

Abstract
Cobalamin riboswitch is a cis-regulatory element widely found in the 5’-UTRs of the vitamin B12-associated genes in bacteria, resulting in modulation and production of a particular protein. Thermoanaerobacter tengcongensis (Tte) AdoCbl riboswitches are the largest of the known riboswitches with 210 nucleotides, partially due to its long peripheral P6-extension, which enable high affinity of AdoCbl. Two structural elements, T-loop/T-looplike motif and kissing loop are key to the global folding of the RNA. While the structure of the TteAdoCbl riboswitch complex is known, we still do not understand the structure and conformation before AdoCbl ligand recognition. In order to delineate the conformational changes and the stabilities of long-range interactions, we have performed extensive all-atom replica-exchange molecular dynamics simulations of the TteAdoCbl riboswitch with a total simulation time of 2296 ns. We found that both the T-loop/T-looplike motif and kissing loop are very stable with ligand binding. The gating conformation changes of P6-extension allow the ligand to bind to the preorganized kissing loop binding pocket. The T-loop/T-looplike motif has much more hydrogen bonds than observed in TteAdoCbl riboswitch complex crystal structure, indicating an allosteric response of the T-loop/T-looplike motif. Our study demonstrated that the conformational ensemble of TteAdoCbl riboswitch provides stable structural elements for conformation selection and population shift in cobalamin recognition.
Graphical Abstract
1. INTRODUCTION
Riboswitches are found in all three domains of life, including bacteria, archaea, and eukaryota. They are mostly spread in bacteria and archaea.1 Riboswitches are commonly used by bacteria to detect a variety of metabolites and ions to regulate gene expression. They are structural RNA elements that are generally located in the 5′ untranslated region of mRNA. Ligand binding to the aptamer domain of a riboswitch triggers a signal to the downstream expression platform.2,3 Riboswitches need to form molecular architectures with sufficient complexity to carry out two main functions: molecular recognition and conformational switching. On the basis of genome-wide bioinformatics analysis, it was suggested that several riboswitches are promising targets for antibacterial drug discovery.4 Cobalamin (vitamin B12) riboswitch is a cis-regulatory element widely found in the 5′-UTRs of the vitamin B12-associated genes in bacteria, resulting in modulation and production of a particular protein. A riboswitch generally consists of an aptamer and an expression platform. The aptameric part is a metabolite-binding moiety whose structure serves as a precise sensor for detecting specific metabolites. Allosteric rearrangement in the mRNA structure is mediated by binding the ligand. Cobalamin riboswitch is found in 5174 species and 36 of which are human pathogens.4
Folding architectures of riboswitches are categorized into classes that are either junctional, pseudoknotted, or combinational of them.5 The adenosylcobalamin (AdoCbl)-sensing riboswitch represents one of the most intricate and complex folding, which includes a kissing loop (KL) and T-loop/T-looplike interactions (Figure 1, Figure S1A).6,7 In Escherichia coli btuB riboswitch, the kissing loop interaction is shown to be not essential for ligand binding. However, kissing loop formation improves ligand binding efficiency and is required to couple ligand binding to the riboswitch conformational changes involved in regulating gene expression.8
Figure 1.
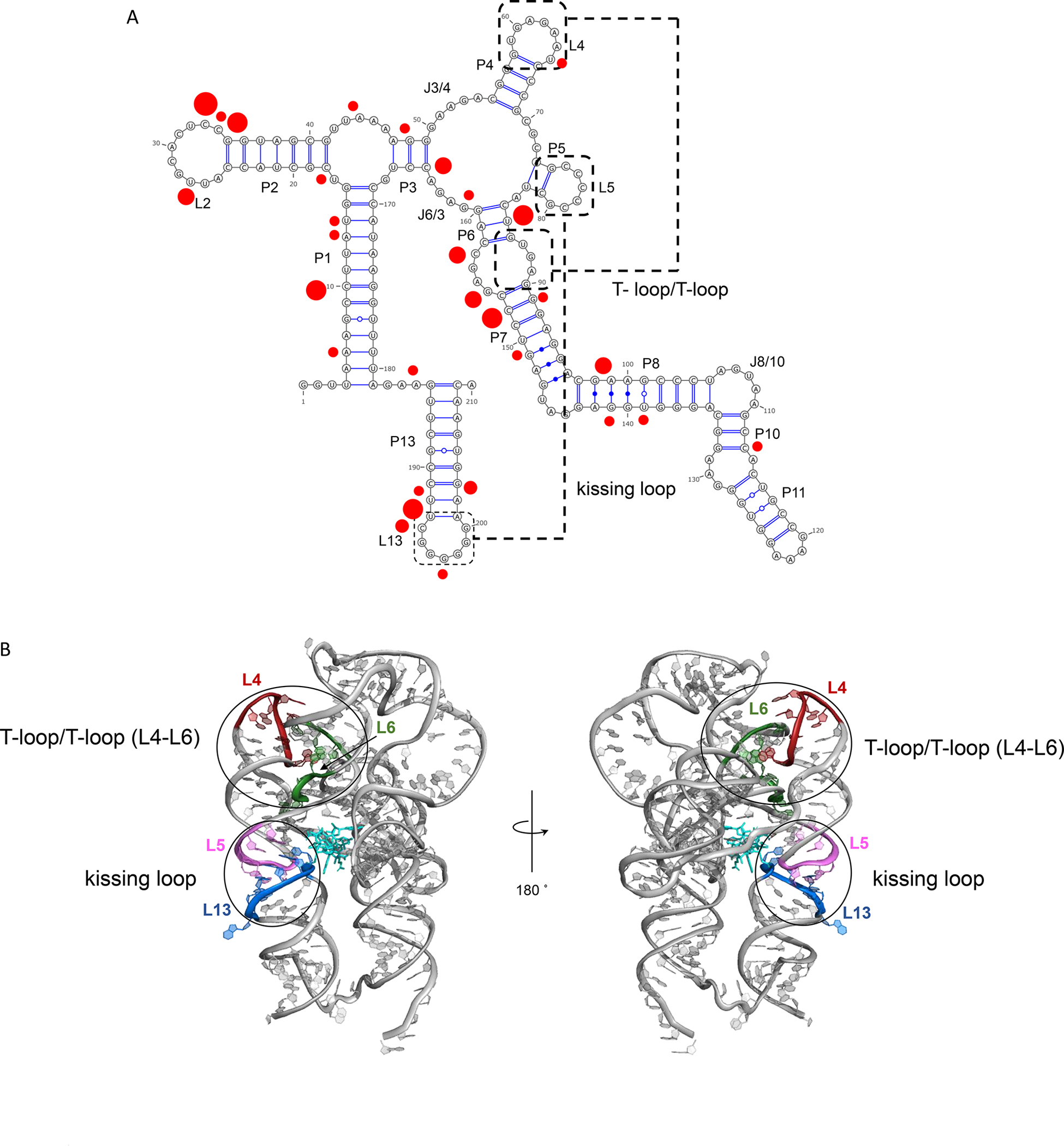
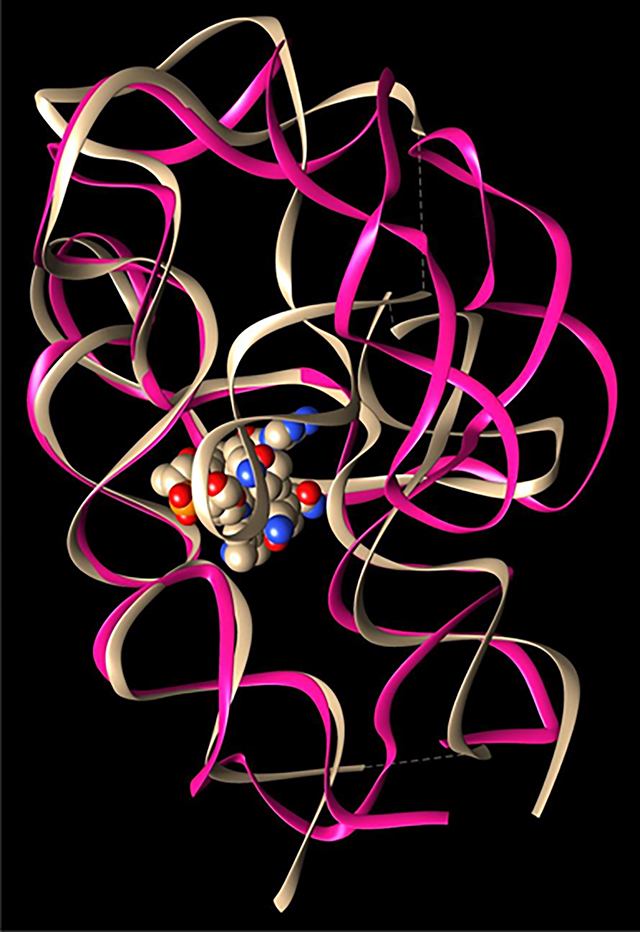
