reduction to reduce the 30-dimensional cell-type fraction vector into a 1D axis representing position along the crypt. We used the cell-type fraction inferred by BayesPrism as input, and used PHATE (Moon et al., 2019) to project this vector onto a 1D axis. We chose PHATE due to its ability to model lighter tails in the kernel function using the α-decaying kernel, which decays faster than the common Gaussian kernel. A faster decaying kernel can be beneficial in creating a step function-like window, with high transition probabilities for spots composed of similar cell types. Such narrow windows prevent spots from distant regions, such as those from the crypt base and top, from having high transition probabilities to induce false edges in the neighbor graph.
We were able to project thousands of spots onto the 1D crypt axis at much higher spatial resolution than the original Visium spots, generating a continuum of values. Moreover, BayesPrism provides cell-type-specific gene expression for each spot, allowing us to plot cell-type-specific gene trends along this axis (see section below for details). While we chose to use BayesPrism for computation of cell type fractions in each spot and PHATE for non-linear dimensionality reduction, alternative spatial deconvolution and dimensionality algorithms can be used within the SpaceFold framework.
To run SpaceFold on our data, we first computed the z-score of the fraction of each cell type by centering to mean zero followed by scaling to standard deviation. This ensures that the distance calculated by the neighbor graph will not be dominated by highly abundant cell types. To run PHATE, we set knn = 10 and ndim = 1, and other parameters at their default values, which includes a high α value, α = 40. The PHATE embedding was rescaled to between 0 and 1 for visualization. Intestinal tissue is known to show heterogeneous gene expression across disparate tissue segments (e.g. proximal or distal) (Parigi et al., 2022), which may violate the assumption of identical repetitive units. This may confound PHATE, causing the 1D axis to model the underlying structure incompletely. Indeed, in the Visium samples of large intestine tissue, two tissue regions displayed differences in cell type abundance from the third region. To ensure the model assumption of SpaceFold holds, we ran SpaceFold separately on spots from each region. We found the predicted 1D axis correlated well between two regions, and hence merged them for visualization in Figure 5C.
Although SpaceFold cartography was developed for mapping gene expression along the crypt axis, it can be easily generalized to other tissue types of repetitive structures. For tissues of more complex structures, projecting onto a 2D plane is also an option.
Visualizing gene expression trends along the projected axis.
BayesPrism outputs a deconvolved 3-dimensional gene expression tensor Unsg (see BayesPrism algorithm) with the number of reads of each gene g in each cell type s from each spot n. To compare gene expression in one particular cell type between different spots, deconvolved reads need to be normalized with respect to the total number of reads in each cell type. One © normalization approach would be to first compute a normalizing factor by summing over the dimension g over Unsg to derive a size factor Uns, which represents the total number of reads contributed by each cell type to each spot, followed by dividing Unsg by the normalizing factor to obtain the normalized expression :
One issue with this normalization is that Uns will approach zero for spots that contain few or no cell types, causing the normalization to be unstable. To circumvent this, we computed normalized gene expression only for spots showing a substantial amount of the cell type of interest. We used the mixture model with 2 components to separate spots that contain cell types of interest from the background. Specifically, we used the Gaussian mixture model to fit the reads contributed by each cell type to each spot, Uns, using the mclust R package (Scrucca et al., 2016), and used the Gamma mixture model to fit cell type fraction, θfns, using the mixtools R package (Benaglia et al.) for each cell type s. Spots from the clusters of higher mean from the Gaussian mixture and the Gamma mixture models were intersected to represent spots containing the cell type of interest. Due to the sparsity of Visium data, we grouped the spots selected for each cell type into 20 bins according to their projected 1D axis, and took the mean and standard error of the normalized gene expression values Unsg over each bin. For visualization, we fitted a loca∣ 2nd order polynomial regression curve for the mean, and mean ± 2 standard error against the mean of 1D coordinates of each bin using the R function loess (Cleveland et al., 2017) with the smoothing parameter “span = 0.75”.
