

Abstract
Second near infrared (NIR-II) window fluorescence imaging between 1000–1700 nm with reduced scattering and autofluorescence, and deep tissue light penetration allows early and non-invasive determination of vascular pathologies. Here, we demonstrate in vivo NIR-II imaging techniques for tracking hyperglycaemia-induced Intracerebral Hemorrhage (ICH) and Blood Brain Barrier (BBB) hyperpermeability in Cerebral Cavernous Malformation (CCM) deficient mice (CCM1+/−). We synthesised PEGylated Ag2S quantum dots (QDs) with a bright fluorescent emission peak centred at 1135 nm under 808-nm NIR light for dynamic imaging of cerebral vasculature in mice and determined the development of ICH and BBB impairment in hyperglycaemic CCM1+/− mice. In vivo optical imaging was verified with micro-CT (including k-mean cluster analysis) as well as in vivo permeability assays using FITC-dextran perfusion and IgG staining, respectively. The increased BBB permeability in CCM1+/− mice was further demonstrated to be associated with high-glucose-caused decrease of CCM1 expression. This study validates that deep-penetrating NIR-II QDs can be used for the tracking of ICH and BBB hyperpermeability in transgenic mice models of cerebral vascular anomalies.
Graphical Abstract
Introduction
In vivo fluorescence imaging in the second near infrared (NIR-II) window between 1000–1700 nm enables higher resolution deep tissue fluorescence imaging compared to the NIR-I window of 700–900 nm due to reduced scattering cross-sections and minimal autofluorescence from endogenous fluorophores in the NIR-II spectrum1–3. A recent study by Yaroslavsky et al.4 confirmed that human brain tissue allows for enhanced NIR-II light penetration in the 1000–1100 nm region. Current NIR fluorescent probes such as organic dyes and inorganic nano-constructs often have minimal or low quantum yield above 1000 nm and/or limited dispersibility in water 5; thus, limiting their use as fluorescent beacons for functional or molecular imaging. Among I-VI semiconductor nanomaterials, Ag2S quantum dots (QDs) have been established as narrow bandgap (~0.9–1.1 eV) materials. They are water dispersible owing to –COOH surface functionalization and possess excellent NIR-II emission properties6. Ag2S QDs with tunable emission bands in both NIR-I and NIR-II windows have been reported for use as exogenous fluorophores and can be synthesized using wet chemistry methods 2, 7. A recent review provided a comprehensive summary of the use of QDs for biomedical applications8.
Hongjie Dai and co-workers demonstrated that due to small size (sub-10nm) and bright NIR-II fluorescence under a lower excitation of light, these QDs can be used for rapid tumor detection, dynamic tumor contrast enhancement, and vascular imaging7. Recently, Awasthi et al.9 encapsulated QDs within dendrimers and demonstrated whole-body imaging in mice. QDs have been demonstrated for lymphatic monitoring and surgical guidance for lymph node dissection as well10. Sensitive non-invasive detection of cerebral vascular pathologies has been rarely reported. There is significant interest in developing research tools for the early detection of cerebral pathologies. Cui et al.11 used an NIR-I dye DANIR-2c excited at 597 nm and monitored the emission at 665 nm after intravenous injection to track the accumulation of β-amyloid plaques in mice brain, a hallmark of Alzheimer’s disease. An identical study was reported by Zhang et al.12 using CRANAD-3 NIR-I fluorophore for tracking Alzheimer’s disease. Despite these efforts made for tracking brain pathology with NIR imaging, in vivo detection of vascular pathology itself with whole-body NIR imaging is not well established; primarily due to the limitations on tissue penetration and spatial resolution imposed by red and NIR-I spectrum imaging. Indocyanine green (ICG), is an established NIR imaging agent due to its FDA-approved status and it has recently been reported for imaging in NIR-II window as well13, 14. However, the NIR-II emission spectra of ICG rapidly declines after 1000 nm, and although emission can be detected up to and exceeding 1300 nm, high excitation laser power densities > 50 mW/cm2 and/or longer sensor exposure times are needed for adequate imaging13. Further, ICG as a negatively charged species has been reported to bind to plasma proteins and serum albumin following injection, potentially resulting in a distribution of fluorescent ICG bound albumin particles in circulation with altered pharmacokinetics14. To track cerebral vascular pathologies including disrupted Blood Brain Barrier (BBB), it is ideal to have a non-interacting homogenous sized contrast agent which is highly soluble and exhibits good signal-to-background ratio in the NIR-II region. In this study, we report the optimization and use of NIR-II emitting QD-enabled dynamic imaging for detection of intracerebral hemorrhage (ICH) and BBB permeability induced by hyperglycemia in living CCM1 heterozygous mice.
Cerebral cavernous malformation (CCM) is a vascular pathology mostly occurring in the central nervous system characterized by single or multiple lumen formation and vascular leakage at the brain capillary level, as well as disruption of BBB15. CCM occurs in 1 out of every 200–600 people in the general population, and 10–50% cases are caused by dysfunctional CCM genes (CCM1/2/3) 15, 16. Functional loss of these genes increases the diameter of blood vessels and decreases their structural integrity due to impairment of endothelial cell (EC) junctions and pericyte recruitment17, 18. Therefore, CCM patients are at increased risk of seizure, headaches, ICH, and consequently neurological deficits18, 19. 20
When mutated, CCM1 is one of three genes causing CCM and nearly 40% of patients in familial cases carry one allele of CCM1 loss-of-function mutation20. CCM1 gene global knockout and endothelial cell specific knockout in mice resulted in severe cerebral cavernous lesion and death21, 22. However, CCM1 heterozygous mice (CCM1+/−) only exhibit a change in BBB permeability23. Even though all the cells in the body have lost one copy of the relevant genes, lesions only occur in cerebral vasculature and not throughout the whole vascular system. Therefore, both somatic mutation of CCM genes and environmental disturbance (second hit) could lead to lesion growth and cause progressive symptoms24. Hyperglycemia, a highly prevalent diabetic condition, is considered as a risk factor for endothelial dysfunction25–27. Patients with diabetes have twice the risk of cerebrovascular disease28. Chronic hyperglycemia affects both macrovascular and microvascular endothelial cells, indicating that hyperglycemia could promote the onset of CCM.