Illustration of fold architecture of TteAdoCbl riboswitch. (A) Secondary structure and long-range interactions. T-loop/T-loop motif and kissing loop are highlighted. The red dots indicated the preferred Mg2+ binding nucleotides with the radius being proportional to the binding frequencies from 70% (small) to 100% (large). (B) Two views of TteAdoCbl riboswitch crystal structure. The AdoCbl is represented as bonds. More structural information can be seen from Figure S1.
Molecular simulations provided snapshots of RNA structural dynamics.9 For example, umbrella sampling and replica exchange molecular dynamics were used to study the thermodynamics of a kissing loop complex.10–12 In this study, in order to delineate the conformational changes and the stabilities of long-range interactions, we have performed extensive all-atom replica-exchange molecular dynamics simulations of the TteAdoCbl riboswitch. Our study demonstrated that the conformational ensemble of TteAdoCbl riboswitch provides stable structural elements for conformation selection and population shift in cobalamin recognition.
2. MATERIALS AND METHODS
The X-ray crystal structure of TteAdoCbl riboswitch (PDB code 4gma) was used as the starting structure in the simulation. The AdoCbl ligand was removed from the complex structure to obtain apo form riboswitch. The missing residues (179–183) were added and minimized to remove covalent violations. Using the Amber Tleap program, the riboswitch is solvated in a TIP3P box with at least 20 Å from each side of solvent atoms. The system was neutralized with minimum number of cations (50 Mg2+ and 109 potassium ions). The final system has a box size of 143.8 × 140.7 × 111.3 Å3 with 62 442 water molecules and total 193 479 atoms.
Amber RNA OL3 force field13–15 was used for simulation. The particle mesh Ewald (PME) method was used to calculate the electrostatic interaction, and the van der Waals interactions were calculated using a cutoff of 8 Å. The system was minimized in 50 000 steps, and heated to 270 K and then 300 K each with 25 000 steps. We first run a 10 ns simulation with NPT ensemble to adjust the system to a proper density at 300 K and 1 bar, resulting in a system with a box size of 138.2 × 135.1 × 106.3 Å3. After the10 ns simulation, the equilibrated system was used as Set I conformers in subsequent replica exchange molecular dynamics (REMD) simulations. The set II conformers are preunfolded riboswitch with different unfolding extent. The riboswitch was unfolded by artificially setting the charges of magnesium and potassium to be zero during the MD simulations. Without countercations, the riboswitch quickly unfolded, including the completely open conformations. No secondary structure constraint was used.
In our REMD simulation of the TteAdoCbl riboswitch, 56 replicas ranging from 300 to 450 K were simulated with Amber18 program.16 The REMD simulation was run using NVT ensemble, with an integration time step of 2 fs. The exchange rate was 20%. We started REMD simulation with two sets of different starting conformations. About one-third starting conformations (18) are the Set I conformer equilibrated from a 10 ns simulation; the rest are the Set II that contains partially unfolded conformers. Each replica was simulated for 41 ns with a total simulation time of 2296 ns. With a mixture of folded and unfolded conformations as starting points, the REMD simulation reflects the relative thermodynamic stabilities of various conformations, avoiding the limitation of producing equilibrated folding/unfolding events during the REMD simulation. The conformers staying at the low temperature can be viewed as the REMD equilibrated ligand-free structures
3. RESULTS AND DISCUSSION
Long-Range Interactions Independent of Ligand Binding.
