

Abstract
Linker histone H1 is an essential regulatory protein for many critical biological processes, such as eukaryotic chromatin packaging and gene expression. Mis-regulation of H1s is commonly observed in tumor cells, where the balance between different H1 subtypes has been shown to alter the cancer phenotype. Consisting of a rigid globular domain and two highly charged terminal domains, H1 can bind to multiple sites on a nucleosomal particle to alter chromatin hierarchical condensation levels. In particular, the disordered H1 amino- and carboxyl-terminal domains (NTD/CTD) are believed to enhance this binding affinity, but their detailed dynamics and functions remain unclear. In this work, we used a coarse-grained computational model, AWSEM-DNA, to simulate the H1.0b-nucleosome complex, namely chromatosome. Our results demonstrate that H1 disordered domains restrict the dynamics and conformation of both globular H1 and linker DNA arms, resulting in a more compact and rigid chromatosome particle. Furthermore, we identified regions of H1 disordered domains that are tightly tethered to DNA near the entry-exit site. Overall, our study elucidates at near-atomic resolution the way the disordered linker histone H1 modulates nucleosome’s structural preferences and conformational dynamics.
Keywords: chromatin, chromatosome, IDP, protein-DNA interaction, MD simulation
Introduction
Eukaryotic chromatin consists of histone proteins and DNA.1 Four types of histone proteins, namely H2A, H2B, H3, and H4, are assembled as an octamer (two H2A-H2B dimers and one H3-H4 tetramer), and wrapped around by ~147 base pairs (bp) of DNA.2 These elementary repeating units, called nucleosomes, are connected by short linker DNA and the linker histone protein H1, forming a “zigzagging ladder” architecture. These nucleosomal arrays are hierarchically folded to form chromatin in the nucleus.3
The fundamental mechanisms of this intricate folding process, still, remain poorly understood. How does chromatin change its condensation levels rapidly and accurately, so that a gene can be precisely transcribed while being well protected from DNA damage?4 The linker histone H1 is believed to play an important role in the fast dynamics of chromatin folding.5–8 As a developmentally regulated protein, H1 has a family of variants that are specific to distinct species or tissues.9 All these H1 variants consist of three parts: a short (20–40 amino acids (AA)) disordered amino-terminal domain (NTD), a highly-conserved globular domain (~80 AA) with rigid structure, and a long (100–125 AA) highly disorganized carboxyl-terminal domain (CTD) with varying lysine and arginine contents, making various binding affinities possible amongst the variants.10–12 The high mobility of H1 when bound to a nucleosome is directly related to many biological processes.13–17 Meanwhile, H1 depletion levels are found to alter global nucleosome spacing and local chromatin compaction.18,19 H1 variants are modified and regulated by various posttranslational modifications (PTMs), which in turn are thought to confer distinct chromatin structures.20 The loss or misregulation of H1 disrupts 3D chromatin structure and leads to many disorders such as lymphoma.21
The first crystal structure of the linker histone globular domain at high-resolution (2.6 Å) discovered its “winged-helix” folding motif over two decades ago.22 However, the high-resolution structural insights of the linker histone-nucleosome complex, referred to as chromatosome, remained elusive until several years ago. Consequently, compared with the four core histone proteins, the structure and dynamics of linker histones are less studied and understood. Interestingly, various H1 subtypes are found to bind on different locations of the nucleosome under different experimental conditions.23 Drosophila H124 and human H1.425 bind to nucleosome near dyad — the center bp defining pseudo-two-fold symmetry axis of the nucleosome — in an asymmetric manner, while chicken linker histone H526,27, Xenopus laevis H1.0b, and human H1.528 bind right on the center of the dyad symmetrically. Zhou et al. proposed these “on-dyad” and “off-dyad” binding modes will lead to distinct chromatin condensation levels.26 A series of atomistic computational studies revealed the conformational selection mechanism of these H1-nucleosome binding modes, as well as the effects of sequence and PTMs.29–32 Further cryoEM studies25,33 and large-scale simulations at ~1 nm spatial resolution34–36 (i.e. mesoscale simulations) of nucleosomal arrays explored how the linker histone’s subtype and concentration determine inter-nucleosome relative positions and resulting distinct chromatin geometries. More experimental studies also discovered other important biological functions of H1 variants besides altering chromatin structure, such as regulating gene expression18 and the overall epigenetic landscape,37 generating epigenetic heterogeneity within tumor cells,38 and directly inhibiting transcriptions.39
Most of the previous studies on linker histones have focused on the globular domain, while the structure and dynamics of the N- and C-termini remain poorly understood, in part because of their intrinsically disordered nature. Recent studies reveal that H1 NTD and CTD are extremely disordered, even when bound with other proteins40 or DNA.41 Their disordered nature, in turn, imparts unique functions of liquid-like glue to promote chromatin folding.42 In particular, the long and highly basic H1 CTD was found to be more essential for the linker histone to bind onto nucleosome with high affinity compared to the NTD.8,43 Similar to the core histone tails, which have been extensively studied by computer simulations,44–46 H1 CTD is also highly disordered and bound to DNA, but much longer than the ~15–40 AA-long core histone tails. A recent cryo-EM experimental study validated the function of CTD in stabilizing the H1-nucleosome complex by primarily binding to one of the linker DNA arms.28 A recent comprehensive study using cryo-EM, NMR, and MD simulation of three human H1 isoforms further confirmed H1 CTD’s stabilizing effect on DNA dynamics and discovered new binding dynamics between H1 and core histone tails.47 FRET experiments48,49 and mesoscale simulations34 demonstrated a variety of H1 CTD conformations depending on the environment of nucleosome arrays and linker DNA length. Furthermore, a series of recent computational studies used both atomistic molecular dynamics simulations and mesoscale Monte Carlo simulations to investigate the H1 disordered domains’ conformation and dynamics in one and multiple chromatosome particles, as well as the regulatory roles of H1 phosphorylation and disorder-toorder transition on the nucleosome asymmetry and chromatin large-scale organization.50,51 Another important recent study, using single-molecule FRET experiments and coarse-grained simulations, revealed that H1 remains disordered when binding with the nucleosome and proposed that the H1-nucleosome dissociation is assisted by another highly disordered chaperone, ProTaα.52
In this work, we studied the structure and dynamics of an H1 variant, Xenopus laevis H1.0b, in complex with a nucleosome particle, via molecular dynamics simulations (we use “H1” below to refer to the Xenopus laevis H1.0b in all the subsequent parts of this study, unless otherwise specified). We used a coarse-grained protein model called AWSEM,53 in combination with the recently developed AWSEM-IDP54 for representing disordered domains, and a coarse-grained DNA force field 3SPN.2,55 to simulate this large protein-DNA complex, the chromatosome. To analyze separate roles of H1 globular and disordered domains in determining chromatosome structure and dynamics, we simulated three combinations of H1-nucleosome systems: (1) nucleosome alone with linker DNA arms; (2) nucleosome with linker DNA arms and the H1 globular domain (GH1); (3) nucleosome with linker DNA arms and the full-length H1 (see Figure 1). Our computational studies showed that the H1 disordered domains restrict conformationally GH1 and linker DNA, shedding new light on H1’s role in chromatosome compaction and chromatin condensation.
Figure 1.
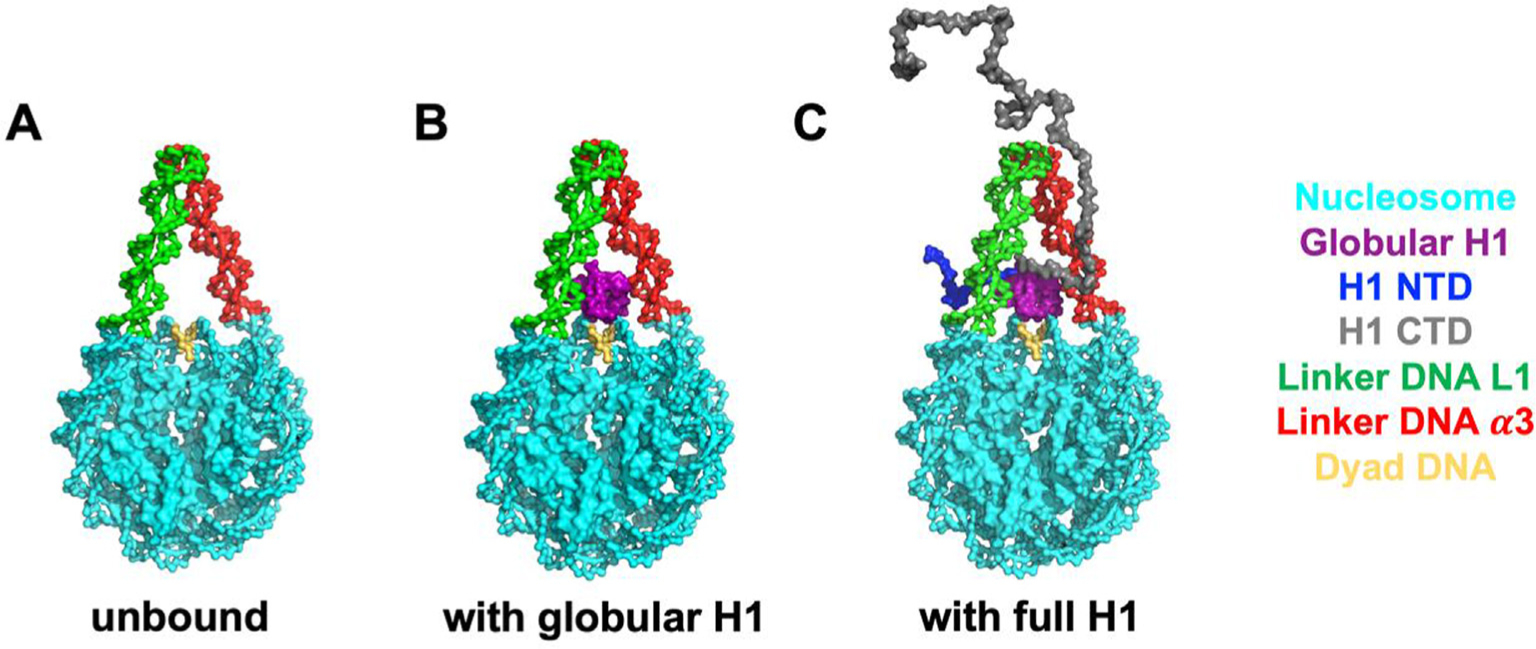
The molecular systems simulated in this study. From left to right: unbound nucleosome without H1 (A), globular H1-nucleosome (B) and full-length H1-nucleosome (C). All the models are based on a recent X-ray crystal structure (PDB: 5NL0) of the Xenopus laevis H1.0b-nucleosome.
