

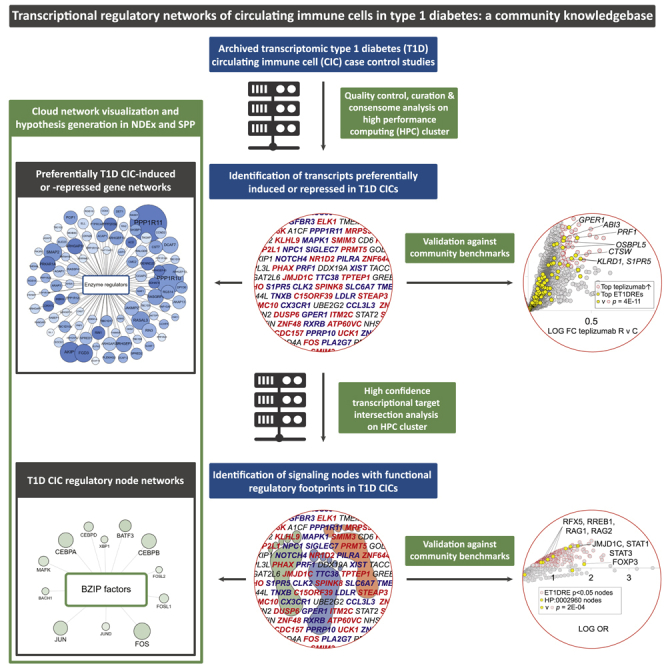
Summary
Investigator-generated transcriptomic datasets interrogating circulating immune cell (CIC) gene expression in clinical type 1 diabetes (T1D) have underappreciated re-use value. Here, we repurposed these datasets to create an open science environment for the generation of hypotheses around CIC signaling pathways whose gain or loss of function contributes to T1D pathogenesis. We firstly computed sets of genes that were preferentially induced or repressed in T1D CICs and validated these against community benchmarks. We then inferred and validated signaling node networks regulating expression of these gene sets, as well as differentially expressed genes in the original underlying T1D case:control datasets. In a set of three use cases, we demonstrated how informed integration of these networks with complementary digital resources supports substantive, actionable hypotheses around signaling pathway dysfunction in T1D CICs. Finally, we developed a federated, cloud-based web resource that exposes the entire data matrix for unrestricted access and re-use by the research community.
Subject areas: Bioinformatics, Biological database, Transcriptomics
Graphical abstract
Highlights
-
•
Re-use of transcriptomic type 1 diabetes (T1D) circulating immune cells (CICs) datasets
-
•
We generated transcriptional regulatory networks for T1D CICs
-
•
Use cases generate substantive hypotheses around signaling pathway dysfunction in T1D CICs
-
•
Networks are freely accessible on the web for re-use by the research community
Bioinformatics; Biological database; Transcriptomics
Introduction
Autoimmune destruction of pancreatic β cells in type 1 diabetes (T1D) is a major global public health concern whose cost in terms of human health and financial expense is immense. A substantial body of evidence indicates that deficient function of circulating immune cells (CICs) makes a significant contribution to the pathogenesis of T1D (Lehuen et al., 2010). Pathways controlling gene expression in normal CIC physiology and T1D comprise a complex network of signaling nodes, encompassing membrane and nuclear receptors, enzymes, transcription factors, and other ancillary factors. Decades of diligent investigation by the immune and inflammatory research communities have assembled a canon of regulatory paradigms for T1D, such as tumor necrosis factor, interleukin, and interferon receptor signaling (Wachlin et al., 2003). These advances notwithstanding, the diverse disciplines represented in the T1D research community are confronted by three interconnected informatic challenges. Firstly, a broader understanding of mechanisms underlying T1D would illuminate opportunities for label expansion of approved drugs. Secondly, existing T1D therapeutics have undesirable contra-indications and side effects, emphasizing the need for a systemic understanding of the diversity of physiological signaling pathways in which specific inflammatory nodes participate to drive T1D. Finally, to expand the range of known immunomodulatory paradigms in T1D, reduced-bias approaches are required to illuminate non-canonical aspects of autoinflammatory signaling that are less extensively characterized in the T1D research literature. In response to these needs, several web-based resources have been developed that aggregate and compute across public T1D genome-wide association studies (GWAS) (Bhattacharya et al., 2018; Mailman et al., 2007; T1DKP, 2021). Despite its use to generate clinically relevant insights in other autoimmune diseases (Haynes et al., 2020), however, the potential for computation across archived public clinical transcriptomic datasets to illuminate mechanisms underlying the pathogenesis of T1D has to date been largely untapped.
The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Information Network, DKNET (Whetzel et al., 2015), recently organized a community challenge, the D-Challenge, in which participants were challenged to leverage digital research resources to generate novel hypotheses around the pathogenesis of T1D. As a contribution to the D-Challenge, we repurposed public clinical T1D transcriptomic datasets to generate sets of genes that are preferentially induced or repressed in T1D CICs. We then computed transcriptional regulatory networks to identify evidence for nodes regulating expression of these genes, as well as those in the original underlying datasets. At each step, we carried out rigorous, systematic validation of the networks against a variety of clinical and mechanistic benchmark datasets. We then developed a set of three use cases that illustrate how researchers for whom no informatics expertise is assumed can generate or refine compelling hypotheses that can be tested at the bench. Finally, we incorporated the networks into a rich web-based visualization environment to ensure unrestricted access for the broader research community for the generation of novel hypotheses around T1D-relevant CIC signaling pathways.
Results
Dataset selection and annotation
Please refer to the Methods section for details on dataset collection and processing. We used Gene Expression Omnibus and ArrayExpress to identify a total of 17 transcriptomic datasets interrogating gene expression between T1D and normoglycemic CICs. The RNA source breakdown for these datasets was as follows: peripheral blood mononuclear cells (PBMCs), 6; CD14+ monocytes, 5; whole blood, 2; and CD3+ T cells, dendritic cells, hematopoietic stem cells, and neutrophils, 1 each. From these datasets, we generated a total of 26 T1D v control contrasts. Table S1 shows an overview dataset- and contrast-level metadata for the 26 experiments. We annotated the T1D CIC datasets with respect to a variety of clinical variables using information provided in the methodological sections of the original published articles. Table S2 shows the full dataset- and contrast-level clinical metadata.
Computation of differential gene expression networks for circulating immune cells in T1D
We previously described transcriptomic consensome meta-analysis, which ranks genes according to their rates of significant differential expression across related but independent expression profiling experiments (Bissig-Choisat et al., 2021; Ochsner et al., 2019, 2020). Here, we adapted the consensome algorithm to identify evidence for genes that were preferentially induced or repressed in T1D CICs relative to healthy controls across the 26 contrasts. We first subjected the 26 T1D case:control contrasts to a uniform differential gene expression pipeline as previously described (Bissig-Choisat et al., 2021; Ochsner et al., 2019, 2020) and committed the gene lists to the consensome algorithm to generate a T1D CIC consensome (Table S3). The consensome assigns a geometric mean fold change (GMFC), p value, and adjusted p value to each gene according to the frequency with which they are significantly differentially expressed across the 26 contrasts.
Because gene-level case v control GMFCs were modest (Table S3), we wished to identify subsets of the consensome transcripts that were preferentially induced or repressed across the independent T1D expression profiling experiments, To do this, we generated a ∼four million data point differential expression data matrix for a total of 72,017 unique features across the 26 independent contrasts (Table S4). In this context, we define feature as either (i) in the case of an expression array experiment, any probeset annotated to a specific gene or (ii) in the case of an RNA-Seq experiment, a unique gene-level annotation. We then isolated a group of features (i) whose mapped gene had a T1D CIC consensome GMFC>1.2, (ii) were not significantly induced in any contrast, and (iii) were significantly (p < 0.05) repressed in one or more contrasts, and designated these exclusively T1D-repressed (ET1DR) features (Table S4, column BM). Similarly, we identified a group of features (i) whose mapped gene had a consensome GMFC>1.2, (ii) were not significantly repressed in any contrast, and (iii) were significantly (p < 0.05) induced in one or more contrasts, and designated these exclusively T1D-induced (ET1DI) features (Table S4, column BN). We then used the hypergeometric test to compare the number of features annotated to a specific gene in these subsets with the total number for that gene in the universe of features (n = 72,017). Using this approach, we identified 2063 genes whose mapped features were significantly enriched (p < 0.05; 625 with p adj <0.05) among the ET1DR features, and designated these ET1DR-enriched genes (ET1DREs; Table S5, columns H–J). We also identified 1229 genes whose mapped features were significantly enriched (p < 0.05; 374 with p adj <0.05) among the ET1DI features, and designated these ET1DI-enriched genes (ET1DIEs; Table S5, columns K–M). We refer to this adaptation of the original consensome pipeline as consensome feature-level enrichment analysis (C-FLEE).
Validation of differential gene expression networks for circulating immune cells in T1D
In order for the C-FLEE-generated ET1DRE and ET1DIE transcriptional signatures to be a reliable resource for generating novel hypotheses around signaling events in T1DCICs, they would be required to assign elevated rankings to genes with established connections to T1D. Accordingly, we designed an approach to benchmarking these signatures in a computationally rigorous manner against three relevant, independent resources. We first pressure-tested the C-FLEE gene sets against a clinical T1D transcriptomic dataset that was not included in our analysis. We identified a study by Xhonneux and colleagues involving longitudinal expression profiling analysis of blood samples from 400 children at high risk for islet autoimmunity and T1D (Xhonneux et al., 2021). This study resolved a gene expression signature, termed IAAsig (n = 64; Table S3, column R), whose expression profile was specific to T1D subjects who initially developed insulin autoantibodies (IAAs). Development of IAAs has been previously shown to be a phenomenon typically associated with earlier and more rapid progression of T1D (Battaglia et al., 2020; Ziegler et al., 2013). Reflecting the accuracy with which C-FLEE analysis assigned elevated rankings to genes implicated in the development of T1D, the IAAsig gene set was strongly enriched among the top-ranked genes in the T1D consensome (ENR = 10, p = 4E-10; Figure 1A), an enrichment that was strikingly increased among the top ET1DRE subset of the top T1D consensome gene set (ENR = 85, 1E-07; Figure 1B).
Figure 1.

Consensome feature level expression enrichment (C-FLEE) analysis identifies genes preferentially repressed or induced in T1D v control circulating immune cells
Selected transcripts at the intersection of the indicated gene sets are labeled. Refer to supplementary text for cut-offs used for hypergeometric tests.
(A) Scatterplot showing enrichment of Xhonneux IAAsig genes among the top-ranked genes in the T1D consensome.
(B) Scatterplot showing super-enrichment of Xhonneux IAAsig genes among the top-ranked ET1DREs in the T1D consensome.
(C) Volcano plot showing enrichment of top ET1DIE genes among top teplizumab responder (R) v control (C)-repressed genes in ABATE T1D cohort.
(E) Scatterplot showing enrichment of strong T1D GWAS candidates among top-ranked ET1DRE genes in the T1D consensome. (E) Selected top 20 ET1DRE (left panel) and ETD1IE (right panel) genes annotated using the three-tier (category, class, and family) SPP-controlled vocabulary {Ochsner, 2019 #83}. Circle size maps to enrichment p value: for reference, TPTEP is 1E-13 and DEFA1 is 4E-07. Light blue – dark blue (ET1DREs) and yellow-orange-red (ET1DIEs) color intensity maps to enrichment odds ratio: for reference in the ET1DREs, ABHD17A is 5.8 and PPP1R11 is 7.7; for reference in the ET1DIEs, IGLC is 7.3 and NAMPT is 11.8. Transcript class borders are color-coded according to their SPP classification category {Ochsner, 2019 #83} as follows: receptors (orange), enzymes (blue), transcription factors (green), and co-nodes (gray).
Because T cells play a key role in the pathophysiology of T1D, induction of T cell unresponsiveness, or tolerance, has been a focus of therapeutic efforts in T1D. CD3 is a T cell co-receptor complex that is involved in activation of both cytotoxic T cells (CD8+ naïve T cells) and T helper cells (CD4+ naïve T cells), and whose blockade has been extensively explored as a therapeutic approach to maintaining β-cell function in T1D. As a second approach to validation of the ET1DRE and ET1DIE gene sets, we located a transcriptomic dataset generated by the AbATE (Autoimmunity-blocking Antibody for Tolerance in Recently Diagnosed Type 1 Diabetes) clinical trial (Long et al., 2016). This dataset profiled whole blood gene expression between T1D subjects who responded to treatment with the anti-CD3 antibody teplizumab and control individuals. We reasoned that the reliability of the ET1DREs would be reflected in their enrichment among genes induced in teplizumab-responsive subjects and, conversely, the reliability of the ET1DIEs would be indicated by their enrichment among genes repressed in teplizumab-responsive subjects. Supporting the trustworthiness of our transcriptional networks, we observed very robust enrichment of ET1DRE transcripts within genes induced in teplizumab-responsive subjects (ENR = 202, p = 4E-11; Figure 1C and Table S6, columns G&H) and, more marginally, of ET1DIE transcripts within genes repressed in teplizumab-responsive subjects (ENR = 28, p = 3E-2; Figure S1A and Table S6, columns I&J).
Although single nucleotide polymorphisms (SNPs) cannot necessarily be assumed to impact expression of the gene in which they are located, it was reasonable to expect some degree of overlaps between our computed gene lists and genes linked to T1D through SNPs generated by GWAS studies. The third approach to validation of the ET1DIEs and ET1DREs therefore involved the Type 1 Diabetes Knowledge Portal (T1DKP, 2021), which computes across independent T1D GWAS cohorts to assign measures of significance of associations between specific SNPs and the incidence of T1D. We retrieved from T1DKP a set of genes mapped to SNPs that had exhibited significant GWAS associations with T1D (Table S5, column N). Reflecting the reliability of the ET1DRE gene set, we observed a robust intersection between genes with the strongest associations with the incidence of T1D and the highest-ranked ET1DRE genes (ENR = 342, p = 1E-05; Figure 1D). A much more marginal intersection was found between top-ranked T1DKP T1D GWAS genes and ET1DIE genes (ENR = 13, p = 1E-02; Figure S1B), which may indicate that the T1DKP-curated T1D SNPs primarily represent loss of function of proteins encoded by genes in which they are located. As an additional level of validation, we used T1DKP to generate a list of 105 genes that had at least one significant GWAS association with T1D and three other autoimmune conditions—Crohn’s disease, ulcerative colitis, and irritable bowel syndrome—and designated these pan-autoimmune GWAS genes (PAGGs; Table S7 & Table S5, column O). Encouragingly, and suggesting the broader significance of the ET1DRE genes across diverse autoimmune conditions, PAGGs were extremely robustly enriched among the highest-ranked consensome ET1DREs (ENR = 1253, p = 8E-07; Figure S1C).