To visualize the expression of zone markers, we plotted the mean z-score of normalized gene expression in enterocytes over the marker genes as follows (Figure 5E). For each zone marker gene g selected for deconvolution, we first summed Unsg over s, for s belonging to bottom zone, mid zone and top zone enterocytes, and divided the sum by the total UMI of each spot to get a normalized expression for gene g in spot n in all enterocytes. We then computed the z score for each gene, and took the mean of the z scores of markers listed for each zone. Taking the mean of z-score makes the expression comparable between different zones. We grouped all spots into 50 bins according to their projected 1D axis, and took the mean and standard error of the mean z-scores computed above over each bin. The local 2nd order polynomial regression curve was computed in the same way as described above.
To quantify total gene expression from a group of cell types with different spatial distributions, such as all Lgr5− cells (as shown in left panels in Figure 5D), we reported the unnormalized sum of Unsg over the cell types from the selected group. We did not normalize by Uns as we found that the difference in the spatial distribution of cell types can sometimes inflate denominators. This will be the case when, for example, a cell type that does not express the gene of interest is present at a high abundance over a region of spots.
Analysis of published human scRNA-seq data
ScRNA-seq data from healthy pediatric and adult human intestinal donor samples (Elmentaite et al., 2021) were downloaded (EMBL-EBI Array Express E-MTAB-9532 and E-MTAB-8901) and analyzed for the expression of candidate lymphatic ligands. Raw counts were normalized by the library size of each cell divided by 104, followed by log2 (X+1) transformation.
Bulk RNA-seq data analysis
RNA from sorted intestinal lymphatic endothelial cells was isolated using the Direct-zol RNA MiniPrep kit (Zymo Research) following manufacturer’s instructions including DNase I treatment. RNA quality was assessed with an Agilent 2100 Bioanalyzer. The cDNA library was generated using the Illumina TrueSeq mRNA sample preparation kit at Weill Cornell Medical College Genomic Core facility (New York, NY). Paired-end sequencing was done on an Illumina NovaSeq instrument. Sequencing reads were aligned to the GRCm38 (mm 10) mouse reference genome using the RSubread package. Raw bulk RNA-seq data of dermal lymphatic and blood endothelial cells were re-analyzed from a published dataset (GSE130976), processed and normalized together with the newly generated intestinal bulk RNA-seq data. Gene expression values were quantified as transcripts per million (TPM) using salmon (Patro et al., 2017). TPM values of skin lymphatic capillaries and collecting vessels were averaged per gene. Differential gene expression analysis was performed using the DESeq2 package in R. A list of selected genes was plotted on a heatmap showing z-scored values.
Quantification and Statistical Analysis
Statistical analyses were performed using the GraphPad Prism 8 software. All in vitro experiments were repeated at least 3 times in duplicates and representative data are shown. Technical replicates were also used for spatial gene expression analysis. For ex vivo tissue staining, tissue from at least two age- and sex-matched mice was utilized per experiment. The number of mice utilized for imaging studies is indicated as n = x mice per timepoint or condition unless specified elsewhere. Phenotypic analyses of organoids were performed independently by 2 experimenters in a blinded manner. Acute lymphatic ablation studies were repeated 3 times for EdU quantification on tissue sections and qPCR, while ablation experiments for EdU quantification via flow cytometry were repeated twice. Unpaired two-tailed Student’s t-tests were used to ascertain statistical significance between two groups. One-way analysis of variance (ANOVA) was used to assess statistical significance between three or more groups with one experimental parameter. See figure legends for more information on statistical tests.
Supplementary Material
Video S1. The vascular architecture of the small intestine. Related to Figure 1 and Figure S1. Whole-mount imaging and 3D reconstruction of the small intestinal vasculature in the villus illustrating LYVE1+ lymphatics (green) and CD31+ blood endothelial cells (red). Volumetric reconstruction of lymphatics (LYVE1+) in yellow and blood vessels (CD31+) in light blue.
