

Abstract
The introduction of sulfur into the phosphate linkage of chemically synthesized oligonucleotides creates the stereocenters on phosphorus atoms. Researchers have valued the nature of backbone stereochemistry and early on investigated drug properties for the individual stereocenters in dimers or short oligomers. Only very recently, it has become possible to synthesize fully stereodefined antisense oligonucleotides in good yield and purity. Non-bridging phosphorodithioate (PS2) introduces second sulfur into the phosphorothioate linkage to remove the chirality of phosphorus atom. Here, we describe the application of symmetrical non-bridging PS2 linkages in the context of stereodefined locked nucleic acids (LNAs) antisense oligonucleotides with the goal of reducing chiral complexity and, ultimately, resulting in single molecules. In addition, we propose a rather simple strategy to rapidly identify stereodefined gapmers, combining PS2 and a preferred stereochemistry motif (RSSR), which supports RNase-H-mediated target knockdown. Pharmacological efficacy and metabolic stability are investigated systematically using ApoB as a target sequence, where in vivo data correlate well to what is observed in vitro.
Keywords: MT: oligonucleotides: therapies and applications, locked nucleic acids, antisense oligonucleotides, phosphorodithioates, stereodefined phosphorodithioates, discovery strategies, pharmacological properties, symmetry
Graphical abstract
Non-bridging phosphorodithioates incorporated into the antisense oligonucleotide (ASO) flanks greatly increase the metabolic stability in vitro and in vivo. The symmetry of non-bridging phosphorodithioates significantly reduces the diastereoisomeric complexity. Taking advantage of the symmetry of phosphorodithioate and chirality, stereodefined phosphorodithioate ASOs with improved pharmacological properties can be rapidly identified.
Introduction
One of the first issues medicinal chemists would address in any given lead optimization program is the understanding of chirality in potential drug candidates that feature unresolved stereocenters. Countless examples have been reported, both for small molecules1 and oligonucleotides,2 where the resolution and inversion of chiral centers demonstrated substantial impact on drug properties, such as potency or safety. As the introduction of sulfur into the backbone of oligonucleotides leads to such unresolved stereocenters, academic groups around Stec et al.3 and Guga and Stec4 have pioneered the enantioselective synthesis of such internucleoside linkages, setting the stage for investigating the impact of “phosphate chirality.” Meanwhile, several approaches are established that do allow the synthesis of fully stereodefined phosphorothioate antisense oligonucleotides in good yield and purity.5,6 Different groups7, 8, 9 have reported the pharmacological profiles of such stereodefined antisense oligonucleotides with a seminal paper published by Verdine et al.,10 clearly describing the impact of chirality on various properties for the individual molecules, such as target binding, lipophilicity, or in vivo efficacy, and therefore, controlling chirality can potentially enhance overall therapeutic profiles of antisense oligonucleotides.
While modern synthetic schemes now allow for the stereocontrolled synthesis of phosphorothioate (PS) linkages and, therefore, provide access to single isomers, given the high complexity of generally 103–105 possibilities, it is unlikely to find single diastereoisomers with improved properties compared with the averaged properties of the overall mixture without a suitable strategy. For example, a conventionally synthesized 20-mer antisense oligonucleotide (ASO) with 19 non-stereodefined PS linkages contains a mixture of 219 = 524,288 diastereoisomers. Given the high number of possible diastereoisomers and the difficulty in finding single diastereoisomers with improved properties, a strategy on rapid identification of fully stereodefined PS-ASOs will offer a new dimension to the RNA therapeutic field. One potential approach to reduce this diastereoisomeric complexity is to insert symmetrical (“non-chiral”) thiophosphate modifications, since every “non-chiral” linkage introduced into the oligonucleotide reduces the overall number of possible diastereoisomers by 50%.11
Here, we report the systematic insertion of non-bridging phosphorodithioate (PS2) into the flank region of locked nucleic acids (LNAs) gapmers and investigate target engagement, mRNA knockdown, lipophilicity, and in vitro toxicity of the resulting PS2-modified ASOs. Furthermore, we suggest a discovery strategy for the rapid identification of single molecules by combining the PS2-modified flanks with fully stereodefined PS linkages in the gap region. A substantially increased in vivo efficacy was observed for an anti-ApoB stereodefined PS2 LNA gapmer, outperforming both its parent (diastereomeric mixture) as well as a fully stereodefined monothioate analog thereof.
Results
Target engagement, lipophilicity, and mRNA knockdown of phosphorodithioate-modified LNA gapmers
Symmetrical PS2, where both non-bridging oxygen atoms within the phosphate group are replaced by sulfur, have been introduced by Caruthers12, 13, 14, 15, 16, 17, 18, 19, 20 and also further investigated by other groups.21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38 Nonetheless, PS2 modifications remained rather unexplored in ASOs, which might be due to somewhat inconsistent and partly unfavorable data reported previously.14,16,20,37 We revisited the PS2 modification and hypothesized that the diligent introduction of only a limited number of PS2 groups at defined positions within AOSs should be well tolerated and may show improved drug properties when compared with fully PS-modified ASOs. Exemplified for RTR3833, a well-characterized anti-ApoB ASO,39 six PS2 analogs thereof were generated (ASO-1–ASO-6), where the PS2 content was increased subsequently in flank regions only. All analogs have been tested for their target knockdown efficacy in mouse primary hepatocytes as represented in Figure 1A. The half-maximal inhibitory concentration (IC50) values were generated by exposing the cells with ASOs for 72 h under gymnotic uptake conditions, i.e., no transfection reagents added (Figure 1B). All compounds performed equally good or better compared with RTR3833. With respect to target engagement (RNA binding), no meaningful change is observed for PS2-modified ASO-1–ASO-6 (see target engagement [Tm] in Figure 1A). As a measure of lipophilicity, we have investigated the impact of PS2 to the retention time (Rt) in reversed phase high performance liquid chromatography (HPLC). Under the given analytical conditions, the reference PS ASO (RTR3833) elutes with a retention time of Rt = 13.2 min. No meaningful difference is observed for ASO-1–ASO-6 (Figure 1A). For reference, we also generated and analyzed the all-PS2 as well as the corresponding phosphate (all-PO) analogs (Figure 1A). While for the full PS2-modified sequence, only a slightly higher retention time is observed (all-PS2: Rt = 13.9 min), the Rt for the all-PO analog is substantially lower (all-PO: Rt = 9.7 min) compared with the reference ASO (RTR3833). This indicates that the increase of lipophilicity is only marginal when introducing PS2 linkages sparingly into otherwise PS-modified ASOs.
Figure 1.

In vitro ApoB mRNA target reduction in primary mouse hepatocytes (PMHs) after 72 h of gapmer ASOs exposure
Reduction of target ApoB mRNA was quantified by qRT-PCR. All experiments are carried out in triplicates. (A) Design, antisense activity, Tm (melting temperature), and Rt (retention time) are shown. Positions of phosphorodithioate (PS2) (yellow o) are shown while the rest of ASO is stereorandom PS modified. ASOs marked with asterisk (∗) were tested in a different experiment following the same screening condition. aMelting temperature to complementary RNA (Tm) is shown; complementary RNA for Tm measurement is as follows: UGAAUACCAAUGC. bRP-HPLC retention time (Rt) is shown. cUpper case designates LNA nucleotide and lower case designates DNA nucleotide, with symbols designating stereorandom PS (•), PS2 (yellow o), and phosphate (red •). dStandard deviation (±) is shown; nd, not determined. (B) Dose-response curves for reducing ApoB mRNA in PMHs are shown.