ET1DRE and ET1DIE T1D consensome subsets are a functionally diverse set of immune effectors
Signal transduction pathways involve functional interactions between cellular signaling nodes (Taniguchi et al., 2006). We previously proposed a classification of these nodes into four high level categories, three of which (receptors, enzymes, and transcription factors) are historically well defined, while a fourth (co-nodes) encompasses a functionally diverse set of nodes that do not fall into the one of the other three categories (Ochsner et al., 2019). Annotation of the ET1DRE and ET1DIE genes using this classification afforded us insight into the diversity of functions impacted in T1D circulating immune cells. Figure 1E shows a Cytoscape-style representation of the top 20 ET1DRE (left panel) and ET1DIE (right panel) genes; the full list is in Table S5. ET1DREs contained numerous genes with known roles in immunosuppression and/or that are functionally depleted in autoimmune disorders. In the context of the receptor category, ET1DREs included a receptor for TGFB, deficiency of which has been implicated in autoimmune disorders (Aoki et al., 2005). Other ET1DREs in the receptor category included CMKLR1 (aka ChemR23), encoding the G-protein coupled receptor chemerin 23, which has been previously shown to mediate the anti-inflammatory effects of chemerin in a rodent model of lung disease (Luangsay et al., 2009), and NOTCH4, a negative regulator of the immune response whose depletion has been shown to correlate with antitumor immunity and immune cell infiltration (Long et al., 2021; Zheng et al., 2018). With respect to encoded ET1DRE enzymes, depletion of the GTPase RAB4B promotes a reduction in Treg count (Gilleron et al., 2018). Moreover, abundant evidence implicates mitogen-activated protein kinases in pathways regulating innate immunity; MAPK1/ERK, for example, has been shown to suppress NFKB signaling in endothelial cells (Maeng et al., 2006). In addition, T cells depleted of PPP1R11 are resistant to Treg-mediated suppression of cytokine expression (Joshi et al., 2019), and the APOBECG3 deaminase inhibits retrotransposition of retroviruses, which have been implicated in autoimmune disorders (Esnault et al., 2005). Among co-nodes, NPC1 has recently been shown to be essential for degradation of STING, a well-known mediator of autoimmune responses (Chu et al., 2021). Of note, in the context of increased interest in non-coding RNAs in regulation of immune responses is the lncRNA XIST, dysfunction of which has been linked to systemic lupus erythematosus (SLE) (Wang et al., 2016). Although none were in the top-ranked ET1DREs, we noted the presence of several transcription factors among the ET1DREs. For example, CEBPB has been shown to stabilize FOXP3 levels in regulatory T cells, whose depletion has been previously linked to T1D pathogenesis (Visperas and Vignali, 2016). In addition, ZNF691 was identified as a hub gene in an epigenome-wide association study for loci whose DNA methylation status was associated with the incidence of T1D (Paul et al., 2016).
Diversity of molecular function was similarly evident in the ET1DIE gene set (Figure 1E). Although the subject of controversy, evidence exists that visfatin, encoded by NAMPT, promotes insulin secretion and insulin receptor signaling in pancreatic beta cells (Brown et al., 2010). Moreover, the progestin and AdipoQ receptor family member MMD has been implicated in inflammatory macrophage differentiation, a phenomenon shown to contribute to the development of T1D (Calderon et al., 2006). ETNK1 was recently shown to promote T cell surface expression of CXCR5, whose ligand CXCL13 has been broadly implicated in autoimmune pathogenesis (Fu et al., 2021). In addition, a considerable volume of research implicates gain of function of the transcription factor HIF1A in autoimmune disorders (Islam et al., 2021). Members of the defensin family of antimicrobial peptides, represented in ET1DIE co-nodes by DEFA1, are an integral component of the innate immune system, and have been suggested to contribute to the pathology of chronic rheumatic diseases (Frasca and Lande, 2012). Finally, although the significance of the presence of three pseudogenes (TPTEP1, OR7E5P, and SMG1P1) among the top ET1DIEs is unclear, existing evidence connects genes in this class, including another olfactory receptor pseudogene, to the regulation of inflammatory responses (Shang et al., 2019).
Computation of transcriptional regulatory networks for circulating immune cells in T1D
We previously developed ChIP-Seq consensome analysis (Ochsner et al., 2019), which ranks genes based on measures of their significant promoter occupancy across publicly archived ChIP-Seq experiments mapped to a specific signaling node IP antigen. We subsequently developed ChIP-Seq high confidence transcriptional target (HCT) intersection analysis to identify nodes that have HCT intersections (i.e. transcriptional footprints) within gene lists of interest (Bissig-Choisat et al., 2021; Ochsner et al., 2020). To gain insight into T1D-specific transcriptional regulatory networks in CICs, we next applied ChIP-Seq HCT intersection analysis to compute intersections between a HCT library comprising a total of 961 unique ChIP-Seq human node antigen HCTs (Table S8) and the ET1DIE or ET1DRE gene sets. We designated nodes that had significant intersections with ET1DRE genes as ET1DRE p < 0.05 nodes (n = 310; Table S9, column K), and those that had significant intersections with ET1DIE genes as ET1DIE p < 0.05 nodes (n = 228; Table S9, column L). To afford users insight into evidence for the role of a specific node in T1D-induced or -repressed genes in a given experiment of interest, we also performed ChIP-Seq HCT intersection analysis on T1D-induced (FC > 1.25 & p < 0.05) and -repressed (FC < 0.8 & p < 0.05) genes at the individual experiment level; these 52 networks are shown in Table S10.
Validation of transcriptional regulatory networks for circulating immune cells in T1D
We designed a three-tier approach to validation of the ET1DRE and ET1DIE HCT intersection analyses. We first mapped our ET1DRE and ET1DIE HCT intersection analyses to genes to which T1DKP had assigned high confidence GWAS associations with the incidence of T1D (Table S9, column M) and to the previously described PAGG gene set (Table S9, column N). Consistent with the reliability of the HCT intersection analysis, we observed significant enrichment among the top-ranked ET1DRE p < 0.05 nodes of nodes encoded by genes with the strongest GWAS associations with T1D (ENR = 12, p = 1E-02; Figure 2A) and the highest-ranked PAGGs (ENR = 6.5, p = 3.5E-02; Figure 2B). No significant intersections were observed for either gene set with the ET1DIE p < 0.05 nodes. We were interested to note that some nodes such as STAT4, NELFE, and FLI had p < 0.05 intersections with both the ET1DRE and ET1DIE gene sets (Table S9, columns K and L). Examples of the presence of both activation and repression functions within the same transcription factor are numerous (Latchman, 2001). As such, our analysis suggests that for certain nodes at least, disruption of both transcriptional activation and repression functions in the same protein might contribute to T1D pathogenesis.
Figure 2.

High confidence transcriptional target (HCT) intersection analysis identifies nodes with transcriptional footprints among ET1DRE and ET1DIE gene sets
Selected transcripts at the intersection of the indicated gene sets are labeled. Refer to supplementary text for cut-offs used for hypergeometric tests.
(A) Nodes encoded by genes that have high confidence GWAS associations with T1D are significantly enriched among the top-ranked ET1DRE nodes.
(B) Nodes encoded by genes that have high confidence GWAS associations across multiple autoimmune diseases are significantly enriched among the top-ranked ET1DRE nodes.
(C) Genes encoding ET1DRE p < 0.05 nodes are enriched among genes whose deletion most negatively impacts T cell function.
(D) Genes encoding ET1DIE p < 0.05 nodes are enriched among genes whose deletion most negatively impacts T cell function.
(E) Human genes mapped to the HPO term “autoimmunity” (HP:0002960) are enriched among the highest-ranked ET1DRE p < 0.05 nodes.
(F) Human genes mapped to the HPO term “autoimmunity” (HP:0002960) are enriched among the highest-ranked ET1DIE p < 0.05 nodes.
(G) Top 20 ET1DRE- and ET1DIE p < 0.05 nodes ranked by intersection p values. Node classes are color-coded according to their SPP classification category {Ochsner, 2019 #83} as follows: receptors (orange), enzymes (blue), transcription factors (green), and co-nodes (gray).
Given that T1D is broadly considered a disease of T cell dysfunction, our second HCT intersection analysis validation strategy was predicated on the anticipation that nodes with significant footprints in the ET1DRE and ET1DIE gene sets would be enriched for nodes with critical roles in T cell function. To address this question in a systematic manner, we sourced a study by Shifrut and colleagues that employed a genome-wide single guide RNA strategy to rank genes according to the essentiality of their roles in supporting primary T cell viability (Shifrut et al., 2018). We then annotated this screen with ET1DRE (Table S11, columns I&J) and ET1DRE (Table S11, columns K&L) p < 0.05 nodes. Supporting our hypothesis, we observed enrichment among the highest negatively ranked genes in the Shifrut screen (i.e. those whose depletion had the greatest negative impact on T cell response to stimulation) with the highest-ranked ET1DRE (ENR = 37, p = 1E-03; Figure 2C and Table S9, column O) and ET1DIE (ENR = 37, p = 3E-02; Figure 2D and Table S9, column O) nodes. These results indicate that our C-FLEE HCT intersection analysis identifies potential roles in T1D pathogenesis for nodes that have critical, non-redundant roles in supporting T cell function.
The Monarch Initiative (Mungall et al., 2017) represents a systematic approach to assigning physiological and clinical annotations to genes using Human Phenotype Ontology (HPO) terms. For our third C-FLEE HCT intersection validation, we retrieved from Monarch a set of genes that mapped to the HPO term “autoimmunity” (HP:0002960). We anticipated that if nodes that had significant HCT footprints within the ET1DRE and ET1DIE gene sets were relevant to autoimmune disease, they would be enriched for nodes encoded by genes mapping to the autoimmunity nodes. Consistent with this notion, nodes encoded by human orthologs of this gene set were strongly enriched among the highest-ranked ET1DRE nodes (ENR = 22, p = 2E-04; Figure 2E and Table S9, column P) and, to a lesser extent, among the highest-ranked ET1DIE nodes (ENR = 11, p = 1E-02; Figure 2F and Table S9, column P). We noted again that several autoimmunity nodes (JMJD1C, STAT3, and RFX5) had strong intersections with both the ET1DRE and ET1DIE gene sets (Table S9, column P). This reiterates the evidence alluded to above that the role of a specific node in T1D potentially encompasses both activation and repression of gene expression in specific CICs.
ET1DRE and ET1DIE genes are regulated by functionally diverse signaling nodes
As with the ET1DRE and ET1DIE transcripts (Figure 1E), annotation of the ET1DRE p < 0.05 and ET1DIE p < 0.05 nodes using SPP’s three-tier controlled vocabulary illuminated the diversity of nodes predicted to regulate these gene sets in T1D CICs. Figure 2G shows the top 20 ET1DRE (upper panel) and ET1DIE (lower panel) p < 0.05 nodes ranked by intersection p value. Transcription factors with the most significant ET1DRE transcriptional footprints include YY1, shown to inhibit differentiation and function of regulatory T cells in a Foxp3-dependent manner (Hwang et al., 2016), and TBX21, shown to be dysregulated in multiple sclerosis (Parnell et al., 2014). Moreover, SMAD2 has been mapped to a murine autoimmune diabetes locus (Hook et al., 2011), and numerous DNA variants associated with increase autoimmune disease susceptibility have been shown to impact binding of the key myeloid transcription factor SPI1, aka PU.1 (Watt et al., 2021). Among top ET1DIE receptor nodes, HMGB1 encodes a member of the high mobility group family that has been characterized as a ligand for both toll-like receptors and the RAGE receptor, and has been previously implicated in the pathogenesis of T1D (Zhang et al., 2009). Moreover, NR4A1 has been identified as a potential therapeutic target in central nervous system autoimmune-based inflammation (Rothe et al., 2017). ET1DIE transcription factor nodes included STAT3, a well-characterized driver of numerous autoimmune conditions (Aqel et al., 2021) and as previously noted, was also identified as a ET1DRE node. They also included RELA, a subunit of the well-known pro-inflammatory mediator NFκB, which has known roles as a driver of autoimmune disorders (Sun et al., 2013). ET1DIE transcription factor nodes also encompass members of the RUNX family, which have been implicated in a number of autoimmune conditions (Alarcón-Riquelme, 2003) and which, like STAT3, also had footprints within ET1DREs. Similarly, the gene encoding AFF1 (ET1DIE p = 2E-11) has been mapped to a susceptibility locus for SLE (Okada et al., 2012). With respect to ET1DIE co-nodes, CREBBP is a coactivator for the transcription factor CREB, which negatively regulates survival of regulatory T cells (Wang et al., 2017).
A federated web-based resource for mining CIC T1D transcriptional regulatory networks
The maximum impact of the CIC T1D transcriptional networks requires that they be made broadly and freely available to the research community. Accordingly, we developed a federated, web-based hypothesis generation environment encompassing two open science informatics resources, namely, NDEx, a Cytoscape-based network visualization resource (Pratt et al., 2015) and the Signaling Pathways Project (SPP), an integrated omics knowledgebase for mammalial cellular signaling pathways (Ochsner et al., 2019). Cloud implementation of both resources maximizes uptime and stability of performance. Table 1 contains links to NDEx versions of the networks associated with our study.
Table 1.
Links to NDEx versions of transcriptional networks developed for this study
| Type | Name | Description |
|---|---|---|
| C-FLEE | p < 0.05 ET1DREs | Transcripts that are preferentially repressed in T1D CICs |
| p < 0.05 ET1DIEs | Transcripts that are preferentially induced in T1D CICs | |
| Top-ranked ET1DREs | Top 20 ET1DREs ranked by enrichment p value (Figure 1E left panel) | |
| Top-ranked ET1DIEs | Top 20 ET1DIEs ranked by enrichment p value (Figure 1E right panel) | |
| HCT intersection | p < 0.05 ET1DRE nodes | Signaling pathway nodes predicted to regulate expression of ET1DREs at p < 0.05 |
| p < 0.05 ET1DIE nodes | Signaling pathway nodes predicted to regulate expression of ET1DIEs at p < 0.05 | |
| Top ET1DRE nodes | Top 20 ET1DRE nodes ranked by intersection p value (Figure 2G, upper panel) | |
| Top ET1DIE nodes | Top 20 ET1DIE nodes ranked by intersection p value (Figure 2G, lower panel) | |
| Expt 6607 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6608 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6609 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D monocytes | |
| Expt 6610 induced | repressed | Nodes predicted to regulate genes differentially expressed in mild T1D CD14+ monocytes | |
| Expt 6611 induced | repressed | Nodes predicted to regulate genes differentially expressed in severe T1D CD14+ monocytes | |
| Expt 6612 induced | repressed | Nodes predicted to regulate genes differentially expressed in mild T1D CD14+ monocytes | |
| Expt 6613 induced | repressed | Nodes predicted to regulate genes differentially expressed in severe T1D CD14+ monocytes | |
| Expt 6614 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D whole blood | |
| Expt 6615 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D whole blood | |
| Expt 6623 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6627 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6628 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6629 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6630 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6631 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6632 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6633 repressed | Nodes predicted to regulate genes repressed in T1D CD14+ monocytes | |
| Expt 6634 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D whole blood | |
| Expt 6642 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D CD34+ HSCs | |
| Expt 6650 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D CD3+ T cells | |
| Expt 6651 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D neutrophils | |
| Expt 6686 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D neutrophils | |
| Expt 6687 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D neutrophils | |
| Expt 6655 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D monocyte-derived DCs | |
| Expt 6656 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs | |
| Expt 6657 induced | repressed | Nodes predicted to regulate genes differentially expressed in T1D PBMCs |
No nodes were identified that had significant HCT intersections with experiment 6633 induced genes.
