

Abstract
Tick-borne encephalitis virus (TBEV) is the aetiological agent of tick-borne encephalitis, an infectious disease of the central nervous system that is often associated with severe sequelae in humans. While TBEV is typically classified into three subtypes, recent evidence suggests a more varied range of TBEV subtypes and lineages that differ substantially in the architecture of their 3ʹ untranslated region (3ʹUTR). Building on comparative genomic approaches and thermodynamic modelling, we characterize the TBEV UTR structureome diversity and propose a unified picture of pervasive non-coding RNA structure conservation. Moreover, we provide an updated phylogeny of TBEV, building on more than 220 publicly available complete genomes, and investigate the molecular epidemiology and phylodynamics with Nextstrain, a web-based visualization framework for real-time pathogen evolution.
Keywords: Tick-borne encephalitis virus, conserved RNA structure, untranslated region, molecular epidemiology
1. Introduction
Tick-borne encephalitis virus (TBEV) is a zoonotic RNA virus of the genus Flavivirus, family Flaviviridae. It is the aetiological agent of tick-borne encephalitis (TBE), an infection of the central nervous system that is considered the most common tick-transmitted disease in Eurasia (Amicizia et al. 2013), where it occurs in risk or endemic areas that are also referred to as foci (Charrel et al. 2004). TBEV is transmitted between haematophagous ticks as vectors and vertebrate hosts. Typical reservoir hosts include wild-living animals such as small rodent species. Large vertebrate species, including humans and ungulates like goats, cows, sheep, swine, and deer, can become infected but appear not to be competent to transmit the virus back to ticks (Labuda et al. 1993). While serological evidence suggests that the majority of human infections are either asymptomatic or subclinical, TBEV is a neurotropic virus that can cause a wide range of life-threatening clinical manifestations comprising febrile, meningeal, meningoencephalitic, poliomyelitic, polyradiculoneuritic, and chronic forms (reviewed in Gritsun, Lashkevich, and Gould 2003), as well as haemorrhagic syndrome (Ternovoi et al. 2003).
1.1. Flavivirus genome organization
TBEV belongs to the group of tick-borne flaviviruses (TBFVs), which, together with mosquito-borne flaviviruses (MBFVs) and no-known-vector flaviviruses, encompass the vertebrate-infecting flaviviruses. On the contrary, insect-specific flaviviruses only replicate in mosquitoes (Blitvich and Firth 2015). Flaviviruses are enveloped, single-stranded (+)-sense viruses that contain a non-segmented, 5ʹ-capped, non-polyadenylated RNA of approximately 11 kb length. The genomic RNA (gRNA) encodes a single open reading frame that is flanked by highly structured untranslated regions (UTRs) of variable length (Rauscher et al. 1997; Ng et al. 2017). Both UTRs are crucially involved in regulating processes that control different aspects of the virus life cycle, including virus replication, genome cyclization and packaging, and immune response (Villordo, Alvarez, and Gamarnik 2010; de Borba et al. 2015; Barrows et al. 2018).
Common architectural traits of flavivirus 3ʹUTRs comprise autonomous RNA structure formation of distinct domains and the presence of evolutionarily conserved RNA elements with specific functional associations. A hallmark of flavivirus biology is their ability to actively dysregulate the host mRNA turnover machinery by stalling endogenous exoribonucleases (Pijlman et al. 2008) at structurally well-defined RNAs in the viral 3ʹUTR (MacFadden et al. 2018). Homologs of these so-called exoribonuclease-resistant RNAs (xrRNAs) are typically found in one or two copies throughout all ecological groups of the genus Flavivirus (Ochsenreiter, Hofacker, and Wolfinger 2019). Another element that is characteristic of flavivirus 3ʹUTRs is the long 3ʹ-terminal stem-loop (3’SL) structure, which is is involved in genome cyclization and panhandle formation during virus replication (Brinton and Basu 2015). As this element is indispensable for the virus life cycle, absence of a 3’SL homolog in sequence data is indicative of incomplete sequencing or truncation. Several other structured RNAs of known and unknown function are found in flavivirus 3ʹUTRs, including dumbbell (DB) elements and various stem-loop (SL) structures that are either conserved throughout the entire genus or selectively found in particular virus species (Wolfinger, Ochsenreiter, and Hofacker 2021).
1.2. TBEV diversity correlates with pathogenicity
TBEV has traditionally been classified into three subtypes, European (TBEV-Eur), Siberian (TBEV-Sib), and Far Eastern (TBEV-FE) (Ecker et al. 1999), based on serology, phylogenetic reasoning, and geographic location of early samples. Increased sampling over the previous decades resulted in the availability of more isolates that did not cluster with the established subtypes (Fig. 1). As such, additional lineages and provisional subtypes have been reported. These include strains that have been isolated in Eastern Siberia and that have accordingly been designated as ‘Baikalean’ isolates, i.e. genotype 4 (TBEV-Bkl-1), represented by the single strain 178–79 (Demina et al. 2012), and genotype 5 (TBEV-Bkl-2), encompassing strain 886–84 and related East-Siberian isolates (Demina et al. 2010; Kovalev and Mukhacheva 2017; Adelshin et al. 2019). Moreover, isolates found in wild rodents in the Quinghai-Tibet Plateau, China, constitute a Himalayan TBEV subtype (TBEV-Him) (Dai et al. 2018). The Siberian subtype, with prototype strain Aina, is considered the most abundant TBEV subtype (Tkachev et al. 2020), and is subdivided into five lineages: Zausaev, Vasilchenko, Baltic, Bosnia, and Obskaya (Gritsun et al. 1993; Golovljova et al. 2004, 2008; Tkachev et al. 2017), with Obsakya also being referenced as TBEV-Ob subtype in the literature (Deviatkin et al. 2020a). The Far-Eastern subtype encompasses three clades that have been referred to as Cluster I–III (Belikov et al. 2014). Each cluster has a prototype strain, i.e. Oshima (Cluster I), Sofijn (Cluster II), and Senzhang (Cluster III). The European subtype comprises lineages containing the Neudoerfl and Hypr stains, as well as a recently described Western European lineage that has been found in the Netherlands (Dekker et al. 2019). The Austrian strain N5-17, isolated from a chamois, shares ancestral roots with the other European clades. Notably, the European TBEV isolates are phylogenetically closer to Louping ill virus (LIV), which is present in the British Isles (Jeffries et al. 2014), than to the Eastern TBEV subtypes.
Figure 1.