Further studies were executed for other target sequences, leading to very similar results, i.e., symmetrical, non-bridging PS2 functionalities introduced in the flank region of LNA gapmers show no significant impact on Tm or in vitro potency (Figures S1 and S2 in supplemental material) and may therefore be used to reduce chiral complexity in any given ASO design.
Assessment of potential toxicity in vitro
In order to assess whether PS2 modifications pose an increased toxicity threat, we have evaluated the potential risk of such phosphate bioisosteres in vitro. The hepatotoxicity potential was assessed after transient transfection of HepG2 cells by monitoring caspase 3/7 activation as reported earlier (Figure 2A).40 None of the modifications induced any increase in caspase activation compared with the parent RTR3833. In addition, we investigated the hepatotoxicity potential in primary mouse hepatocytes after gymnotic uptake (Figure 2B). Again, none of the modifications resulted in worsening the mild toxicity seen with RTR3833 as measured by lactate dehydrogenase (LDH) release into the supernatant.
Figure 2.

In vitro safety profile of phosphorodithioates by caspase 3/7 activation and LDH secretion
(A) In vitro safety profile of PS2 in HepG2 cells 24 h after transient transfection at 100 nM concentration determined by caspase 3/7 activation. Bars (%V) represent mean of the relative caspase activity of tested compounds to the vehicle control (culture medium). Negative control in green bar and positive control in red bar are included. (B) In vitro safety profile of PS2 in primary mouse hepatocytes determined by LDH secretion after 3 days of gymnotic uptake with negative (green) and positive (red) controls tested at 30 μM is shown. Bars (% control) represent mean of the relative secreted LDH of tested compounds to the vehicle control (culture medium).
Discovery strategy for the rapid in vitro identification of stereodefined phosphorodithioate LNA gapmers
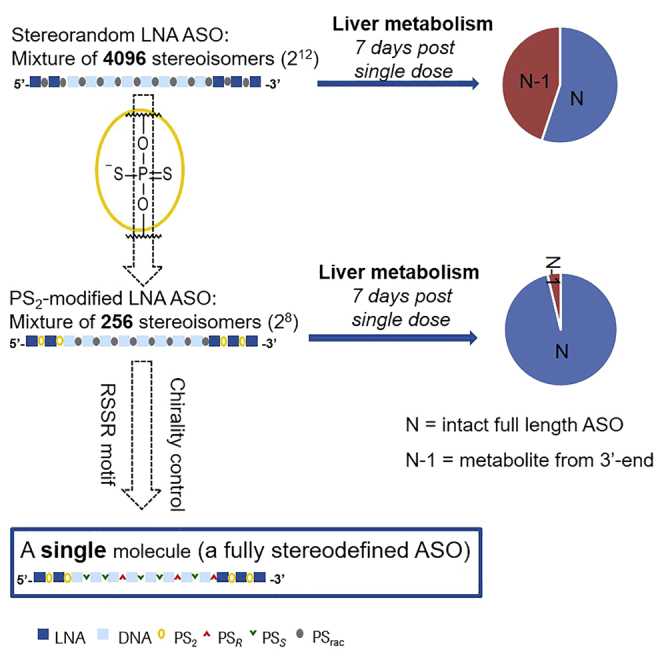
It is well accepted in the ASOs community that primarily the RNA target region, and therefore the drug sequence, as well as the overall ASO architecture (high-affinity entities, i.e., LNA, 2′-O-methoxyethylribose [MOE], etc. versus DNA) are the main drivers for pharmacology.2,41 Insofar as the pharmacokinetics and pharmacodynamics profile of a given, classically synthesized PS ASO can be further improved by evolving diastereoisomeric mixtures into partially or fully stereodefined single molecules is currently under debate.7,8,10 To address this question in the context of LNA gapmers, we investigated and further optimized the anti-ApoB lead sequence RTR3833. Since all PS internucleoside linkages were introduced via state-of-the-art phosphoramidite chemistry, this ASO results in an inseparable mixture of potentially 212 = 4,096 diastereoisomers (DIs). It is a high workload to synthesize and test all 4,096 DIs to identify a single DI with improved properties. Therefore, a rapid strategy for single-molecule identification is necessary. Having demonstrated the tolerability of non-bridging PS2 introduced in the flank region of classical gapmers, we wanted to take advantage of their symmetry and therefore introduced two of these modifications at both ends of LNA sequence of RTR3833, thereby reducing the number of possible DIs from 4,096 to 256 (ASO-6 Figure 3A). Verdine et al.10 have introduced and rationalized the stereo motif 3′-SSR to support RNase-H-mediated target cleavage in stereodefined ASOs. We have made similar observations for a stereo quadruplet RSSR when positioned appropriately in the gap region of the ASO (Figures S3A and S3B in supplemental material). It was observed that RSSR stereo motif in a specific position in DNA gap can enhance the chance of finding a potent fully stereodefined ASO. Based on this finding, we wanted to further exploit the stereo quadruplet in combination with the achiral PS2 introduced in the flank regions. All possible analogues were generated, where this stereo motif was moved in a consecutive manner through the DNA gap region while leaving the remaining PS internucleoside linkages racemic (called RSSR walk). Thus, five partially stereodefined PS2 hybrid ASOs resulted, which now consist of 16 DIs only (Figure 3A). The oligonucleotides were synthesized by means of solid-phase oligonucleotide chemistry similar to those reported in the literatures.6,9 LNA thiophosphoramidite monomers were generated from the correspondingly protected nucleoside alcohols42,43 (also see supplemental material). The in vitro potency of the gapmers were investigated in mouse primary hepatocytes. Cells were exposed to LNAs using gymnotic conditions. ApoB mRNA levels were recorded at 72 h after incubation at eight different concentrations (Figure 3A). Corresponding IC50 values are given in Figure 3B.
Figure 3.

In vitro ApoB mRNA target reduction in primary mouse hepatocytes (PMHs) after 72 h of gapmer ASOs exposure
Reduction of target ApoB mRNA was quantified by qRT-PCR. All experiments are carried out in triplicate. (A) Design, diastereoisomers (DIs), and antisense activity are shown. Positions of PS2 (yellow o) and RSSR motif (red ˄, green ˅, green ˅, red ˄) are shown, while the rest of ASO is stereorandom PS modified. aUpper case designates LNA nucleotide, lower case designates DNA nucleotide, and symbols designate stereorandom PS (•), PS2 (yellow o), R configuration (red ˄), and S configuration (green ˅). bDIs designates diastereoisomers. cStandard deviation (±) is shown. (B) Dose-response curves for reducing ApoB mRNA in PMHs are shown.
The strongest target knockdown with an IC50 = 0.08 μM was observed for ASO-9. We therefore took this partially stereodefined hybrid design forward and used it as the starting point for the second iteration cycle. To stereochemically resolve the remaining four PS positions, all 16 possible stereoisomers were synthesized and tested for ApoB mRNA knockdown (Figure 4A). IC50 values of all possible stereoisomers are shown in Figure 4B.
Figure 4.

In vitro ApoB mRNA target reduction in primary hepatocytes (PMHs) after 72 h of gapmer ASOs exposure
Reduction of target ApoB mRNA was quantified by qRT-PCR. All experiments are carried out in triplicate. (A) Design and antisense activity are shown. Positions of PS2 (yellow o) and stereochemistry of each phosphorothioate linkage (red ˄ and green ˅) are shown. aUpper case designates LNA nucleotide, lower case designates DNA nucleotide, and symbols designate PS2 (yellow o), R configuration (red ˄), and S configuration (green ˅). bStandard deviation (±) is shown. (B) Dose-response curves for reducing ApoB mRNA in PMHs are shown.