To monitor hyperglycemia-increased vascular permeability, we synthesised long circulating neutral charged PEGylated Ag2S QDs, compared the dynamic imaging attributes of these QDs with established agent ICG, and demonstrated the feasibility of dynamic NIR-II window imaging for monitoring the changes in vascular perfusion in hyperglycemic CCM1+/− mouse models injected with a low dose of streptozotocin (STZ) to CCM1+/− mice. Dynamic NIR-II window imaging with Ag2S QDs had advantages over ICG imaging in rapid clearance from brain, and clearly demonstrated the higher cerebral vascular permeability in CCM1+/− mice compared to control mice, both treated with STZ. These results were verified by ex vivo fluorescence and micro-CT imaging. To the best of our knowledge, no study has reported the accurate detection of the changes in BBB integrity in CCM and the pathogenesis of brain hemorrhage in living rodents using a dynamic NIR-II fluorescence imaging approach.
Materials and Methods
Materials
Ethylene glycol anhydrous (EG, 99.8%), silver nitrate ACS reagent (AgNO3, ≥99%), 3-mercaptopropionic acid (MPA, 99%), sodium hydroxide (NaOH, 98%), and sodium chloride (NaCl, ≥99.5%) were purchased from Sigma Aldrich (Milwaukee, WI). Thiol-terminated methyl polyethylene glycol (SH-mPEG, 5k, >95%) was purchased from Nanocs Inc. (Boston, MA), and UltraPure™ DNase/RNase-Free Distilled Water was purchased from Invitrogen (Milwaukee, WI).
Synthesis of Ag2S QDs and Characterization
Ag2S QDs with sub-10nm size were synthesized using a procedure shown in Fig. 1. In a typical synthesis, 0.3 mmol of AgNO3 and 7 mmol of MPA were added to EG (~5 mL) and heated at 198 °C under argon gas flow for 30 min, and the reaction mixture was cooled down to room temperature using a slightly modified procedure by Tang et al.2 using a higher temperature and shorter time. Initially, the color of reaction mixture was white which darkened with time as heating was continued and QDs were formed. At the end of 30-minute heating period, Ag2S QDs were extracted from the suspension by centrifugation (three rounds using water) and re-dispersed in 100 mL water with a drop of NaOH added. PEGylation of the QDs was performed by adding 150 mmol SH-mPEG to Ag2S QDs and stirring for 24 h at room temperature. The QDs were collected by centrifugation and stored at 4 °C in the dark.
Fig. 1.

(A) A schematic illustration of synthesis of PEGylated Ag2S QDs. (B) Size distribution, transmission electron microscopy (TEM) image of Ag2S QDs, zeta potential measurement of COOH-terminated and PEGylated QDs. (C) NIR-II emission spectrum of Ag2S QDs between 900–1700 nm under 808-nm excitation. Inset shows the NIR-II emission from 1.5 mL microcentrifuge tube phantom containing Ag2S QDs. (D, E) Fluorescence kinetic curves for control mouse brain (n = 3 / group) injected with QDs and ICG and imaged with 950 nm or 1100 nm longpass filters respectively (mean ± SEM; two-way ANOVA, ****p <0.0001 in both cases). (F) Comparison between kinetic curves of QDs (n = 3 / group) injected mice imaged with 950 nm and 1100 nm longpass filters (mean ± SEM; two-way ANOVA, ****p <0.0001). The first five principal components (from the first 7 seconds post injection) images for both QDs and ICG injected mice using (G) 950 nm and (H) 1100 nm longpass filters.
TEM imaging was performed using a HITACHI H-600 TEM operating at an acceleration voltage of 70 kV. A drop of diluted colloidal Ag2S QDs was placed over a carbon-coated Cu grid for preparing a TEM grid. The zeta potential of Ag2S QDs was measured using a Malvern Zetasizer Nano ZS operated at 25 °C. NIR-II emission of Ag2S QDs in 1.5 mL microcentrifuge tube was measured under 808-nm excitation (~5 mW/cm2) with an 808 nm notch filter, in combination with longpass filters (rejection OD >6) at four different cut-off wavelengths: 950 nm (Edmund Optics Inc), 1100 nm (Thorlab Inc.), 1200nm (Thorlab Inc.). and 1300nm (Thorlab Inc.). A 10ms exposure was sufficient for imaging and comparing the intensities at different emission wavelengths of concentrated QDs by an InGaAs array (NIRvana 640 camera, Princeton Instruments) with 640 × 512 pixels, sensor cooled to cooled −60 °C. Emission spectrum of the QDs was recorded using a high-performance Acton SpectraPro SP2150 monochromator from Princeton Instruments equipped with a NIRvana 640 camera. A Hg and Ne/Ar dual switchable USB wavelength calibration system (IntelliCal, Princeton Instruments) was used to calibrate the monochromator.
Mouse Model and in vivo NIR-II Dynamic Imaging
CCM1+/− mice were provided by Dr. Douglas Marchuk’s laboratory at the Duke University. The mice were housed in the animal facility of Medical College of Wisconsin, Biological Resource Center. The Institutional Animal Care and Use Committee at the Medical College of Wisconsin approved the procedures performed here. Age matched CCM1+/− mice and WT mice were injected with 50 mg/kg STZ intraperitoneally for five consecutive days according to online DiaComp Protocols29 and monitored until the blood glucose level increased to 250 mg/dL. All animal experiments were conducted in accordance with policies of the NIH Guide for the Care and Use of Laboratory Animals and the Institutional Animal Care and Use Committee (IACUC) of the Medical College of Wisconsin. The animal protocols employed in this study were approved by the Medical College of Wisconsin IACUC.