Video S2. Lymphatic capillaries are highly associated with crypts in the large intestine. Related to Figure 1 and S1. Whole-mount imaging and 3D reconstruction of large intestinal LYVE1+ lymphatic capillaries (white) and CD31+ blood endothelial cells (red) around crypt-based epithelial cells (ECAD+, green). Volumetric reconstruction indicating lymphatics (LYVE1+, yellow) and crypt-based epithelial cells (purple).
Video S3. Lymphatic capillaries are nesting crypt-based stem cells of the human colon. Related to Figure 1. Whole-mount imaging and 3D reconstruction of a human intestinal biopsy, demonstrating association between lymphatics (LYVE1+, red) and epithelial cells of the intestinal crypt base (ECAD+, green) in the distal colon. Volumetric reconstruction indicates lymphatics (LYVE1+, yellow) and crypt-based epithelial cells (purple).
Video S4: Interaction of an intestinal organoid with LECs in coculture conditions. Related to Figure 2 and Figure S2. Representative z-stack intervals of organoids (EpCAM+, green) grown on top of lymphatic endothelial cells (CD31+, white), cultured in 50% LEC:50% organoid medium.
Video S5: Crypt-based lymphatic capillaries highly express REELIN. Related to Figure 6 and Figure S6. Whole-mount imaging and 3D reconstruction of the mouse large intestine showing LGR5+ stem cell-harboring crypts (cyan) sitting atop a network of LYVE1+ lymphatic capillaries (magenta), which highly express RELN (yellow).
Highlights.
Spatial transcriptomics and 3D imaging reveal lymphatics as key crypt niche residents
SpaceFold maps cells and cell-specific transcriptomes to the intestinal crypt-villus axis
Lymphatics serve as a critical signaling hub in the intestinal stem cell niche
Lymphatic factors maintain epithelial stemness and restrict differentiation
Acknowledgments
We thank Rockefeller University’s Resource Centers: Flow Cytometry (S. Mazel, director), Bioinformatics (T. Carroll, W. Wang); Genomics (C. Zhao) and Memorial Sloan Kettering Cancer Center’s Integrated Genomics Operation (N. Mohibullah) and Molecular Cytology Facility (K. Manova-Todorova, Y. Romin, N. Fan). We thank R. Longman for human biopsies and helpful discussions. We thank T. Nawy, R. Chaligné, D. Mucida, S. Josefowicz, S. Tavazoie, A. Hanash, D. Artis for helpful discussions. We thank E.F. lab members for: technical assistance (E. Wong, M. Nikolova, J. Racelis, P. Nasseir); administrative assistance (C. Long); mouse handling (M. Sribour, J. Levorse, L. Polak); assistance with Western blot (N. Guzzi), discussions (N. Guzzi, N. Gomez, S. Ellis, M. Parigi, A. Gola, M.D. Abdusselamoglu, S. Baksh, S. Liu, M. Tierney, J. Novak, C. Xu). We thank J. Shin (Mehrara lab) and A. Mahmoud (Ganesh lab) for technical assistance and G. Oliver (Northwestern University) for sharing tissue from Reelin conditional knockout mice. Some schematics in this manuscript were created with BioRender. R.E.N. is a Burroughs Wellcome CAMS recipient and receives support in part by the National Center for Advancing Translational Sciences, NIH, through Rockefeller University (grants UL1 TR001866 and KL2TR001865). T.C. is a Croucher Foundation and Damon Runyon Quantitative Biology postdoctoral fellow. M.S. is a Rockefeller University Women & Science and Boehringer Ingelheim Fonds graduate fellow. S.G.C. is a Robin Chemers Neustein fellow and former Human Frontier Science Program LT001519/2017 and European Molecular Biology Organization ALTF 1239-2016 fellow. K.A.L. was supported by a T32GM007739 grant. S.R.B. was supported by grant R01HL151388 to G.O. K.G. received funding from R37CA266185, K08CA230213 and U2CCA233284 grants and was supported by a SU2C Convergence Award, Damon Runyon Clinical Investigator Award, Burroughs Wellcome CAMS and AACR NextGen Grant. E.F. and D.P. are Investigators with HHMI. The work was supported by grants from the National Institutes of Health (NIAMS R01-AR050452, E.F.) and the STARR Foundation (2019-009, E.F. and B.J.M.)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests. D.P. is on the scientific advisory board of Insitro. B.J.M is an advisor to and recipient of an investigator-initiated research award from PureTech Corp and recipient of an investigator-initiated research award from Regeneron. E.F. served on the scientific advisory boards of Arsenal Biosciences and L’Oreal.