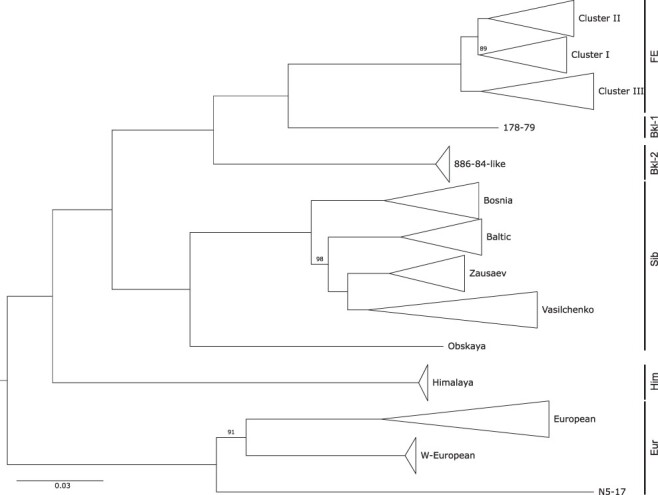
Overview phylogeny of TBEV, depicting the topological arrangement of subtypes TBEV-FE, TBEV-Sib, and TBEV-Eur (all shown sensu stricto here), as well as novel lineages that do not cluster with these subtypes. Strain 178–79 and the Baikalean lineage share ancestral roots with TBEV-FE. The Himalayan lineage is located ancestral to the clades encompassing TBEV-FE and TBEV-Sib subtypes. The European TBEV strains form a separate clade, with the recently detected Western European and N5-17 lineages being clearly separated from the established TBEV-Eur subtype. The midpoint-rooted maximum likelihood tree is based on 256 complete TBEV genomes. Bootstrap values are shown for branches with a support lower than 100.
In a recent study, the genomic diversity of established an provisional TBEV subtypes has
been assessed and classification of TBEV strains into seven subtypes has been proposed
based on a 10 per cent cutoff at nucleotide-level diversity (Deviatkin et al. 2020a). Despite their antigenic similarity, these
viruses differ not only in their phylogeography but also in virulence and pathogenicity.
The Far Eastern subtype is commonly considered the most pathogenic TBEV variant with the
highest number of reported cases of severe central nervous system (CNS) disease. Case
fatality rates of TBEV-FE are typically in the range of 5–35 per cent (Dumpis, Crook, and Oksi 1999), with extreme numbers
reaching up to 60 per cent (Gritsun, Lashkevich, and Gould
2003). On the contrary, the Siberian subtype typically results in less severe
disease with a fatality rate between 1 and 3 per cent (Gritsun, Lashkevich, and Gould 2003), but is more often associated with chronic
TBE (Charrel et al. 2004). Albeit case fatality at
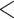 2 per cent is least for the European
subtype (Kaiser 1999), disease induced by TBEV-Eur
infection is typically biphasic with a viraemic phase associated with fever and myalgia,
followed by neurological manifestations of different severity during the second phase,
which occurs in 20–30 per cent of all patients (Gritsun,
Lashkevich, and Gould 2003).
2 per cent is least for the European
subtype (Kaiser 1999), disease induced by TBEV-Eur
infection is typically biphasic with a viraemic phase associated with fever and myalgia,
followed by neurological manifestations of different severity during the second phase,
which occurs in 20–30 per cent of all patients (Gritsun,
Lashkevich, and Gould 2003).
1.3. Functional RNAs in TBEV non-coding regions
The fundamental biological and biochemical traits that govern TBEV pathogenicity are only beginning to be understood. Besides several subtype-specific differences at the amino acid level (Belikov et al. 2014; Kellman et al. 2018), the non-coding portions of the TBEV genome are critical determinants of virulence. The 5ʹUTR is approximately 130 nt long and contains evolutionarily conserved RNA structures that are required for replication through cyclization and panhandle formation. Recently, a cis-acting RNA element in the 5ʹUTR of TBEV has been identified that mediates neurovirulence by hijacking the host mRNA transport system, thus allowing TBEV gRNA to be transported from the cell body to dendrites of neurons, where it replicates locally (Hirano et al. 2014, 2017).
The TBEV 3ʹUTR is longer than the 5ʹUTR and contains RNA elements that are involved in mediating immune escape and pathogenicity. It is characterized by a varied architecture of conserved RNA elements that has been associated with differential pathogenicity of particular subtypes (Belikov et al. 2014; Ternovoi et al. 2019). Although a few TBEV-FE isolates exist in public databases that appear to have extremely shortened 3ʹUTRs of only approximately 50 nt length, typical 3ʹUTRs range from 350 to 760 nt and comprise a 5ʹ-terminal variable region, located immediately downstream of the stop codon, and a 3ʹ-terminal core domain. Earlier comparative studies that have assessed the heterogeneity of these genomic regions at the sequence level have proposed various directed repeat sequences (Wallner et al. 1995; Gritsun et al. 1997). Considering RNA structure formation (Rauscher et al. 1997) allowed for a more fine-grained description and detailed comparative analysis of evolutionarily conserved RNA elements with other TBFVs (Ochsenreiter, Hofacker, and Wolfinger 2019).
An interesting aspect relates to the observation that strains of a particular subtype that have been isolated from ticks or rodents, respectively, are often markedly different from human isolates, particularly in their 3ʹUTRs. This might be the result of selective pressure on the TBEV 3ʹUTR during adaptation from a tick vector to a mammalian host (Asghar et al. 2014). Likewise, spontaneous deletions of parts of the variable region have been observed after repeated passage (Mandl et al. 1998; Ternovoi et al. 2019). This is complicated by the claim that loss of 3ʹUTR elements in infected mammalian cells is not quantitative, leading to a scenario that different portions of the viral population have different 3ʹUTR architectures, each having different replicative fitness when transmitted to ticks (Belikov et al. 2014). Given that strains resulting in different disease manifestations, ranging from subclinical to encephalitic forms, cluster within the same clades or subclades of the TBEV phylogeny (Belikov et al. 2014), the question of functional association and impact on clinical phenotype of different 3ʹUTR elements is emerging. There are contradictory reports in the literature regarding the influence of deletions in the 3ʹUTR variable region on the TBEV phenotype. While earlier works proposed no role of the variable region in virus replication and virulence (Mandl et al. 1998; Hoenninger et al. 2008), recent studies suggest increased virulence induced by partial deletions and polyA insertions in mouse models (Sakai et al. 2014, 2015).
In this study, we investigate the genetic diversity and phylogeographic spread of TBEV, based on a comprehensive set of more than 220 genomes that encompasses all known and provisional subtypes. By considering also strains that do not cluster with the established subtypes, we provide a unified picture of functional RNA conservation in TBEV 3ʹUTRs. Moreover, we contextualize genomic and epidemiological data through Nextstrain, providing an updated online resource to study the molecular evolution of TBEV.
2. Methods
2.1. Taxon and metadata collection
Viral genome and annotation data were downloaded from the public National Center for Biotechnology Information (NCBI) Genbank database (Clark et al. 2016) on 14 January 2021. We obtained 256 TBEV genomes, containing at least full length coding sequences. 221 sequences contained partial or full length 3ʹUTRs. We extracted metadata for these strains, including location and date of collection, subtype and host species from the Genbank record or from the literature. More than two dozen sequences that have been identified as vaccine strains, highly cell-passaged, or that were the result of cloning, mutants, or duplicates, were filtered out and excluded from downstream analyses. Strain identifiers which were not available through online NCBI resources, but obtained by literature research, were added manually.
Countries were assigned to regions according to the M49 Standard of the United Nations Statistic Division (https://unstats.un.org/unsd/methodology/m49/). A categorization of host species was included, which assigns the species of which a particular strain was isolated from to a category according to their role in viral transmission. These categories were vector (mites, ticks), natural host (rodentia, insectivora, aves), non-human dead-end host (large mammalia) or human.