Mice were injected with 200 μL of PEGylated Ag2S QDs (75 μg/mL) by tail vein injection and dynamically imaged using a custom-built imager comprised of a InGaAs array (NIRvana 640 camera, Princeton Instruments) with 640 × 512 pixels, cooled sensor (−60 °C), and 808-nm laser (20 mW/cm2), and images were acquired at 100-ms exposure for 5 min The fluorescence signal was imaged by a combination of filters, which included 808 nm notch based excitation rejection filter, with longpass filters at either 950 nm (Edmund Optics Inc), 1100 nm (Thorlab Inc.) or 1200nm (Thorlab Inc.). For ICG imaging and comparison, same imaging equipment and settings were used except that the mice were injected with 200 μL of ICG (0.05mg/kg) instead of QDs. Brain ROI-time series was extracted and compared between CCM1 heterozygous and control mice using MATLAB.
microCT Scan and H&E staining
After in vivo NIR-II dynamic fluorescence imaging, mice brains were collected. For the verification of compromised BBB and ICH in STZ-treated mice, excised mice brains post NIR imaging were soaked in propidium-iodide contrast agent (60 mg/mL) for 96 h and then washed to remove the excess of contrast agent following a reported procedure30. CT imaging was performed using Triumph Micro SPECT-CT (parameters: 512 views with 4 frames/view, 4X4, 60 kV, magnification of 4.5). The images were further processed using Amira 2019.3 software, Thermo Fisher Scientific, USA. Brains were then embedded and sectioned around the microCT confirmed area. Haematoxylin and eosin (H&E) staining was performed, and imaging was performed using a Leica ICC50 microscope.
BBB Permeability Measurement
A separate cohort of mice was divided into two sub-groups: One sub-group was perfused with PBS, and the other sub-group was perfused with FITC-dextran as previously described31. PBS-perfused mice brains were embedded with paraffin, and FITC-dextran perfused mice brains were embedded in Optimal Cutting Temperature compound. Tissue blocks were cut into 8 μm (PBS-perfused) and 50 μm (FITC-dextran-perfused) sections. IgG staining was applied to paraffin-embedded sections, and images were recorded using a Leica ICC50 microscope. A Nikon Ti confocal microscope was used for imaging FITC-dextran-perfused frozen sections.
Immunofluorescent staining
Immunofluorescent staining was performed on paraffin-embedded sections. Briefly, after dewaxing, hydration and antigen retrieval, tissue sections were incubated with primary antibody Claudin-5 (Invitrogen 34–1600, rabbit, 1:100) and IB4 (Invitrogen, 121412, 1:200) at 4 °C overnight. Then, the tissue sections were washed with PBS and incubated with secondary antibody (Invitrogen A-21206, 1:500) at room temperature for 2 h. After washing with PBS, nuclear staining was carried out with DAPI. After mounting, images were recorded using a Nikon Ti confocal microscope.
Extraction of mice brain endothelial cells
Mice brain microvascular endothelial cells were extracted from CCM1+/− and wild type mice (3–5 week) as previously described with some modification 32. Briefly, mice brains were collected, and the isolated cerebral cortices were minced and homogenised in ice-cold DMEM. After digesting with 10 μg/mL collagenase II and 1000 U/mL DNase I at 37 °C for 2 h, the homogenate was centrifugated at 500 g for 5 min at 4 °C. The lower layer was resuspended in 17% Percoll and centrifuged at 1000 g for 20 min at 4 °C, and then digested with 10 μg/mL collagenase II and 1000 U/mL DNase I at 37 °C for 1 h. After centrifuging at 500 g for 5 min at 4 °C, the precipitate was added into Percoll gradient and centrifuged at 1000 g for 15 min at 4 °C. The cloudy middle layer was harvested and centrifuged at 500 g for 5 min at 4 °C and then seeded into 6-well plates with endothelial cell medium (Sciencell, #1001), incubated at 37 °C in humidified 5% CO2. The cells were divided into control group and high-glucose-treatment group. In the high-glucose-treatment group, the cells were treated with 30 mM D-glucose for seven days.
Endothelial cell permeability assay
Mice brain microvascular endothelial cells were grown to confluence on Transwell filter inserts (24-well format, 0.4 μm pore), and monolayer paracellular permeability was determined as reported 33, 34. Briefly, 100 μg/mL fluorescence dextran, molecular weight 40,000 (FDX-40000; Molecular Probes, Eugene, OR), in an assay buffer (0.1% BSA in DMEM) was added to the upper chamber. Samples (50 μL) were removed from the lower chamber at 0, 30, 60, and 120 min and replaced with equal volumes of assay buffer. The concentration of FDX-40000 applied to the upper chamber was assessed by retrieving a 50 μL sample at 0 min and similarly replacing this with 50 μL assay buffer containing FDX-40,000. The retrieved samples were analyzed using a plate reader with fluorescence-detecting capabilities (excitation λ = 488 nm; emission λ = 510 nm). Permeability coefficient (PC) for FITC-dextran was determined using the following equation: PC (cm/min) = V/(SA x Cd) x (Cr/T), where V is the volume in the receiver (lower) chamber (1 cm3), SA is the surface area of cell monolayer (0.3 cm2), Cd is the concertation of FDX-40000 in the upper chamber at time 0, and Cr is the concentration of FDX-40,000 in the bottom chamber at sampling time T. PC was determined for replicate samples, and the mean value was obtained. Change in permeability following treatment was reported as percent of mean control PC value.
Western blot analysis
An equal amount (30 μg) of protein samples extracted from control or high-glucose-treated cells was loaded and separated on 10% sodium dodecyl sulfate/polyacrylamide gels, then transferred onto nitrocellulose membranes. After blocking non-specific binding with 5% non-fat milk for 1 h, the membranes were incubated with primary antibodies against CCM1 (1:1000, ab196025; Abcam) and GAPDH (1:5000, 60004–1-Ig; Proteintech) overnight at 4 °C. After 40 min washing with TBS-T, the membranes were then incubated with secondary antibodies at room temperature for 2 h and washed with TBS-T. The protein bands were finally visualised using a Bio-rad ChemiDoc machine using SuperSignal West Pico chemiluminescence kit (Thermo Fisher Scientific).
Statistics
The data are expressed as Mean ± SD. Statistical significance was analyzed by ANOVA using GraphPad software, and significance is defined as P <0.05. Western blot image was analyzed using ImageJ software.
