

1. Introduction
The chronic pain state is characterized by changes in somatosensory processing, affect and motivation[28; 31; 36; 42]. Human imaging studies suggest that alterations in mesolimbic circuitry of the brain accompany establishment of this state[1; 2; 44]. The nucleus accumbens (NAc) is a key hub in the mesolimbic circuitry, and the release of dopamine (DA) in the NAc by neurons in the ventral tegmental area (VTA) modulates its functional connectivity with other regions implicated in chronic pain, like the prefrontal cortex, amygdala and hippocampus[5; 8; 25; 35].
Indeed, experimental work in rodent models is consistent with the hypothesis that the NAc and VTA play key roles in the response to peripheral nerve injury. For example, in the mouse spared nerve injury (SNI) model of chronic pain, D2 DA receptor expressing spiny projection neurons (d2SPNs) in the medial shell of the NAc (msNAc) undergo a constellation of adaptations in their intrinsic excitability and functional connectome that appear to be triggered by an alteration in VTA dopaminergic signaling[34]. Surprisingly, SNI-induced adaptations were not observed in adjacent D1 receptor expressing spiny projection neurons (d1SPNs). These cell-specific adaptations modulated the tactile allodynia induced by SNI. Injury-induced adaptations have also been observed in the NAc core (cNAc). Excitatory glutamatergic synapses on d2SPNs (but not d1SPNs) were depressed in rodent model of chronic pain, resulting in decreased motivation, but not allodynia[36]. The functional complexity of the NAc is mirrored in that of the VTA, as it is clear that the projection site of these dopaminergic neurons is a determinant of their response to aversive and rewarding stimuli[11]. Together, these studies underscore the functional heterogeneity of the circuitry linking the VTA and NAc and the distinct roles these circuits play in shaping behavior[30].
The goal of this study was to expand the characterization of adaptations in the cNAc and the VTA in the SNI model of chronic pain[45]. To this end, physiological approaches were used to interrogate VTA and NAc circuits ex vivo and chemogenetic approaches were used to test behavioral inferences drawn from these studies. In agreement with previous work[18; 21], our results suggest that SNI increases DA release in the medial shell of the NAc (msNAc) but decreases it in the cNAc. In the cNAc, the suppression of dopaminergic signaling altered the intrinsic excitability and synaptic connectivity of d2SPNs. These changes appeared to lessen the negative affect associated with SNI, as chemogenetic mimicry alleviated SNI-induced anxiety and social withdrawal. In contrast, chemogenetic excitation of msNAc projecting VTA dopaminergic neurons did not affect anxiety but did significantly diminish tactile allodynia. Taken together, these results argue that regionally specific alterations in dopaminergic signaling induced by SNI are adaptive and shift activity in the cNAc and msNAc in ways that promote re-engagement with the environment. This inference stands in stark contrast to the prevailing view that alterations in dopaminergic signaling promote the maladaptive chronic pain state[50]. Our studies also suggest that homeostatic mechanisms triggered by these adaptive changes in network activity lessen their impact with time, potentially leading to the chronic pain state.
2. METHODS
2.1. Animals
All electrophysiology studies were carried out in 8-week-old male C57BL6 bacterial artificial chromosome (BAC)-transgenic mice (D1 receptor-Td Tomato and D2 receptor–enhanced Green Fluorescent Protein [eGFP]), RV-Cre infected 8-week-old male Ai14-tdTomato-flox transgenic mice or C57BL6 wild type mice. C57Bl/6 bacterial artificial chromosome (BAC)-transgenic mice (D1 receptor-Td tomato and D2 receptor-eGFP) were made in our lab by backcrossing D2-eGFP mice provided by the GENSAT program at Rockefeller University and D1tdTomato mice provided by Dr. Nicole Calakos (Duke University). Ai14-tdTomato-flox transgenic mice were purchased from Jackson Labs (strain ID: 007908). Adora2a-Cre KG139 (A2A:Cre) mice were provided by the GENSAT program at Rockefeller University (Strain ID: 31168-UCD). C57Bl/6 wild type mice were purchased from Jackson Labs (Strain ID: 000664).
Animals were group-housed with littermates, with food and water available ad libitum and with a 12 h light/dark cycle (7 A.M.–7 P.M.). Tactile threshold of each animal was measured one day pre-surgery and on assigned dates post-surgery. 8-week-old male A2A:Cre mice and wild type mice were used in the sets of behavioral experiments. All studies were approved by the Animal Care and Use Committee of Northwestern University and were carried out in accordance with the guidelines of the National Institutes of Health on animal care and with the ethical guidelines for investigation of experimental pain in conscious animals[51].
2.2. Neuropathic pain model: Spared Nerve Injury (SNI)
The SNI model has been described previously[12]. Mice were anesthetized with isoflurane 1.5–2.5% and a mixture of 30% N2O and 70% O2. The left hind leg sciatic nerve was exposed at the level of the trifurcation into the sural, tibial, and common peroneal nerves. The tibial and common peroneal nerves were tightly ligated and severed, leaving the sural nerve intact. Animals in the Sham surgery group had their sciatic nerve exposed as in the SNI procedure, but they received no further manipulations.
2.3. Tactile sensitivity
Paw withdrawal thresholds to Von Frey filament stimulation (VF) were used to assess mechanical sensitivity of the hind-paws. Mice were placed in a Plexiglas box with a wire grid floor and allowed to habituate for 45 min-1h. Then, filaments of various forces (Stoelting) were applied to the plantar surface of each hind-paw. Filaments were applied in a descending and ascending pattern, determined by the response of the animal. Each filament was applied for a maximum of 2 seconds, and paw withdrawal in response to the filament was considered a positive response. Fifty percent thresholds were calculated according to[6].
2.4. Electrophysiology
2.4.1. Brain slices preparation.
Sagittal/coronal brain slices of the cNAc or VTA (250 μm) were obtained from transgenic mice or wild type mice. The mice were anesthetized with ketamine/xylazine and perfused transcardially with ice-cold artificial CSF (aCSF), containing in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 2.0 CaCl2, 1.0 MgCl2, 25 NaHCO3, and 25 glucose, saturated with 95% O2 and 5% CO2. Brains were rapidly removed and sliced in oxygenated, ice-cold, aCSF using a VT1200S vibratome (Leica Microsystems). Slices were transferred to a holding chamber and incubated in aCSF at 35°C for 30 min, and then stored at room temperature until used for patch clamp recordings (1–5 h). The extracellular aCSF was saturated with 95% O2/5% CO2 at all times to maintain oxygenation and a pH ~7.4.
2.4.2. Visualized whole-cell patch-clamp recording ex-vivo.
Slices were visualized using an upright microscope (Nikon) equipped with infrared differential interference contrast (IR-DIC) optics and with a Hitachi CCD camera (KP-M2RN). The recording chamber was superfused with carbogen-saturated aCSF at a flow rate of 2–3 ml min−1. Picrotoxin (100 μM) was added to block GABAA receptor-mediated IPSCs. A combination of CNQX (10 μM) and D-AP5 (50 μM) was used to block glutamatergic transmission during action potential recording. Recordings were made at 32 –34°C using a Multiclamp 700B amplifier with pClamp 10.3 software (Molecular Devices). Signals were filtered at 5 Hz (current clamp) or 2 Hz (voltage clamp) and digitized at 10 kHz with a Digidata 1322A (Molecular Devices). Patch electrodes were pulled on a Flaming-Brown horizontal puller (P-97; Sutter Instruments) from filamented, thick-wall borosilicate-glass (1.5 mm O.D., Sutter Instruments). Pipette resistance was typically ~3–5 MΩ when filled with an internal solution consisting of: 140 mM KMeSO4, 10 mM KCl, 10 mM HEPES, 2 mM Mg2ATP, 0.4 mM NaGTP, 10 mM sodium phosphocreatine and 0.2% (wt/vol) biocytin; pH 7.25–7.30; 300 mOsm.
In each NAc core slice, neuronal somatic eGFP or Td-Tomato expression was verified routinely in cell-attach to confirm cell identity before breaking into whole-cell mode. Current clamp recordings were performed to quantify firing properties. Spikes were evoked using current step injections (800 ms duration at 0.1 Hz,−160 to +320 pA range with 20 pA steps). Rheobase current was defined as the first current step capable of eliciting an action potential. Input resistance (Ri) was monitored on-line with a 40 pA, 150 ms current injections and only cells with a stable Ri (≤20% change for the duration of the recording) were further analyzed. Basal firing frequency of medial VTA neurons was measured over a 5 minutes current-clamp recording after perforated patch was established at the presence of synaptic blockers (20 μM gabazine, 10 μM CNQX and 50 μM D-AP5), and the amplifier bridge circuit were adjusted to compensate for electrode resistance and monitored. In perforated patch mode, the electrical access was obtained using a stock solution of gramicidin (2 mg/ml) in dimethyl sulfoxide (DMSO) which was prepared daily and diluted in the recording solution before use a final concentration of 2 μg/ml. Miniature excitatory postsynaptic currents (mEPSCs) were measured in voltage clamp at a holding potential of −80 mV and in the presence of 500 nM tetrodotoxin (TTX, to block voltage-gated sodium currents). Frequency and amplitude distributions of mEPSCs were analyzed using MiniAnalysis program 6.0.3 (Synaptosoft) over a 5 minutes time interval.