Further studies executed for other target sequences lead to very similar results (Figure S4 in supplemental materials), i.e., RSSR motif in a specific position in DNA gap enhances the in vitro potency, which could be generalized across sequences and targets.
RNase-H-mediated mRNA knockdown in vitro
To assess the impact of PS2 backbone chemistry on RNase-H-mediated target cleavage, we set up a competition assay in which an RNA-ASO heteroduplex formed with the parent compound (RTR3833) is mixed with a heteroduplex formed with a tested, PS2-modified compound (Figure S5 in supplemental materials). The RNA in each of the heteroduplexes in a given reaction was labeled with a distinct fluorophore (FAM or Cy5) to enable independent quantification of the RNA fraction cleaved. Besides the two PS2-modified analogs ASO-6 and ASO-26, we have also investigated a fully stereodefined version of RTR3833, namely RTR28253. This single molecule was identified in earlier studies and turned out to be the most potent ApoB LNA gapmer we have ever tested in our in vitro and in vivo models at that stage. RTR28253 shows the same sequence, LNA/DNA pattern, and PS stereochemistry as ASO-26 but lacking the PS2 modifications in the flank regions, allowing a direct comparison between these two ASOs (Figure 5). Following hydrolysis with recombinant human RNase H1, we have observed no differences in cleavage extent between the parent compound and the PS2-modified racemic analog ASO-6. However, when comparing the two stereodefined versions RTR28253 and ASO-26 with RTR3833, a substantially greater RNA cleavage is observed, as shown in Figure 5 (light gray bars). These data nicely demonstrate that PS2-modified flanks have no negative impact on the mRNA cleavage rate and confirm what was reported earlier, i.e., that chirality motif, such as RSSR, as used in the design of RTR28253 and ASO-26, may well contribute to an increased RNase H activity when positioned accordingly in gapmer designs.
Figure 5.

RNA cleavage extent in the RNase H cleavage competition assay
Fraction of RNA cleaved in each reaction for RNA hybridized with parent compound RTR3833 (dark gray) and for a tested compound (label on x axis; light gray). Left (A) and right (B) plot relate to swapped fluorophore labeling. Bars represent mean of the fractions cleaved for replicates; x symbols indicate individual observation (paired t test based on all four replicates and corrected with Holm’s method: p = 0.03 for both RTR28253 and ASO-26; no significant difference between RNA cleavage extents induced by RTR3833 and ASO-6). RTR3833: G•C•a•t•t•g•g•t•a•t•T•C•A. ASO-6: GoCoa•t•t•g•g•t•a•t•ToCoA. RTR28253: G˄C˄a˅t˅t˄g˅g˅t˄a˅t˄T˅C˅A. ASO-26: GoCoa˅t˅t˄g˅g˅t˄a˅t˄ToCoA. Upper case designates LNA nucleotide, lower case designates DNA nucleotide, and symbols designate R configuration (red ˄), S configuration (green ˅), PS2 linkage (o), and phosphorothioate linkage (•).
In vivo profiling of phosphorodithioate-modified anti-ApoB gapmers
Encouraged by the in vitro activities obtained, we further investigated these PS2 ASOs in vivo. Therefore, animals were treated with a single dose of drug candidates and ApoB mRNA knockdown measured both in liver and kidney. Data points were collected after 7, 21, 28, and 42 days of drug exposure using the stereomixed ASO RTR3833 as a reference. Oligonucleotides were administered intravenously to mice at a dose level of 1 mg/kg (5 mice/group; single administration). Animals were sacrificed at 7, 21, 28, and 42 days and kidneys and livers sampled and analyzed by qPCR (Figure 6).
Figure 6.

In vivo target knockdown in the liver at four different time points (day 7, 21, 28, and 42)
C57BL/6JBomTac female mice (n = 5/group) are singly dosed with 1 mg/kg by intravenous administration. Animals were sacrificed at (A) day 7, (B) day 21, (C) day 28, and (D) day 42. Livers were harvested, and reduction of target ApoB mRNA in the liver was quantified by qRT-PCR relative to GAPDH. One animal in cohort RTR28253/day 28 had to be anesthetized due to exaggerated pharmacology. Statistically significant (one-way ANOVA) data are indicated. ∗∗p < 0.01; ∗∗∗p < 0.001.
For the cohort of animals sacrificed 7 days post-drug administration, a target mRNA knockdown of 30% and 27% was obtained for gapmers RTR3833 and ASO-6, respectively. These two ASOs are racemic in nature and differ only in the PS2 linkages within the flank regions. A substantially increased efficacy was observed for the stereodefined analogs, with RTR28253 showing a target knockdown of 59% and the corresponding PS2 analog (ASO-26) showing 67% target knockdown at the same time point of analysis. For the second animal cohort sacrificed on day 21, the reference ASO RTR3833 shows only a very moderate activity of 13% target knockdown, whereas the PS2 version ASO-6 showed a 20% reduction. For the fully stereodefined RTR28253, a 38% knockdown was observed, while the stereodefined PS2 analog ASO-26 differentiated the most and showed still 51% of target knockdown. Twenty-eight days post-dose, the performance ranking order for the four ASOs was the same as for the other two time points. Again, a particularly strong efficacy was observed for the PS2-stereodefined ASO-26, showing still 49% target knockdown. For the latest time point measured (42 days post-dosing), no statistically significant target knockdown was observed for all ASOs tested.
The mice experiment was conducted in accordance with the European Guidelines for the Care and Use of Laboratory Animals (directive 2010/63/EU) and was approved by the Ethical Committee for Animal Experimentation at Hoffmann-La Roche (Switzerland).
Tissue exposure and PK/PD
Drug concentrations were determined for all four ASOs at 7, 21, 28, and 42 days post-dosing both in liver and kidney. Observations from earlier studies were confirmed, where kidney exposure of the parent molecule (RTR3833) exceeded liver exposure substantially at all the time points measured. Similar profiles were obtained for the chemically modified analogs ASO-6, RTR28253, and ASO-26 as shown in Figures 7A and 7B. Overall, the organ concentrations for RTR3833 and the stereodefined version RTR28253 were almost equal for both organs. Similar profiles were also observed for the two PS2-modified analogs ASO-6 and ASO-26, with the absolute concentrations always exceeding the exposure rates of their corresponding parent molecules. Insofar as higher tissue uptake and/or increased metabolic stability contributes to the beneficial tissue exposure remains to be investigated, but the substantially higher ASO levels observed over time for the two PS2 analogs indicates that increased metabolic stability may indeed be a strong contributor. To address the mechanism of the enhanced tissue accumulation of PS2 ASOs, quantitative metabolite analysis was conducted (Figure 7C). Correlating target knockdown with the actual tissue concentration confirms the superior efficacy for the stereodefined analogs over their corresponding parents. ASO-26 and RTR28253 clearly outperform the stereomixed reference molecules RTR3833 and ASO-6.
Figure 7.

Tissue exposure and PK/PD
Drug concentrations measured in liver (A) and kidney (B) PK/PD relationship for all four drug candidates (C). C57BL/6JBomTac female mice (n = 5/group) are singly dosed with 1 mg/kg by intravenous administration. Animals were sacrificed at day 7, day 21, day 28, and day 42. Livers and kidneys were harvested, and ASO concentrations were measured by hELISA. y axis represents drug concentration (nM, nmols/kg tissue) in liver and kidney separately.