References
- Alexander JS, Chaitanya GV, Grisham MB, and Boktor M (2010). Emerging roles of lymphatics in inflammatory bowel disease. Ann. N. Y. Acad. Sci 1207, E75–E85. [DOI] [PubMed] [Google Scholar]
- Andersson A, Bergenstråhle J, Asp M, Bergenstråhle L, Jurek A, Fernández Navarro J, and Lundeberg J (2020). Single-cell and spatial transcriptomics enables probabilistic inference of cell type topography. Commun. Biol 3, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki R, Shoshkes-Carmel M, Gao N, Shin S, May CL, Golson ML, Zahm AM, Ray M, Wiser CL, Wright CVE, et al. (2016). Foxl1-expressing mesenchymal cells constitute the intestinal stem cell niche. Cell. Mol. Gastroenterol. Hepatol 2, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccin C, Al-Sabah J, Velten L, Helbling PM, Grünschläger F, Hernández-Malmierca P, Nombela-Arrieta C, Steinmetz LM, Trumpp A, and Haas S (2019). Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat. Cell Biol 2019 221 22, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdadi MB, Ayyaz A, Coquenlorge S, Chu B, Kumar S, Streutker C, Wrana JL, and Kim T-H (2021). Enteric glial cell heterogeneity regulates intestinal stem cell niches. Cell Stem Cell 29, 1–15. [DOI] [PubMed] [Google Scholar]
- Barker N, Van Oudenaarden A, and Clevers H (2012). Identifying the Stem Cell of the Intestinal Crypt: Strategies and Pitfalls. Cell Stem Cell 11, 452–460. [DOI] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, Orchard S, Magrane M, Agivetova R, Ahmad S, Alpi E, Bowler-Barnett EH, Britto R, Bursteinas B, et al. (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benaglia T, Chauveau D, Hunter DR, and Young DS mixtools: An R Package for Analyzing Finite Mixture Models. [Google Scholar]
- Bernier-Latmani J, and Petrova TV (2017). Intestinal lymphatic vasculature: structure, mechanisms and functions. Nat. Rev. Gastroenterol. Hepatol 14, 510–526. [DOI] [PubMed] [Google Scholar]
- Beumer J, and Clevers H (2021). Cell fate specification and differentiation in the adult mammalian intestine. Nat. Rev. Mol. Cell Biol 22, 39–53. [DOI] [PubMed] [Google Scholar]
- Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, Tokcan N, Vanderburg CR, Segerstolpe Å, Zhang M, et al. (2021). Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat. Methods 2021 1811 18, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su C-WW, Smillie C, Shekhar K, et al. (2018). T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buettner F, Pratanwanich N, McCarthy DJ, Marioni JC, and Stegle O (2017). f-scLVM: Scalable and versatile factor analysis for single-cell RNA-seq. Genome Biol. 18, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cable DM, Murray E, Zou LS, Goeva A, Macosko EZ, Chen F, and Irizarry RA (2021). Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol 2021 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T, Wang Z, Pe’er D, and Danko CG (2022). Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology. Nat. Cancer 2022 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland WS, Grosse E, and Shyu WM (2017). Local Regression Models. Stat. Model S309–376. [Google Scholar]
- Comazzetto S, Shen B, and Morrison SJ (2021). Niches that regulate stem cells and hematopoiesis in adult bone marrow. Dev. Cell 56, 1848–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Alessio S, Correale C, Tacconi C, Gandelli A, Pietrogrande G, Vetrano S, Genua M, Arena V, Spinelli A, Peyrin-Biroulet L, et al. (2014). VEGF-C–dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J. Clin. Invest 124, 3863–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Scares HD, Morgan JI, and Curran T (1995). A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, and Curran T (1999). Reelin Is a Ligand for Lipoprotein Receptors. Neuron 24, 471–479. [DOI] [PubMed] [Google Scholar]