The global architecture of the TteAdoCbl riboswitch is defined by the organization of the secondary structure into two coaxial stacks, P1–P3–P6 and P4–P5–P13 (Figure 1 and Figure S1A). The P4–P5–P13 stack has a well-organized kissing loop formed by L5 and L13, which is essential for its gene regulatory function. The base pairing of L5 and L13 in the regulatory KL and the minor grooves of the P3–P6 coaxial stack defines the cobalamin binding site. The P1–P3–P6 and P4–P5–P13 stacks are connected by a flexible sequence segment 179–183 (missing in the crystal structure), and nonbonding T-loop interaction of the T-loop (L4) interacts with a T-loop mimic of an internal loop between P6 and P7. Class-specific peripheral P6 extensions (Figure S1A, blue) may gate the entry pocket for cobalamin recognition. From these structural elements, we select four hydrogen bonds to monitor the global conformation changes of the riboswitch. C79N3-G195H1 (C79-G195) for KL, A89H62–U65O4 (A89-U65) for T-loop, C72N3-G156H1 (C72-G156) for packing between P1–P3–P6 and P4–P5–P13 stacks, and A130HO-C161O2 (A130-C161) for P6 extension and core interactions (Figure 1 and Figure S1A).
In Figure 2, we plot the distance distributions of these four hydrogen bonds within different temperature windows, ranging from room temperature region 300–310 K to the artificially high temperature 440–450 K used in the REMD simulations. The hydrogen bond between C72-G156 is extremely stable up to 350 K (Figure 2A), indicating that the core structure of the riboswitch remains to be folded even in the 350 K during the course of simulations. Consistently, the hydrogen bonds in C79-G195 remain intact up to 330 K (Figure 2B), indicating the highly stable kissing-loop interaction. The noncanonical A130-C161 base pair is weak with a broad peak around 5 Å (Figure 2C), resulting in a potential large motion at the end of P6 for the ligand entry of the ligand. In addition, our calculation also suggests that the A89-U65 hydrogen bond for T-loop motif binding is not stable even at room temperature since it has a well-defined second peak around 5 Å. However, further examination of the structures revealed that A89 formed other strong interactions with the T-loop motif (see more structural details in the next section).
Figure 2.
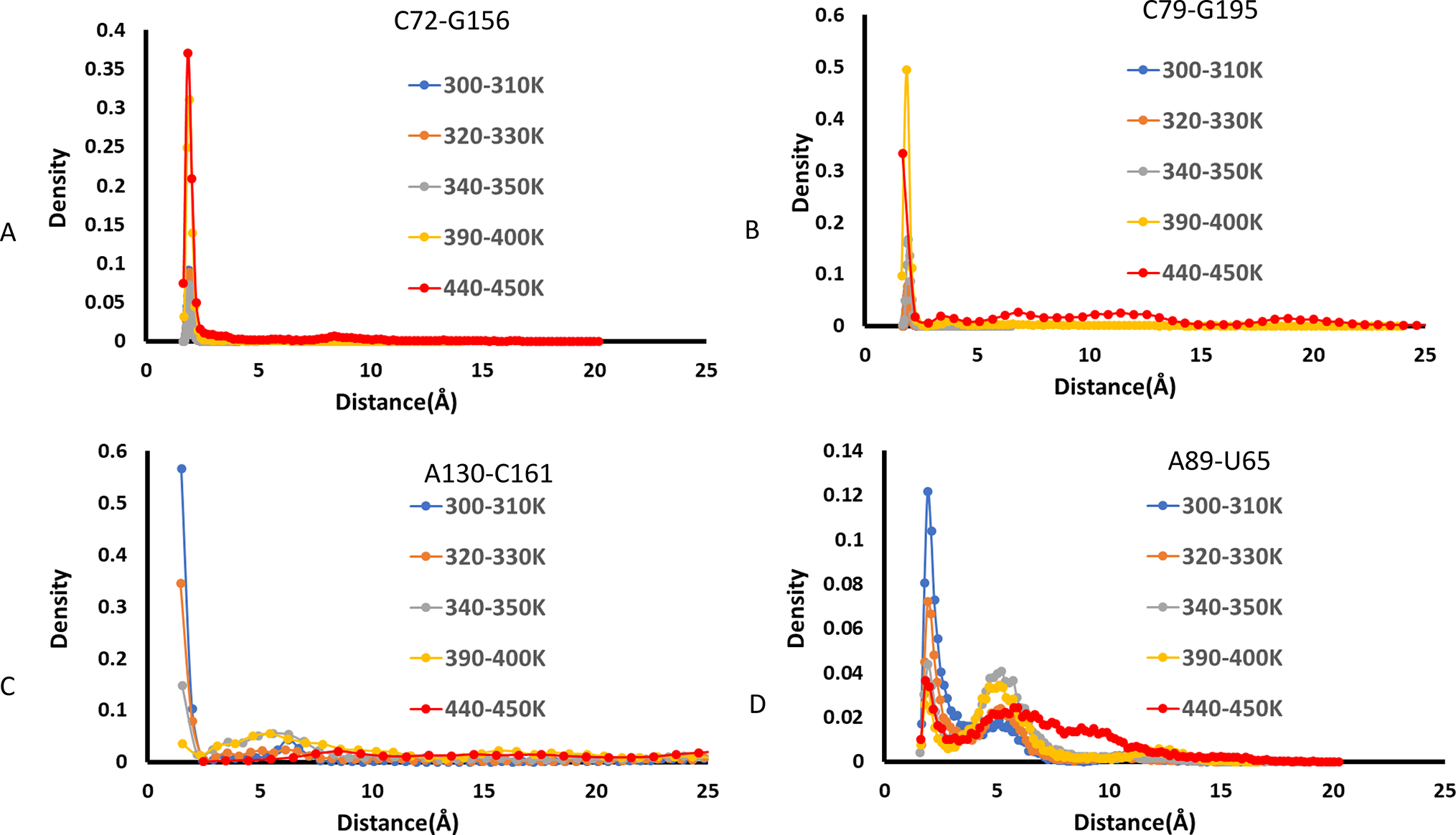
Density distributions of four hydrogen bonds in the TteAdoCbl riboswitch at different temperature regions. (A) C72-G156, (B) C79-G195, (C) A130-C151, and (D) A89-U65.