In vivo metabolite profiling
All four candidates tested in vivo were further investigated with respect to ASO drug metabolism. Therefore, liver and kidney samples of the cohort sacrificed on day 7 were analyzed using an untargeted high-resolution liquid chromatography-mass spectrometry (LC-MS) approach to identify ASO metabolites and to assess their relative abundance (alternative semi-quantification). Data for the two organs are depicted as pie chart format in Figure 8.
Figure 8.

Metabolite formation in liver and kidney after 7 days of drug administration
C57BL/6JBomTac female mice (n = 5/group) are singly dosed with 1 mg/kg by intravenous administration. Liver and kidney were sampled at day 7. The data were measured by LC-MS/MS. Color codes represent the fragmented metabolites. RTR3833: 5′-GCatggtatTCA-3′ ASO-6: 5′-GCatggtatTCA-3′. RTR28253: 5′-GCatggtatTCA-3′. ASO-26: 5′-GCatggtatTCA-3′. Upper case designates LNA nucleotide, lower case designates DNA nucleotide, and symbols designate R configuration (red ˄), S-configuration (green ˅), PS2 linkage ( o), and phosphorothioate linkage (•).
A pronounced formation of the N-1 metabolite was found for RTR3833, resulting from the cleavage of the adenosine nucleotide from the 3′ end. Further cleavage to shorter metabolites was only seen to some extent and at small relative signal intensities in both liver and kidney. We observed a substantial improvement in metabolic stability for the PS2 analog ASO-6, where also the N-1 fragment was detected as the main metabolite, although appearing at much lower abundancy in both organs in comparison to the stereorandom parent sequence RTR3833. Interestingly, when looking at the corresponding stereodefined analogs, the PS2 version of RTR28253 (ASO-26) shows higher relative metabolism than the fully stereodefined gapmer, which is devoid of the PS2 modification. N-1 fragmentation is substantially lower for both analogs compared with parent RTR3833. The main metabolite observed for both stereodefined molecules is the octamer fragment (5'-GCattggt-3').
Discussion
An obvious challenge with respect to stereodefined ASOs is the fact that, with increasing length of the ASOs, the number of stereoisomers increases exponentially, leading to mixtures with 103–105 possible analogs. While the chemical synthesis of all DIs is currently not an attractive option, given the prohibitive number of compounds that would need to be prepared, researchers have focused on the synthesis of a few representatives instead.7, 8, 9, 10 In the context of ASO gapmers (RNase-H-mediated target knockdown), rational design elements, i.e., the 3′-SSR-triplet10 and the RSSR-quadruplet,44 have been reported to facilitate RNase-H-mediated target cleavage when positioned appropriately within the gap region. Having recognized the beneficial drug properties of symmetrical non-bridging PS2, we have set out to implement the concept of reducing stereo-complexity by insertion of such achiral PS2 linkages into the flank regions of a given lead sequence. Furthermore, when combining PS2-modified flanks with the stereo quadruplet RSSR designed within the DNA region of gapmers, the stereo-complexity can be further reduced. For exemplification, we describe our experiments here referencing the anti-ApoB target ASO RTR3833. This molecule is a well-characterized LNA gapmer design based on state-of-the-art phosphoramidite chemistry, which leads to an inseparable mixture of potentially 4,096 DIs. As we demonstrated that PS2 modifications are well tolerated in the flank regions of gapmers, we introduced two PS2 modifications at both ends of this oligonucleotide. This reduces the number of stereocenters to eight and therefore 256 DIs. While this number might be manageable in terms of parallel synthesis capacity, we further simplified the process of identifying stereodefined single molecules, as in a classical drug discovery program, we would usually deal with several lead sequences. Exploiting the beneficial effect of chiral motifs increasing RNA knockdown efficacy, we first performed an “RSSR walk” through the gap region of our PS2-modified lead sequence. Therefore, five ASO analogs, where the RSSR motif was placed at every possible position within the gap, were generated while keeping the remaining gap internucleoside linkages as unresolved PS. ASO-9 was identified as the most potent analog, with the RSSR motif being positioned in the center of the gap region. This partially stereodefined PS2 hybrid was used for the second iteration cycle, where the number of DIs is further reduced to 16 from 256. It is much faster and easier to synthesize and profile 16 ASOs than 4,096 ASOs obtained from the conventional method. These 16 single molecules were synthesized and again tested in vitro for ApoB mRNA knockdown. Compared with the parent gapmer RTR3833, which showed a potency of 0.18 μM, all stereodefined analogs identified via this strategy turned out to be equally potent or better. An approximately 5-fold increase could be obtained for the best-performing and fully stereodefined PS2 analogs. From this set of potential candidates, ASO-26 was selected for in vivo profiling, not only because of its promising in vitro potency but rather because of the identical stereochemistry in the gap region when compared with stereodefined reference RTR28253.
In absolute terms, the stereomixed molecules (RTR3833 and ASO-6) showed ∼30% of ApoB mRNA knockdown while the corresponding stereodefined analogs (RTR28253 and ASO-26) performed significantly better, reducing ApoB mRNA by more than 50% after 7 days of drug administration. Superior efficacy of the single molecules was also confirmed for later time points (21 and 28 days post-dose), where the stereodefined PS2 ASO-26 showed the strongest performance for all three time points measured. No significant effect was observed for the ASOs at the end of the study. When investigating ASO content in liver and kidney, the two PS2 analogs (ASO-6 and ASO-26) show substantially higher organ exposure. Correlating ASO concentration in the liver with efficacy demonstrates again the superiority of the two stereodefined analogues versus their corresponding diastereoisomeric mixtures. How much increased metabolic stability may contribute to superior target knockdown efficacy remains to be further investigated. Nonetheless, from the metabolite profiles generated, it becomes quite obvious that both PS2 modifications as well as S-configured PS linkages are able to reduce metabolic degradation of a given ASO. For the diastereoisomeric mixture RTR3833, the main route of metabolism was induced by exonucleolytic degradation, as confirmed by the generation of the N-1 fragment as the major metabolite. This was clearly reduced for the PS2 analog ASO-6, leading to a much higher overall stability. For the two stereodefined analogs, however, the main route of metabolism seems to be driven by endonucleolytic degradation, as the major metabolite for both molecules was the octamer fragment (5'-GCattggt-3'). This hypothesis is substantiated by the presence of the decamer fragment (5'-GCattggtat-3') as the second most abundant metabolite. For both fragments, the internucleoside linkages cleaved were R configured. As it is very well known that R-configured PS internucleoside linkages are more prone to enzymatic cleavage than their S-configured counterparts, the findings mentioned above are aligned closely to what was reported earlier.10
In summary, we describe herein the impact of PS2-modified LNAs, and we propose a strategy for the identification of stereodefined gapmers that does not require the synthesis of all possible diastereomeric combinations for a given sequence. Our investigations focus on in vitro activity and in vivo efficacy as well as drug metabolism. The data clearly demonstrate superiority of stereodefined LNA gapmers over their racemic parent compounds, with the stereodefined PS2 analog showing strongest in vivo performance. We have further evaluated the metabolic profile of stereodefined ASOs and confirmed the preferred metabolic cleavage for R-configured PS linkages in vivo. The in vivo duration of effect study data described herein strongly suggest that a combination of stereodefined internucleoside linkages with achiral PS2 may lead to significantly superior pharmacology compared with the stereodefined or the stereomixed parent molecules. Further applications for PS2-modified ASOs are currently under investigation and will be reported in due course.