2.2. TBEV subtype and lineage classification
To assign each strain in our data set to a particular TBEV subtype or lineage, we aligned 221 (near) full-length TBEV genomes with MAFFT v7.453 (Katoh and Standley 2013) and inferred a maximum likelihood (ML) phylogeny with iq-tree v1.6.12 (Nguyen et al. 2014) (Fig. 1), employing 1,000 ultrafast bootstrap replicates (Hoang et al. 2018). Strains with initially unknown subtype association were assigned to subtypes via distinguishing subtype- or lineage-specific monophyletic clades in the ML tree. Upon classification of strains, we used the multiple nucleotide sequence alignment (MSA) to identify all unique 3ʹUTR variants within each subtype and lineage, and selected a representative strain for each instance (twenty-eight strains, listed in Supplementary Table S1).
2.3. Characterization of RNA consensus secondary structures
Detection of evolutionary RNA structure conservation builds on the idea of finding homologous RNAs in phylogenetically narrow taxa via RNA family models (Eddy and Durbin 1994). Here, we used infernal (Nawrocki and Eddy 2013) covariance models (CMs) and employed an iterative workflow that has been used recently to characterize evolutionarily conserved RNAs in MBFVs (Wolfinger, Ochsenreiter, and Hofacker 2021). Starting from a set of TBFV CMs described in (Ochsenreiter, Hofacker, and Wolfinger 2019), we performed an initial screen of TBEV 3ʹUTRs. Genomic regions that were not covered by the initial CMs were then realigned with locARNA v2.0.0RC8 (Will et al. 2007) and RNA consensus structures were predicted with RNAalifold and RNALalifold from the ViennaRNA Package v2.4.18 (Lorenz et al. 2011).
2.4. Nextstrain phylogeography
The workflow management system Snakemake v5.32.1 (Köster
and Rahmann 2012) was used to build a pipeline for rapid and reproducible
deployment of Nextstrain (Hadfield et al. 2018)
data sets. augur v11.0.0 (Huddleston et al. 2021)
was used for tracking evolution from nucleotide sequence data, incorporating MAFFT v7.453
(Katoh and Standley 2013), iq-tree v1.6.12 (Nguyen et al. 2014), and TreeTime v0.8.4 (Sagulenko, Puller, and Neher 2018), for MSA, ML
phylogeny, and timed tree inference, respectively, resulting in a JSON file for Nextstrain
visualization. Values for the rate of evolution were chosen according to the molecular
clock rate that has been previously described as 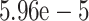 with standard
deviation of
with standard
deviation of 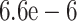 substitutions per
site and year (Bondaryuk et al. 2020).
substitutions per
site and year (Bondaryuk et al. 2020).
2.5. Data availability
Stockholm MSA files of the conserved RNA structures are available at the viRNA GitHub repository (https://github.com/mtw/viRNA). The TBEV Nextstrain build is available at https://nextstrain.org/groups/ViennaRNA/TBEVnext/. Upon display of the TBEV build in a web browser, all data can be downloaded by scrolling down to the bottom of the page and clicking on the ‘Download Data’ link.
3. Results
To obtain an updated view of TBEV subtype diversity, we compared publicly available genome data from a structural and molecular epidemiological perspective. For the structural analysis, we assessed the architectural organization of TBEV 5ʹUTRs and 3ʹUTRs, the latter constituting the most divergent part of the genome. We performed a comparative genomics screen that builds on the concept of predicting consensus structures, i.e. RNA secondary structure that can be formed by all sequences under consideration. We propose a conserved 5ʹUTR organization, as well as the existence of eight distinct, evolutionarily conserved RNA elements in the 3ʹUTR of different TBEV subtypes. The volatile arrangement of these non-coding RNAs (ncRNAs) at inter- and intra-subtype levels accounts for the diverse TBEV 3ʹUTR architectures observed in nature.
3.1. TBEV 5ʹUTRs are structurally conserved
The TBEV 5ʹUTR has a canonical length of 132 nt throughout all subtypes. Despite conservation of 5ʹUTR length, there is considerable sequence variability that accounts for average sequence identity levels between 89 and 95 per cent for TBEV-Sib and TBEV-FE, respectively (Ponomareva et al. 2021). This heterogeneity at the primary sequence level, however, does not translate into structural diversity. Consensus structure prediction yielded a clear picture of uniform 5ʹUTR organization throughout all TBEV subtypes and lineages: The first element of the TBEV genome is a variant of the Y-shaped SLA structure, which has been associated with panhandle formation and recruitment of the viral RNA-dependent RNA polymerase during virus replication (Filomatori et al. 2006). Downstream of the SLA element, a short hairpin (CSA) is located, which is followed by another hairpin (CSB) that overlaps the AUG start codon of the capsid protein coding region. Figure 2 shows the annotated 5ʹUTR of the TBEV reference strain Neudoerfl. Among the three 5ʹUTR-associated RNA structural elements, SLA and CSB exhibit covariation patterns, while CSA is highly sequence-conserved (Supplementary Fig. S1).
Figure 2.
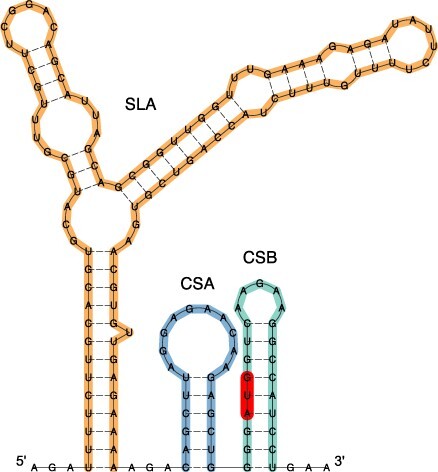
Secondary structure prediction of the 5ʹ-terminal 159 nt of TBEV strain Neudoerfl (NC_001672.1), comprising the 5ʹUTR and the distal portion of the capsid protein coding region. The canonical 5ʹUTR organization of TBEV encompasses a Y-shaped SLA element (orange), as well as the hairpin loops CSA (blue), and CSB (green). The start codon is highlighted in red.
3.2. TBEV 3ʹUTRs comprise up to eight distinct RNA elements
Starting from nucleotide multiple sequence alignments of 221 TBEV genomes that covered all known subtypes and lineages, we set out to construct consensus RNA structures of conserved elements. Following up and building on previous work (Ochsenreiter, Hofacker, and Wolfinger 2019), we identified seven distinguishable families of conserved RNA structures that are found as single, double, or triple copies in a variable arrangement in TBEV strains. Together with the poly-A tract that is found in some TBEV-Eur strains, we characterized eight unique ncRNA elements in TBEV 3ʹUTRs. Inconsistencies with the naming schema of structured TBEV 3ʹUTR elements in the literature have lead us to introduce here a pragmatic naming system for conserved ncRNA elements.
The structured elements encompass four distinct conserved stem-loop (CSL) elements of unknown function, labeled CSL1 through CSL4, starting from the most 3ʹ-terminal elements. Predicted consensus secondary structures of these elements are shown in Supplementary Figs. S2 and S3. CSL1, CSL2, and CSL4 are short SL elements that are found in variable copy numbers in different TBEV strains. We have previously shown evolutionary conservation of CSL2 in other TBFVs, where this element was referred to T.SL6 (Ochsenreiter, Hofacker, and Wolfinger 2019). CSL4, which has a canonical length of approximately 30 nt, has a consensus structure that contains multiple gap characters in the hairpin loop. This is due to the insertion of a poly-A region of variable length in the apical loop of one CSL4 copy in some TBEV-Eur strains. On the contrary, CSL3 is markedly longer than the other CSL elements and is interspersed with several interior loops. Comparison of covariance models with CMCompare revealed that CSL3 is not structurally homologous to SL-III elements that were found in various MBFVs (Wolfinger, Ochsenreiter, and Hofacker 2021).