Figure 3A shows a detail of an NDEx C-FLEE network, in this case, the ET1DRE network. Circles represent transcripts, in which size is proportional to the transcript C-FLEE –log10 p value (ML10P), such that the highest confidence preferentially T1D-repressed transcripts are the largest in size. Similarly, circle color intensity is proportional to C-FLEE enrichment value. The three-tier, color-coded node SPP classification alluded to previously facilitates visual organization and contextualization of T1D-regulated transcripts, guides data mining and fitering decisions, and provides for consistency of the user experience across the networks. Upon landing on the ET1DRE network page, the network info pane orients the user as to the essential biology of the network (Figure 3A, upper inset panel). Clicking on a transcript pulls up a contextual menu listing a set of open access resources that run the gamut from curated information illuminating gene regulation and functional biology, to research resources compiling information on experimental reagents (Figure 3A, lower inset). The SPP transcriptomic Regulation Reports (Figure 3B, upper panel) retrieve significant differential expression data points for a gene of interest across thousands of SPP-curated transcriptomic datasets to give insight into upstream receptor and enzyme nodes regulating expression of that gene (Ochsner et al., 2019). Similarly, the SPP ChIP-Seq Regulation Reports (Figure 3B, lower panel) aggregate ChIP-Seq data originally curated by ChIP-Atlas (Oki et al., 2018) and additionally annotated by SPP curators, that identifies evidence for nodes that bind to the upstream promoter region of a gene of interest (Ochsner et al., 2019). The compact, intuitive format of these interfaces is designed to suggest regulatory relationships of a given target with distinct nodes to provide for pathway hypothesis generation.
Figure 3.

NDEx and SPP integration democratizes access to the T1D CIC transcriptional regulatory networks
(A) Detail of NDEx ET1DRE network.
(B) Detail of SPP transcriptomic (upper panel) and ChIP-Seq Regulation Reports of ET1DREs to show evidence for signaling nodes that regulate their expression.
(C) Detail of NDEx ET1DRE node network. In NDEx networks, transcript (A) and node class edges (B) are color coded as follows: receptors (orange red), enzymes (blue), transcription factors (green), ion channels (mustard), and co-nodes (gray). In both NDEx network types, the network info tab (upper inset) contains a description of the resoure, while clicking on a transcript (A) or node (B) shows the node/ege window (lower inset) containing links to selected contextual web research resources.
Other resources linked to from ET1DREs include the DKNET Discovery Portal, which consolidates curated gene- and protein-level information from over 300 resources to provide for rapid contextualization of gene function, as well as plasmids and antibodies relevant to the gene of interest or its encoded protein (Whetzel et al., 2015). As previously discussed, T1DKP aggregates information across numerous independent studies to summarize the strength of the GWAS connection of a gene to T1D and T1D-related phenotypes. An Entrez GeneID-based link to PubMed limits the displayed research article records to those that have been curated by NCBI staff, rather than the noisier results that would be returned from a free text search using the approved gene symbol. C-FLEE networks also link to Open Targets Genetics, which assembles variant-centric statistical evidence for prioritization of potential drug targets (Mountjoy et al., 2021). As described in our use cases, genome-wide functional screens were of considerable value in fleshing out our T1D network-based research hypotheses. The BioGRID Open Repository of CRISPR Screens curates the rapidly expanding literature on such screens to enable researchers to gather evidence for roles for specific nodes or genes in functional cellular pathways (Oughtred et al., 2021). Finally, links to Monarch (Mungall et al., 2017), which we used to validate the HCT intersection analysis (Figures 2E and 2F), provide for convenient access to information on mammalian phenotypes associated with a particular gene or node of interest. Adding to the value of NDEx integration is the fact that researchers querying their own gene set of interest using the iQuery feature can identify significant intersections between their own gene list and any of the T1D networks without prior knowledge of their existence, resulting in potential novel and unanticipated avenues of T1D research.
The second set of networks implemented in NDEx is the HCT intersection analysis networks, which represent the functional regulatory footprints for DNA-binding signaling nodes among clinical T1D gene sets generated by our study. Figure 3C shows a detail of the NDEx C-FLEE HCT intersection network, in this case, the ET1DRE HCT intersection network. Circles represent nodes, in which size is proportional to the HCT intersection ML10P, such that the nodes with the highest confidence intersections with the ET1DREs are the largest in size. Similarly, circle color intensity is proportional to HCT odds ratio. As with the C-FLEE networks, SPP classification color coding provides for intuitive, biology-driven browsing and searching of the networks. The network info pane conveys essential summary information (Figure 3C, upper inset panel), while clicking on a node opens a window containing node-specific links to the same set of resources that contextualize the transcripts in the C-FLEE networks (Figure 3C, lower inset panel).
Hypothesis generation use cases
We next wished to demonstrate the value of our networks to generate robust in silico research hypotheses around previously unexplored signaling pathways in T1D CICs. A common theme throughout these use cases is the re-use of existing resources and datasets to assemble “bench-ready” hypotheses that themselves withstand validation against independent benchmarks. Collectively, they are intended as a how-to that demonstrates how informed integration of our networks with other freely available resources empowers the user to develop sophisticated, actionable hypotheses around CIC deficits in T1D.
Hypothesis generation use case 1: depletion of a CD56dim CD57+ natural killer cell-specific gene signature links T1D to multiple inflammatory syndrome in children
The first use case began as a demonstration of using ET1DREs to discern specific cell types whose functional depletion contributes to T1D, and concluded with an unexpected connection to another disease state of high current interest, multiple inflammatory syndrome in children (MIS-C). To discern CIC subtype-specific signatures within the ET1DREs, we computed this gene set against a scRNA-Seq study by Travaglini and colleagues that profiled gene expression across 25 immune cell types (Travaglini et al., 2020). In doing so, we observed striking enrichment of high confidence Travaglini natural killer (NK) cell-specific genes among ET1DREs (Travaglini-NK; ENR = 254, p = 1E-07; Figure 4A and Table S5, column P). Although the connection between NK cell dysfunction and T1D pathogenesis is well established (Gardner and Fraker, 2021), the underlying mechanisms are not fully understood. To verify this result, we sourced a recent study by Ota et al. profiling gene expression across 28 immune cell types in a variety of autoinflammatory conditions (Ota et al., 2021). Confirming the NK expression program-enriched character of the ET1DREs, we observed robust over-representation among ET1DRE transcripts of genes preferentially expressed in NK cells from this study (Ota-NK; ENR = 15, p = 1E-11; Figure S2A and Table S5, column Q). Notable intersections of ET1DREs with the two NK gene sets included three members of the granzyme family, whose depletion has been reported in T1D (Mollah et al., 2017), PRF1, encoding a key effector molecule for NK-cell-mediated cytolysis, perforin; an inhibitory killer Ig-like receptor gene, KLRD1; and S1PR5, shown to be essential for NK cell migration toward sphingosine-1 phosphate (Drouillard et al., 2018).
Figure 4.

Depletion of a CD56dim CD57+ natural killer cell-specific gene signature links T1D to multiple inflammatory syndrome in children
Selected transcripts at the intersection of the indicated gene sets are labeled. Refer to supplementary text for cut-offs used for hypergeometric tests.
(A) The highest-ranked ET1DRE genes are enriched for NK cell-specific genes as documented in Table S4 from the Travaglini study.
(B) Top-ranked ET1DRE∩NK genes are strongly enriched among the top 100 ranked genes in cytotoxic (CY-NK) and adaptive (ADNK) NK cells, per snATAC-seq by Chiou and colleagues.
(C) Nodes that have significant transcriptional footprints within the ET1DRE∩NK gene set are enriched for canonical NK transcription factors per Hesslein and colleagues. Nodes with the strongest and most significant intersections with the gene set are distributed in the top right quadrant of the plot.
(D) ET1DRE∩NK genes are further enriched among CD57+-specific CD56dim -induced genes. CD57+-specific genes were obtained from Table S1 of the Collins et al. study.
(E) CD57+-specific, CD56dim ET1DRE∩NK genes are super-enriched among teplizumab-induced genes.
(F) All nine genes in the MIS-C signature are highly ranked ET1DRE∩NK genes.
(G) Five MIS-C signature genes are highly ranked CD57+-specific ET1DRE∩NK genes.
(H) Four MIS-C signature genes are top teplizumab-induced ET1DRE-NK genes.
The robust NK signature in the ET1DREs observation was consistent with the super-enrichment among ET1DREs of the IAAsig gene set (Figure 1B), which itself had been shown to possess a strong NK character (Xhonneux et al., 2021). As an additional confirmation using an alternative technical platform, we identified a recent study that generated a single-nucleus assay for transposase-accessible chromatin with sequencing (snATAC-seq)-based accessible chromatin reference map of 28 peripheral blood and pancreas cell types from donors without diabetes (Chiou et al., 2021). We then defined a set of 77 transcripts representing the intersection (∩) of the ET1DREs and the two previously referenced scRNA-Seq-defined NK-specific gene sets (Travaglini-NK and Ota-NK). We designated these ET1DRE∩NK genes, and ranked them in ascending order of their ET1DRE enrichment and Travaglini NK-specificity p values (Table S5, column R). Reiterating the NK specificity of the ET1DRE∩NK gene set, the top 20 ranked ET1DRE∩NK genes were most strongly enriched (per hypergeometric test) among the top 100 snATAC-seq ranked genes in cytotoxic NK cells (CY-NK: ENR = 46, p = 6E-08; Figure 4B), less appreciably in adaptive NK cells (ADNK: ENR = 28, p = 2E-04; Figure 4B), and were not significantly enriched in the top 100 ranked genes from any other cell type (Figure 4B and Table S12, column AE). These data indicated to us that genes that were preferentially repressed across the independent T1D cohorts were enriched for genes that are preferentially expressed in NK cells.
Based on this analysis, we anticipated that ET1DRE∩NKs would contain transcriptional footprints for nodes with known roles in NK cell differentiation and function. To evaluate this, we performed HCT intersection analysis on the gene set (see Table S9, column Q for nodes that have p < 0.05 intersections with this gene set). In order to validate this analysis, we retrieved two sets of nodes identified in independent, comprehensive reviews of transcriptional regulators of NK cell development and differentiation, and designated these Hesslein (Hesslein and Lanier, 2011) and Brillantes (Brillantes and Beaulieu, 2019) nodes (Table S9, columns R & S respectively). Consistent with the accuracy of our analysis, both the Hesslein (ENR = 120, p = 6E-05; Figure 4C) and Brillante (ENR = 107, p = 8E-05; Figure S2B) node sets were robustly enriched among the most highly ranked ET1DRE∩NK nodes. The top ranking was assigned to the canonical NK cell transcription factor TBX21/T-BET, whose encoding gene we had earlier identified as an ET1DRE (Table S5). Nodes with prominent ET1DRE∩NK footprints included several others with known roles in NK cell differentiation, including CBFB and RUNX factors, SREBPs, GATA2, and STAT5B.
Human NK cells are classified into two major subsets based on the expression of the adhesion molecule CD56, namely, CD56bright and CD56dim (Poli et al., 2009). The former generate abundant quantities of cytokines, whereas the latter are cytotoxic effectors expressing inhibitory killer immunoglobulin-like receptor (KIR) genes, as well as PRF1. The presence in the ET1DRE∩NK signature of PRF1 and KLRD1 suggested to us that depletion of a CD56dim NK cell-specific gene set might contribute to the pathogenesis of T1D. To explore this question further, we identified a study by Hanna and colleagues (Hanna et al., 2004) that compared gene expression profiles between CD56bright and CD56dim NK cells and evaluated the distribution within this dataset of the ET1DRE∩NK gene set (Table S13, columns E–G). Consistent with our hypothesis, the highest-ranked ET1DRE∩NK transcripts were strongly enriched among the top induced genes in CD56dim relative to CD56bright cells (ENR = 94, p = 8E-08; Figure S2C), but not among CD56bright-induced genes. The CD56dim NK population is further partitioned into two subsets, CD57− and CD57+, the latter having more potent lytic activity. To further define the CD56dim subset of origin of the ET1DRE∩NK genes, we used a transcriptional census of NK subtypes by Collins and colleagues (Collins et al., 2019) to define CD57+- and CD57−-specific subsets of the Hanna CD56dim-induced gene set (Table S13, columns H&I). We observed enrichment of the top-ranked ET1DRE∩NK genes within the CD57+ subset of the CD56dim-induced genes that was over 10-fold higher than their enrichment in the broader CD56dim gene set (ENR = 990, p = 2E-09; Figure 4D), but not within the CD57− subset. This result resonates with reports of reduced levels of circulating CD57+ NK cells in other autoimmune diseases (Nielsen et al., 2013) and is consistent with specific depletion of a CD56dim CD57+ NK signature in CIC transcriptomes of T1D subjects.
CD56dim cells have been shown to promote CD8+ T cell exhaustion, and this phenomenon has been linked to improved prognosis in autoimmune diseases (McKinney et al., 2015). To validate our original C-FLEE analysis, we had previously shown (Figure 1C) that ET1DREs were strongly enriched among genes induced by administration of teplizumab in clinical T1D, resolution of which was accompanied by CD8+ T cell exhaustion (Long et al., 2016). We speculated therefore that if the ET1DRE∩NK CD56dim-induced CD57+-specific gene subset (Table S5, column T) was relevant to the resolution of clinical T1D, we would expect to see enhanced enrichment of these genes within the teplizumab-induced gene set. Remarkably, enrichment of the ET1DRE CD56dim-induced CD57+-specific genes among teplizumab-induced genes was over 3-fold higher than that of the ET1DRE gene set as a whole (ENR = 687, p = 3E-06; Figure 4E and Table S6).
MIS-C is a recently identified Kawasaki disease-related complication of COVID-19 characterized by progressive inflammatory responses across multiple organs (Galeotti and Bayry, 2020). While our own study was nearing completion, a report was published (Beckmann et al., 2021) that identified a nine-gene signature that was repressed in MIS-C and other inflammatory diseases and was highly expressed in CD8+ T cells and NK cells. Given the recently reported evidence for an autoimmune component of MIS-C (Porritt et al., 2021), we speculated whether there was any overlap between the MIS-C signature and the gene sets we had defined in this use case that might point to common NK-centric mechanisms between MIS-C and T1D. Strikingly, all nine genes in the MIS-C signature were highly ranked ET1DRE∩NKs (ENR = 704, p = 7E-27; Figure 4F and Table S5, column U) and five (PRF1, TGFBR3, C1orf21, S1PR5, and MYBL1) were among the highest-ranked CD57+-specific ET1DRE∩NKs (ENR = 895, p = 5E-15; Figure 4G and Table S5, column U). Finally, suggesting the potential therapeutic application of teplizumab in MIS-C and Kawasaki disease, four genes in the MIS-C signature (PRF1, GZMA, S1PR5, and KLRD1) were among the highest-ranked ET1DRE∩NKs that were induced in T1D subjects in response to teplizumab treatment in the study by Long and colleagues (ENR = 3175, p = 9E-15; Figure 4H and Table S6, column O). Underscoring the relevance of this study to our own analysis, one of the nine MIS-C genes was TBX21, encoding the node we previously identified with the strongest transcriptional footprint in the ET1DRE∩NK cells (Figures 4C and 4D). In summary, this use case represents strong evidence for common NK-specific mechanisms underlying T1D and MIS-C.