2.4.3. Optically-evoked synaptic responses.
EPSCs of cNAc SPNs were evoked by ChR2 / Chronos stimulation using 473 nm wavelength light pulses (1ms, 0.33Hz LED, CoolLED), and were recorded in voltage clamp (Vh= −80 mV). The pipette internal solution contained (in mM): 120 CsMeSO3, 5 NaCl, 10 TEA-Cl, 10 Hepes, 3 QX-314, 4 ATP-Mg, 0.3 GTP-Na, and 0.25 EGTA. The access resistance was monitored by a hyperpolarizing step of −10 mV with each sweep. For recordings designed to determine the AMPA receptor/NMDA receptors current ratios, AMPAR-mediated currents were obtained with holding at −80 mV in the presence of picrotoxin (100 μM). The Cells were then held at +40 mV to isolate NMDAR-mediated current in the presence of AMPAR antagonist CNQX (10 μM). To induce the state-dependent LTD, a physiologically realistic induction protocol was used in which 0.5 s postsynaptic depolarizations from −70 to −50 mV (mimicking the down- to up-state transition) was paired with 10 stimuli delivered at 25 Hz (mimicking synchronous excitatory input excitation) and repeated every second for 5 min.
2.5. Drugs and drug application
Naspm, WIN 55,212–2, quinpirole, sulpiride and the electrophysiological tool compounds were obtained from Tocris, USA. PSEM89s was obtained from Apex Scientific Inc. (Stony Brook, NY). Drugs were bath applied for at least 15 min to establish equilibrium in the tissue.
2.6. Confocal imaging and anatomical reconstruction
For anatomical reconstruction, 0.2% biocytin (Sigma, USA) was included in the internal solution and neurons were recorded in whole-cell mode for at least 30 min. Slices were fixed in 4% paraformaldehyde overnight. After several washes, slices were reacted for 6 hours with streptavidin-Cy5 (1:200, Invitrogen, catalog number: SA1011) in 0.5% Triton-X, 1% BSA, 10% normal goat serum (in PBS) at 20–22 °C in the dark. Sections were then washed and coverslipped with prolong gold antifade reagent (Invitrogen). A cell was rejected if the soma was not intact or any dye was seen in neighboring cells. For anatomical reconstruction, serial optical sections (Z-stacks) were acquired using a 0.415×0.415×0.5 μm3 voxel size on a laser-scanning confocal microscope (LSM 510; Olympus) with a 60x/1 NA oil-immersion objective (Zeiss). Z-series of the same cell were combined using Fiji Software (VIAS; Mt. Sinai Computational Neurobiology and Imaging Center), and subsequently reconstructed and analyzed using the Neurolucida/Neuroexplorer suite (MicroBrightField). No correction was applied for tissue shrinkage. For spine counting, serial Z-stacks were acquired using a 0.069×0.069×0.1 μm3 voxel size. Images were deconvoluted using Autoquata (MediaCybernetics). Counting was performed on 45- to 105-μm long segments of the principal dendrite. Spines were counted only if they had both a punctuate head and visible neck.
2.7. Stereotaxic injection of AAV-ChR2, AAV-Chronos and RV-Cre
Mice were anesthetized with isoflurane and the stereotaxic guided surgeries were performed for all virus injections. A glass filled with viral solution was lowered through a small hole drilled through the skull and two unilateral injections were made. The injection pipette was withdrawn 5 min after the infusion and the scalp was then sealed. For the injection of AAV-ChR2 or AAV-Chronos, AAV9-CAG-hChR2-mCherry-WPRE.SV40 (titre: 5.16 × 1012 genomic copies per mL) or AAV8- Synapsin-Chronos-GFP (titre: 3.2 × 1012 genomic copies per mL)were injected into prelimbic prefrontal cortex (100nl ChR2-AAV at coordinate of bregma 1.90, lateral 0.30, ventral 2.25) or basolateral amygdala (100nl ChR2-AAV at coordinate of bregma −1.50, lateral 3.40, ventral 4.75) of BAC transgenic mice. For RV-Cre virus injection, RV-Cre (titre: 1.19 × 1010 genomic copies per mL) were injected into Ai14-td Tomato-flox mice at coordinates of cNAc (bregma 1.18, lateral 1.10, ventral 4.35) or msNAc (bregma 1.48, lateral 0.50, ventral 4.55). Animals were used for patch-clamp recording at 3 weeks after injection and the injection site was verified post hoc.
2.8. Chemogenetic approaches
Stereotaxic guided surgeries were performed as same as the injection of AAV-ChR2. To activate cNAc d2SPNs, A2A::Cre mice were used and 100nl of AAV9-Syn-DIO-rev-PSAML141F,Y115F-5HT3HC-IRES-GFP-WPRE (titre:2.04 × 1013 genomic copies per mL) were injected into bilateral cNAc at coordinate of bregma 1.18, lateral 1.10, ventral 4.35. 4 weeks after PSAM-AAV virus injection, SNI surgeries were performed. On assigned days after SNI surgery, PSEM89s (30 mg/kg) or saline (0.1 ml/10g) were given intraperitoneally (i.p.) and the effect of treatments were assessed by open field and sociability tests at 1 hour post injection. To specifically inhibit VTA neurons innervating cNAc and activate VTA neurons projecting to msNAc, wild-type and D2-GFP mice were firstly infected with RV-Cre (100nl RV-Cre at bilateral cNAc or bilateral msNAc). 2 weeks later, RV-Cre infected mice were stereotaxically injected with 200nl of AAV9-Syn-DIO-rev-PSAML141F,Y115F-GlyR-IRES-GFP-WPRE (titre:2.10 × 1013 genomic copies per mL) or 200nl of AAV9-Syn-DIO-rev-PSAML141F,Y115F-5HT3HC-IRES-GFP-WPRE (titre:2.04 × 1013 genomic copies per mL) into bilateral VTA (bregma −3.10, lateral 0.65, ventral 4.55) to modulate VTA neurons projecting to cNAc and msNAc respectively. 4 weeks after second virus injection, mice underwent SNI surgeries. From Day 5 after SNI surgery, PSEM89s (30 mg/kg) or saline (0.1 ml/10g) were intraperitoneally injected (twice a day) until sociability test and electrophysiological recording were performed on assigned days at 1 hour post i.p. injection. The injection site was verified post hoc.
2.9. Open field test
Behavior was evaluated during its exploration of an open field (OF), made up of an opaque 45 × 45 × 45 cm Plexiglas box. Locomotor activity (distance traveled in the box) and emotional status (time spent in the center of the field and in the periphery of the field) was recorded during 5 minutes and analyzed using the video ANY-maze Visual Tracking System and Software (v4.99, Stoelting Co.). The animal activity was assessed at 1 h post i.p. injection of either PSEM89s (30 mg/kg) or saline (0.1 ml/10g) on Day 10 or 20 after SNI/Sham surgery. Three groups were compared: Sham mice, PSEM89S-treated with PSAM-injected SNI mice (30 mg/kg) and saline-treated with PSAM-injected SNI mice (0.1 ml/10g).
2.10. Sociability test
The Sociability Test, adapted from[49], assessed an animal’s preference for social versus non-social interaction. Briefly, the apparatus consisted of an opaque Plexiglas box with two wired cups (A and B) placed directly across from one another; these cups allowed the experimental animals to approach and interact with confined conspecifics/objects without direct contact. The box was placed in a quiet room, and a camera was fixed over the box to record behavior. The animals received 10-minute habituation (both cups were empty) and twenty-four hours later, another 10-minute habituation was received. Sociability test was performed after another 5-min home cage stay. In each test, a male mouse was placed in the box and exposed to a juvenile female mouse (Cup A) and an object (Cup B) for 10 min. The object used was approximately the same size and the same color as the conspecific to control for any different visual cues. Using ANY-maze visual tracking software (version 4.99), we measured the time mice spent interacting with each cup (interaction is defined as mouse’s head being within the zone surrounding each cup), and the ratio of exploration time for Cup A over that for Cup B was calculated. The box, cups, and objects were cleaned with soapy water and wiped with bleach and alcohol before each animal’s tests. The animal sociability was assessed at 1 h post i.p. injection of either PSEM89s (30 mg/kg) or saline (0.1 ml/10g) on Day 10 or 20 after SNI/Sham surgery. Three groups were compared: Sham mice, PSEM89S-treated with PSAM-injected SNI mice (30 mg/kg) and saline-treated with PSAM-injected SNI mice (0.1 ml/10g).