Besides the CSL elements, TBEV 3ʹUTRs contain previously described ncRNAs with known functional associations, such as three-way junction elements that form exoribonuclease-resistant RNAs (xrRNAs). These elements, which are ubiquitous in the viral world (Steckelberg, Vicens, and Kieft 2018; Dilweg, Gultyaev, and Olsthoorn 2019), provide quantitative protection of downstream nucleotides against degradation by endogenous exoribonucleases and have been structurally and mechanistically characterized in different TBFVs (MacFadden et al. 2018; Harima et al. 2021). A Y-shaped structure (Y1), together with the the 3ʹ-terminal part of TBEV 3ʹUTRs, encompassing a small hairpin loop and a long stem-loop element, labelled 3’SL here, comprise the promoter element, which is essential for virus replication. Screening for structural homology of the above elements in other, i.e. non-TBFVs corroborated earlier knowledge that xrRNA and 3’SL elements are ubiquitously conserved throughout the genus Flavivirus. While the CSL elements do not appear to be universally conserved outside TBFVs, a structurally homologous Y1 element was found in the 3ʹUTR of Modoc virus (MODV), a tick-borne related no-known vector flavivirus.
A structural annotation of TBEV 3ʹUTRs is shown in Fig. 3, which highlights the differences in architectural organization among and within TBEV subtypes. Selection of strains featured in Fig. 3 was based on subtype diversity, i.e. coverage of all established and prospective subtypes, as well as within-subtype variability of 3ʹUTR organization. We included at least one representative isolate of each 3ʹUTR variant that has been present in our data set. Full-length 3ʹUTRs (where available) are plotted to scale in Fig. 3, with coloured boxes representing evolutionarily conserved RNA elements. Homologous elements are shown in the same colour for each strain.
Figure 3.
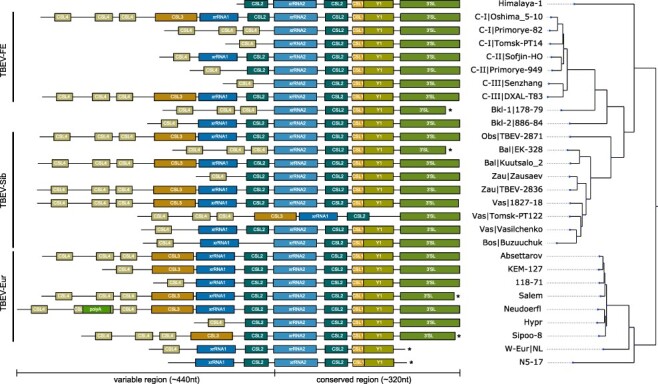
Annotated 3ʹUTRs of representative TBEV strains. Strains were selected to cover the complete 3ʹUTR diversity found in publicly available genome data. 3ʹUTR schemes are plotted to scale, highlighting the variable length and architectural organization of individual strains. Coloured boxes represent evolutionarily conserved, structured RNAs. Truncated sequences from incomplete sequencing are marked by an asterisk. The phylogenetic tree on the right has been computed from full genome nucleotide multiple sequence alignments of 28 TBEV strains. Identifiers show strain name, and lineage association where available, i.e. TBEV-FE Clusters I/II/III (C-I/C-II/C-III), the two Baikalean subtypes TBEV-Bkl-1 (Bkl-1), and TBEV-Bkl-2 (Bkl-2), and TBEV-Sib lineages Baltic (Bal), Zausaev (Zau), Vasilchenko (Vas), Bosnia (Bos), Obskaya (Obs). Accession numbers and 3ʹUTR lengths are listed in Supplementary Table S1.
Our comparative analysis of TBEV 3ʹUTRs suggests a common pattern of structural conservation, revealing that both core and variable regions contain specific RNA elements that are characteristic of their location within the 3ʹUTR. This is particularly marked in the core region, which contains five highly conserved structured RNA elements within approximately 320 nt. These include an exoribonuclease-resistant RNA (xrRNA2), CSL2, CSL1, Y1, and the 3ʹ-terminal long SL structure (3’SL). Consensus secondary structures of core region elements are shown in Supplementary Fig. S2. The well-conserved architecture of potentially functional RNAs in the core region is observed in all strains considered here, except Tomsk-PT122 (TBEV-Sib).
The variable region ranges between approximately 80 (Tomsk-PT-14) and 440 nt (Neudoerfl), and contains up to five different types of RNA elements, i.e. CSL4, poly-A, CSL3, xrRNA1, and CSL2. Consensus secondary structures are shown in Supplementary Fig. S3. Some of these RNAs occur in multiple copies at particular loci. Most strains contain between one and three copies of CSL4 at the 5ʹ-terminal portion of their 3ʹUTRs. There are only two exceptions to this observation within our set of strains, i.e. Himalaya-1 and N5-17. The internal poly-A region observed in some European strains is inserted into the hairpin loop of the second CSL4 copy. While a variable number of CSL4 homologs is found in almost all strains, a long variant of the remaining portion of the variable region, containing CSL3, an exoribonuclease-resistant RNA (xrRNA1), and CSL2, is only conserved in some strains. These long 3ʹUTR variants are found in all subtypes, e.g. Oshima 5–10 (TBEV-FE), 1827–18 (TBEV-Sib), and Absettarov (TBEV-Eur). The structured elements CSL3, xrRNA1 and CSL2 in the variable region appear to be dispensable for virus replication, as other strains either lack a CSL3 element (e.g. Sofjin-HO or Vasilchenko), CSL3 and xrRNA1 (e.g. Hypr or Zausaev), or CSL3, xrRNA1 and CSL2 (e.g. Senzhang or Tomsk PT14).
To illustrate the differences in 3ʹUTR architectures due to alternative layout of the variable region, we show secondary structure plots of four complete 3ʹUTRs in Fig. 4. With all strains exhibiting a common core region architecture, we depict representative strains of the European subtype (Neudoerfl, panel A) and the Obskaya lineage of the Siberian subtype (TBEV-2871, panel B), that both exhibit long variants of the variable region. These comprise three CSL4 copies, CSL3, xrRNA1 and one CSL2 element. Insertion of a poly-A stretch results in the formation of a large hairpin loop in the second CSL4 copy of the Neudoerfl strain. The 886–84 strain, representing the Baikalean subtypes, (panel C) has a considerably shorter variable region that contains a single CSL4 copy, xrRNA1, and CSL2, with additional CSL4 structures and CSL3 missing. The Shenzhang strain of the Far Eastern subtype (depicted in panel D) has the shortest 3ʹUTR in our data set, and contains just one CSL4 element in the variable region.
Figure 4.