Hypothesis generation use case 2: Transcriptional induction in neutrophils of TRPC6 supports calcineurin/STAT2-dependent type 1 interferon signaling in early onset T1D
Inspecting the ET1DIEs, we noted that one of the highest ranked (ENR = 12, p = 5E-05) was TRPC6, which encodes a member of the transient receptor potential cation channel gene family. Studying the underlying experimental gene expression data, we were interested to note that TRPC6 was strongly induced (T1D v C LFC = 1.74; Table S4, column AB) in a dataset generated by a study of neutrophils in a cohort of subjects with early onset T1D (Table S2, dataset 1274, experiment 6651). Supportive of its identification as an ET1DIE gene, gain of function single nucleotide polymorphisms (SNPs) in TRPC6 have been previously linked to the autoimmune disorder familial focal segmental glomerusclerosis (Winn et al., 2005) and Trpc6 deletion has been previously shown to be protective at early age in the Akita mouse model of T1D (Wang et al., 2019a). Although the canonical pathway of TRPC6 involves activation of Ca2+ signaling, the mechanisms underlying its potential role in autoimmune disorders are not clearly understood. Accordingly, we selected TRPC6 for further characterization of its potential role in neutrophils in driving pathogenesis in early onset T1D.
Because TRPC6 was transcriptionally induced in neutrophils in early onset T1D, we hypothesized that the encoded TRPC6 protein might in turn participate in the T1D-induced transcriptional program in these cells. To address this question, we retrieved a set of genes that were transcriptionally repressed in response to a TRPC6 loss of function mutation (TRPC6M↓)(Griesi-Oliveira et al., 2015) and evaluated their distribution within the neutrophil early onset T1D-induced (NEOT1DI) gene set (Table S14, columns F&G). Consistent with a role for TRPC6 in neutrophil signaling events in early onset T1D, we observed robust enrichment of TRPC6M↓ genes among the most prominent NEOT1DI transcripts (ENR = 54, p = 8E-07; Figure 5A). Scrutinizing the intersecting genes, we noted many of the effectors of the classical type 1 interferon pathway that the authors of the original study had shown to be induced in the T1D neutrophils, including MXI, members of the 2'-5'-oligoadenylate synthetase (OAS1, OAS2, and OAS3) and interferon-induced transcript (IFIT1 and IFIT3) families, the RIG-I cosensor DDX60, and ESTI1 (Figure 5A). Based on this observation, we hypothesized that TRPC6 might be specifically involved in regulation of the type 1 interferon response in early onset T1D neutrophils. To pressure test this idea, we retrieved a set of genes shown to be transcriptionally induced in response to infection of neutrophils by influenza A virus (IAVN↑)(Malachowa et al., 2018), a well-characterized cue for the type 1 interferon response. Confirming the strong type 1 interferon character of the NEOT1DI genes, the neutrophil IAV-induced signature was strongly enriched among the NEOT1DI genes (ENR = 254, p = 6E-08; Figure 5B and Table S14, columns H&I). Remarkably, and strongly suggestive of a role for TRPC6 in induction of the type 1 interferon response in early onset T1D neutrophils, this enrichment was increased 10-fold in the TRPC6M↓ subset of the NEOT1DI genes (TRPC6M↓∩T1DI: ENR = 2936, p = 1E-07; Figure 5C).
Figure 5.

Transcriptional induction of TRPC6 in neutrophils supports calcineurin/STAT2-dependent type 1 interferon signaling in new onset T1D
Selected transcripts at the intersection of the indicated gene sets are labeled. Refer to supplementary text for cut-offs used for hypergeometric tests.
(A) Genes that are transcriptionally repressed in response to TRPC6 depletion are induced in new onset T1D neutrophils.
(B) A type 1 interferon signature is strongly enriched in neutrophil T1D-induced genes. The IAV signature was retrieved from Malachowa et al. Table S1, Mex09 IAV vs. Mock RPMI-18h p < 0.05.
(C) The type 1 interferon signature is super-enriched in neutrophil T1D-induced genes that are regulated by TRPC6.
(D) Canonical transcriptional drivers of the type 1 interferon response are consolidated among nodes with robust transcriptional footprints among TRPC6M↓∩NEOT1DI genes. TRPC6M↓∩NEOT1DI cut-offs: NEOT1DI LFC>0.5 & p < 0.05, TRPC6M LFC < −1 & p < 0.01. Nodes with the strongest and most significant intersections with the gene set are distributed in the top right quadrant of the plot.
(E) Genes downregulated in response to Ppp3r1/CNB1 depletion in dendritic cells are enriched among NEOT1DI genes.
(F) Genes downregulated in response to Ppp3r1/CNB1 depletion in dendritic cells are further enriched among TRPC6M↓∩NEOT1DI genes.
(G) STAT2 HCTs downregulated in response to Ppp3r1/CNB1 depletion in dendritic cells are most strongly enriched among TRPC6M↓∩NEOT1DI genes.
(H) Reactome interferon alpha/beta signaling pathway targets are very strongly enriched among TCS-NEOT1DI genes relative to NEOT1DI genes. Refer to Table S14 for the underlying data.
We next wished to define candidate transcription factor nodes downstream of TRPC6 in the early onset T1D neutrophils. We noted that consistent with the strong type 1 interferon character of the NEOT1DI transcripts (Figure 5B), HCT intersection analysis of these genes assigned elevated rankings to numerous nodes with well characterized roles as transcriptional drivers of the type 1 interferon response, most notably STAT2 (p = 8E-22; Table S10, column AB). Confirming this, a set of canonical α/β (i.e. type 1) interferon response nodes retrieved from the Reactome knowledgebase (Joshi-Tope et al., 2005) was robustly enriched within the most highly ranked NEOT1DI nodes (ENR = 72, p = 3E-06; Figure S3). We hypothesized that given its similarly sizeable footprint within the NEOT1DI genes (Figure 5A), TRPC6 might signal through STAT2 to support the NEOT1DI transcriptional program. To investigate this further, we defined a set of genes representing the intersection of the TRPC6M↓ and NEOT1DI genes (TRPC6M↓∩NEOT1DI: n = 30; Table S14, column J). We then subjected this gene set to HCT intersection analysis and compared the results with those for HCT intersection analysis of the broader NEOT1DI gene set (Table S15). Consistent with consolidation of type 1 interferon signaling targets within the TRPC6M↓∩NEOT1DI gene set, the three nodes with the largest TRPC6M↓∩NEOT1DI enrichments and the largest fold-increases in enrichment between the two gene sets—STAT2, IRF1, and STAT1—were members of the Reactome canonical type 1 interferon pathway (ENR = 192, p = 7E-08; Figure 5D, Table S15, column L and Table S9, column T).
The canonical TRPC6 pathway is known to signal through calcineurin (Kuwahara et al., 2006); indeed, of relevance to T1D, calcineurin inhibitors are standard immunosuppressive therapeutics (Park et al., 2020). We hypothesized therefore that the candidate TRPC6-STAT2 pathway driving induction of NEOT1DI genes might also encompass calcineurin. To test this hypothesis, we sourced a transcriptomic dataset that examined the effect of calcineurin loss of function in dendritic cells—represented by deletion of the calcineurin CNB1 subunit (encoded by Ppp3r1)—on their inflammatory response to A. fumigatus challenge (Zelante et al., 2017). We retrieved a set of genes from this dataset that were repressed in Ppp3r1 KO cells (CNB1KO↓) and evaluated their distribution among the NEOT1DI genes. Supportive of a role for calcineurin in the NEOT1DI transcriptional program, CNB1KO↓ genes were enriched among NEOT1DI genes (ENR = 6.5, p = 3E-03; Figure 5E & Table S14, columns K&L). Moreover, specifically implicating a TRPC6-calcineurin pathway in the regulation of NEOT1DI genes, CNB1KO↓ genes were further enriched among the TRPC6M↓∩NEOT1DI subset (ENR = 28, p = 2E-03; Figure 5F). We next hypothesized that, given its top ranking in the TRPC6M↓∩NEOT1DI HCT intersection analysis (Figure 5D), STAT2 was a primary downstream target of the candidate TRPC6-calcineurin signaling axis. To test this hypothesis, we defined a subset of calcineurin targets that were candidate STAT2 HCTs (STAT2-HCTs∩CNB1KO↓; Table S14, column M). Consistent with our hypothesis, the STAT2-HCTs∩CNB1KO↓ genes were over 10-fold further enriched among the TRPC6M↓∩NEOT1DI gene set (ENR = 315, p = 2E-05; Figure 5G).
As a final step in this use case, we wished to place our results in the context of canonical downstream effectors of interferon signaling, widely considered a critical trigger of T1D pathogenesis. Specifically, if our candidate TRPC6-calcineurin-STAT2 pathway were valid, we expected to see consolidation within its putative target gene set of transcriptional effectors of type 1 interferon signaling. To do this, we defined a set of seven genes at the intersection of the TRPC6M↓, CNB1KO↓, STAT2-HCT, and NEOT1DI gene sets (TCS-NEOT1DI; RSAD2, OAS2, DDX60, EPSTI1, IFIT1, IFIT3, and MX1). We then used Reactome pathway analysis (RPA) (Joshi-Tope et al., 2005) to compute enrichment statistics of functional pathways within this gene set and compared them to their enrichment within the entire NEOT1DI gene set (Table S16). Consonant with our hypothesis, the pathway with the most significant enrichment among TCS-NEOT1DI genes (p = 1E-16) and the largest increase in enrichment between the two gene sets (11 orders of magnitude) was for interferon alpha/beta (i.e. type 1 interferon) signaling (Figure 5H and Table S16). In contrast, enrichment of the gamma-interferon pathway, classically associated with IRF1 and STAT1 as target transcription factors, was considerably reduced in the TCS-NEOT1DI genes relative to the NEOT1DI genes as a whole (Figure 5H and Table S16). Collectively, our results represent evidence for the existence of a neutrophil TRPC6-calcineurin-STAT2 pathway driving induction of a type 1 interferon expression signature in new onset T1D.
Hypothesis generation use case 3: A PCF11-PP1R10-GTF2I regulatory module connects alternative splicing in monocyte activation to severity of T1D
The monocyte-macrophage lineage is a key axis in the innate immune system and has well established links to autoimmune diseases (Ma et al., 2019). Broadly speaking, monocyte differentiation into macrophages results in the adoption of the pro-inflammatory M1 phenotype, or the anti-inflammatory M2 phenotype (Martinez and Gordon, 2014). Although monocyte and macrophage activation have well-established connections to T1D pathogenesis (Josefsen et al., 1994), the underlying mechanisms are not fully understood. One of the datasets included in our resource compared peripheral blood monocyte gene expression between recent-onset T1D and healthy controls in two separate cohorts (Table S2, datasets 1256 & 1257, experiments 6610-6613). The investigators found that monocyte expression profiles clustered into two distinct subgroups, mild (MT1D) and severe (ST1D) deviation of T1D subjects from healthy controls, on the basis of HbA1c levels 3 and 6 months after diagnosis (Table S2). We set out to use this dataset to identify candidate inflammatory transcriptional complexes that distinguished ST1D and MT1D monocytes.
We first retrieved HCT intersection analysis data for the eight gene sets within the GSE33440 dataset (ST1D- and MT1D-induced or repressed in both array cohorts; Table S10). We then computed the ratio of aggregate significant MT1D-induced and ST1D-induced ORs and -log10p (ML10P) values for each node across the two cohorts (Table S17). For the purposes of subsequent analyses, we designated a set of 20 nodes as candidate ST1D drivers (ST1DDs; Figure S4 & Table S17, column W; see Figure S4 legend for ST1DD cut-off criteria). We reasoned that if our analysis to this point were valid, ST1DDs would be enriched for known regulators of macrophage activation. To investigate this in a more statistically systematic manner, we sourced a recent genome-wide CRISPR screen by Covarrubias and colleagues that identified genes with essential roles in macrophage viability (Table S18) (Covarrubias et al., 2020). This study assigned Mann-Whitney (MW) scores to 21,379 mouse genes, such that the gene with the lowest MW score was designated the most essential for macrophage viability. Validating our list of ST1DDs, genes encoding top-ranked ST1DD nodes were strongly enriched among the top-ranked hits in the CRISPR macrophage viability screen (ENR = 61, p = 4E-04; Figure 6A and Table S18, column F).
Figure 6.

Combined HCT intersection analysis and hierarchical clustering defines a novel splicing regulatory module in T1D
Selected transcripts at the intersection of the indicated gene sets are labeled. Refer to supplementary text for cut-offs used for hypergeometric tests.
(A) Mouse orthologs of genes encoding ST1DDs are enriched among the top-ranked hits in a CRISPR screen for macrophage viability.
(B) Hierarchical clustering of nodes with significant HCT intersections defines candidate regulatory clades among ST1DDs.
(C) Table showing results of hypergeometric test of clade members with respect to ST1DDs and MΦ viability screen.
(E) Enrichment for hypergeometric test; p: p value for hypergeometric test (D) Clade 150c nodes are enriched among ST1DDs. Nodes with the strongest and most significant intersections with the gene set are distributed in the top right quadrant of the plot.
(E) Genes encoding clade 150c nodes are enriched among the top-ranked hits in the CRISPR macrophage viability screen.
(F) Green-IR genes are enriched among cross-cohort monocyte ST1D v MT1D-induced genes.
(G) Green-IR genes are super-enriched among cross-cohort monocyte ST1D v MT1D-induced clade 150c SCTs.
(H) ST1D-induced predicted clade 150c targets are strongly enriched among the top-ranked hits in the CRISPR macrophage viability screen.