Synthesis of PEGylated Ag2S QDs for determining the change of blood perfusion
Sub-10nm PEGylated Ag2S QDs were synthesised by modifying the scheme reported by Tang et al.2 as depicted in Fig. 1A. TEM image indicated Ag2S QDs were ~8 ± 2 nm in size with uniform spherical shape (inset of Fig. 1B(i)). Zeta potential of COOH-terminated QDs was found to be −20 mV, and PEGylation reduced the zeta potential to −6.7 mV as depicted in the inset of Fig. 1B(ii). Emission spectrum of Ag2S QDs showed a range spanning from 950–1700 nm upon 808-nm excitation as depicted in Fig. 1C. Gaussian curve fitting to emission spectrum of QDs confirmed that the peak is centred at 1135 mm, and the intensity exponentially decreased until 1600 nm, with Full width at half maximum (FWHM) of approximately 170 nm, R2 = 0.9823 (Table S1, Fig. S1(A), see ESI). To further clarify the emission intensity distribution, the spectrum was also fitted with two peaks of Gaussian profile, which confirmed that the first peak is centred at 1110 nm, FWHM = 129 nm, and second peak is centred at 1213 nm, FWHM = 212 nm with R2 = 0.9984 (Table S1, Fig. S1(B), see ESI). Thus, 52.5% of emission intensity of Ag2S QDs was centred around 1110 nm, and 47.5% of emission intensity of Ag2S QDs was centred around 1213 nm. Inset of Fig. 1C shows the emission from Ag2S QDs in 1.5 mL Eppendorf tubes under 808-nm excitation ~20 mW/cm2 and 950 nm long-pass filter with a high signal-to-background ratio suitable for in vivo imaging. To select optimum imaging filters for mice, first the QDs in quartz vials were excited with 808 nm light (5 mW/cm2) and 10 ms exposure with sequentially longer cut-off wavelength long-pass filters (950, 1100, 1200, and 1300 nm). This data is depicted in Fig S2. The signal was superior with 950 nm longpass filter and fell off sharply with 1200 nm longpass filters.
In vivo NIR-II imaging of QDs was assessed and compared with ICG in control mice with tail vein bolus injections. Fluorescence kinetic data in the brian ROI for the QDs (n = 3 / group) and ICG (n = 3 / group) acquired using 950 nm, 1100 nm, 1200 nm longpass filter is depicted in Fig. 1D, Fig. 1E, and Fig. S3, respectively. The comparison between emission kinetics profiles at 950 and 1100 nm longpass filters in QD injected mice is depicted in Fig. 1F. First five principal component image decomposition (PC1, PC2, PC3, PC4, and PC5) corresponding to both QDs and ICG injected mice using 950 nm, 1100 nm and 1200 nm longpass filter is depicted in Fig. 1G, Fig. 1H, and Fig S4, respectively. These studies indicated differences in washout of QDs vs ICG independent of imaging wavelengths. QD kinetics in brain ROI was similar at 950 nm and 1100 nm longpass with higher signal at 950 nm, and no differences in image quality. Thus, QD imaging with 950 nm longpass filter was chosen for subsequent experiments.
Determination of increased blood perfusion in the brain of STZ-injected CCM1+/− mice
After five days of consecutive administration of a low dose of STZ, both wild type and CCM1+/− mice reached a similar glucose level which was much higher than the normal range and near diabetes criteria. However, the glucose level of mice receiving the vehicle control remained around the base line (Fig. S5, seeESI). Dynamic NIR-II fluorescence images of the control and CCM1+/− mice in STZ-treated group at post-5s injection of PEGylated Ag2S QDs are shown in Figs. 2A–B and show significantly increased higher wash-in in STZ-treated CCM1+/− mice, indicating the disruption of BBB around brain micro-capillaries.
Fig. 2.

(A, B) NIR-II fluorescence emission from brain at 5 s post-injection of PEGylated Ag2S QDs in both control (n = 6) and CCM1+/− group (n = 6) mice. (C) A typical whole-body PCA overlaid image using dynamic NIR-II fluorescence images. (D) Dynamic NIR-II image intensity from both control and CCM1+/− group mice brain for post-15sec of QD injection (mean ± SEM; two-way ANOVA, ****p <0.0001).
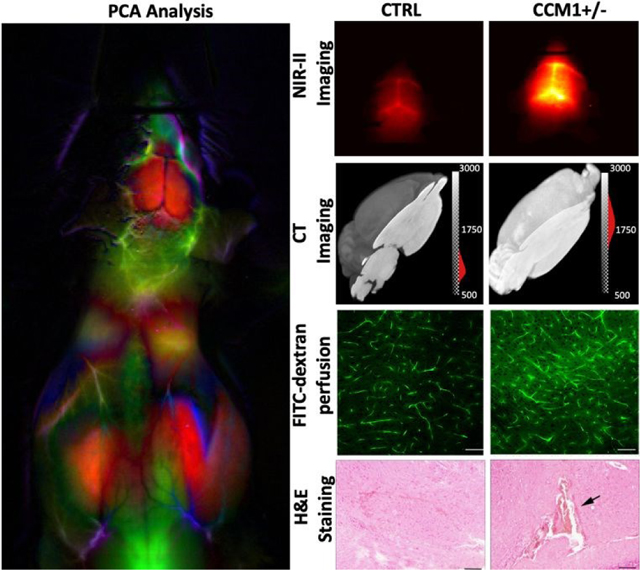
Principal component analysis (PCA) on the imaging time series for each pixel was performed by applying singular value decomposition algorithm in MATLAB to clearly identify the brain ROI using the first few PCA components as shown in Figs. 2C and S6 (see ESI). The average dynamic NIR-II fluorescence intensity from both STZ-treated WT (n = 6) and STZ-treated CCM1+/− (n = 6) group brain ROI is shown in Fig. 2D. Statistically significant differences were observed in contrast kinetics (****p <0.0001, ANOVA2 in MATLAB). We observed that after bolus injection, the QDs permeate in higher amounts in the (CCM1+STZ) mice brains compared to the control group mice (CTRL+STZ). The brain ROI averaged dynamic NIR-II fluorescence intensity from the brain ROIs for individual animals of control and CCM1 group mice is shown in Figs. S7(A–B), see ESI, overlaid with group mean, and in Fig. S7C, see ESI, CCM1+/− group animals are plotted overlaid with the control group mean. These data illustrate the increased heterogeneity in cerebrovascular disruption in the CCM1+/− mice when hyperglycemia is induced with STZ treatment. A pseudo-real time movie of dynamic fluorescence imaging in NIR-II window from the mice brain of control (left) and CCM1 +/− (right) group mice following the tail vein injection of PEGylated Ag2S QDs is shown in Fig. S8, see ESI, for the first 60s post-injection time-lapse imaging (movie link: https://figshare.com/s/d2ffba8c9d87b006b972 (DOI: 10.6084/m9.figshare.12672389). These results are supported by previous observations by Chiu et al.35, wherein the authors reported that rats with hyperglycemia exhibited a higher fluid content (Cerebral Edema) in brain and demonstrated severe BBB destruction. Similarly, in our studies reported here, the increased BBB disturbance in hyperglycemic CCM1+/− mice potentially results in a higher initial uptake of sub-10nm Ag2S QDs in brain vasculature compared to the control hyperglycemic mice. To validate this imaging-derived finding, we performed multiple BBB disturbance characterization studies in both CCM1+/− and control mice treated