Statistics:
Data analyses were done with ClampFit 9 (Molecular Devices), MiniAnalysis 6.0.3 (Synaptosoft) and GraphPad Prism 6.0 (GraphPad Software). Distribution free statistics were used for all data sets regardless of sample size and variance. For plasticity experiments, the magnitude of LTD was calculated as a percentage of the baseline where the baseline is defined as the average of minutes 0–10 of recording time. Intensity-response, LTD recording and morphological Sholl analysis data are presented as median with shaded interquartile (quartile 1 to quartile 3). Box plots are used to represent all the other data: the central line represents the median, the edges represent the interquartile ranges, and the whiskers represent the overall distribution. Statistical analysis was performed with GraphPad Prism 6.0 (GraphPad Software) using non–parametric tests (two-tailed Mann–Whitney rank sum Test or two-tailed Wilcoxon matched-pairs signed rank Test). LTD data were analyzed with Wilcoxon signed rank test. Kolmogorov-Smirnov tests were used for cumulative distribution analysis of miniature EPSCs. Statistical tests are indicated in each figure’s legend. Data collection and analysis were not performed blind to the condition of the experiments, and no randomization was used. No data points were removed from statistical analysis except specified. In all experiments no statistical methods were used to predetermine sample sizes, but sample size is similar to sample sizes routinely used in the field for similar experiments[36]. Neurons or brain slices for each experimental group were collected from at least five mice, and one or two brain slice per mouse. In behavioral test, the animal number of each group is between 5–9 animals from at least 2 litters. Probability threshold for statistical significance was P<0.05. No significant difference (n.s.) P>0.05, *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001.
3. Results
3.1. SNI selectively altered the functional connectome of cNAc d2SPNs
Previous work in a model of chronic pain found a reduction in excitatory synaptic connectivity of cNAc d2SPNs[36]. However, given the heterogeneity in excitatory connections of cNAc SPNs[13], this SNI-induced change could be specific to a particular input. To address this question, optogenetic approaches were used to selectively activate axons arising from basolateral amygdala (BLA) neurons or pyramidal neurons in prelimbic cortex (PL) – two of the most prominent glutamatergic inputs to cNAc SPNs implicated in chronic pain states[13]. Stereotaxic injection of an adenoassociated virus (AAV) carrying a synapsin-driven channelrhodopsin2 (ChR2)-mCherry expression construct into the BLA of mice expressing enhanced green fluorescent protein (eGFP) in d2SPNs and tdTomato in D1 receptor expressing SPNs (d1SPNs)[34] led to robust labeling of NAc projecting axons (Fig. 1a). In ex vivo brain slices prepared from these so-called ‘Christmas’ (Xmas) mice three weeks after AAV injection, full-field optical stimulation of BLA axons evoked robust glutamatergic excitatory postsynaptic currents (EPSCs) in both cNAc d2SPNs and d1SPNs measured using whole cell voltage clamp techniques with a Cs+-based internal solution[34]. EPSCs mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) were measured by holding the cell at −80 mV, whereas N-methyl-D-aspartate receptor (NMDAR)-mediated EPSCs were isolated by recording in the presence of selective AMPAR antagonist CNQX (10 μM) and holding at + 40 mV[34; 36]. The relative strength the synaptic connection was gauged by the ratio of AMPAR and NMDAR EPSCs (A/N ratio). In agreement with an earlier report[36], the A/N ratio of at BLA synapses on d2SPNs was significantly reduced by SNI, whereas that at synapses on d1SPNs was unchanged (Fig. 1b–e). In contrast, using the same approach to examine the functional connectivity of PL pyramidal neurons with cNAc SPNs led to a different outcome (Fig. 1f). In d2SPNs from SNI-mice, the A/N ratio of PL-evoked EPSCs was significantly larger than that in sham-lesioned controls (Fig. 1g,h). The A/N ratio of PL-evoked EPSCs in neighboring d1SPNs was unchanged by SNI (Fig. 1i,j). These results clearly indicate that SNI led to strengthening of PL synapses and weakening of BLA synapses with d2SPNs, leaving synaptic connections of d1SPNs unchanged.
Figure 1. The strength of BLA synapses on cNAc d2SPNs was reduced whereas the strength of PL synapses on cNAc d2SPNs was increased in SNI animals.

a, Sample slices displaying the expression of ChR2-mCherry (red) in BLA (larger image) and cNAc (smaller image) after virus injection in the BLA. Scale bars represent 0.5 mm. b-e, Optical stimulation-evoked d2SPN AMPA/NMDA receptor responses (A/N ratio) were inhibited in SNI (n = 7–8 neurons from 5 mice per group; U = 9, p = 0.0289), but no change was detected in d1SPNs (j, n = 6 neurons from 5 mice per group with U = 17, p = 0.8983). b and d AMPA and NMDA receptor-mediated currents elicited in the cNAc SPNs with optical stimulation of BLA inputs (calibration: 50 pA, 100 ms). f, Sample images displaying the expression of ChR2-mCherry (red) in prelimbic (PL)-PFC and cNAc after virus injection in BLA (scale bar: 0.5 mm). g-j, Optically evoked d2SPNs AMPA/NMDA receptor responses were enhanced in SNI, but no change was detected in d1SPNs (n = 6 neurons from 5 mice per group, Mann–Whitney U test with U = 2, p = 0.0087 for c and U = 16, p = 0.8182 for e). g and i showed the representative AMPA and NMDA receptor-mediated currents elicited in the cNAc SPNs with optical stimulation of PL inputs (calibration: 50 pA, 100 ms). Data are presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
In dorsal striatum d2SPNs, the strength of corticostriatal connectivity is bi-directionally controlled by DA[40]. D2 receptors oppose the induction of long-term potentiation (LTP) by inhibiting adenosine type 2a receptor (A2aR) stimulation of adenylyl cyclase and promote the induction of endocannabinoid (eCb) dependent, long-term depression (LTD). As in this region, bath application of the type 1 eCb receptor (CB1R) agonist WIN 55,212–2 (5 μM) led to a robust LTD at PL synapses on cNAc d2SPNs (Fig. 2a–d). In contrast, WIN had no discernible effect on BLA synapses (Fig. 2d). This result is consistent with previous work showing that synaptic depression at d2SPN synapses following SNI, which our data suggest is occurring at BLA synapses, is not mediated by dopamine, but rather by galanin signaling[36].
Figure 2. D2 receptor signaling modulated state-dependent LTD at PL-cNAc d2SPN synapses.

a-b, Representative images showing Chronos-GFP (green) expression in PL-PFC and BLA at 4-week post injection (scale bar: 0.5 mm). c-d, Bath application of CB1 receptor agonist WIN 55,212–2 induced LTD of AMPAR EPSC amplitude in PL-cNAc d2SPN synapses, but had no effect on BLA-d2SPN synapses (n = 5 neurons from 5 mice per group; Wilcoxon test with W = −4543, p < 0.0001; calibration for c: 50 pA, 10 ms). e, Diagram of D2R-dependence of the eCb-dependent LTD. f, The representative recording traces showing the EPSCs of PL-cNAc d2SPNs synapses before and after state-dependent LTD induction in the presence of D2 receptor agonist quinpirole (10 μM, calibration: 50 pA, 10 ms). g, State-dependent LTD induction protocol (calibration: 300 pA, 500 ms). h, At PL-cNAc d2SPNs synapses, the induction protocol elicited a robust LTD in the presence of quinpirole but not sulpiride (10 μM of quinpirole and sulpiride, n = 5 neurons from 5 mice per group; Wilcoxon test with W = −5130, p < 0.0001). Data are presented as median with shaded interquartile (quartile 1 to quartile 3).
To verify the D2R-dependence of the eCb-dependent LTD (Fig. 2e), two experiments were performed. First, PL axons were optogenetically stimulated and evoked EPSCs measured in d2SPNs held at −50 mV; bath application of the D2R agonist quinpirole (10 μM) produced a robust decrease in evoked EPSC amplitude, consistent with D2R-mediated eCb release and presynaptic CB1R activation (Fig. 2f). Next, PL axons were optogenetically stimulated while d2SPNs were episodically depolarized by somatic current injection to −50 mV[20] (Fig. 2g). In the presence of quinpirole (10 μM), this protocol induced a robust LTD (Fig. 2h). As expected, antagonizing D2Rs with sulpiride (10 μM) completely blocked LTD induction (Fig. 2h). These results establish a clear parallel in the mechanisms controlling synaptic plasticity at PL synapses on cNAc d2SPNs and those described at corticostriatal synapses on d2SPNs in dorsal striatum.
There is compelling evidence that peripheral injury suppresses the activity of a subpopulation of VTA dopaminergic neurons, leading to a suppression of DA release in NAc[18]. Indeed, loss of D2R signaling is a ready explanation for the strengthening of PL synapses on d2SPNs. Adding further weight to this conclusion, the frequency of miniature EPSCs in cNAc d2SPNs, but not d1SPNs, was significantly elevated in SNI mice (Fig. S1). Again, this is consistent with the loss of a tonic inhibitory presynaptic modulation of PL synapses by CB1Rs activated by eCb production in d2SPNs.
SNI also has been reported to selectively increase the proportion of GluA1 subunits in AMPARs at cNAc synapses, leading to increases Ca2+ permeability[17]. Indeed, bath application of the GluA1-specific AMPAR antagonist NASPM (200 μM), more potently reduced the amplitude of EPSCs at PL-PFC synapses on d2SPNs from SNI mice than controls (Fig. S2).
Taken together, our results point to a coordinated modulation of PL synapses on d2SPNs following SNI that is attributable to hypodopaminergic signaling, paralleling galanin-mediated changes in BLA synapses[36].