Materials and methods
Oligonucleotide synthesis and purification
Oligonucleotides were synthesized using a MerMade 12 or MerMade 192 automated DNA synthesizer by Bioautomation. Syntheses were conducted on a 1-μmol scale using a controlled pore glass support (500 Å, LGC-Biosearch Technologies, UK) bearing a universal linker. Standard phosphoramidite synthesis procedures were used for unmodified LNA- and DNA-phosphoramidites (Sigma-Aldrich), i.e., 3% dichloroacetic acid in dichloromethane (CH2Cl2) for deblocking, 1 M 4,5-dicyanoimidazole (DCI)/0.1 M N-methylimidazole in acetonitrile (MeCN) as activator, 10% acetic anhydride in tetrahydrofuran (THF) and 10% N-methylimidazole in THF/pyridine for capping, and 0.1 M xanthane hydride in pyridine: MeCN 1:1 (v/v) for thiolation. Standard and chiral phosphoramidites were dissolved at 0.1 M MeCN (+3.5% pyridine for chiral monomers) and incorporated using 3-min coupling times, while thiophosphoroamidites were prepared at 0.15 M in CH2Cl2: MeCN 1:9 (v/v) using 10-min coupling times. Removal of the nucleobase-protecting groups and cleavage from the solid support were achieved under standard conditions using 25% aqueous ammonia at 55°C for 12–16 h. Crude dimethoxytrityl(DMT)-on oligonucleotides were purified either using a solid-phase extraction cartridge (Oasis HLB 6 cc extraction cartridges from waters) or by preparative reverse-phase HPLC (RP-HPLC) purification (C18 column, NH4OAc/CH3CN buffer system) followed by DMT removal with 80% aqueous acetic acid. Following HPLC purification, oligonucleotides were desalted using HiPrep 26/10 desalting column on ÄKTA pure 25 M and lyophilized. Oligonucleotides were characterized by reversed phase ultra-performance liquid chromatography coupled to high-resolution electrospray mass spectrometry. Oligonucleotides used as capture (5'-[5bio]-[HEG]-TGAATAC-3′) and detection probe (5′-CAATGC-[HEG]-[digc63]-3') were synthesized as phosphate analogs by incorporation Biotin-CE phosphoramidite at 5′ end of LNA sequence (5'-TGAATAC-3') and conjugation with digoxigenin-NHS ester at 3′ end of LNA sequence (5'-CAATGC-3'). The labeling agents were separated by the introduction of a hexaethylene glycol (HEG) linker (Spacer-CE phosphoramidite 18).
Tm measurements
LNA gapmers and complementary RNAs were added to 10 mM phosphate buffer, 100 mM NaCl, and 0.1 nM EDTA (pH 7), resulting in a final concentration of 1.5 μM. Samples were heated to 95°C for 3 min and then slowly cooled to room temperature over a period of 30 min. Thermal melting curves were recorded at 260 nm on a Agilent Cary 3500 equipped with a Peltier Temperature Programmer using a temperature gradient that was increased by 5°C/min from 25°C to 95°C and then decreased to 25°C. The first derivatives of both curves were used to determine the melting temperature (Tm). The values are averaged over three heating and cooling curves and are reported as value ± standard deviation.
HPLC analysis
Retention times generated for Figure 1A have been executed on an Agilent 1290 Infinity II instrument (column: Waters Acquity UPLC BEH C-18, 2.1 × 50 mm, 1.7 μM; flow: 0.8 mL/min; buffer A: 1,900 mL water/50 mL MeOH, 42 mL hexafluoroisopropanol [HFIP], 4.2 mL triethanolamine [TEA]; buffer B: 350 mL water/1,600 mL MeOH, 42 mL HFIP, 4.2 mL TEA; gradient: 0.0 min - 97% A/3% B; 0.1 min - 97% A/3% B; 22.0 min - 85% A/15% B; 30.0 min - 0% A/100% B).
Cell culture, oligonucleotide treatment, mRNA isolation, and qPCR
Hepatocytes were isolated from C57BL/6 mice by a two-step collagenase liver perfusion method as previously described. Freshly isolated primary mouse hepatocytes were plated in collagen-l-coated 96-well plates and treated in Williams Medium E containing 10% fetal bovine serum without antibiotics. Cells were treated with LNA solutions in the indicated concentrations in full cell culture medium. After an incubation time of 72 h, the cells were lysed with 125 μL PureLink Pro lysis buffer and total RNA isolated using the PureLink Pro 96 RNA Kit from Thermo Fisher according to the manufacturer’s instructions. Expression ApoB mRNA was evaluated in a One Step RT-qPCR using Thermo Fisher TaqMan assay Mm01545150_m1 normalized to expression of Gapdh (Mm99999915_g1). All data points were performed in triplicate and IC50 values determined using GraphPad Prism.
RNase H cleavage competition assay
Heteroduplex RNase H substrates were prepared by mixing 20 volumes of 10× annealing buffer (200 mM Tris-HCl [pH 7.5], 200 mM KCl, 200 mM 2-mercaptoethanol, and 1 mM EDTA), two volumes of 100 μM fluorescently labeled RNA target site (5′-ACUGAAUACCAAUGCUG-3′, 5′ labeled with FAM or Cy5; manufactured by Integrated DNA Technologies), and three volumes of 100 μM ASO and 175 volumes of H2O, followed by strand annealing in thermal cycler with a program: 2 min at 95°C, 10 min at 55°C, 10 min at 30°C, cool down to 4°C, followed by placing samples on ice. Ten microliters of the reference heteroduplex (annealed with RTR3833) was mixed with 10 μL of the tested heteroduplex (annealed with a tested compound), which was labeled with a different fluorophore. Enzymatic solution was prepared by mixing one volume of 10× RNH1-2× Mg buffer (the same composition as 10× annealing buffer supplemented with 40 mM MgCl2), 8.97 volumes of H2O, and 0.03 volumes of 0.2 mg/mL recombinant human RNase H1 isoform 2 (NP_001273763.1 preceded with glutathione [GSH] triamino acid; manufactured by Leaderna Biostructures based on a protocol kindly shared by Dr. Marcin Nowotny laboratory). Twenty microliters of the enzymatic solution were added to the mixed heteroduplexes, and the samples were incubated at 4°C for 1 min and 37°C for 10 min, followed by cooling down to 4°C. No enzyme control employed RTR3833 for both reference and tested compound. Reactions were terminated by addition of 80 μL of the stop solution (8 M urea, 1× Tris-borate-EDTA [TBE], and 5 mM EDTA). Samples were heat denatured (2 min at 95°C) and resolved on 15% polyacrylamide urea-TBE gel. Gels were visualized for FAM and Cy5 channels using iBright FL1000 Imaging System (Thermo Fisher Scientific), and bands for full length and cleaved fractions were quantified using ImageJ. The experiment was performed in triplicate for FAM-labeled reference and Cy5-labeled tested substrate and for the opposite labeling. We have observed a general trend that FAM-labeled compounds gave higher cleavage efficiency, which could be a real effect or an artifact; however, the paired t test used equal number of pairs from each labeling order (FAM-Cy5 or Cy5-FAM for reference-tested pairs), hence the impact of this confounding factor is eliminated.
Caspase activation assay
HepG2 cells were cultivated at app. 70% confluence in minimum essential medium (MEM) with GlutaMax (Gibco, no. 41090), supplemented with 10% heat-inactivated fetal calf serum. Cells were detached with 0.25% Trypsin-EDTA solution (Gibco, no. 25200056) and seeded into black, clear 96-well plates (Corning Life Sciences, no. 3904, NY, USA) at a density of 1 × 104 cells/well. Twenty-four hours post-seeding, HepG2 cells were transiently transfected with Lipofectamine 2000 (Life Technologies, no. 11668019) using 100 nM oligonucleotides dissolved in Opti-MEM (Gibco, no. 31985). Caspase 3/7 activity was determined using the Caspase-Glo 3/7 Assay (Promega, Madison, WI, USA). Reconstituted Caspase-Glo 3/7 reagent was added to the cells 24 h post-transfection, incubated for 60 min, and cell lysates were transferred into opaque 96-well plates (Corning Life Sciences, no. 3600, NY, USA) before luminescence was determined on an Enspire multi-mode plate reader (PerkinElmer) according to the manufacturer’s instructions. Background readings determined from wells containing culture medium only were subtracted. Relative caspase activity was calculated as 100%× luminescence reading of a mock-treated control. All experiments were performed in triplicate.