Results
H1 disordered domains confine GH1 dynamics
To quantify how the presence of H1 disordered domains affects the dynamics of GH1, we defined a 3D coordinate system to track the position of the chromatosome and the trajectories of GH1’s COM in our simulations (Figure 2). The trajectories in the absence of H1 disordered domains demonstrate that GH1 is very dynamic (Figure 2 (B)). Starting from the initial position on dyad, GH1 not only swings near the dyad but also may drift away from the nucleosome. By contrast, the presence of H1 NTD/CTD significantly restricts GH1’s range of activity (Figure 2(C)). As a part of the highly basic and disordered full-length H1, the globular domain still explores multiple conformations but all adjacent to the nucleosome. We also computed the radius of gyration (Rg) of each individual GH1 COM trajectory to assess their range of motion. The Rg without H1 NTD/CTD (13.6 ± 4.2 Å) is larger than that with H1 NTD/CTD (10.4 ± 3.2 Å). The Kolmogorov–Smirnov test on these two datasets also shows that they arise from distinct distributions (p-value = 0.0016). Altogether, these data strongly suggest that H1 disordered domains inhibit GH1 dynamics.
Figure 2.
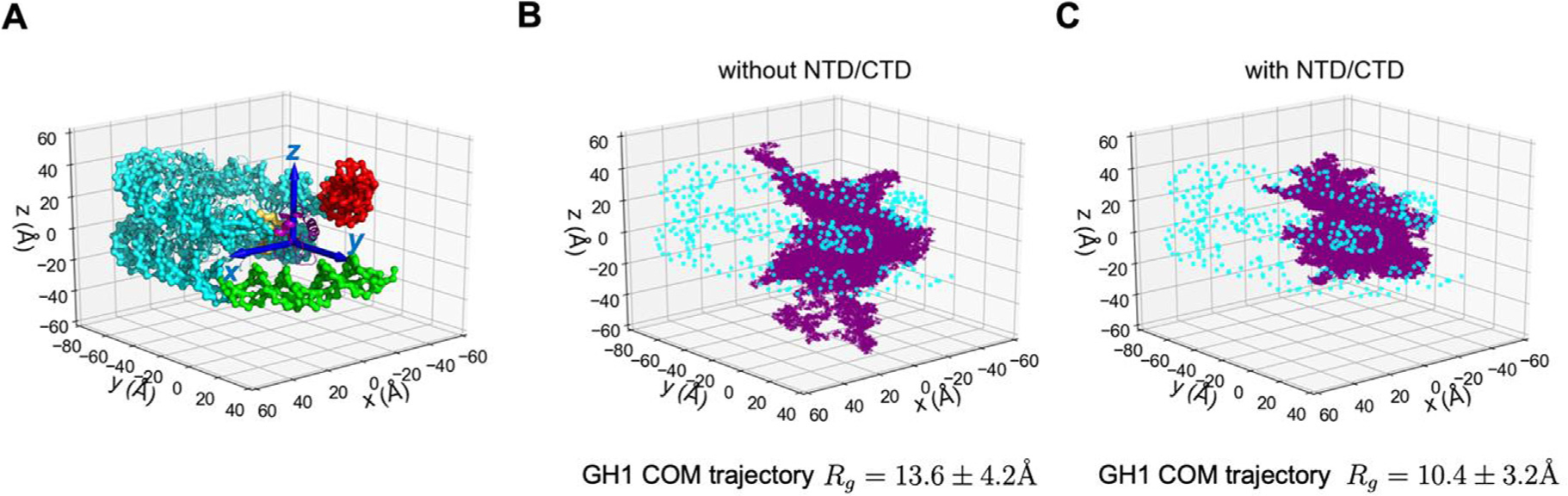
GH1 dynamics is constrained by H1 NTD/CTD. (A) Coordinates of the chromatosome are defined by a 3D cartesian coordinate system (axes shown in blue). Different regions of the chromatosome are represented using the same color scheme as in Figure 1. The trajectories of GH1 COM in the H1-nucleosome system without H1 NTD/CTD (B) and with H1 NTD/CTD (C) are plotted as purple dots. The DNA beads are represented as cyan circles. The core histone octamer and H1 NTD/CTD are not shown in (B-C) for clarity.
The scatter plots of trajectories serve as an overview of GH1 conformations and dynamics, but the preferred GH1-nucleosome binding locations are not clearly identifiable. Therefore, we set up a three-dimensional reference coordinate system (x, y, z), and define the location of GH1 COM relative to the nucleosome core particle’s COM in spherical coordinates (r, θ, ϕ) (see Figure 3(A) for detailed definition). Then we computed 2D histograms of all three spherical coordinates to identify the most probable GH1 binding modes. Here we found the effects brought by H1 disordered domains are best represented by the (ϕ, r) histogram (Figure 3(B) and (C), the other two histograms of (θ, ϕ) and (ϕ, r) are shown in Figure S5). For GH1 without the disordered domains, the (ϕ, r) distribution, especially ϕ values, is very dispersed (Figure 3(B)). This means GH1 can almost freely rotate around the vertical y-axis across the dyad, whereas in the full-length H1-nucleosome simulations, we found the 2D distribution is more concentrated (Figure 3(C)). Almost all the GH1 conformations with H1 disordered domains have positive ϕ values, indicating that GH1 is only allowed to be located on one side of the nucleosomal disk. This analysis further demonstrates that the disordered domains reduce the variability of GH1’s binding modes.
Figure 3.
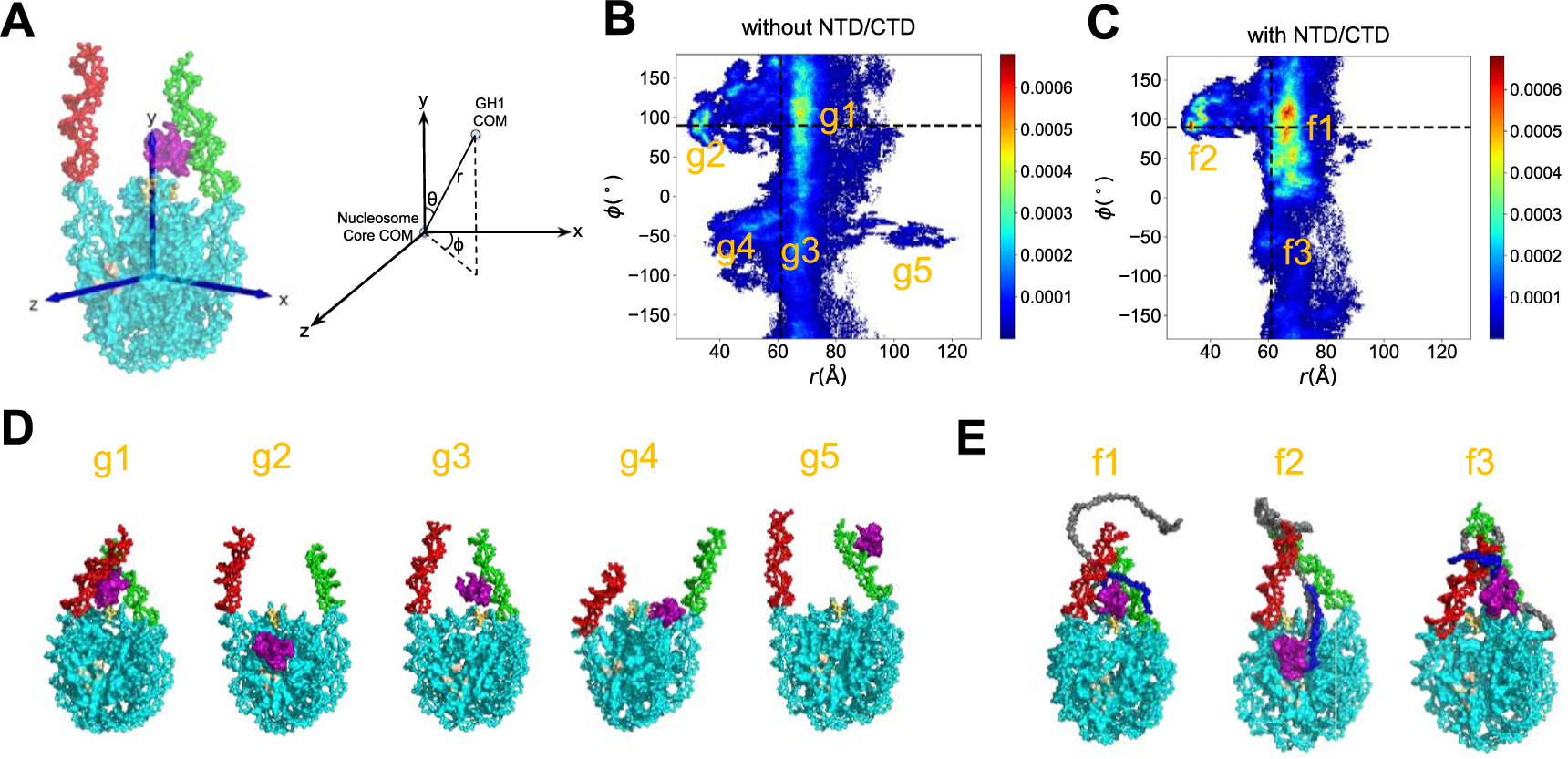
GH1-nucleosome relative conformations. (A) Definition of the 3D reference coordinate system and GH1 COM spherical coordinates (r, θ, ϕ). The 2D histogram of (ϕ, r) for GH1-nucleosome (B) and full-length H1-nucleosome (C) are shown as heat maps. The color bar from blue to red indicates a probability from low to high. The ϕ and r values at the beginning of simulations are labeled as black dashed lines. The representative snapshots of all the major basins labeled in (B-C) are shown in (D) and (E). The color code for different parts of molecules is the same as Figure 1, except for the region in light brown for the histone acidic patch.