3.2. Chemogenetic activation of cNAc d2SPNs attenuated SNI-induced anxiety and social interaction deficits
A fundamental question is whether the alteration in synaptic connectivity is adaptive or maladaptive. Previous optogenetic studies have shown that stimulating PL inputs to cNAc alleviates the anxiety produced by SNI in mice[24]. Thus, potentiation of PL synapses on cNAc d2SPNs could in principle have the same effect. On the other hand, it is widely believed that hypodopaminergic signaling drives the chronic pain state[34]. As a first step toward testing the hypothesis that enhancing the activity of d2SPNs is adaptive, chemogenetic approaches were used[39]. An AAV carrying Cre-dependent PSAM-5HT3 HC-green fluorescent protein (PSAM-5HT3) expression construct was bilaterally injected into the cNAc of Adora2a-Cre mice (Fig. 3a). When activated, this 5-HT3 derived receptor produces a depolarizing, cation current[39]. In ex vivo brain slices taken from mice 4 weeks after injection, the PSAM ligand - PSEM89S - induced a reversible depolarization and spiking of d2SPNs (Fig. 3b–c). In PSAM-5HT3 expressing mice, the SNI procedure was performed; five days after SNI, mice received an intraperitoneal injection of PSEM89S (30 mg/kg) or saline (10 ml/kg) (Fig. 3d). Activating cNAc d2SPNs with PSEM89S did not alter mechanical allodynia (Fig. 3e). However, PSEM89S treatment did significantly diminish SNI-induced anxiety as manifested in open-field assays. Specifically, SNI mice spent less time in the center of the open field than did control mice (Fig. 3f–g). Increasing the excitability of cNAc d2SPNs with PSAM treatment normalized this behavior (Fig. 3f–g). This was not a consequence of any generalized effect on locomotion (Fig. S3). To complement this assay, mice were subjected to a social recognition test. As previously reported[34], SNI diminished the time a male mouse spent interacting with a female mouse compared to an object (Fig. 3h,i). As with the open field test, PSAM-mediated excitation of cNAc d2SPNs reversed this deficit (Fig. 3h,i). Thus, directly enhancing the excitability of cNAc d2SPNs had behavioral effect similar to those produced by directly stimulating PL inputs to them.
Figure 3. Chemogenetically activating cNAc d2SPNs relieved anxiety and social recognition deficits in SNI mice.
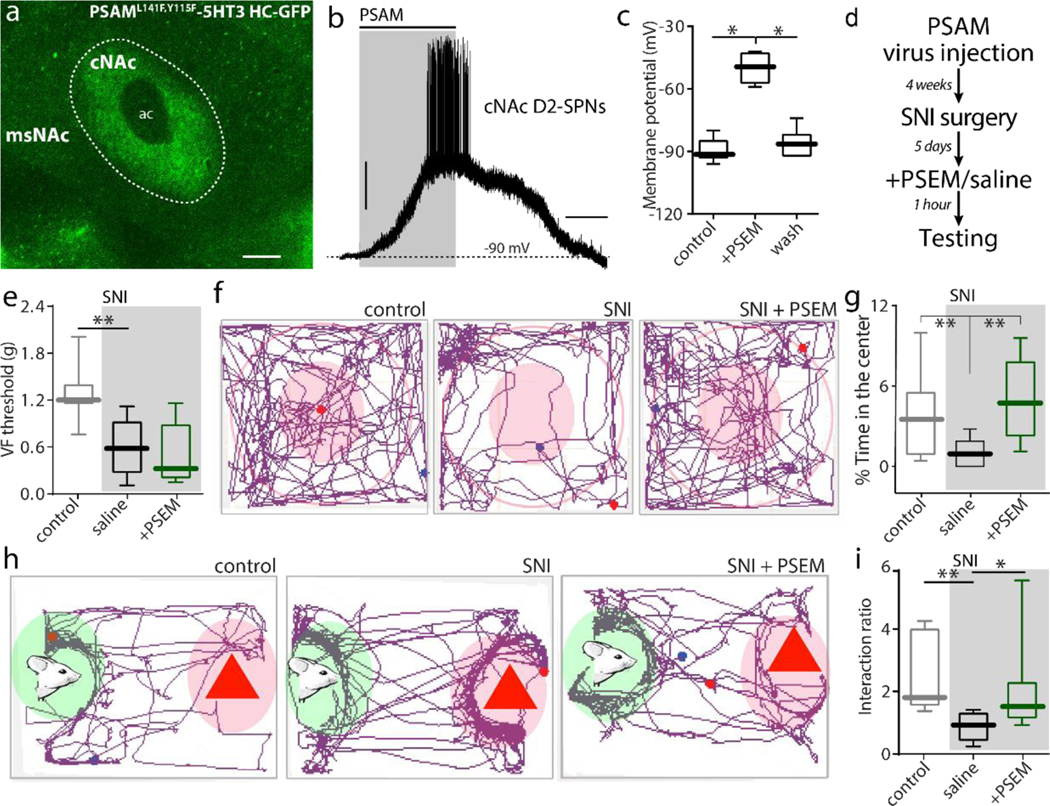
a, In AAV-PSAML141F,Y115F-5HT3 HC-GFP transduced A2A::Cre mice, GFP fluorescence was exclusively expressed in cNAc. b-c, PSEM89S induced firing in PSAM-5HT3-GFP expressing cNAc d2SPNs from A2a-Cre mice (n = 7 neurons from 5 mice per group in c, Wilcoxon test with W = 28, p = 0.0156 for PRE versus SPEM and W = 28, p = 0.0156 for PSEM versus WASH). d, The timeline for behavioral tests in the PSAM-5HT3 infected mice (n = 7 in Sham, n = 5–6 in Saline-SNI, n = 8 in PSEM-SNI; Mann–Whitney U test for data statistics). e, Activating cNAc d2SPNs by i.p. injection of PSEM89S had no effect on SNI-induced tactile allodynia (U = 1, p = 0.0051 for Sham versus Saline-SNI; U = 15.50, p = 0.5462 for Saline-SNI versus PSEM-SNI).f-g, In open field test, PSAM-5HT3 expressing SNI mice receiving PSEM89S treatment spent more time in the center zone of open field than those receiving saline treatment (U = 2, p = 0.0062 for Sham versus Saline-SNI; U = 2, p = 0.0062 for Saline-SNI versus PSEM-SNI). h-i, In social recognition task, PSEM89S treatment abolished the deficits in social recognition associated with SNI (n = 7 in Sham, n = 6 in Saline-SNI, n = 8 in PSEM-SNI; U = 1, p = 0.0023 for Sham versus Saline-SNI; U = 6, p = 0.02 for Saline-SNI versus PSEM-SNI). Data are presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
3.3. Chemogenetic suppression of cNAc projecting VTA dopaminergic neurons mimicked d2SPN activation
The SNI-induced changes seen in d2SPNs were attributable in large measure to a suppression in the activity of VTA dopaminergic neurons innervating them. Many lines of evidence, including electrophysiological studies, suggest that aversive stimuli suppress the activity of a sub-population of lateral VTA dopaminergic neurons[18; 48]. VTA neurons in this region preferentially innervate the cNAc[21].
To test the hypothesis that suppressing the activity of these cNAc-projecting VTA neurons would reproduce the behavioral effects of exciting cNAc d2SPNs, an intersectional chemogenetic approach was used. First, a non-toxic rabies virus carrying a Cre recombinase expression construct (RV–Cre) was injected into the cNAc to infect VTA neurons that innervated the cNAc (Fig. 4a). Next, an AAV carrying a Cre-dependent PSAML141F,Y115F-glycine receptors-GFP construct (PSAM-GlyR) was injected into the VTA of wild-type or D2-eGFP mice; this intersectional strategy restricted expression of the inhibitory PSAM to just those VTA neurons innervating cNAc. Indeed, the eGFP reporter in these mice was restricted to those VTA regions previous anatomical studies had shown to innervate the cNAc[21]. In ex vivo brain slices from infected mice, PSAM-GlyR ligand -PSEM89S- reversibly reduced the excitability of infected VTA neurons, verifying functional expression of the PSAM (Fig. 4b–c). Mice were then subjected to SNI surgery and PSEM89S was administered by intraperitoneal injection (30 mg/kg, twice a day from SNI Day 1 to Day 5). Chemogenetic inhibition of VTA neurons innervating cNAc had similar behavioral effect as increasing the excitability of cNAc d2SPNs (Fig. 4d). That is, administration of PSEM89S reversed the social recognition deficits produced by SNI (Fig. 4e), but had no effect on SNI-induced tactile allodynia (Fig. S4a).
Figure 4. VTA neurons innervating cNAc and msNAc differentially affected pain-associated behaviors in SNI mice.