LDH assay
LDH released into the culture media was determined using a Cytotoxicity Detection Kit (Roche 11,644,793,001, Roche Diagnostics, Roche Applied Science, Mannheim, Germany) according to the manufacturer’s protocol. All experiments were performed in triplicate.
In vivo study
In vivo experiments were conducted according to the European standards, and protocols were approved by the Danish National Committee for Ethics in Animal Experiments. Inbred C57BL/6JBomTac female mice weighing 20 ± 2 g (arithmetic mean ± standard deviation) were obtained from Taconic (Denmark). The animals were housed in groups of five, and water and a standard diet were supplied ad libitum. The vivarium was maintained at a constant temperature (23°C ± 1°C) and humidity (40% ± 5%) under a 12 h light: 12 h dark cycle (lights on at 08:00 h) throughout the study. Mice were dosed intravenously on day 0 and anesthetized (70% CO2 and 30% O2) before termination by cervical dislocation on day 7. The treatment groups (n = 5) received either 0.9% saline or saline-formulated gapmer administered by intravenous injection. At the end of the study, livers and kidneys were snap frozen for subsequent analysis.
mRNA isolation and qPCR measurements from tissue samples
Total RNA from liver and kidney was isolated using the RNeasy kit (Qiagen), and quantification of messenger RNA (mRNA) was done using TaqMan assays (Applied Biosystems). The reverse transcription reaction was carried out with random decamers, 0.5 mg total RNA, and the Moloney murine leukemia virus (M-MLV) reverse-transcriptase enzyme (Ambion) according to protocol for first-strand complementary DNA (cDNA) synthesis. Depending on expression levels, 10 ng cDNA per reaction was subsequently diluted five times in nuclease-free water before addition to the RT-PCR reaction mixture. The Applied Biosystems 7500/7900/ViiA real-time PCR instruments were used for amplification. Within each study, mRNA levels were normalized to actin, beta (Actb), or glyceraldehyde-3-phosphate dehydrogenase (Gapdh) and presented as fold changes relative to average levels in saline controls.
Quantification of intracellular LNA content by hELISA
LNA content was determined by hELISA, using a biotinylated capture probe and a digoxigenin-conjugated detection probe as described previously.39 Sixty milligrams of tissue samples are lysed in 750 μL MagnaPure Lysis buffer. The resulted lysates were diluted and incubated with 35 nM biotinylated capture probe and 30 nM digoxigenin-coupled detection probe for 30 min at room temperature in SSCT buffer (5× saline sodium citrate buffer [SSC Buffer 20× Concentrate, Sigma-Aldrich, no. 6639] containing 0.05% Tween 20 [Sigma-Aldrich, no. P9416]) in a 96-well plate. The assembled complex is then captured on a streptavidine-coated ELISA plate (Nunc 436014) for 1 h, and after three washing steps with 2 x SSCT buffer, each well is incubated with an anti-digoxigenin-alkaline phosphatase (AP)-Fab fragment (Roche, no. 11093274910) for 1 h at room temperature. After three additional washing steps, BluePhos substrate (Kirkegaard & Perry Labs [KPL], no. 50-88-00) was added to the plates, and color development was measured spectrophotometrically at 615 nm after 20 min. For the oligo content analysis, several dilutions of each sample (50×, 100×, 200×, 400×, 800×, and 1,600×) are measured. Calculating back to the respective tissue weight and making an average of the valid values from different dilutions (i.e., the readouts within the linear range of the standard curve) generate the drug concentration.
Metabolite profiling in liver and kidney tissues
Liver and kidney tissue samples were collected from the above-described in vivo study and were stored at −80°C prior to analysis. To prepare tissue homogenates, 100 mg of liver or kidney tissue was placed into a CK28 tube (Precellys) and 500 μL (for liver tissue) or 300 μL (for kidney tissue) of guanidine thiocyanate (4 M in 0.1 M Tris buffer at pH 7.5) was added before mixing in the Precellys homogenizer for 10 min. The analysis of biotransformation of the oligonucleotide compounds in tissue homogenates was performed as described previously.45,46 In brief, 50 μL of homogenate was mixed with 250 μL of guanidine thiocyanate 4 M in 0.1 M Tris buffer (pH 7.5) for 15 min at 25°C in a Thermomixer. Then 700 μL H2O/HFIP/DIPEA 100/2/0.2 (v/v/v) was added and mixed for 1 h at 25°C. Then the samples were centrifuged for 5 min at 14,000 rpm, and the supernatant was subjected to a solid-phase extraction (SPE) OASIS HLB 1 cc 30 mg cartridge (Waters, Wexford, Ireland) followed by the analysis with LC-MS. A Thermo Scientific Dionex UltiMate NCP-3200RS Binary Rapid Separation HPLC system was used in combination with a Pal autosampler (CTC Analytics AG, Zwingen, Switzerland) and a Thermo Scientific Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Scientific, Bremen, Germany) equipped with an electrospray ionization source. The oligonucleotide metabolites were analyzed in negative ionization mode using a full-scan MS experiment combined with two parallel MS2 experiments, one data-dependent scan, and an untargeted all-ion-fragmentation (AIF) experiment applying high collision energy. In the AIF scan, a diagnostic fragment originating from the PS backbone (O2PS−: m/z 94.936) was formed efficiently upon collisional activation. Based on this fragment, an accurate determination of metabolites of oligonucleotides was achieved, independent of their sequence in an untargeted but highly selective manner. MS data were analyzed using XCalibur software (Thermo Scientific, Bremen Germany). MS intensities were recorded as the sum of the most intense charge states of the most intense isotopes of the respective analyte, applying an m/z window of 5 ppm.
Acknowledgments
We gratefully acknowledge Heidii Mazur for her help running the in vivo study as well as Charlotte Øverup, Sidsel Boesen, and Christian Weile, who performed the bioanalytic analysis of in vivo samples. We also thank Jon Bodnar for proofreading the manuscript. This research was supported by Roche Postdoc Fellowship (RPF) program, Switzerland.
Author contributions
J.D. and M.L. designed and performed experiments, analyzed data, and wrote the manuscript. A.S. supported oligo synthesis. M.D. performed the in vitro evaluation experiments and analyzed data. K.J. and M.R.M. performed the in vivo study. S. Schmidt and H.N. performed in vitro evaluation experiments and analyzed data, analyzed the in vivo data, and reviewed and edited the manuscript. T.M. performed caspase induction experiment. S. Sewing analyzed the caspase data and reviewed and edited the paper. C.H. performed in vitro metabolic stability experiment and in vivo metabolite identification study and analyzed data. S. Schadt and A.B. analyzed in vitro metabolic stability and in vivo metabolite identification data and reviewed and edited the manuscript. L.J.K. performed RNase H cleavage competition assay, analyzed data, and reviewed and edited the manuscript. E.K. coordinated in vivo study and reviewed and edited the manuscript. E.F. provided valuable historical data and advice. T.K. and J.W. provided support to the paper. K.B. conceived the project idea, supervised the work, and wrote the manuscript.