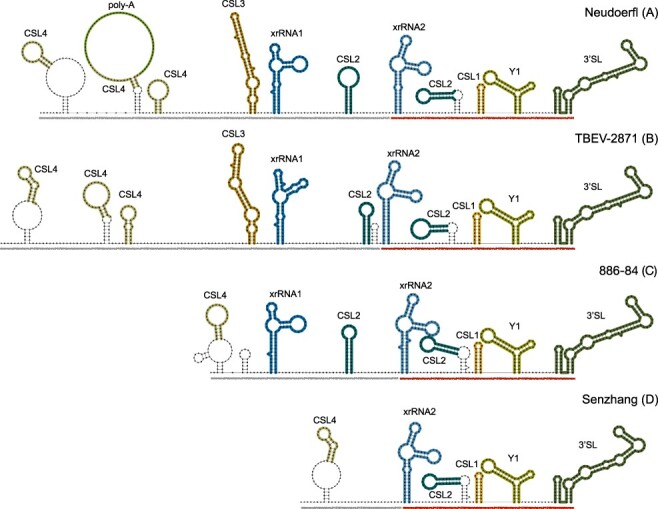
Diverging 3ʹUTR architecture in four representative TBEV strains, Neudoerfl (A), TBEV-2871 (B), 886–84 (C), and Senzhang (D). Core and variable regions are underlined in red and grey, respectively. Structural homologs of evolutionarily conserved RNA elements are depicted in the same colour for all strains (matching the colours used in Fig. 3). Structural components that are not found in all homologs, such as closing stems of some CSL4 and CSL2 elements, or additional short stem loops that are only predicted in particular strains, are not considered conserved.
Some of the recently described strains have a 3ʹUTR organization that is also found in other lineages. The Western European strain NL and N5-17, while both being truncated at the 3ʹ end, show similar patterns e.g. to strain 886–84. On the contrary, other strains exhibit alternative 3ʹUTR architectures that are manifested by unique organizational patterns in the variable region that are not observed elsewhere. For example, the 3ʹUTR of Himalaya-1 does not have a CSL4 element at the 5ʹ-end, but begins with a CSL2 structure. The Buzuuchuk strain, representing the Bosnian lineage of TBEV-Sib, does not have a CSL2 element in the variable region. Sipoo-8, collected in Finland, lacks the exoribonuclease-resistant RNA element in the variable region, although it has three CSL4 elements and CSL3. The most unconventional 3ʹUTR organization is found in Tomsk-PT122 (Vasilchenko lineage, TBEV-Sib), which is the only example of a strain that exhibits deletions the core region. Tomsk-PT122 is predicted to contain the long version of the variable region described above, while xrRNA2, CSL2, CSL1 and Y elements are are not found in the core region.
3.3. TBEVnext: TBEV molecular epidemiology
To get a comprehensive picture of TBEV phylogeography, considering all publicly available isolates, we sought to to investigate the molecular epidemiology and phylodynamics of TBEV. To this end, we compiled an interactive visualization of the global TBEV spread in Nextstrain, termed TBEVnext, which is publicly available at https://nextstrain.org/groups/ViennaRNA/TBEVnext. At the time of writing, TBEvnext v1.0 has been made available with 225 TBEV strains, encompassing all subtypes and lineages.
The overall topology of our TBEV phylogeny (Fig. 5A) is in agreement with the literature, featuring Eastern (TBEV-FE, TBEV-Sib), and Western (TBEV-Eur) types as major clades (Heinze, Gould, and Forrester 2012). Strains that are not assigned to one of the larger clades are 178–79 (TBEV-Bkl-1), which is located ancestral to the TBEV-FE clade, and N5-17, which appears ancestral to all other European clades. The East-Siberian/Baikalean/886-84-like (TBEV-Bkl-2) lineage, as well as the Western European and Himalayan lineages are smaller monophyletic groups that comprise between two and eight isolates. TBEV-Bkl-2 clusters with the Far-Eastern clade, Western European isolates cluster with TBEV-EUR, and the Himalayan isolates share ancestral roots with the clade that comprises all Eastern TBEV isolates. Divergence of the different TBEV subtypes and lineages is exposed in the TBEVnext data set through an unrooted phylogenetic tree (Fig. 5B).
Figure 5.
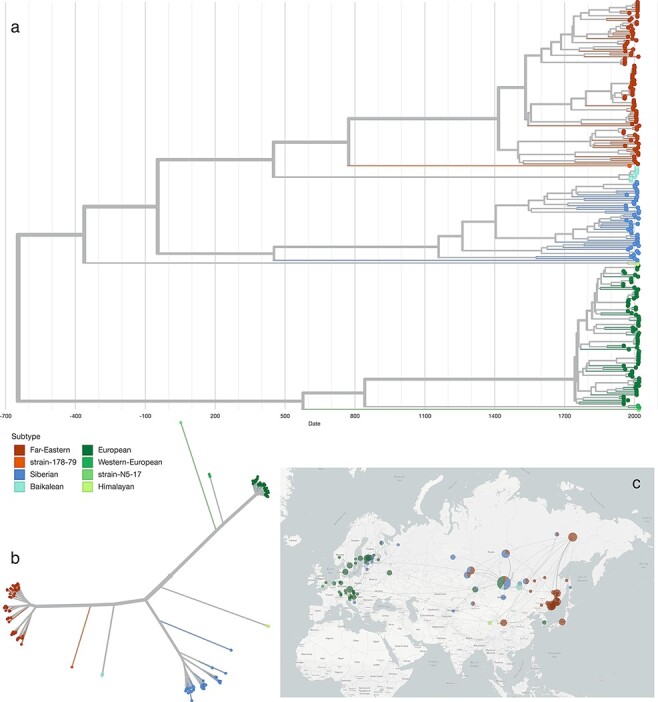
Panel a: Visualization of the TBEV phylodynamics in Nextstrain and Panel b: Unrooted phylogenetic tree, exposing the divergence of TBEV subtypes and novel lineages. c Geographic spread of TBEV across Eurasia.
The Nextstrain framework is particularly useful for studying phylogeographic traits. As such, we established a fine-grained geographic location labelling of strains that had relevant location information (i.e. place of isolation) in their metadata. While an exact place of isolation could not be inferred for all strains, sub-national geo-locations are available for many isolates in TBEVnext. This is particularly relevant for studying the spread of different subtypes across geographically extended regions, such as presence of various subtypes in the Russian Federation. An example is Irkutsk Oblast, which shows an accumulation of different subtypes: Western, East-Siberian, Siberian, Far-Eastern and strain 178–79 were collected in this region.
The TBEV Nextstrain data set features a timed tree, which provides estimates for the time of the most recent common ancestor (TMRCA) associated with each clade (Table 1). The timed tree has been inferred from a ML tree with TreeTime, assuming a constant evolutionary rate throughout the entire tree. Diverging nucleotide substitution rates have been proposed for different TBEV subtypes (Subbotina and Loktev 2012; Bondaryuk et al. 2020; Deviatkin et al. 2020b), hence our numbers should be seen as a rough estimate.
Table 1.