a, RV-Cre was stereotaxically injected into cNAc and AAV-DIO- PSAM-GlyR constructs were injected into the VTA respectively. Representative image demonstrating 4-week expression of PSAM-GlyR-GFP in lateral VTA (l-VTA)-cNAc neurons (GFP expression). Robust PSAM-GlyR-GFP expression was routinely detected in the VTA, but no attempt was made to rigorously estimate the percentage of the neurons labeled. b-c, PSEM89S (The activator of PSAM) lowered spontaneous firing in these PSAM-GlyR expressing VTA neurons (n = 12 neurons from 6 mice, Wilcoxon test with W = 12, p = 0.0024). d-e, Diagram showing chemogenetic inhibition of VTA neurons innervating cNAc subsequently elevated the excitability of cNAc d2SPNs (d), and administration of PSEM89S in these virus-infected mice normalized the SNI-induced social recognition deficits (n = 6 in control group, n = 6 in PSEM group; U = 4, p = 0.026). f, By the similar virus strategy (msNAc with RV-Cre injection and VTA with PSAM-5HT3 injection), the PSAM-5HT3-GFP neurons are detected in medial VTA (m-VTA). g-h, In the slices from SPAM-5HT3 infected mice, PSEM increased the excitability of VTA-msNAc neurons (GFP positive neurons, n = 11 neurons from 6 mice, Wilcoxon test with W = 66, p = 0.00100). i-j, In SNI mice, chemogenetic activation of VTA neurons projecting to msNAc reversed tactile allodynia (n = 5 in control group, n = 6 in PSEM group; U = 0, p = 0.0043). Data are analyzed by Mann–Whitney U test and presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
As other studies suggest that aversive stimuli excite, rather than inhibit, VTA dopaminergic neurons projecting to the msNAc[11], the impact of chemogenetically manipulating this population of neurons was examined next. A similar intersectional genomics strategy was used to accomplish this goal. RV-Cre was stereotaxically injected into the msNAc and then an AAV carrying the Cre-dependent excitatory PSAM (5-HT3R-GFP) was injected into the VTA (Fig. 4f). In the slices from virus infected mice, PSEM excited GFP-positive VTA neurons (Fig. 4g–h), attesting to the expression of the PSEM. Intraperitoneal injections of PSEM (30 mg/kg, twice a day from SNI Day 1 to Day 5) reversed SNI-induced tactile allodynia (Fig. 4i,j), but left performance in the social recognition task unaffected (Fig. S4b). These results are consistent with our previous work showing that chemogenetic inhibition of msNAc d2SPNs selectively relieved SNI-induced tactile allodynia in mice[34].
3.4. cNAc and VTA neurons manifested compensatory homeostatic changes following SNI
Sustained perturbations in the activity of neurons has been shown to engage both synaptic and intrinsic mechanisms that serve to restore activity to a cell-specific set-point[43]. For example, dopamine-depleting lesions of the dorsal striatum trigger both intrinsic and synaptic forms of homeostatic plasticity in SPNs[15]. In particular, the intrinsic excitability of d2SPNs falls. If there was a sustained reduction in the activity of VTA dopaminergic neurons projecting to the cNAc and a reduction in DA release that dis-inhibited d2SPNs, then there should be a homeostatic reduction in their intrinsic excitability. To test this possibility, the intrinsic excitability and somatodendritic anatomy of d2SPNs and d1SPNs was examined in ex vivo brain slices from Xmas mice that had undergone SNI (or sham) surgery 5 days prior to sacrifice. In each brain slice, both a d2SPN and a d1SPN was patched to help control for animal-to-animal variability. Unlike the situation in msNAc d2SPNs[34], the intrinsic excitability of cNAc d2SPNs was significantly lower after SNI. Following SNI, d2SPNs required more current to evoke a given level of spiking (Fig. 5a,b). Moreover, first spike latency at rheobase was greater following SNI (Fig. 5c–d). Antagonizing D2Rs with sulpiride (10 μM) had no effect on the measures of excitability, demonstrating that the effects of SNI were not mediated by a change in dopaminergic signaling in the ex vivo preparation (data not shown). In contrast to d2SPNs, the excitability of neighboring d1SPNs was unchanged by SNI (Fig. 5e–h).
Figure 5. SNI selectively decreased the intrinsic excitability of cNAc d2SPNs.

a, Sample recordings from Sham and SNI slices (calibration bars: 20 mV, 200 ms). 5 days after Sham or SNI surgery, acute slices of cNAc were collected and input/output responses of intrinsic d2SPN excitability were obtained with depolarizing current injections (current injections: −40, 60, 120 and 160 pA). b-d, In SNI slices, intrinsic d2SPN excitability was reduced (n = 10–11 neurons from 5 mice per group; b, U = 12717, p = 0.0008) and accompanied by a longer first spike latency (c, U = 139, p < 0.0001) and elevated rheobase current (d, U = 18, p = 0.0065) in cNAc d2SPNs from SNI. e-h, the excitability of cNAc d1SPNs (input/output curve, first-spike latency and rheobase current) was unchanged by SNI (each group n=10–11 neurons from 5 mice; current injection for e : −40, 100, 220 and 260 pA; U = 15534, p = 0.6657 for f, U = 484, p = 0.8831 for g, U = 44.5, p = 0.4714 for h). Data for b,c,f,g are shown as median with shaded interquartile (quartile 1 to quartile 3); data for d,h are presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data. Data are analyzed by Mann–Whitney U test.
To determine if there were morphological changes accompanying the elevation in intrinsic excitability, cNAc SPNs were filled with biocytin and then reconstructed using standard methods [15] (Fig. S5). Dendritic branching, the total dendritic length and spine density of cNAc d2SPNs, as well as d1SPNs, were unchanged by SNI (Fig. 6; Fig. S6). The lack of a significant change in dendritic morphology suggests that the increase in PL connectivity is balanced by the decrease in BLA connectivity.
Figure 6. SNI had no effect on the morphological properties.

a: Representative reconstructed d2SPNs from sham and SNI groups. b, A three-dimensional Sholl analysis of reconstructed cNAc d2SPNs showed that the dendritic complexity of cNAc d2SPNs was unchanged by SNI (n = 8 neurons from 5 mice in each group; Mann–Whitney U test with U = 13647, p = 0.6000). c-e, SNI had no effect on the total dendritic length or average spine density of cNAc d2SPNs (n=8 neurons from 5 mice in each group with Mann–Whitney U test; U = 28, p = 0.7023 for c and U = 32, p > 0.9999 for e). Data for b are shown as median with shaded interquartile (quartile 1 to quartile 3); whisker box plots show median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
The alterations in intrinsic excitability of d2SPNs could have been produced by a sustained reduction in dopaminergic signaling in the cNAc following SNI. This conclusion is consistent with a number of other studies pointing to hypodopaminergic signaling following SNI[18]. If this were the case, VTA dopaminergic neurons should manifest homeostatic changes in their intrinsic excitability, just as did d2SPNs. VTA dopaminergic neurons are slow autonomous pacemakers[27], making an assessment of their intrinsic excitability straightforward in ex vivo brain slices bathed in blockers of ionotropic receptors for glutamate and GABA. To this end, RV-Cre was injected into the cNAc or the msNAc of Ai14 reporter mice in which a floxed tdTomato expression construct has been inserted into the Rosa locus[26]. One week after injection of RV-Cre into the cNAc, tdTomato expression was found almost exclusively in the lateral VTA, as expected[21] (Fig. 7a–c). Having validated this labeling approach, mice were subjected to SNI or sham surgery and then sacrificed 5 days later. Visually identified VTA neurons in ex vivo brain slices were recorded from using perforated patch methods in the presence of ionotropic receptor blockers. In VTA neurons projecting to the cNAc, the autonomous spiking rate was elevated in slices from SNI mice (Fig. 7d,e), consistent with the proposition that these neurons had been chronically inhibited in vivo. Injection of RV-Cre into the msNAc resulted in td-Tomato expression primarily in the medial VTA[21] (Fig. 7f–h). In contrast to lateral VTA neurons, in VTA neurons projecting to the msNAc, the autonomous spiking rate was depressed in slices from SNI mice (Fig. 7i,j), consistent with the proposition that these medial VTA neurons had been chronically excited in vivo.
Figure 7. SNI differentially modulated VTA dopaminergic innervation of cNAc and msNAc.