Declaration of interests
J.D. is a member of a shareholder group with pooled voting rights of Roche as well as one of their representatives on the board of directors. J.W. is an academic collaborator. All the rest of the authors are or were employees of F. Hoffmann-La Roche AG.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.06.010.
Supplemental information
References
- 1.Nguyen L.A., He H., Pham-Huy C. Chiral drugs: an overview. Int. J. Biomed. Sci. 2006;2:85–100. [PMC free article] [PubMed] [Google Scholar]
- 2.Hagedorn P.H., Persson R., Funder E.D., Albaek N., Diemer S.L., Hansen D.J., Moller M.R., Papargyri N., Christiansen H., Hansen B.R., et al. Locked nucleic acid: modality, diversity, and drug discovery. Drug Discov. Today. 2018;23:101–114. doi: 10.1016/j.drudis.2017.09.018. [DOI] [PubMed] [Google Scholar]
- 3.Stec W.J., Grajkowski A., Koziolkiewicz M., Uznanski B. Novel route to oligo(deoxyribonucleoside phosphorothioates). Stereocontrolled synthesis of P-chiral oligo(deoxyribonucleoside phosphorothioates) Nucleic Acids Res. 1991;19:5883–5888. doi: 10.1093/nar/19.21.5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guga P., Stec W.J. Synthesis of phosphorothioate oligonucleotides with stereodefined phosphorothioate linkages. Curr. Protoc. Nucleic Acid Chem. 2003;Chapter 4:Unit 4.17. doi: 10.1002/0471142700.nc0417s14. [DOI] [PubMed] [Google Scholar]
- 5.Knouse K.W., DeGruyter J.N., Schmidt M.A., Zheng B., Vantourout J.C., Kingston C., Mercer S.E., Mcdonald I.M., Olson R.E., Zhu Y., et al. Unlocking P(V): reagents for chiral phosphorothioate synthesis. Science. 2018;361:1234–1238. doi: 10.1126/science.aau3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oka N., Yamamoto M., Sato T., Wada T. Solid-phase synthesis of stereoregular oligodeoxyribonucleoside phosphorothioates using bicyclic oxazaphospholidine derivatives as monomer units. J. Am. Chem. Soc. 2008;130:16031–16037. doi: 10.1021/ja805780u. [DOI] [PubMed] [Google Scholar]
- 7.Li M., Lightfoot H.L., Halloy F., Malinowska A.L., Berk C., Behera A., Schumperli D., Hall J. Synthesis and cellular activity of stereochemically-pure 2'-O-(2-methoxyethyl)-phosphorothioate oligonucleotides. Chem. Commun. (Camb.) 2017;53:541–544. doi: 10.1039/c6cc08473g. [DOI] [PubMed] [Google Scholar]
- 8.Ostergaard M.E., De Hoyos C.L., Wan W.B., Shen W., Low A., Berdeja A., Vasquez G., Murray S., Migawa M.T., Liang X.H., et al. Understanding the effect of controlling phosphorothioate chirality in the DNA gap on the potency and safety of gapmer antisense oligonucleotides. Nucleic Acids Res. 2020;48:1691–1700. doi: 10.1093/nar/gkaa031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wan W.B., Migawa M.T., Vasquez G., Murray H.M., Nichols J.G., Gaus H., Berdeja A., Lee S., Hart C.E., Lima W.F., et al. Synthesis, biophysical properties and biological activity of second generation antisense oligonucleotides containing chiral phosphorothioate linkages. Nucleic Acids Res. 2014;42:13456–13468. doi: 10.1093/nar/gku1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwamoto N., Butler D.C.D., Svrzikapa N., Mohapatra S., Zlatev I., Sah D.W.Y., Meena Standley S.M., Lu G., Apponi L.H., et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017;35:845–851. doi: 10.1038/nbt.3948. [DOI] [PubMed] [Google Scholar]
- 11.Duschmale J., Hansen H.F., Duschmale M., Koller E., Albaek N., Moller M.R., Jensen K., Koch T., Wengel J., Bleicher K. In vitro and in vivo properties of therapeutic oligonucleotides containing non-chiral 3' and 5' thiophosphate linkages. Nucleic Acids Res. 2020;48:63–74. doi: 10.1093/nar/gkz1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brill W.K.D., Nielsen J., Caruthers M.H. Synthesis of deoxydinucleoside phosphorodithioates. J. Am. Chem. Soc. 1991;113:3972–3980. [Google Scholar]
- 13.Brill W.K.D., Tang J.Y., Ma Y.X., Caruthers M.H. Synthesis of oligodeoxynucleoside phosphorodithioates via thioamidites. J. Am. Chem. Soc. 1989;111:2321–2322. [PubMed] [Google Scholar]
- 14.Caruthers M.H., Beaton G., Cummins L., Dellinger D., Graff D., Ma Y.X., Marshall W.S., Sasmor H., Shankland P., Van Wu J., et al. Chemical and biochemical studies with dithioate DNA. Nucleosides Nucleotides. 1991;10:47–59. [Google Scholar]
- 15.Tonkinson J.L., Guvakova M., Khaled Z., Lee J., Yakubov L., Marshall W.S., Caruthers M.H., Stein C.A. Cellular pharmacology and protein binding of phosphoromonothioate and phosphorodithioate oligodeoxynucleotides: a comparative study. Antisense Res. Dev. 1994;4:269–278. doi: 10.1089/ard.1994.4.269. [DOI] [PubMed] [Google Scholar]
- 16.Cummins L., Graff D., Beaton G., Marshall W.S., Caruthers M.H. Biochemical and physicochemical properties of phosphorodithioate DNA. Biochemistry. 1996;35:8734–8741. doi: 10.1021/bi960318x. [DOI] [PubMed] [Google Scholar]
- 17.Vaughn J.P., Stekler J., Demirdji S., Mills J.K., Caruthers M.H., Iglehart J.D., Marks J.R. Inhibition of the erbB-2 tyrosine kinase receptor in breast cancer cells by phosphoromonothioate and phosphorodithioate antisense oligonucleotides. Nucleic Acids Res. 1996;24:4558–4564. doi: 10.1093/nar/24.22.4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wiesler W.T., Caruthers M.H. Synthesis of phosphorodithioate DNA via sulfur-linked, base-labile protecting groups(1) J. Org. Chem. 1996;61:4272–4281. doi: 10.1021/jo960274y. [DOI] [PubMed] [Google Scholar]
- 19.Cheng X., DeLong R.K., Wickstrom E., Kligshteyn M., Demirdji S.H., Caruthers M.H., Juliano R.L. Interactions between single-stranded DNA binding protein and oligonucleotide analogs with different backbone chemistries. J. Mol. Recognit. 1997;10:101–107. doi: 10.1002/(SICI)1099-1352(199703/04)10:2<101::AID-JMR344>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 20.Grandas A., Marshall W.S., Nielsen J., Caruthers M.H. Synthesis of deoxycytidine oligomers containing phosphorodithioate linkages. Tetrahedron Lett. 1989;30:543–546. [Google Scholar]
- 21.Bjergarde K., Dahl O. Solid phase synthesis of oligodeoxyribonucleoside phosphorodithioates from thiophosphoramidites. Nucleic Acids Res. 1991;19:5843–5850. doi: 10.1093/nar/19.21.5843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Capaldi D.C., Cole D.L., Ravikumar V.T. Highly efficient solid phase synthesis of oligonucleotide analogs containing phosphorodithioate linkages. Nucleic Acids Res. 2000;28:E40. doi: 10.1093/nar/28.9.e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li N.S., Frederiksen J.K., Piccirilli J.A. Automated solid-phase synthesis of RNA oligonucleotides containing a nonbridging phosphorodithioate linkage via phosphorothioamidites. J. Org. Chem. 2012;77:9889–9892. doi: 10.1021/jo301834p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang X. Solid-phase synthesis of oligodeoxynucleotide analogs containing phosphorodithioate linkages. Curr. Protoc. Nucleic Acid Chem. 2016;66:4–71. doi: 10.1002/cpnc.13. 1–4.71.14. [DOI] [PubMed] [Google Scholar]