We next examine the couplings among these four hydrogen bonds by examining the two-dimensional density distributions. In Figure 3, we show that the distributions of C79-G195 and C72-G156 hydrogen bonds are not affected by the distances of A89-U65 and A130-C161. Because of their high stabilities, both the C79-G195 and C72-G156 stay around their hydrogen bond distance when the A89-U65 distances change from 2 to 15 Å and those of A130-C161 vary from 2 to 50 Å. However, there are interesting allosteric interactions between the A89-U65 and A130-C160. As can be seen in Figure 3D, there are two highly populated groups for the distances of A89-U65 and A130-C160. The first one is the low A130-C160 separation, where A89-U65 tends to have a longer distance (region I). The second group has two distribution strips (II and III, Figure 3D). Region II has an A89-U65 pairing that falls within the hydrogen bond distance, and Region III corresponds to the 5 Å peak in Figure 2D. The distance in the A89-U65 pair remains relatively constant in all of the conformers in these two clusters (II and III), whereas the distance between A130-C160 varies from 15 to 50 Å (for example, one conformer shown in Figure S1C). The two-dimensional distributions of these four pairs at higher temperature were reported in Figures S2–S5 for temperatures up to the 450 K. It can be seen the riboswitch displays a very large separation of A130-C160. However, there are still considerable number of conformers with intact kissing loop and T-loop interactions.
Figure 3.
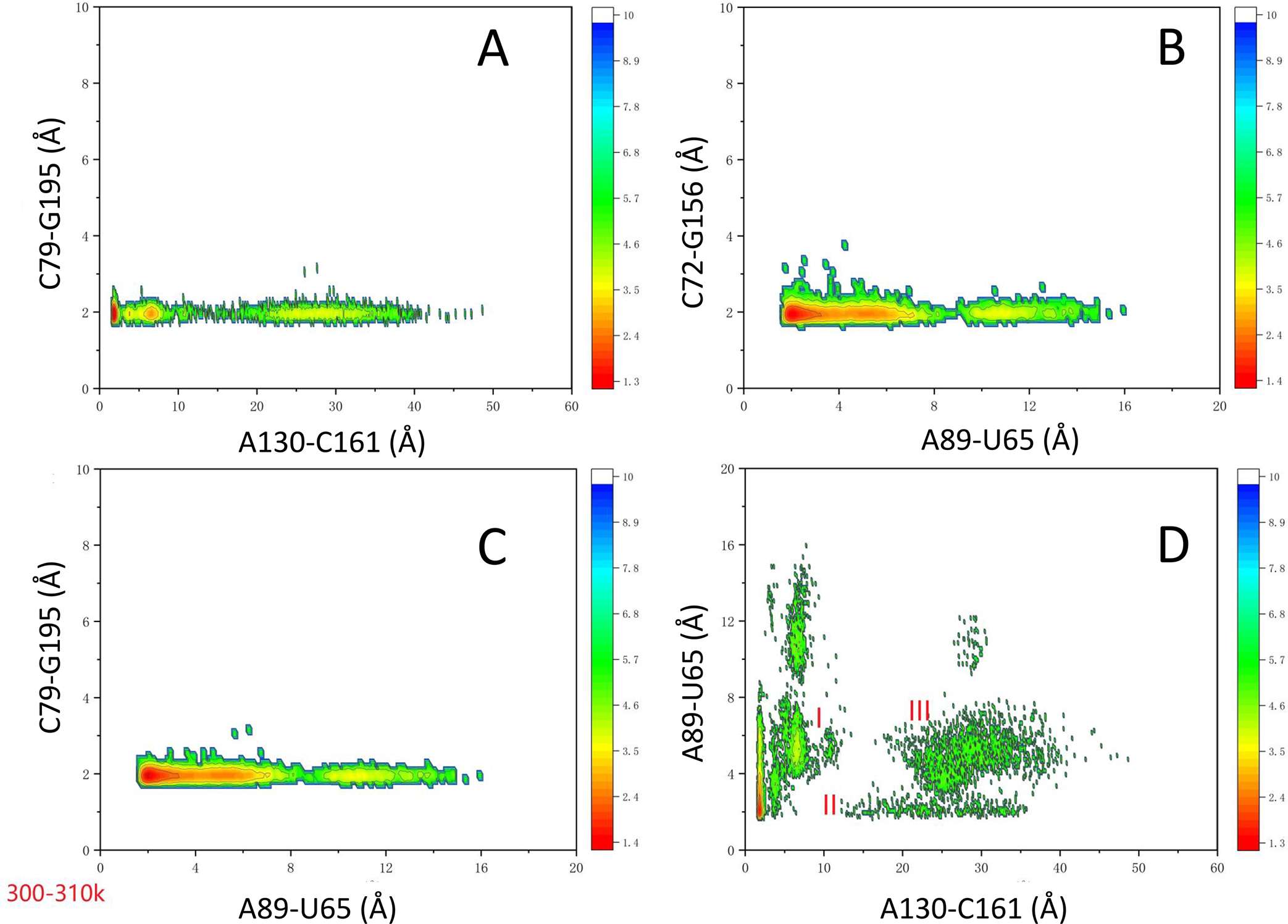
Two-dimensional density distributions of four hydrogen bonds in the TteAdoCbl riboswitch at the room temperature region. The color scale is kcal/mol. Three regions (I, II, and III) are label in (D): I, low A130-C160 separation; II, A89-U65 hydrogen bond region; III, 5 Å peak region in Figure 2D.
T-loop Is More Organized and May Have a Stronger Interaction with Intercalating A89 in the Ligand-Free Riboswitch state.