a-c, After the injection of RV-Cre into the cNAc of an Ai14-tdTomato-flox mouse, tdTomato signal was restricted to the cNAc (a) and retrogradely labeling was detected in the lateral VTA (lVTA, b-c). Robust tdTomato expression was routinely detected in the VTA, but no attempt was made to rigorously estimate the percentage of the neurons labeled. d-e, The intrinsic neuronal excitability of VTA-DA neurons innervating the cNAc was upregulated following SNI (n = 17 neurons from 6 mice per group, U = 76, p = 0.0173). d, Sample traces of VTA neurons innervating cNAc recorded from Sham and SNI slices. f-h, The VTA neurons projecting to NAc medial shell were dominantly located in medial VTA (mVTA) of the rostral midbrain. i-j, Medial VTA neurons projecting to msNAc from SNI mice had slower firing than that from Sham mice (n = 15 neurons from 6 mice per group, U = 61, p = 0.0319). ml: medial lemniscus; PBP: parabrachial pigmented nucleus of the VTA; SNr: substantia nigra, reticular part; VS: ventral subiculum; VTA: ventral tegmental area. Data are analyzed by Mann–Whitney U test and presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
4. Discussion
There is a growing literature implicating the mesoaccumbal circuitry in the response to peripheral nerve injury and the establishment of a chronic pain state[29; 41; 47]. Our studies provide new insight into these mechanisms, suggesting that persistent SNI-induced suppression of dopaminergic signaling to the cNAc is adaptive. There are three observations in the SNI mouse model of chronic pain that support this conclusion. First, SNI induced a remodeling of the functional connectome of cNAc d2SPNs, increasing their synaptic coupling with PL neurons and decreasing their coupling with BLA neurons. As PL stimulation in SNI models alleviates pain symptoms[24] and blocking BLA activity decreases neuropathic pain [37], this shift in connectivity should be adaptive. Second, chemogenetically enhancing the excitability of cNAc d2SPNs alleviated the anxiety and social interaction deficits accompanying SNI, demonstrating the potential adaptive value of enhanced excitatory PL connectivity with d2SPNs and decreased stimulation of D2Rs that suppress intrinsic excitability[16]. Third, chemogenetically suppressing the excitability of VTA dopaminergic neurons projecting to the cNAc in SNI mice mimicked the beneficial effects of increasing the excitability of cNAc d2SPNs on anxiety and social interactions. In contrast, enhancing the excitability of VTA dopaminergic neurons projecting to the msNAc diminished SNI-induced tactile allodynia, but did not alter anxiety, mimicking the effects of suppressing the excitability of msNAc d2SPNs[34]. Taken together, these results suggest that in the days following peripheral nerve injury, regionally specific alterations in the activity of VTA dopaminergic neurons trigger adaptive shifts in cNAc and msNAc circuitry that promote re-engagement with the environment by lessening both allodynia and anxiety. However, alterations in the intrinsic excitability of NAc and VTA neurons, both of which appear to be driven by homeostatic mechanisms, may compromise the ability to sustain these adaptive alterations in network activity, and in so doing, could promote the chronic pain state.
VTA DA neurons are heterogeneous in their anatomy and in their response to appetitive and aversive stimuli[11; 48]. Specifically, aversive stimuli inhibit parabrachial VTA neurons that project to the cNAc and excite paranigral neurons projecting to the msNAc[11; 22]. Our studies are consistent with this body of work if interpreted through the lens of cellular homeostatic plasticity. Wherever it has been closely examined, neurons manifest compensatory mechanisms aimed at restoring spike rate to a ‘set-point’[43]. Neurons can change the weighting of synaptic inputs or their intrinsic excitability to achieve this end. The inferred goal of this form of plasticity is to ensure the global stability of network activity in the face of perturbation. In our ex vivo work, the autonomous spike rate of cNAc-projecting, VTA DA neurons from SNI mice was elevated, as would be predicted by engagement of a homeostatic correction for sustained inhibition in vivo. Conversely, the autonomous spike rate of msNAc-projecting, VTA DA neurons was depressed, as one would expect if they were tonically excited in vivo. In addition, the intrinsic excitability of cNAc d2SPNs was depressed in slices from SNI mice, just as expected if they had been tonically dis-inhibited by reduced DA release[15]. In msNAc d2SPNs, SNI induces precisely the opposite intrinsic compensation[34] – as predicted.
SNI also triggered an alteration in the functional connectome of d2SPNs. At PL glutamatergic synapses on d2SPNs, but not those on d1SPNs, the AMPAR/NMDAR ratio was significantly increased following SNI, indicating that they had undergone a form of LTP involving postsynaptic AMPAR trafficking into the synapse[7]. This change can be attributed to a shift in DA signaling. DA not only controls the intrinsic excitability of NAc SPNs, it also modulates synaptic plasticity[40]. At PL synapses on cNAc d2SPNs, D2R signaling readily induced an eCb-dependent form of LTD, as in the dorsal striatum[20]. Previous work employing electrical stimulation of PL inputs to cNAc SPNs is consistent with this finding[4; 32; 46]. In the dorsal striatum, D2R signaling not only promotes eCb-LTD, it also suppresses metabotropic A2a adenosine receptor (A2aR) stimulation of adenylyl cyclase and protein kinase A (PKA), which drive a postsynaptic form LTP at cortical glutamatergic synapses[38]. Thus, the loss of D2R signaling in the cNAc following SNI should bias synaptic plasticity at PL synapses toward LTP.
In contrast, the AMPA/NMDA ratio at BLA glutamatergic synapses on d2SPNs decreased after SNI, suggesting they had undergone a form of postsynaptic LTD. Unlike PL synapses, BLA synapses were insensitive to eCB signaling[14]. Previous work by Schwartz et al. found that galanin receptor mediated signaling induced LTD at an unidentified glutamatergic synapse on d2SPNs following peripheral injury. Given that BLA neurons express galanin[19], it may be that the plasticity they observed was at these synapses.
It is widely held that peripheral nerve injury suppresses dopaminergic signaling in the NAc, triggering maladaptive network activity that underlies the chronic pain state[18]. While this model has plausibility, there are several reasons to question it. First, this model fails to take into account the heterogeneity in response properties of VTA dopaminergic neurons[30]. Our data suggest that peripheral nerve injury triggers region-specific alteration in dopaminergic signaling. In agreement with previous studies [11], our work suggests that SNI suppresses VTA dopaminergic neurons projecting to the cNAc and excites those projecting to the msNAc. Second, this prevailing model assumes that d2- and d1SPNs act in a ‘push-pull’ manner, such that the activity of d2SPNs simply decreases motivation, positive affect and vigor. To the contrary, in vivo recording in the dorsal striatum argues that d2- and d1SPNs work in concert [9]. There is no reason to think the situation is any different in the ventral striatum.
From this perspective, the remodeling of the functional connectome of d2SPNs takes on a different significance. The consequences of changing the strength of specific synaptic connections cannot be determined by simple algebra (that is, the sum of changes in input strength). Rather, this remodeling must shift what information drives d2SPNs spiking, how this activity is coordinated with other brain regions and how it affects behavior. SNI strengthened PL inputs to d2SPNs and weakened BLA inputs. The activity of PL pyramidal neurons is commonly linked to goal-directed behavior and executive function[33]. In line with this literature, optogenetic stimulation of PL promotes interactive behavior following SNI [24]. In contrast, the activity of many BLA neurons is linked to fear and anxiety [3; 23]. Thus, the synaptic remodeling seen here should bias the activity of d2SPNs away from fear and anxiety and toward interactive, goal-directed actions. In this regard, it is important to note that chemogenetic approaches, in contrast to optogenetic or other pharmacological approaches, allow naturally occurring network activity to control spiking in targeted neuronal populations. Thus, chemogenetically enhancing the excitability of d2SPNs should simply amplify the effects of altering the connectome and bring about a stronger correlation with PL activity – resulting in diminished anxiety and more interactive behavior.
In humans, enhanced connectivity between the prefrontal cortices and the NAc is correlated with chronic pain [1; 2]. What is not clear is whether the enhanced connectivity promotes the chronic pain state or whether it is an attempt to ameliorate it. Our data suggest the latter (Fig. 8). That said, there are obvious caveats attached to this conclusion. One is that our studies have focused on the initial (5 days post SNI) response to peripheral nerve injury. It will be critical to determine whether the cellular and network adaptations persist. Another caveat is that it is difficult with current human imaging approaches to readily distinguish between connections with the cNAc and the adjacent msNAc. In mice, SNI reduces the strength of synaptic connections between IL pyramidal neurons and msNAc d2SPNs [34]. Precisely mapping the functional connectomes between these regions in humans will require more advanced imaging strategies.
Figure 8. Schematic summarizing the adaptive synaptic and behavioral changes after pain induction.

Sustained aversive stimulation by SNI injury elevated the dopamine signaling in msNAc whereas decreased the dopamine in cNAc. This alterations of dopamine signaling in NAc appears to be adaptive – lessening the negative affect and enhancing the suppression of escape behavior, implying that cellular ‘homeostatic’ plasticity in NAc that comes with SNI is maladaptive at the network level, providing a novel therapeutic target for chronic pain.
If the persistent changes in VTA dopaminergic signaling and changes in the functional connectome of NAc d2SPNs are adaptive, then why do mice continue to manifest behavioral deficits following SNI? One possibility is that cellular homeostatic mechanisms reduce the effectiveness of the changes in network function. In both d2SPNs and in cNAc-projecting VTA dopaminergic neurons, there were significant shifts in baseline excitability that were opposite in sign from the change in activity inferred to be present in vivo. In cNAc d2SPNs, intrinsic excitability fell following SNI, in agreement with the proposition that these neurons had been dis-inhibited by falling D2R signaling in vivo. Conversely, basal spiking in cNAc projecting VTA dopaminergic neurons rose, in agreement with the hypothesis that they were tonically inhibited by nociceptive signaling in vivo. Precisely the opposite set of adaptations was seen in msNAc d2SPNs and msNAc-projecting VTA dopaminergic neurons following SNI. These homeostatic changes would appear to be counter-productive and lessen the impact of adaptive changes in network activity (Fig. 8). One way to test this hypothesis would be to blunt homeostatic plasticity by inhibiting Cav1 (L-type) Ca2+ channels [10].