Cell signaling involves the organization of individual regulatory nodes into higher order complexes (McKenna et al., 1998; McKenna and O'Malley, 2002). Having identified and validated the ST1DDs, we next wished to gather evidence for the extent to which they clustered into regulatory modules representing such complexes. To do this, we carried out hierarchical clustering of nodes that had at least two significant (p < 0.05) intersections across the eight independent gene sets. This generated a total of 600 nodes organized into 100 clades (k = 100 cutoff), many of which were further subdivided into discrete subclades at lower k cutoffs (Figure S5). We designed a two-tier approach to prioritizing clusters whose members were most strongly implicated as drivers of monocyte activation in ST1D. We first shortlisted a set of seven clades (40, 40b, 56, 150, 150c, 177, and 177a; Figure 6B) whose constituent nodes were significantly enriched among the ST1DDs (Figure 6C, ST1DDs & Table S17, column X). We then further refined this set of clades to identify those whose members were significantly enriched among the most highly ranked genes in the macrophage viability screen (Figure 6C, MΦ viability). After the second prioritization step, only clades 40b, 150, 150c, and 177a remained. At each prioritization step, the clade with the strongest enrichment statistics was clade 150c (Figure 6C). Figure 5 panels D&E represent scatterplots depicting enrichment of clade 150c members among, respectively, ST1DDs (ENR = 30, p = 3E-05; Figure 6D) and among the top-ranked genes in the Covarrubias macrophage viability screen (ENR = 41, p = 8E-03; Figure 6E and Table S18, column G). To validate clade 150c, we retrieved three sets of proteins from BioGRID (Oughtred et al., 2021) representing curated protein-protein interaction partners of PCF11, GTF2I, and PPP1R10. We then used the hypergeometric test to compare the frequency of these proteins within nodes that had strong (r > 0.95) HCT intersection correlations with each node, relative to the entire set of nodes included the analysis. In all three cases (Figure S6A, GTF2I; Figure S6B, PCF11; and Figure S6C, PPP1R10), we observed appreciable enrichment of BioGRID-curated interacting partners of each clade 150c node among nodes whose intersections correlated most strongly with that node.
Clade 150c comprised three nodes with known roles as components of the cellular splicing apparatus, namely, the pre-mRNA cleavage complex member PCF11; the human ortholog of Drosophila PNUTS, PPP1R10; and the general transcription factor GTF2I (Cvitkovic and Jurica, 2013). This was intriguing given the expanding canon of evidence linking aberrant splicing to disruption of immune homeostasis (Newman et al., 2017) and, in the case of GTF2I, numerous GWAS-based connections to the autoimmune condition, Sjögren’s syndrome (Li et al., 2013). Of specific relevance to the macrophage-monocyte lineage, splicing has been shown to play a central in activation of these cells in the innate immune response (Liu et al., 2018; Wagner et al., 2021). In this context, we noted with interest a study by Green and colleagues (Green et al., 2020) showing that decreased intron retention (IR) in monocytes and macrophages is coupled to increased expression of targets encoding key regulators of phagocytosis and inflammatory signaling. Placing these observations in the context of our own findings, we hypothesized that (i) IR genes would be enriched among genes expressed at higher levels in ST1D monocytes relative to MT1D monocytes; and (ii) this enrichment would be enhanced in the case of genes regulated by members of clade 150c. To test this hypothesis, we generated a probeset-level ST1D v MT1D cross-cohort analysis (Table S19). We then retrieved a set of IR transcripts that were upregulated in inflammatory monocyte differentiation at the protein level per Green and colleagues (Green et al., 2020) (designated Green-IR; Table S19, column H). Reflecting a strong relationship between monocyte inflammatory IR and ST1D monocyte-induced transcriptional programs, Green-IR transcripts were appreciably enriched among cross-cohort ST1D-induced genes (ST1D↑; ENR = 7.5, p = 2E-03; Figure 6F), but not among cross-cohort ST1D-repressed genes. ST1D↑∩GREEN-IR genes common to both cohorts included several with known roles in macrophage activation and inflammatory responses. Among the overlapping genes, FMNL1 is an essential component of podosomes, which are adhesive structures required for tissue invasion by inflammatory macrophages (Mersich et al., 2010). Moreover, ATG16L1 encodes an effector of autophagy, which has been shown to be an essential process in monocyte-macrophage activation and phagocytosis (Zhang et al., 2012), and has been previously implicated in Crohn disease (Rioux et al., 2007). In addition, MOV10 (Chertova et al., 2006) and ZNFX1 (Wang et al., 2019b) have been shown to be involved in viral infection of macrophages, an event identified as a potential trigger for autoimmune responses.
Next, to explore evidence for a specific role for clade 150c in ST1D-specific IR splicing events, we annotated the probeset-level ST1D v MT1D cross-cohort analysis with SPP ChIP-Seq consensome percentiles for PCF11, PPP1R10, and GTF2I (Table S19, columns I–K). These values indicate the percentilized relative strength of the node-target gene regulatory relationship computed across all publicly archived ChIP-Seq experiments in which that node is the IP antigen (Ochsner et al., 2019). As a sanity check, we firstly validated the PCF11 ChIP-Seq consensome against a set of genes differentially expressed in response to depletion of PCF11 in cultured cells (Kamieniarz-Gdula et al., 2019) (Figure S6D). Consistent with a role for members of clade 150c in modulating IR in ST1D monocytes, the already strong enrichment of Green-IR transcripts among ST1D↑ genes (Figure 6A) was strikingly enhanced when we limited the ST1D↑ genes to those that were strong confidence transcriptional targets (SCTs; defined as the 75th %ile of a consensome) for at least two of the three members of clade 150c (ENR = 96, p = 4E-06; Figure 6G). As a final step in our use case, we surmised that clade 150c SCTs would themselves be enriched among genes with essential roles in macrophage viability. Consistent with this notion, the highest-ranked ST1D↑ clade 150c SCTs —which included several genes with known roles in macrophage activation—were considerably enriched among genes with elevated rankings in the macrophage viability screen (ENR = 49; p = 6E-04; Figure 6H and Table S18, column H & Table S19, column L). Collectively, our results are a strong basis for the hypothesis that members of clade 150c represent a multi-node module that whose regulation of monocyte alternative splicing events distinguishes severe and mild type 1 diabetes.
Comparison of informatic methods with other approaches
To complete our study, we compared the performance of the informatic strategies underlying this study against existing approaches. We first compared ET1DREs with T1D gene expression signatures selected on the basis of three distinct criteria. Firstly, to demonstrate the value that C-FLEE meta-analysis adds to the underlying stand-alone studies, we selected a list of genes that were downregulated in one of the underlying T1D case:control studies (Kaizer GSE9006 gene set; T1D v C LFC < -0.3 & p < 0.05). Secondly, to compare our approach to a case:control cohort study not included in our analysis, we retrieved a list of genes shown by Mehdi and colleagues (Mehdi et al., 2018) to be downregulated in T1D subjects relative to control individuals. Thirdly, to demonstrate the value of C-FLEE over other network informatic approaches, we identified a set of 1003 candidate T1D genes from a recent gene expression profiling meta-analysis (De Silva et al., 2022). As an independent, objective benchmark, we compared enrichment of ET1DREs and these three gene sets against the Type 1 Diabetes Knowledge Portal list of 1000 genes that exhibit significant (p < 5E-03) associations with the incidence of type 1 diabetes (Table S20, columns A&B). Reflecting the accuracy with which our analysis identifies genes with strong causative associations with T1D, we observed significant enrichment among the top-ranked T1DKP genes of ET1DREs (ENR = 4, & p = 1E-02 hypergeometric test; Table S19, column C), but not of the Kaizer (Table S20, column D), Mehdi (Table S20, column E), or De Silva (Table S20, column F) gene sets. Finally, we had previously shown that a set of literature-curated NK cell transcription factors (Hesslein and Lanier, 2011) was strongly enriched among nodes that had significant HCT intersections with ET1DRE∩NK genes (Hesslein NK nodes, Figure 4C). We wished to compare the performance of this analysis with the popular ENRICHR (Xie et al., 2021) and GSEA (Subramanian et al., 2005) enrichment analysis tools. Again consistent with the superior accuracy of our analytics over these approaches, we failed to observe significant enrichment of the Hesslein NK nodes among p < 0.05 intersections for either the ENRICHR or GSEA analyses (Table S21). In summary, this analysis demonstrates the value that the informatic approaches supporting this study add to existing resources.
Discussion
Datasets profiling gene expression in CICs have been of considerable value in demonstrating the transcriptome-wide impact of deficits in T1D CIC signaling in individual clinical cohorts (Table S1). Independently archived, however, these datasets are isolated both from each other, and from a broader universe of datasets that document regulatory relationships between signaling nodes and their transcriptional targets in a reduced-bias manner (Ochsner et al., 2019). Here, we extended the value of these T1D CIC datasets along three dimensions. We firstly surveyed across the datasets in aggregate to identify genes that exhibited significant tendencies toward preferential induction or repression in T1D CICs. Secondly, computation of these datasets against millions of archived transcriptomic and ChIP-Seq data points relevant to cellular signaling nodes enabled the prediction of T1D transcriptional regulatory networks. Thirdly, we leveraged a widely popular web-based visualization environment to promote engagement of these networks by the broader research community.
In designing our study, we took into account the fact that rigorous validation against independent cross-disciplinary indices would be critical to its reliability as a hypothesis generation resource. To this end, we compiled a set of orthogonal validation strategies that ran the gamut of clinical, population genetic, and mechanistic facets of T1D. The performance of our networks against gene sets generated by a recent independent clinical T1D cohort analysis was highly encouraging (Figures 1A and 1B). Moreover, the availability of a clinical transcriptomic dataset for the anti-CD3 antibody teplizumab represented a critical, independent “reversal-of-function” benchmark that demonstrated alignment of our predictive networks with resolution of clinical T1D (Figure 1C). Similarly, the degree of convergence between our predicted T1D genes and those generated by large cohort GWAS studies was an additional strong source of validation (Figure 1D). On a mechanistic level, we leveraged a CRISPR screen for genes with critical roles in T cell function and viability to demonstrate the important role of ET1DRE and ET1DIE nodes as non-redundant points of failure in T cell function and viability (Figures 2C and 2D).
Our study introduces a previously unpublished informatic method, C-FLEE, which addresses the inherent heterogeneity of transcriptomic datasets that can arise from, for example, differential regulation of alternative splice forms mapped to different probesets. Adding to this technical heterogeneity in our study was the fact that the dataset source biosample was often multicellular in nature. In technical terms, this heterogeneity manifested itself in the modest gene level aggregate GMFCs of the original consensome analysis (Table S3). It was only when we applied modified consensome analysis to include the FLEE module that meaningful subsets of T1D-repressed or -induced transcripts were resolved that withstood subsequent validation. Although heterogeneity of transcriptomic datasets has been typically cited as an obstacle to their prognostic and diagnostic value (Bonifacio, 2015), other studies have demonstrated that such heterogeneity can actually be an asset in gleaning insights into fundamental mechanisms underlying human disease (Cahan and Khatri, 2020). Indeed, our discovery of a compelling degree of identity between the top-ranked NK-cell specific ET1DREs and a nine-gene signature that was consistently repressed in MIS-C (Figures 4F–4H) would not have been possible without the use of C-FLEE analysis to resolve the original set of ET1DREs. We noted with interest the fact that the performance of the ET1DREs against validation gene sets was much more robust than that of the ET1DIEs. This discrepancy may to some extent reflect bias in the validation gene sets, but can also be interpreted as evidence that transcriptional dysfunction of CICs in T1D is dominated by depletion of gene expression programs in cell types with critical roles in tolerance. Although this is a necessarily speculative interpretation, evidence exists that loss of function of specific CIC populations is a contributing factor in T1D (Jeker et al., 2012; Oras et al., 2019; Visperas and Vignali, 2016).
A consistent computational theme across our use cases is the isolation of discrete groups of genes to resolve deficits in CIC signaling pathways that were less apparent in the broader universe of T1D-regulated genes from each dataset. Predicates for this strategy encompassed selective expression in specific immune cell lineages, high confidence connections to downstream pathways indicated by genome-wide functional screens, and preferential regulatory relationships with upstream signaling nodes predicted by our own consensome analysis (Ochsner et al., 2019). In use case 1, for example, computation of ET1DREs against scRNA-Seq immune cell expression atlases (Ota et al., 2021; Travaglini et al., 2020) drew our attention to NK cells, from which additional community datasets (Collins et al., 2019; Hanna et al., 2004) enabled progressively finer resolution of the CD56dim CD57+ ET1DRE gene subset (Figure 3). In use case 3, although we observed a modest enrichment of IR genes in the ST1D-induced gene set, it was not until we applied HCT intersection clustering and consensome annotations that the potential role of clade 150c in connecting alternative splicing events in macrophages to severity of type 1 diabetes became apparent (Figure 6). Although scRNA-Seq approaches have exciting potential in type 1 diabetes, and would certainly add value to our resource, they have not yet been archived in sufficient numbers to support meaningful meta-analysis. Our use cases demonstrate however that while this situation persists it need not necessarily be an impediment to discerning cell-type-specific signaling deficits contributing to T1D.
A critical component of our study is the availability of the predictive networks in a freely accessible, federated web resource. The broad, cross-domain userbases of NDEx and SPP will give investigators across diverse fields the opportunity to bring their own domain- and paradigm-specific expertise to bear upon the complexity of T1D. Making the data matrix available in two resources that place a high prioroty on ease of use enables bench researchers to benefit from the results of analytical approaches that are more typically the preserve of laboratories with advanced informatics expertise and computational infrastructure. That said, although researchers lacking informatics expertise are the primary focus of our resource, sophisticated informaticians may develop alternative approaches to computing against the T1D gene expression matrix that complement the networks generated by C-FLEE and HCT intersection analyses. Supported by appropriate levels of funding, our resource will be supplemented with future T1D case control datasets that will serve to iteratively improve their predictive capacity. Placed in a broader context, our approach demonstrates the role that judicious selection, biocuration, and computation of existing archived omics-scale datasets can make to supporting hypothesis-driven research into human disease states.