TMRCA estimates of distinct clades and associated subtypes of the TBEV phylogeny extracted from the TBEV Nextstrain data set (all dates are given as year CE).
| Clade | Subtype | TMRCA | Confidence interval | First isolation |
|---|---|---|---|---|
| Siberian | TBEV-Sib | 452 | 121–703 | 1963 |
| Far-Eastern | TBEV-FE | 1416 | 1287–1512 | 1937 |
| Himalayan | TBEV-Him | 1971 | 1960–1985 | 2013 |
| Strain 178–79 | TBEV-Bkl-1 | 773 | 493–961 | 1979 |
| East-Siberian/Baikalean/886-84-like | TBEV-Bkl-2 | 1933 | 1917–1947 | 1984 |
| European | TBEV-Eur | 1744 | 1703–1786 | 1951 |
| Western European | TBEV-Eur | 1973 | 1962–1987 | 2015 |
| Strain N5–17 | TBEV-Eur | 576 | 274–809 | 2017 |
Animation of historic dispersal events across Eurasia within Nextstrain suggests that TBEV originated in Central Russia almost 2,700 years ago, and subsequently moved both eastwards, forming the TBEV-Sib and TBEV-FE lineages, and westwards into Europe. While this observation is in agreement with earlier reports (Heinze, Gould, and Forrester 2012), the deep splits of the Western types with inferred TMRCAs approximately 1,000–1,500 years ago raises the question whether TBEV has arrived in Central Europe earlier than previously reported.
One of Nextstrain’s strengths is the possibility to augment the data set with custom traits, in addition to default attributes like date and place of collection. Adopting this feature, we maintain in TBEVnext a mapping of species from which a particular strain has been isolated. It has been previously described that different TBEV subtypes are specialized to particular vector species of Ixodidae. The Eastern subtypes (Far-Eastern, Siberian) are typically transmitted via Ixodes persulcatus whereas the Western subtypes (European) are usually transmitted by Ixodes ricinus ticks. While this is broadly supported by our data, metadata analysis of 225 full genome isolates also discloses counter-examples: Nine isolates collected from Ixodes persulcatus ticks between 1971 and 2009 in Irkutsk Oblast and the Altai region in Russia carried the European subtype, while two Ixodes ricinus ticks carried the Siberian subtype. (Data shown in the interactive visualization of TBEVnext).
4. Discussion
Increased sample collection and availability of large amounts of genome data in public databases has stipulated research on flaviviruses in recent years. TBEV, being endemic in large parts of Europe and Asia, has been intensively studied, resulting in numerous publications focusing on the functional associations of the variable 3ʹUTR region in particular strains and viral (neuro)tropism. Some of these studies reported a variety of sequence repeats in the 3ʹUTR of TBFVs, based on qualitative comparisons at the nucleotide level. Other studies performed computational modelling of RNA secondary structure at the level of individual sequences, yielding a varied picture of stem-loop structures that makes it difficult to compare structured entities. A unified, reproducible picture of ncRNA structure conservation in different TBEV subtypes has not been available. Here, we set out to obtain an updated view of RNA structure conservation in TBEV 3ʹUTRs, aiming at capturing a unified picture of the architectural diversity found in different subtypes. To this end, we performed a comparative genomics screen in TBEV 3ʹUTRs that cover all known subtypes and lineages. The computational approach employed here is based on a thermodynamic model of RNA structure formation, as implemented in the ViennaRNA package.
TBEV 3ʹUTRs comprise two domains, a core domain at the 3ʹend of the viral genome and a variable region upstream of the core domain. The 3ʹ-terminal core domain constitutes the most conserved portion of TBEV 3ʹUTRs, and harbors five different structured ncRNA elements. Two of these, an exoribonuclease-resistant RNA (xrRNA2) and the terminal 3ʹ stem-loop element are also known in other ecological groups of flaviviruses and have been associated with mediating virulence and virus replication (Wolfinger, Ochsenreiter, and Hofacker 2021). The variable region, on the other hand, contains between two and four distinct structural elements. Observed differences in the length of TBEV genomes that belong to different subtypes is primarily due to heterogeneity of the variable region in TBEV 3ʹUTRs. Here, another xrRNA copy (xrRNA1) constitutes the only element with known functional association, while simple and extended stem-loop structures of unknown function appear in variable copy numbers. Moreover, an internal poly-A tract is found in several European isolates (Mandl, Kunz, and Heinz 1991).
TBEV fits well into the picture of selective conservation of RNA structure in the viral 3ʹUTR, with core and variable regions containing functional RNA elements, i.e. xrRNAs, that are also found in other arboviruses. Unlike other viruses that exhibit strictly lineage-specific patterns of 3ʹUTR architecture, such as Chikungunya virus (de Bernardi Schneider et al. 2019; Spicher et al. 2021), TBEV shows a large degree of 3ʹUTR variability within particular subtypes and lineages. Our data highlight that there is no common 3ʹUTR architecture associated with certain TBEV subtypes besides the core region. On the other side, we show that the TBEV 3ʹ architectural diversity is realized by combination of a restricted pool of eight structural and one non-structural elements, suggesting that the observed 3ʹUTR variability is driven by structural, rather than sequence conservation.
Beyond studying evolutionary traits of TBEV at the level of RNA structure conservation, we address the issue also from the perspective of molecular epidemiology. To this end, we provide TBEVnext, a Nextstrain dataset that reveals spatiotemporal and phylodynamic characteristics of the global TBEV spread. Our dataset comprises 225 complete genomes from Eurasia that have been sampled between 1951 and 2019, thereby representing the most comprehensive view of TBEV phylogeographics so far.
In addition to the three major TBEV subtypes TBEV-Eur, TBEV-Sib, and TBEV-Eur, TBEVnext encompasses provisional subtypes like TBEV-Bkl1, TBEV-Bkl-2, and TBEV-Him. The Obskaya lineage of TBEV-Sib, deemed an independent subtype in the recent literature, is considered part of the Siberian clade here, while the strain N5-17 is considered part of TBEV-Eur. Analysis of the geographic dispersion of these clades in TBEVnext immediately reveals that they are not specific for particular regions of the world. Lack of an unique association between TBEV subtypes (clades) and geographic occurrence is particularly evident in Irkutsk Oblast, where different strains of Siberian, Far-Eastern, Western, and Baikalean subtypes have been isolated. Likewise, the presence of Western strains in the Republic of Korea strains has been reported (Yun et al. 2011).
TBEVnext features a timed phylogenetic tree that has been inferred from a maximum-likelihood phylogeny under the assumption of a constant evolutionary rate, effectively neglecting the possibility of divergent rates in different parts of the tree. Therefore, our timed tree should be seen as a proxy that is in agreement with proposed TBEV tree topologies and divergence patterns. Importantly, the order of TMRCAs of different clades is in overall agreement with published data that were based on computationally more demanding Bayesian approaches (Adelshin et al. 2019).
Availability of global vector/host associations of many strains in TBEVnext yields a comprehensive view on ecological aspects of the global virus spread and makes unique properties of strains accessible.
In summary, we provide here an updated picture of the different evolutionary aspects of TBEV. Our data expose patterns of pervasive conservation of individual RNA elements in TBEV 3ʹUTRs and provide an interactive view of TBEV molecular epidemiology that yields novel insight into the global virus spread.
Supplementary Material
Acknowledgements
We thank Gabor Erdo Mate for fruitful discussions during the preparation of the manuscript.
Contributor Information
Lena S Kutschera, Department of Theoretical Chemistry, University of Vienna, Währinger Straße 17, Vienna 1090, Austria.
Michael T Wolfinger, Department of Theoretical Chemistry, University of Vienna, Währinger Straße 17, Vienna 1090, Austria; Research Group Bioinformatics and Computational Biology, Faculty of Computer Science, University of Vienna, Währinger Straße 29, Vienna 1090, Austria.
Supplementary data
Supplementary data are available at Virus Evolution online.
Conflict of interest:
None declared.
