

Abstract
The use of monoclonal antibodies represents an important and efficient diagnostic and therapeutic tool in disease management and modern science but remains limited by several factors including the uneven distribution in diseased tissues as well as undesired activation of side immune reactions. Major scientific advancements including Recombinant DNA Technology, Hybridoma Technology, and Polymerase Chain Reaction have considerably impacted the use of monoclonal antibodies providing technical and effective solutions to overcome the shortcomings encountered with conventional antibodies. Initially, the introduction of antibody fragments allowed a more uniform and deeper penetration of the targeted tissue and reduced unwanted activation of Fc-mediated immune reactions. On another level, the immunogenicity of murine-derived antibodies was overcome by humanizing their encoding genes with specific sequences of human origin andtransgenic mice able to synthesize fully human antibodies were successfully created. Moreover, the advancement of genetic engineering techniques supported by the modular structure of antibody coding genes paved the way for the development of a new generation of antibody fragments with a wide spectrum of monospecific and bispecific agents. These later could be monovalent, bivalent, or multivalent, and either expressed as a single chain, assembled in multimeric forms or stringed in tandem. This has conferred improved affinity, stability, and solubility to antibody targetting. Lately, a new array of monoclonal antibody fragments was introduced with the engineering of nanobody and antibody mimetics as non-immunoglobulin-derived fragments with promising diagnostic and therapeutic applications. In this review, we decipher the molecular basis of monoclonal antibody engineering with a detailed screening of the antibody derivatives that provides new perspectives to expand the use of monoclonal fragments into previously unexplored fields.
Keywords: Monoclonal antibodies, MABS fragments, Humanized antibodies, Xenomice, Single chain fragment variable, Diabodies, Minibodies, Nanobodies, Antibodies mimetics
1. Introduction
The very first report denoting the term «antibodies» emanated from Emil von Behring and Shibasabura Kitasato's work in 1890[1]. In their milestone publication, they established the concept of passive immunization. describing the use of serum drained from diphtheria-inoculated horses that provides a back then novel treatment modality for infected animals. It was an early insight in the potential use of antibody-rich serum as neutralizing agents (antitoxins) in clinical applications.One decade later von Behring was awarded the Nobel Prize in Physiology and Medicine in 1901for his scientific contributions[1], [2], [3].
Despite a long century of focused research The chemical nature and the mode of action of the so called «antibodies» (Fig. 1) remained ambiguous for many years to come after that. In 1959, Edelman demonstrated that antibodies are composed of separate chains of proteins joined together by sulfur bonds forming crosslinks. Porter showed that when these chains were treated enzymatically with papain, theycould be separated into three fragments Two of these fragments maintain their capacity to bind the antigens (weighing between 50 and 55 KDa each), while the third (about 80 KDa) lacks this ability[4], [5], [6]. In 1963, Porter's team projected a new feature of the multi-subunit polypeptide forming a Y-shaped complex assembled through five inter-chain disulphide bridges maintaining the tetrameric protein[7].
Fig. 1.

Schematic representation of a full length antibody composed of a pair of two identical heavy chains (H) assembled with two identical light chains (L). Heavy chains are composed of one variable domain (VH) and three constant domains (CH1, CH2, and CH3). The light chains consist of one constant domain (CL) and one variable domain (VL). CH1 and CH2 domains are separated by a flexible hinge region connected by disulphide bridges.
Another significant milestone in our understanding and use of antibodies comes from Dreyer and Bennett.They described the molecular concept of the variable (V) and constant (C) domains production as a result of the genetic expression of two similar but not identical genes assembled together by the previously identified disulphide bonds[8]. Further details were provided by Hood and Ein who established the famous statement “two genes, one polypeptide chain”, demonstrating that the immunoglobulin Lambda light chain is encrypted by two distinct genes whose expression results in the synthesis of a single polypeptide chain[9]. Based on the comparison of aminoacid termini of about 64 light chains, Hood and Talmage suggested that much of the antibodies’ diversity could be rooted back the germ line variability in addition to the genetic recombination at the level of the gene families[10]. The complete sequence of a gamma-globulin was first reported by Edelman showing the presence of the variable (V) and constant (C) regions in both structures of the Heavy (H) and Light (L) antibody chains[5]. In order to acquire the functional sequence of the immunoglobulin gene, Hozumi and Tonegawa demonstrated that both the variable and constant gene segments, were permissive to undergo a process of somatic hypermutation leading to the rearrangements of the complete functional gene[11,12].
The last three decades of the twentieth century witnessed major scientific breakthroughs that marked that start of unprecedented advancement of both science and medicine. In 1972, Stanley Cohen and Herbert Boyer were first to apply for a patent on «Recombinant DNA Technology» a discovery that paved the road to the genetic engineering of many therapeutic proteins, including antibodies[13]. Later, in 1984, Jerne, Milstein and Kohler were jointly awarded the Nobel Prize in Physiology and Medicine for their development of the so-called «Hybridoma Technology» which allowed the in vitro mass production of «Monoclonal Antibodies» in cell cultures. The leading purpose of this technology is to maintain a constant supply of pure and highly specific monoclonal antibodies[14,15]. In 1985, Karry Mullis shared the Nobel Prize in Chemistry with Michael Smith for their pioneer invention of Polymerase Chain Reaction» or PCR molecular technique. This allowed the enzymatic synthesis of a large number of DNA sequences starting from a limited number of copiesusing a thermocycler in the presence of free nucleotides, a pair of forward and reverse primers and a thermostable DNA polymerase [16,17].
With the establishment of the various modern molecular techniques, the need for «Monoclonal Antibodies» as a very efficient tool in the molecular diagnosis and the treatment of a large spectrum of illnesses and diseases has increased [18], [19], [20], [21]. The first FDA approved therapeutic monoclonal antibody (mAb) was designed to function as an immuno-suppressive agent used to manage post-transplantation rejections of kidney transplants[22]. Muromonab-CD3, the so-named Orthoclone (OKT3) is an IgG-2a fully murine monoclonal antibody. It is directed against CD3 which isa cluster of differentiation of about 20 KDa, representing a part of a larger macromolecular complex expressed at the surface of mature T cells and the medullary thymic lymphocytes in the proximity of the TCR chains. However, due to its increased immunogenicity, and clinical side effects (Fever, thrombotic disorders, dermatitis, and anaphylactic shock), the commercial marketing and production of murine OKT3 hawas discontinued as of July 30th, 2011[23,24].
The modular nature of the antibody gene clusters coding for the constant and variable domains of the heavy and light chains allowed molecular biotechnologists to efficiently manipulate the murine and human gene segments. This has resulted in constructing hybrid entities that maintain the ability to specifically bind antigens with a lower immunogenicity and higher therapeutic efficiency. Novel monoclonal antibody “designers” apportioned the utmost level of consideration to reduce immunogenicity, and promote the therapeutic effect at the best of specificity, safety, and optimal distribution[25]. The modern improvements in genetic engineering initiated the emergence of a spectrum of immunoglobulins conjugates, such as immunotoxins, multivalent fragments and multispecific complexes that conferred an extended efficacy as therapeutic immunoglobulins. Hundreds of newly developed therapeutic antibodies and their derivatives are currently under clinical investigations before being approved by FDA and industrialized for global marketing[26].
The design of therapeutic monoclonal antibodies is optimized to target a large spectrum of oncological illnesses and malignancies. Besides, a number of these antibodies are currently used for the management of hematological disorders, inflammatory diseases, organ transplantation, autoimmune diseases, prevention of migraine and recently for the treatment of COVID-19 patients[27]. Since the discovery and the approval of murine Muromonab (OKT3) in 1986, scientific and industrial efforts were directed towards reducing the side effects of murine antibodies characterized by their high immunogenicity[28]. Consequently, humanized antibodies appeared for the first time in 1997 with an FDA approved anti-IL-2 Receptor antibody (Daclizumab) designed for the prevention of organ transplant rejection[29]. Moreover, fully humanized antibodies generated by transgenic mice marked a revolution in mAbs production when the first anti-Epidermal Growth factor Receptor (EGFR) was approved by FDA (Panitumumab) to become available in the market as of 2006 and recommended for the treatment of metastatic colorectal cancer[30,31]. A fully humannized anti-TNF-α monoclonal antibody (Adalimumab, Humira) was designed to block the interaction between the pro-inflammatory cytokine and its transmembrane and/or soluble receptor. The subcutaneously administrated mAb (Humira) represents a disease modifier with its ability to block the signaling pathway reducing the TNF-mediated reactions associated with rheumatoid arthritis and other inflammatory conditions such as IBD[32]. Adalimumab was FDA approval in 2002 as an effective drug recommended, not only for RA, but also for the management of a spectrum of chronic inflammatory diseases (i.e. psoriatic arthritis, ankylosing spondylitis, Crohn's disease, ulcerative colitis, psoriasis, hidradenitis suppurativa, uveitis, and juvenile idiopathic arthritis)[33,34]. Nivolumab and Pembrolizumab (Opdivo and Keytuda respectively) are both directed against the PD-1 receptor intended to block the interaction with its ligand underlying the immune checkpoints involved in the inhibition of tumor progression[35,36]. Monoclonal antibodies which block the Calcitonin Gene-Related Peptide (CGRP) were known to be effective in reducing migraine episodes. For that purpose, three different constructs, namely Erenumab (Aimovig), Galcanezumab (Emgality), and Fremaezumab (Ajovy) were approved to prevent migraine-related pain episodes[37].
2. Adverse effects of full-length monoclonal antibodies
Several factors contribute to immunogenicity of monoclonal antibodies. These include the chemical nature of the relatively conserved and variable amino acids in the primary structure, the three dimensional conformation, the animal species or the hybridoma host cells used the molecular mass of the protein complex, and the diversity of the heavy and light chains binding sites [38].
IgG antibodies are perceived as large multimeric complexes (dimers of dimers) of about 150 KDa which limits their penetration capacity into solid tissues and tumor masses. The molecular size of these immunoglobulins hinders their ability to cross the blood-brain barrier which restricts their interaction with the cellular targets of the Central Nervous System[39]. Functionally, these antibodies possess two highly specific antigen binding sites (bivalent and monospecific) carried by the uppermost variable domains of the heavy and light chains held together by a highly conserved, biologically active fragment, named Fc region. The presence of this later maintains an extended circulation time with penury of target tissue concentration which leads to the reduction of its therapeutic effectiveness[19,39,40]. Although monoclonal antibodies are commonly tolerated by the human body, their adverse effects were reported at many instances. This mounts significant side effects after instillation and/or injection of monoclonal antibodies, some of which are summarized in Table 1. For instance, the anti-TNF-α antibodies were shown to induce an autoimmune activity leading to a lupus-like syndrome associated with anti-nuclear antibodies and anti-dsDNA antibodies[41,42]. Yet another monoclonal antibody directed towards the cytotoxic T-Lymphocyte associated antigen 4 (CTLA4) could stand behind the development of autoimmune colitis[43,44]. Cardio-toxicity has been clinically documented in patients treated with ERBB2 monoclonal antibodies[45,46]. The antithrombotic agents directed against the antiplatelet glycoprotein IIb and IIIa caused an immune thrombocytopenia in patients with high risk of developing spontaneous thrombosis[47,48]. Furthermore, an intense cytokine storm has been released following the infusion of an anti-CD28SA antibody subsequently leading to a full revision of the treatment protocols and the adoption of additional measures aiming at improving the safety of the clinically approved monoclonal antibodies[49].
Table 1.
Clinical side effects associated with the more commonly used monoclonal antibodies.
| Monoclonal Antibody | Trade Name | Product's Type | Hypersensitivity | Immunogenicity | Immunosuppression | Infusion Reactions | Infections | Anemia | Cytopenia | Autoimmunity | Coagulation Disorders | Heart Failure | Malignancy | Others |
| Infliximab | Remicade | Chimeric | + | + | + | + | + | + | + | + | + | Increased Liver Enzymes | ||
| Basiliximab | Simulect | Chimeric | +++ | +++ | + | + | + | Skin Rash | ||||||
| Rituximab | Rituxan | Chimeric | + | + | +++ | Hypotension & Serum Sckness | ||||||||
| Cetuximab) | Erbitux | Chimeric | + | +++ | Urticaria & Bronchospasm | |||||||||
| Abciximab | ReoPro | Chimeric Fragment (Fab) | + | + | + | + | Bleeding | |||||||
| Adalimumab | Humira | Fully Human | + | + | + | + | + | + | + | + | + | Increased Liver Enzymes | ||
| Panitumumab | Vectibix | Fully human | + | + | Skin Rash & Diarrheae | |||||||||
| Alemtuzumab | Campath | Humanized | + | + | + | + | + | Thyroid Disorders | ||||||
| Daclizumab | Zenapax | Humanized | +++ | +++ | + | + | + | Cytokine Release Syndrome | ||||||
| Bevacizumab | Avastin | Humanized | + | + | + | + | Slow Wound Healing | |||||||
| Eculizumab | Soliris | Humanized | + | + | Meningitis | |||||||||
| Efalizumab | Raptiva | Humanized | + | +++ | + | + | Encephalitis | |||||||
| Natalizumab | Tysabri | Humanized | + | + | + | Hepatotoxicity | ||||||||
| Omalizumab | Xolair | Humanized | + | + | + | Injection Site Reactions | ||||||||
| Palivizumab | Synagis | Humanized | + | Apnea & Fever | ||||||||||
| Trastuzumab | Herceptin | Humanized | + | + | Skin Reactions & Cardiotoxicity | |||||||||
| Tocilizumab | Actemra | Humanized | + | +++ | + | Anaphylais & Headache | ||||||||
| Ranibizumab | Lucentis | Humanized Fragment (Fab) | + | Ocular Inflammation | ||||||||||
| Muromonab | CD3 | Mouse | + | + | + | + | Hepatitis | |||||||
| Certolizumab | Cimzia | Pegylated Humanized | + | + | + | + | + | + | + | + | + | Increased Liver Enzymes |
The glycosylation of monoclonal antibodies which involves a multi-layered system of poly-enzymatic reactions, has also proved to impact the antigen binding ability, biosafety, and biological activity of monoclonal antibodies[50]. This has shown to affect the immunogenicity of the antibodies, their vascular half-life and subsequently the purposes for which they are produced. In contrast to the hyper-variable domains of the Fab region involved in the recognition and binding of the antigenic determinants, the Fc region has a highly conserved structure with limited diversity[51]. This consensus sequence of the heavy chain encloses two conserved N-glycosylation sites located ate the Asn297 on each of the paired chains. Site directed modifications of these N-glycans have been shown to hinder the conformation leading to the hampering of the IgG with molecules of the immune system and may also hamper the activation of the complement system[52]. Glycation, deamination, acetylation, and/or oxidation are among the more common post-translational biochemical mechanisms that alter the immunogenicity of monoclonal antibodies[53,54].