Limitations of the study
Our study has a number of limitations. Abundant literature evidence emerging from both clinical cohorts and model systems points to the complex, polygenic basis of T1D and its associated clinical complications. As has been previously pointed out (Bonifacio, 2015), it is unrealistic for a meta-analysis of diverse, heterogeneous datasets to arrive at prognostic or diagnostic principles that apply across all cohorts, and no such assertion is implied by our study. To assist users in resolving the heterogeneity of the underlying datasets, we provided the complete feature-level differential data matrix (Table S4) such that users can identify specific patterns of differential expression in the underlying contrasts. Another limitation relates to the design of the available studies; since most archived T1D are based upon peripheral blood samples, our study is limited to the CIC compartment and does not address the important roles known to be played by local tissue-resident immune cell populations in T1D. Moreover, since the transcriptomic datasets and ChIP-Seq-based HCT intersection analysis are fundamentally transcriptional methodologies, the roles in T1D of deficits at the protein and metabolite levels are not directly addressed by our study. Finally, although extensively validated and statistically robust, our use cases are inferential in nature, and discerning specific underlying mechanisms is beyond the scope of our informatic analysis. For example, it is unclear whether enrichment of the CD56dim CD57+ NK cell genes among ET1DREs represents either reduced absolute numbers of these cells, or selective repression of transcriptional programs within this cell type. Although hypotheses such as those resolved by our analysis must be pressure-tested before their true clinical value can be established, the value for laboratories with limited budgets to avoid the informatic heavy lifting required to arrive at such predictions can be nonetheless be appreciated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited Data | ||
| Gene Expression Omnibus | SCR_007303 | |
| ArrayExpress | SCR_002964 | |
| Type 1 Diabetes Knowledge Portal | SCR_020936 | |
| ChIP-Atlas | SCR_015511 | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/vA3dd7GjOJ | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human CD14+ monocytes isolated from peripheral blood mononuclear cells (PBMCs) | 10.1621/UVifeAYAbV | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human CD14+ monocytes isolated from peripheral blood mononuclear cells (PBMCs) | 10.1621/k7BYeTib2R | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human CD14+ monocytes isolated from peripheral blood mononuclear cells (PBMCs) | 10.1621/3HLF1qydpY | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human whole blood | 10.1621/ooDpatg6cJ | |
| Analysis of the type 1 diabetic (T1D), type 2 diabetic (T2D), and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/8j3AQeLmFm | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/GbS5YGB6aX | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/o93ke5Km3V | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/3LVkDefCXZ | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood mononuclear cells (PBMCs) | 10.1621/VosI2QKTZb | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human CD14+ monocytes isolated from peripheral blood mononuclear cells (PBMCs) | 10.1621/vYTTxDS6RR | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human whole blood | 10.1621/fYxnk3NySG | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human hematopoietic stem cells | 10.1621/VenFStgTOS | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human peripheral blood T-cells | 10.1621/SQhBjEhdCZ | |
| Analysis of the type 1 diabetic (T1D), at-risk pre-symptomatic, and non-diabetic transcriptomes in human peripheral blood neutrophils | 10.1621/aJT6THuICG | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human monocyte-derived dendritic cells | 10.1621/1O6M5NThYy | |
| Analysis of the type 1 diabetic (T1D) and non-diabetic transcriptome in human CD14+ monocytes isolated from peripheral blood mononuclear cells (PBMCs) | 10.1621/ulQrgbGNvi | |
| Signaling Pathways Project | SCR_018412 | |
| Network Data Exchange | SCR_003943 | |
| Software and algorithms | ||
| Signaling Pathways Project | SCR_018412 | |
| Bioconductor GeneOverlap | SCR_018419 | |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Neil McKenna (nmckenna@bcm.edu).
Materials availability
This study did not generate any new reagents.
Method details
Dataset collection
Searching the Gene Expression Omnibus and Array Express transcriptomic dataset repositories, we identified a total of 21 clinical studies comparing gene expression profiles between subjects diagnosed with T1D and normoglycemic controls. A confirmatory literature search of PubMed identified a number of studies that did not archive their full datasets but instead provided abridged gene lists as supplementary data. To avoid introducing any bias associated with the criteria applied to generate these gene lists, these studies were excluded in our study. Four datasets compared gene in T1D and control donor pancreas samples; due to their limited number these datasets were not included in our study. Likely reflecting the relative ease with which these samples can be collected from subjects, the vast majority (n = 17) of the available archived T1D v control datasets were based upon RNA prepared from CICs. Of the 17 CIC datasets, 13 were expression array and four were high-throughput RNA-Seq-based (Table S1). One of the CIC datasets included a type 2 diabetes v control contrast that was not included in our study. IRB approval was not required since the original datasets were de-identified and archived in a public repository.
Gene expression analysis
To process microarray expression data, we utilized the log2 summarized and normalized array feature expression intensities provided by the investigator and housed in GEO. These data are available in the corresponding “Series Matrix Files(s)”. The full set of summarized and normalized sample expression values were extracted and processed using R software (http://cran.r-project.org). To calculate differential gene expression for investigator-defined experimental contrasts, we used the linear modeling functions from the Bioconductor limma analysis package. Initially, a linear model was fitted to a group-means parameterization design matrix defining each experimental variable. Subsequently, we fitted a contrast matrix that recapitulated the sample contrasts of interest, in this case viral infection versus mock infection, producing fold-change and significance values for each array feature present on the array. The current BioConductor array annotation library was used for annotation of array identifiers. p values obtained from limma analysis were not corrected for multiple comparisons prior to consensome analysis. To process RNA-Seq expression data, we utilized the aligned, un-normalized, gene summarized read count data provided by the investigator and housed in GEO. These data are available in the corresponding “Supplementary file” section of the GEO record. The full set of raw aligned gene read count values were extracted and processed in the statistical program R using the limma and edgeR analysis packages. Read count values were initially filtered to remove genes with low read counts. Gene read count values were passed to downstream analysis if all replicate samples from at least one experimental condition had cpm >1. Sequence library normalization factors were calculated to apply scale normalization to the raw aligned read counts using the TMM normalization method implemented in the edgeR package followed by the voom function to convert the gene read count values to log2-cpm. The log2-cpm values were initially fit to a group-means parameterization design matrix defining each experimental variable. This was subsequently fit to a contrast design matrix that recapitulates the sample contrasts of interest (case versus control contrasts) producing fold-change and significance values for each aligned sequenced gene. If necessary, the current BioConductor human organism annotation library was used for annotation of investigator-provided gene identifiers. p values obtained from limma analysis were not corrected for multiple comparisons prior to consensome analysis.
Consensome-FLEE analysis
Differential expression values from individual case:control contrasts were committed to the consensome analysis pipeline as previously described (Ochsner et al., 2019). Briefly, the consensome algorithm surveys each experiment across all datasets and ranks genes according to the frequency with which they are significantly differentially expressed. For each transcript, we counted the number of experiments where the significance for differential expression was ≤0.05, and then generated the binomial probability, referred to as the consensome p value (CPV), of observing that many or more nominally significant experiments out of the number of experiments in which the transcript was assayed, given a true probability of 0.05. p values were adjusted using the p.adj function in R to generate q values (CQV). Genes were ranked firstly by CQV, then by GMFC. Please refer to the main text for details on the FLEE component of this analysis.
High confidence transcriptional target intersection analysis
High confidence transcript intersection analysis (Ochsner et al., 2020) was used to compute overlap between T1D gene lists and ChIP-Seq consensome sets of 961 human signaling pathway nodes. Briefly, ChIP-Seq consensomes rank genes based upon the average peak height of a gene over all public ChIP-Seq datasets in which a specific node was an IP antigen (Ochsner et al., 2019). Source data were obtained from the ChIP-Atlas resource (Oki et al., 2018). Genes in the top 5% of these consensomes were designated as high confidence transcriptional targets (HCTs) for a given node (Ochsner et al., 2020). We then input gene lists into the Bioconductor GeneOverlap analysis package implemented in R. Given a whole set I of IDs and two sets A ∈ I and B ∈ I, and S = A ∩ B, GeneOverlap calculates the significance of obtaining S. With specific reference to this study, S represents the overlap between node HCTs and ET1DREs, ET1DIEs and subsets thereof, or significantly induced or repressed gene sets from the underlying individual case:control contrasts. The enrichment (ENR) number represents the ratio of the number of observed intersecting genes and the number of expected intersecting genes. The problem was formulated as a hypergeometric distribution or contingency table and solved by Fisher’s exact test. p values were adjusted for multiple testing by using the method of Benjamini & Hochberg to control the false discovery rate as implemented with the p.adjust function in R, to generate q values. The universe for the intersection was set at a conservative estimate of the total number of transcribed (protein and non-protein-coding) genes in the human genome (25,000).
Pan-autoimmune GWAS genes
We retrieved the top 1000 common variant gene-level associations for (p ≤ 0.05) for type 1 diabetes, Crohn’s disease, ulcerative colitis and irritable bowel syndrome from T1DKP. To generate the list of PAGGs, we ranked genes in each of the four categories then generated an average ranking
Hierarchical clustering
Hierarchical cluster analysis of HCTI results was done using the hclust function in R with “complete” agglomeration to evaluate dissimilarities. Pearson correlation coefficients between MLP values were used as a distance measure (1-coefficient). Functions from the dendextend and corrplot libraries were used to visualize clustering results as dendrograms. All functions were run in R 3.6.3.
Quantification and statistical analysis
The hypergeometric test was used to calculate the significance of enrichment of a given gene set within another gene set. In the interests of full transparency of our statistical approaches, gene set cut-offs for all hypergeometric tests are itemized in the supplementary text. All hypergeometric test functions were run in R 3.6.3.
Acknowledgments
We thank Drs. Maryann Martone and Ko-Wei Lin of DKNET, and Dr. Monica Westley of the Sugar Science, for their support in devising the D-Challenge.
This work was supported by: National Institute of Diabetes and Digestive and Kidney Diseases grant U24DK097771 (JSG, NJM)
Cloud migration and maintenance of SPP was supported by NIH Office of Data Science Strategy (ODSS) High-Value Datasets program (JSG, NM)
Institute For Collaboration In Health grant (NJM)
National Cancer Institute grant CA184427 (RTP).
Author contributions
NJM conceived and designed the study and generated the use cases. SAO carried out dataset identification, quality control and processing, as well as all downstream regulatory analyses. RTP created the NDEx networks. JSG and DR performed migration and maintenance of SPP in the AWS cloud.
Declaration of interests
The authors declare no competing interests
Published: July 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104581.
Supplemental information
Contains original publication PubMed ID (where available), repository accession number, dataset name, description and underlying experiments & their descriptions.
Detailed information on study cohorts curated from the original publication, biosamples (RNA source) and comments regarding principal component analysis and verification of fold changes against the original publication where available.
The consensome algorithm surveys each experiment across all datasets and ranks genes according to the frequency with which they are significantly differentially expressed. For each transcript, we counted the number of experiments where the significance for differential expression was <0.05, and then generated the binomial probability, referred to as the consensome p-value (CPV), of observing that many or more nominally significant experiments out of the number of experiments in which the transcript was assayed, given a true probability of 0.05. Genes were ranked firstly by CPV, then by geometric mean fold change (GMFC).
Experiment differentially expressed features (in expression arrays, probesets; in RNA-Seq experiments, annotated gene symbol) at p < 0.05 across the 26 T1D v C experiments.
"Exclusively type 1 diabetes repressed-enriched (ET1DRE) genes are those whose features were significantly enriched among features that were significantly repressed in at least one experiment and significantly induced in zero. Exclusively type 1 diabetes induced-enriched (ET1DIE) genes are those whose features were significantly enriched among features that were significantly induced in at least one experiment and significantly repressed in zero.
Dataset GSE84934 by Long and colleagues was generated from the AbATE (Autoimmunity-blocking Antibody for Tolerance in Recently Diagnosed Type 1 Diabetes) clinical trial. It profiled whole blood gene expression between T1D subjects who responded to treatment with the anti-CD3 antibody teplizumab and control individuals.
We used T1DKP to generate a list of 105 genes that had at least one significant GWAS association with T1D and three other autoimmune conditions: Crohn’s disease, ulcerative colitis and irritable bowel syndrome and designated these pan-autoimmune GWAS genes (PAGGs). PAGGs were ranked by the average rank of the p-value for their association with each condition.
All intersections between (i) ET1DRE genes or (ii) ET1DRE genes and node high confidence transcriptional targets (HCTs) derived from consensomes based upon archived ChIP-Seq experiments.
p < 0.05 intersections between (i) ET1DRE genes or (ii) ET1DRE genes and node high confidence transcriptional targets (HCTs) derived from consensomes based upon archived ChIP-Seq experiments.
p < 0.05 intersections between experiment differentially expressed gene sets (case v control FC > 1.25 induced or repressed, p < 0.05) and node high confidence transcriptional targets (HCTs) derived from archived ChIP-Seq experiments.
Genome-wide single guide RNA (sgRNA) screen that ranks genes according to the essentiality of their roles in supporting primary T cell viability.
Single-nucleus assay for transposase-accessible chromatin with sequencing (snATAC–seq)-based accessible chromatin reference map of 28 peripheral blood and pancreas cell types from donors without type 1 diabetes.
Pooled purified peripheral blood derived CD56dimCD16+ and CD56brightCD16- NK subsets were obtained from 9 healthy donors. Each pooled NK subset sample was hybridized in replicates (A and B).
Full gene list from dataset 1274, experiment 6651, generated by a study of neutrophils in a cohort of subjects with early onset T1D.
Comparison of HCT intersection analysis results for TRPC6M-NEOT1DI and NEOT1DI gene sets.
Enrichment statistics of Reactome functional pathways within TCS-NEOT1DI and NEOT1DI gene sets.
Ratio of aggregate significant MT1D-induced and ST1D-induced HCT intersection analysis ORs and -log10p (ML10P) values for each node across the two cohorts (Table S16).
Genome-wide CRISPR screen by Covarrubias that assigns Mann-Whitney (MW) scores to 21379 mouse genes, such that the gene with the lowest MW score is designated the most essential for macrophage viability.
Analysis demonstrating performance of this analysis against T1D gene lists from other studies
Analysis demonstrating performance of HCTI against other transcription factor enrichment tool.
Data and code availability
-
•
This paper analyzes existing, publicly available datasets. The accession numbers for the datasets are listed in the Key Resources table.
-
•
All secondary data generated this study are available from the Signaling Pathways Project and Network Data Exchange resources. RRIDs for these resources are provided in the Key Resources table.