References
- Adelshin R. V. et al. (2019) ‘‘886-84-Like’ Tick-Borne Encephalitis Virus Strains: Intraspecific Status Elucidated by Comparative Genomics’, Ticks and Tick-borne Diseases, 10: 1168–72. [DOI] [PubMed] [Google Scholar]
- Amicizia D. et al. (2013) ‘Epidemiology of Tick-Borne Encephalitis (TBE) in Europe and Its Prevention by Available Vaccines’, Human Vaccines & Immunotherapeutics, 9: 1163–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar N. et al. (2014) ‘Tick-Borne Encephalitis Virus Sequenced Directly from Questing and Blood-Feeding Ticks Reveals Quasispecies Variance’, PLoS One, 9: e103264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrows N. J. et al. (2018) ‘Biochemistry and Molecular Biology of Flaviviruses’, Chemical Reviews, 118: 4448–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belikov S. I. et al. (2014) ‘The Relationship between the Structure of the Tick-Borne Encephalitis Virus Strains and Their Pathogenic Properties’, PLoS One, 9: e94946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitvich B., and Firth A. (2015) ‘Insect-Specific Flaviviruses: A Systematic Review of Their Discovery, Host Range, Mode of Transmission, Superinfection Exclusion Potential and Genomic Organization’, Viruses, 7: 1927–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondaryuk A. N. et al. (2020) ‘Reporting of New Tick-borne Encephalitis Virus Strains Isolated in Eastern Siberia (Russia) in 1960–2011 and Explaining Them in an Evolutionary Context Using Bayesian Phylogenetic Inference’, Ticks and Tick-borne Diseases, 11: 101496. [DOI] [PubMed] [Google Scholar]
- Brinton M. A., and Basu M. (2015) ‘Functions of the 3’ and 5’ Genome RNA Regions of Members of the Genus Flavivirus’, Virus Research, 206: 108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charrel R. N. et al. (2004) ‘Tick-Borne Virus Diseases of Human Interest in Europe’, Clinical Microbiology and Infection, 10: 1040–55. [DOI] [PubMed] [Google Scholar]
- Clark K. et al. (2016) ‘GenBank’, Nucleic Acids Research, 44: D67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X. et al. (2018) ‘A New Subtype of Eastern Tick-Borne Encephalitis Virus Discovered in Qinghai-Tibet Plateau, China’, Emerg Microb Infect, 7: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bernardi Schneider A. et al. (2019) ‘Updated Phylogeny of Chikungunya Virus Suggests Lineage-Specific RNA Architecture’, Viruses, 11: 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Borba L. et al. (2015) ‘Overlapping Local and Long-Range RNA-RNA Interactions Modulate Dengue Virus Genome Cyclization and Replication’, Journal of Virology, 89: 3430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker M. et al. (2019) ‘Emergence of Tick-Borne Encephalitis (TBE) in the Netherlands’, Ticks and Tick-borne Diseases, 10: 176–9. [DOI] [PubMed] [Google Scholar]
- Demina T. V. (2010) ‘Genotyping and Characterization of the Geographical Distribution of Tick-Borne Encephalitis Virus Variants with a Set of Molecular Probes’, Journal of Medical Virology, 82: 965–76. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2012) ‘Genotypes 4 and 5 of the Tick-Borne Encephalitis Virus: Features of the Genome Structure and Possible Scenario for Its Formation’, Voprosy Virusologii, 57: 13–9. [PubMed] [Google Scholar]
- Deviatkin A. A. (2020a) ‘TBEV Subtyping in Terms of Genetic Distance’, Viruses, 12: 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2020b) ‘Tick-Borne Encephalitis Virus: An Emerging Ancient Zoonosis?’ Viruses, 12: 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilweg I. W., Gultyaev A. P., and Olsthoorn R. C. (2019) ‘Structural Features of an Xrn1-Resistant Plant Virus RNA’, RNA Biology, 16: 838–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumpis U., Crook D., and Oksi J. (1999) ‘Tick-Borne Encephalitis’, Clinical Infectious Diseases, 28: 882–90. [DOI] [PubMed] [Google Scholar]
- Ecker M. et al. (1999) ‘Sequence Analysis and Genetic Classification of Tick-Borne Encephalitis Viruses from Europe and Asia’, Journal of General Virology, 80: 179–85. [DOI] [PubMed] [Google Scholar]
- Eddy S. R., and Durbin R. (1994) ‘RNA Sequence Analysis Using Covariance Models’, Nucleic Acids Research, 22: 2079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filomatori C. V. et al. (2006) ‘A 5’ RNA Element Promotes Dengue Virus RNA Synthesis on A Circular Genome’, Genes & Development, 20: 2238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovljova I. (2008) ‘Unique Signature Amino Acid Substitution in Baltic Tick-Borne Encephalitis Virus (TBEV) Strains within the Siberian TBEV Subtype’, International Journal of Medical Microbiology, 298: 108–20. [Google Scholar]
- ——— et al. (2004) ‘Characterization of Tick-Borne Encephalitis Virus from Estonia’, Journal of Medical Virology, 74: 580–8. [DOI] [PubMed] [Google Scholar]
- Gritsun T. S. et al. (1993) ‘Nucleotide and Deduced Amino Acid Sequence of the Envelope Gene of the Vasilchenko Strain of TBE Virus; Comparison with Other Flaviviruses’, Virus Research, 27: 201–9. [DOI] [PubMed] [Google Scholar]
- Gritsun T. S., Lashkevich V. A., and Gould E. A. (2003) ‘Tick-Borne Encephalitis’, Antiviral Research, 57: 129–46. [DOI] [PubMed] [Google Scholar]
- Gritsun T. S. et al. (1997) ‘Complete Sequence of Two Tick-Borne Flaviviruses Isolated from Siberia and the UK: Analysis and Significance of the 5’ and 3’-UTRs’, Virus Research, 49: 27–39. [DOI] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘Nextstrain: Real-Time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harima H. et al. (2021) ‘An African Tick Flavivirus Forming an Independent Clade Exhibits Unique Exoribonuclease-Resistant RNA Structures in the Genomic 3’-Untranslated Region’, Scientific Reports, 11, Article number: 4883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinze D. M., Gould E. A., and Forrester N. L. (2012) ‘Revisiting the Clinal Concept of Evolution and Dispersal for the Tick-Borne Flaviviruses by Using Phylogenetic and Biogeographic Analyses’, Journal of Virology, 86: 8663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M. (2017) ‘Dendritic Transport of Tick-Borne Flavivirus RNA by Neuronal Granules Affects Development of Neurological Disease’, Proceedings of the National Academy of Sciences, 114: 9960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘Tick-Borne Flaviviruses Alter Membrane Structure and Replicate in Dendrites of Primary Mouse Neuronal Cultures’, Journal of General Virology, 95: 849–61. [DOI] [PubMed] [Google Scholar]
- Hoang D. T. et al. (2018) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenninger V. M. et al. (2008) ‘Analysis of the Effects of Alterations in the Tick-Borne Encephalitis Virus 3’-Noncoding Region on Translation and RNA Replication Using Reporter Replicons’, Virology, 377: 419–30. [DOI] [PubMed] [Google Scholar]