The basic architecture of the T-loop has base-pairing between the first and fifth nucleotides and an intercalating nucleobase stacked between the fourth and fifth T-loop positions.17 In the crystal structure, A89 sits between A63 and G62, but A63 and U59 are not on the same plane (Figure 4A). We found that the REMD equilibrated ligand-free structure restores to a better T-loop motif with strong Hoogsteen base pair (Figure 4B,C). The U59H3-A63N7 has the highest density among all hydrogen bonds among the T-loop motif interactions (Figure 4D). When the P6 extension is completely opened up (Figure S1C), A89 can maximize its interaction with the L4 T-Loop. As depicted in Figure 4C, while A89 is still able to keep its hydrogen bond with U65 (even though it is weaker than in the bound state), it forms new interactions with G60 by forming the G60H22-A89N1 hydrogen bond, and its CH group (A89H2) also interacts with G60N3. In addition, its NH group may also form a hydrogen bond with ribose oxygen of A63. The network of these hydrogen bonds and intercalating van de Waals forces makes the A89 to be able to adjust its interaction with the L4 T-loop with different P6 extension conformations (Figure 3D).
Figure 4.
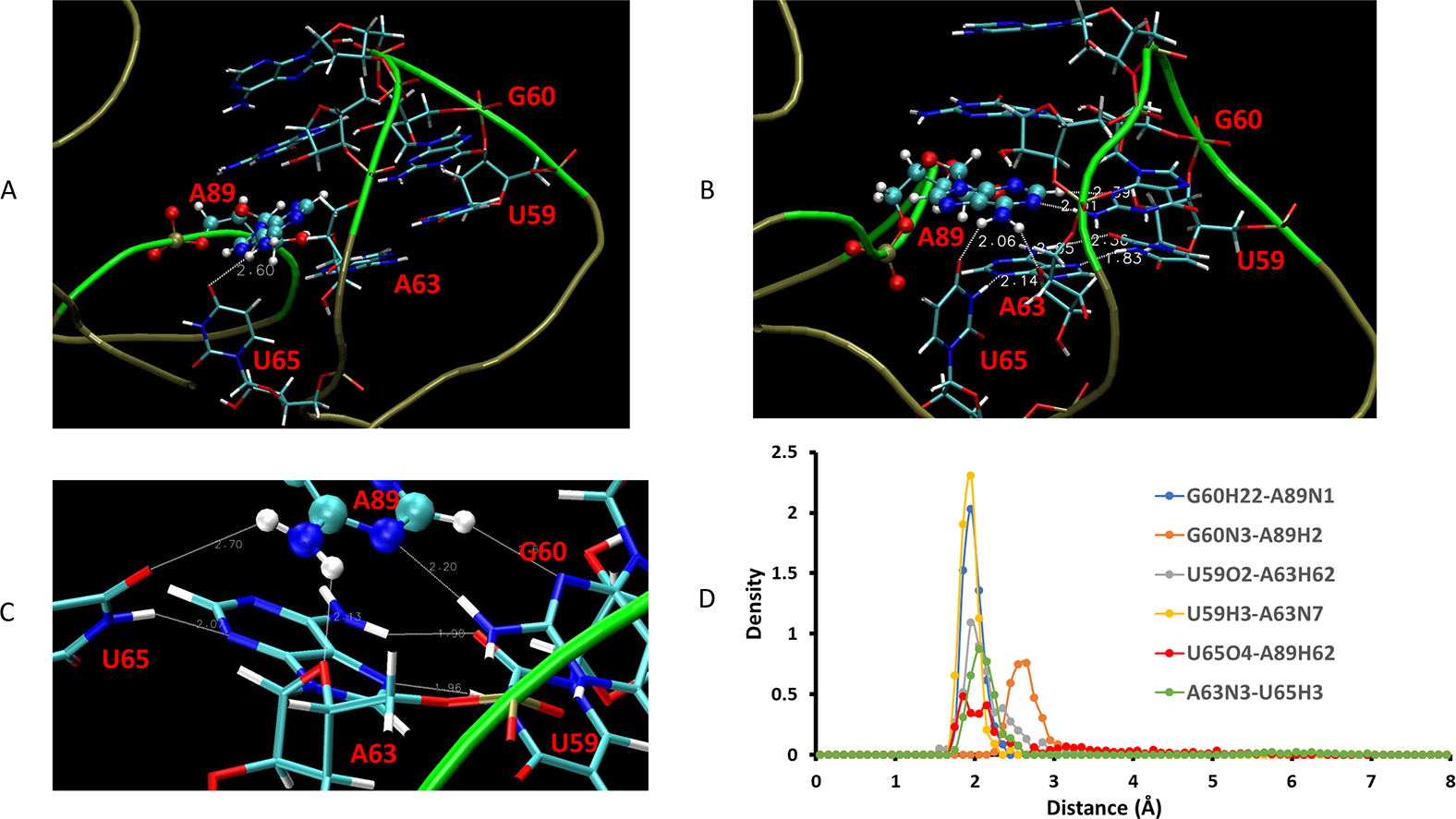
Comparison of hydrogen bonds of T-loop in the crystal structure and in the ligand-free state. (A) A89/T-loop interaction in crystal. (B) A89/T-loop interaction in the ligand-free state from REMD simulation. (C) Highlighting the hydrogen bond network in the ligand-free state. (D) Density distribution of the hydrogen bond network in the T-loop interactions.
Our observation of the L4 T-loop dynamics is consistent with a previous experimental finding that cobalamin riboswitches have a broad spectrum of preference for the two biological forms of cobalamin. This selectivity is primarily mediated by the interaction between a peripheral element of the RNA that forms a T-loop module and a subset of nucleotides in the cobalamin-binding pocket.18
Clustering of the Conformational Ensemble Reveals Possible Dynamics Conformational Selection of Ligand Binding.