- Dann E, Henderson NC, Teichmann SA, Morgan MD, and Marioni JC (2021). Differential abundance testing on single-cell data using k-nearest neighbor graphs. Nat. Biotechnol doi: 10.1038/s41587-021-01033-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David MB, Valenta T, Fazilaty H, Hausmann G, and Basler K (2020). Distinct populations of crypt-associated fibroblasts act as signaling hubs to control colon homeostasis. PLOS Biol. 18, e3001032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirmenci B, Valenta T, Dimitrieva S, Hausmann G, and Basler K (2018). GLI1-expressing mesenchymal cells form the essential Wnt-secreting niche for colon stem cells. Nature 558, 449–453. [DOI] [PubMed] [Google Scholar]
- Dulabon L, Olson EC, Taglienti MG, Eisenhuth S, McGrath B, Walsh CA, Kreidberg JA, and Anton ES (2000). Reelin Binds α3β1 Integrin and Inhibits Neuronal Migration. Neuron 27, 33–44. [DOI] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, and Huber W (2005). BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440. [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, and Huber W (2009). Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc 2009 48 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, Kleshchevnikov V, Dabrowska M, Pritchard S, Bolt L, et al. (2021). Cells of the human intestinal tract mapped across space and time. Nat. 2021 5977875 597, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elosua-Bayes M, Nieto P, Mereu E, Gut I, and Heyn H (2021). SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res. 49, e50–e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, and Von Heijne G (2000). Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol 300, 1005–1016. [DOI] [PubMed] [Google Scholar]
- Esterházy D, Canesso MCC, Mesin L, Muller PA, de Castro TBR, Lockhart A, ElJalby M, Faria AMC, and Mucida D (2019). Compartmentalized gut lymph node drainage dictates adaptive immune responses. Nat. 2019 5697754 569, 126–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: A flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming SJ, Marioni JC, and Babadi M (2019). CellBender remove-background: a deep generative model for unsupervised removal of background noise from scRNA-seq datasets. BioRxiv 791699. [Google Scholar]
- Fu YY, Egorova A, Sobieski C, Kuttiyara J, Calafiore M, Takashima S, Clevers H, and Hanash AM (2019). T Cell Recruitment to the Intestinal Stem Cell Compartment Drives Immune-Mediated Intestinal Damage after Allogeneic Transplantation. Immunity 51, 90–103.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayoso A, and Shor J (2020). JonathanShor/DoubletDetection: doubletdetection v3.0. [Google Scholar]
- Gehart H, and Clevers H (2019). Tales from the crypt: new insights into intestinal stem cells. Nat. Rev. Gastroenterol. Hepatol 16, 19–34. [DOI] [PubMed] [Google Scholar]
- Gonzales KAU, and Fuchs E (2017). Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 43, 387–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorieff A, Pinto D, Begthel H, Destrée O, Kielman M, and Clevers H (2005). Expression Pattern of Wnt Signaling Components in the Adult Intestine. Gastroenterology 129, 626–638. [DOI] [PubMed] [Google Scholar]
- Greicius G, Kabiri Z, Sigmundsson K, Liang C, Bunte R, Singh MK, and Virshup DM (2018). PDGFRα+ pericryptal stromal cells are the critical source of Wnts and RSPO3 for murine intestinal stem cells in vivo. Proc. Natl. Acad. Sci. U. S. A 115, E3173–E3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur-Cohen S, Yang H, Baksh SC, Miao Y, Levorse J, Kataru RP, Liu X, de la Cruz-Racelis J, Mehrara BJ, and Fuchs E (2019). Stem cell-driven lymphatic remodeling coordinates tissue regeneration. Science (80-. ). 366, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nat. 2017 5517680 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He XC, Zhang J, Tong WG, Tawfik O, Ross J, Scoville DH, Tian Q, Zeng X, He X, Wiedemann LM, et al. (2004). BMP signaling inhibits intestinal stem cell self-renewal through suppression of Wnt-beta-catenin signaling. Nat. Genet 36, 1117–1121. [DOI] [PubMed] [Google Scholar]
- Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, and Nakai K (2007). WoLF PSORT: protein localization predictor. Nucleic Acids Res. 35, W585–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston SA, Cerovic V, Thomson C, Brewer J, Mowat AM, and Milling S (2015). The lymph nodes draining the small intestine and colon are anatomically separate and immunologically distinct. Mucosal Immunol. 2016 92 9, 468–478. [DOI] [PubMed] [Google Scholar]
- Hsu Y-C, Li L, and Fuchs E (2014). Emerging interactions between skin stem cells and their niches. Nat. Med 20, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacomy M, Venturini T, Heymann S, and Bastian M (2014). ForceAtlas2, a Continuous Graph Layout Algorithm for Handy Network Visualization Designed for the Gephi Software. PLoS One 9, e98679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang JY, Koh YJ, Lee S-H, Lee J, Kim KH, Kim D, Koh GY, and Yoo OJ (2013). Conditional ablation of LYVE-1+ cells unveils defensive roles of lymphatic vessels in intestine and lymph nodes. Blood 122, 2151–2161. [DOI] [PubMed] [Google Scholar]
- Jin S, Guerrero-Juarez CF, Zhang L, Chang I, Ramos R, Kuan CH, Myung P, Plikus MV, and Nie Q (2021). Inference and analysis of cell-cell communication using CellChat. Nat. Commun 2021 121 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen J, Chen HH, Parikh K, Antanaviciute A, Jagielowicz M, Fawkner-Corbett D, Ashley N, Cubitt L, Mellado-Gomez E, Attar M, et al. (2018). Structural Remodeling of the Human Colonic Mesenchyme in Inflammatory Bowel Disease. Cell 175, 372–386.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, Elmentaite R, Lomakin A, Kedlian V, Gayoso A, et al. (2022). Cell2location maps fine-grained cell types in spatial transcriptomics. Nat. Biotechnol 2022 1–11. [DOI] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, and Regev A (2015). Single cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kruiningen HJ, and Colombel J-F (2008). The forgotten role of lymphangitis in Crohn’s disease. Gut 57, 1–4. [DOI] [PubMed] [Google Scholar]
- Lay K, Kume T, Fuchs E, Christiano A, Coulombe PA, and Horsley V (2016). FOXC1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc. Natl. Acad. Sci. U. S. A 113, E1506–E1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine JH, Simonds EF, Bendall SC, Davis KL, Amir EAD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER, et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindemans CA, Calafiore M, Mertelsmann AM, O’Connor MH, Dudakov JA, Jenq RR, Velardi E, Young LF, Smith OM, Lawrence G, et al. (2015). Interleukin-22 promotes intestinal-stem-cell-mediated epithelial regeneration. Nature 528, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, De la Cruz E, Gu X, Balint L, Oxendine-Burns M, Terrones T, Ma W, Kuo HH, Lantz C, Bansal T, et al. (2020). Lymphoangiocrine signals promote cardiac growth and repair. Nature 588, 705–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez R, Li B, Keren-Shaul H, Boyeau P, Kedmi M, Pilzer D, Jelinski A, Yofe I, David E, Wagner A, et al. (2022). DestVI identifies continuums of cell types in spatial transcriptomics data. Nat. Biotechnol 2022 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, Marioni JC, Ji H, duVerle D, Rausell A, and Descartes P (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research 2016 52122 5, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutter S, Xie S, Tatin F, and Makinen T (2012). Smooth muscle–endothelial cell communication activates Reelin signaling and regulates lymphatic vessel formation. J. Cell Biol 197, 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah AT, Yan KS, and Kuo CJ (2016). Wnt pathway regulation of intestinal stem cells. J. Physiol 594, 4837–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy N, Manieri E, Storm EE, Saadatpour A, Luoma AM, Kapoor VN, Madha S, Gaynor LT, Cox C, Keerthivasan S, et al. (2020). Distinct Mesenchymal Cell Populations Generate the Essential Intestinal BMP Signaling Gradient. Cell Stem Cell 26, 391–402.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke J, Ji M, Liang H-E, and Locksley RM (2016). Tuft-cell-derived IL-25 regulates an intestinal ILC2–epithelial response circuit. Nature 529, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, van den Elzen A, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol 2019 3712 37, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moor AE, Harnik Y, Ben-Moshe S, Massasa EE, Rozenberg M, Eilam R, Bahar Halpern K, and Itzkovitz S (2018). Spatial Reconstruction of Single Enterocytes Uncovers Broad Zonation along the Intestinal Villus Axis. Cell 175, 1156–1167.e15. [DOI] [PubMed] [Google Scholar]
- Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y, Elci B, Brandenberg N, Kolotuev I, Gjorevski N, et al. (2020). Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nat. 2020 5857826 585, 574–578. [DOI] [PubMed] [Google Scholar]
- O’Rourke K, Ackerman S, Dow L, and Lowe S (2016). Isolation, Culture, and Maintenance of Mouse Intestinal Stem Cells. Bio-Protocol 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogasawara R, Hashimoto D, Kimura S, Hayase E, Ara T, Takahashi S, Ohigashi H, Yoshioka K, Tateno T, Yokoyama E, et al. (2018). Intestinal Lymphatic Endothelial Cells Produce R-Spondin3. Sci. Rep 8, 10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parigi SM, Larsson L, Das S, Ramirez Flores RO, Frede A, Tripathi KP, Diaz OE, Selin K, Morales RA, Luo X, et al. (2022). The spatial transcriptomic landscape of the healing mouse intestine following damage. Nat. Commun 2022 131 13, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson DS, and Lichtman JW (2015). Clarifying Tissue Clearing. Cell 162, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulis M, and Flavell RA (2016). Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differentiation 92, 116–131. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, Van De Wetering M, Barker N, Stange DE, Van Es JH, Abo A, Kujala P, Peters PJ, et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nat. 2009 4597244 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Sato T, Van Es JH, Snippert HJ, Stange DE, Vries RG, Van Den Born M, Barker N, Shroyer NF, Van De Wetering M, and Clevers H (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrucca L, Fop M, Murphy TB, and Raftery AE (2016). mclust 5: Clustering, Classification and Density Estimation Using Gaussian Finite Mixture Models. R J. 8, 289. [PMC free article] [PubMed] [Google Scholar]
- Setty M, Tadmor MD, Reich-Zeliger S, Angel O, Salame TM, Kathail P, Choi K, Bendall S, Friedman N, and Pe’Er D (2016). Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol 2016 346 34, 637–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoshkes-Carmel M, Wang YJ, Wangensteen KJ, Tóth B, Kondo A, Massassa EE, Itzkovitz S, and Kaestner KH (2018). Subepithelial telocytes are an important source of Wnts that supports intestinal crypts. Nature 557, 242–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven Alexander J, Chaitanya GV, Grisham MB, and Boktor M (2010). Emerging roles of lymphatics in inflammatory bowel disease. Ann. N. Y. Acad. Sci 1207, E75–E85. [DOI] [PubMed] [Google Scholar]