H1 disordered domains restrict GH1-nucleosome interaction sites
Besides describing GH1 dynamics, the 2D histograms in Figure 3 also identify the preferred GH1-nucleosome binding modes. Here we found five major basins (g1, g2, g3, g4, g5) for GH1-nucleosome and three (f1, f2, f3) for full-length H1-nucleosome. The representative snapshots of each basin are shown in Figure 3(D) and (E) (see Table S2 and Supporting Information for definitions and population percentages of each basin, and the detailed procedure to select representative snapshots). In two major conformations g1 and g3 of H1-nucleosome without disordered domains, GH1 is still located near the dyad DNA minor groove (labeled as yellow). In basins g2 and g4, however, GH1 tends to slip off the dyad region and move towards the histone octamer (see Supplemental Movie 1 to demonstrate this behavior). With a small probability (~0.9%), GH1 even moves to the tip of one linker DNA arm to escape away from the dyad in basin g5. Meanwhile, four out of these five major basins (25.4%) have very different ϕ and r values from the on-dyad initial binding mode (black dashed lines). For full-length H1-nucleosome, two out of three basins (f1 and f3) are located more proximal to the initial binding mode. This contrast clearly shows that the presence of H1 NTD/CTD shifts the preferable GH1 binding positions closer to the dyad. Similar to g2, there is also a basin f2 representing a faraway-from-dyad binding conformation even with NTD/CTD. One possible reason might be the competitive electrostatic attraction from the acidic patch region on histone core (shown as brown in Figure 3(D) and (E), also see Figure S6 for snapshots with a zoomed-in view). We would like to note here that despite these non-native conformations being of great kinetic interest, the on-dyad binding mode is the most preferable thermodynamic state for both systems, consistent with previous hydroxyl radical experiments.56 Overall, these analyses indicate that H1 disordered domains tend to stabilize the GH1’s position near the DNA entry-exit site.
Previous experiments have found that various types of linker histones recognize nucleosomal DNA via different parts of their “winged-helix” folding motif.26,28 To further probe the DNA-binding preferences of these secondary structure elements, we divided GH1 and DNA into several regions, and computed the contact probability of two arbitrary beads belonging to a certain pair of regions (Figure 4). For the GH1-nucleosome without NTD/CTD, the main regions of H1 in contact with dyad DNA are L1/β1 and β2 (Figure 4(B), 2nd row). By contrast, when NTD/CTD are present, α2 and α3 helical regions form stronger contacts to the dyad DNA (Figure 4(C), 2nd row). These two helices also have more residues proximal to the dyad DNA in the X-ray crystal structure28 (labeled with purple stars), suggesting that our results with NTD/CTD have a similar near-dyad binding pattern compared to the experimental results. On the other hand, GH1’s binding probabilities with the α3- and L1- linker DNA (Figure 4(B) and (C), 1st, 3rd row) are mostly low with or without the disordered domains. But when NTD/CTD are present (Figure 4 (C), 1st row), the α3 helix forms significantly more contacts with the α3 DNA, consistent with the crystal structure as well. Our contact map analyses provide a detailed description of the GH1-DNA interface in the presence of H1 NTD/CTD: GH1 recognizes nucleosome mainly through dyad DNA by α2/α3 helix. But instead of being located in the exact dyad center, GH1 is tilted towards α3 DNA. The absence of H1 disordered domains, on the contrary, changes this stable interaction network and makes GH1 more flexible.
Figure 4.
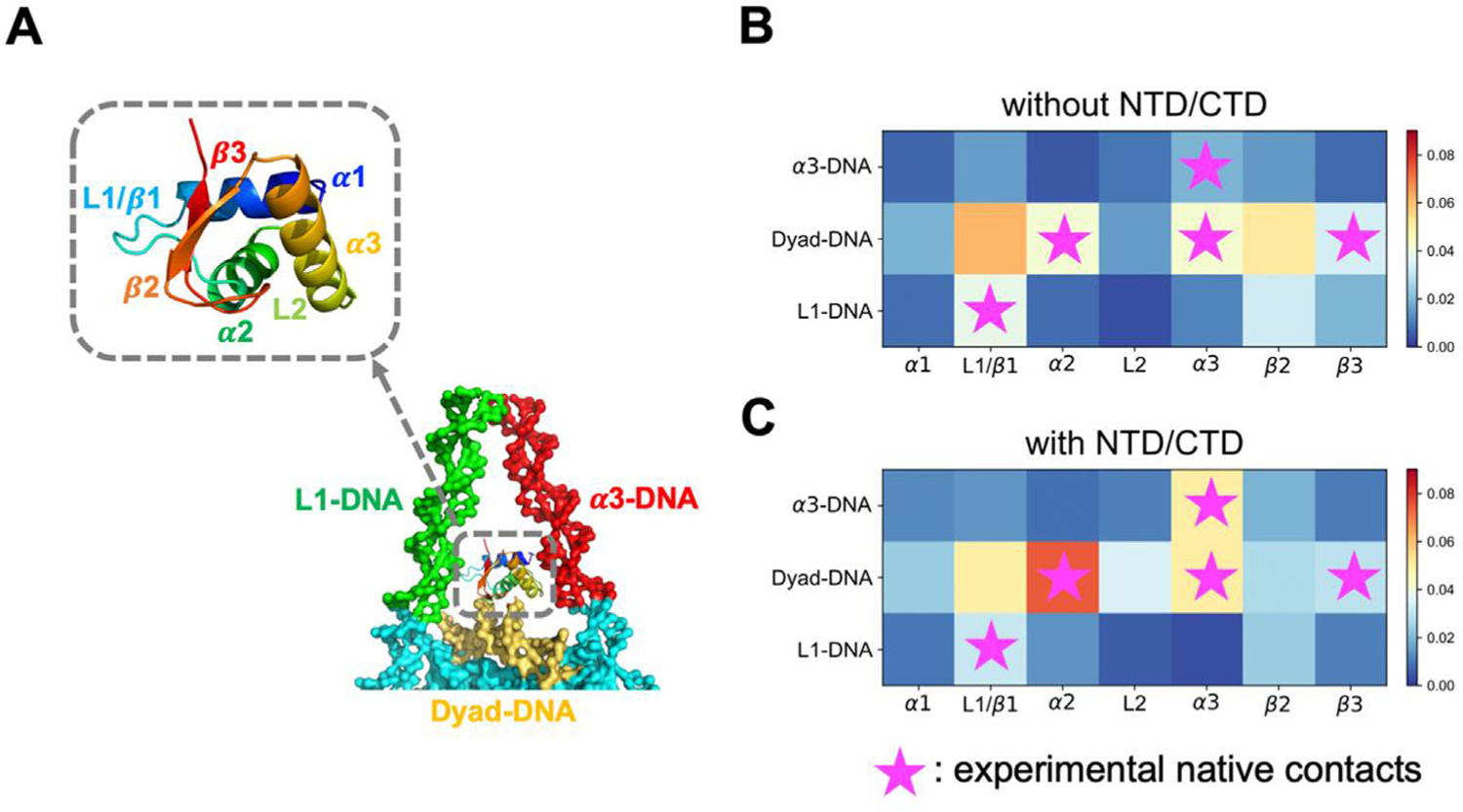
H1 disordered domains regulate and stabilize GH1-DNA binding interface. GH1 and nearby DNA structures are represented in (A), where colors represent different protein or DNA regions. (B-C) GH1-DNA contact maps without and with H1 NTD/CTD. Horizontal and vertical axes represent GH1 and DNA regions respectively. Color code from blue to red indicates the contact probability of a protein-DNA beads pair in this region from low to high. Purple stars represent the experimental “native contact regions”, where more than two residues in this GH1 region are close to a DNA region.