3. Transition from murine to humanized monoclonal antibodies
The continuous expansion of molecular manipulation, DNA sequencing and recombinant DNA technologies allowed the design of a novel system, including a combination of rodent and human antibody coding sequences and resulting in the synthesis of the so-called Chimeric Monoclonal Antibodies (Fig. 2B)[55]. Chimerization, first proposed by Morrison in 1980′s, aimed at decreasing the mAbs immunogenicity by grafting murine variable sequences of the heavy and light chains into human antibody coding sequences generating novel proteins with about 70% of human content[56]. Before being amplified by PCR, the hybridoma extracted genes, encoding for the rodent variable regions are separated. The amplification is further used to yield multiple copies of the human fraction of the heavy and light chains constant regions. Resulting PCR products, including murine and human sequences are inserted into a shuttling plasmid used to transfect a prokaryotic host strain. The chimeric proteins are collected in the form of cytoplasmic inclusions that are purified and tested in vivo following cell lysis. Limiting factors include aggregation of cytoplasmic recombinant proteins in the form of inclusion bodies as well as the limited growth rate of transformed bacteria [57]. Anti-GPIIb-IIIa antibody (abciximab), directed against integrin receptors of the platelets surface was approved by FDA as an antithrombotic agent that inhibits platelet aggregation in patients with a high risk of cardiovascular illnesses[58,59]. In oncology, Rituximab, a chimeric recombinant IgG1 antibody, targeting the CD20 at the surface of cancer T cells was FDA approved and indicated for the management of non-Hodgkin's Lymphoma by the end of 1997[60,61]. Although chimerization is a major advancement in the optimization of monoclonal antibodies, they still hold a considerable fraction of the rodent immunoglobulins which clinically imposes further focus on reducing their immunogenicity.
Fig. 2.

(A) Murine Antibody compared to genetically engineered antibodies shown in B and C. Chimeric Antibody (B): The coding genes of the murine heavy and light chains variable regions were replaced by the human sequences yielding a chimeric protein with rodent binding sites. Humanized Antibody (C): The murine derived Complementary Determining Regions (CDR1, CDR2, and CDR3) are used to replace the human CDRs generating a protein with human properties and murine antigen binding site. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The molecular illustration of the variable region holding within the antigen binding site demonstrated the presence of two anti-parallel β-sheets assembled together by a disulphide bridge. The comparison of the heavy and light chains sequences revealed the presence of three distinct hypervariable regions termed Complementary Determining Regions (CDRs) surrounded by four relatively conserved framework regions (FRs) that constitute the majority of the β-sheets[62,63]. It has been suggested that the Framework Regions establish a scaffolding platform on which the proximate CDRs build up the antigen binding sites outside the context of the FRs[64,65]. Jones and his colleagues published the first work that elaborated the protocol of human CDRs substitution with those of the murine antibodies. The algorithm of antibody humanization encompasses the grafting of heavy and light chain murine-based CDRs into a human myeloma protein. They reported that the novel protein acquired the same affinity towards the hapten concluding that the CDR substitution offered an efficient mean to construct a “Humanized Antibody” (Fig. 2C)[65].
4. Advantages of antibody fragments and their derivatives
A substantial advancement in the arena of immunotherapy and immunoassays designed for diagnostic purposes has been evoked by the development of smaller, but more efficient, antibody-derived fragments that maintained the antigen binding capacity of conventional antibodies. Facilitated by the modular structure of immunoglobulin coding genes, a large spectrum of antibody fragments emerged in the last decades including the Fragment for Antigen Binding (Fab), the Single Chain Fragment Variable (scFv), the Crystallizable Fragment (Fc), and the Single-Domain Immunoglobulins[66]. Anticipated with great hope, the cloning techniques in conjunction with genetic engineering of therapeutic proteins allowed biotechnologists to puzzle the gene segments resulting in a wide range of agents with dual or multiple specificities (bispecific and multi-specific), composed of paired or multimeric domains and providing a diverse spectrum of biological activities[67]. Antibody fragments, unlike conventional full length monoclonal antibodies, are not glycosylated which facilitated their synthesis in bacteria[68]. The ease of their production in prokaryotic host cells used as microbial expression machineries, added to the fast growing rate of the microbes in inexpensive growth media results in a larger yield at a less expensive cost and makes them more advantageous than approved conventional antibodies[23]. Clinically, the use of small antibody fragments resulted in a relatively higher tissue penetration but then a faster renal clearance which increases the requirement for higher effective doses and boosting the frequency of their administration unless alleviated by pegylation, polysialylation, or by fusion and conjugation with serum albumin[66,69]. The small size of such recombinant therapeutics has been shown to be very advantageous. Some of them acquired a more efficient binding capacity to the receptors of their targets especially those that are inaccessible to the full length antibodies encumbered by their large size and three dimensional conformations. Some derivatives are designed to have multitude of identical binding sites while some others are potentially able to bind two or even more antigenic determinants[20,70].
5. Molecular mechanisms of antibodies and antibody fragments production
Conventionally, antibody fragments are produced using recombinant DNA technologies to transform special bacterial strains with shuttling plasmid(s) bearing the coding genes. The antibody fragments encoding genes are cloned on autonomous, self-replicating plasmids inserted at a relatively high copy number. The genes of interest are traditionally placed under the control of strong, regulated promoter in order to control the timing of their induction and the level of their expression. Lac promoter, Trp-promoter, the hybrid Tac promoter (Lac & Trp) and bacteriophage T7 promoter are commonly used as they are highly manageable and fully controllable[68]. Much work was directed during the last years towards optimizing the protocols of protein production at the industrial level. Optimal strategies were not limited to upgrading of the transcription process but encompassed also the tuning of the translation and the post-translational modifications required for a protein to acquire its biological activity. A number of modifications were proposed to promote the stability of nascent proteins either in the cytoplasm or the periplasm. A number of strains were genetically modified in order to secrete the native form of the antibody fragments as membrane-bound or extracellular which facilitated their separation and then their purification[71], [72], [73]. For that purpose, a number of “optimal host cell” models were developed using prokaryotic cells (especially Escherichia coli), yeast cell strains (Saccharomyces cerevisiae, Pichia pastoris), or mammalian cell lines (Chinese Hamster Ovary cells (CHO), Human Embryonic Kidney cells (HEK), and Murine Myeloma Cells)[68,74,75]. For these later, many molecular aspects were subject to continuous improvement in the aim of increasing the success rate of their use at the industrial level. Among each other, one can include the position of the ribosomal binding site and its affinity to the translation factors, the number of antibody fragments cloned genes, the localization of the nascent peptide fragment either in the cytoplasm, or in the periplasm, at the surface of the cell membrane or even secreted in the medium which affects their intrinsic stability and purity. Some host cells showed an overwhelmed metabolism altering the efficiency of the antibody production. Metabolic burden has been related to a multitude of factors including the high plasmid copy number, the large-sized vectors, the overproduction of the proteins, the synthesis of degrading enzymes and the depletion of oxygen[76,77]. Technical and molecular adjustments are continuously being adopted to increase translation efficiency, facilitate the folding of the different subunits, enhance the antibody fragment stability, decrease the degradation of cytoplasmic aggregates, reduce the formation of inclusion bodies, encourage protein secretion and facilitate purification[71,77,78]. Recently, insight gained from cell lysates (E. coli, CHO cells, insect cells) was used to develop the well-established Cell-Free systems. These later were shown to be more flexible and reliable providing fast and scalable machinery for antibody fragments production. The total absence of the phospholipid bilayer of the plasma membrane allowed the direct addition of molecular resources facilitating the synthesis of antibody peptides such as ATP molecules, free amino acids, the bacterial disulfide bridge isomerase C (dsbC) and the chaperone proteins. Although still not fully functional, these cell free systems are expected to be, once fully optimized, preferably adopted for antibody fragments production[79,80]. An alternative technology to generate antibody fragments, known as «Phage Display» of combinatorial antibody fragments genes has been explored in the early 1990s. For that purpose, a wide range of variable cDNA coding sequences collected from antibody producing cells or engineered scFv combinatorial sequences of VH and VL are expressed at the surface of M13 filamentous bacteriophage. The expressed scFvs are panned after reacting with antigen-coated plates to select the chains of interest that have their affinity apt to increase through a chain of recurrent mutations of the CDR coding sequences and the subsequent panning steps which also allows the bypassing of technical difficulties encountered with hybridoma techniques[81], [82], [83].
6. Mechanism of action elaborated by different approved monoclonal antibodies
Monoclonal antibody mediated cancer immunotherapy is generally based on apoptotic activation induced by a number of pro-apoptotic factors mediating cancer cell death or through a number of diverse factors blocking the survival signaling pathways hijacked by dividing tumor cells[84,85]. For example, Trastuzumab is directed against HER2 receptors which are expressed by breast cancer cells, inhibiting the cellular proliferation and challenging their survival[86] while Cetuximab blocks the Epidermal Growth factor Receptor (EGFR) signaling pathways in colorectal lung tumor cells, blocking pro-growth signals and retarding tumor growth[87,88]. Moreover, a multitude of mechanisms of antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC) and antibody-dependent cellular phagocytosis (ADCP) are induced by the targeted cancer cells (Table 2)[89]. Relapses in therapeutic antibody treatment and ineffective outcomes are related to a number of prevalent cross-talks among intra and intercellular signaling pathways that hinder the expected immune functions[90,91]. In contrast, bispecific antibodies are bivalent molecules able to crosslink two different epitopes simultaneously at the surface of the targeted tumor cell and the effector cytotoxic cell which reduces the needed dosage and alleviates the subsequent side effects[92,93]. Blinatumomab is an example of an approved bispecific antibody in cancer therapy, which targets two different clusters, namely CD3 and CD19, and is intended to treat the Philadelphia Chromosome negative B cell acute lymphoblastic leukemia (ALL)[94]. Moreover, Emicizumab is another bispecific mAbs which conjugates the clotting factor IXa and X and is envisioned for the treatment of Hemophilia A[95]. In order to overcome the incapacity of full length antibodies to recruit effector T cells, BiTEs were designed to target expressed tumor markers simultaneously with TCR-associated CD3 which efficiently contributes to the transduction of specific signals leading to the activation and proliferation of CD669+ and CD25+ T cells[96]. The aforementioned activation of T cells promotes the so-called BiTE-mediated tumor lysis[97,98]. Induced T cells secrete substantial amount of cytolytic proteins termed perforin and granzymes at the level of the immunological synapses resulting in the perforation of cancer cell membranes. Endocytosed perforins and granzymes are sequestered in cytosolic endosomes whose membranes are disrupted by the cytolytic activity of perforins leading to the release of granzymes and the subsequent lysis of the cancer cells[99,100].
Table 2.
Examples of FDA Approved Monoclonal Antibodies along with their Clinical Indications and Mechanism of Action.
| Monoclonal Antibody (Trade Name) | mAbs & Target Ag | Tumor disease | ADCP | ADCC | CDC | RND | CDD | SB |
|---|---|---|---|---|---|---|---|---|
| Rituximab (Rituxan) | Chimeric anti-CD20 | Chronic & Non-Hodgkin's lymphoma | + | + | + | |||
| Ofatumumab (Arzerra) | Human anti-CD20 | Chronic lymphocytic leukemia | + | + | + | |||
| Ibritumomab tiuxetan (Zevalin) | Murine anti-CD20 | Non-Hodgkin's lymphoma | + | |||||
| Tositumomab-I131 (Bexxar) | Murine anti-CD20 | Non-Hodgkin's lymphoma | + | |||||
| Obinutuzumab (Gazyvaro) | Humanized anti-CD20 | Chronic lymphocytic leukemia | + | + | ||||
| Alemtuzumab (Campath) | Humanized anti-CD52 | B-cell chronic lymphocytic leukemia | + | + | + | |||
| Tafasitamab (Monjuvi) | Humanized anti-CD19 | Diffuse large B-cell lymphoma | + | + | + | |||
| Loncastuximab tesirine (Zynlonta) | Humanized anti-CD19 | Diffuse large B-cell lymphoma | + | |||||
| Polatuzumab vedotin (Polivy) | Humanized anti-CD79b | Diffuse large B-cell lymphoma | + | |||||
| Daratumumab (Darzalex) | Human anti-CD38 | Multiple Myeloma | + | + | + | |||
| Isatuximab (Sarclisa) | Chimeric anti-CD38 | Multiple Myeloma | + | + | + | |||
| Mogamuizumab (Poteligeo) | Humanized anti-CCR4 | Cutaneous T cell lymphoma | + | + | + | |||
| Gemtuzumab ozogamicin (Mylotarg) | Humanized anti-CD33 | Acute myeloic leukemia (AML) | + | |||||
| Inotuzumab ozogamicin (Besponsa) | Humanized anti-CD22 | Acute lymphoblastic leukemia | + | |||||
| Moxetumomab pasudotox (Lumoxiti) | Murine anti-CD22 | Hairy cell leukemia | + | |||||
| Belantamab mafodotin (BLENREP) | Humanized anti-BCMA | Multiple Myeloma | + | |||||
| Brentuximab vedotin (Adcetris) | Chimeric anti-CD30 | Hodgkin lymphoma (HL) | + | |||||
| Elotuzumab (Elotuzumab) | Humanized anti-SLAMF7 | Multiple Myeloma | + | + | + | |||
| Trastuzumab (Herceptin) | Humanized anti-HER2 | Breast cancer; metastatic adenocarcinoma | + | + | ||||
| Ado-Trastuzumab emtansine (Kadcyla) | Humanized anti-HER2 | Breast cancer | + | |||||
| [fam]-trastuzumab deruxtecan (Enhertu) | Humanized anti-HER2 | Breast cancer | + | |||||
| Pertuzumab (Perjeta) | Humanized anti-HER2 | Breast cancer | + | + | + | |||
| Margetuximab (Margenza) | Chimeric anti-HER2 | Breast cancer | + | + | ||||
| Cetuximab (Erbitux) | Chimeric anti-EGFR | Head and neck cancer; colorectal cancer | + | + | + | |||
| Panitumumab (Vectibix) | Human2 anti-EGFR | Metastatic colorectal carcinoma | + | |||||
| Necitumumab (Portrazza) | Human anti-EGFR | Carcinoma, non-small-cell lung | + | + | ||||
| Dinutuximab (Unituxin) | Chimeric anti-GD2 | Neuroblastoma | + | + | ||||
| Naxitamab (Danyelza) | Humanized anti-GD2 | Neuroblastoma | + | + | + | |||
| Enfortumab vedotin (Padcev) | Human anti-Nectin-4 | Urothelial cancer | + | |||||
| Sacituzumab govitecan (Trodelvy) | Humanized anti-TROP-2 | Breast cancer | + |
ADCP: Antibody-Dependent-Cellular-Phagocytosis; ADCC: Antibody-Dependent-Cellular-Cytotoxicity; CDC: Complement Dependent Cytotoxicity; RND: Radio-Nucleotide Delivery; CDD: Cytotoxic Drug Delivery; SB: Signal Blockade.
7. Antibody drug conjugates
In order to compensate the inefficiency of monoclonal antibody-mediated cell toxicity using naked mAbs, a novel design, termed Antibody-Drug Conjugate (ADC) has been developed [101,102]. Each ADC is composed of a target-specific monoclonal antibody, chemically linked to a highly potent cytotoxic payload which synergistically enhances the therapeutic ratio of the construct[103]. Conventionally, an effective ADC should maintain its serum stability, readily reach its target cell, and ultimately release the associated cytotoxic payload in the proximity of the target cell[102,104]. Ideally, the target tumor marker should be expressed at the surface of the cell rather than being intracellular, soluble, or secreted in the blood circulation which risks inciting the cytolysis to take place outside the context of the tumor site. In order to engender the highest cytotoxic effect, the ADCs should be internalized by receptor mediated endocytosis followed by the release of the cytotoxic payload causing the cell death by apoptosis to be induced as a result of the cytoskeletal microtubules and DNA alteration[105], [106], [107]. During the last two decades, the FDA approved 14 different ADCs designed for the treatment of a wide spectrum of hematological and non-hematological malignancies[108,109]. Recently, a novel format of the ADCs termed Antibody-Antibiotic Conjugates has been designed to counteract against bacterial infections as a novel modality to limit the emergence of antimicrobial resistance[110]. Beside their immunogenicity, first generation ADCs such as BR96-Doxorubincin failed to engender a significant rate of tumor cell death [111,112]. A second generation of ADCs has been developed to overcome the drawbacks of the first line of products. These novel designs (Bretuximab-Vedotin and Adotrastuzumab-Emtansine) carry smaller but more toxic, water soluble payloads with optimized linkers and enhanced tumor cell targeting capacity[113], [114], [115]. Although, significant improvements were reached, many purposes were unmet; essentially, ADC biased toxicity, aggregation and rapid clearance which paved the road for the third generation. Based on the technology of Site-Specific conjugation, innovative consistent formats were introduced in the market (Polatuzumab vedotin, Enfortumab vedotin, Fam-trastuzumab deruxtecan). These molecules provoke high anticancer activity with less off-target toxicity, establish improved pharmacokinetic features, and cause lower immunogenicity based on the use of fully human rather murine or chimeric entities[116].