- Stzepourginski I, Nigro G, Jacob JM, Dulauroy S, Sansonetti PJ, Eberl G, and Peduto L (2017). CD34+ mesenchymal cells are a major component of the intestinal stem cells niche at homeostasis and after injury. Proc. Natl. Acad. Sci. U. S. A 114, E506–E513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science (80-. ). 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst. 8, 281–291.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Ding J, Porter CBM, Wallrapp A, Tabaka M, Ma S, Fu S, Guo X, Riesenfeld SJ, Su C, et al. (2019). Transcriptional Atlas of Intestinal Immune Cells Reveals that Neuropeptide α-CGRP Modulates Group 2 Innate Lymphoid Cell Responses. Immunity 51, 696–708.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan KS, Janda CY, Chang J, Zheng GXY, Larkin KA, Luca VC, Chia LA, Mah AT, Han A, Terry JM, et al. (2017). Non-equivalence of Wnt and R-spondin ligands during Lgr5+ intestinal stem-cell self-renewal. Nat. 2017 5457653 545, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Zarkada G, Han J, Li J, Dubrac A, Ola R, Genet G, Boyé K, Michon P, Künzel SE, et al. (2018). Lacteal junction zippering protects against diet-induced obesity. Science (80-. ). 361, 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. The vascular architecture of the small intestine. Related to Figure 1 and Figure S1. Whole-mount imaging and 3D reconstruction of the small intestinal vasculature in the villus illustrating LYVE1+ lymphatics (green) and CD31+ blood endothelial cells (red). Volumetric reconstruction of lymphatics (LYVE1+) in yellow and blood vessels (CD31+) in light blue.
Video S2. Lymphatic capillaries are highly associated with crypts in the large intestine. Related to Figure 1 and S1. Whole-mount imaging and 3D reconstruction of large intestinal LYVE1+ lymphatic capillaries (white) and CD31+ blood endothelial cells (red) around crypt-based epithelial cells (ECAD+, green). Volumetric reconstruction indicating lymphatics (LYVE1+, yellow) and crypt-based epithelial cells (purple).
Video S3. Lymphatic capillaries are nesting crypt-based stem cells of the human colon. Related to Figure 1. Whole-mount imaging and 3D reconstruction of a human intestinal biopsy, demonstrating association between lymphatics (LYVE1+, red) and epithelial cells of the intestinal crypt base (ECAD+, green) in the distal colon. Volumetric reconstruction indicates lymphatics (LYVE1+, yellow) and crypt-based epithelial cells (purple).
Video S4: Interaction of an intestinal organoid with LECs in coculture conditions. Related to Figure 2 and Figure S2. Representative z-stack intervals of organoids (EpCAM+, green) grown on top of lymphatic endothelial cells (CD31+, white), cultured in 50% LEC:50% organoid medium.
Video S5: Crypt-based lymphatic capillaries highly express REELIN. Related to Figure 6 and Figure S6. Whole-mount imaging and 3D reconstruction of the mouse large intestine showing LGR5+ stem cell-harboring crypts (cyan) sitting atop a network of LYVE1+ lymphatic capillaries (magenta), which highly express RELN (yellow).
Data Availability Statement
All data that support the findings of this study are available within the paper and its supplementary files. Single-cell and spatial transcriptomic data have been deposited in the Gene Expression Omnibus (GEO) repository with the accession code GSE190037. Newly generated bulk RNA sequencing data are associated with the accession code GSE199082. All original code used to analyze the data is available at https://github.com/dpeerlab/SpaceFold_paper.git. The SpaceFold R package along with tutorials is available at https://github.com/dpeerlab/SpaceFold.git as of the date of publication. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.