H1 globular and disordered domains converge linker DNA
The geometry of linker DNA arms is another important feature to evaluate chromatosome structural compaction. Previous cryo-EM experiments28 show that H1 induces a more compact and rigid nucleosome conformation by keeping the linker DNA arms convergent or “closed”. Here we computed angles α and β of both linker DNA arms to quantify the linker DNA’s geometry parallel with and perpendicular to the nucleosomal disk (shown in Figure 5, also see Figure S7 for the same analyses on each individual linker DNA arm, which show that α3-DNA and L1-DNA arms sample roughly similar α and β angles, especially in the presence of the full-length H1). We found the α angle distribution of full-length H1-nucleosome (29.3° ± 14.8°) shifts toward higher values compared to the unbound results (15.6° ± 17.7°) (Figure 5(A)), while β angle (0.9° ± 16.7°) moves to lower values than the unbound (9.6° ± 16.3°) (Figure 5(B)). Both trends show that full-length H1 keeps the linker DNA collapsed in our simulations, consistent with previous experimental measurements.28 In particular, we found that both α (20.0° ± 16.7°) and β (5.2° ± 16.3°) distributions in GH1-nucleosome simulations are in between unbound and full-length H1 results. This demonstrates that while globular H1 by itself can bring the linker DNA together, the disordered domains will significantly reinforce this converging effect.
Figure 5.
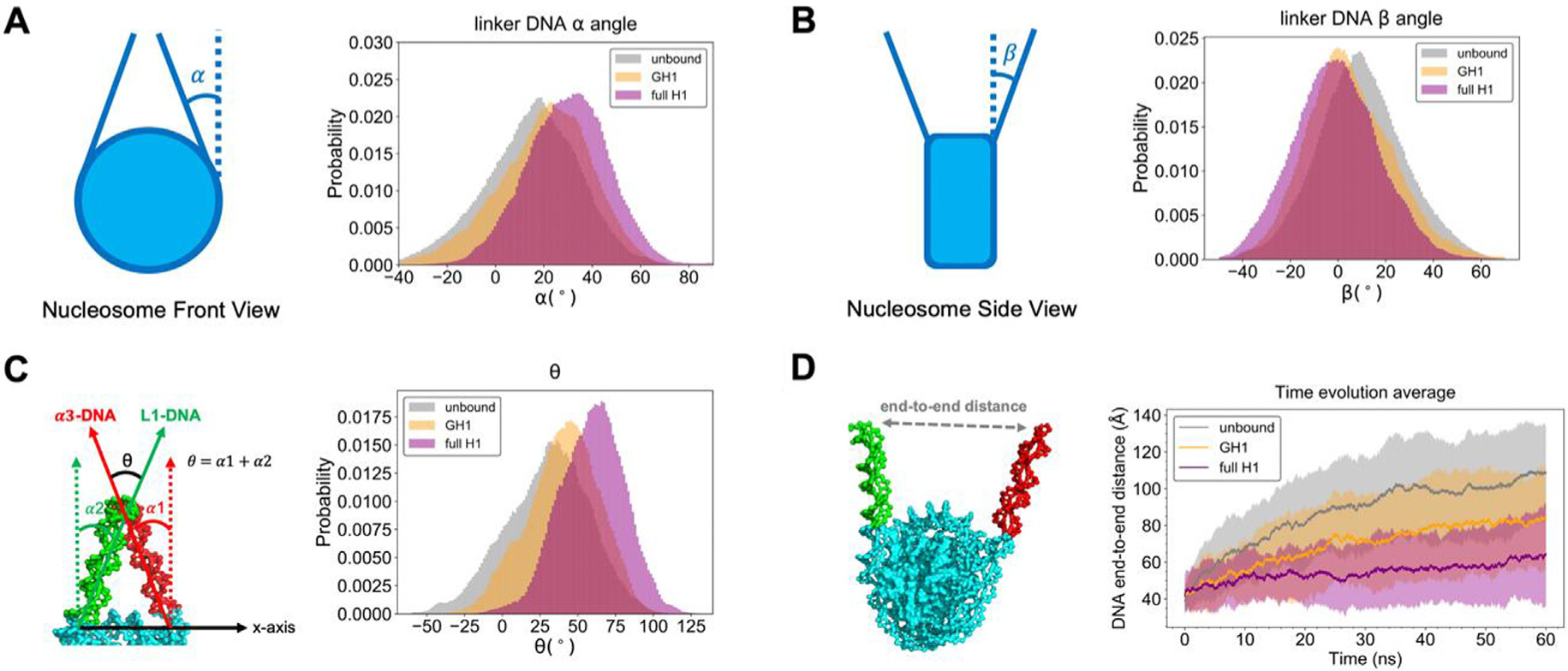
Linker DNA arms are more convergent with full-length H1 than globular H1 or unbound. The definition and histogram of α, β, and θ angles are shown in (A, B, C). (D) The time evolution of DNA end-to-end distance, where solid lines with shaded regions represent average values with standard deviations. The same set of legends is used in all the subset figures above (gray: unbound nucleosome; yellow: GH1-nucleosome; purple: full-length H1-nucleosome).
Analyses of individual linker DNA arms allow us to measure their relative conformations and distances to describe the compaction of chromatosome with further accuracy. Here we define an angle θ = α1 + α2 between the two linker DNA arms and plot its distribution (Figure 5(C)). Its histogram demonstrates that the θ of GH1-nucleosome (40.1° ± 23.5°) only increases by a small amount compared with the unbound nucleosome result (30.2° ± 27.0°). While the binding of full-length H1 increases θ significantly (58.6° ± 21.4°). This comparison reveals the disordered domains are more crucial than the GH1 in compacting chromatosome’s structure.
We also calculated the end-to-end distance between the tips of linker DNA, and plotted its average and standard deviation over all runs along the simulation time (Figure 5(D)). We found in absence of H1, the linker DNA arms start to separate from each other after 20 ns. The end-to-end distance reaches ~100 Å at the end of simulations. Globular H1 slows down the linker DNA’s separation but cannot fully prevent it. The end-to-end distance still rises to ~80 Å by the end of simulations. On the other hand, the binding of full-length H1 remarkably inhibits this separation, as found in previous experiments.28 The end-to-end distance does not show any apparent increasing trend beyond fluctuations. These results demonstrate that globular H1 alone is not sufficient to fully compact the chromatosome as found in previous experiments, highlighting the crucial role of disordered domains.
As a more comprehensive comparison between our simulation results and cryo-EM experiments, we also ran simulations of the H1.5ΔC50 system, another chromatosome with H1.5 linker histone, where the last 50 residues on CTD were removed, following Bednar et al.28 (see Figure S8 for its sequence and structure, the simulation details are elaborated in Supporting Information). The statistics of all the DNA metrics used above (α, β, and θ angles and end-to-end distances) of H1.5ΔC50 are very similar to the full-length H1.0 results (Figure S9). This similarity of DNA dynamics reveals that H1.5ΔC50, even though belonging to a different H1 subtype and having a shorter CTD, also leads to relatively compact linker DNA geometry and dynamics. These outcomes further indicate that the function of H1 disordered domains is to restrict linker DNA. We also measured GH1-nucleosome relative conformations of H1.5ΔC50 (Figure S10), using the same spherical coordinates as in Figure 3. We found that H1.5’s NTD and CTD, similar to H1.0’s disordered domains, also restrict the GH1 to bind preferably near the nucleosomal dyad. This observation is consistent with the experimental findings in Bednar et al.28 and implies that the compaction of the chromatosome is primarily driven by H1′s disordered tails rather than its globular domain.
H1 NTD/CTD are tethered to both linker DNA arms
We next computed a regional contact map for H1 NTD/CTD and DNA (Figure 6), using the same definitions as in Figure 4 and Table S1. For H1 CTD, the GH1-proximal C1, C2, and C3 regions have higher contact probabilities (Figure 6(B), 3rd-5th column), while the GH1-distal regions form very minimal contacts with DNA (Figure 6(B), 6th-10th column). A similar trend is found for NTD, where the GH1-proximal N2 region is much more tightly bound than the distal N1 region (Figure 6 (B), 1st-2nd column). Both CTD and NTD have closely bound regions with the dyad DNA on N2, C1, and C2 (Figure 6(B), 2nd-4th column, 2nd row). On the other hand, CTD prefers binding with L1-DNA via its C1 and C3 regions (Figure 6(B), 3rd and 5th column, 1st row), while NTD is particularly bound with α3-DNA via the N2 region (Figure 6 (B), 2nd column, 3rd row). This contact map elucidates how the full-length H1 is steadily bound with DNA near the entry-exit site: the two disordered domains, especially the GH1-proximal parts, act as “hands” of GH1 to grasp the dyad and linker DNA. Such an entanglement between H1 disordered domains and DNA precludes GH1 from escaping far away from the dyad region and confines linker DNA dynamics.
Figure 6.