8. Transgenic xenomice for human antibody production
In order to overcome the molecular difficulties associated with the Phage Display technology and to avoid the time-consuming conventions yielding low-affinity antibodies, a genetically engineered strain of mice in which the murine antibody coding genes were inactivated while a set of human heavy and light chains encoding sequences were incorporated into the transgenic mouse genome. This novel success owes in much to the success of the transgenesis technology using the Yeast Artificial Chromosome (YAC) as a shutter due to their extremely high transfer capacity into the murine germ line cells (Fig. 3). The YAC bearing human antibody coding genes are introduced into the mouse germ cells following the fusion of yeast spherocytes(devoid of cell wall) with embryonic stem cells (ESC). The aforementioned manipulation generates a large number of transgenic ESC where human coding genes are found to be integrated in a stable alliance with the chromosomal material. Some of the transfected cells used to produce mice carried a combination of the human and the murine immunoglobulin coding genes capable of producing both types of antibodies. Later, two of the resulting transgenic progeny, one carrying the two types of genes, the other with only the knocked-out murine DNA were crossbred producing a transgenic strain able to synthesize fully human immunoglobulins with great potential for approval as therapeutic monoclonal antibodies. Ongoing research is focused on growing up the repertoire of human antibodies and antibody fragments to be generated by this promising technology. Similarly, efforts are being devoted to introduce lambda coding genes into the mouse genome or inferring this technique in cattle using microcell-mediated gene transfer technology.
Fig. 3.

Genetic engineering of Xenomice. Murine antibody coding genes are disabled by site-directed mutagenesis. The mutated stem cells are used to create transgenic disarmed strains (left-up) that are unable to synthesize antibodies. Human heavy and light chains coding sequences were then cloned into Yeast Artificial Chromosome (YAC) and introduced into ESC used to yield another strain able to produce both, murine and human antibodies (right-up). These strains are crossbred and Xenomice able to synthesize human antibodies are screened and selected. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
9. Antibody fragments and related derivatives
9.1. Fragments of antigen binding (Fab & F(ab’)2)
The Fragment of Antigen Binding (Fab) was the first molecular format adopted as a therapeutic antibody fragment followed by a dozen of novel molecules that were respectively introduced in clinical trials starting from mid-1990s where they represented about 49% of therapeutic fragments under investigation[67]. Originally, Fab fragments could be easily generated by enzymatic cleavage of IgG using papain, a protease that cleaves the heavy chains at the sequence located just above the so-called “hinge region” resulting in the production of two Fab fragments in addition to a stretch of CH2-CH3-HR (Fig. 4)[117]. Each Fab fragment weighing about 50 KDa is the result of the association between a full length light chain (CL & VL) joined by a disulfide bridge to the cleaved heavy chain comprising a variable domain (VH) with the first constant domain (CH1) where the protease has cleaved the chain[118,119]. Fab fragments are perceived as “Natural Fragments” since they are generated by enzymatic cleavage of the immunoglobulins which alleviates the need for genetic engineering required for the production of their counterparts with enhanced stability gained from reciprocal stabilization naturally existing between the variable and constant domains of the heavy and light chains[23,120]. The lack of the crystallizable fragment (Fc) hinders the ability of these fragments to cause the bystander activation of undesirable “opsonized” immune reactions mediated by the effector function of the antibodies and their subsequent cytotoxicity (namely the Antibody Dependent Cell Mediated Cytotoxicity (ADCC), Antibody Dependent Cellular Mediated Phagocytosis (ADCP), and Complement Mediated Cytotoxicity (CDC))[39].
Fig. 4.

Antibody Fragments produced by enzymatic digestion of the immunoglobulin IgG with papain and pepsin generating Fab and F(ab’)2 respectively. Papain digests the sequence just above the hinge region and generates two monovalent monospecific fragments whereas pepsin cuts in the region beneath the hinge region yielding a large monospecific bivalent fragment.
In contrast to the Fab, the F(ab’)2 is a bivalent, monospecific fragment (Fig. 4) essentially composed of two individual Fab fragments assembled together by the means of the flexible hinge region. The production of F(ab’)2 is easy, natural and straightforward. A direct treatment of IgG with Pepsin cleaves the full length antibody, just beneath the hinge region, generating combined Fabs with a detectable Fc fragment[121,122]. Their molecular mass is about 110 KDa, double that of a separate Fab which reduces their tissue penetration compared to Fab; but still higher than a full length antibody[123]. Since they possess two binding sites for the same epitope, therefore, their avidity is amplified giving them an advantage over other monovalent fragment in terms of their binding capacity to the antigens[120,124]. Size reduction comes with a shorter Fab serum half-life that could be addressed by their pegylation or the traditional conjugation to albumin and XTEN fusion proteins[69,125]. A number of antigen binding fragments were successfully designed and introduced into clinical trials. Ranibizumab (Fab) and Certolizumab Pegol (Fab’) were produced in their definitive forms in the E.coli expression system while Abciximab has been first generated as a full-length IgG before being enzymatically digested with Papain to cleave the Fc region and generate the Fab fragment[126]. These fragments were mostly used for the treatment of acute conditions; some patterns however were designated for the management of the chronic inflammatory diseases. For example, the Fab Certolizumab Pegol is indicated in the framework of rheumatoid arthritis and Crohn's disease. In order to prolong their serum half-life, the cysteine residue located in the proximity of the C-terminal end is adapted as the specific site for pegylation[127].
9.2. Single chain fragment variable (scFv)
In the issue released on October 21st, 1988, Bird and his colleagues described the Single Chain Fragment variable (scFv) for the first time. The recombinant polypeptides are antigen binding agents composed of the variable regions of both, light (VL) and the heavy (VH) chains, tethered by the mean of a linker peptide sequence (conventionally rich in glycine and serine with scattered hydrophilic residues) that joins the carboxyl terminal ends of VL and VH (Fig. 5)[128]. In order to optimize their thermal stability and their affinity towards the antigen, the number of amino acid residues making the linker peptide may differ from one scFv into another. It has been postulated that a linker peptide must spatially extend over a distance of 3.5 nm between the VL and VH domains outside the context of their binding site formation[129]. Stretches of thirty to thirty eight residues were engineered in such a way the cysteine residues involved in creating disulfide bridges between constant domains of the heavy (CH1) and light chains (CL) were deleted[130,131].
Fig. 5.
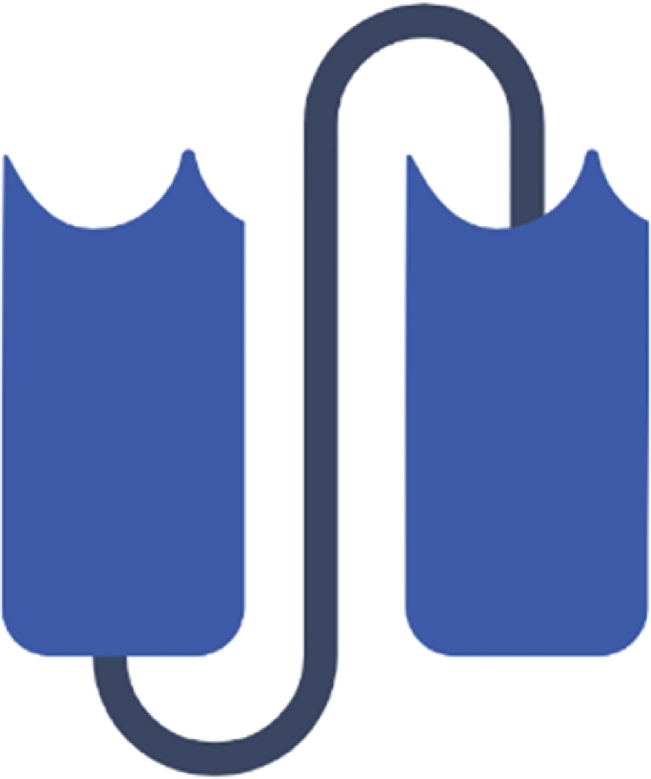
Schematic representation of the Single Chain Fragment Variable (scFv). This monospecific monovalent fragment is composed of the variable regions of light (VL) and heavy (VH) chains, connected by the mean of a linker peptide sequence.
The scFv maintains the same antigenic specificity with equivalent affinity to the epitopes for which the parental full length antibody has been generated. Their simple structure allows their production by expressing individual genes in the phage or in the form of combinatorial library of surface protein within a yeast cell surface display. This molecular modality permits in vitro screening and selection of target chains amongst a full repertoire of proteins. The genes of interest could be expressed inside the cell where they are potentially subject for a substantial change of conformation at the issue of their folding and various subcellular localization of the target fragments[132]. Compared to the full length antibodies, the scFv confer several advantages structurally, functionally and clinically. Their largely reduced size (25–30 KDa) makes them optimal for mass production in microbial bioreactors systems[133]. Since VL and VH are designed in series in the same recombinant system, the scFv would be produced faster, in greater yield, and at reduced costs when compared to conventional mAbs traditionally produced in higher expression systems. The clinical efficiency of the immunotherapy, especially in oncology, witnessed a substantial success with the advent of the scFv that facilitated a greater access to the malignant tissues and enhanced the tumor penetrance and receptor binding[134]. The physical absence of the Fc region from scFv reduces the risk of bystander activation of the immune cells but allows the agent to bind the specific receptor on the target cell without activation of the immune reaction. Although the lack of the Fc region might be often advantageous for tumor access, yet it decreases the thermostability, increases the propensity for aggregation, which promotes their immunogenicity and reduces their circulatory half-life[135,136]. In such cases, the required effective dose is conventionally increased and the pharmaceutical must be administered more frequently. This drawback is consistently addressed by scFv pegylation[125] or their fusion with albumin[135]. A humanized scFV, anti-VEGF (Vascular Endothelial Growth) factor (Brolucizumab) has been designed for the treatment of Age Related Macular Degeneration (AMD). Instead of the conventional direct injection of Anti-VEGF drugs, Brolucizumab showed an extended durability with great effectiveness based on a 12 weeks treatment scheme[137]. In order to enhance their therapeutic efficacy for oncological applications and their in vivo retention frequency, scFv antibody fragments were conjugated with toxins and/or other immune simulators to target malignant cells. Oportuzumab montax, an anti-neoplastic scFv intended to bind an Epithelial tumor cell-adhesion protein (EpCAM), fused with truncated Pseudomonas aerugenosa Endotoxin A has been approved for the treatment of BCG resistant urothelial bladder carcinoma[138,139]. An anti-CD22 (Highly expressed by B cell lymphomas and Non-Hodgkin's lymphomas) fusion protein conjugated with apoptin has been designed to bind the lymphoma cells, cross their plasma membrane, and induce their apoptosis[140].
9.3. Tandem single chain fragment variable (Ta-scFv)
Designed to string on tandem, two, three or even more scFv bodies are linked together by a flexible linker composed of a helical strand of amino acids. The tandem construct (Fig. 6) may include identical subunits, generating mono-specific, bivalent (two subunits), or multivalent (three subunits or more) fragments, or non-identical subunits, generating bispecific (two subunits), or multispecific, multivalent (three subunits or more) fragments[141]. The order of the cloned gene segments defines the format of the tandem scFv based on the orientation of VH and VL cloned sequence (VH-VL or VL-VH), the number of subunits, and the length of the linker peptide that would affect the folding, and the antigen binding capability of the tandem chain[142].
Fig. 6.

Schematic representation of the Tandem Single Chain Fragment Variable (Ta-scFv). This monospecific bivalent or multivalent fragment is composed of two (or even more) variable regions of light (VL) and heavy (VH) chains (n number of subunits), connected by the mean of a linker peptide sequence.
Homopolymers of Alanine (Ala-3), hydrophilic amino acid based stretches, Glycine-Serine-Rich spacers, and sequences derived from conventional immunoglobulins or other globular proteins were used to scheme the linkers that join the tandem repeats of single chains providing flexibility and resistance to protease degradation[142,143]. The length of connectors did not show a significant difference in their in vitro bioactivity demonstrating that short linkers retain adequate flexibility to intensely bind the nearby accessible antigens. Connectors composed of 12 residues and above define the native orientation of the antigen binding sites. The genetic reduction of the scFv linkers peptides by reducing the length of their coding sequences leads to a spontaneous assembly of the subunits in the form of multimeric complexes such as diabodies (see next paragraph)[144], [145], [146], [147]. The resulting products are used to target one, two or even more antigenic determinants with improved avidity and extended half-life when compared to single chains [148]. Similarly, this technique opened the way to produce bispecific full length antibodies able to recognize two different epitopes or antigenic determinants (Fig. 7). These products are featured by the acquisition of novel functions that do not initially exist in their parental antibodies[149].
Fig. 7.

Schematic representation of bispecific antibodies derived from two different parental immunoglobulins directed against antigen A and antigen B. The resulting molecule is able to bind antigenic determinants on the surface of targets A and B.
In 2018, Eggenreich and his collaborators[150] introduced a recombinant Ta-scFv designed to manage Celiac Disease. The consumption of Prolamins triggers the secretion of many autoantibodies, pro-inflammatory cytokines, and tissue trans-glutaminases leading to the development of a chronic inflammation associated with long term immune complications of the gut[151,152]. The unique effective treatment of Celiac Disease has been limited to a strict gluten-free diet that might be hindered by the traces of prolamins in the consumed food products[153]. The Ta-scFv serves as a neutralizing agent able to form a complex with Prolamins in the lumen of the gut, outside the context of the epithelium, preventing the formation of immunogenic agents and lead to their excretion[150,154].
9.4. Multimeric scFv fragments - Diabodies, triabodies, & tetrabodies
Different strategies were devised to improve the pharmacokinetic attributes and enhance the residual affinity towards antigenic determinants of the scFvs introduced in preclinical studies and clinical trials. The most effective approach resides in multimerizing the single scFvs bivalents dimers (Diabodies, 55–60 KDa) (Fig. 8A), trivalent trimmers (Triabodies, 80–90 KDa) (Fig. 8B), and/or tetravalent tetramers (Tetrabodies, 110–120 KDa) (Fig. 8C)[146,147].
Fig. 8.

Schematic representation of the Diabodies (A), Triabodies (B), and Tetrabodies (C).