- 25.Yang X. Solid-phase synthesis of RNA analogs containing phosphorodithioate linkages. Curr. Protoc. Nucleic Acid Chem. 2017;70:4–77. doi: 10.1002/cpnc.40. 1–4.77.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jaroszewski J.W., Clausen V., Cohen J.S., Dahl O. NMR investigations of duplex stability of phosphorothioate and phosphorodithioate DNA analogues modified in both strands. Nucleic Acids Res. 1996;24:829–834. doi: 10.1093/nar/24.5.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furrer P., Billeci T.M., Donati A., Kojima C., Karwowski B., Sierzchala A., Stec W., James T.L. Structural effect of complete [Rp]-phosphorothioate and phosphorodithioate substitutions in the DNA strand of a model antisense inhibitor-target RNA complex. J. Mol. Biol. 1999;285:1609–1622. doi: 10.1006/jmbi.1998.2305. [DOI] [PubMed] [Google Scholar]
- 28.Krieg A.M., Matson S., Fisher E. Oligodeoxynucleotide modifications determine the magnitude of B cell stimulation by CpG motifs. Antisense Nucleic Acid Drug Dev. 1996;6:133–139. doi: 10.1089/oli.1.1996.6.133. [DOI] [PubMed] [Google Scholar]
- 29.Yang X., Sierant M., Janicka M., Peczek L., Martinez C., Hassell T., Li N., Li X., Wang T., Nawrot B. Gene silencing activity of siRNA molecules containing phosphorodithioate substitutions. ACS Chem. Biol. 2012;7:1214–1220. doi: 10.1021/cb300078e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pallan P.S., Yang X., Sierant M., Abeydeera N.D., Hassell T., Martinez C., Janicka M., Nawrot B., Egli M. Crystal structure, stability and Ago2 affinity of phosphorodithioate-modified RNAs. RSC Adv. 2014;4:64901–64904. [Google Scholar]
- 31.Wu S.Y., Yang X., Gharpure K.M., Hatakeyama H., Egli M., McGuire M.H., Nagaraja A.S., Miyake T.M., Rupaimoole R., Pecot C.V., et al. 2′-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat. Commun. 2014;5:3459. doi: 10.1038/ncomms4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang X., Fennewald S., Luxon B.A., Aronson J., Herzog N.K., Gorenstein D.G. Aptamers containing thymidine 3'-O-phosphorodithioates: synthesis and binding to nuclear factor-kappaB. Bioorg. Med. Chem. Lett. 1999;9:3357–3362. doi: 10.1016/s0960-894x(99)00600-9. [DOI] [PubMed] [Google Scholar]
- 33.Yang X., Bassett S.E., Li X., Luxon B.A., Herzog N.K., Shope R.E., Aronson J., Prow T.W., Leary J.F., Kirby R., et al. Construction and selection of bead-bound combinatorial oligonucleoside phosphorothioate and phosphorodithioate aptamer libraries designed for rapid PCR-based sequencing. Nucleic Acids Res. 2002;30:e132. doi: 10.1093/nar/gnf132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abeydeera N.D., Egli M., Cox N., Mercier K., Conde J.N., Pallan P.S., Mizurini D.M., Sierant M., Hibti F.E., Hassell T., et al. Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016;44:8052–8064. doi: 10.1093/nar/gkw725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang X., Dinuka Abeydeera N., Liu F.W., Egli M. Origins of the enhanced affinity of RNA–protein interactions triggered by RNA phosphorodithioate backbone modification. Chem. Commun. (Camb.) 2017;53:10508–10511. doi: 10.1039/c7cc05722a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dolot R., Lam C.H., Sierant M., Zhao Q., Liu F.W., Nawrot B., Egli M., Yang X. Crystal structures of thrombin in complex with chemically modified thrombin DNA aptamers reveal the origins of enhanced affinity. Nucleic Acids Res. 2018;46:4819–4830. doi: 10.1093/nar/gky268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghosh M.K., Ghosh K., Dahl O., Cohen J.S. Evaluation of some properties of a phosphorodithioate oligodeoxyribonucleotide for antisense application. Nucleic Acids Res. 1993;21:5761–5766. doi: 10.1093/nar/21.24.5761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okruszek A., Sierzcha-La A., Fearon K.L., Stec W.J. Synthesis of oligo(deoxyribonucleoside phosphorodithioate)s by the dithiaphospholane approach. J. Org. Chem. 1995;60:6998–7005. [Google Scholar]
- 39.Straarup E.M., Fisker N., Hedtjärn M., Lindholm M.W., Rosenbohm C., Aarup V., Hansen H.F., Ørum H., Hansen J.B., Koch T. Short locked nucleic acid antisense oligonucleotides potently reduce apolipoprotein B mRNA and serum cholesterol in mice and non-human primates. Nucleic Acids Res. 2010;38:7100–7111. doi: 10.1093/nar/gkq457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sewing S., Boess F., Moisan A., Bertinetti-Lapatki C., Minz T., Hedtjaern M., Tessier Y., Schuler F., Singer T., Roth A.B. Establishment of a predictive in vitro assay for assessment of the hepatotoxic potential of oligonucleotide drugs. PLoS One. 2016;11:e0159431. doi: 10.1371/journal.pone.0159431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seth P.P., Swayze E.E. In: Advances in Nucleic Acid Therapeutics. Agrawal S., Gait M.J., editors. The Royal Society of Chemistry; 2019. The medicinal chemistry of RNase H-activating antisense oligonucleotides; pp. 32–61. [Google Scholar]
- 42.Bleicher K., Duschmale J., Duschmale M.B., Hansen H.F., Funder E., Koch T., Li M., Schaeublin A., Shu X., Wu Y. 2019. Gapmer Oligonucleotides Comprising a Phosphorodithioate Internucleoside Linkage. WO 2019122282 A1. [Google Scholar]
- 43.Bleicher K., Duschmale J., Duschmale M.B., Hansen H.F., Koch T., Li M., Schaeublin A., Shu X., Wu Y. 2019. Novel Thiophosphoramidites. WO 2019122277 A1. [Google Scholar]
- 44.Bleicher K., Hansen H.F., Koch T., Albaek N., Funder E.D. 2019. Methods for Identifying Improved Stereodefined Phosphorothioate Oligonucleotide Variants of Antisense Oligonucleotides Utilising Sub-libraries of Partially Stereodefined Oligonucleotides. WO 2019073018 A1. [Google Scholar]
- 45.Husser C., Brink A., Zell M., Muller M.B., Koller E., Schadt S. Identification of GalNAc-conjugated antisense oligonucleotide metabolites using an untargeted and generic approach based on high resolution mass spectrometry. Anal. Chem. 2017;89:6821–6826. doi: 10.1021/acs.analchem.7b01244. [DOI] [PubMed] [Google Scholar]
- 46.Husser C., Koller E., Brink A., Schadt S. Studying the biotransformation of phosphorothioate-containing oligonucleotide drugs by LC-MS. Methods Mol. Biol. 2019;2036:307–315. doi: 10.1007/978-1-4939-9670-4_18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.