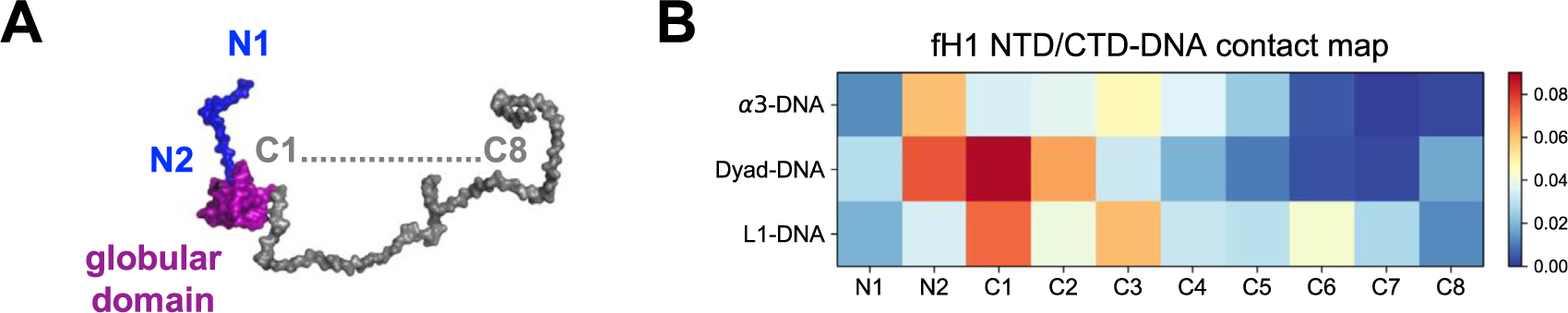
H1 NTD and CTD are tightly bound with DNA mainly via the residues proximal to the globular domain. (A) H1 NTD (blue) is divided into two regions (N1-N2, from GH1-distal to GH1-proximal). H1 CTD (orange) is divided into eight regions (C1-C8, from GH1-proximal to GH1-distal). (B) shows the contact map of DNA with NTD and CTD. The horizontal axis represents NTD (N1-N2) and CTD (C1-C8) regions. The vertical axis and color bar are the same as in Figure 4(B).
We also observed that the contact probability of the GH1-distal region of the CTD (especially C4-C8) is much smaller, indicating that it is only transiently bound to DNA. Interestingly, the entire NTD and CTD remain highly disordered in the simulation (see Figure S10). This observation is consistent with a previous NMR study, which revealed that H1 CTD remains disordered in the DNA-bound state41 and may explain the absence of H1 tails in almost all chromatosome X-ray crystal structures. Interestingly, a recent atomistic-MD study51 pointed out that H1 NTD can undergo disorder-to-order transition upon binding with the nucleosome. This phenomenon, however, was not observed either in our study, or in some recent experimental and computational works.47,52 Therefore, further exploration of these discrepancies may be warranted.
Discussion
In this work, we investigated binding dynamics of the H1-nucleosome complex using extensive computer simulations using a state-of-art protein-DNA model. Our focus was on the regulatory effect of H1 disordered domains on the chromatosome structure. By quantitative comparisons among different molecular systems, we found that H1 disordered domains, especially GH1-proximal regions, compact chromatosome structure by constraining the conformation and dynamics of linker DNA arms and GH1. By contrast, without the disordered domains, the binding affinity between the H1 globular domain and DNA is notably reduced. In this case, linker DNA arms become more separated, and GH1 may even escape away from the nucleosome, resulting in a more “open” chromatosome conformation.
Previous studies suggested that H1 can bind with a nucleosome on- or off-dyad to regulate distinct chromatin higher-order structure.26 A recent review23 pointed out that H1-nucleosome binding modes depend on H1 species and experimental conditions, and the resulting chromatosome structures should be viewed as an ensemble. Interestingly, our simulations not only replicate H1-nucleosome on/off-dyad binding modes found in previous experiments and all-atom simulations, but also discover new H1 binding modes far from the dyad. One commonly observed new binding mode in our simulation corresponds to GH1 completely escaping from the dyad and reaching close to the acidic patch on histone core. Our findings suggest that the disordered domains play a balancing role to prevent the chromatosome from being too rigid or too flexible. The resulting structural plasticity of the chromatosome enables H1’s rapid interconversion among different modes, including binding/unbinding to the nucleosome, which, in turn, may allow timely remodeling and adjustment of chromatin compaction levels.
In the “zigzagging ladder” model of chromatin folding, H1 acts as the “rung” between nucleosome particles. Based on our findings, we propose that H1 NTD/CTD may affect the location and strength of the connection between H1 and neighboring nucleosomes, thus regulating nucleosome array organization and chromatin higher-order structure. Compared with the globular domain, H1 disordered domains are more prone to various PTMs.20 These PTMs would enable many important biological functions, including not only chromatin folding,57 but also apoptosis58 and DNA transcription.59 In particular, the phosphorylations on H1 CTD have been proved to change the secondary structure of CTD and regulate chromatin condensation level.60,61 We expect that the phosphorylation will drive the dynamics of H1 to a moderate level between the two systems in this study (globular H1 and full-length H1) because its binding affinity to the linker DNA would be stronger than the former, but weaker than the latter.
We also observed that GH1 can even escape from the dyad region and move towards the acidic patch (Figure 3). The reason this behavior is both novel and potentially biologically meaningful, is that it might explain the mechanistic basis for decades-old observations that in vitro, H1 can reposition nucleosomes without the use of ATP or remodelers.62,63 The spacing function of H1s remains mysterious, but our observation of H1 breaking symmetry at the center of the nucleosome might subtly lower the energetic barrier at the pseudo-dyad, allowing nucleosomal looping in advance of sliding and repositioning along the DNA fiber. Our contact analyses in Figure 4 reveal that α2 and α3 helices are the major GH1-dyad binding regions, no matter whether H1 disordered domains are present or not. Thus, it will be promising for further experiments to mutate some key residues in these regions, such as S49, K53, K69, and K74, to test how they affect this H1 escaping-dyad motion.
Shortly after the observation of nucleosome ladders by Hewish and Burgoyne,64 coupled with classic EM experiments visualizing regularly spaced beads on a string by Woodcock65 and the Olinses,66 early workers in the chromatin field67 discovered the still inexplicable phenomenon of species-specific nucleosome repeat lengths. There NRLs were thought to derive from species-specific H1 variants. Our work here provides a testable theoretical framework to explore how species-specific H1 residue changes over evolutionary time,68 in the NTD, the CTD and the GD might subtly alter the motions of H1 we observed in this study, thereby potentially contributing to the global spacing of nucleosomes. We note with excitement that the advent of high-speed AFM69–73 provides precisely the kinetic handle needed to complement H1 fast dynamics observed in FRAP studies5,74 and H1 off/on dyad classic steady-state biochemistry experiments75,76 glean insights into linker histone biology as it relates to chromatin spacing and folding. A comprehensive review of powerful experimental tools for probing linker histones in a chromatin environment can be found in Melters et al.,77 where the authors successfully visualized the dynamics of nucleosome arrays with H1.5 in real time by high-speed AFM.
One limitation of our study is the lack of individual H1 NTD/CTD’s functions. Previous FRAP experiments found deletion of the longer and less conserved CTD reduces H1-nucleosome binding affinity to a much greater extent than removal of NTD.43 On the other hand, NTD has been found to contribute more to nucleosome specificity rather than to DNA/chromatin condensation.56,78 Hence, it would be interesting to computationally model an H1-nucleosome system with only NTD or CTD and investigate their independent effects in regulating chromatosome structure and dynamics. Meanwhile, for different variants, H1 NTD and CTD have much less conserved sequences and distinct lengths. Our study already shows that H1.0 and H1.5ΔC50 have similar structural properties, but it would be more interesting to conduct a systematic investigation on how the sequence and length of H1 disordered domains regulate chromatosome structure and dynamics.
Methods
Hybrid coarse-grained model for the H1-nucleosome complex
To investigate binding dynamics of the H1-nucleosome more efficiently and accurately, we modeled this large molecular complex (~950 protein AA + 193 DNA bp) with a coarse-grained protein force field AWSEM53 and a DNA force field 3SPN.2,55 which is a different computational approach from previous landmark atomistic MD studies of nucleosome systems.79,80 Inspired by the funneled energy landscape theory,81 AWSEM uses three beads (Cα, Cβ, and O) to represent one residue and adopts both physical and bioinformatic potentials to account for amino acid interactions. Compared with a more coarse force field used recently for H1-nucleosome simulations52 which represents a residue with only one bead, AWSEM simulations operate at a near-atomic resolution.53,82 AWSEM has been applied to study many different types of proteins,54,83–85 including histones82,86 successfully. Similarly, 3SPN.2 uses three sites phosphate, deoxyribose sugar, and nitrogenous base to represent a nucleotide and was carefully calibrated by structural and thermodynamic properties of single- and double-strand DNA. With comparable length scale and implicit solvent assumption, these two models have been combined to study several protein-DNA systems,87–90 including the nucleosome.91
Here, we elaborate on the detailed Hamiltonian of this protein-DNA force field. For protein–protein interactions, we used a strategy similar to Zhang et al.,91 which is mostly consistent with the original AWSEM but also introduces two modifications: an explicit Debye-Hückel (DH) electrostatic interactions between charged Cβ atoms (+1 for arginine and lysine, −1 for aspartic acid and glutamic acid), and a weak Gō potential with fine-tuned parameters for the entire histone octamer to bias it towards the crystal structure. Additionally, we applied the newly developed AWSEM-IDP force field54 to model the disordered H1 CTD and NTD. We reduced the helical potential weights only for these disordered domains to avoid artificial propensity to form helices. We also performed extensive atomistic simulations for the structural segments of these disordered domains and used the resulting trajectories to bias local tail segments in AWSEM simulations, similar to Lin et al.92 (see Supporting Information and Figure S1–S3 for details of the atomistic simulations, sanity checks, and related structure bias setup). DNA-DNA interactions followed the 3SPN.2 model.