Diabodies (Dbs) (Fig. 8A) are bivalent conjugates, that could be either monospecific or bispecific whether their scFv forming units are identical or not. With a five residue long spacer (G4S), the VH and VL of each variable chain subunit are interconnected together. Instead of flattering intrachain assembly of the antigen binding sites, the shortness of the linker peptide brings about the dimerization of the two chains in a head-to-tail format generating a compact configuration [145,155,156]. The transfected cells are supposed to yield two different chains formats (VH-A–VL-B & VH-B–VL-A) or (VL-A–VH-B & VL-B–VH-A) (where letters A and B correspond to the different antigenic specificities)[155,157]. Since the different chains of the bispecific diabodies are simultaneously expressed within the same cell, it is very likely that identical single chains assemble together yielding unproductive homodimers that lose their specificities and might be more susceptible to instability[158]. Molecular studies demonstrated that the linker's extension is critical for the conformation of the multimeric complex. When the latter is reduced to less than three amino acid residues, higher amount of triabodies (Fig. 8B) is generated in the periplasm since the shortness of this sequence affects the variable domain's flexibility and prolongs the multimeric assembly time what brings about additional subunit yielding more triabodies conjugates[145]. When it comes to the antigen binding capacity, the spacial distribution of the individual binding sites and the orientation of the variable modules affect the accessibility of the diabodies and triabodies to the surface antigens[145,159].
10. DART, bite, & tandem antibodies
Over the last decade, a number of molecular modifications were devised to the diabodies format to improve thermal stability, clinical efficacy, and tumor availability. Based on their structural configuration, the bispecific diabodies could be classified into either “Fc-less antibody fragments” with a minimalistic antigen binding domain or Immunoglobulin-like antibodies[155]. Therefore, they are either conventional IgG-like or unorthodox non-conventional IgG-like molecules. Cross-Mab®[160,161], Knobs-in-Holes[162], Dock-&-Lock[163], Quadroma®[164], Duobody®[165], and Dual-Variable-Domain[166] were developed by a number of research groups in the aim to harvest different IgG-like formats, namely the Dual Affinity Re-Targeting (DART) proteins, the Bi-specific T Cell Engagers (BiTE), and Tandem Diabodies (Tand-Ab)[167,168]. These synthetic fragments are manufactured by assembling a number of building blocks (i.e. Fab, Single Chain Fragment Variable (scFv), Single Domains Antibodies (sd-Abs)) merged together by the mean of a peptide linker[169].
11. DART antibodies
Dual Affinity Re-Targeting proteins (Fig. 9) consist of a pair of engineered variable chains in which the VH domains are swapped with the opposite one. More explicitly, VH from anti-A antibody is exchanged with the VH from anti-B antibody. The first binding site will consists of VH-A paired with VL-B and the opposed binding site will consists of VH-B paired with VL-A[170]. The interchange of the variable domains yields a number of diverse fragments as a result of the three dimensional conformation resulting from individual peptides interaction affected by the shortened connecting linker similar to the natural interaction occurring in conventional antibodies. A promising T cell recruiting diabody-like molecule (Flotetuzumab, MGD006) has been designed to bind CD3 and CD123 clusters of the Acute Myeloid Leukemia cells or in the case of refractory myelodysplastic syndrome inducing the engagement and the activation of T cell killing of the malignant clones[171]. Besides, MGD007 is directed against GPA33 protein that characterizes gastrointestinal malignancies. The fusion with Fc region prolongs the serum half-life of MGD007 in contrast to the Fc-less format of MGD006 which is cleared more rapidly from the patients’ serum[172]. DARTs were shown to yield an enhanced B and T cell in addition to their contribution in recruiting the CD16 Natural Killer cells engendering the secretion of potent cytokines resulting in an effective cytolytic activity of targeted malignant cells[173]. In 2020, Tebotelimab (MGD013) has been investigated in a Phase-I clinical trial as a new format in the class of DART designed as a bispecific tetravalent to target PD-1 and LAG-3. The tested novel DART has been shown to provoke efficient and coordinated blocking attributes of PD-1 and LAG-3 leading to the enhancement of the T cell activity[174]. Clinical data shows that MDG013 demonstrates adequate safety attributes with promising anti-tumor efficacy[175].
Fig. 9.

Genetic engineering of Dual Affinity Re-Targeting proteins (DART).
Characterized by a relatively small size, DARTs are amenable to rapid renal excretion, high resistance to aggregation, and extreme potential following their administration in vitro and in vivo[170,176]. To overcome their short serum half-life, Fc bearing DARTs were developed providing the clinicians a wider spectrum of amended protocols with different drug dosage and some products were stabilized by introducing a disulfide bridge[168].
12. BiTE antibodies
One of the key immune reactions directed towards eliminating the cancer cells involves the enhancement of T cells interactions with the tumor cell receptors[177]. BiTEs were designed to bridge the cancer cells surface antigens to the T cell Cluster of Differentiation (CD3) situated in the close proximity of the TCR which potentiates the unreactive T lymphocytes and reinforces their cytotoxic activity against the targeted tumor cells (Fig. 10). This way, the opsonizing BiTEs bring about a more specific and efficient T cell mediated immune response[167]. To manufacture BiTEs, a first scFv, directed against the CD3, is coupled to another single chain directed towards the tumor cells surface antigens (BiTEs could be expanded by adding a third scFv to become trivalent and multispecific). This conformation stands behind the proliferation of T lymphocytes inducing a T-cell mediated cytotoxicity[178].
Fig. 10.

Genetic engineering of Bi-specific T Cell Engagers (BiTEs).
Engineered Recombinant DNA carried by a plasmid is used to transfect CHO or HEK cells that synthesize the fragments to be selected for the desired pharmaceutical properties. Like many other recombinant antibody fragments, the defective Fc and the reduced molecular size of BiTEs shorten their serum half-life. However, this does not hinder their efficiency in inducing an extremely potent cytotoxic effect against tumor cells even if used at picomolar concentrations[167]. Blinatumomab (AMG103, MT103), an anti-CD19 is one of the most promising Bispecific T Cell Engagers (BiTE) in clinical trials, has garnered FDA approval for the treatment and eradication of B-Cell Precursor Acute Lymphoblastic Leukemia (ALL)[94,179]. Cases of Chronic Lymphoblastic Leukemia (CLL) showed a good prognosis when treated with Blinatumomab. Tumor regression has been also documented in the cases of Non-Hodgkin's Lymphomas[92,180]. Further clinical studies are conducted to assess the efficiency of using a multitude of BiTEs designed to target EpCAM (Colorectal Cancer), CEA (Gastric Carcinoma), HER-2 (Breast Cancer), PsMA (Prostate Cancer) and other tumor markers[180]. In 2019, a bicistronic model has been constructed to overcome the limitation of Chimeric Antigen Receptor (CAT) T Cell treatment of solid gliobastoma. The construct involves a CAR expression system for EGFRvIII antigen with a bispecific T Cells Engager (BiTE) directed against EGFR. The combined system (CAR-T # BiTE) has been shown to be more effective than either separate treatment modalities[181].
13. Tandem antibodies
Bearing four antigen binding sites directed against two different antigenic determinants, the Tandem Antibodies (TanAbs) are tetravalent bispecific complexes of about 100–110 KDa. The genetically engineered constructs encode a single chain of tandem subunits consisting of four different variable modules assembled together by a short peptide linker (Fig. 11). Compared to their analogues (BiTEs and DARTs), the TanAb bridges the molecular pattern of target cells to the receptors expressed at the surface of the Natural Killers (NKs) and/or CD8+ cytotoxic T cells. Owing to their bivalent domains, TanAbs preserve a comparable avidity as their conventional antibodies with the optimal affinity to the target antigens. Their relatively larger size prolongs their serum half-life that can reach up to 24 h, comparatively higher than the half-life of all other antibody fragments[159,178].
Fig. 11.

Schematic representation of tandem antibodies consisting of consisting of four different variable modules assembled together by a short peptide linker.
Tandem Antibody platform has been used to design a CD19-CD3-AFM11 bispecific humanized tetravalent construct, extremely powerful immune cell recruiter in the case of B cells malignancies[182]. AFM11 exhibits substantial avidity to the highly expressed CD3 which turns it into a serial-killer of CD19 expressing B cell lineage. Through its anti-CD3 domains, AFM11 recruits effector T cells stimulating the so-called “Immunological Synapse” which results into effective elimination of malignant cells in the cases Relapsed B-Cell Non-Hodgkin Lymphoma and Refractory B-Cell Non-Hodgkin Lymphoma[183,184]. Besides, the tetravalent bispecific Tandem construct directed against CD30 and CD16A was shown to represent a potent therapeutic agent able to effectively recruit the Natural Killer cells and induce the destruction and lysis of Hodgkin's lymphoma cancer cells[185]. In 2021, Watanabe et al., from University of Tokyo, developed a series of constructs including a tandem scFv-Fc, a diabody-Fc, and a fusion protein scFv-Fc-scFv, each of which possess four scFv domains and able to recognize and bind the extracellular domains of ROBO1[186].
14. Minibodies
Pairs of single chain variable fragments joined together by the mean of their CH3 heavy chain's constant domains (2 x scFv-CH3) were coined the term Minibodies (Fig. 12)[187]. These substitutes of diabodies, are bivalent dimers with an intermediate molecular weight of about 80 KDa. Minibodies could be designed to be either monospecific or bispecific according to whether their binding sites are identical or not[188]. The multivalence of Minibodies allows them to readily bind their targets, with a rather higher affinity than the monovalent counterparts. Due to their moderately small size, the Minibodies could be easily expressed at a high yield in prokaryotic host cells and therefore, when clinically used, they show a good tumor-to-normal tissue ratio as a result of their eased penetration into cancerous tissues[189].
Fig. 12.

Schematic representation of Minibodies composed of two single chain variable fragments linked together by the mean of a CH3 heavy chain's constant domains (2 x scFv-CH3).
Although widely used in clinical trials, the approval of Minibodies is restricted by their low thermostability and their propensity for aggregation and denaturation in vivo due to the weakness of the variable chains’ interaction[190]. The extension of the linkers joining the scFvs from 15 to 18 aa improved the strength of the thermal stability in the 48 h that follow their injection[191]. Their solubility has been also affected by their variable domains orientation where VH-VL and shows an increased solubility when compared to the opposite orientation (VL-VH)[192].
Many different formats of minibodies were engineered to target a number of tumor markers in oncology. Among these, the scFv-CH3γ1 dimer, was designed to target and block the Carcinoembryonic Antigen (CEA). This molecule showed a high specificity in CEA binding and short serum half-life[193]. A11 minibody has been engineered to target the prostate stem cells specific antigen. The minibody-conjugated form combined to polypeptide-based gold nanoshells has been tested in the context of thermal therapy. The minibody-conjugate demonstrates a substantial localized reduction in metastatic prostate cancer cells[193,194]. Tositumomab, a radionuclide-coupled anti-CD20 antibody (131I labeled) and Ibritumomab (90Y conjugated) have been FDA approved for the management of Non-Hodgkin's Lymphoma[195].
15. Heavy chain antibodies and nanobodies
In 1989, an unexpected finding, discovered at the issue of the whole and fractionated serum analysis derived from Camelidae (camels, llamas and alpacas), has perpetually changed the conceptual organization of conventional antibodies, primarily perceived as “dimers of dimers” composed of two heavy chains assembled to two identical light chains[196]. Hereinafter, a new format of non-canonical antibodies has been found to lack the light chain while only two heavy chains (lacking CH1 domains) are revealed. The expanded forms give rise to additional peaks featuring the electrophoretic profile of the serum Camelid globulin fractions. These latter were coined the term Heavy Chain Antibodies (HCAbs) containing VHH domains with relatively lower molecular weight (90 KDa) compared to conventional IgGs (150 KDa)[197]. The DNA coding sequences of Camelid HCAbs were deciphered using molecular sequencing techniques. It has been demonstrated that HCAbs are encoded by the same conventional locus but with a different set of gene segments. The camelidae genome consists of about 40 genes, assuming the same genetic organization of conventional antibody coding sequences. Although present on the sequence of coding genes, the CH1 domain coding exon bears a point mutation located at the boundary between CH1 and the proximate hinge region intron which hinders the consensus splice site between the G and T and omits the coding sequence[198]. The diversification of HCAbs antigen binding repertoire is triggered by a set of molecular mechanisms including the formation of disulfide bridges, the extended surface area of the hypervariable domains and the formal reshaping of paratopes of the binding groove. It has been postulated that a number of signal sequences stretched over the VHH stand behind the multitude of diversity mechanisms. The lack of the light chain enables such changes to take place generating novel specificities[199]. Structurally, VHHs are composed of four conserved framework regions (FRs) neighboring three contiguous CDRs (emphasis on CDR3) that contribute to the conformation of the antigenic paratopes (Fig. 13).
Fig. 13.

Comparative representation of Conventional Antibody (A) structure composed of heavy and light chains to the Camelid Heavy Chain Antibody (B) lacking light chains and deprived of CH1 constant domain. The Variable Domains of the Heavy Chain VHH are shown in C.
Derivatives of the camelid, the variable domains of the HCAbs, establishing the antigen binding sites, the naturally occurring Nanobodies are miniaturized fragments with extended CDR3 loops and low molecular weight of only about 15 KDa[200]. Alternatively, the Nbs could be engineered using a convenient recombinant system and optimal host cell which facilitate their synthesis and multimerization to generate multivalent therapeutic agents. The three dimensional conformation of Nbs paratopes allows them to easily access the hidden epitopes or cryptic determinants on the surface of antigen. Nanobodies are characterized by a substantial stability, higher solubility and a significantly improved affinity, with a critically reduced immunogenicity[200,201]. Their small size facilitates their tissue penetration, distribution, and targeting. Owing to these strategic attributes, Nbs are considered as one of the main promising diagnostic and therapeutic agents with a great potential use for a diversity of illnesses (oncology, neurological disorders, inflammatory, and infectious diseases)[200]. Nanobodies were proven to be amenable for protein fusion and conjugation with toxins or even radioactive elements transforming them into clinically efficient drug delivery systems for active drugs, enzymes, toxins, or inactive prodrugs which promoted their selectivity and efficacy[200,202]. An ongoing multicentric Phase I/II evaluates the therapeutic efficacy and safety of Zenocutuzumab (MCLA-128) an Anti-HER-2 x Anti-HER-3 Bispecific Heavy Chain antibody designed to treat the patients harboring solid tumors with NRG1 fusion. The molecule is able to mediate antibody-dependent cellular cytotoxicity as it binds and blocks HER-3 as the NRG-1 and NRG-1 fusion proteins counterparts[203,204].
Among those nanobodies that showed a significant clinical efficacy, the FDA-approved Caplacizumab, is an anti-von Willebrand factor designed to manage the Aquired Thomobotic Thrombocytopenic Purpura (aTTP) by blocking the A-1 domain of the coagulation factor and inhibits their interaction with the platelets by 67% the risk to develop an aTTP[205,206]. Ga-HER2-Nanobody, directed against HER-2 tumor marker of breast carcinoma, showed high kidney filtration, fast blood clearance after a short serum half-life (2.5 h), and a substantial capacity to identify the majority of the tumor cells locations and distribution[207,208].