- Huddleston J. et al. (2021) ‘Augur: A Bioinformatics Toolkit for Phylogenetic Analyses of Human Pathogens’, Journal of Open Source Software, 6: 2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffries C. L. et al. (2014) ‘Louping Ill Virus: An Endemic Tick-Borne Disease of Great Britain’, Journal of General Virology, 95: 1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser R. (1999) ‘The Clinical and Epidemiological Profile of Tick-Borne Encephalitis in Southern Germany 1994–98: A Prospective Study of 656 Patients’, Brain, 122: 2067–78. [DOI] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellman E. M. et al. (2018) ‘Viral Determinants of Virulence in Tick-Borne Flaviviruses’, Viruses, 10: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster J., and Rahmann S. (2012) ‘Snakemake-a Scalable Bioinformatics Workflow Engine’, Bioinformatics, 28: 2520–2. [DOI] [PubMed] [Google Scholar]
- Kovalev S. Y., and Mukhacheva T. A. (2017) ‘Reconsidering the Classification of Tick-Borne Encephalitis Virus within the Siberian Subtype Gives New Insights into Its Evolutionary History’, Infection, Genetics and Evolution, 55: 159–65. [DOI] [PubMed] [Google Scholar]
- Labuda M. et al. (1993) ‘Non-Viraemic Transmission of Tick-Borne Encephalitis Virus: A Mechanism for Arbovirus Survival in Nature’, Experientia, 49: 802–5. [DOI] [PubMed] [Google Scholar]
- Lorenz R. et al. (2011) ‘ViennaRNA Package 2.0’, Algorithms for Molecular Biology, 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFadden A. et al. (2018) ‘Mechanism and Structural Diversity of Exoribonuclease-Resistant RNA Structures in Flaviviral RNAs’, Nature Communications, 9: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl C. W. et al. (1998) ‘Spontaneous and Engineered Deletions in the 3’ Noncoding Region of Tick-Borne Encephalitis Virus: Construction of Highly Attenuated Mutants of a Flavivirus’, Journal of Virology, 72: 2132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandl C. W., Kunz C., and Heinz F. X. (1991) ‘Presence of Poly(A) in a Flavivirus: Significant Differences between the 3’Noncoding Regions of the Genomic RNAs of Tick-Borne Encephalitis Virus Strains’, Journal of Virology, 65: 4070–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki E. P., and Eddy S. R. (2013) ‘Infernal 1.1: 100-Fold Faster RNA Homology Searches’, Bioinformatics, 29: 2933–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng W. et al. (2017) ‘The 5’ and 3’ Untranslated Regions of the Flaviviral Genome’, Viruses, 9: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2014) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochsenreiter R., Hofacker I. L., and Wolfinger M. T. (2019) ‘Functional RNA Structures in the 3’UTR of Tick-Borne, Insect-Specific and No-Known-Vector Flaviviruses’, Viruses, 11: 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijlman G. P. et al. (2008) ‘A Highly Structured, Nuclease-Resistant, Noncoding RNA Produced by Flaviviruses Is Required for Pathogenicity’, Cell Host & Microbe, 4: 579–91. [DOI] [PubMed] [Google Scholar]
- Ponomareva E. P. et al. (2021) ‘Genetic Variability of Tick-Borne Encephalitis Virus Genome 5’-UTR from Northern Eurasia’, Molecular Biology, 55: 372–80. [DOI] [PubMed] [Google Scholar]
- Rauscher S. et al. (1997) ‘Secondary Structure of the 3’-Noncoding Region of Flavivirus Genomes: Comparative Analysis of Base Pairing Probabilities’, RNA, 3: 779–91. [PMC free article] [PubMed] [Google Scholar]
- Sagulenko P., Puller V., and Neher R. A. (2018) ‘TreeTime: Maximum-Likelihood Phylodynamic Analysis’, Virus Evolution, 4: vex042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai M. (2015) ‘Virulence of Tick-Borne Encephalitis Virus Is Associated with Intact Conformational Viral RNA Structures in the Variable Region of the 3’-UTR’, Virus Research, 203: 36–40. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2014) ‘The Variable Region of the 3’untranslated Region Is a Critical Virulence Factor in the Far-Eastern Subtype of Tick-Borne Encephalitis Virus in Mouse Model’, Journal of General Virology, 95: 823–35. [DOI] [PubMed] [Google Scholar]
- Spicher T. et al. (2021) ‘Dynamic Molecular Epidemiology Reveals Lineage-Associated Single-Nucleotide Variants that Alter RNA Structure in Chikungunya Virus’, Genes, 12: 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steckelberg A.-L., Vicens Q., and Kieft J. S. (2018) ‘Exoribonuclease-Resistant RNAs Exist within Both Coding and Noncoding Subgenomic RNAs’, MBio, 9: e02461–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbotina E. L., and Loktev V. B. (2012) ‘Molecular Evolution of the Tick-Borne Encephalitis and Powassan Viruses’, Molecular Biology, 46: 75–84. [PubMed] [Google Scholar]
- Ternovoi V. A. (2019) ‘Variability in the 3’ Untranslated Regions of the Genomes of the Different Tick-Borne Encephalitis Virus Subtypes’, Virus Genes, 55: 448–57. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2003) ‘Tick-Borne Encephalitis with Hemorrhagic Syndrome, Novosibirsk Region, Russia, 1999’, Emerging Infectious Diseases, 9: 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkachev S. E. (2020) ‘Genetic Diversity and Geographical Distribution of the Siberian Subtype of the Tick-Borne Encephalitis Virus’, Ticks and Tick-borne Diseases, 11: 101327. [DOI] [PubMed] [Google Scholar]
- ——— et al. (2017) ‘New Genetic Lineage within the Siberian Subtype of Tick-Borne Encephalitis Virus Found in Western Siberia, Russia’, Infection, Genetics and Evolution, 56: 36–43. [DOI] [PubMed] [Google Scholar]
- Villordo S. M., Alvarez D. E., and Gamarnik A. V. (2010) ‘A Balance between Circular and Linear Forms of the Dengue Virus Genome Is Crucial for Viral Replication’, RNA, 16: 2325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallner G. et al. (1995) ‘The Flavivirus 3’-Noncoding Region: Extensive Size Heterogeneity Independent of Evolutionary Relationships among Strains of Tick-Borne Encephalitis Virus’, Virology, 213: 169–78. [DOI] [PubMed] [Google Scholar]
- Will S. et al. (2007) ‘Inferring Noncoding RNA Families and Classes by Means of Genome-Scale Structure-Based Clustering’, PLoS Computational Biology, 3: e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfinger M. T.Ochsenreiter R., and Hofacker I. L. (2021) ‘Functional RNA Structures in the 3’UTR of Mosquito-Borne Flaviviruses’. In: Frishman, D., and Marz, M. (eds) Virus Bioinformatics, pp. 65–100. Chapman: Hall/CRC Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S.-M. et al. (2011) ‘First Complete Genomic Characterization of Two Tick-Borne Encephalitis Virus Isolates Obtained from Wild Rodents in South Korea’, Virus Genes, 42: 307–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Stockholm MSA files of the conserved RNA structures are available at the viRNA GitHub repository (https://github.com/mtw/viRNA). The TBEV Nextstrain build is available at https://nextstrain.org/groups/ViennaRNA/TBEVnext/. Upon display of the TBEV build in a web browser, all data can be downloaded by scrolling down to the bottom of the page and clicking on the ‘Download Data’ link.