As we have shown in the last two sections, the kissing loop and the packing of the two coaxial stacks (P1–P3–P6 and P4–P5–P13) are mostly well maintained for most low-temperature stable conformers, including those with a large opening of P6 extension. We examined the conformers with a replica temperature of 301.7 K obtained during the last 4 ns of REMD simulation. We cluster these conformers using a small RMSD cutoff value of 2.0 Å. Then we superimpose these clusters onto the crystal structure of TteAdoCbl riboswitch to examine if these conformations are compatible to AdoCbl binding. As illustrated in Figures 5 and 6, all of them have the pockets to allow the AdoCbl binding. As can be seen in the figures, the AdoCbl riboswitch binding site is the most stable region, which can preserve near native binding conformation even though the other part unfolds. In Figure 6, the first and second clusters with 8% and 6% of the population, respectively, show a continuous variation of the binding pocket and P6 extension conformations. Within the continuous variation of conformation, each conformer has a RMSD value less than 2.0 Å with at least one conformer in the cluster indicating the highly dynamic nature of TteAdoCbl riboswitch conformation transition. The third cluster, ~3.5% of the population, has a conformation very similar to that of the crystal complex, indicating a possible conformation selection in AdoCbl binding. Figure 6H shows a conformer with a complete open binding pocket. When AdoCbl diffuses into the open riboswitch pocket, subsequent conformational dynamics and conformation equilibration may lead to a close conformation. It is known that aptamers can be structurally preorganized to various extents in the absence of a ligand in the lock-and-key model,19 but ligand binding induces at least some structural reorganization or stabilization of aptamer substructures that consequently influences the folding and function of the adjoining expression platform.
Figure 5.
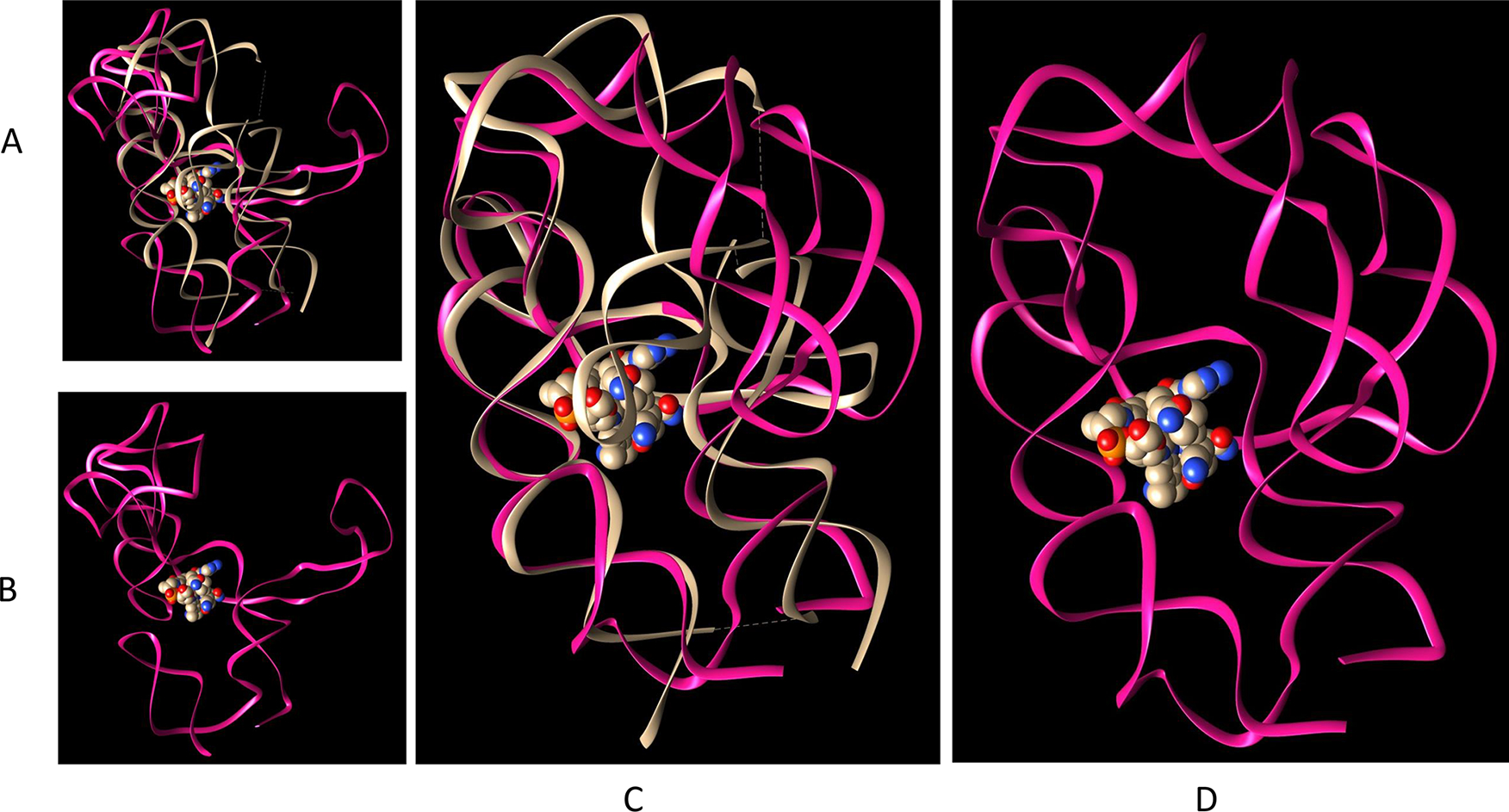
TteAdoCbl riboswitch binding site is the most stable region. (A) Superimposition of the crystal structure and one unfolded conformer. (B) The unfolded conformer in (A). (C) Superimposition of the crystal structure and one unfolded conformer with a preserved binding site. (D) The unfolded conformer in (C).
Figure 6.