16. Antibody mimetics
The structural and functional limitations encountered with the full length antibodies and many of their derivatives triggered research to develop alternative platforms to create novel agents, able to “mimic” the binding capacity of the conventional antibodies. For that purpose, the monovalent “Antibody Mimetics”, a new class of minimalistic binding proteins, were manufactured outside the context of the immune system panels, generally composed of non-immunoglobulin alpha helices and beta sheets or even randomly looped stretches and are intended to include a range of antigenic binding sites[209]. A number of protein engineering strategies were devoted to yield and then select these antibody mimetics including site-directed mutagenesis, multisite directed mutagenesis, and random mutagenesis. The rationale behind designing antibody mimetics depends on the accurate selection of the proper framework suitable for the integration of recognition domains. Conventionally, this framework is a folded protein domain amenable for mutational manipulations. It should possess a substantial level of flexibility tolerating the insertion of mutations without affecting the secondary structure of the protein, without deterring the antigenic specificity and affinity to the target epitopes[210]. The isolation of the suitable mimetic could be achieved using a directed evolution cycle. Presumably, the designated mutation sites of the adopted binding domain are subject to a process of mutagenesis to construct the DNA library that will be displayed on the surface of the expression system. An error-prone PCR along with DNA shuffling process are adopted to promote the diversification of the yield and mature the desired antibody mimetics[209,211]. Commonly, the selected products should be soluble, stable, resistant to degradation by proteases, thermostable, tolerating extreme pH values, and composed of a single domain peptide without disulfide bridges or need for a process of glycosylation. Preferably, the antibody mimetics are to be synthesized in the cytoplasm of the host recombinant bacteria without being transformed into inclusion bodies or being degraded[212,213]. Despite the fact that the recognition and binding of the epitope to the conventional antibodies (and most of their fragments) involves the contribution of the loops protruding from the six different complementary determining regions of the heavy and light chains, it has been proposed that CDRs-FRs-based mimetic peptides (about 3 KDa) may be manufactured and used for a large number of medical purposes. The CDR-FR peptides are connected by alternating two CDRs by the means of a framework region selected from VH and VL coding sequences. The CDR3 is the most obvious component of the peptide mimetic due to its great accessibility, diversity, and its critical contribution to the antigen binding. The C-terminal end of CDR1/2 loops and the N-terminal end of the CDRH/L3 are interconnected with a trailed FR randomly chosen from VH and VL coding sequences[210]. A number of various versions of antibody mimetics were developed, namely the Nanofitins (Nanofitins), Anticalins (Lipocalins), Affibodies (B-domain of SPA), DARPins (Designed Ankyrin Repeat Proteins), Avimers (A-domain), and Fynomers (SH3 Domain of Fyn Kinase). Many of these later are currently under rigorous clinical investigation suggesting a very promising future with unlimited application tenacity of such human proteins derivatives for diagnostic and therapeutic purposes[214]. Unlike conventional antibodies, the antibody mimetics exhibit a short half-life associated with a high kidney clearance rate due to their reduced molecular weight (5–10 KDa). The absence of the Fc region alters their ability to bridge and activate the immune system cells. This drawback could be overcome by combining the mimetics to free Fc domains and reestablish their functional activity. Alternatively, antibody mimetics could be further restructured by PEGylation, PASylation (Proline-Alanine-Serine), or by fusion to proteins conjugates (Albumin Binding Domain) without affecting their affinity and their tumor infiltration power[215], [216], [217]. The management of Acute Myeloid Leukaemia (AML) has revolutionized during the last decades. A number of drugs were clinically tested and FDA approved. Among these, is Venetoclax, an orally administered antibody mimetic that selectively targets BCL-2 but not the BCL-XL. Preclinical studies revealed a promising efficacy of Venetoclax in the management of AML when combined with hypomethylating agents (HMA) and chemotherapeutic drugs taking it to clinical trials. Data collected from Venetoclax-based therapy combined to HMA and Cytrabine in elderly (75 years and beyond) with AML (unfit for conventional intensive chemotherapy) in addition to its safety profile secured a fast FDA approval and widespread incorporation of such antibody-mimetic based therapy in the clinical context[218,219].
17. Conclusions
It is commonly said that “The smaller pieces and details make up the big broader picture”. When it comes to the mosaic puzzle of therapeutic antibodies it can be fairly said “The smaller pieces make the larger ones look better”. The great potential of antibody fragments and their mimetics has no limits with wide range of formats and the unprecedented access to uncharted purposes and overcome the new challenges coming upon in with the fast and limitless technological breakthroughs and new era of medicine. This review provides summary of the molecular mechanisms governing the genetic engineering of antibodies and their derivatives, starting with murine, chimeric and humanized antibodies to the different fragments generated in vitro using either prokaryotic host cells, eukaryotic expression systems, cell free systems, or transgenic mice. Conventional antibodies show a number of disadvantages due to their large size and immunogenicity. Antibody derivatives and modifications aim to increase tissue penetration and reduce undesired activation of autoimmunity or “side-immunity” for therapeutic purposes. On a side note, tissue penetration and yet quick enough body clearance represent a challenge to balance effectiveness and toxicity. Developing antibodies with multiple targets and/or acting via multiple mechanisms would definitely enhance treatment of highly mutating tumors. This can include activation of T-cells, better engagement of innate and adaptive immune system components and suppressing multiple oncogenic pathways in cancer treatment. The advancement of therapeutic options provided with antibody fragments and modifications can be further elevated with use of combination treatments along with immunomodulators such as immune checkpoint blockers. Individual assessment of each particular antibody treatment remains a “best” practice to reduce side effects. The use of suitable in vitro and in vivo and perhaps 3D organoid systems to assess toxicity can drive more rigorous exploration of new mimetics or derivatives prior to clinical trials and human administration.
Disclosures
The authors declare no potential conflict of interest regarding the content of this manuscript.
Funding
The authors received no financial grants for the research or the publication of this paper.
FDA approved mAbs data available on
https://www.antibodysociety.org/resources/approved-antibodies https://www.accessdata.fda.gov/scripts/cder/daf
Declaration of Competing Interest
None
References
- 1.Behring Kitasato. Ueber das Zustandekommen der Diphtherie-Immunitüt und der Tetanus-Immunitüt bei Thieren. Deutsche Medizinische Wochenschrift. 1890 Dec 4;16(49):1113–1114. [PubMed] [Google Scholar]
- 2.Paul Ehrlich - Biographical [Internet]. [cited 2021 Jun 18]. Available from: https://www.nobelprize.org/prizes/medicine/1908/ehrlich/biographical/.
- 3.Kaufmann S H.E. Ehrlich contributions. Nat. Immunol. 2008;9(7):705–712. doi: 10.1038/ni0708-705. [DOI] [PubMed] [Google Scholar]
- 4.Edelman G.M. Dissociation of Γ-globulin. J. Am. Chem. Soc. 1959;81:3155–3156. [Google Scholar]
- 5.Ribatti D. Edelman's view on the discovery of antibodies. Immunol. Lett. 2015;164:72–75. doi: 10.1016/j.imlet.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Porter R.R. The hydrolysis of rabbit y-globulin and antibodies with crystalline papain. Biochem. J. 1959 Sep;73(1):119–126. doi: 10.1042/bj0730119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleischman J.B., Porter R.R. Press em.the arrangement of the peptide chains in gamma-globulin. Biochem. J. 1963;88(2):220–228. doi: 10.1042/bj0880220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dreyer W.J., Bennett J.C. The Molecular Basis of Antibody Formation: A Paradox*. [DOI] [PMC free article] [PubMed]
- 9.Hood L., Ein D. Immunoglobulin lambda chain structure: two genes, one polypeptide chain. Nature. 1968;220(5169):764–767. doi: 10.1038/220764a0. [DOI] [PubMed] [Google Scholar]
- 10.Hood L., Talmage D.W. Mechanism of antibody diversity: germ line basis for variability. Science. 1970;168(3929):325–334. doi: 10.1126/science.168.3929.325. [DOI] [PubMed] [Google Scholar]
- 11.Sakano H., Rogers J.H., Hüppi K., Brack C., Traunecker A., Maki R., et al. Domains and the hinge region of an immunoglobulin heavy chain are encoded in separate DNA segments. Nature. 1979 Feb 1;277(5698):627–633. doi: 10.1038/277627a0. [DOI] [PubMed] [Google Scholar]
- 12.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 13.Cohen S.N., Chang A.C.Y., Boyer H.W. Helling RB. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. U.S.A. 1973;70(11):3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raju T.N. The Nobel chronicles. 1984: niels Kai Jerne, (1911-94); César Milstein (b 1926); and Georges Jean Franz Köhler (1946-95) Lancet. 2000;355(9197):75. doi: 10.1016/s0140-6736(05)72025-0. [DOI] [PubMed] [Google Scholar]
- 15.[The 1984 Nobel Prize in Medicine (Cesar Milstein, George Köhler, Niels Jerne)] - PubMed [Internet]. [cited 2021 Jun 20]. Available from: https://pubmed.ncbi.nlm.nih.gov/6394268/.
- 16.Mullis K., Faloona F., Scharf S., Saiki R., Horn G., Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986;51(1):263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 17.Mullis K., Faloona F., Scharf S., Saiki R., Horn G., Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Biotechnology (Reading, Mass) 1986;24:17–27. 1992. [PubMed] [Google Scholar]
- 18.Banker DD. Monoclonal antibodies A review. Indian J. Med. Sci. 2001;55(12):651–654. [PubMed] [Google Scholar]
- 19.Lu R.M., Hwang Y.C., Liu I.J., Lee C.C., Tsai H.Z., Li H.J., et al. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020;27(1):1–30. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goulet D.R., Atkins W.M. Considerations for the design of antibody-based therapeutics. J. Pharm. Sci. 2020;109(1):74–103. doi: 10.1016/j.xphs.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Posner J., Barrington P., Brier T., Datta-Mannan A. Monoclonal antibodies: past, present and future. Handb. Exp. Pharmacol. 2019:81–141. doi: 10.1007/164_2019_323. [DOI] [PubMed] [Google Scholar]
- 22.Starzl T.E., Fung J.J. ORTHOCLONE OKT3 in treatment of allografts rejected under cyclosporine-steroid therapy. Transplant. Proc. 1986;18(4):937–941. [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandes J.C. Vol. 23. Elsevier Ltd; 2018. Therapeutic application of antibody fragments in autoimmune diseases: current state and prospects; pp. 1996–2002. (Drug Discovery Today). [DOI] [PubMed] [Google Scholar]
- 24.D A., A C., M G. Anaphylactic shock after retreatment with OKT3 monoclonal antibody. N. Engl. J. Med. 1992 Sep 3;327(10):736. doi: 10.1056/NEJM199209033271018. –736. [DOI] [PubMed] [Google Scholar]
- 25.Adair J.R. Engineering antibodies for therapy. Immunol. Rev. 1992;130(1):5–40. doi: 10.1111/j.1600-065x.1992.tb01519.x. [DOI] [PubMed] [Google Scholar]
- 26.Presta L. Engineering antibodies for therapy. Curr. Pharm. Biotechnol. 2005 Mar 25;3(3):237–256. doi: 10.2174/1389201023378256. [DOI] [PubMed] [Google Scholar]
- 27.Houen G. Therapeutic antibodies: an overview. Methods Mol. Biol. 2022;2313:1–25. doi: 10.1007/978-1-0716-1450-1_1. [DOI] [PubMed] [Google Scholar]
- 28.Zlabinger G.J., Ulrich W., Pohanka E., Kovarik J. [OKT 3 treatment of kidney transplant recipients] Wien Klin Wochenschr. 1990 Mar 2;102(5):142–147. [PubMed] [Google Scholar]
- 29.Tsurushita N., Hinton P.R., Kumar S. Design of humanized antibodies: from anti-Tac to Zenapax. Methods. 2005;36(1):69–83. doi: 10.1016/j.ymeth.2005.01.007. May. [DOI] [PubMed] [Google Scholar]
- 30.Gibson T.B., Ranganathan A., Grothey A. Randomized phase III trial results of panitumumab, a fully human anti-epidermal growth factor receptor monoclonal antibody, in metastatic colorectal cancer. Clin. Colorectal Cancer. 2006;6(1):29–31. doi: 10.3816/CCC.2006.n.01. May. [DOI] [PubMed] [Google Scholar]
- 31.Moroni M., Veronese S., Benvenuti S., Marrapese G., Sartore-Bianchi A., Di Nicolantonio F., et al. Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol. 2005;6(5):279–286. doi: 10.1016/S1470-2045(05)70102-9. May. [DOI] [PubMed] [Google Scholar]
- 32.Zamora-Atenza C., Diaz-Torne C., Geli C., Diaz-Lopez C., Ortiz M.A., Moya P., et al. Adalimumab regulates intracellular TNFα production in patients with rheumatoid arthritis. Arthritis Res. Ther. 2014 Jul 18;16(4):R153. doi: 10.1186/ar4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burmester G.R., Panaccione R., Gordon K.B., McIlraith M.J., Lacerda A.P.M. Adalimumab: long-term safety in 23 458 patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis and Crohn's disease. Ann. Rheum. Dis. 2013 Apr;72(4):517–524. doi: 10.1136/annrheumdis-2011-201244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sator P. Safety and tolerability of adalimumab for the treatment of psoriasis: a review summarizing 15 years of real-life experience. Ther. Adv. Chronic Dis. 2018 Aug;9(8):147–158. doi: 10.1177/2040622318772705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connors J.M., Jurczak W., Straus D.J., Ansell S.M., Kim W.S., Gallamini A., et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin's lymphoma. N. Engl. J. Med. 2018 Jan 25;378(4):331–344. doi: 10.1056/NEJMoa1708984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C., Thudium K.B., Han M., Wang X.T., Huang H., Feingersh D., et al. In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res. 2014 Sep;2(9):846–856. doi: 10.1158/2326-6066.CIR-14-0040. [DOI] [PubMed] [Google Scholar]