-
•
All code relevant to this study is available in the SPP GitHub account under a Creative Commons CC BY 4.0 license at https://github.com/signaling-pathways-project.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Alarcón-Riquelme M.E. A RUNX trio with a taste for autoimmunity. Nat. Genet. 2003;35:299–300. doi: 10.1038/ng1203-299. [DOI] [PubMed] [Google Scholar]
- Aoki C.A., Borchers A.T., Li M., Flavell R.A., Bowlus C.L., Ansari A.A., Gershwin M.E. Transforming growth factor beta (TGF-beta) and autoimmunity. Autoimmun. Rev. 2005;4:450–459. doi: 10.1016/j.autrev.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Aqel S.I., Yang X., Kraus E.E., Song J., Farinas M.F., Zhao E.Y., Pei W., Lovett-Racke A.E., Racke M.K., Li C., Yang Y. A STAT3 inhibitor ameliorates CNS autoimmunity by restoring Teff:Treg balance. JCI insight. 2021;6:142376. doi: 10.1172/jci.insight.142376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M., Ahmed S., Anderson M.S., Atkinson M.A., Becker D., Bingley P.J., Bosi E., Brusko T.M., DiMeglio L.A., Evans-Molina C., et al. Introducing the endotype concept to address the challenge of disease heterogeneity in type 1 diabetes. Diabetes Care. 2020;43:5–12. doi: 10.2337/dc19-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann N.D., Comella P.H., Cheng E., Lepow L., Beckmann A.G., Tyler S.R., Mouskas K., Simons N.W., Hoffman G.E., Francoeur N.J., et al. Downregulation of exhausted cytotoxic T cells in gene expression networks of multisystem inflammatory syndrome in children. Nat. Commun. 2021;12:4854. doi: 10.1038/s41467-021-24981-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S., Dunn P., Thomas C.G., Smith B., Schaefer H., Chen J., Hu Z., Zalocusky K.A., Shankar R.D., Shen-Orr S.S., et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data. 2018;5:180015. doi: 10.1038/sdata.2018.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig-Choisat B., Alves-Bezerra M., Zorman B., Ochsner S.A., Barzi M., Legras X., Yang D., Borowiak M., Dean A.M., York R.B., et al. A human liver chimeric mouse model for non-alcoholic fatty liver disease. JHEP Rep. 2021;3:100281. doi: 10.1016/j.jhepr.2021.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio E. Predicting type 1 diabetes using biomarkers. Diabetes Care. 2015;38:989–996. doi: 10.2337/dc15-0101. [DOI] [PubMed] [Google Scholar]
- Brillantes M., Beaulieu A.M. Transcriptional control of natural killer cell differentiation. Immunology. 2019;156:111–119. doi: 10.1111/imm.13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.E.P., Onyango D.J., Ramanjaneya M., Conner A.C., Patel S.T., Dunmore S.J., Randeva H.S. Visfatin regulates insulin secretion, insulin receptor signalling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J. Mol. Endocrinol. 2010;44:171–178. doi: 10.1677/jme-09-0071. [DOI] [PubMed] [Google Scholar]
- Cahan E.M., Khatri P. Data heterogeneity: the enzyme to catalyze translational bioinformatics? J. Med. Internet Res. 2020;22:e18044. doi: 10.2196/18044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon B., Suri A., Unanue E.R. In CD4+ T-cell-induced diabetes, macrophages are the final effector cells that mediate islet beta-cell killing: studies from an acute model. Am. J. Pathol. 2006;169:2137–2147. doi: 10.2353/ajpath.2006.060539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertova E., Chertov O., Coren L.V., Roser J.D., Trubey C.M., Bess J.W., Jr., Sowder R.C., 2nd, Barsov E., Hood B.L., Fisher R.J., et al. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 2006;80:9039–9052. doi: 10.1128/jvi.01013-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou J., Geusz R.J., Okino M.L., Han J.Y., Miller M., Melton R., Beebe E., Benaglio P., Huang S., Korgaonkar K., et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature. 2021;594:398–402. doi: 10.1038/s41586-021-03552-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu T.T., Tu X., Yang K., Wu J., Repa J.J., Yan N. Tonic prime-boost of STING signalling mediates Niemann-Pick disease type C. Nature. 2021;596:570–575. doi: 10.1038/s41586-021-03762-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P.L., Cella M., Porter S.I., Li S., Gurewitz G.L., Hong H.S., Johnson R.P., Oltz E.M., Colonna M. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. 2019;176:348–360.e12. doi: 10.1016/j.cell.2018.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias S., Vollmers A.C., Capili A., Boettcher M., Shulkin A., Correa M.R., Halasz H., Robinson E.K., O'Briain L., Vollmers C., et al. High-throughput CRISPR screening identifies genes involved in macrophage viability and inflammatory pathways. Cell Rep. 2020;33:108541. doi: 10.1016/j.celrep.2020.108541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvitkovic I., Jurica M.S. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41:D132–D141. doi: 10.1093/nar/gks999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva K., Demmer R.T., Jönsson D., Mousa A., Forbes A., Enticott J. Marker genes of incident type 1 diabetes in peripheral blood mononuclear cells of children: a machine learning strategy for large-p, small-n scenarios. medRxiv. 2022 doi: 10.1101/2022.02.07.22270652. Preprint at. [DOI] [Google Scholar]
- Drouillard A., Mathieu A.L., Marçais A., Belot A., Viel S., Mingueneau M., Guckian K., Walzer T. S1PR5 is essential for human natural killer cell migration toward sphingosine-1 phosphate. J. Allergy Clin. Immunol. 2018;141:2265–2268.e1. doi: 10.1016/j.jaci.2017.11.022. [DOI] [PubMed] [Google Scholar]
- Esnault C., Heidmann O., Delebecque F., Dewannieux M., Ribet D., Hance A.J., Heidmann T., Schwartz O. APOBEC3G cytidine deaminase inhibits retrotransposition of endogenous retroviruses. Nature. 2005;433:430–433. doi: 10.1038/nature03238. [DOI] [PubMed] [Google Scholar]
- Frasca L., Lande R. Role of defensins and cathelicidin LL37 in auto-immune and auto-inflammatory diseases. Curr. Pharm. Biotechnol. 2012;13:1882–1897. doi: 10.2174/138920112802273155. [DOI] [PubMed] [Google Scholar]
- Fu G., Guy C.S., Chapman N.M., Palacios G., Wei J., Zhou P., Long L., Wang Y.D., Qian C., Dhungana Y., et al. Metabolic control of T(FH) cells and humoral immunity by phosphatidylethanolamine. Nature. 2021;595:724–729. doi: 10.1038/s41586-021-03692-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti C., Bayry J. Autoimmune and inflammatory diseases following COVID-19. Nat. Rev. Rheumatol. 2020;16:413–414. doi: 10.1038/s41584-020-0448-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner G., Fraker C.A. Natural killer cells as key mediators in type I diabetes immunopathology. Front. Immunol. 2021;12:722979. doi: 10.3389/fimmu.2021.722979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleron J., Bouget G., Ivanov S., Meziat C., Ceppo F., Vergoni B., Djedaini M., Soprani A., Dumas K., Jacquel A., et al. Rab4b deficiency in T cells promotes adipose Treg/Th17 imbalance, adipose tissue dysfunction, and insulin resistance. Cell Rep. 2018;25:3329–3341.e5. doi: 10.1016/j.celrep.2018.11.083. [DOI] [PubMed] [Google Scholar]
- Green I.D., Pinello N., Song R., Lee Q., Halstead J.M., Kwok C.T., Wong A.C.H., Nair S.S., Clark S.J., Roediger B., et al. Macrophage development and activation involve coordinated intron retention in key inflammatory regulators. Nucleic Acids Res. 2020;48:6513–6529. doi: 10.1093/nar/gkaa435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesi-Oliveira K., Acab A., Gupta A.R., Sunaga D.Y., Chailangkarn T., Nicol X., Nunez Y., Walker M.F., Murdoch J.D., Sanders S.J., et al. Modeling non-syndromic autism and the impact of TRPC6 disruption in human neurons. Mol. Psychiatry. 2015;20:1350–1365. doi: 10.1038/mp.2014.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J., Bechtel P., Zhai Y., Youssef F., McLachlan K., Mandelboim O. Novel insights on human NK cells' immunological modalities revealed by gene expression profiling. J. Immunol. 2004;173:6547–6563. doi: 10.4049/jimmunol.173.11.6547. [DOI] [PubMed] [Google Scholar]
- Haynes W.A., Haddon D.J., Diep V.K., Khatri A., Bongen E., Yiu G., Balboni I., Bolen C.R., Mao R., Utz P.J., et al. Integrated, multicohort analysis reveals unified signature of systemic lupus erythematosus. JCI insight. 2020;5:e122312. doi: 10.1172/jci.insight.122312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesslein D.G.T., Lanier L.L. Transcriptional control of natural killer cell development and function. Adv. Immunol. 2011;109:45–85. doi: 10.1016/b978-0-12-387664-5.00002-9. [DOI] [PubMed] [Google Scholar]
- Hook S.M., Phipps-Green A.J., Faiz F., McNoe L., McKinney C., Hollis-Moffatt J.E., Merriman T.R. Smad2: a candidate gene for the murine autoimmune diabetes locus Idd21.1. J. Clin. Endocrinol. Metab. 2011;96:E2072–E2077. doi: 10.1210/jc.2011-0463. [DOI] [PubMed] [Google Scholar]
- Hwang S.S., Jang S.W., Kim M.K., Kim L.K., Kim B.S., Kim H.S., Kim K., Lee W., Flavell R.A., Lee G.R. YY1 inhibits differentiation and function of regulatory T cells by blocking Foxp3 expression and activity. Nat. Commun. 2016;7:10789. doi: 10.1038/ncomms10789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S.T., Won J., Khan M., Mannie M.D., Singh I. Hypoxia-inducible factor-1 drives divergent immunomodulatory functions in the pathogenesis of autoimmune diseases. Immunology. 2021;164:31–42. doi: 10.1111/imm.13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeker L.T., Bour-Jordan H., Bluestone J.A. Breakdown in peripheral tolerance in type 1 diabetes in mice and humans. Cold Spring Harb Perspect. Med. 2012;2:a007807. doi: 10.1101/cshperspect.a007807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsen K., Nielsen H., Lorentzen S., Damsbo P., Buschard K. Circulating monocytes are activated in newly diagnosed type 1 diabetes mellitus patients. Clin. Exp. Immunol. 1994;98:489–493. doi: 10.1111/j.1365-2249.1994.tb05517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi-Tope G., Gillespie M., Vastrik I., D'Eustachio P., Schmidt E., de Bono B., Jassal B., Gopinath G.R., Wu G.R., Matthews L., et al. Reactome: a knowledgebase of biological pathways. Nucleic Acids Res. 2005;33:D428–D432. doi: 10.1093/nar/gki072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi R.N., Fernandes S.J., Shang M.M., Kiani N.A., Gomez-Cabrero D., Tegnér J., Schmidt A. Phosphatase inhibitor PPP1R11 modulates resistance of human T cells toward Treg-mediated suppression of cytokine expression. J. Leukoc. Biol. 2019;106:413–430. doi: 10.1002/jlb.2a0618-228r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamieniarz-Gdula K., Gdula M.R., Panser K., Nojima T., Monks J., Wiśniewski J.R., Riepsaame J., Brockdorff N., Pauli A., Proudfoot N.J. Selective roles of vertebrate PCF11 in premature and full-length transcript termination. Mol. Cell. 2019;74:158–172.e9. doi: 10.1016/j.molcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara K., Wang Y., McAnally J., Richardson J.A., Bassel-Duby R., Hill J.A., Olson E.N. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J. Clin. Invest. 2006;116:3114–3126. doi: 10.1172/jci27702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latchman D.S. Transcription factors: bound to activate or repress. Trends Biochem. Sci. 2001;26:211–213. doi: 10.1016/s0968-0004(01)01812-6. [DOI] [PubMed] [Google Scholar]
- Lehuen A., Diana J., Zaccone P., Cooke A. Immune cell crosstalk in type 1 diabetes. Nat. Rev. Immunol. 2010;10:501–513. doi: 10.1038/nri2787. [DOI] [PubMed] [Google Scholar]
- Li Y., Zhang K., Chen H., Sun F., Xu J., Wu Z., Li P., Zhang L., Du Y., Luan H., et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren's syndrome at 7q11.23. Nat. Genet. 2013;45:1361–1365. doi: 10.1038/ng.2779. [DOI] [PubMed] [Google Scholar]
- Liu H., Lorenzini P.A., Zhang F., Xu S., Wong M.M., Zheng J., Roca X. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res. 2018;46:6069–6086. doi: 10.1093/nar/gky401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long J., Wang D., Yang X., Wang A., Lin Y., Zheng M., Zhang H., Sang X., Wang H., Hu K., Zhao H. Identification of NOTCH4 mutation as a response biomarker for immune checkpoint inhibitor therapy. BMC Med. 2021;19:154. doi: 10.1186/s12916-021-02031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S.A., Thorpe J., DeBerg H.A., Gersuk V., Eddy J.A., Harris K.M., Ehlers M., Herold K.C., Nepom G.T., Linsley P.S. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci. Immunol. 2016;1:eaai7793. doi: 10.1126/sciimmunol.aai7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luangsay S., Wittamer V., Bondue B., De Henau O., Rouger L., Brait M., Franssen J.D., de Nadai P., Huaux F., Parmentier M. Mouse ChemR23 is expressed in dendritic cell subsets and macrophages, and mediates an anti-inflammatory activity of chemerin in a lung disease model. J. Immunol. 2009;183:6489–6499. doi: 10.4049/jimmunol.0901037. [DOI] [PubMed] [Google Scholar]
- Ma W.T., Gao F., Gu K., Chen D.K. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front. Immunol. 2019;10:1140. doi: 10.3389/fimmu.2019.01140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeng Y.S., Min J.K., Kim J.H., Yamagishi A., Mochizuki N., Kwon J.Y., Park Y.W., Kim Y.M., Kwon Y.G. ERK is an anti-inflammatory signal that suppresses expression of NF-κB-dependent inflammatory genes by inhibiting IKK activity in endothelial cells. Cell Signal. 2006;18:994–1005. doi: 10.1016/j.cellsig.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Mailman M.D., Feolo M., Jin Y., Kimura M., Tryka K., Bagoutdinov R., Hao L., Kiang A., Paschall J., Phan L., et al. The NCBI dbGaP database of genotypes and phenotypes. Nat. Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malachowa N., Freedman B., Sturdevant D.E., Kobayashi S.D., Nair V., Feldmann F., Starr T., Steele-Mortimer O., Kash J.C., Taubenberger J.K., et al. Differential ability of pandemic and seasonal H1N1 influenza A viruses to alter the function of human neutrophils. mSphere. 2018;3 doi: 10.1128/mSphereDirect.00567-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez F.O., Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep. 2014;6:13. doi: 10.12703/p6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna N.J., Nawaz Z., Tsai S.Y., Tsai M.J., O'Malley B.W. Distinct steady-state nuclear receptor coregulator complexes exist in vivo. Proc. Natl. Acad. Sci. U S A. 1998;95:11697–11702. doi: 10.1073/pnas.95.20.11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna N.J., O'Malley B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- McKinney E.F., Lee J.C., Jayne D.R.W., Lyons P.A., Smith K.G.C. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdi A.M., Hamilton-Williams E.E., Cristino A., Ziegler A., Bonifacio E., Le Cao K.A., Harris M., Thomas R. A peripheral blood transcriptomic signature predicts autoantibody development in infants at risk of type 1 diabetes. JCI insight. 2018;3:98212. doi: 10.1172/jci.insight.98212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersich A.T., Miller M.R., Chkourko H., Blystone S.D. The formin FRL1 (FMNL1) is an essential component of macrophage podosomes. Cytoskeleton. 2010;67:573–585. doi: 10.1002/cm.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollah Z.U.A., Quah H.S., Graham K.L., Jhala G., Krishnamurthy B., Dharma J.F.M., Chee J., Trivedi P.M., Pappas E.G., Mackin L., et al. Granzyme A deficiency breaks immune tolerance and promotes autoimmune diabetes through a type I interferon-dependent pathway. Diabetes. 2017;66:3041–3050. doi: 10.2337/db17-0517. [DOI] [PubMed] [Google Scholar]