Supplementary Material
Figure S1. SNI increased synaptic input in cNAc d2SPNs but had no effect on cNAc d1SPNs. a-c, In SNI, the d2SPN mEPSC frequency was augmented without detectable change in the amplitude of d2SPN mEPSC (n= 6 neurons from 6 mice per group; Kolmogorov-Smirnov test for representative cumulative probability plots with D = 0.5000, P = 0.0366 for b and D = 0.2632, p = 0.1439 for c; Mann-Whitney U test for box plots with U = 3, P = 0.0152 for b and U = 18, p > 0.9999 for c). d-f, there is no difference in mEPSCs frequency or amplitude of d1SPNs between SNI and Sham animals (n = 8 from 6 mice in each group, Kolmogorov-Smirnov test for representative cumulative probability plots with D =0.1462, p = 0.9891 for e and D =0.1600, p = 0.2923 for f; Mann–Whitney test for box plots with U = 20, p = 0.2317 for e and U = 29, p = 0.7768 for f). Whisker box plots are displayed as median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
Figure S2. SNI increased the proportion of calcium-permeable AMPARs at PL-cNAc synapses. a-c, Calcium-permeable AMPAR antagonist Naspm decreased AMPAR EPSC amplitudes in sham and SNI mice (n = 7 neurons from 5 mice per group; Wilcoxon test with W = −28, p = 0.0156 for b W = −28, p = 0.0156 for c). d, Naspm had bigger effect on the d2SPNs from SNI mice than those from sham mice (Mann–Whitney U test with U = 7, p = 0.0262). e-f: Naspm reversed enhanced A/N ratio at PL-cNAc d2SPNs synapse in SNI mice (n = 6 neurons from 5 mice per group; Mann–Whitney U test with U = 5, p = 0.0411; calibration: 50 pA, 100 ms). Data are presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
Figure S3. In open field test, chemogenetic activation of cNAc d2SPNs in PSAM-5HT3 expressing SNI mice by PSEM89S treatment unchanged locomotor activities (U = 17, p = 0.9773 for Sham versus Saline-SNI; U = 19, p = 0.9433 for Saline-SNI versus PSEM-SNI). Data are reported as median, first and third quartiles (box plots), and minimum and maximum of data set (whiskers), and are analyzed by Mann–Whitney test.
Figure S4. a, The Chemogenetic inhibition of cNAc projection VTA neurons by intraperitoneal injections of PSEM in RV-Cre (in cNAc) and AAV-PSAM-GlyR (VTA) viruses infected mice had no effect on SNI-induced tactile allodynia (n = 6 for each group; U = 15.50, p = 0.7359). b, activation msNAc projecting VTA neurons by the same virus strategy unchanged the social recognition deficits in SNI mice (n = 5 in control and n = 6 in PSEM; U = 20, p = 0.2159). Data are reported as median, first and third quartiles (box plots), and minimum and maximum of data set (whiskers), and are analyzed by Mann–Whitney test.
Figure S5. Biocytin visualized the SPNs’ dendritic morphology in the cNAc slices from Christmas (Xmas) transgenic mice. a, The d2SPNs (eGFP-positive, green) and d1SPNs (Td tomato-positive, red) are identified in the same cNAc slice taken from BAC transgenic mouse (Calibration: 20 μm). b, The biocytin-filled neuron overlapped with populations of d2SPNs. c, Another biocytin-filled neuron overlapped with populations of d1SPNs.
Figure S6. Morphological analysis in reconstructed d1SPNs from SNI and Sham animals. a-c, For d1SPNs, no significant difference in Sholl analysis or total tree length was detected between SNI and Sham (n = 8 from 5 mice in each group, U = 13639 and p = 0.5946 for b and U = 31 and p = 0.9319 for c, Mann–Whitney test). d: Representative images of d1SPN dendrites/spines from SNI/Sham animals. e, Spine density of d1SPNs were similar between SNI and Sham (n = 8 from 5 mice in each group, Mann–Whitney test with U = 21, p = 0.2747). Data for b are shown as median with shaded interquartile (quartile 1 to quartile 3); whisker box plots are displayed as median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
Acknowledgements:
This work was supported by NIH NIDA grant P50DA044121 (VA).
Footnotes
Competing financial interest:
The authors declare no competing financial interests.
REFERENCES
- [1].Baliki MN, Geha PY, Fields HL, Apkarian AV. Predicting value of pain and analgesia: nucleus accumbens response to noxious stimuli changes in the presence of chronic pain. Neuron 2010;66(1):149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Baliki MN, Petre B, Torbey S, Herrmann KM, Huang L, Schnitzer TJ, Fields HL, Apkarian AV. Corticostriatal functional connectivity predicts transition to chronic back pain. Nature neuroscience 2012;15(8):1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Beyeler A, Namburi P, Glober GF, Simonnet C, Calhoon GG, Conyers GF, Luck R, Wildes CP, Tye KM. Divergent Routing of Positive and Negative Information from the Amygdala during Memory Retrieval. Neuron 2016;90(2):348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Brady AM, O’Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. The Journal of neuroscience : the official journal of the Society for Neuroscience 2004;24(5):1040–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bromberg-Martin ES, Matsumoto M, Hikosaka O. Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 2010;68(5):815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 1994;53(1):55–63. [DOI] [PubMed] [Google Scholar]
- [7].Chater TE, Goda Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci 2014;8:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai HC, Pomeranz L, Christoffel DJ, Nectow AR, Ekstrand M, Domingos A, Mazei-Robison MS, Mouzon E, Lobo MK, Neve RL, Friedman JM, Russo SJ, Deisseroth K, Nestler EJ, Han MH. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature 2013;493(7433):532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Costa RM. Plastic corticostriatal circuits for action learning: what’s dopamine got to do with it? Annals of the New York Academy of Sciences 2007;1104:172–191. [DOI] [PubMed] [Google Scholar]
- [10].Day M, Wang Z, Ding J, An X, Ingham CA, Shering AF, Wokosin D, Ilijic E, Sun Z, Sampson AR, Mugnaini E, Deutch AY, Sesack SR, Arbuthnott GW, Surmeier DJ. Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nature neuroscience 2006;9(2):251–259. [DOI] [PubMed] [Google Scholar]
- [11].de Jong JW, Afjei SA, Pollak Dorocic I, Peck JR, Liu C, Kim CK, Tian L, Deisseroth K, Lammel S. A Neural Circuit Mechanism for Encoding Aversive Stimuli in the Mesolimbic Dopamine System. Neuron 2019;101(1):133–151 e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000;87(2):149–158. [DOI] [PubMed] [Google Scholar]
- [13].Deroche MA, Lassalle O, Castell L, Valjent E, Manzoni OJ. Cell-Type- and Endocannabinoid-Specific Synapse Connectivity in the Adult Nucleus Accumbens Core. The Journal of neuroscience : the official journal of the Society for Neuroscience 2020;40(5):1028–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ding J, Peterson JD, Surmeier DJ. Corticostriatal and thalamostriatal synapses have distinctive properties. The Journal of neuroscience : the official journal of the Society for Neuroscience 2008;28(25):6483–6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fieblinger T, Graves SM, Sebel LE, Alcacer C, Plotkin JL, Gertler TS, Chan CS, Heiman M, Greengard P, Cenci MA, Surmeier DJ. Cell type-specific plasticity of striatal projection neurons in parkinsonism and L-DOPA-induced dyskinesia. Nat Commun 2014;5:5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 2011;34:441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Goffer Y, Xu D, Eberle SE, D’Amour J, Lee M, Tukey D, Froemke RC, Ziff EB, Wang J. Calcium-permeable AMPA receptors in the nucleus accumbens regulate depression-like behaviors in the chronic neuropathic pain state. The Journal of neuroscience : the official journal of the Society for Neuroscience 2013;33(48):19034–19044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Huang S, Borgland SL, Zamponi GW. Peripheral nerve injury-induced alterations in VTA neuron firing properties. Mol Brain 2019;12(1):89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kawa L, Barde S, Arborelius UP, Theodorsson E, Agoston D, Risling M, Hokfelt T. Expression of galanin and its receptors are perturbed in a rodent model of mild, blast-induced traumatic brain injury. Experimental neurology 2016;279:159–167. [DOI] [PubMed] [Google Scholar]
- [20].Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. The Journal of neuroscience : the official journal of the Society for Neuroscience 2005;25(45):10537–10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology 2014;76 Pt B:351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. Input-specific control of reward and aversion in the ventral tegmental area. Nature 2012;491(7423):212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee H, Kim JJ. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 1998;18(20):8444–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee M, Manders TR, Eberle SE, Su C, D’Amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, Wang J. Activation of corticostriatal circuitry relieves chronic neuropathic pain. The Journal of neuroscience : the official journal of the Society for Neuroscience 2015;35(13):5247–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Luscher C. The Emergence of a Circuit Model for Addiction. Annual Review of Neuroscience, Vol 39 2016;39:257–276. [DOI] [PubMed] [Google Scholar]