- 37.Noseda R., Bedussi F., Gobbi C., Zecca C., Ceschi A. Safety profile of erenumab, galcanezumab and fremanezumab in pregnancy and lactation: analysis of the WHO pharmacovigilance database. Cephalalgia. 2021 Jun;41(7):789–798. doi: 10.1177/0333102420983292. [DOI] [PubMed] [Google Scholar]
- 38.Dalum I., Jensen M.R., Gregorius K., Thomasen C.M., Elsner H.I., Mouritsen S. Induction of cross-reactive antibodies against a self protein by immunization with a modified self protein containing a foreign T helper epitope. Mol. Immunol. 1997 Nov 1;34(16–17):1113–1120. doi: 10.1016/s0161-5890(97)00147-8. [DOI] [PubMed] [Google Scholar]
- 39.Kholodenko R.V., Kalinovsky D.V., Doronin I.I., Ponomarev E.D., Kholodenko I.V. Antibody fragments as potential biopharmaceuticals for cancer therapy: success and limitations. Curr. Med. Chem. 2019 Apr 1;26(3):396–426. doi: 10.2174/0929867324666170817152554. [DOI] [PubMed] [Google Scholar]
- 40.Ahmad Z.A., Yeap S.K., Ali A.M., Ho W.Y., Alitheen N.B.M., Hamid M. ScFv antibody: principles and clinical application. Clin. Dev. Immunol. 2012 doi: 10.1155/2012/980250. 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsia E.C., Ruley K.M., Rahman M.U. Infliximab (Remicade®): from bench to clinical practice. A paradigm shift in rheumatology practice. APLAR J. Rheumatol. 2006;9:107–118. [Google Scholar]
- 42.Ramos-Casals M., Brito-Zerón P., Muñoz S., Soria N., Galiana D., Bertolaccini L., et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore) 2007 Jul;86(4):242–251. doi: 10.1097/MD.0b013e3181441a68. [DOI] [PubMed] [Google Scholar]
- 43.Weber J. Review: anti–CTLA-4 antibody ipilimumab: case studies of clinical response and immune-related adverse events. Oncologist. 2007;12(7):864–872. doi: 10.1634/theoncologist.12-7-864. Jul. [DOI] [PubMed] [Google Scholar]
- 44.Peggs K.S., Quezada S.A., Korman A.J., Allison J.P. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 2006;18:206–213. doi: 10.1016/j.coi.2006.01.011. Curr Opin Immunol. [DOI] [PubMed] [Google Scholar]
- 45.Guglin M., Cutro R., Mishkin J.D. Trastuzumab-induced cardiomyopathy. J. Cardiac Failure. 2008;14:437–444. doi: 10.1016/j.cardfail.2008.02.002. J Card Fail. [DOI] [PubMed] [Google Scholar]
- 46.Hansel T.T., Kropshofer H., Singer T., Mitchell J.A., George A.J.T. Vol. 9. Nature Publishing Group; 2010. The safety and side effects of monoclonal antibodies; pp. 325–338. (Nature Reviews Drug Discovery). [DOI] [PubMed] [Google Scholar]
- 47.Knight D.M., Wagner C., Jordan R., McAleer M.F., DeRita R., Fass D.N., et al. The immunogenicity of the 7E3 murine monoclonal Fab antibody fragment variable region is dramatically reduced in humans by substitution of human for murine constant regions. Mol. Immunol. 1995;32(16):1271–1281. doi: 10.1016/0161-5890(95)00085-2. [DOI] [PubMed] [Google Scholar]
- 48.Tamhane U.U., Gurm H.S. Vol. 7. Expert Opin Drug Saf; 2008. The chimeric monoclonal antibody abciximab: a systematic review of its safety in contemporary practice; pp. 809–819. (Expert Opinion on Drug Safety). [DOI] [PubMed] [Google Scholar]
- 49.Suntharalingam G., Perry M.R., Ward S., Brett S.J., Castello-Cortes A., Brunner M.D., et al. Cytokine storm in a phase 1 trial of the Anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 2006 Sep 7;355(10):1018–1028. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 50.Milstein C.P., Deverson E.V. Primary structure of ϰ light chain from a human myeloma protein. Eur. J. Biochem. 1974;49(2):377–391. doi: 10.1111/j.1432-1033.1974.tb03843.x. [DOI] [PubMed] [Google Scholar]
- 51.Hossler P., Khattak S.F., Li Z.J. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. 2009;19:936–949. doi: 10.1093/glycob/cwp079. [DOI] [PubMed] [Google Scholar]
- 52.Zhang P., Woen S., Wang T., Liau B., Zhao S., Chen C., et al. Vol. 21. Elsevier Ltd; 2016. Challenges of glycosylation analysis and control: an integrated approach to producing optimal and consistent therapeutic drugs; pp. 740–765. (Drug Discovery Today). [DOI] [PubMed] [Google Scholar]
- 53.Eggleton P., Haigh R., Winyard P.G. Consequence of neo-antigenicity of the ‘altered self. Rheumatol. Rheumatol. (Oxford) 2008;47:567–571. doi: 10.1093/rheumatology/ken014. [DOI] [PubMed] [Google Scholar]
- 54.Doyle H.A., Gee R.J., Mamula M.J. Altered immunogenicity of isoaspartate containing proteins. Autoimmunity. 2007 Mar;40(2):131–137. doi: 10.1080/08916930601165180. [DOI] [PubMed] [Google Scholar]
- 55.Morrison S.L., Johnson M.J., Herzenberg L.A., Oi V.T. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. U.S.A. 1984;81(21 I):6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrison S.L., Johnson M.J., Herzenberg L.A., Oi V.T. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. U.S.A. 1984;81(21 I):6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Antibodies and In Vivo Therapy. Elsevier; 2012. Immunology For Pharmacy; pp. 102–109. [Google Scholar]
- 58.Morrison S.L., Johnson M.J., Herzenberg L.A., Oi V.T. Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. U.S.A. 1984;81(21 I):6851–6855. doi: 10.1073/pnas.81.21.6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diamos A.G., Hunter J.G.L., Pardhe M.D., Rosenthal S.H., Sun H., Foster B.C., et al. High Level Production of Monoclonal Antibodies Using an Optimized Plant Expression System. Front. Bioeng. Biotechnol. 2020;7(January) doi: 10.3389/fbioe.2019.00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maloney D.G., Grillo-López A.J., White C.A., Bodkin D., Schilder R.J., Neidhart J.A., et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin's lymphoma. Blood. 1997 Sep 15;90(6):2188–2195. [PubMed] [Google Scholar]
- 61.Maloney D.G., Grillo-López A.J., Bodkin D.J., White C.A., Liles T.M., Royston I., et al. Idec-c2b8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin's lymphoma. J. Clin. Oncol. 1997 Sep 21;15(10):3266–3274. doi: 10.1200/JCO.1997.15.10.3266. [DOI] [PubMed] [Google Scholar]
- 62.Poljak R.J., Amzel L.M., Avey H.P., Chen B.L., Phizackerley R.P., Saul F. Three dimensional structure of the Fab’ fragment of a human immunoglobulin at 2.8Å resolution. Proc. Natl. Acad. Sci. U.S.A. 1973;70(12):3305–3310. doi: 10.1073/pnas.70.12.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marquart M., Deisenhofer J., Huber R., Palm W. Crystallographic refinement and atomic models of the intact immunoglobulin molecule Kol and its antigen-binding fragment at 3.0Å and 1.9Å resolution. J. Mol. Biol. 1980;141(4):369–391. doi: 10.1016/0022-2836(80)90252-1. [DOI] [PubMed] [Google Scholar]
- 64.Segal D.M., Padlan E.A., Cohen G.H., Rudikoff S., Potter M., Davies D.R. The three dimensional structure of a phosphorylcholine binding mouse immunoglobulin Fab and the nature of the antigen binding site. Proc. Natl. Acad. Sci. U.S.A. 1974;71(11):4298–4302. doi: 10.1073/pnas.71.11.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones P.T., Dear P.H., Foote J., Neuberger M.S., Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321(6069):522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- 66.Bates A. Power CA.David vs. Goliath: the structure, function, and clinical prospects of antibody fragments. Antibodies. 2019 Apr 9;8(2):28. doi: 10.3390/antib8020028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nelson A.L. Antibody fragments: hope and hype. MAbs. 2010;2:77–83. doi: 10.4161/mabs.2.1.10786. MAbs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Spadiut O., Capone S., Krainer F., Glieder A., Herwig C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014;32:54–60. doi: 10.1016/j.tibtech.2013.10.002. Trends Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Antibody fragments as therapeutics. Therapeutic Antibody Eng. 2012:265–595. [Google Scholar]
- 70.Milgrom L.R. Towards recombinant antibody-fragment targeted photodynamic therapy. Sci. Prog. 2008;91(3):241–263. doi: 10.3184/003685008X361415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schulman K.A., Glick H.A., Rubin H., Eisenberg J.M. Cost-effectiveness of HA-1A monoclonal antibody for gram-negative sepsis: economic assessment of a new therapeutic agent. JAMA: The J. Am. Med. Assoc. 1991 Dec 25;266(24):3466–3471. [PubMed] [Google Scholar]
- 72.Casadevall A., Goldman D.L., Feldmesser M. Vol. 95. Elsevier Masson SAS; 1997. Antibody-based therapies for infectious diseases: renaissance for an abandoned arsenal? pp. 247–257. (Bulletin De L'institut Pasteur). [Google Scholar]
- 73.Molecular biotechnology : Principles and Applications of Recombinant DNA - NLM Catalog - NCBI [Internet]. [cited 2021 Jul 2]. Available from: https://www.ncbi.nlm.nih.gov/nlmcatalog/101758497.
- 74.Katsuda T., Sonoda H., Kumada Y., Yamaji H. Production of antibody fragments in escherichia coli. Methods in Mol. Biol. 2012;907:305–324. doi: 10.1007/978-1-61779-974-7_18. [DOI] [PubMed] [Google Scholar]
- 75.Zarschler K., Witecy S., Kapplusch F., Foerster C., Stephan H. High-yield production of functional soluble single-domain antibodies in the cytoplasm of Escherichia coli. Microb. Cell Fact. 2013 Oct 27;12(1) doi: 10.1186/1475-2859-12-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferndahl C., Bonander N., Logez C., Wagner R., Gustafsson L., Larsson C., et al. Increasing cell biomass in Saccharomyces cerevisiae increases recombinant protein yield: the use of a respiratory strain as a microbial cell factory. Microb. Cell Fact. 2010 Jun 17;9 doi: 10.1186/1475-2859-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jahic M., Rotticci-Mulder J., Martinelle M., Hult K., Enfors S.O. Modeling of growth and energy metabolism of Pichia pastoris producing a fusion protein. Bioprocess Biosyst. Eng. 2002 Mar 1;24(6):385–393. [Google Scholar]
- 78.Molecular Biotechnology: Principles and Applications of Recombinant DNA, 5th Edition Wiley [Internet]. [cited 2021 Jul 2]. Available from: https://www.wiley.com/en-us/Molecular+Biotechnology%3A+Principles+and+Applications+of+Recombinant+DNA%2C+5th+Edition-p-9781555819361.
- 79.Stech M., Nikolaeva O., Thoring L., Stöcklein W.F.M., Wüstenhagen D.A., Hust M., et al. Cell-free synthesis of functional antibodies using a coupled in vitro transcription-Translation system based on CHO cell lysates. Sci. Rep. 2017 Dec 1;7(1) doi: 10.1038/s41598-017-12364-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oh I.S., Lee J.C., Lee M.S., Chung J.H., Kim D.M. Cell-free production of functional antibody fragments. Bioprocess Biosyst. Eng. 2010;33(1):127–132. doi: 10.1007/s00449-009-0372-3. [DOI] [PubMed] [Google Scholar]
- 81.JD M., HR H., TP B., J M., AD G., G W. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J. Mol. Biol. 1991 Dec 5;222(3):581–597. doi: 10.1016/0022-2836(91)90498-u. [DOI] [PubMed] [Google Scholar]
- 82.A P., L B. Phage display of antibody fragments. Curr. Protein Pept. Sci. 2000 Mar 25;1(2):155–169. doi: 10.2174/1389203003381397. [DOI] [PubMed] [Google Scholar]
- 83.Alfaleh M.A., Alsaab H.O., Mahmoud A.B., Alkayyal A.A., Jones M.L., Mahler S.M., et al. Phage display derived monoclonal antibodies: from bench to bedside. Front. Immunol. 2020;11(August 2020) doi: 10.3389/fimmu.2020.01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011 Mar 4;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 85.Witsch E., Sela M., Yarden Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010 Apr;25(2):85–101. doi: 10.1152/physiol.00045.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tai W., Mahato R., Cheng K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control Release. 2010 Sep 15;146(3):264–275. doi: 10.1016/j.jconrel.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pabla B., Bissonnette M., Konda V.J. Colon cancer and the epidermal growth factor receptor: current treatment paradigms, the importance of diet, and the role of chemoprevention. World J. Clin. Oncol. 2015 Oct 10;6(5):133–141. doi: 10.5306/wjco.v6.i5.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ponz-Sarvisé M., Rodríguez J., Viudez A., Chopitea A., Calvo A., García-Foncillas J., et al. Epidermal growth factor receptor inhibitors in colorectal cancer treatment: what's new? World J. Gastroenterol. 2007 Nov 28;13(44):5877–5887. doi: 10.3748/wjg.v13.i44.5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kholodenko R.V., Kalinovsky D.V., Doronin I.I., Ponomarev E.D., Kholodenko I.V. Antibody fragments as potential biopharmaceuticals for cancer therapy: success and limitations. Curr. Med. Chem. 2019;26(3):396–426. doi: 10.2174/0929867324666170817152554. [DOI] [PubMed] [Google Scholar]
- 90.Speirs C.K., Hwang M., Kim S., Li W., Chang S., Varki V., et al. Harnessing the cell death pathway for targeted cancer treatment. Am. J. Cancer Res. 2011;1(1):43–61. [PMC free article] [PubMed] [Google Scholar]
- 91.Yang F., Wen W., Qin W. Bispecific antibodies as a development platform for new concepts and treatment strategies. Int. J. Mol. Sci. 2016 Dec 28;18(1):E48. doi: 10.3390/ijms18010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brinkmann U., Kontermann R.E. The making of bispecific antibodies. MAbs. 2017 Mar;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sedykh S.E., Prinz V.V., Buneva V.N., Nevinsky G.A. Bispecific antibodies: design, therapy, perspectives. Drug Des Devel Ther. 2018;12:195–208. doi: 10.2147/DDDT.S151282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Przepiorka D., Ko C.W., Deisseroth A., Yancey C.L., Candau-Chacon R., Chiu H.J., et al. FDA Approval: blinatumomab. Clin. Cancer Res. 2015 Sep 15;21(18):4035–4039. doi: 10.1158/1078-0432.CCR-15-0612. [DOI] [PubMed] [Google Scholar]
- 95.Scott L.J., Kim E.S. Emicizumab-kxwh: first Global Approval. Drugs. 2018 Feb;78(2):269–274. doi: 10.1007/s40265-018-0861-2. [DOI] [PubMed] [Google Scholar]