For protein-DNA interactions, we first tested the ansatz from Potoyan et al.87 and Zhang et al.,91 which consists of a non-residue-specific Lennard-Jones (LJ) potential and Debye-Hückel electrostatic interactions. We found these two forces are still too weak to keep a stable nucleosome structure within our standard simulation time, where DNA tends to unwrap from the histone octamer (Figure S4). In the nucleosome crystal structure, 14 arginine side chains from histone octamer insert deeply into the DNA minor grooves to further stabilize the nucleosome.2 This important effect was not included in the previous AWSEM treatments of the nucleosome. To mimic this special interaction, we added additional site-specific Lennard-Jones forces between these pairs of arginine Cβ and DNA phosphate beads. With this new potential, nucleosomal DNA stays wrapped around the histone core during an entire simulation (Figure S4). Detailed formulas and parameters of this nucleosome-specific potential are elaborated in the Supporting Information and Figure S4.
In summary, the formulae of the applied AWSEM-DNA force field are listed below, where V stands for the potential energy of each term:
Simulation details
All the coarse-grained simulations were performed with the open-source simulation package LAMMPS93 (version 31Mar17), in which our modification of AWSEM-DNA was implemented.53,55,87,91 The initial conformation of the H1-nucleosome was taken from a recent X-ray crystal structure28 (Xenopus laevis H1.0b, PDB: 5NL0). Three different molecular combinations were constructed to test the function of H1, including the nucleosome with linker DNA and (1) without H1 (unbound); (2) with the H1 globular domain; (3) with the full-length H1 (Figure 1). Note that the disordered H1 NTD and CTD are missing in the crystal structure. Hence we used MODELLER.94 (version 9.19) to generate their initial conformations. Tails of core histones were not included in this study, because most of them are located far away from H1 or the nucleosome dyad — the main region of interest in this study, and unlikely to interact with H1 or linker DNA. Moreover, including these disordered tails would have slowed down the convergence of our simulations.
We set the simulation time step to 5 fs and used non-periodic shrink-wrapped boundary conditions. The Langevin thermostat was applied, with the damping parameter = 500 fs. We used a parameter set to mimic electrostatic interactions in 150 mM NaCl solution, which is close to a physiological cellular environment (see Supporting Information for detailed parameters). With five different random initial velocity distributions, we heated up and annealed the system shortly (500 ps heating from 300 K to 330 K, 500 ps annealing from 330 K to 300 K) to generate five different initial configurations. Then for each initial configuration, we ran 10 independent simulations for 60 ns at 300 K and constant volume, each with different initial velocity distributions. In effect, for each molecular system we obtained simulation trajectories summing up to 3 μs. This coarse-grained timescale approximately corresponds to milliseconds of simulation time when explicit solvent atomistic molecular dynamics are carried out for the same systems.95–97
Analysis
Linker DNA geometry.
We used several different metrics to quantify the conformation and dynamics of linker DNA arms. We computed α and β angles between the linker DNA arms and the vertical dyad axis to quantify compaction of the chromatosome particle (see Figure 5(A) and (B) for graphical illustrations), following similar definitions from Bednar et al.28 and Woods et al.98 Vectors representing linker DNA are defined as the center of mass (COM) of the beginning bp pointing to COM of the terminal bp. The vector representing the vertical dyad axis is defined as COM of bp at the center of nucleosome bottom pointing to the dyad. The “front” and “side” plane for angle α and β are defined as parallel with and perpendicular to the nucleosome core disk. Positive α indicates linker DNA arms bend “inward” while positive β means bending “outward”. Similarly, we also defined and calculated angle θ and end-to-end distance between the linker DNA to quantify their relative geometry (see Figure 5(C) and (D) for graphical illustrations). We used VMD to conduct all the analyzes mentioned above.
Protein-DNA regional contact map.
We computed regional contact maps between H1 and DNA near the entry-exit site to describe their binding sites. We first divided H1 and DNA into small regions based on their secondary structure elements or location (see Table S1 for detailed region definitions). A pair of protein-DNA beads in coarse-grained representation will be determined as “contact” if their inter-bead distance is smaller than a predetermined threshold (=8 Å, around 1.5 times of protein + DNA site radius for excluded volume effects). Based on this definition, we computed the contact probability by counting contact number within a certain H1/DNA region and normalizing it with the total number of protein-DNA bead pairs:
Supplementary Material
Acknowledgements
Garegin Papoian’s efforts were supported by the Amazon AWS Machine Learning Research Award. Yamini Dalal’s work is supported by NIH. Computational resources are provided by the Deepthought2 HPC cluster at the University of Maryland.
Abbreviations:
- NTD/CTD
N/C terminal domain
- PTMs
post-translational modifications
- GH1
H1 globular domain
- COM
center of mass
- AWSEM
associated memory, water-mediated, structure and energy model
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Hao Wu: Conceptualization, Methodology, Investigation, Visualization, Writing - original draft, Writing - review & editing. Yamini Dalal: Supervision, Conceptualization, Writing - original draft, Writing - review & editing. Garegin A. Papoian: Supervision, Conceptualization, Writing - original draft, Writing - review & editing.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2021.166881.
References
- 1.Kornberg RD, (1974). Chromatin structure: A repeating unit of histones and DNA. Science (80-.), 184, 868–871. 10.1126/science.184.4139.868. [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ, (1997). Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature, 389, 251–260. 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 3.Woodcock CL, Ghosh RP, (2010). Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol, 2, 10.1101/cshperspect.a000596a000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takata H, Hanafusa T, Mori T, Shimura M, Iida Y, Ishikawa K, Yoshikawa K, Yoshikawa Y, Maeshima K, (2013). Chromatin compaction protects genomic DNA from radiation damage. PLoS One, 8, 1–11. 10.1371/journal.pone.0075622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT, (2000). Dynamic binding of histone H1 to chromatin in living cells. Nature, 408, 877–881. 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 6.Catez F, Brown DT, Misteli T, Bustin M, (2002). Competition between histone H1 and HMGN proteins for chromatin binding sites. EMBO Rep, 3, 760–766. 10.1093/embo-reports/kvf156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M, (2004). Network of dynamic interactions between histone H1 and high-mobility-group proteins in chromatin. Mol. Cell. Biol, 24, 4321–4328. 10.1128/mcb.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown DT, Izard T, Misteli T, (2006). Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol, 13, 250–255. 10.1038/nsmb1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Izzo A, Kamieniarz K, Schneider R, (2008). The histone H1 family: Specific members, specific functions?. Biol. Chem, 389, 333–343. 10.1515/BC.2008.037. [DOI] [PubMed] [Google Scholar]
- 10.Allan J, Hartman PG, Crane-robinson C, Aviles FX, (1980). The structure of histone H1 and its location in chromatin. Nature, 288, 675–679. 10.1038/288675a0. [DOI] [PubMed] [Google Scholar]
- 11.Hergeth SP, Schneider R, (2015). The H1 linker histones: Multifunctional proteins beyond the nucleosomal core particle. EMBO Rep, 16, 1439–1453 10.15252/embr.201540749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyubitelev AV, Nikitin DV, Shaytan AK, Studitsky VM, Kirpichnikov MP, (2016). Structure and functions of linker histones. Biochem, 81, 213–223. 10.1134/S0006297916030032. [DOI] [PubMed] [Google Scholar]