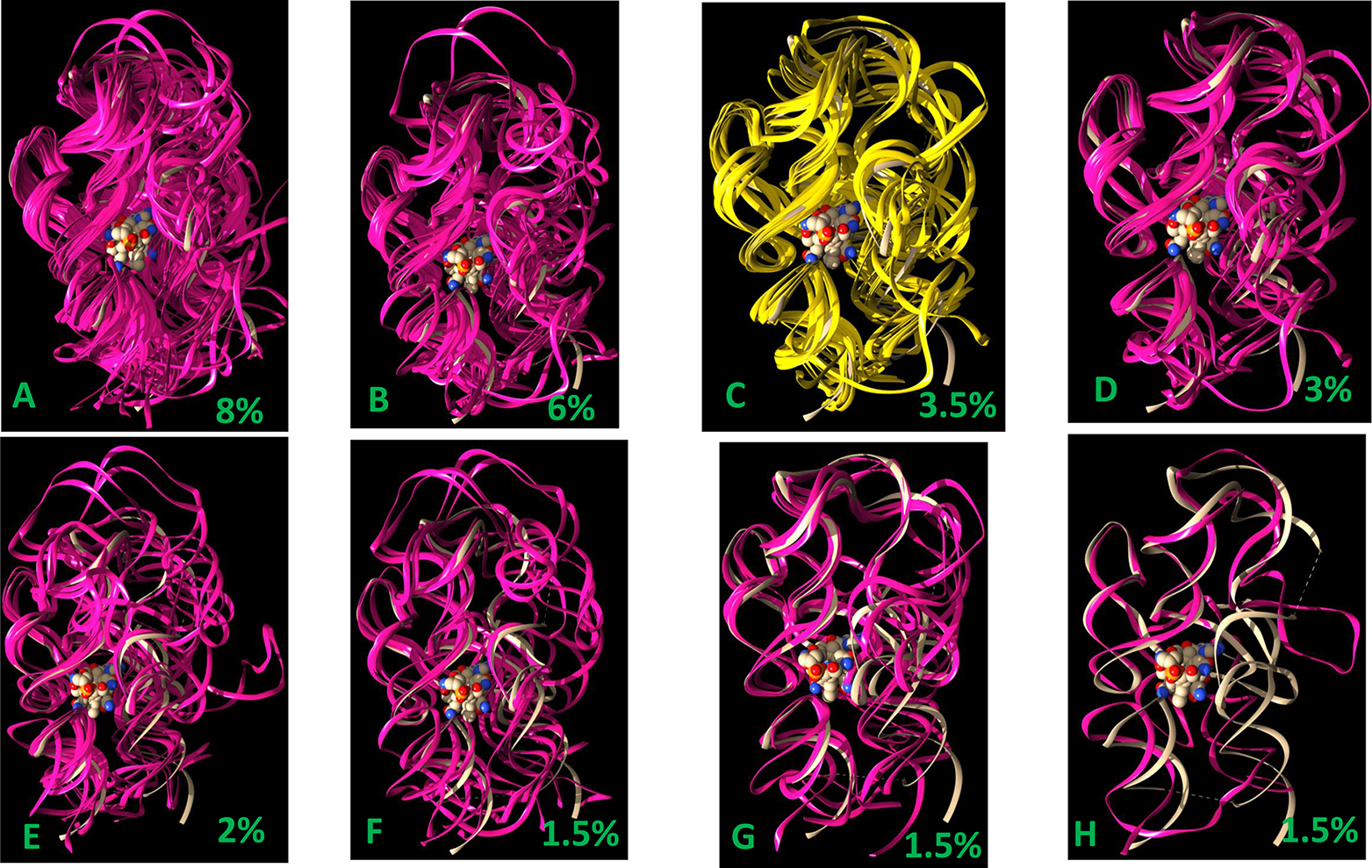
Comparison of riboswitch conformation in the crystal structure and the clusters of ligand-free state obtained from REMD simulation. The deep purple and yellow ribbons represent conformers from REMD simulations. The light gray ribbon represents the crystal structure conformer, and the AdoCbl is represented as vdw spheres.
While the general principle of the structural dynamics and hierarchical folding–unfolding of RNA20 governs the dynamics and population distribution of riboswitch conformational ensembles, this type of stepwise conformational selection and population shift can also be seen in proteins as well.21–25 Despite their inherent differences, this may not be surprising20 since polypeptides and polynucleotides are governed by similar physicochemical principles.25,26
It is known that the kissing-loop interaction defines substrate recognition in other riboswitches.8,27,28 For TteAdoCbl riboswitch, the requirement for the preformed stable kissing loop binding site is the Mg2+ ion concentration. Previously, it was found that in the manganese riboswitch the addition of physiological Mg2+ to a nonselective metal ion binding site induces a ligand-free folded structure.29 In our simulations, the addition of Mg2+ as well as K+ stabilizes key structural elements. We analyzed the association of the riboswitch with Mg2+ and K+. All Mg2+ cations were 100% bound to RNA at 310 K while having lower frequencies at 350 K. We labeled the nucleotides with a high rate of Mg2+ binding in Figure 1. As can be seen in Figure 1, there are several hot spots for Mg2+ binding: L2, L13, and the junction connecting P6/P7. L13 is the kissing loop and junction connecting P6/P7 is one portion of T-loop/T-loop motif. K+ cations have 50–100% association rate with RNA and were unspecific. In a future study, we will investigate the riboswitches’ conformational dynamics at different ionic conditions.
4. CONCLUSIONS
In order to delineate the conformational changes and the stabilities of long-range interactions, we have performed extensive all-atom replica-exchange molecular dynamics simulations of the TteAdoCbl riboswitch. We found that both the T-loop/T-looplike motif and kissing loop are very stable in the absence of a ligand. The gating of conformation changes by P6-extension allows the ligand to bind to the preorganized kissing loop binding pocket. We found that the P6-extension gating motion is coupled with the L4 T-loop motif, indicating an allosteric response. In the ligand-free state, the T-loop/T-looplike motif has much more extensive hydrogen bonds than observed in the ligand bound state especially when the P6-extension is unfolded from the core of the riboswitch. Our study demonstrates that the conformational ensemble of TteAdoCbl riboswitch provides stable structural elements for conformation selection and population shift in cobalamin recognition.
Supplementary Material
ACKNOWLEDGMENTS
Simulations were performed at the NIH Biowulf HPC system and HPC system in Shanghai Jiaotong University. This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. BM and GB thanks support from Shanghai Jiaotong University.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.1c00038.
Structural elements and two-dimensional density distributions of hydrogen bonds at high temperatures (Figures S1–S5) (PDF)
Contributor Information
Buyong Ma, Engineering Research Center of Cell and Therapeutic Antibody, MOE, School of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China; Basic Science Program, Leidos Biomedical Research, Inc. Laboratory of Cancer ImmunoMetabolism, National Cancer Institute, Frederick, Maryland 21702, United States.
Ganggang Bai, Engineering Research Center of Cell and Therapeutic Antibody, MOE, School of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China.