- 96.Brischwein K., Parr L., Pflanz S., Volkland J., Lumsden J., Klinger M., et al. Strictly target cell-dependent activation of T cells by bispecific single-chain antibody constructs of the BiTE class. J. Immunother. 2007 Dec;30(8):798–807. doi: 10.1097/CJI.0b013e318156750c. [DOI] [PubMed] [Google Scholar]
- 97.Dustin M.L. The immunological synapse. Cancer Immunol. Res. 2014 Nov;2(11):1023–1033. doi: 10.1158/2326-6066.CIR-14-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Stinchcombe J.C., Bossi G., Booth S., Griffiths G.M. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001 Nov;15(5):751–761. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 99.Thiery J., Keefe D., Boulant S., Boucrot E., Walch M., Martinvalet D., et al. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol. 2011 Jun 19;12(8):770–777. doi: 10.1038/ni.2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Trapani J.A. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust. N Z J. Med. 1995 Dec;25(6):793–799. doi: 10.1111/j.1445-5994.1995.tb02883.x. [DOI] [PubMed] [Google Scholar]
- 101.Chofflon I., Dietrich P.Y., Thang N.N.T. [Antibody-drug conjugates: a new therapeutic class?] Rev. Med. Suisse. 2013 May 22;9(387):1080–1082. 1084–6. [PubMed] [Google Scholar]
- 102.Sievers E.L., Senter P.D. Antibody-drug conjugates in cancer therapy. Annu. Rev. Med. 2013;64:15–29. doi: 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- 103.Lambert J.M., Berkenblit A. Antibody-drug conjugates for cancer treatment. Annu. Rev. Med. 2018 Jan 29;69:191–207. doi: 10.1146/annurev-med-061516-121357. [DOI] [PubMed] [Google Scholar]
- 104.Fu Z., Li S., Han S., Shi C., Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022 Mar 22;7:93. doi: 10.1038/s41392-022-00947-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Birrer M.J., Moore K.N., Betella I., Bates R.C. Antibody-drug conjugate-based therapeutics: state of the science. J. Natl. Cancer Inst. 2019 Jun 1;111(6):538–549. doi: 10.1093/jnci/djz035. [DOI] [PubMed] [Google Scholar]
- 106.Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs. 2016 Jun;8(4):659–671. doi: 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ritchie M., Tchistiakova L., Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs. 2013 Feb;5(1):13–21. doi: 10.4161/mabs.22854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.El Khatib S., Omar M. Nutritional considerations in childhood acute lymphoblastic leukemia. Cancer and Oncol. Res. 2020 Jan;6(1):11–25. [Google Scholar]
- 109.Norsworthy K.J., Ko C.W., Lee J.E., Liu J., John C.S., Przepiorka D., et al. FDA approval summary: mylotarg for treatment of patients with relapsed or refractory CD33-positive acute myeloid leukemia. Oncologist. 2018 Sep;23(9):1103–1108. doi: 10.1634/theoncologist.2017-0604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cavaco M., Castanho M.A.R.B., Neves V. The use of antibody-antibiotic conjugates to fight bacterial infections. Front. Microbiol. 2022;13 doi: 10.3389/fmicb.2022.835677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Abdollahpour-Alitappeh M., Lotfinia M., Gharibi T., Mardaneh J., Farhadihosseinabadi B., Larki P., et al. Antibody-drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol. 2019 May;234(5):5628–5642. doi: 10.1002/jcp.27419. [DOI] [PubMed] [Google Scholar]
- 112.Dosio F., Brusa P., Cattel L. Immunotoxins and anticancer drug conjugate assemblies: the role of the linkage between components. Toxins (Basel) 2011 Jul;3(7):848–883. doi: 10.3390/toxins3070848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Erickson H.K., Widdison W.C., Mayo M.F., Whiteman K., Audette C., Wilhelm S.D., et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010 Jan;21(1):84–92. doi: 10.1021/bc900315y. [DOI] [PubMed] [Google Scholar]
- 114.Katz J., Janik J.E., Younes A. Brentuximab Vedotin (SGN-35) Clin. Cancer Res. 2011 Oct 15;17(20):6428–6436. doi: 10.1158/1078-0432.CCR-11-0488. [DOI] [PubMed] [Google Scholar]
- 115.Lambert J.M., Chari R.V.J. Ado-trastuzumab Emtansine (T-DM1): an antibody-drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014 Aug 28;57(16):6949–6964. doi: 10.1021/jm500766w. [DOI] [PubMed] [Google Scholar]
- 116.Hoffmann R.M., Coumbe B.G.T., Josephs D.H., Mele S., Ilieva K.M., Cheung A., et al. Antibody structure and engineering considerations for the design and function of antibody drug conjugates (ADCs) Oncoimmunology. 2018;7(3) doi: 10.1080/2162402X.2017.1395127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang A.C., Wang I.Y. Cleavage sites of human IgGl immunoglobulin by papain. Immunochemistry. 1977;14(3):197–200. doi: 10.1016/0019-2791(77)90194-x. [DOI] [PubMed] [Google Scholar]
- 118.Chothia C., Novotný J., Bruccoleri R., Karplus M. Domain association in immunoglobulin molecules. The packing of variable domains. J. Mol. Biol. 1985 Dec 5;186(3):651–663. doi: 10.1016/0022-2836(85)90137-8. [DOI] [PubMed] [Google Scholar]
- 119.Mitsue K., Shoji O., Teruo O. An unusual papain cleavage of a human IgG1 (λ) myeloma protein (Mot) Mol. Immunol. 1982;19(9):1095–1103. doi: 10.1016/0161-5890(82)90320-0. [DOI] [PubMed] [Google Scholar]
- 120.Bazin-Redureau M.I., Renard C.B., Scherrmann J.M.G. Pharmacokinetics of heterologous and homologous immunoglobulin G, F(ab’)2 and Fab after intravenous administration in the rat. J. Pharmacy and Pharmacol. 1997 Mar 1;49(3):277–281. doi: 10.1111/j.2042-7158.1997.tb06795.x. [DOI] [PubMed] [Google Scholar]
- 121.Jones R.G.A., Landon J. Enhanced pepsin digestion: a novel process for purifying antibody F(ab′)2 fragments in high yield from serum. J. Immunol. Methods. 2002 May 1;263(1–2):57–74. doi: 10.1016/s0022-1759(02)00031-5. [DOI] [PubMed] [Google Scholar]
- 122.Jones R.G.A., Landon J. A protocol for ‘enhanced pepsin digestion’: a step by step method for obtaining pure antibody fragments in high yield from serum. J. Immunol. Methods. 2003 Apr 1;275(1–2):239–250. doi: 10.1016/s0022-1759(03)00005-x. [DOI] [PubMed] [Google Scholar]
- 123.Li Z., Krippendorff B.F., Sharma S., Walz A.C., Lavé T., Shah D.K. Influence of molecular size on tissue distribution of antibody fragments. MAbs. 2016 Jan 1;8(1):113–119. doi: 10.1080/19420862.2015.1111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tiller K.E., Tessier P.M. Vol. 17. Annual Reviews Inc.; 2015. Advances in Antibody Design; pp. 191–216. (Annual Review of Biomedical Engineering). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jain A., Jain S.K. Vol. 25. Begell House Inc.; 2008. PEGylation: an approach for drug delivery. A review; pp. 403–447. (Critical Reviews in Therapeutic Drug Carrier Systems). [DOI] [PubMed] [Google Scholar]
- 126.Sheridan C. Ablynx's nanobody fragments go places antibodies cannot. Nat. Biotechnol. 2017 Dec 8;35(12):1115–1117. doi: 10.1038/nbt1217-1115. [DOI] [PubMed] [Google Scholar]
- 127.Dozier J.K., Distefano M.D. Site-specific PEGylation of therapeutic proteins. Int. J. Mol. Sci. 2015 Oct 28;16(10):25831–25864. doi: 10.3390/ijms161025831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Bird R.E., Hardman K.D., Jacobson J.W., Johnson S., Kaufman B.M., Lee S.M., et al. Single-chain antigen-binding proteins. Science. 1988;242(4877):423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- 129.Huston J.S., Levinson D., Mudgett-Hunter M., Tai M.S., Novotny J., Margolies M.N., et al. Protein engineering of antibody binding sites: recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1988;85(16):5879–5883. doi: 10.1073/pnas.85.16.5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.HS L., L S., R D.P., M G., M Z., EA P., et al. Generation and characterization of a novel single-gene-encoded single-chain immunoglobulin molecule with antigen binding activity and effector functions. Mol. Immunol. 1999 Jan;36(1):61–71. doi: 10.1016/s0161-5890(98)00109-6. [DOI] [PubMed] [Google Scholar]
- 131.JT K., MJ H., JA W. An improved single-chain Fab platform for efficient display and recombinant expression. J. Mol. Biol. 2015 Jan 30;427(2):576–586. doi: 10.1016/j.jmb.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Miller T.W., Messer A. Gene Therapy of the Central Nervous System: From Bench to Bedside. Elsevier Inc.; 2006. Gene therapy for CNS diseases using intrabodies; pp. 133–149. [Google Scholar]
- 133.Montoliu-Gaya L., Esquerda-Canals G., Bronsoms S., Villegas S. Production of an anti-Aβ antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic effect. PLoS ONE. 2017 Aug 1;12(8) doi: 10.1371/journal.pone.0181480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yokota T., Milenic D.E., Whitlow M., Schlom J. Rapid Tumor Penetration of a Single-Chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992;52(12) [PubMed] [Google Scholar]
- 135.Cumber A.J., Ward E.S., Winter G., Parnell G.D., Wawrzynczak E.J. Comparative stabilities in vitro and in vivo of a recombinant mouse antibody FvCys fragment and a bisFvCys conjugate. The J. Immunol. 1992;149(1) [PubMed] [Google Scholar]
- 136.Sanz L., Cuesta Á.M., Compte M., Álvarez-Vallina L. Vol. 26. Nature Publishing Group; 2005. Antibody engineering: facing new challenges in cancer therapy; pp. 641–648. (Acta Pharmacologica Sinica). [DOI] [PubMed] [Google Scholar]
- 137.Yannuzzi N.A., Freund K.B. Brolucizumab: evidence to date in the treatment of neovascular age-related macular degeneration. Clin. Ophthalmol. 2019 Jul 24;13:1323–1329. doi: 10.2147/OPTH.S184706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eyvazi S., Farajnia S., Dastmalchi S., Kanipour F., Zarredar H., Bandehpour M. Antibody based EpCAM targeted therapy of cancer, review and update. Curr. Cancer Drug Targets. 2018;18(9):857–868. doi: 10.2174/1568009618666180102102311. [DOI] [PubMed] [Google Scholar]
- 139.Vlachostergios P.J., Jakubowski C.D., Niaz M.J., Lee A., Thomas C., Hackett A.L., et al. Antibody-drug conjugates in bladder cancer. Bladder Cancer. 2018 Jul 30;4(3):247–259. doi: 10.3233/BLC-180169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Amiri S.A., Zarei N., Enayati S., Azizi M., Khalaj V., Shahhosseini S. Expression optimization of anti-cd22 scfv-apoptin fusion protein using experimental design methodology. Iran Biomed J. 2018 Jan;22(1):66–69. doi: 10.22034/ibj.22.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brinkmann U., Kontermann R.E. The making of bispecific antibodies. MAbs. 2017;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.JU S., MK G., PR D., JS M., DA V. Heterodimeric bispecific single chain variable fragments (scFv) killer engagers (BiKEs) enhance NK-cell activity against CD133+ colorectal cancer cells. Target Oncol. 2016 Jun 1;11(3):353. doi: 10.1007/s11523-015-0391-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Korn T., Nettelbeck D.M., Völkel T., Müller R., Kontermann R.E. Recombinant bispecific antibodies for the targeting of adenoviruses to CEA-expressing tumour cells: a comparative analysis of bacterially expressed single-chain diabody and tandem scFv. J. Gene Med. 2004 Jun 1;6(6):642–651. doi: 10.1002/jgm.555. [DOI] [PubMed] [Google Scholar]
- 144.Atwell J.L., Breheney K.A., Lawrence L.J., McCoy A.J., Kortt A.A., Hudson P.J. scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. Design and Selection. 1999 Jul 1;12(7):597–604. doi: 10.1093/protein/12.7.597. [DOI] [PubMed] [Google Scholar]
- 145.A T., RC R., O D., AA K., HR H., PJ H. Design and application of diabodies, triabodies and tetrabodies for cancer targeting. J. Immunol. Methods. 2001 Feb 1;248(1–2):47–66. doi: 10.1016/s0022-1759(00)00342-2. [DOI] [PubMed] [Google Scholar]
- 146.Iliades P., Kortt A.A., Hudson P.J. Triabodies: single chain Fv fragments without a linker form trivalent trimers. FEBS Lett. 1997 Jun 16;409(3):437–441. doi: 10.1016/s0014-5793(97)00475-4. [DOI] [PubMed] [Google Scholar]
- 147.JL A., KA B., LJ L., AJ M., AA K., PJ H. scFv multimers of the anti-neuraminidase antibody NC10: length of the linker between VH and VL domains dictates precisely the transition between diabodies and triabodies. Protein Eng. 1999;12(7):597–604. doi: 10.1093/protein/12.7.597. [DOI] [PubMed] [Google Scholar]
- 148.M M., G R., P K. A small bispecific antibody construct expressed as a functional single-chain molecule with high tumor cell cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 1995;92(15):7021–7025. doi: 10.1073/pnas.92.15.7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discovery. 2019 Jun 7;18(8):585–608. doi: 10.1038/s41573-019-0028-1. 2019 18:8. [DOI] [PubMed] [Google Scholar]
- 150.Eggenreich B., Scholz E., Wurm D.J., Forster F., Spadiut O. The production of a recombinant tandem single chain fragment variable capable of binding prolamins triggering celiac disease. BMC Biotechnol. 2018 May 29;18:30. doi: 10.1186/s12896-018-0443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bascuñán K.A., Vespa M.C., Araya M. Celiac disease: understanding the gluten-free diet. Eur. J. Nutr. 2017 Mar;56(2):449–459. doi: 10.1007/s00394-016-1238-5. [DOI] [PubMed] [Google Scholar]
- 152.Schuppan D., Junker Y., Barisani D. Celiac disease: from pathogenesis to novel therapies. Gastroenterology. 2009 Dec;137(6):1912–1933. doi: 10.1053/j.gastro.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 153.Hundsberger H., Koppensteiner A., Hofmann E., Ripper D., Pflüger M., Stadlmann V., et al. A screening approach for identifying gliadin neutralizing antibodies on epithelial intestinal caco-2 cells. SLAS Discov. 2017 Sep;22(8):1035–1043. doi: 10.1177/2472555217697435. [DOI] [PubMed] [Google Scholar]
- 154.Stadlmann V., Harant H., Korschineck I., Hermann M., Forster F., Missbichler A. Novel avian single-chain fragment variable (scFv) targets dietary gluten and related natural grain prolamins, toxic entities of celiac disease. BMC Biotechnol. 2015 Dec 1;15:109. doi: 10.1186/s12896-015-0223-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.P H., T P., G W. Proceedings of the National Academy of Sciences of the United States of America. Vol. 90. 1993 Jul 15. Diabodies’: small bivalent and bispecific antibody fragments; pp. 6444–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Diabodies: Molecular Engineering and Therapeutic Applications - PubMed [Internet]. [cited 2021 Jul 7]. Available from: https://pubmed.ncbi.nlm.nih.gov/20016854/.