with STZ. Even though the NIR-II dynamic fluorescence imaging significantly discriminated between control and CCM1+/− group mice, the surface-weighted nature of these measurements required confirmation with ex vivo methods. To verify the increased permeability of contrast agents throughout the cerebral vasculature, microCT imaging was performed after treating mice brain with an iodinated CT contrast for 96 h followed by repeated washing to remove the free contrast agent. The MicroCT-reconstructed images were analysed using AMIRA software (2019.3; Thermo Fisher Scientific, USA). The images indicated uniformly increased contrast in brains from CCM1+/− mice treated with STZ. A three-dimensionally (3D) reconstructed and consistently intensity-thresholded maximum intensity projection of a representative control and CCM1+/− group mice brain overlaid with YZ axial plane is shown in Figs. 3A–B. A colorbar with “gray” scale intensity overlaid histogram is shown in the figure itself. Bright contrast in the volume-rendering image corresponds to a higher uptake of Iodine contrast in CCM1+/− group compared to a lower contrast in the control group of mice brain. To thoroughly study the differences in iodine contrast uptake in these brains, a data-driven approach was developed to study the separation between control and CCM+/− groups by using k-means clustering. Voxel intensity data pooled from n = 3 mice from each group were classified using the squared Euclidean distance metric and the k-means++ algorithm in MATLAB R2020a software. This algorithm classified the micro-CT data into two different clusters (K = 2) corresponding to CTRL+STZ and CCM1+STZ groups with clearly separated centroids as shown in Fig. 3C. Thus, the increased BBB permeability indicated by NIR-II window surface measurements was validated by 3D micro-CT imaging to be present over the entire brain.
Fig. 3.

(A) Visualization of intensity-thresholded volume-rendering reconstruction of micro-CT data of (A) CTRL + STZ, (B) CCM1+STZ mice brain treated with iodine contrast under similar condition overlaid with a corresponding YZ oriented image plane crosses across the centre of the mice brain. (C) A clear separation between CTRL+ STZ (n = 3; Cluster 1 – Left Side) and CCM1 + STZ (n = 4; Cluster 2 – Right Side) was obtained by applying squared Euclidean distance metric and k-means++ algorithm that applies heuristic to find the centroid seeds for k-means clustering (number of total clusters, K = 2). (D) FITC-dextran perfusion images of vehicle and STZ-injected WT and CCM1+/− group demonstrate a significant permeable BBB in hyperglycemic CCM1+/− group (scale bar, 50 μm).(E) IgG staining of vehicle and STZ-treated WT and CCM1+/− group (scale bar, 100 μm). (F) microCT images of STZ-treated CCM1+/− mice indicate hemorrhage spots (red arrows) in brain. (G) H&E staining images sectioned at hemorrhagic location (indicated by CT data) confirmed intracerebral hemorrhage (black arrows, scale bar, 100 μm).
Results and Discussion
Hyperglycemic CCM1+/− mice exhibit hyperpermeable BBB and intracerebral hemorrhage
As a second gold-standard confirmatory measurement of BBB disruption, FITC-dextran perfusion assay and IgG staining were used for further confirmation of BBB permeability change in each group. The brain tissue sections for each group were analyzed histologically. Vehicle-treated CCM1+/− mice exhibited moderately increased FITC-dextran extravasation compared to vehicle-treated WT mice, consistent with a previous report23. While in the vehicle-treated CCM1+/− group FITC-dextran extravasation dramatically increased compared to the STZ-treated CCM1+/− group, indicating a much more permeable BBB. As depicted in the fourth panel of Fig. 3D, the diffusion of FITC-dextran signal outside the capillaries can be attributed to the extravasation of FITC-dextran beyond the BBB. Interestingly, STZ-treated WT group had more FITC-dextran extravasation than vehicle-treated WT group, i.e., chronic hyperglycemia itself can increase BBB permeability at moderate levels. IgG staining showed no dramatic difference between vehicle-treated CCM1+/− group and vehicle-treated WT group, neither STZ-treated WT group and vehicle-treated WT group. However, much stronger IgG staining was observed in the vicinity of the vasculature in STZ-treated CCM1+/− group compared to other groups (Figs. 3D–E). This is probably due to vascular dysfunction in the brain of STZ-treated CCM1+/− group which allows for the penetration of circulating FITC-dextran and IgG into parenchyma via BBB.
Besides the finding of BBB disruption, MicroCT scan also indicates hemorrhage locations, marked as red arrows in STZ-treated CCM1+/− brain shown in Fig. 3F. H&E staining image sectioned at hemorrhagic location indicated by CT data further confirmed intracerebral hemorrhage, which is marked with black arrow as shown in Fig. 3G. These data suggest that the microvasculature in the brain of CCM1+/− is more vulnerable to hyperglycemia induced vascular pathology.
Tight junction protein Claudin-5 decreases in hyperglycemic CCM1+/− mice
Tight junction proteins between endothelial cells, including Claudin-5, Occludin, and ZO-1, are crucial for maintaining BBB integrity. Among those tight junction proteins, Claudin-5 is specifically expressed in brain endothelial cells and has unique importance in BBB integrity36–38. According to previous reports, tight junctions in CCM patients lesion sites decrease compared to normal brain tissue39, 40. Chronic hyperglycemia also induces the reduction of tight junction Claudin-5 in brain endothelial cells. To further elucidate the pathological change in BBB permeability in hyperglycemic CCM1+/− mice, we examined the tight junction protein Claudin-5 in each group using immunofluorescent staining. Both hyperglycemia and CCM1 heterozygous mutation exhibited a decrease in Claudin-5 in brain vasculature. However, hyperglycemia exacerbated the decrease of Claudin-5 in the brain of CCM1 heterozygous mice (Fig. 4), further contributing to the impairment of BBB integrity in hyperglycemic CCM1+/− mice.