- 13.Bustin M, Catez F, Lim JH, (2005). The dynamics of histone H1 function in chromatin. Mol. Cell, 17, 617–620. 10.1016/j.molcel.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Catez F, Ueda T, Bustin M, (2006). Determinants of histone H1 mobility and chromatin binding in living cells. Nat. Struct. Mol. Biol, 13, 305–310. 10.1038/nsmb1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bednar J, Hamiche A, Dimitrov S, (2016). H1-nucleosome interactions and their functional implications. Biochim. Biophys. Acta - Gene Regul. Mech, 1859 (3), 436–443. 10.1016/j.bbagrm.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 16.Flanagan TW, Brown DT, (2016). Molecular dynamics of histone H1. Biochim. Biophys. Acta - Gene Regul. Mech, 1859 (3), 468–475. 10.1016/j.bbagrm.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Roque A, Ponte I, Suau P, (2016). Interplay between histone H1 structure and function. Biochim. Biophys. Acta - Gene Regul. Mech, 1859 (3), 444–454. 10.1016/j.bbagrm.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, Stein A, Woodcock CL, Skoultchi AI, (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell, 123, 1199–1212. 10.1016/j.cell.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 19.Routh A, Sandin S, Rhodes D, (2008). Nucleosome repeat length and linker histone stoichiometry determine chromatin fiber structure. Proc. Natl. Acad. Sci. U. S. A, 105, 8872–8877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fyodorov DV, Zhou BR, Skoultchi AI, Bai Y, (2018). Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol, 19, 192–206. 10.1038/nrm.2017.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yusufova N, Kloetgen A, Teater M, Osunsade A, Camarillo JM, Chin CR, Doane AS, Venters BJ, Portillo-Ledesma S, Conway J, Phillip JM, Elemento O, Scott DW, Béguelin W, Licht JD, Kelleher NL, Staudt LM, Skoultchi AI, Keogh MC, Apostolou E, Mason CE, Imielinski M, Schlick T, David Y, Tsirigos A, Allis CD, Soshnev AA, Cesarman E, Melnick AM, (2020). Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature, 10, 26. 10.1038/s41586-020-3017-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramakrishnan V, Finch JT, Graziano V, Lee PL, Sweet RM, (1993). Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature, 362, 219–223. 10.1038/362219a0. [DOI] [PubMed] [Google Scholar]
- 23.Öztürk MA, Cojocaru V, Wade RC, (2018). Toward an ensemble view of chromatosome structure: A paradigm shift from one to many. Structure, 26, 1050–1057. 10.1016/j.str.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Zhou B-R, Feng H, Kato H, Dai L, Yang Y, Zhou Y, Bai Y, (2013). Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. U. S. A, 110, 19390–19395. 10.1073/pnas.1314905110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, Li G, (2014). Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science (80-.), 344, 376–380. 10.1126/science.1251413. [DOI] [PubMed] [Google Scholar]
- 26.Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, Bai Y, (2015). Structural mechanisms of nucleosome recognition by linker histones. Mol. Cell, 59, 628–638. 10.1016/j.molcel.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou BR, Feng H, Ghirlando R, Li S, Schwieters CD, Bai Y, (2016). A small number of residues can determine if linker histones are bound on or off dyad in the chromatosome. J. Mol. Biol, 428, 3948–3959. 10.1016/j.jmb.2016.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, Menoni H, Papin C, Skoufias DA, Kurumizaka H, Lavery R, Hamiche A, Hayes JJ, Schultz, Angelov D, Petosa C, Dimitrov S, (2017). Structure and dynamics of a 197 bp nucleosome in complex with linker histone H1. Mol. Cell, 66, 384–397. e8. 10.1016/j.molcel.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pachov GV, Gabdoulline RR, Wade RC, (2011). On the structure and dynamics of the complex of the nucleosome and the linker histone. Nucleic Acids Res, 39, 5255–5263. 10.1093/nar/gkr101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Öztürk MA, Pachov GV, Wade RC, Cojocaru V, (2016). Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome. Nucleic Acids Res, 44, 6599–6613. 10.1093/nar/gkw514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Öztürk MA, Cojocaru V, Wade RC, (2018). Dependence of chromatosome structure on linker histone sequence and posttranslational modification. Biophys. J, 114, 2363–2375. 10.1016/j.bpj.2018.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Öztürk MA, De M, Cojocaru V, Wade RC, (2020). Chromatosome structure and dynamics from molecular simulations. Annu. Rev. Phys. Chem, 71, 101–119. 10.1146/annurev-physchem-071119-040043. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, et al. , (2018). Structure of an H1-Bound 6-nucleosome array reveals an untwisted two-start chromatin fiber conformation. Mol. Cell, 72, 902–915.e7. 10.1016/j.molcel.2018.09.027. [DOI] [PubMed] [Google Scholar]
- 34.Perišić O, Schlick T, (2017). Dependence of the linker histone and chromatin condensation on the nucleosome environment. J. Phys. Chem. B, 121, 7823–7832. 10.1021/acs.jpcb.7b04917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perišić O, Portillo-Ledesma S, Schlick T, (2019). Sensitive effect of linker histone binding mode and subtype on chromatin condensation. Nucleic Acids Res, 47, 4948–4957. 10.1093/nar/gkz234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Portillo-Ledesma S, Schlick T, (2020). Bridging chromatin structure and function over a range of experimental spatial and temporal scales by molecular modeling. WIREs Comput. Mol. Sci, 10 10.1002/wcms.1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willcockson MA, Healton SE, Weiss CN, Bartholdy BA, Botbol Y, Mishra LN, Sidhwani DS, Wilson TJ, et al. , (2020). H1 histones control the epigenetic landscape by local chromatin compaction. Nature,, 1–6. 10.1038/s41586-020-3032-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torres CM, Biran A, Burney MJ, Patel H, Henser-Brownhill T, Cohen AHS, Li Y, Ben-Hamo R, et al. , (2016). The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science (80-.), 353 10.1126/science.aaf1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada M, Chen WY, Nakadai T, Onikubo T, Guermah M, Rhodes D, Roeder RG, (2019). Gene-specific H1 eviction through a transcriptional activator→ p300→NAP1→H1 pathway. Mol. Cell, 74, 268–283.e5. 10.1016/j.molcel.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, et al. , (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature, 555, 61–66. 10.1038/nature25762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Turner AL, Watson M, Wilkins OG, Cato L, Travers A, Thomas JO, Stott K, (2018). Highly disordered histone H1–DNA model complexes and their condensates. Proc. Natl. Acad. Sci. U. S. A, 115, 11964–11969. 10.1073/pnas.1805943115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibbs EB, Kriwacki RW, (2018). Linker histones as liquid-like glue for chromatin. Proc. Natl. Acad. Sci. U. S. A, 115, 11868–11870. 10.1073/pnas.1816936115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hendzel MJ, Lever MA, Crawford E, Th’Ng JPH, (2004). The C-terminal domain is the primary determinant of histone H1 binding to chromatin in vivo. J. Biol. Chem, 279, 20028–20034. 10.1074/jbc.M400070200. [DOI] [PubMed] [Google Scholar]
- 44.Potoyan DA, Papoian GA, (2011). Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J. Am. Chem. Soc, 133, 7405–7415. 10.1021/ja1111964. [DOI] [PubMed] [Google Scholar]
- 45.Potoyan DA, Papoian GA, (2012). Regulation of the H4 tail binding and folding landscapes via Lys-16 acetylation. Proc. Natl. Acad. Sci. U. S. A, 109, 17857–17862. 10.1073/pnas.1201805109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winogradoff D, Echeverria I, Potoyan DA, Papoian GA, (2015). The acetylation landscape of the H4 histone tail: Disentangling the interplay between the specific and cumulative effects. J. Am. Chem. Soc, 137, 6245–6253. 10.1021/jacs.5b00235. [DOI] [PubMed] [Google Scholar]
- 47.Zhou BR, Feng H, Kale S, Fox T, Khant H, de Val N, Ghirlando R, Panchenko AR, Bai Y, (2021). Distinct structures and dynamics of chromatosomes with different human linker histone isoforms. Mol. Cell, 81, 166–182.e6. 10.1016/j.molcel.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fang H, Clark DJ, Hayes JJ, (2012). DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res, 40, 1475–1484. 10.1093/nar/gkr866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fang H, Wei S, Lee TH, Hayes JJ, (2016). Chromatin structure-dependent conformations of the H1 CTD. Nucleic Acids Res, 44, 9131–9141. 10.1093/nar/gkw586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sridhar A, Farr SE, Portella G, Schlick T, Orozco M, Collepardo-Guevara R, (2020). Emergence of chromatin hierarchical loops from protein disorder and nucleosome asymmetry. Proc. Natl. Acad. Sci. U. S. A, 117, 7216–7224. 10.1073/pnas.1910044117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sridhar A, Orozco M, Collepardo-Guevara R, (2020). Protein disorder-to-order transition enhances the nucleosome-binding affinity of H1. Nucleic Acids Res,, 1–14. 10.1093/nar/gkaa285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heidarsson P, Mercadante D, Sottini A, Nettels D, Borgia M, Borgia A, Kilic S, Fierz B, et al. , (2020). Disordered proteins enable histone chaperoning on the nucleosome 2020.04.17.046243 BioRxiv,. 10.1101/2020.04.17.046243. [DOI] [Google Scholar]
- 53.Davtyan A, Schafer NP, Zheng W, Clementi C, Wolynes PG, Papoian GA, (2012). AWSEM-MD: Protein structure prediction using coarse-grained physical potentials and bioinformatically based local structure biasing. J. Phys. Chem. B, 116, 8494–8503. 10.1021/jp212541y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu H, Wolynes PG, Papoian GA, (2018). AWSEM-IDP: A coarse-grained force field for intrinsically disordered proteins. J. Phys. Chem. B, 122, 11115–11125. 10.1021/acs.jpcb.8b05791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hinckley DM, Freeman GS, Whitmer JK, De Pablo JJ, (2013). An experimentally-informed coarse-grained 3-site-per-nucleotide model of DNA: Structure, thermodynamics, and dynamics of hybridization. J. Chem. Phys, 139 10.1063/1.4822042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, et al. , (2010). Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc. Natl. Acad. Sci. U. S. A, 107, 9620–9625. 10.1073/pnas.1000309107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, et al. , (2014). Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature, 507, 104–108. 10.1038/nature12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim K, Jeong KW, Kim H, Choi J, Lu W, Stallcup MR, An W, (2012). Functional interplay between p53 acetylation and H1.2 phosphorylation in p53-regulated transcription. Oncogene, 31, 4290–4301. 10.1038/onc.2011.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Talasz H, Sarg B, Lindner HH, (2009). Site-specifically phosphorylated forms of H1.5 and H1.2 localized at distinct regions of the nucleus are related to different processes during the cell cycle. Chromosoma, 118, 693–709. 10.1007/s00412-009-0228-2. [DOI] [PubMed] [Google Scholar]