- Mountjoy E., Schmidt E.M., Carmona M., Schwartzentruber J., Peat G., Miranda A., Fumis L., Hayhurst J., Buniello A., Karim M.A., et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat. Genet. 2021;53:1527–1533. doi: 10.1038/s41588-021-00945-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall C.J., McMurry J.A., Köhler S., Balhoff J.P., Borromeo C., Brush M., Carbon S., Conlin T., Dunn N., Engelstad M., et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2017;45 doi: 10.1093/nar/gkw1128. D712–d722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman J.R.B., Conesa A., Mika M., New F.N., Onengut-Gumuscu S., Atkinson M.A., Rich S.S., McIntyre L.M., Concannon P. Disease-specific biases in alternative splicing and tissue-specific dysregulation revealed by multitissue profiling of lymphocyte gene expression in type 1 diabetes. Genome Res. 2017;27:1807–1815. doi: 10.1101/gr.217984.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C.M., White M.J., Goodier M.R., Riley E.M. Functional significance of CD57 expression on human NK cells and relevance to disease. Front. Immunol. 2013;4:422. doi: 10.3389/fimmu.2013.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner S.A., Abraham D., Martin K., Ding W., McOwiti A., Kankanamge W., Wang Z., Andreano K., Hamilton R.A., Chen Y., et al. The signaling pathways project, an integrated 'omics knowledgebase for mammalian cellular signaling pathways. Sci. Data. 2019;6:252. doi: 10.1038/s41597-019-0193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsner S.A., Pillich R.T., McKenna N.J. Consensus transcriptional regulatory networks of coronavirus-infected human cells. Sci. Data. 2020;7:314. doi: 10.1038/s41597-020-00628-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y., Shimane K., Kochi Y., Tahira T., Suzuki A., Higasa K., Takahashi A., Horita T., Atsumi T., Ishii T., et al. A genome-wide association study identified AFF1 as a susceptibility locus for systemic lupus eyrthematosus in Japanese. PLoS Genet. 2012;8:e1002455. doi: 10.1371/journal.pgen.1002455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki S., Ohta T., Shioi G., Hatanaka H., Ogasawara O., Okuda Y., Kawaji H., Nakaki R., Sese J., Meno C. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 2018;19:e46255. doi: 10.15252/embr.201846255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oras A., Peet A., Giese T., Tillmann V., Uibo R., OrAs A., Peet A. A study of 51 subtypes of peripheral blood immune cells in newly diagnosed young type 1 diabetes patients. Clin. Exp. Immunol. 2019;198:57–70. doi: 10.1111/cei.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M., Nagafuchi Y., Hatano H., Ishigaki K., Terao C., Takeshima Y., Yanaoka H., Kobayashi S., Okubo M., Shirai H., et al. Dynamic landscape of immune cell-specific gene regulation in immune-mediated diseases. Cell. 2021;184:3006–3021.e17. doi: 10.1016/j.cell.2021.03.056. [DOI] [PubMed] [Google Scholar]
- Oughtred R., Rust J., Chang C., Breitkreutz B.J., Stark C., Willems A., Boucher L., Leung G., Kolas N., Zhang F., et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021;30:187–200. doi: 10.1002/pro.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.J., Yoo S.A., Kim M., Kim W.U. The role of calcium-calcineurin-NFAT signaling pathway in health and autoimmune diseases. Front. Immunol. 2020;11:195. doi: 10.3389/fimmu.2020.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnell G.P., Gatt P.N., Krupa M., Nickles D., McKay F.C., Schibeci S.D., Batten M., Baranzini S., Henderson A., Barnett M., et al. The autoimmune disease-associated transcription factors EOMES and TBX21 are dysregulated in multiple sclerosis and define a molecular subtype of disease. Clin. Immunol. 2014;151:16–24. doi: 10.1016/j.clim.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Paul D.S., Teschendorff A.E., Dang M.A., Lowe R., Hawa M.I., Ecker S., Beyan H., Cunningham S., Fouts A.R., Ramelius A., et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016;7:13555. doi: 10.1038/ncomms13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli A., Michel T., Thérésine M., Andrès E., Hentges F., Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126:458–465. doi: 10.1111/j.1365-2567.2008.03027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt R.A., Binek A., Paschold L., Rivas M.N., McArdle A., Yonker L.M., Alter G., Chandnani H.K., Lopez M., Fasano A., et al. The autoimmune signature of hyperinflammatory multisystem inflammatory syndrome in children. J. Clin. Invest. 2021;131:e151520. doi: 10.1172/jci151520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt D., Chen J., Welker D., Rivas R., Pillich R., Rynkov V., Ono K., Miello C., Hicks L., Szalma S., et al. NDEx, the network data Exchange. Cell Syst. 2015;1:302–305. doi: 10.1016/j.cels.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rioux J.D., Xavier R.J., Taylor K.D., Silverberg M.S., Goyette P., Huett A., Green T., Kuballa P., Barmada M.M., Datta L.W., et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothe T., Ipseiz N., Faas M., Lang S., Perez-Branguli F., Metzger D., Ichinose H., Winner B., Schett G., Krönke G. The nuclear receptor Nr4a1 acts as a microglia rheostat and serves as a therapeutic target in autoimmune-driven central nervous system inflammation. J. Immunol. 2017;198:3878–3885. doi: 10.4049/jimmunol.1600638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang W., Gao Y., Tang Z., Zhang Y., Yang R. The pseudogene Olfr29-ps1 promotes the suppressive function and differentiation of monocytic MDSCs. Cancer Immunol. Res. 2019;7:813–827. doi: 10.1158/2326-6066.cir-18-0443. [DOI] [PubMed] [Google Scholar]
- Shifrut E., Carnevale J., Tobin V., Roth T.L., Woo J.M., Bui C.T., Li P.J., Diolaiti M.E., Ashworth A., Marson A. Genome-wide CRISPR screens in primary human T cells reveal key regulators of immune function. Cell. 2018;175:1958–1971.e15. doi: 10.1016/j.cell.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S.C., Chang J.H., Jin J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013;34:282–289. doi: 10.1016/j.it.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- T1DKP . 2021. The Type 1 Diabetes Knowledge Portal. https://t1d.hugeamp.org/ [Google Scholar]
- Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cel. Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Travaglini K.J., Nabhan A.N., Penland L., Sinha R., Gillich A., Sit R.V., Chang S., Conley S.D., Mori Y., Seita J., et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. 2020;587:619–625. doi: 10.1038/s41586-020-2922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visperas A., Vignali D.A.A. Are regulatory T cells defective in type 1 diabetes and can we fix them? J. Immunol. 2016;197:3762–3770. doi: 10.4049/jimmunol.1601118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wachlin G., Augstein P., Schröder D., Kuttler B., Klöting I., Heinke P., Schmidt S. IL-1β, IFN-γ and TNF-α increase vulnerability of pancreatic beta cells to autoimmune destruction. J. Autoimmun. 2003;20:303–312. doi: 10.1016/s0896-8411(03)00039-8. [DOI] [PubMed] [Google Scholar]
- Wagner A.R., Scott H.M., West K.O., Vail K.J., Fitzsimons T.C., Coleman A.K., Carter K.E., Watson R.O., Patrick K.L. Global transcriptomics uncovers distinct contributions from splicing regulatory proteins to the macrophage innate immune response. Front. Immunol. 2021;12:656885. doi: 10.3389/fimmu.2021.656885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Syrett C.M., Kramer M.C., Basu A., Atchison M.L., Anguera M.C. Unusual maintenance of X chromosome inactivation predisposes female lymphocytes for increased expression from the inactive X. Proc. Natl. Acad. Sci. U S A. 2016;113:E2029–E2038. doi: 10.1073/pnas.1520113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Chang J.H., Buckley A.F., Spurney R.F. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019;95:321–332. doi: 10.1016/j.kint.2018.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ni L., Chang D., Lu H., Jiang Y., Kim B.S., Wang A., Liu X., Zhong B., Yang X., Dong C. Cyclic AMP-responsive element-binding protein (CREB) is critical in autoimmunity by promoting Th17 but inhibiting Treg cell differentiation. EBioMedicine. 2017;25:165–174. doi: 10.1016/j.ebiom.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Yuan S., Jia X., Ge Y., Ling T., Nie M., Lan X., Chen S., Xu A. Mitochondria-localised ZNFX1 functions as a dsRNA sensor to initiate antiviral responses through MAVS. Nat. Cel. Biol. 2019;21:1346–1356. doi: 10.1038/s41556-019-0416-0. [DOI] [PubMed] [Google Scholar]
- Watt S., Vasquez L., Walter K., Mann A.L., Kundu K., Chen L., Sims Y., Ecker S., Burden F., Farrow S., et al. Genetic perturbation of PU.1 binding and chromatin looping at neutrophil enhancers associates with autoimmune disease. Nat. Commun. 2021;12:2298. doi: 10.1038/s41467-021-22548-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whetzel P.L., Grethe J.S., Banks D.E., Martone M.E. The NIDDK information network: a community portal for finding data, materials, and tools for researchers studying diabetes, digestive, and Kidney diseases. PLoS One. 2015;10 doi: 10.1371/journal.pone.0136206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn M.P., Conlon P.J., Lynn K.L., Farrington M.K., Creazzo T., Hawkins A.F., Daskalakis N., Kwan S.Y., Ebersviller S., Burchette J.L., et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. doi: 10.1126/science.1106215. [DOI] [PubMed] [Google Scholar]
- Xhonneux L.P., Knight O., Lernmark Å., Bonifacio E., Hagopian W.A., Rewers M.J., She J.X., Toppari J., Parikh H., Smith K.G.C., et al. Transcriptional networks in at-risk individuals identify signatures of type 1 diabetes progression. Sci. Transl Med. 2021;13:eabd5666. doi: 10.1126/scitranslmed.abd5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Bailey A., Kuleshov M.V., Clarke D.J.B., Evangelista J.E., Jenkins S.L., Lachmann A., Wojciechowicz M.L., Kropiwnicki E., Jagodnik K.M., et al. Gene set knowledge discovery with enrichr. Curr. Protoc. 2021;1:e90. doi: 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelante T., Wong A.Y.W., Mencarelli A., Foo S., Zolezzi F., Lee B., Poidinger M., Ricciardi-Castagnoli P., Fric J., MencArelli A. Impaired calcineurin signaling in myeloid cells results in downregulation of pentraxin-3 and increased susceptibility to aspergillosis. Mucosal Immunol. 2017;10:470–480. doi: 10.1038/mi.2016.52. [DOI] [PubMed] [Google Scholar]
- Koprivica I., Vujičić M., Gajić D., Saksida T., Stojanović I. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int. J. Clin. Exp. Pathol. 2009;3:24. doi: 10.3389/fimmu.2018.03130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Morgan M.J., Chen K., Choksi S., Liu Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood. 2012;119:2895–2905. doi: 10.1182/blood-2011-08-372383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R., Liu H., Zhou Y., Yan D., Chen J., Ma D., Feng Y., Qin L., Liu F., Huang X., et al. Notch4 negatively regulates the inflammatory response to Mycobacterium tuberculosis infection by inhibiting TAK1 activation. J. Infect. Dis. 2018;218:312–323. doi: 10.1093/infdis/jix636. [DOI] [PubMed] [Google Scholar]
- Ziegler A.G., Rewers M., Simell O., Simell T., Lempainen J., Steck A., Winkler C., Ilonen J., Veijola R., Knip M., et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA. 2013;309:2473. doi: 10.1001/jama.2013.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains original publication PubMed ID (where available), repository accession number, dataset name, description and underlying experiments & their descriptions.
Detailed information on study cohorts curated from the original publication, biosamples (RNA source) and comments regarding principal component analysis and verification of fold changes against the original publication where available.
The consensome algorithm surveys each experiment across all datasets and ranks genes according to the frequency with which they are significantly differentially expressed. For each transcript, we counted the number of experiments where the significance for differential expression was <0.05, and then generated the binomial probability, referred to as the consensome p-value (CPV), of observing that many or more nominally significant experiments out of the number of experiments in which the transcript was assayed, given a true probability of 0.05. Genes were ranked firstly by CPV, then by geometric mean fold change (GMFC).
Experiment differentially expressed features (in expression arrays, probesets; in RNA-Seq experiments, annotated gene symbol) at p < 0.05 across the 26 T1D v C experiments.
"Exclusively type 1 diabetes repressed-enriched (ET1DRE) genes are those whose features were significantly enriched among features that were significantly repressed in at least one experiment and significantly induced in zero. Exclusively type 1 diabetes induced-enriched (ET1DIE) genes are those whose features were significantly enriched among features that were significantly induced in at least one experiment and significantly repressed in zero.
Dataset GSE84934 by Long and colleagues was generated from the AbATE (Autoimmunity-blocking Antibody for Tolerance in Recently Diagnosed Type 1 Diabetes) clinical trial. It profiled whole blood gene expression between T1D subjects who responded to treatment with the anti-CD3 antibody teplizumab and control individuals.
We used T1DKP to generate a list of 105 genes that had at least one significant GWAS association with T1D and three other autoimmune conditions: Crohn’s disease, ulcerative colitis and irritable bowel syndrome and designated these pan-autoimmune GWAS genes (PAGGs). PAGGs were ranked by the average rank of the p-value for their association with each condition.
All intersections between (i) ET1DRE genes or (ii) ET1DRE genes and node high confidence transcriptional targets (HCTs) derived from consensomes based upon archived ChIP-Seq experiments.
p < 0.05 intersections between (i) ET1DRE genes or (ii) ET1DRE genes and node high confidence transcriptional targets (HCTs) derived from consensomes based upon archived ChIP-Seq experiments.
p < 0.05 intersections between experiment differentially expressed gene sets (case v control FC > 1.25 induced or repressed, p < 0.05) and node high confidence transcriptional targets (HCTs) derived from archived ChIP-Seq experiments.
Genome-wide single guide RNA (sgRNA) screen that ranks genes according to the essentiality of their roles in supporting primary T cell viability.
Single-nucleus assay for transposase-accessible chromatin with sequencing (snATAC–seq)-based accessible chromatin reference map of 28 peripheral blood and pancreas cell types from donors without type 1 diabetes.
Pooled purified peripheral blood derived CD56dimCD16+ and CD56brightCD16- NK subsets were obtained from 9 healthy donors. Each pooled NK subset sample was hybridized in replicates (A and B).
Full gene list from dataset 1274, experiment 6651, generated by a study of neutrophils in a cohort of subjects with early onset T1D.
Comparison of HCT intersection analysis results for TRPC6M-NEOT1DI and NEOT1DI gene sets.
Enrichment statistics of Reactome functional pathways within TCS-NEOT1DI and NEOT1DI gene sets.
Ratio of aggregate significant MT1D-induced and ST1D-induced HCT intersection analysis ORs and -log10p (ML10P) values for each node across the two cohorts (Table S16).
Genome-wide CRISPR screen by Covarrubias that assigns Mann-Whitney (MW) scores to 21379 mouse genes, such that the gene with the lowest MW score is designated the most essential for macrophage viability.
Analysis demonstrating performance of this analysis against T1D gene lists from other studies
Analysis demonstrating performance of HCTI against other transcription factor enrichment tool.
Data Availability Statement
-
•
This paper analyzes existing, publicly available datasets. The accession numbers for the datasets are listed in the Key Resources table.
-
•
All secondary data generated this study are available from the Signaling Pathways Project and Network Data Exchange resources. RRIDs for these resources are provided in the Key Resources table.
-
•
All code relevant to this study is available in the SPP GitHub account under a Creative Commons CC BY 4.0 license at https://github.com/signaling-pathways-project.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.