Fig. 4.

Claudin-5 and IB4 staining in vehicle and STZ-treated WT and CCM1+/− mice demonstrates a significant decrease of tight junction protein Claudin-5 expression in hyperglycemic CCM1+/− mice (scale bar, 25 μm).
High-glucose treatment increases endothelial cell permeability in the brain of CCM1+/− mice through downregulating CCM1 expression
CCM1 knockout mice exhibit obvious CCM lesions in the brain 22. However, CCM1+/− mice exhibit increased BBB permeability but no notable CCM lesions because the expression of CCM1 in the brain and lung of CCM1+/− mice is only lost at approximately half of level as in WT mice, i.e., a remarkable reduction of CCM1 expression is the key to trigger the onset of CCM. We isolated endothelial cells from the brain of WT and CCM1+/− mice brain and used FITC-dextran assay to evaluate the permeability changes in response to high-glucose treatment (30 mM). As shown in Fig. 5A, the basal permeability of CCM1+/− brain endothelial cells is higher than WT endothelial cells, consistent with the previously reported results23. High-glucose treatment further increased the permeability of CCM1+/− brain endothelial cells, i.e., high glucose could further impair endothelial layer integrity consistent with the in vivo results (Fig. 5A). Western blot results show that high-glucose treatment decreases CCM1 protein levels, especially in CCM1+/− brain endothelial cells (Fig. 5B), which is less than 35% of CCM1 protein levels in WT endothelial cells.
Fig. 5.

(A) Basal permeability of endothelial cells isolated from the brain of CCM1+/− mice is higher than that of WT endothelial cells. Permeability further increases after high glucose treatment. Relative endothelial permeability in CCM1+/− normal glucose group is 1.344 ± 0.021, in WT high-glucose group is 1.272 ± 0.045, and in CCM1+/− high-glucose group is 1.75 ± 0.059 as normalized to WT normal glucose group, n = 3. *P <0.05 vs. WT normal glucose group. ΔP <0.05 vs. CCM1+/− normal glucose group. (B) CCM1 expression in endothelial cells isolated from the brain of CCM1+/− mice decreases after 7 d high-glucose treatment. Endothelial cells were isolated from 10 WT and 10 CCM1+/− mice.
We have shown that PEGylated Ag2S QDs provide promising NIR-II fluorescence contrast for deep tissue non-invasive and relatively low power brain imaging at an excitation wavelength of 808 nm (20 mw/cm2) with strong NIR-II window emission. Dynamic fluorescent imaging characterized brain perfusion differences between STZ-treated WT and CCM1 +/− mice with statistically significant value ****P <0.0001. These results also provide evidence that NIR-II fluorescent emission from mice brains has advantages compared to traditional NIR-I fluorescent window imaging, including lower autofluorescence and background signal. This imaging technique also exploits widely separated excitation (808 nm) and emission (>950 nm) windows reducing the leakage of spurious light through optical filters and its detrimental effect on signal to noise ratio.
In vivo tracking of BBB integrity is crucial for the pathological discovery of cerebrovascular diseases. Impaired BBB leads to the accumulation of contrast agents via enhanced permeability and retention (EPR) effect. NIR-II fluorescence imaging is a non-invasive tool with ambulatory and relatively affordable characteristics without the compulsion of subjects; therefore, it has attracted much attention41. We synthesised PEGylated Ag2S QDs, ~8 nm in size and neutral surface charge, with fluorescence in NIR-II window (around 1135 nm) upon 808-nm light excitation. These Ag2S QDs were intravenously injected in both hyperglycemic WT (n = 6) and hyperglycemic CCM1+/− group (n = 6) mice under similar conditions, and in vivo dynamic fluorescence imaging in NIR-II window from mice brain ROI was compared. The average fluorescence counts of QDs are significantly higher in hyperglycemic CCM1+/− mice compared to hyperglycemic WT mice group with P value of <0.0001 as determined using ANOVA analysis in MATLAB. NIR-II fluorescence signal in mouse brains post-5s injection in the brain of hyperglycemic CCM1+/− mice is four times higher than hyperglycemic WT mice. Furthermore, the washout rate of QDs CCM1+/− group mice brain is ~200% higher than hyperglycemic WT group, whereas the washout rate in hyperglycemic CCM1+/− is slightly lower compared to hyperglycemic WT group within the limits of error bar. A higher retention of NIR-II fluorescence intensity in CCM1 +/− group can be attributed to impaired vascular junctions. The most important finding is that the observed increase in BBB permeability via NIR imaging is consistent with the results obtained by traditional biological approaches such as FICT-dextran extravasation assay and IgG staining. Further histology analysis elucidated BBB integrity impairment associated with the decrease of tight junction Claudin-5. Mechanistically, we demonstrate that long-term hyperglycemia induces the loss of CCM1 expression in brain endothelial cells. It can be concluded that hyperglycemia-induced ICH and BBB hyperpermeability in the brain of CCM1+/− mice can be monitored non-invasively using NIR-II fluorescence QDs.
Recently, interesting results have been published by Wu et al.,42 using BTPPA molecules encapsulated in L1013 (DSPE-PEG2k construct) nano-particles with emission at 1013 nm on excitation at808-nm light. They used blood in situ injection hemorrhage stroke model, which causes brain parenchyma damage and hyperpermeable BBB. The results showed a great enhancement (~190%) in fluorescence intensity in the right hemorrhage side of the brain. The results are consistent with our observation on ICH and BBB breakdown in hyperglycemic CCM1+/− mice. Similar fluorescence imaging methods have been used clinically using NIR-I ICG contrast agent to track the differences in subcutaneous microcirculation43, 44. Thus, Ag2S-enabled dynamic imaging might be helpful to find vascular pathologies across the whole body45. In addition to vascular imaging, image-guided brain tumour surgical resection has also been reported46 and is a further application for nano-particles.