Ruth Nussinov, Basic Science Program, Leidos Biomedical Research, Inc. Laboratory of Cancer ImmunoMetabolism, National Cancer Institute, Frederick, Maryland 21702, United States; Sackler Institute of Molecular Medicine, Department of Human Genetics and Molecular Medicine, Sackler School of Medicine, Tel Aviv University, Tel Aviv 69978, Israel.
Jienyu Ding, Protein-Nucleic Acid Interaction Section, Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702, United States.
Yun-Xing Wang, Protein-Nucleic Acid Interaction Section, Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute, Frederick, Maryland 21702, United States.
REFERENCES
- (1).Pavlova N; Kaloudas D; Penchovsky R Riboswitch distribution, structure, and function in bacteria. Gene 2019, 708, 38–48. [DOI] [PubMed] [Google Scholar]
- (2).Breaker RR Riboswitches and the RNA world. Cold Spring Harb Perspect Biol. 2012, 4 (2), a003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).McCown PJ; Corbino KA; Stav S; Sherlock ME; Breaker RR Riboswitch diversity and distribution. RNA 2017, 23 (7), 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pavlova N; Penchovsky R Genome-wide bioinformatics analysis of FMN, SAM-I, glmS, TPP, lysine, purine, cobalamin, and SAH riboswitches for their applications as allosteric antibacterial drug targets in human pathogenic bacteria. Expert Opin. Ther. Targets 2019, 23 (7), 631–643. [DOI] [PubMed] [Google Scholar]
- (5).Peselis A; Serganov A Themes and variations in riboswitch structure and function. Biochim. Biophys. Acta, Gene Regul. Mech 2014, 1839 (10), 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Johnson JE Jr; Reyes FE; Polaski JT; Batey RT B12 cofactors directly stabilize an mRNA regulatory switch. Nature 2012, 492 (7427), 133–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Peselis A; Serganov A Structural insights into ligand binding and gene expression control by an adenosylcobalamin riboswitch. Nat. Struct. Mol. Biol 2012, 19 (11), 1182–4. [DOI] [PubMed] [Google Scholar]
- (8).Lussier A; Bastet L; Chauvier A; Lafontaine DA A kissing loop is important for btuB riboswitch ligand sensing and regulatory control. J. Biol. Chem 2015, 290 (44), 26739–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sponer J; Bussi G; Krepl M; Banas P; Bottaro S; Cunha RA; Gil-Ley A; Pinamonti G; Poblete S; Jurecka P; Walter NG; Otyepka M RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev 2018, 118 (8), 4177–4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Di Palma F; Bottaro S; Bussi G Kissing loop interaction in adenine riboswitch: insights from umbrella sampling simulations. BMC Bioinf. 2015, 16 (9), S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Allner O; Nilsson L; Villa A Loop-loop interaction in an adenine-sensing riboswitch: a molecular dynamics study. RNA 2013, 19 (7), 916–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Girard N; Dagenais P; Lacroix-Labonte J; Legault P A multi-axial RNA joint with a large range of motion promotes sampling of an active ribozyme conformation. Nucleic Acids Res. 2019, 47 (7), 3739–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cornell WD; Cieplak P; Bayly CI; Gould IR; Merz KM; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197; [Google Scholar]; J. Am. Chem. Soc 1996, 118 (9), 2309–2309. [Google Scholar]
- (14).Cheatham TE 3rd; Cieplak P; Kollman PA; et al. A modified version of the Cornell force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn 1999, 16 (4), 845–62. [DOI] [PubMed] [Google Scholar]
- (15).Wang J; Cieplak P; Kollman PA How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem 2000, 21 (12), 1049–1074. [Google Scholar]
- (16).Case DA; Cheatham TE 3rd; Darden T; Gohlke H; Luo R; Merz KM Jr; Onufriev A; Simmerling C; Wang B; Woods RJ The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26 (16), 1668–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chan CW; Chetnani B; Mondragon A Structure and function of the T-loop structural motif in noncoding RNAs. Wiley Interdiscip. Rev.: RNA 2013, 4 (5), 507–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Polaski JT; Webster SM; Johnson JE Jr; Batey RT Cobalamin riboswitches exhibit a broad range of ability to discriminate between methylcobalamin and adenosylcobalamin. J. Biol. Chem 2017, 292 (28), 11650–11658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Binas O; Schamber T; Schwalbe H The conformational landscape of transcription intermediates involved in the regulation of the ZMP-sensing riboswitch from Thermosinus carboxydivorans. Nucleic Acids Res. 2020, 48 (12), 6970–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Ganser LR; Kelly ML; Herschlag D; Al-Hashimi HM The roles of structural dynamics in the cellular functions of RNAs. Nat. Rev. Mol. Cell Biol 2019, 20 (8), 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ma B; Kumar S; Tsai CJ; Nussinov R Folding funnels and binding mechanisms. Protein Eng., Des. Sel 1999, 12 (9), 713–20. [DOI] [PubMed] [Google Scholar]
- (22).Ma B; Shatsky M; Wolfson HJ; Nussinov R Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002, 11 (2), 184–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ma B; Nussinov R Enzyme dynamics point to stepwise conformational selection in catalysis. Curr. Opin. Chem. Biol 2010, 14 (5), 652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Nussinov R; Ma B; Tsai CJ Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem 2014, 186, 22–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wei G; Xi W; Nussinov R; Ma B Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev 2016, 116 (11), 6516–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nussinov R; Wolynes PG A second molecular biology revolution? The energy landscapes of biomolecular function. Phys. Chem. Chem. Phys 2014, 16 (14), 6321–2. [DOI] [PubMed] [Google Scholar]
- (27).Bouchard P; Legault P A remarkably stable kissing-loop interaction defines substrate recognition by the Neurospora Varkud Satellite ribozyme. RNA 2014, 20 (9), 1451–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Holmstrom ED; Polaski JT; Batey RT; Nesbitt DJ Single-molecule conformational dynamics of a biologically functional hydroxocobalamin riboswitch. J. Am. Chem. Soc 2014, 136 (48), 16832–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sung HL; Nesbitt DJ Single-Molecule FRET Kinetics of the Mn(2+) Riboswitch: Evidence for Allosteric Mg(2+) Control of “Induced-Fit” vs “Conformational Selection” Folding Pathways. J. Phys. Chem. B 2019, 123 (9), 2005–2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.