- 157.DG D., CY X., XB S., GL D., SJ D. Anti-HLA-DR/anti-DOTA diabody construction in a modular gene design platform: bispecific antibodies for pretargeted radioimmunotherapy. Cancer Biother. Radiopharm. 2001;16(6):525–535. doi: 10.1089/10849780152752128. [DOI] [PubMed] [Google Scholar]
- 158.SM K., G M., M B., U R., B C., F L.G., et al. Effect of domain order on the activity of bacterially produced bispecific single-chain Fv antibodies. J. Mol. Biol. 2003 Jun 27;330(1):99–111. doi: 10.1016/s0022-2836(03)00526-6. [DOI] [PubMed] [Google Scholar]
- 159.A P., P P. New protein engineering approaches to multivalent and bispecific antibody fragments. Immunotechnol. : An Int. J. Immunol. Eng. 1997;3(2):83–105. doi: 10.1016/s1380-2933(97)00067-5. [DOI] [PubMed] [Google Scholar]
- 160.Klein C., Schaefer W., Regula J.T. The use of CrossMAb technology for the generation of bi- and multispecific antibodies. MAbs. 2016 Aug 17;8(6):1010–1020. doi: 10.1080/19420862.2016.1197457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.W S., JT R., M B., J S., R C., H D., et al. Proceedings of the National Academy of Sciences of the United States of America. Vol. 108. 2011 Jun 20. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies; pp. 11187–11192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.C S., M M., A H., Z Z., NY Y., J P., et al. Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies. Nat. Biotechnol. 2013 Jul 7;31(8):753–758. doi: 10.1038/nbt.2621. [DOI] [PubMed] [Google Scholar]
- 163.EA R., DM G., TM C., WJ M., RM S., CH C. Proceedings of the National Academy of Sciences of the United States of America. Vol. 103. 2006 May 2. Stably tethered multifunctional structures of defined composition made by the dock and lock method for use in cancer targeting; pp. 6841–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lindhofer H., Mocikat R., Steipe B., Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. The J. Immuno. 1995;155(1) [PubMed] [Google Scholar]
- 165.Schuurman J., Graus Y.F., Labrijn A.F., Ruuls S.R., Parren P.W. Opening the door to innovation. MAbs. 2014;6(4):812. doi: 10.4161/mabs.29004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.C W., H Y., C G., S B., R M., A C., et al. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 2007 Nov;25(11):1290–1297. doi: 10.1038/nbt1345. [DOI] [PubMed] [Google Scholar]
- 167.PA B., P K., R B. BiTE: teaching antibodies to engage T-cells for cancer therapy. Curr. Opin. Mol. Ther. 2009 Feb 1;11(1):22–30. [PubMed] [Google Scholar]
- 168.PA M., W Z., GJ R., S B., H L., L H., et al. Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood. 2011 Apr 28;117(17):4542–4551. doi: 10.1182/blood-2010-09-306449. [DOI] [PubMed] [Google Scholar]
- 169.H A., GL D., SJ D. Monospecific bivalent scFv-SH: effects of linker length and location of an engineered cysteine on production, antigen binding activity and free SH accessibility. J. Immunol. Methods. 2006 Mar 20;310(1–2):100–116. doi: 10.1016/j.jim.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 170.RE K., U B. Bispecific antibodies. Drug Discov. Today. 2015 Jul 9;20(7):838–847. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 171.Uy G., Stewart S., Baughman J., Rettig M., Chichili G., Bonvini E., et al. A Phase I trial of MGD006 in patients with relapsed acute myeloid leukemia (AML) J. Immunother Cancer. 2014 Nov 6;2(Suppl 3):P87. [Google Scholar]
- 172.MacroGenics A. clinicaltrials.gov; 2022 Feb. Phase 1, First-in-Human, Open Label, Dose Escalation Study of MGD007, A Humanized gpA33 x CD3 DART® Protein in Patients With Relapsed/Refractory Metastatic Colorectal Carcinoma [Internet]https://clinicaltrials.gov/ct2/show/NCT02248805 [cited 2022 Apr 28]. Report No.: NCT02248805. Available from. [Google Scholar]
- 173.Gleason M.K., Verneris M.R., Todhunter D.A., Zhang B., McCullar V., Zhou S.X., et al. Bispecific and trispecific killer cell engagers directly activate human NK cells through CD16 signaling and induce cytotoxicity and cytokine production. Mol. Cancer Ther. 2012 Dec;11(12):2674–2684. doi: 10.1158/1535-7163.MCT-12-0692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Luke J.J., Patel M.R., Hamilton E.P., Chmielowski B., Ulahannan S.V., Kindler H.L., et al. A phase I, first-in-human, open-label, dose-escalation study of MGD013, a bispecific DART molecule binding PD-1 and LAG-3, in patients with unresectable or metastatic neoplasms. JCO. 2020 May 20;38(15_suppl):3004. –3004. [Google Scholar]
- 175.Jiang H., Ni H., Zhang P., Guo X., Wu M., Shen H., et al. PD-L1/LAG-3 bispecific antibody enhances tumor-specific immunity. Oncoimmunology. 2021;10(1) doi: 10.1080/2162402X.2021.1943180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Müller D., Karle A., Meißburger B., Höfig I., Stork R., Kontermann R.E. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J. Biol. Chem. 2007 Apr 27;282(17):12650–12660. doi: 10.1074/jbc.M700820200. [DOI] [PubMed] [Google Scholar]
- 177.El Khatib S, M Omar. Nutritional considerations in childhood acute lymphoblastic leukemia. Cancer and Oncol. Res. 2020;6(1):11–25. [Google Scholar]
- 178.D M., RE K. Bispecific antibodies for cancer immunotherapy: current perspectives. BioDrugs : Clin. Immunotherapeutics, Biopharmaceuticals and Gene Therapy. 2010;24(2):89–98. doi: 10.2165/11530960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 179.clinicaltrials.gov; 2020 Jan. Amgen Research (Munich) GmbH. A Confirmatory Multicenter, Single-arm Study to Assess the Efficacy, Safety, and Tolerability of the BiTE® Antibody Blinatumomab in Adult Patients With Minimal Residual Disease (MRD) of B-precursor Acute Lymphoblastic Leukemia (BLAST) [Internet]https://clinicaltrials.gov/ct2/show/NCT01207388 [cited 2022 Apr 28]. Report No.: NCT01207388. Available from. [Google Scholar]
- 180.Kontermann R.E., Brinkmann U. Bispecific antibodies. Drug Discov. Today. 2015 Jul 1;20(7):838–847. doi: 10.1016/j.drudis.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 181.Choi B.D., Yu X., Castano A.P., Bouffard A.A., Schmidts A., Larson R.C., et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019 Sep;37(9):1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]
- 182.Kipriyanov S.M., Moldenhauer G., Schuhmacher J., Cochlovius B., Von der Lieth C.W., Matys E.R., et al. Bispecific tandem diabody for tumor therapy with improved antigen binding and pharmacokinetics. J. Mol. Biol. 1999 Oct 15;293(1):41–56. doi: 10.1006/jmbi.1999.3156. [DOI] [PubMed] [Google Scholar]
- 183.GmbH Affimed. clinicaltrials.gov; 2019 Jun. A Pharmacodynamically-guided, Dose-escalation, Phase I Study to Assess the Safety of AFM11 (Recombinant Antibody Construct Against Human CD19 and CD3) in Patients With Relapsed and/Or Refractory CD19 Positive B-cell NHL. [Internet]https://clinicaltrials.gov/ct2/show/NCT02106091 [cited 2022 May 3]. Report No.: NCT02106091. Available from: [Google Scholar]
- 184.Reusch U., Duell J., Ellwanger K., Herbrecht C., Knackmuss S.H., Fucek I., et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19+ tumor cells. MAbs. 2015 May 4;7(3):584–604. doi: 10.1080/19420862.2015.1029216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Reusch U., Burkhardt C., Fucek I., Le Gall F., Le Gall M., Hoffmann K., et al. A novel tetravalent bispecific TandAb (CD30/CD16A) efficiently recruits NK cells for the lysis of CD30+ tumor cells. MAbs. 2014 Jun;6(3):728–739. doi: 10.4161/mabs.28591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Watanabe Y., Tanabe A., Hamakubo T., Nagatoishi S., Tsumoto K. Development of biparatopic bispecific antibody possessing tetravalent scFv-Fc capable of binding to ROBO1 expressed in hepatocellular carcinoma cells. J. Biochem. 2021 Oct 11;170(2):307–315. doi: 10.1093/jb/mvab049. [DOI] [PubMed] [Google Scholar]
- 187.zhen Hu S, L Shively, Raubitschek A., Sherman M., Williams L.E., Wong J.Y.C., et al. Minibody: a novel engineered anti-carcinoembryonic antigen antibody fragment (Single-Chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996;56(13) [PubMed] [Google Scholar]
- 188.Shahied L.S., Tang Y., Alpaugh R.K., Somer R., Greenspon D., Weiner L.M. Bispecific minibodies targeting HER2/neu and CD16 exhibit improved tumor lysis when placed in a divalent tumor antigen binding format*. 2004;. [DOI] [PubMed]
- 189.A W., A P. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001 Feb 2;305(5):989–1010. doi: 10.1006/jmbi.2000.4265. [DOI] [PubMed] [Google Scholar]
- 190.Y T., N J., C P., D H. Selection of linkers for a catalytic single-chain antibody using phage display technology. J. Biol. Chem. 1996;271(26):15682–15686. doi: 10.1074/jbc.271.26.15682. [DOI] [PubMed] [Google Scholar]
- 191.Kim Y.P., Park D., Kim J.J., Choi W.J., Lee S.H., Lee S.Y., et al. Effective therapeutic approach for head and neck cancer by an engineered minibody targeting the EGFR receptor. PLoS ONE. 2014;9(12) doi: 10.1371/journal.pone.0113442. Dec 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.M A., RF B., L F de C., JW C.H., JW L., JV G. Variable region sequence modulates periplasmic export of a single-chain Fv antibody fragment in Escherichia coli. BioTechniques. 1995 May 1;18(5):832. 835–8, 840. [PubMed] [Google Scholar]
- 193.Hu S., Shively L., Raubitschek A., Sherman M., Williams L.E., Wong J.Y., et al. Minibody: a novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-CH3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996 Jul 1;56(13):3055–3061. [PubMed] [Google Scholar]
- 194.Mayle K.M., Dern K.R., Wong V.K., Chen K.Y., Sung S., Ding K., et al. Engineering A11 minibody-conjugated, polypeptide-based gold nanoshells for prostate stem cell antigen (PSCA)–targeted photothermal therapy. SLAS Technol.: Translating Life Sci. Innovation. 2017 Feb 1;22(1):26–35. doi: 10.1177/2211068216669710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Song H., Du Y., Sgouros G., Prideaux A., Frey E., Wahl R.L. Therapeutic potential of 90Y- and 131I-labeled anti-CD20 monoclonal antibody in treating non-hodgkin's lymphoma with pulmonary involvement: a monte carlo–based dosimetric analysis. J. Nucl. Med. 2007 Jan;48(1):150–157. [PMC free article] [PubMed] [Google Scholar]
- 196.C H.C., T A., S M., G R., C H., EB S., et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363(6428):446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- 197.BP W., LG F., P van der L., PJ N. The structure of the llama heavy chain constant genes reveals a mechanism for heavy-chain antibody formation. Immunogenetics. 1999;50(1–2):98–101. doi: 10.1007/s002510050694. [DOI] [PubMed] [Google Scholar]
- 198.VK N., R H., L W., S M. Loss of splice consensus signal is responsible for the removal of the entire C(H)1 domain of the functional camel IGG2A heavy-chain antibodies. Mol. Immunol. 1999 Jun;36(8):515–24. [DOI] [PubMed]
- 199.Nguyen V.K., Hamers R., Wyns L., Muyldermans S. Camel heavy-chain antibodies: diverse germline VHH and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 2000 Mar 1;19(5):921. doi: 10.1093/emboj/19.5.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bannas P., Hambach J., Koch-Nolte F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies As Antitumor Therapeutics. Front. Immunol. 2017 Nov 22;8(NOV):1603. doi: 10.3389/fimmu.2017.01603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Singh P., Gao F., Bernat A. Nanobodies and their in vivo applications. Adv. Biosensors for Health Care Appl. 2019 Jan 1:263–277. [Google Scholar]
- 202.Siontorou C.G. Nanobodies as novel agents for disease diagnosis and therapy. Int. J. Nanomed. 2013 Nov 1;8:4215. doi: 10.2147/IJN.S39428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.de Vries Schultink A.H.M., Bol K., Doornbos R.P., Murat A., Wasserman E., Dorlo T.P.C., et al. Population pharmacokinetics of MCLA-128, a HER2/HER3 bispecific monoclonal antibody, in patients with solid tumors. Clin. Pharmacokinet. 2020 Jul;59(7):875–884. doi: 10.1007/s40262-020-00858-2. [DOI] [PubMed] [Google Scholar]
- 204.Schram A.M., Odintsov I., Espinosa-Cotton M., Khodos I., Sisso W.J., Mattar M.S., et al. Zenocutuzumab, a HER2xHER3 bispecific antibody, is effective therapy for tumors driven by NRG1 gene rearrangements. Cancer Discov. 2022 May 2;12(5):1233–1247. doi: 10.1158/2159-8290.CD-21-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gómez-Seguí I., Fernández-Zarzoso M., de la Rubia J. A critical evaluation of caplacizumab for the treatment of acquired thrombotic thrombocytopenic purpura. Expert Rev. Hematol. 2020 Nov;13(11):1153–1164. doi: 10.1080/17474086.2020.1819230. [DOI] [PubMed] [Google Scholar]
- 206.Scully M., Cataland S.R., Peyvandi F., Coppo P., Knöbl P., Kremer Hovinga J.A., et al. Caplacizumab treatment for acquired thrombotic thrombocytopenic purpura. N. Engl. J. Med. 2019 Jan 24;380(4):335–346. doi: 10.1056/NEJMoa1806311. [DOI] [PubMed] [Google Scholar]
- 207.D'Huyvetter M., Vos J.D., Caveliers V., Vaneycken I., Heemskerk J., Duhoux F.P., et al. Phase I trial of 131I-GMIB-Anti-HER2-VHH1, a new promising candidate for her2-targeted radionuclide therapy in breast cancer patients. J. Nucl. Med. 2021 Aug 1;62(8):1097–1105. doi: 10.2967/jnumed.120.255679. [DOI] [PubMed] [Google Scholar]
- 208.Keyaerts M., Xavier C., Heemskerk J., Devoogdt N., Everaert H., Ackaert C., et al. Phase I study of 68Ga-HER2-Nanobody for PET/CT assessment of HER2 expression in breast carcinoma. J. Nucl. Med. 2016 Jan;57(1):27–33. doi: 10.2967/jnumed.115.162024. [DOI] [PubMed] [Google Scholar]
- 209.Yu X., Yang Y.P., Dikici E., Deo S.K., Daunert S. Beyond antibodies as binding partners: the role of antibody mimetics in bioanalysis. https://doi.org/101146/annurev-anchem-061516-045205. 2017 Jun 12;10:293–320. [DOI] [PMC free article] [PubMed]
- 210.Qiu X.Q., Wang H., Cai B., Wang L.L., Yue S.T. Small antibody mimetics comprising two complementarity-determining regions and a framework region for tumor targeting. Nat. Biotechnol. 2007 Aug 5;25(8):921–929. doi: 10.1038/nbt1320. 2007 25:8. [DOI] [PubMed] [Google Scholar]
- 211.Schönfeld D., Matschiner G., Chatwell L., Trentmann S., Gille H., Hülsmeyer M., et al. An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proceedings of the Nat. Acad. Sci. 2009 May 19;106(20):8198–8203. doi: 10.1073/pnas.0813399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.J L., FY F., S S. Non-immunoglobulin based protein scaffolds. Curr. Opin. Biotechnol. 2011 Dec;22(6):843–848. doi: 10.1016/j.copbio.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 213.HK B., P A., A P. Engineering novel binding proteins from nonimmunoglobulin domains. Nat. Biotechnol. 2005 Oct;23(10):1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 214.Yu X., Yang Y.P., Dikici E., Deo S.K., Daunert S. Beyond antibodies as binding partners: the role of antibody mimetics in bioanalysis. Ann. Rev. analytical Chem. (Palo Alto, Calif) 2017 Jun 12;10(1):293. doi: 10.1146/annurev-anchem-061516-045205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Baloch A.R., Baloch A.W., Sutton B.J., Zhang X. Antibody mimetics: promising complementary agents to animal-sourced antibodies. http://dx.doi.org/103109/073885512014958431. 2014 Mar 3;36(2):268–75. [DOI] [PubMed]
- 216.AS G., D A., M H., A H., EC M., MF F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature. 1995;374(6518):168–173. doi: 10.1038/374168a0. [DOI] [PubMed] [Google Scholar]
- 217.J H., N H., KA Z., A S., N F., D M., et al. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng. Design & Selection : PEDS. 2010 Nov;23(11):827–834. doi: 10.1093/protein/gzq058. [DOI] [PubMed] [Google Scholar]
- 218.Samra B., Konopleva M., Isidori A., Daver N., DiNardo C. Venetoclax-based combinations in acute myeloid leukemia: current evidence and future directions. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.562558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Wei A.H., Strickland S.A., Hou J.Z., Fiedler W., Lin T.L., Walter R.B., et al. Venetoclax Combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J. Clin. Oncol. 2019 May 20;37(15):1277–1284. doi: 10.1200/JCO.18.01600. [DOI] [PMC free article] [PubMed] [Google Scholar]