One of the weaknesses of whole-body NIR imaging is the surface-weighted nature of these images, with potential questions about sensitivity to deep tissue vascular changes. Here, we thoroughly tested in vivo imaging with another gold standard ex vivo imaging method of iodine contrast enhanced micro-CT. To validate the presence of hemorrhage and BBB disruption, brain tissues were soaked in propidium iodide contrast agent, and 3D reconstruction of micro-CT data confirmed that CCM1+/− mice had uniformly higher iodine contrast uptake compared to hyperglycemic WT animals. A clear intraventricular hemorrhage location identified on hyperglycemic CCM1+/− mice was also confirmed by H&E staining, which is around the micro-CT confirmed hemorrhage location. Owing to the intact vasculature in the normal brain, FICT-dextran and IgG do not leak in cortical vasculature47. As shown in the results of FITC-dextran extravasation assay and IgG staining, the vessel walls in hyperglycemic CCM1+/− mice show the leakage of FICT-dextran and IgG into the locations surrounding vessels, whereas only minimal FITC-dextran leaking was observed in hyperglycemic WT and normal CCM1+/− group. No such leakage was observed in normal control group animals. These results confirm that an increase in ICH and BBB permeability occurs in hyperglycemic CCM1+/− mice. These vascular lesions were further confirmed by decreased staining of tight junction protein Claudin-5 in hyperglycemic CCM1+/− mice.
Even though previous reports suggested that no significant association with lesion count and abnormal white matter hyperintensities was observed for diabetes in familial CCM1 cases48, 49, the association between hyperglycemia and the etiology of CCM is still unclear. Furthermore, many evidences clearly show the statistical association between hyperglycemia and the incidence of hemorrhagic stroke28, 50. To investigate the underlying mechanism of hyperglycemia effect on CCM1+/− mice, we extracted endothelial cells from CCM1+/− mice and treated with high glucose for seven days. A decrease in the CCM1 protein levels in high-glucose-treated endothelial cells indicates that hyperglycemia can induce a further decrease of CCM1 in endothelial cells of CCM1+/− mice and exacerbate the damage of vascular junctions in CCM1+/− mice.
Conclusions
In summary, this study demonstrates that NIR-II fluorescence imaging with PEGylated Ag2S QDs is an effective non-invasive method for detecting brain vascular lesions such as ICH and BBB hyperpermeability. With NIR imaging and rigorous gold standard validation of brain vascular integrity, we also demonstrate that hyperglycemia further decreases the CCM1 expression in CCM1+/− mice and diminishes endothelial tight junctions. This impairs BBB integrity and can lead to vascular rupture and brain hemorrhage.
Supplementary Material
Acknowledgements
Dr. Douglas Marchuk at the Duke University generously provided CCM1 heterozygous mice. The research was supported by R01HL141733 and AHA 17GRNT33671180 to Q.M. and MCW Cancer Center and NIH R01CA193343 support to Dr. A. Joshi. The authors declare no conflict of interest.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Bhavane R, Starosolski Z, Stupin I, Ghaghada KB and Annapragada A, Sci Rep, 2018, 8, 14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang R, Xue J, Xu B, Shen D, Sudlow GP and Achilefu S, ACS Nano, 2015, 9, 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Upputuri PK and Pramanik M, J Biomed Opt, 2019, 24, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yaroslavsky AN, Schulze PC, Yaroslavsky IV, Schober R, Ulrich F. and Schwarzmaier HJ, Phys Med Biol, 2002, 47, 2059–2073. [DOI] [PubMed] [Google Scholar]
- 5.Zhu S, Yang Q, Antaris AL, Yue J, Ma Z, Wang H, Huang W, Wan H, Wang J, Diao S, Zhang B, Li X, Zhong Y, Yu K, Hong G, Luo J, Liang Y. and Dai H, Proc Natl Acad Sci U S A, 2017, 114, 962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang P, Zhu CN, Zhang ZL, Tian ZQ and Pang DW, Biomaterials, 2012, 33, 5130–5135. [DOI] [PubMed] [Google Scholar]
- 7.Hong G, Robinson JT, Zhang Y, Diao S, Antaris AL, Wang Q. and Dai H, Angew Chem Int Ed Engl, 2012, 51, 9818–9821. [DOI] [PubMed] [Google Scholar]
- 8.Chinnathambi S. and Shirahata N, Sci Technol Adv Mater, 2019, 20, 337–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Awasthi P, An X, Xiang J, Kalva N, Shen Y. and Li C, Nanoscale, 2020, 12, 5678–5684. [DOI] [PubMed] [Google Scholar]
- 10.Li C, Zhang Y, Wang M, Zhang Y, Chen G, Li L, Wu D. and Wang Q, Biomaterials, 2014, 35, 393–400. [DOI] [PubMed] [Google Scholar]
- 11.Cui M, Ono M, Watanabe H, Kimura H, Liu B. and Saji H, J Am Chem Soc, 2014, 136, 3388–3394. [DOI] [PubMed] [Google Scholar]
- 12.Zhang X, Tian Y, Zhang C, Tian X, Ross AW, Moir RD, Sun H, Tanzi RE, Moore A. and Ran C, Proc Natl Acad Sci U S A, 2015, 112, 9734–9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carr JA, Franke D, Caram JR, Perkinson CF, Saif M, Askoxylakis V, Datta M, Fukumura D, Jain RK, Bawendi MG and Bruns OT, Proc Natl Acad Sci U S A, 2018, 115, 4465–4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Z, Fang C, Li B, Zhang Z, Cao C, Cai M, Su S, Sun X, Shi X, Li C, Zhou T, Zhang Y, Chi C, He P, Xia X, Chen Y, Gambhir SS, Cheng Z. and Tian J, Nat Biomed Eng, 2020, 4, 259–271. [DOI] [PubMed] [Google Scholar]