- 60.Roque A, Ponte I, Arrondo JLR, Suau P, (2008). Phosphorylation of the carboxy-terminal domain of histone H1: Effects on secondary structure and DNA condensation. Nucleic Acids Res, 36, 4719–4726. 10.1093/nar/gkn440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lopez R, Sarg B, Lindner H, Bartolomé S, Ponte I, Suau P, Roque A, (2015). Linker histone partial phosphorylation: Effects on secondary structure and chromatin condensation. Nucleic Acids Res, 43, 4463–4476. 10.1093/nar/gkv304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stein A, Bina M, (1984). A model chromatin assembly system: Factors affecting nucleosome spacing. J. Mol. Biol, 178, 341–363. 10.1016/0022-2836(84)90148-7. [DOI] [PubMed] [Google Scholar]
- 63.Gunjan A, Alexander BT, Sittman DB, Brown DT, (1999). Effects of H1 histone variant overexpression on chromatin structure. J. Biol. Chem, 274, 37950–37956. 10.1074/jbc.274.53.37950. [DOI] [PubMed] [Google Scholar]
- 64.Hewish DR, Burgoyne LA, (1973). Chromatin substructure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem. Biophys. Res. Commun, 52, 504–510. 10.1016/0006-291X(73)90740-7. [DOI] [PubMed] [Google Scholar]
- 65.Woodcock CLF, (1973). Ultrastructure of inactive chromatin. J. Cell Biol, 59, 368a. [Google Scholar]
- 66.Olins AL, Olins DE, (1974). Spheroid chromatin units (v bodies). Science (80-.), 183, 330–332. 10.1126/science.183.4122.330. [DOI] [PubMed] [Google Scholar]
- 67.Van Holde KE, (1989). Chromatin. Springer-Verlag, New York. 10.1007/978-1-4612-3490-6. [DOI] [Google Scholar]
- 68.Talbert PB, Ahmad K, Almouzni G, Ausió J, Berger F, Bhalla PL, Bonner WM, Cande W, et al. , (2012). A unified phylogeny-based nomenclature for histone variants. Epigenetics Chromatin, 5, 7. 10.1186/1756-8935-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miyagi A, Ando T, Lyubchenko YL, (2011). Dynamics of nucleosomes assessed with time-lapse high-speed atomic force microscopy. Biochemistry, 50, 7901–7908. 10.1021/bi200946z. [DOI] [PubMed] [Google Scholar]
- 70.Lyubchenko YL, (2014). Nanoscale nucleosome dynamics assessed with time-lapse AFM. Biophys. Rev, 6, 181–190. 10.1007/s12551-013-0121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stumme-Diers MP, Banerjee S, Hashemi M, Sun Z, Lyubchenko YL, (2018). Nanoscale dynamics of centromere nucleosomes and the critical roles of CENP-A. Nucleic Acids Res, 46, 94–103. 10.1093/nar/gkx933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vlijm R, Lee M, Lipfert J, Lusser A, Dekker C, Dekker NH, (2015). Nucleosome assembly dynamics involve spontaneous fluctuations in the handedness of tetrasomes. Cell Rep, 10, 216–225. 10.1016/j.celrep.2014.12.022. [DOI] [PubMed] [Google Scholar]
- 73.Kim SH, Vlijm R, van der Torre J, Dalal Y, Dekker C, (2016). CENP-A and H3 nucleosomes display a similar stability to force-mediated disassembly. PLoS One, 11, 10.1371/journal.pone.0165078e0165078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Phair RD, Scaffidi P, Elbi C, Vecerova J, Dey A, Ozato K, Brown DT, Hager G, et al. , (2004). Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol, 24, 6393–6402. 10.1128/mcb.24.14.6393-6402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vermaak D, Steinbach OC, Dimitrov S, Rupp RAW, Wolffe AP, (1998). The globular domain of histone H1 is sufficient to direct specific gene repression in early Xenopus embryos. Curr. Biol, 8, 533–536. 10.1016/s0960-9822(98)70206-4. [DOI] [PubMed] [Google Scholar]
- 76.Vermaak D, Wolffe AP, (1998). Chromatin and chromosomal controls in development. Dev. Genet, 22, 1–6. . [DOI] [PubMed] [Google Scholar]
- 77.Melters D, Dalal Y, (2021). Nano-surveillance: Tracking individual molecules in a sea of chromatin. J. Mol. Biol, 433 (6), 166720. 10.1016/j.jmb.2020.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Öberg C, Belikov S, (2012). The N-terminal domain determines the affinity and specificity of H1 binding to chromatin. Biochem. Biophys. Res. Commun, 420, 321–324. 10.1016/j.bbrc.2012.02.157. [DOI] [PubMed] [Google Scholar]
- 79.Materese CK, Savelyev A, Papoian GA, (2009). Counterion atmosphere and hydration patterns near a nucleosome core particle. J. Am. Chem. Soc, 131, 15005–15013. 10.1021/ja905376q. [DOI] [PubMed] [Google Scholar]
- 80.Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, Panchenko AR, (2016). Coupling between histone conformations and DNA geometry in nucleosomes on a microsecond timescale: Atomistic insights into nucleosome functions. J. Mol. Biol, 428, 221–237. 10.1016/j.jmb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Onuchic JN, Luthey-Schulten Z, Wolynes PG, (1997). Theory of protein folding: The energy landscape perspective. Annu. Rev. Phys. Chem, 48, 545–600. 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 82.Zhao H, Winogradoff D, Bui M, Dalal Y, Papoian GA, (2016). Promiscuous histone mis-assembly is actively prevented by chaperones. J. Am. Chem. Soc, 138, 13207–13218. 10.1021/jacs.6b05355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zheng W, Schafer NP, Davtyan A, Papoian GA, Wolynes PG, (2012). Predictive energy landscapes for protein–protein association. Proc. Natl. Acad. Sci, 109, 19244–19249. 10.1073/PNAS.1216215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zheng W, Schafer NP, Wolynes PG, (2013). Frustration in the energy landscapes of multidomain protein misfolding. Proc. Natl. Acad. Sci. U. S. A, 110, 1680–1685. 10.1073/pnas.1222130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim BL, Schafer NP, Wolynes PG, (2014). Predictive energy landscapes for folding α-helical transmembrane proteins. Proc. Natl. Acad. Sci. U. S. A, 111, 11031–11036. 10.1073/pnas.1410529111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao H, Winogradoff D, Dalal Y, Papoian GA, (2019). The oligomerization landscape of histones. Biophys. J, 116, 1845–1855. 10.1016/j.bpj.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Potoyan DA, Zheng W, Komives EA, Wolynes PG,(2016). Molecular stripping in the NF-κB/IκB/DNA genetic regulatory network. Proc. Natl. Acad. Sci. U. S. A, 113, 110–115. 10.1073/pnas.1520483112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Potoyan DA, Bueno C, Zheng W, Komives EA, Wolynes PG, (2017). Resolving the NFjB heterodimer binding paradox: Strain and frustration guide the dinding of dimeric transcription factors. J. Am. Chem. Soc, 139, 18558–18566. 10.1021/jacs.7b08741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tsai M-Y, Zhang B, Zheng W, Wolynes PG, (2016). Molecular mechanism of facilitated dissociation of Fis protein from DNA. J. Am. Chem. Soc, 138, 13497–13500. 10.1021/jacs.6b08416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tsai M-Y, Zheng W, Chen M, Wolynes PG, (2019). Multiple binding configurations of Fis protein pairs on DNA: Facilitated dissociation versus cooperative dissociation. J. Am. Chem. Soc,. 10.1021/jacs.9b08287. [DOI] [PubMed] [Google Scholar]
- 91.Zhang B, Zheng W, Papoian GA, Wolynes PG, (2016). Exploring the free energy landscape of nucleosomes. J. Am. Chem. Soc, 138, 8126–8133. 10.1021/jacs.6b02893. [DOI] [PubMed] [Google Scholar]
- 92.Lin X, Roy S, Jolly MK, Bocci F, Schafer NP, Tsai MY, Chen Y, He Y, Grishaev A, Weninger K, Orban J, Kulkarni P, Rangarajan G, Levine H, Onuchic JN, (2018). PAGE4 and conformational switching: Insights from molecular dynamics simulations and implications for prostate cancer. J. Mol. Biol, 430, 2422–2438. 10.1016/j.jmb.2018.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Plimpton S, (1995). Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys, 117, 1–19. 10.1006/jcph.1995.1039. [DOI] [Google Scholar]
- 94.Webb B, Sali A, (2016). Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinforma, 2016, 5.6.1–5.6.37. 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Habibi M, Rottler J, Plotkin SS, (2016). As simple as possible, but not simpler: Exploring the fidelity of coarse-grained protein models for simulated force spectroscopy. PLoS Comput. Biol, 12, 1005211. 10.1371/journal.pcbi.1005211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A, (2016). Coarse-grained protein models and their applications. Chem. Rev, 116, 7898–7936. 10.1021/acs.chemrev.6b00163. [DOI] [PubMed] [Google Scholar]
- 97.Perlmutter JD, Drasler WJ, Xie W, Gao J, Popot JL, Sachs JN, (2011). All-atom and coarse-grained molecular dynamics simulations of a membrane protein stabilizing polymer. Langmuir, 27, 10523–10537. 10.1021/la202103v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Woods DC, Wereszczynski J, (2020). Elucidating the influence of linker histone variants on chromatosome dynamics and energetics. Nucleic Acids Res, 48, 3591–3604. 10.1093/nar/gkaa121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.