- [26].Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature neuroscience 2010;13(1):133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol 2006;577(Pt 3):907–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller LR, Cano A. Comorbid Chronic Pain and Depression: Who Is at Risk? Journal of Pain 2009;10(6):619–627. [DOI] [PubMed] [Google Scholar]
- [29].Mitsi V, Zachariou V. Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience 2016;338:81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Morales M, Margolis EB. Ventral tegmental area: cellular heterogeneity, connectivity and behaviour. Nature reviews Neuroscience 2017;18(2):73–85. [DOI] [PubMed] [Google Scholar]
- [31].Nicholson B, Verma S. Comorbidities in chronic neuropathic pain. Pain Medicine 2004;5:S9–S27. [DOI] [PubMed] [Google Scholar]
- [32].O’Donnell P, Grace AA. Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Res 1994;634(1):105–112. [DOI] [PubMed] [Google Scholar]
- [33].Pascoli V, Turiault M, Luscher C. Reversal of cocaine-evoked synaptic potentiation resets drug-induced adaptive behaviour. Nature 2011;481(7379):71–75. [DOI] [PubMed] [Google Scholar]
- [34].Ren W, Centeno MV, Berger S, Wu Y, Na X, Liu X, Kondapalli J, Apkarian AV, Martina M, Surmeier DJ. The indirect pathway of the nucleus accumbens shell amplifies neuropathic pain. Nature neuroscience 2016;19(2):220–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schultz W. Multiple dopamine functions at different time courses. Annu Rev Neurosci 2007;30:259–288. [DOI] [PubMed] [Google Scholar]
- [36].Schwartz N, Temkin P, Jurado S, Lim BK, Heifets BD, Polepalli JS, Malenka RC. Chronic pain. Decreased motivation during chronic pain requires long-term depression in the nucleus accumbens. Science 2014;345(6196):535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Seno MDJ, Assis DV, Gouveia F, Antunes GF, Kuroki M, Oliveira CC, Santos LCT, Pagano RL, Martinez RCR. The critical role of amygdala subnuclei in nociceptive and depressive-like behaviors in peripheral neuropathy. Sci Rep 2018;8(1):13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science 2008;321(5890):848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci 2014;37:387–407. [DOI] [PubMed] [Google Scholar]
- [40].Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends in neurosciences 2007;30(5):228–235. [DOI] [PubMed] [Google Scholar]
- [41].Taylor AM, Mehrabani S, Liu S, Taylor AJ, Cahill CM. Topography of microglial activation in sensory- and affect-related brain regions in chronic pain. J Neurosci Res 2017;95(6):1330–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Turk DC, Audette J, Levy RM, Mackey SC, Stanos S. Assessment and Treatment of Psychosocial Comorbidities in Patients With Neuropathic Pain. Mayo Clinic Proceedings 2010;85(3):S42–S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Turrigiano GG. Homeostatic plasticity in neuronal networks: the more things change, the more they stay the same. Trends Neurosci 1999;22(5):221–227. [DOI] [PubMed] [Google Scholar]
- [44].Vachon-Presseau E, Tetreault P, Petre B, Huang L, Berger SE, Torbey S, Baria AT, Mansour AR, Hashmi JA, Griffith JW, Comasco E, Schnitzer TJ, Baliki MN, Apkarian AV. Corticolimbic anatomical characteristics predetermine risk for chronic pain. Brain 2016;139(Pt 7):1958–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Volkow ND, McLellan AT. Mitigation Strategies for Opioid Abuse. N Engl J Med 2016;375(1):96. [DOI] [PubMed] [Google Scholar]
- [46].Wang W, Dever D, Lowe J, Storey GP, Bhansali A, Eck EK, Nitulescu I, Weimer J, Bamford NS. Regulation of prefrontal excitatory neurotransmission by dopamine in the nucleus accumbens core. The Journal of physiology 2012;590(16):3743–3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Watanabe M, Narita M. Brain Reward Circuit and Pain. Adv Exp Med Biol 2018;1099:201–210. [DOI] [PubMed] [Google Scholar]
- [48].Yang H, de Jong JW, Tak Y, Peck J, Bateup HS, Lammel S. Nucleus Accumbens Subnuclei Regulate Motivated Behavior via Direct Inhibition and Disinhibition of VTA Dopamine Subpopulations. Neuron 2018;97(2):434–449 e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Yang M, Scattoni ML, Zhodzishsky V, Chen T, Caldwell H, Young WS, McFarlane HG, Crawley JN. Social approach behaviors are similar on conventional versus reverse lighting cycles, and in replications across cohorts, in BTBR T+ tf/J, C57BL/6J, and vasopressin receptor 1B mutant mice. Front Behav Neurosci 2007;1:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang H, Qian YL, Li C, Liu D, Wang L, Wang XY, Liu MJ, Liu H, Zhang S, Guo XY, Yang JX, Ding HL, Koo JW, Mouzon E, Deisseroth K, Nestler EJ, Zachariou V, Han MH, Cao JL. Brain-Derived Neurotrophic Factor in the Mesolimbic Reward Circuitry Mediates Nociception in Chronic Neuropathic Pain. Biol Psychiatry 2017;82(8):608–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain 1983;16(2):109–110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. SNI increased synaptic input in cNAc d2SPNs but had no effect on cNAc d1SPNs. a-c, In SNI, the d2SPN mEPSC frequency was augmented without detectable change in the amplitude of d2SPN mEPSC (n= 6 neurons from 6 mice per group; Kolmogorov-Smirnov test for representative cumulative probability plots with D = 0.5000, P = 0.0366 for b and D = 0.2632, p = 0.1439 for c; Mann-Whitney U test for box plots with U = 3, P = 0.0152 for b and U = 18, p > 0.9999 for c). d-f, there is no difference in mEPSCs frequency or amplitude of d1SPNs between SNI and Sham animals (n = 8 from 6 mice in each group, Kolmogorov-Smirnov test for representative cumulative probability plots with D =0.1462, p = 0.9891 for e and D =0.1600, p = 0.2923 for f; Mann–Whitney test for box plots with U = 20, p = 0.2317 for e and U = 29, p = 0.7768 for f). Whisker box plots are displayed as median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
Figure S2. SNI increased the proportion of calcium-permeable AMPARs at PL-cNAc synapses. a-c, Calcium-permeable AMPAR antagonist Naspm decreased AMPAR EPSC amplitudes in sham and SNI mice (n = 7 neurons from 5 mice per group; Wilcoxon test with W = −28, p = 0.0156 for b W = −28, p = 0.0156 for c). d, Naspm had bigger effect on the d2SPNs from SNI mice than those from sham mice (Mann–Whitney U test with U = 7, p = 0.0262). e-f: Naspm reversed enhanced A/N ratio at PL-cNAc d2SPNs synapse in SNI mice (n = 6 neurons from 5 mice per group; Mann–Whitney U test with U = 5, p = 0.0411; calibration: 50 pA, 100 ms). Data are presented as whisker box plots displaying median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.
Figure S3. In open field test, chemogenetic activation of cNAc d2SPNs in PSAM-5HT3 expressing SNI mice by PSEM89S treatment unchanged locomotor activities (U = 17, p = 0.9773 for Sham versus Saline-SNI; U = 19, p = 0.9433 for Saline-SNI versus PSEM-SNI). Data are reported as median, first and third quartiles (box plots), and minimum and maximum of data set (whiskers), and are analyzed by Mann–Whitney test.
Figure S4. a, The Chemogenetic inhibition of cNAc projection VTA neurons by intraperitoneal injections of PSEM in RV-Cre (in cNAc) and AAV-PSAM-GlyR (VTA) viruses infected mice had no effect on SNI-induced tactile allodynia (n = 6 for each group; U = 15.50, p = 0.7359). b, activation msNAc projecting VTA neurons by the same virus strategy unchanged the social recognition deficits in SNI mice (n = 5 in control and n = 6 in PSEM; U = 20, p = 0.2159). Data are reported as median, first and third quartiles (box plots), and minimum and maximum of data set (whiskers), and are analyzed by Mann–Whitney test.
Figure S5. Biocytin visualized the SPNs’ dendritic morphology in the cNAc slices from Christmas (Xmas) transgenic mice. a, The d2SPNs (eGFP-positive, green) and d1SPNs (Td tomato-positive, red) are identified in the same cNAc slice taken from BAC transgenic mouse (Calibration: 20 μm). b, The biocytin-filled neuron overlapped with populations of d2SPNs. c, Another biocytin-filled neuron overlapped with populations of d1SPNs.
Figure S6. Morphological analysis in reconstructed d1SPNs from SNI and Sham animals. a-c, For d1SPNs, no significant difference in Sholl analysis or total tree length was detected between SNI and Sham (n = 8 from 5 mice in each group, U = 13639 and p = 0.5946 for b and U = 31 and p = 0.9319 for c, Mann–Whitney test). d: Representative images of d1SPN dendrites/spines from SNI/Sham animals. e, Spine density of d1SPNs were similar between SNI and Sham (n = 8 from 5 mice in each group, Mann–Whitney test with U = 21, p = 0.2747). Data for b are shown as median with shaded interquartile (quartile 1 to quartile 3); whisker box plots are displayed as median, lower and upper quartiles, and whiskers representing minimum and maximum of the data.