- 15.Kim J, BMB Rep, 2016, 49, 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zafar A, Quadri SA, Farooqui M, Ikram A, Robinson M, Hart BL, Mabray MC, Vigil C, Tang AT, Kahn ML, Yonas H, Lawton MT, Kim H. and Morrison L, Stroke, 2019, 50, 1294–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tanriover G, Sozen B, Seker A, Kilic T, Gunel M. and Demir N, Clin Neurol Neurosurg, 2013, 115, 438–444. [DOI] [PubMed] [Google Scholar]
- 18.Draheim KM, Fisher OS, Boggon TJ and Calderwood DA, J Cell Sci, 2014, 127, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Curling O Jr., Kelly DL Jr., Elster AD and Craven TE, J Neurosurg, 1991, 75, 702–708. [DOI] [PubMed] [Google Scholar]
- 20.Cavalcanti DD, Kalani MY, Martirosyan NL, Eales J, Spetzler RF and Preul MC, J Neurosurg, 2012, 116, 122–132. [DOI] [PubMed] [Google Scholar]
- 21.Whitehead KJ, Plummer NW, Adams JA, Marchuk DA and Li DY, Development, 2004, 131, 1437–1448. [DOI] [PubMed] [Google Scholar]
- 22.Mleynek TM, Chan AC, Redd M, Gibson CC, Davis CT, Shi DS, Chen T, Carter KL, Ling J, Blanco R, Gerhardt H, Whitehead K. and Li DY, Hum Mol Genet, 2014, 23, 6223–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockton RA, Shenkar R, Awad IA and Ginsberg MH, J Exp Med, 2010, 207, 881–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leblanc GG, Golanov E, Awad IA, Young WL and Biology NWC of Vascular Malformations of the Brain, Stroke, 2009, 40, e694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia Soriano F, Virag L, Jagtap P, Szabo E, Mabley JG, Liaudet L, Marton A, Hoyt DG, Murthy KG, Salzman AL, Southan GJ and Szabo C, Nat Med, 2001, 7, 108–113. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y. and Vanhoutte PM, J Diabetes, 2017, 9, 434–449. [DOI] [PubMed] [Google Scholar]
- 27.Eelen G, de Zeeuw P, Simons M. and Carmeliet P, Circ Res, 2015, 116, 1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hill MD, Handb Clin Neurol, 2014, 126, 167–174. [DOI] [PubMed] [Google Scholar]
- 29.Low-Dose Streptozotocin Induction Protocol (Mouse), https://www.diacomp.org/shared/showFile.aspx?doctypeid=3&docid=19, (accessed 1/2/2018, 2018). [Google Scholar]
- 30.Girard R, Zeineddine HA, Orsbon C, Tan H, Moore T, Hobson N, Shenkar R, Lightle R, Shi C, Fam MD, Cao Y, Shen L, Neander AI, Rorrer A, Gallione C, Tang AT, Kahn ML, Marchuk DA, Luo ZX and Awad IA, J Neurosci Methods, 2016, 271, 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Natarajan R, Northrop N. and Yamamoto B, Curr Protoc Neurosci, 2017, 79, 9 58 51–59 58 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mark KS and Davis TP, Am J Physiol Heart Circ Physiol, 2002, 282, H1485–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams JE and Cavanaugh DC, J Wildl Dis, 1983, 19, 154–155. [DOI] [PubMed] [Google Scholar]
- 34.Song L, Ge S. and Pachter JS, Blood, 2007, 109, 1515–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiu CD, Chen CC, Shen CC, Chin LT, Ma HI, Chuang HY, Cho DY, Chu CH and Chang C, Stroke, 2013, 44, 1682–1689. [DOI] [PubMed] [Google Scholar]
- 36.Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N, Furuse M. and Tsukita S, J Cell Biol, 2003, 161, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kluger MS, Clark PR, Tellides G, Gerke V. and Pober JS, Arterioscler Thromb Vasc Biol, 2013, 33, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daneman R. and Prat A, Cold Spring Harb Perspect Biol, 2015, 7, a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schneider H, Errede M, Ulrich NH, Virgintino D, Frei K. and Bertalanffy H, J Neuropathol Exp Neurol, 2011, 70, 417–429. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Ramirez MA, Fonseca G, Zeineddine HA, Girard R, Moore T, Pham A, Cao Y, Shenkar R, de Kreuk BJ, Lagarrigue F, Lawler J, Glass CK, Awad IA and Ginsberg MH, J Exp Med, 2017, 214, 3331–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H, Hong G, Luo Z, Chen J, Chang J, Gong M, He H, Yang J, Yuan X, Li L, Mu X, Wang J, Mi W, Luo J, Xie J. and Zhang XD, Adv Mater, 2019, DOI: 10.1002/adma.201901015., e1901015 [DOI] [PubMed] [Google Scholar]
- 42.Wu W, Yang YQ, Yang Y, Yang YM, Wang H, Zhang KY, Guo L, Ge HF, Liu J. and Feng H, Int J Nanomedicine, 2019, 14, 3571–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tipirneni KE, Warram JM, Moore LS, Prince AC, de Boer E, Jani AH, Wapnir IL, Liao JC, Bouvet M, Behnke NK, Hawn MT, Poultsides GA, Vahrmeijer AL, Carroll WR, Zinn KR and Rosenthal E, Ann Surg, 2017, 266, 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupree A, Riess H, Detter C, Debus ES and Wipper SH, Innov Surg Sci, 2018, 3, 193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma G, Jagtap JM, Parchur AK, Gogineni VR, Ran S, Bergom C, White SB, Flister MJ and Joshi A, Theranostics, 2020, 10, 5368–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cho SS, Salinas R. and Lee JYK, Front Surg, 2019, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi M, Ku T, Chong K, Yoon J. and Choi C, Proc Natl Acad Sci U S A, 2011, 108, 9256–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Golden MJ, Morrison LA, Kim H. and Hart BL, AJNR Am J Neuroradiol, 2015, 36, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choquet H, Nelson J, Pawlikowska L, McCulloch CE, Akers A, Baca B, Khan Y, Hart B, Morrison L. and Kim H, Cerebrovasc Dis, 2014, 37, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Munoz-Rivas N, Mendez-Bailon M, Hernandez-Barrera V, de Miguel-Yanes JM, Jimenez-Garcia R, Esteban-Hernandez J. and Lopez-de-Andres A, J Stroke Cerebrovasc Dis, 2016, 25, 1431–1443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.